The cell surface leucine-rich repeat receptor forAtPep1, an endogenous peptide elicitor inArabidopsis, is functional in transgenic tobacco cellsYube Yamaguchi*, Gregory Pearce, and Clarence A. Ryan†
Institute of Biological Chemistry, Washingston State University, Pullman, WA 99164-6340
Contributed by Clarence A. Ryan, May 8, 2006
AtPep1 is a 23-aa endogenous peptide elicitor from Arabidopsisleaves that signals the activation of components of the innateimmune response against pathogens. Here, we report the isolationof an AtPep1 receptor from the surface of Arabidopsis suspension-cultured cells. An 125I-labeled AtPep1 analog interacted with sus-pension-cultured Arabidopsis with a Kd of 0.25 nM, and an 125I-labeled azido-Cys-AtPep1 photoaffinity analog specifically labeleda membrane-associated protein of �170 kDa. The labeled proteinwas purified to homogeneity, and its tryptic peptides were iden-tified as gene At1g73080, which encodes a leucine-rich repeatreceptor kinase, here called PEPR1. Verification of the bindingprotein as the receptor for AtPep1 was established by demonstrat-ing the loss of function of microsomal membranes of two SALKinsertional mutants and by a gain in function of the alkalinizationresponse to AtPep1 by tobacco suspension-cultured cells express-ing the At1g73080 transgene. Synthetic homologs of AtPep1,deduced from the C termini of six known paralogs of PROPEP1,were biologically active and were competitors of the interaction ofan AtPep1 radiolabeled analog with the receptor. The data areconsistent with a role for PEPR1 as the receptor for AtPep1 toamplify innate immunity in response to pathogen attacks.
A 23-aa peptide recently isolated from Arabidopsis called At-Pep1, when supplied to Arabidopsis leaves at nanomolar
concentrations, activates expression of the AtPep1 precursor genePROPEP1 and defensin, PDF1.2, and induces the production ofH2O2 (1). Arabidopsis plants transformed with the AtPep1 precur-sor gene PROPEP1 overexpress defense genes, producing a phe-notype with enhanced resistance toward Pythium irregulare, a rootpathogen (1).
The innate immune response in animals and plants is the first lineof defense against microbial pathogens and is more ancient thanadaptive immunity (2–6). Innate immunity in plants exhibits sim-ilarities with those of Drosophila and higher animals, and it appearsthat these responses have ancient evolutionary relationships (2).The immune responses are initiated by the interaction of conservedmolecules derived from the pathogens. In animals, these moleculesare called pathogen-associated molecular patterns, and in plants,they are called elicitors. Pathogen-associated molecular patternsand elicitors are recognized at the cell surface by receptors that aretypically composed of an extracellular leucine-rich repeat (LRR)domain flanked by disulfide pairs, a transmembrane domain, andan intracellular signaling domain. Although endogenous peptidesfrom plants have been shown to activate defenses against herbivores(7–9) and pathogen-derived peptides have been shown to activatedefense responses against pathogens (10–14), AtPep1 is a uniqueexample of an endogenous peptide signal in plants that activatesdefense genes targeted specifically against pathogens.
Here, we report the isolation of an AtPep1-binding protein fromArabidopsis suspension-cultured cells. Characterization of trypticdigests of the purified receptor protein has led to the identificationof the gene in the Arabidopsis Information Resource as At1g73080,
a LRR receptor kinase. The identity of this gene as the AtPep1receptor gene was confirmed by using two SALK insertionalmutants of At1g73080 that lost their ability to be photoaffinity-labeled and by a gain of AtPep1 binding to the cell surface oftobacco cells transformed with At1g73080. The receptor appears tobe a key component of AtPep1 signaling for the amplification of theinnate immune response in Arabidopsis in response to pathogenattacks.
