Contents lists available at SciVerse ScienceDirect
Biochimica et Biophysica Acta
j ourna l homepage: www.e lsev ie r .com/ locate /bba l ip
The human liver fatty acid binding protein (FABP1) gene is activated byFOXA1 and PPARα; and repressed by C/EBPα: Implications in FABP1down-regulation in nonalcoholic fatty liver disease
Carla Guzmán a, Marta Benet a,d, Sandra Pisonero-Vaquero b, Marta Moya a, M. Victoria García-Mediavilla b,d,M. Luz Martínez-Chantar c,d, Javier González-Gallego b,d, José Vicente Castell a,d,e,Sonia Sánchez-Campos b,d, Ramiro Jover a,d,e,⁎a Experimental Hepatology Unit, IIS Hospital La Fe, Valencia, Spainb Institute of Biomedicine (IBIOMED), Universidad de León, Spainc CIC bioGUNE, Technology Park of Bizkaia, Spaind CIBERehd, Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas, Spaine Department of Biochemistry and Molecular Biology, University of Valencia, Spain
Liver fatty acid binding protein (FABP1) prevents lipotoxicity of free fatty acids and regulates fatty acid traffickingand partition. Our objective is to investigate the transcription factors controlling the human FABP1 gene and theirregulation in nonalcoholic fatty liver disease (NAFLD). Adenovirus-mediated expression of multiple transcriptionfactors in HepG2 cells and cultured human hepatocytes demonstrated that FOXA1 and PPARα are among themosteffective activators of human FABP1, whereas C/EBPα is amajor dominant repressor. Moreover, FOXA1 and PPARαinduced re-distribution of FABP1 protein and increased cytoplasmic expression. Reporter assays demonstrated thatthemajor basal activity of the human FABP1 promoter locates between−96 and−229 bp, where C/EBPα binds toa composite DR1-C/EBP element.Mutation of this element at−123 bp diminished basal reporter activity, abolishedrepression by C/EBPα and reduced transactivation by HNF4α. Moreover, HNF4α gene silencing by shRNA in HepG2cells caused a significant down-regulation of FABP1 mRNA expression. FOXA1 activated the FABP1 promoterthrough binding to a cluster of elements between −229 and −592 bp, whereas PPARα operated through aconserved proximal element at−59 bp. Finally, FABP1, FOXA1 and PPARαwere concomitantly repressed in animalmodels of NAFLD and in human nonalcoholic fatty livers, whereas C/EBPα was induced or did not change. Weconclude that human FABP1 has a complexmechanism of regulationwhere C/EBPα displaces HNF4α and hampersactivation by FOXA1 and PPARα. Alteration of expression of these transcription factors in NAFLD leads to FABP1 genrepression and could exacerbate lipotoxicity and disease progression.
and inhibiting the action of multiple receptors and enzymes [1]. Toprevent FA lipotoxicity, mammals have evolved a family of smallcytosolic fatty acid binding proteins to maintain the intracellularconcentration of unbound free FAs and fatty acyl CoAs at a low level(i.e. nM range) [2]. The liver has a very active role in long-chain fattyacid (LCFA) uptake and metabolism and in agreement has a high levelof a fatty acid binding protein, FABP1 or L-FABP, which represents 2–5%of the cytosolic protein (0.1–0.4 mM) [2]. FABP1 has a broad range/promiscuity for a variety of ligands, and high affinity for LCFAs andlong-chain fatty acyl CoAs (LCFA-CoAs) [3].
Studies performed in vitro and in vivo show that FABP1 facilitatesuptake of LCFA from membranes, binds LCFAs and LCFA-CoAs tominimize toxic effects, enhances intracellular transport/diffusion, func-tions as a donor for both peroxisomal and mitochondrial LCFAβ-oxidation, and targets LCFA and LCFA-CoA to the endoplasmicreticulum for esterification into membrane lipids (phosphatidic acid,phospholipids) or into lipids for storage and secretion in very low
804 C. Guzmán et al. / Biochimica et Biophysica Acta 1831 (2013) 803–818
density lipoproteins (triglycerides, cholesteryl esters) [4]. Moreover,FABP1 transfers and channels lipidic ligands into nuclei for ini-tiating nuclear receptor transcriptional activity and activating newlipid signaling pathways that affect lipid and glucose metabolism[5].
The physiological significance of FABP1 has been extended andconfirmed by numerous studies using FABP1 deficient mice. It hasbeen found that FABP1 plays a role not only in hepatic FA metabo-lism, but also in hepatic cholesterol metabolism [6]. Indeed, FABP1seems to protect against gallstone in lithogenic diet-fed mice [7].Moreover, studies with FABP1 deficient mice [8] and with a Thr94Alamutation in human FABP1 [9] also suggest that FABP1 is able to influ-ence glucose homeostasis. In this regard, it has also been shown thatFABP1 can bind glucose and glucose-1-phosphate resulting in alteredFABP1 conformation and increased affinity, uptake and distributionof lipidic ligands [10].
