Nano Res
1
The behaviors of ultra-low-gold loaded catalysts
(Au/CeO2) for CO oxidation in the presence of water on
the catalysts
Qiaoling Li1 (), Wei Xie1, Guoqin Chen1, Yuefeng Li1, Yanjun Huang1, and Xiaodong Chen2 ()
Nano Res., Just Accepted Manuscript • DOI 10.1007/s12274-015-0813-z
http://www.thenanoresearch.com on May 13, 2015
© Tsinghua University Press 2015
Just Accepted
This is a “Just Accepted” manuscript, which has been examined by the peer-review process and has been
accepted for publication. A “Just Accepted” manuscript is published online shortly after its acceptance,
which is prior to technical editing and formatting and author proofing. Tsinghua University Press (TUP)
provides “Just Accepted” as an optional and free service which allows authors to make their results available
to the research community as soon as possible after acceptance. After a manuscript has been technically
edited and formatted, it will be removed from the “Just Accepted” Web site and published as an ASAP
article. Please note that technical editing may introduce minor changes to the manuscript text and/or
graphics which may affect the content, and all legal disclaimers that apply to the journal pertain. In no event
shall TUP be held responsible for errors or consequences arising from the use of any information contained
in these “Just Accepted” manuscripts. To cite this manuscript please use its Digital Object Identifier (DOI®),
which is identical for all formats of publication.
Nano Research
DOI 10.1007/s12274-015-0813-z
TABLE OF CONTENTS (TOC)
The Behaviors of Ultra-Low-Gold Loaded Catalysts
(Au/CeO2) for CO Oxidation in the Presence of Water
on the Catalysts
Qiaoling Li1*, Wei Xie1, Guoqin Chen1, Yuefeng Li1, Yan
jun Huang1, and Xiaodong Chen2*
1 Technology Center of China Tobacco Fujian Industrial
Co., Ltd, China
2 Xiamen University, China
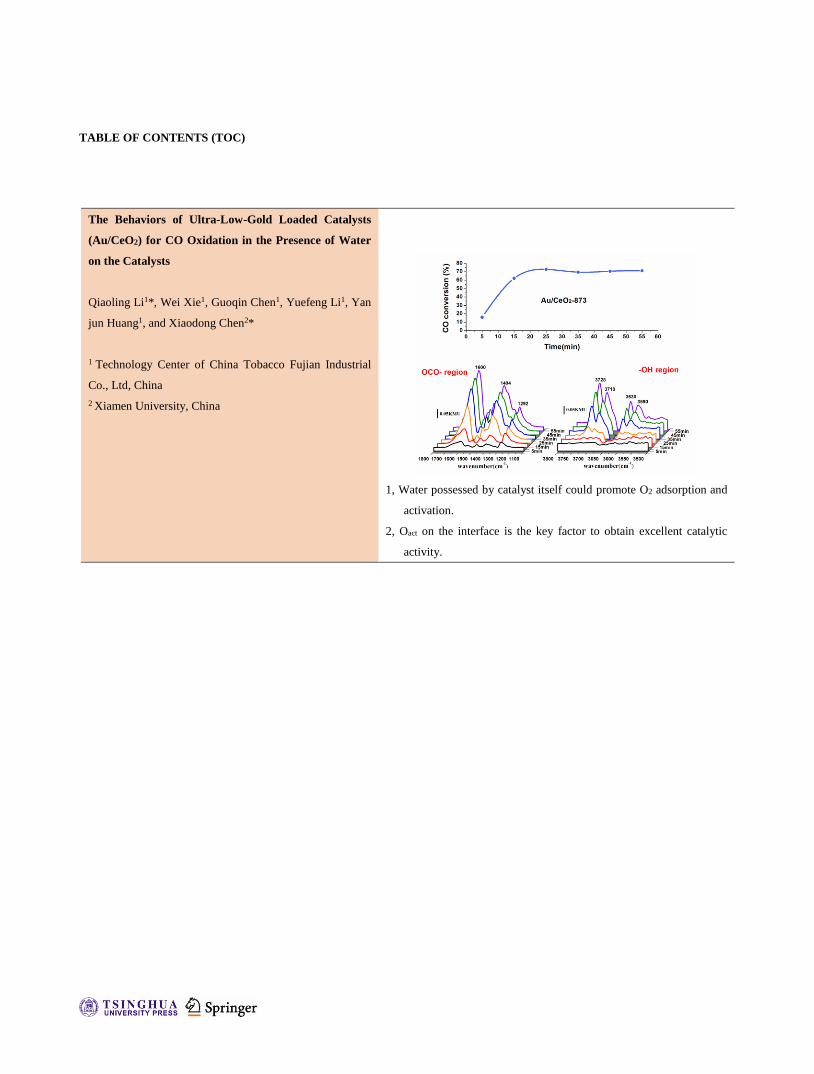
1, Water possessed by catalyst itself could promote O2 adsorption and
activation.