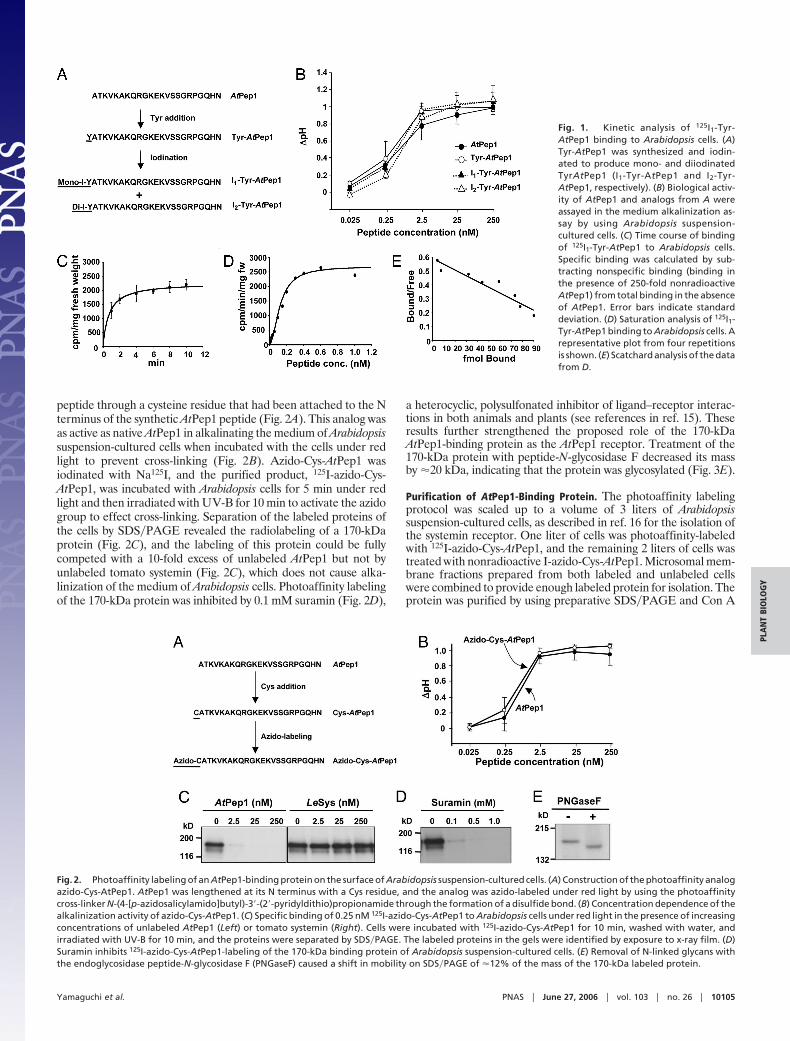
Results and DiscussionBiological Activity of Tyr-AtPep1 Analogs. To radiolabel AtPep1 toquantify binding to Arabidopsis suspension-cultured cells, a tyrosineresidue was attached to the N terminus of synthetic AtPep1 formodification with 125I (Fig. 1A). This addition of a single tyrosineto the N terminus of the peptide did not affect its mediumalkalinization activity (Fig. 1B). Tyr-AtPep1 was iodinated, and theseparated monoiodinated and diiodinated products, I1-Tyr-AtPep1and I2-Tyr-AtPep1, were found to be as fully active as AtPep1 incausing the alkalinization of Arabidopsis suspension-cultured cells(1) (Fig. 1B). A half-maximal alkalinization response occurred at�2.5 nM, which is within the concentration range for half-maximalactivity of AtPep1 and Tyr-AtPep1. Tyr-AtPep1 was iodinated withNa125I as above, and monoiodinated 125I1-Tyr-AtPep1 was recov-ered as 95% of the total label with a specific activity of �2mCi�nmol (1 Ci � 37 GBq). This species was used for the followingbinding studies.
Kinetic Analysis of 125I1-Tyr-AtPep1 Binding. Arabidopsis suspension-cultured cells were incubated with 0.25 nM 125I1-Tyr-AtPep1 andwashed with cold Murashige and Skoog medium containing 3%sucrose, and the total bound radioactivity was quantified. Toestimate the contribution of nonspecific binding, a 250-fold excessof unlabeled AtPep1 was added to the cells along with 125I1-Tyr-AtPep1, and the remaining bound radioactivity was determined.Specific binding was calculated by subtracting nonspecific bindingfrom total binding. Binding of 125I1-Tyr-AtPep1 to the cells could bedetected within 1 min, and the initial rapid binding was completewithin �4 min (Fig. 1C). The specific binding of 125I1-Tyr-AtPep1was saturated at a level of �0.6 nM (Fig. 1D). From the data, a Kdof 0.25 nM for the binding of 125I1-Tyr-AtPep1 was estimated (Fig.1E), which is in agreement with the concentration of AtPep1required for half-maximal medium alkalinization (Fig. 1B).
Photoaffinity Labeling of an AtPep1-Binding Protein. An azido-Cys-AtPep1 analog was constructed (see Materials and Methods) byusing the photoaffinity cross-linker N-(4-[p-azidosalicylamido]butyl)-3�-(2�-pyridyldithio)propionamide, which was coupled to the
Conflict of interest statement: No conflicts declared.
Abbreviations: LRR, leucine-rich repeat; T-DNA, transfer DNA.
*Permanent address: Graduate School of Biological Sciences, Nara Institute of Science andTechnology, Nara 630-0192, Japan.
†To whom correspondence should be addressed. E-mail: [email protected].
peptide through a cysteine residue that had been attached to the Nterminus of the synthetic AtPep1 peptide (Fig. 2A). This analog wasas active as native AtPep1 in alkalinating the medium of Arabidopsissuspension-cultured cells when incubated with the cells under redlight to prevent cross-linking (Fig. 2B). Azido-Cys-AtPep1 wasiodinated with Na125I, and the purified product, 125I-azido-Cys-AtPep1, was incubated with Arabidopsis cells for 5 min under redlight and then irradiated with UV-B for 10 min to activate the azidogroup to effect cross-linking. Separation of the labeled proteins ofthe cells by SDS�PAGE revealed the radiolabeling of a 170-kDaprotein (Fig. 2C), and the labeling of this protein could be fullycompeted with a 10-fold excess of unlabeled AtPep1 but not byunlabeled tomato systemin (Fig. 2C), which does not cause alka-linization of the medium of Arabidopsis cells. Photoaffinity labelingof the 170-kDa protein was inhibited by 0.1 mM suramin (Fig. 2D),
a heterocyclic, polysulfonated inhibitor of ligand–receptor interac-tions in both animals and plants (see references in ref. 15). Theseresults further strengthened the proposed role of the 170-kDaAtPep1-binding protein as the AtPep1 receptor. Treatment of the170-kDa protein with peptide-N-glycosidase F decreased its massby �20 kDa, indicating that the protein was glycosylated (Fig. 3E).