FABP1 function is also relevant for body weight and, dependingon the diet type and themouse strain, weight gain [11–14] or protec-tion against obesity [8,15,16] has been observed in FABP1 deficientmice.
In the liver, FABP1 could influence the level of triglycerides. A lowerconcentration of triglycerides have been found in FABP1 deficient miceunder different diets [6,8,15,16] and in response to 48-h fasting [17].Lipid analysis of FABP1 gene-ablated hepatocytes has also revealedlower triglyceride levels [18]. Moreover, the Thr94Ala mutation in theFABP1 gen, which supposedly contributes negatively to FA binding,caused decreased triglycerides compared to the wild-type cells, whenincubated with extracellular FAs [19].
A different question is whether FABP1 levels are altered or not bymetabolic conditions such asNAFLD. Very few studies have investigatedthis point. A clinical study has shown that FABP1 is overexpressed insimple steatosis, but paradoxically underexpressed in nonalcoholicsteatohepatitis [20]. FABP1 levels were also decreased in a steatoticrat model established by administration of 17α-ethynylestradiol [21].Lower FABP1 level/function could lead to a lower capacity to attenuatethe cytotoxic detergent effect of free FAs and potentiate lipotoxicity inNAFLD [20]. The underlying mechanism for FABP1 deregulation inNAFLD has not yet been investigated.
Given the key role of FABP1 in lipid metabolism and its potentialinfluence in metabolic disorders [22], the study of FABP1 gene regu-lation is of particular relevance. FABP1 is induced by both fibratehypolipidemic drugs and LCFA, and a functional peroxisomeproliferatoractivated receptor (PPAR)-responsive element has been identified inthe proximal 5′-flanking region of the murine FABP1 [23,24]. FABP1 isalso induced by statins in rodents through a mechanism involvingPPARα upregulation and FABP1 activation via its PPAR-responsiveelement [25]. Other major transcription activator of FABP1 in the liveris HNF1α, and mice with HNF1α gene null mutation exhibit completeloss of FABP1 expression [26,27]. In addition, several liver enrichedtranscription factors including C/EBPs [28], FOXAs [27], and HNF4α[29] have been found to be capable of transactivating rodent FABP1.Thus, the regulation of FABP1 gene has been investigated in rodentsbut, despite substantial differences in the regulation of tissue-specificgenes between rodents and humans [30], very few studies have extrap-olated rodent data and investigated the regulation of the FABP1 gene inthe human liver.
Here we present a comprehensive study of the regulation ofhuman FABP1 and demonstrate that FOXA1 and HNF4α contributeto the high expression of FABP1 in the liver through newly identifiedcognate elements. We have also confirmed the regulation of humanFABP1 by PPARα and uncovered C/EBPα as a dominant transcrip-tional repressor of FABP1 in human hepatocytes. We also show thatFABP1 is repressed in NAFLD and that this repression correlates withan altered expression of its transcription factors, thus suggesting thatthemechanisms of FABP1 regulation could have potential clinical impli-cations in NAFLD.
2. Materials and methods
2.1. Animals
To induce NAFLD, male C57BL/6J mice (8–10 weeks old) were fed aMCD diet for 5 weeks (cat. no. 960439, ICN, Aurora, OH, USA). Controlmice received the same MCD diet supplemented with DL-methionine(3 g/kg) and choline chloride (2 g/kg) (cat. no. 960441, ICN). Mice(five per group) were allowed food and water ad libitum up to5 weeks. At the end of the feeding experiment, mice were euthanizedand livers were excised. A part of the right posterior lobe was fixedin 10% formalin. The presence of steatohepatitis was proven histologi-cally in the MCD diet-fed mice. The remaining liver was snap frozen.
To examine the function of PPARα on liver FABP1 in vivo, mice feda MCD diet for 5 weeks received a daily gavage dose of either thePPARα agonist Wy-14,643 (50 mg/kg/day) or the carrier methylcellu-lose (0.1%) for 4 days. Twenty-four hours after the last dose, animalswere deeply anesthetized and liver and plasma were collected.
As an alternative NAFLDmodel we usedmice deficient in methionineadenosyltransferase 1A (MAT1A), which spontaneously developnonalcoholic steatohepatitis [31–33]. The 3- and 9-month-old maleMAT1A-KO mice and their wild-type (WT) C57BL/6 littermates wereobtained from the animal facility of CIC bioGUNE (Derio, Bizkaia,Spain). Mice were housed at 22 °C with a 12-hour light–dark cycleand allowed food (Teklad Global 18% Protein Rodent Diet 2018S,Madison, WI, USA) and water ad libitum. Animals were anesthetized(sodium pentobarbital, 60 mg/kg body weight, given intraperitoneally)and liver collected.