2, Oact on the interface is the key factor to obtain excellent catalytic
activity.
The Behaviors of Ultra-Low-Gold Loaded Catalysts
(Au/CeO2) for CO Oxidation in the Presence of Water on
the Catalysts
Qiaoling Li1 (), Wei Xie1, Guoqin Chen1, Yuefeng Li1, Yanjun Huang1, and Xiaodong Chen2 ()
Received: day month year
Revised: day month year
Accepted: day month year
(automatically inserted by
the publisher)
© Tsinghua University Press
and Springer-Verlag Berlin
Heidelberg 2014
KEYWORDS
ultra-low-gold catalysts,
water addition on the
catalysts, hydroxyl and
carbonate-like species,
reaction pathways
ABSTRACT
The catalytic behaviors of ultra-low-gold loaded catalysts Au/CeO2 during CO
oxidation have been studied using HRTEM, TGA, in-situ FTIR, CO-TPD,
O2-TPD and O2-TPO. Herein, water is present on the catalysts rather than from
the reactant gas to facilitate CO oxidation. Experimental results give direct
evidence that the presence of water on catalysts enhances the adsorption and
activation of O2 to produce Oact on the catalyst surface, resulting in excellent
catalytic activities. Different reaction pathways for CO oxidation over Au/CeO2
catalysts may be at work, but which one is dominant is influenced by the
species on the catalyst surface.
1 Instruction
Gold-based catalysts are commonly used in CO
oxidation due to their superior activity [1-11], but the
high price of gold and low availability limit their
application. Previously, we have reported our
ultra-low-gold loaded catalysts are quite reactive,
some of which could oxidize 70% CO at ambient
temperature even though the gold loaded were only
0.06 wt% [12]. The main reason can be attributed to
the contribution of water. Herein, water is present on
the catalysts rather than from the humid reactant gas
to facilitate CO oxidation. As we known, many
reported studies focused on the catalytic roles of
Nano Research
DOI (automatically inserted by the publisher)
Address correspondence to Q. L. Li, [email protected]; X. D. Chen, [email protected]
Research Article
| www.editorialmanager.com/nare/default.asp
2 Nano Res.
water from the humid reactant gas [13-15], while few
studies mentioned the situation of the water
contained in catalyst. Haruta et al. have reported that
the amount of water absorbed on the catalyst
influences the activity rather than the water content
in the gas phase [13]. Consequently, we investigated
the cases where the catalysts possess water by
themselves, which omits the necessary step of water
adsorption on the catalysts from the humid gas
phase.
Even though extensive experimental studies have
also shown that the presence of water is crucial to
obtain excellent catalytic activity for CO oxidation
reaction, but the intrinsic role of water in this process
is still unclear. Haruta et al. considered the water not
only activate the O2 molecular but also benefit the
decomposition of carbonate-like species via the
transformation of stable species to unstable species
[16]. In contrast, Goodman et al. proposed that the
promotional effect of water can only be rationalized
by its effects on O2 adsorption and activation, and the
appearance of carbonate-like species inhibit CO
oxidation [17]. From the active debate, the consensus
of the adsorption and activation of O2 by water has
been reached. Liu et al. provided the calculation
results by DFT that -OH groups play a key role in O2
adsorption on TiO2(110) [18]. Bongiorno et al. used
the first-principles calculations to investigate and
found that an adsorbed H2O molecular serves as an
“attractor” of O2 to its vicinity [19]. Based on DFT
and kinetic isotope effects study, a conclusion was
obtained that H2O mediated O2 activation via the
formation of hydroperoxy intermediates (-OOH),
which readily reacts with CO and account for the
remarkable reactivity [20, 21]. However, to date no
direct experimental evidence could be obtained to
validate the persistent speculation, which was just
obtained by theoretical studies [18-21] and exclusive
method [17]. How the water change during the
activation process, and how to directly prove the real
existence of the proposed active -OH or -OOH
groups? Furthermore, the biggest difference between
the two opinions is the role of carbonate-like species
resulting from the reaction between CO and -OH or
-OOH groups. Do the carbonate-like species
contribute to the activity or inhibit? What is the
rate-determining step for CO oxidation, the
appearance of carbonate species or the activation of
O2?
In-situ FTIR (fourier transform infrared
spectroscopy) is the most widely used technique for
studying the reaction pathways. In order to directly
shed light on how the water improve the catalytic
activity, the reaction pathways for CO oxidation in
the presence of water on the catalysts were
monitored by in-situ FTIR. It is important to note
that the choice of the water on the catalyst rather
than the water from the reactant gas has played an
important role in detecting the delicately changed
signals of -OH groups by in-situ FTIR. In the case of
the water coming from the reactant gas, it is very
difficult to apply in-situ FTIR to investigate the
changes occurring in the -OH (hydroxyl) region,
because the water content in the environment is
much higher than that adsorbing on the catalyst,
which would cover the delicately changed signals
of -OH groups on the catalyst surface. For this
reason, our study about the water possessed by the
catalyst itself avoids the above problem.