Purification of AtPep1-Binding Protein. The photoaffinity labelingprotocol was scaled up to a volume of 3 liters of Arabidopsissuspension-cultured cells, as described in ref. 16 for the isolation ofthe systemin receptor. One liter of cells was photoaffinity-labeledwith 125I-azido-Cys-AtPep1, and the remaining 2 liters of cells wastreated with nonradioactive I-azido-Cys-AtPep1. Microsomal mem-brane fractions prepared from both labeled and unlabeled cellswere combined to provide enough labeled protein for isolation. Theprotein was purified by using preparative SDS�PAGE and Con A
Fig. 1. Kinetic analysis of 125I1-Tyr-AtPep1 binding to Arabidopsis cells. (A)Tyr-AtPep1 was synthesized and iodin-ated to produce mono- and diiodinatedTyrAtPep1 (I1-Tyr-AtPep1 and I2-Tyr-AtPep1, respectively). (B) Biological activ-ity of AtPep1 and analogs from A wereassayed in the medium alkalinization as-say by using Arabidopsis suspension-cultured cells. (C) Time course of bindingof 125I1-Tyr-AtPep1 to Arabidopsis cells.Specific binding was calculated by sub-tracting nonspecific binding (binding inthe presence of 250-fold nonradioactiveAtPep1) from total binding in the absenceof AtPep1. Error bars indicate standarddeviation. (D) Saturation analysis of 125I1-Tyr-AtPep1 binding to Arabidopsis cells. Arepresentative plot from four repetitionsis shown. (E) Scatchard analysis of the datafrom D.
Fig. 2. Photoaffinity labeling of an AtPep1-binding protein on the surface of Arabidopsis suspension-cultured cells. (A) Construction of the photoaffinity analogazido-Cys-AtPep1. AtPep1 was lengthened at its N terminus with a Cys residue, and the analog was azido-labeled under red light by using the photoaffinitycross-linker N-(4-[p-azidosalicylamido]butyl)-3�-(2�-pyridyldithio)propionamide through the formation of a disulfide bond. (B) Concentration dependence of thealkalinization activity of azido-Cys-AtPep1. (C) Specific binding of 0.25 nM 125I-azido-Cys-AtPep1 to Arabidopsis cells under red light in the presence of increasingconcentrations of unlabeled AtPep1 (Left) or tomato systemin (Right). Cells were incubated with 125I-azido-Cys-AtPep1 for 10 min, washed with water, andirradiated with UV-B for 10 min, and the proteins were separated by SDS�PAGE. The labeled proteins in the gels were identified by exposure to x-ray film. (D)Suramin inhibits 125I-azido-Cys-AtPep1-labeling of the 170-kDa binding protein of Arabidopsis suspension-cultured cells. (E) Removal of N-linked glycans withthe endoglycosidase peptide-N-glycosidase F (PNGaseF) caused a shift in mobility on SDS�PAGE of �12% of the mass of the 170-kDa labeled protein.
Yamaguchi et al. PNAS � June 27, 2006 � vol. 103 � no. 26 � 10105
PLA
NT
BIO
LOG
Y
affinity Sepharose (Fig. 3A). The photoaffinity-labeled proteinbound so tightly to the Con A Sepharose that it could not be elutedby 0.5 M �-methyl-D-glucoside. However, the proteins were solu-bilized from the Sepharose by boiling in 5% SDS and separated bySDS�PAGE. After electrophoresis, the section of the gel contain-ing the radiolabeled protein was treated with trypsin, and thepeptide fragments were identified by mass spectroscopy (Fig. 3B)
as being derived from the gene At1g73080. Tryptic digests of thedeglycosylated protein confirmed the identity of the protein.
At1g73080, which we call PEPR1, encodes a protein of 1,124 aathat includes a signal peptide, 26 LRR motifs with flanking cysteinepairs, a transmembrane region, and a kinase domain. Based onphylogenetic analysis of all Arabidopsis receptor-like kinases (17),At1g73080 belongs to a 28-member subfamily of LRR XI genes thatincludes HAESA (18), CLV1 (19), and three CLV1-related LRRreceptor-like kinase genes, BAM1, BAM2, and BAM3 (20) (Fig. 3C).These putative receptors are the only members of this 28-membersubfamily that have been identified as having signaling roles inArabidopsis, and only CLV1 and PEPR1 have known ligands, bothbeing peptides.