All animal studies were approved by the local Animal Ethics Com-mittees in accordancewith the guidelines of EuropeanResearch Councilfor animal care and use.
2.2. Human nonalcoholic fatty liver (NAFL) samples
NAFL (n=18) and non-steatotic liver (n=25) samples wereobtained from the Human Liver Bank of the Hospital La Fe-CIBERehd(SPAIN). This is a collection of donor livers which were primarilyassigned to liver transplantation but, due to failings in some of theinclusion criteria (e.g. NAFL) they were donated to research. Thisstudy was approved by the Research Ethics Committee at the HospitalLa Fe (Valencia, Spain), and family's informed consent was obtainedby the National Transplant Organization Coordination. The anthropo-metric and analytical characteristics of these liver donors have beendescribed elsewhere [34].
2.3. Isolation and culture of human hepatocytes and cell lines
Human liver samples (1–4 g) without steatosis (pathologist con-firmation) were obtained from cadaveric liver donors after family'sinformed consent. Hepatocytes were isolated using a two-stepperfusion technique [35]. The study was approved by the InstitutionalReview Board (Ethics Committee) at the Hospital La Fe (Valencia,Spain). None of the donors (4 men and 2 women aged between 17and 74, BMIb30) were regular consumers of alcohol or of otherdrugs (including lipid-lowering drugs), and were not suspected ofharboring any infectious disease. Isolated hepatocytes were seededon fibronectin-coated plastic dishes (3.5 μg/cm2) at a density of8×104 viable cells/cm2. Hepatocytes were cultured at 37 °C withHam's F-12/Williams' E medium (1:1) (Gibco BRL, Life Technologies,Madrid, Spain) supplemented as described elsewhere [35]. The medi-um was changed 1 h later to remove unattached hepatocytes. By24 h, the cells were shifted to serum-free hormone-supplementedmedium (100 nM dexamethasone and 10 nM insulin).
Human hepatoma HepG2 cells (ATCC HB-8065) were cultured inHam's F-12/Leibovitz L-15 (1:1, v/v) medium (Gibco BRL, Life Tech-nologies) and cervical cancer HeLa cells (ATCC CCL-2) were cultured
805C. Guzmán et al. / Biochimica et Biophysica Acta 1831 (2013) 803–818
in DMEM (Gibco BRL, Life Technologies), both supplemented with 7%fetal bovine serum and 2 mM L-glutamine. Cultures were routinelysupplemented with 50 U/ml penicillin and 50 μg/ml streptomycin.
The ligands to activate nuclear receptors were dissolved in DMSOand added to culture media at the following concentrations: 50 μMRifampicin (PXR), 500 nM 6-(4-chlorophenyl) imidazo[2,1-b] [1,3]-thiazole-5-carbaldehyde-O-(3,4-ichlorobenzyl)oxime (CITCO) (CAR),1 μM TO-901317 (LXRα), 1 μM 9-cis retinoic acid (RXRα) and 1 μMGW7647 (PPARα). Control cultures received 0.1% DMSO.
2.4. Plasmid constructs
Chimeric luciferase reporter constructs with different lengths of the5′ flanking region of the human FABP1 gene were obtained by cloningFABP1-5′ flanking sequences into the enhancerless, promoterlesspGL3-Basic vector (Promega Biotech Iberica, Madrid, Spain). The tran-scription start site was assigned according to the full-lengthFABP1 cDNA described by Lowe et al. [36]. Four DNA fragmentsspanning nucleotides +45 to −2183, −1205, −592 and −96 bpof the FABP1 5′ flanking regionwere PCR-cloned fromhuman genomicDNA (Roche Applied Science, Barcelona, Spain) using the Expand HighFidelity PCR System (Roche Applied Science). The PCR primers usedare described in Supplemental Table 1. The resulting amplicons weredigested with the restriction enzymes shown in the Supplemental Table1, and ligated into the pGL3-Basic vector previously digested with thesame enzymes. All the pGL3-B-FABP1 constructs were confirmed byrestriction digestion and double-strand DNA sequencing.
The −229 bp FABP1-5′ flanking sequence and the different mutantconstructs were synthesized by GenScript Technologies (Piscataway,NJ, USA). FABP1 5′-flanking sequences from +45 to −229 bp andfrom +45 to −592 bp with base-substitutions in the predicted DR1-C/EBP composite site, FOXA1 binding sites and in the DR1 sequence homol-ogous to the mouse PPARα response element [23] (see Fig. 7A) werecloned in the pUC57 plasmid and excised by restriction enzymes to besubcloned in pGL3-Basic.
2.5. Adenoviral vectors
The recombinant adenoviruses encoding CAR, PXR, C/EBPα, C/EBPβ,FOXA1, HNF4α and PGC1α were developed in our laboratory asdescribed elsewhere: CAR [37,38], PXR [35], C/EBPα [39], C/EBPβ[40,41], FOXA1 [34], HNF4α [42], and PGC1α [42].