Furthermore, we used CO-TPD
(temperature-programmed desorption), O2-TPD
and O2-TPO (temperature-programmed oxidation)
to characterize the catalysts to investigate the role of
carbonate species and the correlation between water
and O2.
2 Experimental
2.1 Catalyst preparation
In this work, different support structures of
CeO2 were prepared by direct thermal
decomposition of Ce(NO3)3.6H2O at 873 and 1073 K
under atmosphere.
0.1 wt% gold was then introduced to above
CeO2 supports by deposition precipitation method.
Appropriate quantity of CeO2 support was
suspended in de-ionized water to produce a
suspension. The pH value of the suspension was
raised to 9 by NaOH and then the base suspension
was heated to 343 K, followed by introducing
www.theNanoResearch.com∣www.Springer.com/journal/12274 | Nano Research
3 Nano Res.
HAuCl4 aqueous solution into it and reacted for 60
min. The resulted suspension was cooled and
filtered. The solid obtained was washed thoroughly
and just dried at 333 K for 16 h without further
calcinations to produce the catalysts. Such a low
heat-treating temperature could result in some
water residual on/in the catalysts. By using different
CeO2 supports, different catalysts can be obtained,
named Au/CeO2-873 and Au/CeO2-1073
respectively.
For comparison, similar pretreatment were
performed on support CeO2-873 and CeO2-1073,
which was stirred in alkaline water (pH=9) at 343 K
for 60 min, but without the addition of HAuCl4, and
also just dried at 333 K for 16 h with some water
presenting on/in the support, named h-CeO2-873
and h- CeO2-1073.
2.2 Catalytic activity measurement
Activity measurements were performed in a
quartz tubular fixed–bed reactor with 0.2 g of the
catalyst per pass under the reaction conditions of
atmosphere pressure, 298 K and GHSV=18000 h-1;
the composition of the reactant gas includes 1% CO,
1% O2 and 98% N2.
2.3 Catalyst characterization
The HRTEM images of the samples were obtained
using a Phillips Analytical FEI Tecnai 30 electron
microscope operating at 300 kV. The samples were
ground to fine powder, which was then mixed with
acetone to make a suspension. A drop of the
suspension was placed on a lacey carbon nickel grid,
subsequently dried at room temperature for
measurement. In order to examine the size of Au
particles, the same method was used to prepare the
above two catalysts but the gold content was
increased to 1 wt% rather than 0.1 wt%.
The water contents in studied catalysts were
evaluated by using SDT-Q600 instrument. Samples of
approximately 6.5 mg were used in TGA tests. The
temperature was increased to 850 K at a heating rate
of 10 K/min. Weight and temperature data were
recorded using TGA software.
In-situ FTIR investigations were performed in a
Nicolet Nexus. This setup allows measurement in a
continuous flow of gas mixtures, equaling to those
used in the activity measurements (1% CO, 1% O2
and 98% N2), and at 298 K. The samples were in
self-supported pellets without KBr and putted in the
in-situ reaction cell unit. The samples surfaces were
cleaned by vacuum pumping and then exposed to
the reactant gas. The FTIR data were reported as in
Kubelka-Munk unit, and 400 scans (nominal
resolution 8 cm-1) were co-added for one spectrum. It
is worth mentioning that the background spectra
were measured in the 3rd minute after the reactant
gas had flowed through the catalyst rather than in
the vacuum system, therefore any peak or valley
were related to the amount of corresponding species
increasing or decreasing during the reaction period.
If the surface species don’t change, the FTIR spectra
will be a straight line.
CO-TPD and O2-TPD: Temperature-programmed
desorption of CO and O2 experiments were
performed in a quartz microreactor connected to a
MS (Hiden QIC-20). The fresh catalyst was first
pretreated under CO or O2 at room temperature for
1 h (30 mL/min), and purged with Ar for 1 h. The
temperature was then ramped from room
temperature to 1073 K linearly (10 K/min) in a
carrier Ar gas introduced at a rate of 10 mL/min.
O2-TPO: Temperature-programmed oxidation of
O2 experiments was performed in a quartz
microreactor connected to a MS (Hiden QIC-20).
The catalyst after reacting 1 hour was treated under
O2, and the temperature was ramped from room
temperature to 1073 K linearly (10 K/min) in a
carrier Ar gas introduced at a rate of 10 mL/min.