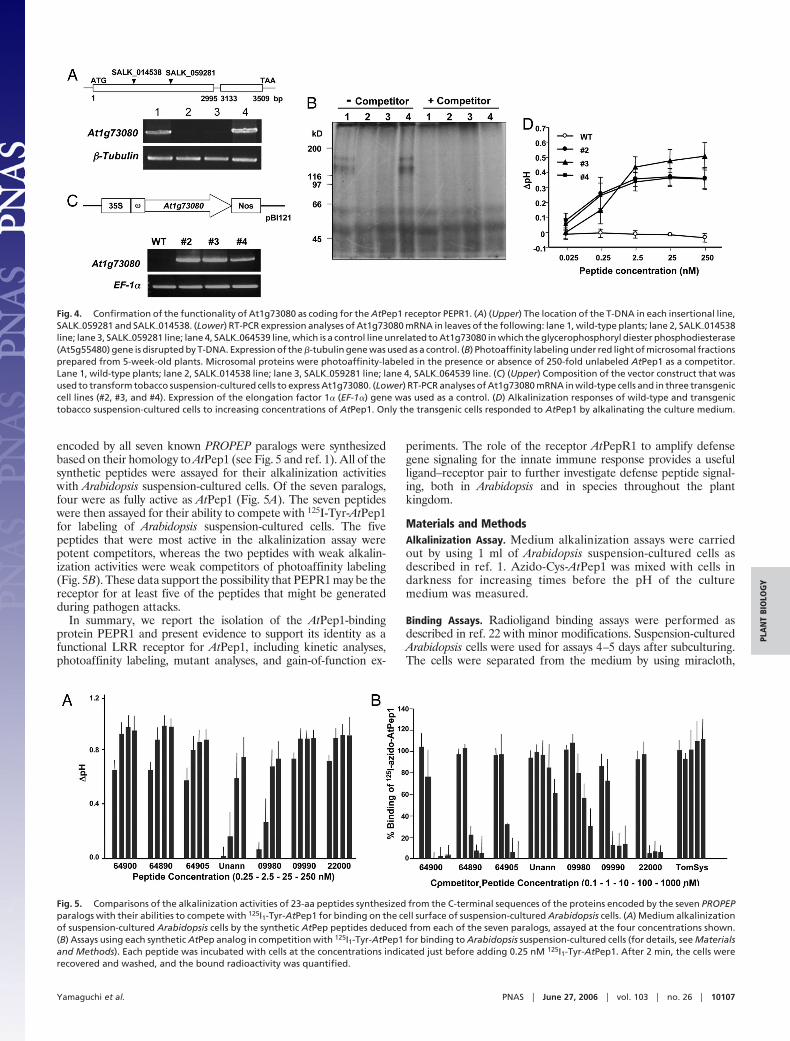
Functionality of PEPRI as the AtPep1 Receptor Gene. Total RNA wasisolated from leaves of each of two SALK insertional mutant linesof PEPR1 (At1g73080), SALK�059281 and SALK�014538 (Fig. 4AUpper), and assayed by RT-PCR (Fig. 4A Lower), establishing thatthe gene was not expressed in either line. Microsomal membranesprepared from the two SALK mutant lines were subjected tophotoaffinity labeling with 125I-azido-Cys-AtPep1 and comparedwith labeling of membranes from wild-type plants and from theSALK insertional line SALK�064539, which encodes a phospho-diesterase gene that is unrelated to PEPRI. A protein of 170 kDaand a lower molecular mass protein were labeled in membranesfrom the wild type and SALK�064539 but not in membranes of thetwo mutant lines (Fig. 4B). This labeling suggested that the 170-kDaprotein was the same protein that was identified and isolated fromArabidopsis cells, with the lower molecular mass species likely beinga degradation product of the 170-kDa species because no labelingof this protein was found in either of the two insertional mutants.Possible loss of resistance against pathogens and changes in theability to activate defensin and other defense genes in response towounding and elicitors in the two insertional mutants have yet to bestudied.
To further investigate the role of PEPR1 as the AtPep1 receptor,gain-of-function experiments were carried out with tobacco sus-pension-cultured cells that do not respond to AtPep1 by alkalinatingthe cell medium. Tobacco cells were transformed with a CaMV-35S-PEPR1 gene (Fig. 4C Upper) to determine whether the foreignreceptor protein was targeted to the cell surface, where, in responseto AtPep1, it would activate intracellular signaling to effect thealkalinization of the medium of the transgenic tobacco cells.Expression of the PEPR1 gene was confirmed by RT-PCR analysisof RNA from three independently transformed tobacco cell lines(Fig. 4C Lower). All three of the tobacco lines responded to theaddition of nanomolar concentrations of AtPep1 by producing astrong, reproducible alkalinization of the cell culture medium,whereas no alkalinization was detected in the culture medium ofwild-type cells challenged with AtPep1 (Fig. 4D). These results notonly confirm the identity of PEPR1 as the AtPep1 receptor, but theyindicate that the receptor-mediated intracellular signaling pathwayin tobacco cells that regulates alkalinization in response to peptideligands can accommodate signaling by PEPR1 and its peptideligand. This type of accommodation by suspension cultured cells toa foreign receptor and its peptide ligand has been reported for thetomato systemin receptor in tobacco cells (21), the tomato xylanasereceptor in tobacco cells (22), the Arabidopsis flagellin receptor intomato cells (23), and the receptor EFR in Nicotiana benthmianacells (24). Kinase domains of receptors are known to possess highlyconserved regions, and it may be that these regions in foreignreceptors are recognized by interacting signaling components,perhaps through phosphorylation events. It will be of interest todetermine the phylogenetic distance from Arabidopsis where thePEPR1 gene can still initiate intracellular signaling in response toAtPep1 when transformed into foreign plant species.
PEPR1 may also be the receptor for peptides related to AtPep1generated from PROPEP1 paralogs (1). The putative peptides
Fig. 3. Purification and identification of an AtPep1-binding protein. (A)(Left) SDS�PAGE analysis of photoaffinity-labeled proteins during purification(described in Materials and Methods). Protein staining is as follows: lane 1,crude extract; lane 2, microsomal fraction; lane 3, the eluate from preparativeSDS�PAGE; lane 4, eluate from Con A Sepharose. (Right) Radioautography ofgels from Left. (B) Mass spectral analysis of peptides resulting from trypticdigest of the purified, radiolabeled AtPep1-binding protein from lane 4 in A(probability-based molecular weight search score � 609; P � �0.05). Peptidefragments marked with an asterisk matched predicted amino acid sequencesof Arabidopsis gene At1g73080. (C) The phylogenetic relationships are basedon comparison of the amino acid sequence of PEPR1 with the gene productsof other members of the LRR XI subfamily of Arabidopsis LRR receptor-likeprotein kinases.