The recombinant adenoviruses encoding LXRα, RXRα and PPARαwere constructed as follows:Human LXRα andRXRα cDNAswere kindlyprovided by Dr. D. Mangelsdorf (UT Southwestern Medical Center,Dallas, Texas). The coding sequences were released from the plasmidpCMX with KpnI-BamHI (LXRα) or with EcoRI (RXRα), and subclonedin the adenoviral shuttle vector pAC/CMVpLpA [43] pre-digested withthe same restriction enzymes.
The cDNA for human PPARα was PCR amplified from a human livercDNA with primers PPARα-151: CTC GGC GGT ACC ACC AGC ACC ATC(forward) and PPARα-1564: AAC TCA GGA TCC GTC CCT GTA GATCTC CT (reverse). The amplicon was digested with KpnI-BamHI andsubcloned in the adenoviral vector pAC/CMVpLpA pre-digested withthe same restriction enzymes. The right sequence in the pACC/CMV-PPARα plasmid was confirmed by double-strand DNA sequencing.
The shuttle plasmids for LXRα, RXRα or PPARα, and the pJM17containing the dl309 adenovirus-5 genome, were co-transfected into293 with FuGENE HD Transfection Reagent (Roche Applied Sciences).The homologous recombination between these two plasmids resultedin replication defective viruses (Ad-LXRα, Ad-RXRα and Ad-PPARα),which were plaque-cloned, amplified and purified, with the VivapureAdenoPACKTM100Kit fromSartorius (Goettingen, Germany), into stocksof 1.7–2.5×107 plaque-forming units/μl.
Cultured cells were infected with recombinant adenovirusesencoding transcription factors or with an insertless adenoviral vector
(Ad-CONT) for 24 h at a multiplicity of infection (MOI) ranging from 2to 45 plaque-forming units/cell. Thereafter, cells were washed andfresh medium was added. Ligands for nuclear receptors were added at24 h post-infection and at 48 h post-infection cells were harvestedand analyzed. Adenoviral vectors achieved nearly 100% transfectionefficiency as assessed by Ad-LacZ infection and β-galactosidase staining[44].
2.6. Transfection and reporter gene assays
Plasmid DNAs were purified with PureYield™ Plasmid MaxiprepSystem kit columns (Promega Biotech Iberica) and quantified byNanoDrop (Thermo Fisher Scientific, Madrid, Spain). The day beforeinfection, cells were plated in 12-well plates to reach 50% confluence.Cells were infected with recombinant adenoviruses and transfectedthe next day with 0.132 pmoles of firefly luciferase expressionconstructs (pGL3-Basic and pGL3-FABP1-LUC) and X-tremeGENE HPDNA Transfection Reagent (Roche Applied Science). In parallel,70 ng of pRL-SV40 (a plasmid expressing Renilla reniformis luciferase)was cotransfected to correct the variations in transfection efficiency.Forty-eight hour post-transfection luciferase activities were mea-sured using the Dual-Luciferase® Reporter Kit (Promega BiotechIberica). Ligands for nuclear receptors were present in the culturemedium for the last 24 h.
2.7. RNA interference experiments
We knocked-down HNF4α expression in HepG2 cells with lentiviralshort hairpin RNAs (shRNAs). The lentiviral shRNA Target Gene Set,containing four different pLKO.1 clones against human HNF4α, waspurchased from OPEN Biosystems (RHS4533-EG3172, Thermo Scientif-ic). As a negative control we used the lentiviral vector pLKO.1-shRNA-LUC. The shRNA vectors were co-transfected with the packagingplasmid psPAX2 (pCMV-Gag-Pol, the Broad Institute, Cambridge, MA)and the envelope plasmid pMDG.2 (pCMV-VSV-G-poly-A, the BroadInstitute) into 293 T cells using the calcium phosphate method. Forty-eight hours post-transfection, medium containing the lentivirus washarvested and filtered, and was used to infect HepG2 cells. Afterlentiviral shRNA infection, cells were cultured for 72 h and total RNAand protein were extracted and analyzed.
2.8. Reverse transcription and real-time quantitative PCR
Total RNA fromhuman cultured cells or fromhuman liver tissuewasextracted with the RNeasy Mini Kit (Qiagen, Madrid, Spain). Total RNA(1 μg) was reverse transcribed as described [35] and real-time quanti-fied using SYBRGreen IMaster (RocheApplied Sciences) and the appro-priate primers (Supplemental Table 2) in a LightCycler 480 Instrument.In parallel, the housekeeping porphobilinogen deaminase (PBGD) wasanalyzed for normalization. Relative mRNA levels were calculatedwith the LightCycler Relative Quantification Analysis software. Briefly,the efficiency of each PCR reactionwas estimated from a serially dilutedhuman liver cDNA curve. This curve allows to determine the PCRefficiency of each pair of primers. Based on these PCR efficiencies andthe cycle thresholds (Ct), the software calculates the relative concentra-tion of target and reference (PBGD) cDNAs and their ratio.