3 Results and discussion
3.1 Physicochemical properties of ultra-low-gold
loaded catalysts
We have reported that the introduced Au (0.1
wt%) remained 0.06 wt% on the surface or the
subsurface layers of Au/CeO2-873 and
Au/CeO2-1073; the gold nano-particle size and the
specific surface areas are: 4.8 nm and 66 m2/g for
Au/CeO2-873, and 5.1 nm and 6 m2/g for
Au/CeO2-1073 [12]. The respective gold particle size
| www.editorialmanager.com/nare/default.asp
4 Nano Res.
is close to each other but the specific surface areas
dropped significantly with increasing the support
calcination temperature (Fig. 1). Since the catalysts
were prepared by depositing gold on an already
preformed ceria support and just dried at 333 K
without any further heat-treatment, the effect of
particle size on the catalytic activity is negligible.
The water contents in studied catalysts were
evaluated by using TGA. The results show that the
catalysts Au/CeO2-873 and Au/CeO2-1073 dried at
333 K contain 2.39 and 0.53 wt% water (Fig. 2). The
relative higher water content in Au/CeO2-873 can be
attributed to the higher specific surface areas.
3.2 Reaction pathway of ultra-low-gold loaded
catalyst Au/CeO2-873
Herein, the special reaction pathways of the
ultra-low-gold loaded catalysts for CO oxidation in
the presence of water on the catalysts were
monitored by in-situ FTIR. In the OCO- (carbonyl)
region, 1588 cm-1 is assigned to bidentate formate,
1369 and 1292 cm-1 are related to carbonate-like
species in WGS (water-gas shift) reaction [22]. For
the pure h-CeO2-873 support, the peaks 1588, 1369
and 1292 cm-1 were observed and remained about
constant (Fig. 3b), indicating that CO could adsorb
on the hydroxylated support surface and form
formate and carbonate-like species even though
without gold. In contrast, for the catalyst
Au/CeO2-873, the intensity of 1588, 1369 and 1292
cm-1 increased rapidly with time during the first
35min reaction, but leveled off after that (Fig. 4b).
Such formation rates are identical to CO conversion
rate during the activity test (Fig. 4a), inferring that
they are probably the reaction intermediates during
CO oxidation. If these formate and carbonate-like
species were formed by reaction product CO2
reabsorbing on the surface [23], the corresponding
peaks of them could not be detected on the pure
h-CeO2-873 support, because which shows no
activity for CO oxidation at 298 K (Fig. 3a).
The adsorption behavior of CO on h-CeO2-873
support and Au/CeO2-873 catalyst can be further
investigated with CO-TPD experiments (Fig. 5). The
main signal was CO2 rather than CO, suggesting
CO can be oxidized to CO2 during the desorption
process. The results show that h-CeO2-873 has the
ability to absorb CO, but the adsorption capacity is
much lower than Au/CeO2-873. In addition, Au
could weaken Ce-O bond to increase lattice oxygen
mobility [24], so CO2 desorption peak at 663 K on
h-CeO2-873 migrated to 613 K on Au/CeO2-873.
Moreover, even though the hydroxylated support
h-CeO2-873 could adsorb CO to form formate and
carbonate-like species as Au/CeO2-873 did, it shows
no activity for CO oxidation at 298 K. Therefore we
speculate that the excellent activity of the
Au/CeO2-873 catalyst was governed by other key
factors, and less concerns with the formate and
carbonate-like species.
In the -OH (hydroxyl) region, the peaks of
Au/CeO2-873 at 3590, 3630, 3710 and 3728 cm-1 (Fig.
4c), representing active -OH groups [22,25], also
had similar appearance rates with formate and
carbonate-like species, but not much changes could
be observed for h-CeO2-873 in the -OH region (Fig.
3c). Meantime, the water bending mode at 3650 cm-1
decreased and formed a valley for Au/CeO2-873. By
comparing the FTIR and CO-TPD results between
h-CeO2-873 and Au/CeO2-873, it can be found that a
small amount of gold loaded on the surface not
only enhanced the adsorption of CO on the catalyst,
but also activate H2O to produce some new -OH
groups locating at different position.
How H2O can produce the new -OH species in
the conditions of the system being studied, reacting
with CO or O2? Although CO and H2O were present
simultaneously, water gas shift reaction required
significantly higher temperature to convert CO to
CO2, thereby excluding the possibility of the WGSR
during CO oxidation in the presence of H2O at
room temperature. The only way to produce the
active -OH group just left the reaction between H2O
and O2.There are two kinds of way for H2O to
activate O2. One was proposed by Haruta et al [16]:
O2 + H2O → 2-OH + Oact, another was reported by
Ojeda et al [20]: O2 + H2O → -OOH + -OH. O2 could
be activated by H2O to produce active -OH groups,
hydroperoxy groups -OOH and Oact at the
perimeter interfaces between gold and support.
www.theNanoResearch.com∣www.Springer.com/journal/12274 | Nano Research
5 Nano Res.
More facile oxygen adsorption in the presence of
H2O is also corroborated by DFT calculations [18].