10106 � www.pnas.org�cgi�doi�10.1073�pnas.0603729103 Yamaguchi et al.
encoded by all seven known PROPEP paralogs were synthesizedbased on their homology to AtPep1 (see Fig. 5 and ref. 1). All of thesynthetic peptides were assayed for their alkalinization activitieswith Arabidopsis suspension-cultured cells. Of the seven paralogs,four were as fully active as AtPep1 (Fig. 5A). The seven peptideswere then assayed for their ability to compete with 125I-Tyr-AtPep1for labeling of Arabidopsis suspension-cultured cells. The fivepeptides that were most active in the alkalinization assay werepotent competitors, whereas the two peptides with weak alkalin-ization activities were weak competitors of photoaffinity labeling(Fig. 5B). These data support the possibility that PEPR1 may be thereceptor for at least five of the peptides that might be generatedduring pathogen attacks.
In summary, we report the isolation of the AtPep1-bindingprotein PEPR1 and present evidence to support its identity as afunctional LRR receptor for AtPep1, including kinetic analyses,photoaffinity labeling, mutant analyses, and gain-of-function ex-
periments. The role of the receptor AtPepR1 to amplify defensegene signaling for the innate immune response provides a usefulligand–receptor pair to further investigate defense peptide signal-ing, both in Arabidopsis and in species throughout the plantkingdom.
Materials and MethodsAlkalinization Assay. Medium alkalinization assays were carriedout by using 1 ml of Arabidopsis suspension-cultured cells asdescribed in ref. 1. Azido-Cys-AtPep1 was mixed with cells indarkness for increasing times before the pH of the culturemedium was measured.
Binding Assays. Radioligand binding assays were performed asdescribed in ref. 22 with minor modifications. Suspension-culturedArabidopsis cells were used for assays 4–5 days after subculturing.The cells were separated from the medium by using miracloth,
Fig. 4. Confirmation of the functionality of At1g73080 as coding for the AtPep1 receptor PEPR1. (A) (Upper) The location of the T-DNA in each insertional line,SALK�059281 and SALK�014538. (Lower) RT-PCR expression analyses of At1g73080 mRNA in leaves of the following: lane 1, wild-type plants; lane 2, SALK�014538line; lane 3, SALK�059281 line; lane 4, SALK�064539 line, which is a control line unrelated to At1g73080 in which the glycerophosphoryl diester phosphodiesterase(At5g55480) gene is disrupted by T-DNA. Expression of the �-tubulin gene was used as a control. (B) Photoaffinity labeling under red light of microsomal fractionsprepared from 5-week-old plants. Microsomal proteins were photoaffinity-labeled in the presence or absence of 250-fold unlabeled AtPep1 as a competitor.Lane 1, wild-type plants; lane 2, SALK�014538 line; lane 3, SALK�059281 line; lane 4, SALK�064539 line. (C) (Upper) Composition of the vector construct that wasused to transform tobacco suspension-cultured cells to express At1g73080. (Lower) RT-PCR analyses of At1g73080 mRNA in wild-type cells and in three transgeniccell lines (#2, #3, and #4). Expression of the elongation factor 1� (EF-1�) gene was used as a control. (D) Alkalinization responses of wild-type and transgenictobacco suspension-cultured cells to increasing concentrations of AtPep1. Only the transgenic cells responded to AtPep1 by alkalinating the culture medium.
Fig. 5. Comparisons of the alkalinization activities of 23-aa peptides synthesized from the C-terminal sequences of the proteins encoded by the seven PROPEPparalogs with their abilities to compete with 125I1-Tyr-AtPep1 for binding on the cell surface of suspension-cultured Arabidopsis cells. (A) Medium alkalinizationof suspension-cultured Arabidopsis cells by the synthetic AtPep peptides deduced from each of the seven paralogs, assayed at the four concentrations shown.(B) Assays using each synthetic AtPep analog in competition with 125I1-Tyr-AtPep1 for binding to Arabidopsis suspension-cultured cells (for details, see Materialsand Methods). Each peptide was incubated with cells at the concentrations indicated just before adding 0.25 nM 125I1-Tyr-AtPep1. After 2 min, the cells wererecovered and washed, and the bound radioactivity was quantified.
Yamaguchi et al. PNAS � June 27, 2006 � vol. 103 � no. 26 � 10107
PLA
NT
BIO
LOG
Y
washed once with 40 ml of culture medium, and adjusted to a freshweight of 0.2 mg�ml with fresh medium. A 2-ml aliquot of cells waspipeted into a well of a 12-well culture plate and allowed toequilibrate for 1 h at room temperature while agitated on an orbitalshaker (160 rpm). 125I1-Tyr-AtPep1 was added to the medium togive a final concentration of 0.02–1.0 nM. To assay binding, 500 �lof cells was removed and filtered through a 2.5-cm Type A�E GlassFiber Filter (Pall) by using a 12-well vacuum filtration manifold(Millipore). The cells were washed three times with 5 ml of coldMurashige and Skoog (MS) medium containing 3% sucrose, sus-pended in 1 ml of MS medium containing 3% sucrose, transferredto a test tube, and then analyzed for total radioactivity in agamma-ray counter (Isodata 2020; Isodata, Palatine, IL). Specificbinding was calculated by subtracting nonspecific binding (bindingin the presence of 250-fold native AtPep1) from total binding.