Gene expression analysis of mouse liver genes was performed byRT-PCR as follows: Total RNA was obtained by using a Trizol reagent(Life Technologies, Madrid, Spain). First-standard cDNA was synthesizedusing High-Capacity cDNA Archive Kit (Applied Biosystems, Weiterstadt,Germany). For gene expression assays, cDNAs were amplified usingmultiplex real-time PCR reactions on a StepOne Plus (AppliedBiosystems) [45]. TaqMan primers and probes were derived fromthe commercially available TaqMan® Gene Expression Assays(Applied Biosystems) (Supplemental Table 2). Relative changes ingene expression levels were determined using the 2−ΔΔCt method.
806 C. Guzmán et al. / Biochimica et Biophysica Acta 1831 (2013) 803–818
The housekeeping, glyceraldehyde-3-phosphate dehydrogenase(GAPDH) was analyzed for normalization. PCR efficiency was de-termined by TaqMan analysis on a standard curve for targets andcontrol (GAPDH), which were highly similar.
2.9. Cell fractionation and immunoblotting analysis
Cultured cells or liver tissue were homogenized with 140 mMNaCl, 15 mM EDTA, 10% glycerol, 20 mM Tris–HCl (pH=7.5) and aprotease inhibitor cocktail. The mixture was centrifuged for 30 minat 13,000 g and 4 °C and the supernatant was kept as “whole proteinextracts”. Nuclear and cytoplasmic fractionation of HepG2 cells wasalso conducted using the NE-PER Nuclear and Cytoplasmic ExtractionReagents kit (Thermo Fisher Scientific, Madrid, Spain) according tothe manufacturer's protocol. Protein concentration was determinedby a Bradford assay.
Western blotting analysis of whole protein extracts or cytoplasmic/nuclear fractions was performed as described [46], using rabbit poly-clonal antibodies against FABP1 (Abcam, Cambridge, UK) or β-actin(Sigma-Aldrich, Madrid, Spain). Bound primary antibody was detectedwith horseradish peroxidase-conjugated anti-rabbit antibody (DAKO,Glostrup, Denmark), and blots were developed using an enhancedchemiluminescence detection system (ECL kit, Amersham Pharmacia,Uppsala, Sweden). The intensity of specific bands was quantified withan imaging densitometer (Scion Image, Maryland, MA).
2.10. Immunofluorescence analysis
Immunofluorescence analysis was performed as described [47].Coverslips were incubated with rabbit anti-FABP1 antibody (Abcam)at 4 °C overnight. Thereafter, the secondary antibody donkey anti-rabbit conjugated with FITC (Jackson ImmunoResearch, Baltimore, PA,USA)was applied. Nuclei were stainedwith DAPI (blue). After washing,the coverslips were mounted on DakoCytomation Fluorescent Mount-ing Medium (DAKO). The preparations were analyzed with an invertedfluorescence microscope (Nikon Eclipse Ti-U).
2.11. Chromatin immunoprecipitation assay
Cells were infected with adenoviral vectors and incubated thenext day with nuclear receptor ligands or vehicle for another 24-hperiod. Then, cells were treated with 1% formaldehyde in PBS bufferby gentle agitation for 10 min at room temperature to crosslinkproteins to DNA. Next, cells were washed, resuspended in lysis bufferand sonicated on ice for 8×15 s steps at a 20% output in a BransonSonicator. Sonicated samples were centrifuged to clear supernatants.DNA content was quantified by picogreen (Life Technologies) andproperly diluted to maintain an equivalent amount of DNA in all thesamples (input DNA). For the immunoprecipitation of protein-DNAcomplexes, 4 μg of specific antibodies (anti-C/EBPα sc-61, anti-FOXA1sc-101058 and anti-PPARα sc-9000, Santa Cruz Biotechnology) wereadded. Samples were incubated overnight at 4 °C on a 360° rotator(antibody-bound DNA fraction). For each cell preparation, an additionalmock immunoprecipitationwith rabbit preimmune IgG (sc-2027, SantaCruz Biotechnology) was performed in parallel (background DNAfraction). Immunocomplexes were affinity absorbed with 60 μl ofprotein G agarose/Salmon Sperm DNA (Millipore, Billerica, MA, USA)(pre-washed with lysis buffer for 1 h at 4 °C by gentle rotation), andcollected by centrifugation (1000 rpm, 1 min). The antibody-boundand background DNA fractions were washed as described [38].Cross-links were reversed by adding 100 μl of 10% Chelex (Bio-RadLaboratories) and boiling for 10 min. The Chelex/protein G beadsuspensions were incubated with proteinase K (20 mg/ml) for30 min at 55 °C while shaking, followed by another 10 min boiling. Sus-pensions were centrifuged and supernatants were collected. The eluateswere used directly (input 1/10, bound 1/2) as a template for Q-PCR
with a LightCycler 480 instrument. Amplification was real-time moni-tored, stopped in the exponential phase of amplification and analyzedby agarose gel electrophoresis. Amplifications of the FABP1 gene se-quences (5′-flanking) among the pull of DNA were performed withspecific primers flanking these regions (Supplemental Table 3).