Oact could directly react with the Au bonded CO
(Au-CO) to produce CO2, while the active -OH
groups and the hydroperoxy groups -OOH could
react with Au-CO to form the intermediates, such as
formate (Au-COOH)interface and carbonate-like
(Au-CO3H)interface species. These intermediates
forming at the perimeter interfaces are easily
converted into CO2 with the attack of Oact. Boccuzzi
et al, performed CO oxidation over Au/TiO2 with
C16O and 18O2 in the presence of H216O and observed
only the formation of C16O18O. This excluded the
participation of oxygen from the support or H2O
but indicates that the promotional effect of H2O
may be associated with the dissociation of O2 [26].
Despite Oact did not be detected in this work, our
experimental results from the water side to shed
light on the relationship between water and O2.
However, the formate (-COOH)support and
carbonate-like (-CO3H)support species forming on the
support, which were observed in the OCO- region
of h-CeO2-873, kept stable. This is due to the lack
of Au to produce Oact. In WGS reaction, the
decomposition of the formate and carbonate-like
species needs a much higher temperature, which is
also due to the lack of the source of Oact in the
reactant gas. In addition, because without the
addition of O2 in the CO-TPD experiment system,
the desorption peak of CO2 at 403 K also did not
shift to room temperature as the activity test shown
over the catalyst Au/CeO2-873.
Consequently, there are two kinds of formate and
carbonate-like species on the surface of the catalyst
Au/CeO2-873: (1) forming on the interfaces between
gold and support; and (2) forming solely on the
support. The former could act as reaction
intermediates to produce CO2 by the attack of Oact.
The latter would keep stable on the support and
reduce O2 adsorption capacity of CeO2, finally
inhibit the catalytic activity. In order to prove our
argument, a comparative experiment was designed.
CO-TPD and CO2-TPD were used for the fresh
catalyst Au/CeO2-873, and O2-TPO was applied for
the catalyst Au/CeO2-873 after reacting 1 hour (Fig.
6). CO2 signal are detected in the above three cases.
The peak around at 400 K is detected in all cases,
but there are a small shoulder peak at 335 K and
two peaks at 527 and 605 K for O2-TPO, which are
not detected in CO2-TPD and CO-TPD. These three
special CO2 desorption peaks can be attributed to
the decomposition of formate (-COOH) and
carbonate-like (-CO3H) species, which produced by
the co-adsorption of the active -OH, -OOH groups
and CO on the catalyst surface. In the cases of
CO-TPD and CO2-TPD without O2 addition, these
three CO2 desorption peaks cannot be found,
because H2O in the catalyst has no chance to react
with O2 to produce active -OH groups and
hydroperoxy groups -OOH. However, the peak at
335 K represents the decomposition of
intermediates (Au-COOH)interface and
(Au-CO3H)interface species by the attack of O2, and the
peaks at 527 and 605 K represent the thermal
decompositions of the formate (-COOH)support and
carbonate-like (-CO3H)support species at high
temperature.
This comparative experiment supply the
information that during the reaction process, some
formate and carbonate-like species can be produced
on the catalyst surface because of co-adsorption of
the active -OH, -OOH groups and CO, therefore the
formation rates of the species in the OCO- region
keep synchronization with those in the -OH region.
Meantime, the excellent CO activity is decided by
the promotion of O2 activation by H2O (O2 + H2O →
2-OH + Oact and O2 + H2O → -OOH + -OH), since the
formation rates of the species in the OCO- region
and in the -OH region are also identical to CO
conversion rate during the activity test. But just the
formate (Au-COOH)interface and carbonate-like
species (Au-CO3H)interface forming on the interfaces
are reaction intermediates, and the others would
keep stable on the support, such as (-COOH)support
and (-CO3H)support.
3.3 Reaction pathway of ultra-low-gold loaded
catalyst Au/CeO2-1073
Figure 7 and 8 show the in-situ FTIR results of
h-CeO2-1073 and Au/CeO2-1073. It can be found
| www.editorialmanager.com/nare/default.asp
6 Nano Res.
that no significant changes happened on
h-CeO2-1073 in the OCO- region (Fig. 7b) and -OH
region (Fig. 7c), therefore showed no activity for CO
oxidation (Fig. 7a). Figure 2 shows the catalysts
Au/CeO2-873 and Au/CeO2-1073 dried at 333 K
contain 2.39 and 0.53 wt% water, speculating the
water content in h-CeO2-1073 would also be much
lower than h-CeO2-873, therefore the FTIR intensity
of formate (-COOH)support and carbonate-like
(-CO3H)support species forming on the support
h-CeO2-1073 was also weaker than h-CeO2-873.
Meanwhile, without the addition of gold in
h-CeO2-1073, H2O and O2 lacked the interface to
produce some new -OH group and Oact.