AtPep1 Analogs. AtPep1, Cys-AtPep1, Tyr-AtPep1, and AtPep1homologs were synthesized by using solid-phase chemistry with anApplied Biosystems model 431 synthesizer. Cys-AtPep1 was cou-pled in darkness to the azido-photoaffinity cross-linker, N-(4-[p-azidosalicylamido]butyl)-3�-(2�-pyridyldithio)propionamide(Pierce), and azido-Cys-AtPep1 was purified by HPLC as describedin ref. 25. Iodination of Tyr-AtPep1 (50 nmol) was achieved by usingIODO-GEN Pre-Coated Iodination Tubes (Pierce) by the indirectmethod as described by the manufacturer. Iodinated forms ofTyr-AtPep1 were purified by HPLC as described in ref. 25 andquantified by using the bicinchoninic acid method (Pierce) withAtPep1 as a standard. All analogs were analyzed with an LCQ iontrap mass spectrometer (Finnigan, San Jose, CA). 125I iodinationwas performed by using 2 mCi of Na125I with 12.5 nmol ofazido-Cys-AtPep1 and Tyr-AtPep1, followed by purification byHPLC as described in ref. 25. The specific radiolabel of each analogwas calculated to be �2 mCi�nmol.
Photoaffinity Labeling and Purification of AtPep1-Binding Protein.The procedures for photoaffinity labeling were identical to thosedescribed in ref. 25 except that 125I-azido-Cys-AtPep1 was addedunder red light to a final concentration of 0.25 nM (1.4 � 105 cpm).The plate containing the cells was maintained on ice while it wasirradiated with a UV-B lamp (F15T8.UV-B, 15 W) for 10 min. Thecells were recovered from the well, washed at room temperaturewith 1 ml of Murashige and Skoog medium, and sedimented bycentrifugation at 12,000 � g, and the pellet was mixed with 400 �lof 5% SDS while in the microfuge tube. The cells were disruptedby placing the tube in a boiling water bath for 30 min, and the celldebris was removed by centrifugation in a microfuge at 12,000 � g.Proteins in the supernatant were precipitated by addition of 400 �lof methanol and 100 �l of chloroform and pelleted in a microfuge.The pellet was dissolved in 100 �l of Laemmli sample buffercontaining 5% SDS at 65°C, and 10 �l was separated by 8%SDS�PAGE. The gels were dried and exposed to x-ray film for 50 h.
Large-scale photoaffinity labeling of cell surface AtPep1-bindingproteins was performed according to the method of Scheer andRyan (16). Briefly, from 3 liters of Arabidopsis cells, 6 days aftersubculturing, the 1-liter and 2-liter sets of cells were placed inseparate large, shallow plastic dishes (33 cm in diameter). Both setsof cells were incubated under red light while shaking on an orbitalshaker (90 rpm) for 10 min. To the 1-liter set of cells was added125I-azido-Cys-AtPep1 (to a final concentration of 0.25 nM), and tothe 2-liter set of cells was added nonradiolabeled I-azido-Cys-AtPep1 (also to a final concentration of 0.25 nM). Both sets of cellswere exposed to UV irradiation for 10 min while shaking on anorbital shaker, and each set of cells was collected by filtration asdescribed in ref. 16. Each set of cells was dispersed in 300 ml ofextraction buffer, the cells were disrupted in a Parr Cell DisruptionBomb (Parr Instruments, Moline, IL), and microsomal fractionswere prepared by differential centrifugation (16). Radiolabeled andunlabeled microsomal proteins (25 and 55 mg, respectively), were
mixed together, and the proteins were introduced into singlehorizontal wells of two 7.5% SDS�PAGE gels (0.6 � 14 � 9 cm)and separated during 16 h at a constant 37 V at room temperature.The gel was sliced horizontally into 5-mm widths, and the radio-activity was monitored with a gamma counter. The single gel piecethat contained the most radioactivity from each gel was maceratedto allow for protein extraction by using a 20-ml syringe (26). Theprotein was extracted with 25 ml of Con A binding buffer (20 mMTris�HCl, pH 7.5�0.5 M NaCl) by gently shaking at room temper-ature for 30 min. The gel material was centrifuged at 2,000 � g for10 min, and the supernatant was recovered. The extraction wasrepeated twice, and the supernatants were pooled (50 ml total). A75-�l aliquot of Con A Sepharose 4B (Amersham PharmaciaBioscience), previously equilibrated in the Con A binding buffer,was added to the extracted proteins and incubated at room tem-perature with constant mixing for 6 h on an orbital shaker (100rpm). Con A Sepharose was recovered by centrifugation at 500 �g for 10 min at 4°C and washed three times with 50 ml of 0.5 M�-methyl-D-glucoside, followed by three washes with distilled water.The proteins were released from the Con A Sepharose by boilingin 500 �l of 5% SDS, centrifuging as above, and repeating theboiling procedure. The proteins in the supernatants were pooledand separated into 500-�l samples, and each was mixed with 500 �lof methanol and 100 �l of chloroform to precipitate proteins. Onesample was subjected to 7.5% SDS�PAGE (0.15 � 14 � 8.5 cm) ata constant 100 V for 8 h at room temperature. The gel was slicedhorizontally into 1-mm widths, and the radioactivity was monitoredwith a gamma counter. One gel piece containing 59% of theradioactivity added to the gel was treated with modified trypsin(Promega) (27). The second sample was treated with peptide-N-glycosidase F (Prozyme, San Leandro, CA), separated by 7.5%SDS�PAGE, and digested with trypsin as above. The peptides fromboth trypsin-digested proteins were analyzed by MALDI-TOFmass spectrometry (Voyager-DE RP Biospectrometry Worksta-tion; Applied Biosystems). The gene encoding the AtPep1-bindingprotein was identified by searching in the National Center forBiotechnology Information database with MASCOT.