2.12. Statistical analysis
Differences among groups were analyzed by one-way ANOVA andTukey's pairwise comparisons. Differences between groups of datawere evaluated by the Student's t-test or the nonparametric Mann–Whitney test, as indicated. Positive and negative interaction coefficientsfrom a fitted linear model were used to determine statistically signifi-cant effects when two or more factors were combined. A p value ofless than 0.05 was considered to be statistically significant. Values areexpressed as mean±SEM.
3. Results
3.1. The human FABP1 gene is induced by PPARα and FOXA1, and repressedby C/EBPα
Human hepatoma HepG2 cells were transduced with adenoviralvectors encoding 10 transcription factors. The selection of these factorswas based on two criteria: 1) a proven role in transcription regulation ofenergy/metabolism genes (particularly fatty acid metabolism genes)and 2) an important expression level in the liver. Based on these criteria,we selected four lipid-sensor nuclear receptors: PPARα, LXRα, PXR andCAR; their heterodimeric partner: RXRα; one liver-enriched nuclearreceptor: HNF4α; one nuclear receptor coactivator: PGC1α; and threeliver-enriched transcription factors: C/EBPα, C/EBPβ and FOXA1/HNF3α[48–50].
Expression analysis of the selected transcription factors revealedthat all of them but HNF4α had a decreased mRNA expression inHepG2 when compared with human hepatocytes (Supplemental Fig. 1).All adenoviral vectors caused a significant increase in the mRNA of theencoded transcription factors, reaching levels on average 20-fold higherthan in cultured hepatocytes (Supplemental Fig. 1) but within the widerange of expression found in human liver.
Expression analysis of human FABP1 under these experimentalconditions revealed that the native FABP1 gene was significantlyinduced by PPARα and FOXA1, whereas C/EBPα caused a markedrepression (Fig. 1A).
We studied in more detail the response of FABP1 to these three fac-tors and found that the induction of FABP1 mRNA by Ad-PPARα andAd-FOXA1, and its repression by Ad-C/EBPα, were dose-dependent(Fig. 1B). Ad-C/EBPαwas able to down-regulate FABP1 to 13% of controlat 25 MOI.
Next we investigated the combined effect of these three factors. Theinduction of FABP1 by the combination of FOXA1 and PPARα (3.8-fold)was significantly higher than the induction by each factor individually.On the other hand, C/EBPα was able to blunt the positive effects ofPPARα and FOXA1 (Fig. 1C). The coefficients of a fitted linear modelwith up to third-order interactions confirmed that C/EBPα had adominant negative effect over the positive effects of FOXA1 andPPARα (C/EBPα: FOXA1, p=0.00472; C/EBPα: PPARα, p=0.000015).However, the interaction coefficient of FOXA1 and PPARα evidencedthat their combined effect on FABP1 gene transcription was just addi-tive (FOXA1: PPARα, p=0.39).
C/EBPα is a well-characterized transcription activator, but occasion-ally it has also demonstrated negative effects, most of them associatedto an alternative translational initiation isoform of 30 kDa (p30). Weinvestigated whether our Ad-C/EBPα vector was expressing significantamounts of p30. Immunoblotting analysis, however, demonstratedthat only p42 was expressed after Ad-C/EBPα infection in HepG2 cells(data not shown).
Fig 1. Response of human FABP1 gene to multiple transcription factors in HepG2 cells. A, Effect of 10 adenoviral vectors encoding transcriptional regulators on FABP1 mRNA ex-pression. HepG2 cells were infected with selected effective doses of the different adenoviruses (10–35 MOI) for 48 h. The ligands to activate nuclear receptors (see the Materialsand methods section) were present in the cultures during the last 24 h. Control cells were infected with an insertless adenoviral vector (Ad-CONT, 30 MOI). Determination of FABP1mRNA level was performed as described in the Materials and methods section. The results for 4 independent experiments are presented as mean±SEM. ⁎pb0.05, ⁎⁎pb0.01,⁎⁎⁎pb0.001 vs. Ad-CONT. B, Dose–response of human FABP1 mRNA to Ad-C/EBPα, Ad-FOXA1 and Ad-PPARα. Adenoviral vectors were added for 48 h. The ligand for PPARα,1 μM GW7647, was present during the last 24 h. Data represent mean±SEM from 3 independent experiments. ⁎pb0.05, ⁎⁎pb0.01, ⁎⁎⁎pb0.001 vs. 0 MOI. C, Response of FABP1mRNA to combinations of C/EBPα (20 MOI), FOXA1 (12 MOI) and PPARα (20 MOI). Data represent mean±SEM from 3 to 4 independent experiments. ⁎⁎⁎pb0.001 vs. conditionwithout Ad-C/EBPα; #pb0.001 vs. Ad-FOXA1 alone; ¶pb0.05 vs. Ad-PPARα alone.