Interestingly, no obvious peaks were detected for
Au/CeO2-1073 in the OCO- region (Fig. 8b), while
the intensities of peaks in the -OH region slightly
increased and kept stable after 15 min (Fig. 8c),
suggesting this catalyst experienced different
reaction pathway, in which formate or
carbonate-like species forming on the interface are
not necessary and Au-CO directly react with Oact to
produce CO2. Based on the XPS results [12], only
Au0 could be detected on Au/CeO2-1073 but 16%
Auδ+ was found on Au/CeO2-873. An inference can
be drawn that Auδ+ are the requisite species to form
the formate and carbonate-like species, which is
consistent with the WGS reaction mechanism. No
matter Au0 species act as spectators or contribute to
the WGS activity, Auδ+ species have been accepted
as active species to form reaction intermediates [22,
27].
We also reported the catalyst Au/CeO2-1073
showed poor activity to oxidize CO after drying at
373 K (CO conversion decreased from 35% to 10%)
[12]. TGA results show that the water content
decreased from 0.53 wt% to 0.35 wt% (Fig. 2). Lack
of the water-derived species makes it difficult to
exhibit the promotional effects on molecular oxygen
adsorption and activation. Au/CeO2-873 is definitely
more active than Au/CeO2-1073, however based on
the XPS results, it can be inferred that a large
amount of Auδ+-OH still remained on the surface of
Au/CeO2-873, and drying these catalysts at 373 K
will result in the thermal decomposition of Auδ+-OH
species or the recombination of -OH groups, in such
a way that Auδ+ species transformed to Au0, thus the
changes of the activity could not only be attributed
to the effect of water or the Au0/Auδ+ ratio. On the
contrary, Au0 was the main gold species on the
surface of Au/CeO2-1073, and the sintering of Au
particle does not occur at so low temperatures,
therefore the change of the activity can be chiefly
attributed to the amount of water absorbed on the
surface [12]. In order to understand the specific
effect of O2 and H2O on CO conversion, O2-TPD
experiments were performed over the catalyst
Au/CeO2-1073 dried at 333 K and 373 K. The mass
spectra of O2 and H2O in process O2-TPD are
illustrated in Figure 9 respectively.
When the catalysts were dried from 333 to 373 K,
the amount of H2O on/in catalysts decreased
significantly. Especially for the H2O peak at 673 K
nearly disappeared in the case of Au/CeO2-1073
dried at 373 K, which represents the chemisorbed
water or the recombination of -OH groups. During
the O2 adsorption process, the following reactions
happened: O2 + H2O → 2-OH + Oact and O2 + H2O →
-OOH + -OH. Oact could be stored by CeO2 and then
desorbed as the form O2 when the temperature
increased. With the decrease of H2O content, the
amount of the adsorbed O2 also reduced. In
addition, when the catalyst was dried at 333 K, most
of O2 tend to desorb at lower temperature,
attributing to the adsorption of O2 at metal support
interfacial sites [28]. On the contrary, most of O2
desorbed at higher temperature when the catalyst
was dried at 373 K, which is due to the desorption
of chemisorbed O2 molecule in the pores of catalyst
[26]. These results further proved the promotional
effect of water on the adsorption and activation of
O2.
3.4 Effect of Oact,H2O and -OH groups on the
catalyst activity
Most of reported catalysts applied in low
temperature CO oxidation had higher gold loadings,
nearly above 1 wt%, but the reaction activity in this
study could be improved when the catalyst with
own water on the surface, even though the gold
www.theNanoResearch.com∣www.Springer.com/journal/12274 | Nano Research
7 Nano Res.
content was decreased to less than 0.1 wt%. These
findings lead to the conclusion that Oact greatly
contribute to the activity, and the interfaces between
gold and support supply a platform to produce Oact.
The addition of H2O accelerated the adsorption and
activation of O2 and -OH groups are the byproducts
during the process. Even though -OH groups can
react with Au-CO to form intermediates, such as
formate (Au-COOH)interface and carbonate-like
(Au-CO3H)interface species, which will keep stable
rather than decompose to CO2 without the attack of
Oact. From these studies, they just suggest the
participation of -OH groups in CO oxidation, but
Oact is the most important active species. Saavedra
et al. evaluated the reaction kinetics and also found
that weakly adsorbed water increased the effective
number of active sites rather than changing their
inherent reactivity [21]. It is worth to note that these
studies just show us that the product CO2 may be
obtained by two reaction pathways, but which
reaction pathway is more effective still keeps
unclear. Because of the difference of surface areas,
water amounts and Au0/Auδ+ ratio between
Au/CeO2-873K and Au/CeO2-1073K, the activities of
them were difficult to compare. Therefore, it is
difficult to analyze that Oact attack the formate and
carbonate-like species to produce CO2 is more easier
or Oact directly react with Au-CO to produce CO2 is
more easier. In addition, a suitable amount of water
on the surface is crucial to obtain excellent activity,
because too much water also could cause the
coverage of active sites and the accumulation of
stable carbonate-like species on the support, which
all negatively effect on the activity.