Analysis of Transfer DNA (T-DNA) Insertional Lines. Arabidopsis thali-ana T-DNA insertional lines, SALK�014538, SALK�059281, andSALK�064539, were obtained from the Arabidopsis BiologicalResource Center (Columbus, OH) (28). Homogeneous lines werescreened by two sets of PCR analyses, one using the gene-specificprimer pair and the other using a gene-specific primer and theT-DNA left border of the vector as a primer. Microsomal proteinswere prepared from 1-month-old wild-type and SALK mutantplants by differential centrifugation as described in ref. 16. Themicrosomal proteins were adjusted to 1 �g��l in binding buffer (10mM Hepes-KOH, pH 7.4�100 mM NaCl�100 mM sucrose�5 mMMgCl2), and 250 �l was added under red light to ice-cold 0.25 nM125I-azido-Cys-AtPep1 with or without 250-fold unlabeled AtPep1for 60 min on ice. After incubation, the reaction mixture wasirradiated with UV-B (lamp F15T8.UV-B, 15 W) for 15 min. Themicrosomal proteins were precipitated by centrifugation at12,000 � g for 5 min after mixing with 250 �l of methanol and 50�l of chloroform, dissolved in 20 �l of Laemmli sample buffercontaining 5% SDS at 65°C, and then separated by SDS�PAGE.
Transgenic Tobacco Cells. The �-glucuronidase gene (excised be-tween its XbaI site and its SacI site) in a pBI121 vector was replacedwith a PCR fragment consisting of an XbaI site, a �-prime leadersequence (29), and KpnI, SmaI, BamHI, and SacI sites to enhancethe translation and further construction. PCR primers for thisconstruction were as follows: �-XbaI, 5�-CCG TCT AGA TTTTTA CAA CAA TTA CCA ACA ACA ACA AAC AAC AAACAA CAT TAC AAT TA-3�; �-BamHI, 5�-CCG AGC TCG GATCCC GGG GTA CCA TGG TAA TTG TAA ATA GTA ATTGTA ATG TTG TTT-3�. A 3,604-bp fragment consisting of a KpnI
10108 � www.pnas.org�cgi�doi�10.1073�pnas.0603729103 Yamaguchi et al.
site, the coding region of At1g73080 including an intron, and aBamHI site was amplified by PCR using 73080-KpnI (5�-CGGGGT ACC TCG ATC TTT AAA CTC AGA TGA AGA A-3�)and 73080-BamHI (5�-CGC GGA TCC TAC CGG TAA GCCCAG TTA CG-3�) primers. This PCR fragment was ligated intoKpnI and BamHI sites of pBI121 vector containing �-prime leadersequence and then introduced into tobacco suspension-culturedcells by using Agrobacterium tumefaciens strain GV3850 by themethod of Nakayama et al. (30). Transgenic cells were assayed inthe alkalinization assay as described above.
RT-PCR Analyses. Total RNA from leaves of T-DNA insertion linesand transgenic tobacco cells were extracted by using TRIzol totalRNA isolation reagent (Invitrogen) according to the manufactur-er’s instructions. Reverse transcription was performed by usingSuperscript III Reverse Transcriptase (Invitrogen) and oligo(dT)primer according to the manufacturer’s instructions. To amplifyAt1g73080, 73080-KpnI and 73080-BamHI primers were used.Arabidopsis �-tubulin gene and the tobacco elongation factor 1�
(EF-1�) gene were amplified as internal controls by using �-Tub1(5�-CAA CGC TAC TCT GTC TGT CC-3�), �-Tub2 (5�-TCTGTG AAT TCC ATC TCG TC-3�), EF-1�-1 (5�-TTT GGC CCTACT GGT TTG AC-3�), and EF-1�-2 (5�-CTC ATG TCC CTCACA GCA AA-3�).
Phylogenic Analysis. Full-length amino acid sequences of all mem-bers in subfamily LRR XI (18) were analyzed by using CLUSTAL W(31), and a phylogenic tree was produced by using TREEVIEW (32).
We thank Julia Gothard and Sue Vogtmann for growing our plants,Guido Barona for maintaining the cell cultures, Dr. Jerry Reeves and Dr.David Deavila (Washington State University) for help and advice inradiolabeling, Dr. William Siems for advice and assistance in obtainingmass spectrometric analyses, and Dr. Gerhard Munske (WashingtonState University) for peptide synthesis. This work was supported by theToyobo Biotechnology Research Foundation of Osaka, Japan (Y.Y.),National Science Foundation Grant IBN 0090766, the Charlotte Y.Martin Foundation, and the Washington State University College ofAgriculture, Human, and Natural Resources Sciences.