807C. Guzmán et al. / Biochimica et Biophysica Acta 1831 (2013) 803–818
In order to confirm the negative effect of p42 C/EBPα on FABP1we transduced cultured human hepatocytes from different liverdonors with Ad-C/EBPα. The lack of effect of an insert-less vector(Ad-CONT) in parallel cultures ruled out any adenovirus-dependentnegative effects. Results undoubtedly demonstrated that C/EBPαrepresses FABP1 to a large extent in humanhepatocytes (26%of controlat 10–25 MOI) (Fig. 2A). Cell transduction with selected doses ofAd-FOXA1 and Ad-PPARα also confirmed the positive effects of thesefactors on FABP1 in human hepatocytes (Fig. 2B).
3.2. FABP1 protein expression and cellular localization are modulated byC/EBPα, FOXA1 and PPARα
FABP1 protein in HepG2 was first analyzed by immunoblotting.As expected, FABP1 protein was repressed by C/EBPα and inducedby FOXA1 and PPARα (Fig. 3A and B). There was a good correlationbetween mRNA and protein levels. However, the 3-fold increase inFABP1 mRNA induced by Ad-PPARα resulted only in a 2-fold in-crease in FABP1 protein.
Fig. 2. FABP1 expression is modulated by C/EBPα, FOXA1 and PPARα in human hepato-cytes. Twenty-four hour cultured hepatocytes were infected with increasing doses ofAd-C/EBPα or Ad-CONT (A) or with 5 MOI Ad-FOXA1, 3 MOI Ad-PPARα or 5 MOIAd-CONT (B) and 48 h later the mRNA level of FABP1 was determined by quantitativeRT-PCR. The ligand for PPARα, 1 μM GW7647, or 0.1% DMSO was present during thelast 24 h. Data represent mean±SEM from 3 to 4 independent experiments. ⁎pb0.05,⁎⁎pb0.01 vs. Ad-CONT; #pb0.05, ##pb0.01 vs. DMSO.
Fig. 3. FABP1 protein is induced by FOXA1 and PPARα; and repressed by C/EBPα. HepG2cells were infected with adenoviral vectors as indicated (Ad-C/EBPα: 20 MOI, Ad-CONT:20 MOI, Ad-FOXA1: 12 MOI, or Ad-PPARα 20 MOI+1 μM GW7647). Forty-eight hourpost-infection the level of FABP1 protein was determined by immunoblotting (A). Thedensity of the specific bands was quantified with an imaging densitometer (B). Datarepresent mean±SEM from 5 independent experiments, expressed as % of Ad-CONT.⁎⁎pb0.01, ⁎⁎⁎pb0.001 vs. Ad-CONT.
808 C. Guzmán et al. / Biochimica et Biophysica Acta 1831 (2013) 803–818
Immunofluorescence microscopy confirmed the repression ofFABP1 protein by C/EBPα and its induction by FOXA1 and PPARα(Fig. 4A). Moreover, microphotographs suggested that FABP1 proteinwas primarily localized in the nuclear/perinuclear region in controlHepG2 cells, as well as in cells transduced with Ad-C/EBPα. However,FOXA1 and particularly PPARα induced re-distribution of FABP1 proteinand increased cytoplasmic expression (Fig. 4A).
To confirm this possibility we determined FABP1 protein levels innuclear and cytoplasmic extracts by immunoblotting and found againthat FOXA1 and PPARα favor cytoplasmic expression of FABP1 (Fig. 4Band Supplemental Fig. 2). After FOXA1 and PPARα the cytoplasmic/nuclear FABP1 expression ratios were between 4.3 and 5.0, whereas incontrol or Ad-C/EBPα cells cytoplasmic/nuclear ratios were between1.8 and 3.7.
Taken together, our results confirm that C/EBPα is an effective re-pressor of both basal and induced FABP1 expression, whereas FOXA1and PPARα trigger an increase in FABP1 levels and enhance its cytoplas-mic localization, where it regulates hepatic FA trafficking and partition.
3.3. Analysis of FABP1 5′-flanking region and identification of C/EBPα,PPARα and FOXA1 responsive regions
To delineate the cis-acting elements of the FABP1 gene, which aredirectly responsible for the transcription regulation by C/EBPα, FOXA1and PPARα, several FABP1-LUC chimeric constructs were generated(from+45 to−96,−229,−592,−1205 and−2183 bp) and reporterassays were performed.