4 Conclusions
In summary, in-situ FTIR combined with HRTEM,
TGA, CO-TPD, O2-TPO and O2-TPD experiments
presented herein provide a more direct way to
understand the catalytic performance of
ultra-low-gold loaded catalysts with own water for
CO oxidation at ambient temperature. It is the first
time to detect the delicately changed signals of -OH
groups on the catalyst surface by using in-situ FTIR,
which supply the experimental evidence of the
changes of water during the reaction process. The
superior catalytic activity could be rationally
explained by that the appropriate water content in
catalysts themselves could promote O2 adsorption
and activation to produce Oact species. The product
CO2 may be obtained by two reaction pathways: (1)
the decomposition of reaction intermediates on the
perimeter interface by the attack of Oact, such as
formate (Au-COOH)interface and carbonate-like
(Au-CO3H)interface species; (2) and the direct reaction
between Au-CO and Oact. Auδ+ species are requisite
to form the formate and carbonate-like species on
the interface, therefore the contribution of each is
dependent on the catalyst surface composition.
Au/oxide catalysts also have been attracted much
attention in PEM fuel cells field duo to their
excellent catalytic activities for the selective
oxidation of CO. In the presence of H2, what will
happen on the catalyst surfaces which possess
water by itself? In future, an interesting research
about the behavior of Au/oxide catalysts with water
for selective oxidation of CO will be conducted.
Acknowledgements
The authors are grateful for the financial support of
Technology Center of China Tobacco Fujian
Industrial Co., Ltd.
References
[1] Bond, G. C.; Louis, C.; Thompson, D. T. Catalysis by Gold,
London, 2006.
[2] Carabineiro, S. A. C.; Bogdanchikova, N.; Avalos-Borja, M.;
Pestryakov, A.; Tavares, P. B.; Figueiredo, J. L. Gold
supported on metal oxides for carbon monoxide oxidation.
Nano. Res. 2011, 4, 180–193.
[3] Wu, B. H.; Zhang, H.; Chen, C.; Lin, S. C.; Zheng, N. F.
Interfacial activation of catalytically inert Au (6.7
nm)-Fe3O4 dumbbell nanoparticles for CO oxidation. Nano.
Res. 2009, 2, 975–983.
[4] Peng, S.; Lee, Y. M.; Wang, C.; Yin, H. F.; Dai, S.; Sun, S.
H. A facile synthesis of monodisperse Au nanoparticles and
their catalysis of CO oxidation. Nano. Res. 2008, 1,
229–234.
[5] Kast, P.; Kučerová, G.; Behm, R. J. On the nature of the
active Au species: CO oxidation on cyanide leached
| www.editorialmanager.com/nare/default.asp
8 Nano Res.
Au/TiO2 catalysts. Catal. Today. 2015, 244, 146–160.
[6] Gavril, D.; Georgakab, A.; Loukopoulosb, V.; Karaiskakisb,
G.; Nieuwenhuysa, B. E. On the mechanism of selective
CO oxidation on nanosized Au/γ-Al2O3 catalysts. Gold.
Bull. 2006, 39, 192–199.
[7] Gavril, D. CO oxidation on nanosized Au/Al2O3 by surface
hydroxyl groups and in the absence of O2, studied by
inverse gas chromatography. Catal. Today. 2015, 244,
36–46.
[8] Hutchings, G. J. Catalysis by gold. Catal. Today. 2005, 100,
55–61.
[9] Chen, M. S.; Goodman, D. W. The structure of catalytically
active gold on titania. Science. 2004, 306, 252–255.
[10] Vijay, A.; Mills, G.; Metiu, H. Adsorption of gold on
stoichiometric and reduced rutile TiO2 (110) surfaces. J.
Chem. Phys. 2003, 118, 6536–6551.
[11] Varganov, S. A.; Olson, R. M.; Gordon, M. S.; Metiu, H.
The interaction of oxygen with small gold clusters. J.
Chem. Phys. 2003, 119, 2531–2537.
[12] Li, Q. L.; Zhang, Y. H.; Chen, G. X.; Fan, J. Q.; Lan, H. Q.;
Yang, Y. Q. Ultra-low-gold loading Au/CeO2 catalysts for
ambient temperature CO oxidation: Effect of preparation
conditions on surface composition and activity. J. Catal.
2010, 273, 167–176.
[13] Date, M.; Haruta, M. Moisture effect on CO oxidation
over Au/TiO2 catalyst. J. Catal. 2001, 201, 221–224.
[14] Kung, H. H.; Kung, M. C.; Costello, C. K. Supported Au
catalysts for low temperature CO oxidation. J. Catal. 2003,
216, 425–432.
[15] Calla, J. T.; Davis, R. J. Oxygen-exchange reactions during
CO oxidation over titania- and alumina-supported Au
nanoparticles. J. Catal. 2006, 241, 407–416.
[16] Date, M.; Okumura, M.; Tsubota, S.; Haruta, M. Vital Role
of Moisture in the Catalytic Activity of Supported Gold
Nanoparticles. Angew. Chem. Int. Ed. 2004, 43,
2129–2132.