1. Huffaker, A., Pearce, G. & Ryan, C. A. (2006) Proc. Natl. Acad. Sci. USA 103,10098–10103.
2. Hoffman, J. A., Kafatos, F. C., Janeway, C. A., Jr., & Ezekowitz, R. A. B. (1999)Science 284, 1313–1318.
3. Menezes, H. & Jared, C. (2002) Comp. Biochem. Physiol. C Toxicol. Pharmocol.132, 1–7.
4. Nurnberger, T., Brunner, F., Kemmerling, B. & Piater, L. (2004) Immunol. Rev.198, 249–266.
5. Parker, J. (2003) Trends Plant. Sci. 8, 245–247.6. Zipfel, C., Robatzek, S., Navarro, L., Oakeley, E. J., Jones, J. D. G., Felix, G.
& Boller, T. (2004) Nature 428, 764–767.7. Pearce, G., Strydom, D., Johnson, S. & Ryan, C. A. (1991) Science 253,
895–897.8. Pearce, G., Moura, D. S., Stratmann, J. & Ryan, C. A. (2001) Nature 411,
817–820.9. Pearce, G. & Ryan, C. A. (2003) J. Biol. Chem. 278, 30044–30050.
10. Hahlbrock, K., Scheel, D., Logemann, E., Nurnberger, T., Parniske, M.,Reinnold, S., Sacks, W. R. & Schmelzer, E. (1995) Proc. Natl. Acad. Sci. USA92, 4150–4157.
11. van den Askerveken, G. G. J. M., Vassen, J. A. M. J. & De Wit, P. J. G. M.(1993) Plant Physiol. 103, 91–96.
12. Kammpren, S. (2001) Curr. Opin. Plant Biol. 4, 295–300.13. Kunze, G., Zipfel, C., Robatzek, S., Niehaus, K., Boller, T. & Felix, G. (2004)
Plant Cell 16, 3496–3507.14. Navarro, L., Zipfel, C., Rowland, R., Keller, I., Robatzek, S., Boller, T. & Jones,
J. D. G. (2004) Plant Physiol. 135, 1113–1128.15. Stratmann, J., Scheer, J. & Ryan C. A. (2000) Proc. Natl. Acad. Sci. USA 97,
8862–8867.
16. Scheer, J. M. & Ryan, C. A. (2002) Proc. Natl. Acad. Sci. USA 99, 9585–9590.17. Shiu, S. H. & Bleecker, A. B. (2001) Proc. Natl. Acad. Sci. USA 98, 10763–
10768.18. Jinn, T.-L., Stone, J. M. & Walker, J. C. (2000) Genes Dev. 14, 108–117.19. Clark, S. E., Williams, R. W. & Meyerowitz, E. M. (1997) Cell 89, 575–585.20. DeYoung, B. J., Bickle, K. L., Schrage, K. J., Muskett, P., Patel, K. & Clark,
S. E. (2006) Plant J. 45, 1–6.21. Scheer, J. M., Pearce, G. & Ryan, C. A. (2003) Proc. Natl. Acad. Sci. USA 100,
10114–10117.22. Ron, M. & Avini, A. (2004) Plant Cell 16, 1604–1615.23. Chinchilla, D., Bauer, Z., Ragenass, M., Boller, T. & Felix, G. (2006) Plant Cell
18, 465–476.24. Zipfel, C., Kunze, G., Chinchilla, C., Caniard, A., Boller, T. & Felix, G. (2006)
Cell 125, 749–760.25. Scheer, J. M. & Ryan, C. A. (1999) Plant Cell 11, 1525–1535.26. Scheer, J. M. & Ryan, C. A. (2001) Anal. Biochem. 298, 130–132.27. Shevchenko, A., Wilm, M., Vorn, O. & Mann, M. (1996) Anal. Chem. 68,
850–858.28. Alonso, J. M., Stepanova, A. N., Leisse, T. J., Kim, C. J., Chen, H., Shinn, P.,
Stevenson, D. R., Zimmerman, K., Barajas, P., Cheuk, R., et al. (2003) Science301, 653–657.
29. Gallie, D. R., Sleat, D. E., Watts, J. W., Turner, P. C. & Wilson, T. M. (1987)Nucleic Acids Res. 15, 3257–3273.
30. Nakayama, H., Yoshida, K., Ono, H., Murooka, Y. & Shinmyo, A. (2000) PlantPhysiol. 122, 1239–1248.
31. Thompson, J. D., Higgins, D. G. & Gibson, T. J. (1994) Nucleic Acids Res. 22,4673–4680.
32. Page, R. D. (1996) Comput. Appl. Biosci. 12, 357–358.
Yamaguchi et al. PNAS � June 27, 2006 � vol. 103 � no. 26 � 10109










![Structural model of the dimeric Parkinson s protein LRRK2 reveals … · in the leucine-rich repeat kinase 2 (LRRK2) gene [ PARK8;Online Mendelian Inheritance in Man (OMIM) no. 609007]](https://static.documents.pub/doc/80x56/5ecd8296609bc861f02d5245/structural-model-of-the-dimeric-parkinson-s-protein-lrrk2-reveals-in-the-leucine-rich.jpg)