[17] Gao, F.; Wood, T. E.; Goodman, D. W. The effects of water
on CO oxidation over TiO2 supported Au catalysts. Catal.
Lett. 2010, 134, 9–12.
[18] Liu, L. M.; McAllister, B.; Ye, H. Q.; Hu, P. Identifying an
O2 supply pathway in CO oxidation on Au/TiO2(110): A
density functional theory study on the intrinsic role of
water. J. Am. Chem. Soc. 2006, 128, 4017–4022.
[19] Bongiorno, A. Water-enhanced catalysis of CO oxidation
on free and supported gold nanoclusters. Phys. Rev. Lett.
2005, 95, 106102-106105.
[20] Ojeda, M.; Zhan, B. Z.; Lglesia, E. Mechanistic
interpretation of CO oxidation turnover rates on supported
Au clusters. J. Catal. 2012, 285, 92–102.
[21] Saavedra, J.; Doan, H. A.; Pursell, C. J.; Grabow, L. C.;
Chandler, B. D. The critical role of water at the
gold-titania interface in catalytic CO oxidation. Science.
2014, 345, 1599–1602.
[22] Karpenko, A.; Leppelt, R.; Plzak, V.; Behm, R. J. The role
of cationic Au3+ and nonionic Au0 species in the low
temperature water-gas shift reaction on Au/CeO2 catalysts.
J. Catal. 2007, 252, 231–242.
[23] Ntho, T. A.; Anderson, J. A.; Scurrell, M. S. CO oxidation
over titanate nanotube supported Au: Deactivation due to
bicarbonate. J. Catal. 2009, 261, 94-100.
[24] Fu, Q.; Saltsburg, H. Flytzani-Stephanopoulos, M. Active
nonmetallic Au and Pt species on ceria-based water-gas
shift catalysts. Science. 2003, 301, 935–938.
[25] Karpenko, A.; Denkwitz, Y.; Plzak, V.; Cai, J.; Leppelt, R.;
Schumacher, B.; Behm, R. J. Low-temperature water-gas
shift reaction on Au/CeO2 catalysts-the influence of
catalyst pre-treatment on the activity and deactivation in
idealized reformate. Catal. Lett. 2007, 116, 105–115.
[26] Boccuzzi, F.; Chiorino, A.; Manzoli, M.; Lu, P.; Akita, T.;
Ichikawa, S.; Haruta, M. Au/TiO2 nanosized samples: A
catalytic, TEM, and FTIR study of the effect of
calcination temperature on the CO oxidation. J. Catal.
2001, 202, 256–267.
[27] Kim, C. H.; Thompson, L T. On the importance of
nanocrystalline gold for Au/CeO2 water–gas shift catalysts.
J. Catal. 2006, 244, 248–250.
[28] Parida, K. M.; Sahu, N.; Mohapatra, P.; Scurrell, M. S.
Low temperature CO oxidation over gold supported
mesoporous Fe-TiO2. J. Molecular. Catal. A: Chem. 2010,
319, 92–97.
www.theNanoResearch.com∣www.Springer.com/journal/12274 | Nano Research
9 Nano Res.
FIGURES.
Figure 1 High-resolution transmission electron microscopic
images of the catalysts with 1 wt% gold content: (a)
Au/CeO2-873, (b) Au/CeO2-1073.
Figure 2 TGA test for the catalysts Au/CeO2-873, Au/CeO2-1073
dried at 333 K, and the catalyst Au/CeO2-1073 dried at 373 K.
Figure 3 Activity (a) and series of in-situ FTIR spectra (b and c)
obtained during the CO oxidation at 298 K over 55 min on
h-CeO2-873.
Figure 4 Activity (a) and series of in-situ FTIR spectra (b and c)
obtained during the CO oxidation at 298 K over 55 min on
Au/CeO2-873.
Figure 5 Mass spectra of CO2 during CO-TPD on Au/CeO2-873
and h-CeO2-873.
Figure 6 Mass spectra of CO2 during CO-TPD, CO2-TPD for the
fresh catalyst Au/CeO2-873 and O2-TPO for the catalyst
Au/CeO2-873 after reacting 1 hour.
| www.editorialmanager.com/nare/default.asp
10 Nano Res.
Figure 7 Activity (a) and series of in-situ FTIR spectra (b and c)
obtained during the CO oxidation at 298 K over 55 min on
h-CeO2-1073.
Figure 8 Activity (a) and series of in-situ FTIR spectra (b and c)
obtained during the CO oxidation at 298 K over 55 min on
Au/CeO2-1073.
Figure 9 Mass spectra of O2 and H2O during O2-TPD on the
catalyst Au/CeO2-1073 dried at 333 K and 373 K.
www.theNanoResearch.com∣www.Springer.com/journal/12274 | Nano Research
Nano Res.