The largest difference in basal LUC activity in HepG2 cells was foundbetween the −96 and the −229 bp constructs (142-fold; Fig. 5),demonstrating that most of the constitutive transcription activity ofthe FABP1 gene lays within this 133 bp fragment. An HNF1α functionalresponse element has been reported in this 133 bp fragment in the
mouse [26]. Nevertheless, other transcription factors can also be actingthrough this fragment to sustain the high constitutive FABP1 activity.
We next investigated the effect of each transcription factor onthe reporter activity of this series of constructs. Transactivationby C/EBPα had a positive effect on the smallest −96 FABP1-LUCconstruct, but an opposite negative effect of C/EBPα was observedon all the longer constructs (Fig. 6A), thus suggesting that therepressive effect of C/EBPα also locates in the 133 bp fragment between−96 and −229 bp. FOXA1 only transactivated the −592, −1250and −2183 constructs (Fig. 6A), pointing to the 363 bp fragment be-tween −229 and −592 bp as the putative target region of FOXA1on the human FABP1 promoter. Finally, PPARα transactivated all theFABP1-LUC constructs (Fig. 6A), thus indicating that the PPARα re-sponse element is located within the first 96 bp of the FABP1 promoter,in agreement with previous reports showing a functional PPARαresponse element in the proximal promoter (−55 bp) of the murineFABP1 gene [24]. Altogether our results point to three different regionsin the FABP1 promoter, the more proximal and distal would mediatethe activating effects by PPARα and FOXA1, respectively, whereas themiddle one, where the high constitutive activity of the gene is located,would mediate the repressive effect by C/EBPα.
Finally we tested the combined effects of these three factorson those constructs containing the three responsive regions (i.e. −592,−1205 and −2183 FABP1-LUC). We found that, in agreement withFABP1 mRNA and protein results, FOXA1 and PPARα cause additivetransactivation, whereas C/EBPα is able to blunt the increase in reporteractivity induced by these two factors (Fig. 6B).
3.4. Search for C/EBPα, PPARα and FOXA1 responsive elements in the humanFABP1 promoter and functional analysis by site-specific mutagenesis
We used the MatInspector 8.0 Software [51] and defined frequencymatrices from PAZAR database [52] to analyze the new identifiedregions for potential C/EBPα, PPARα and FOXA1 binding sites. A high
Fig. 4. PPARα and FOXA1 induce cytoplasmic FABP1 protein. HepG2 cells were infected with adenoviral vectors as indicated in the legend to Fig. 3, and 48 h later cells were fixed,permeabilized and processed for immunostaining using an anti-FABP1 antibody and a secondary antibody conjugated with FITC (green). Nuclei were stained with DAPI (blue). Themerged image with green and blue fluorescence is also shown (lower panels). Photographs are representative results of six independent experiments (A). Parallel cultures wereseparated into subcellular fractions as described in the Materials and methods section. FABP1 protein was analyzed in cytoplasmic and nuclear extracts by immunoblotting andthe density of the specific bands was quantified with an imaging densitometer. Data represent mean±SEM from 3 independent experiments, expressed as % of cytoplasmicFABP1 in Ad-CONT cells (B).
809C. Guzmán et al. / Biochimica et Biophysica Acta 1831 (2013) 803–818
density cluster of consensus FOXA1 response elementswas identified inthe 363 bp fragment between−229 and−592 bp (4 sites with perfectmatch and 30 sites with 1 mismatch; Supplemental Fig. 3). The highconcentration of potential FOXA1 sites was restricted to this 363 bp
Fig. 5. Basal reporter activity of constructs with FABP1 5′-flanking sequences in human hepainto the firefly luciferase pGL3-basic reporter vector. HepG2 cells were transfected with thepost-transfection, cells were lysed, and both firefly and Renilla reniformis luciferase activiexpressed as firefly/Renilla luciferase activity ratios. ⁎⁎⁎pb0.001 vs. the −96 FABP1-LUC co
region. Analysis of the first 1205 bp revealed only 4 FOXA1 sites outsidethis region against 34 sites inside (Supplemental Fig. 3). In the 133 bpfragment, between −96 and −229 bp, with high constitutive activity,where C/EBPα is acting, we found an imperfect C/EBP binding site
toma cells. Sequential deletion fragments of the FABP1 5′-flanking region were clonedFABP1 reporter constructs and the normalization plasmid pRL-SV40. Forty-eight hoursties were determined. Bars represent mean±SEM of 4–6 independent experiments,nstruct.
Fig. 6. Transactivation of FABP1 reporter constructs by C/EBPα, FOXA1 and PPARα. HepG2 cells were infected with the three adenoviral vectors individually (A) or combined (B)(Ad-C/EBPα: 20 MOI, Ad-CONT: 20 MOI, Ad-FOXA1: 12 MOI, or Ad-PPARα 20 MOI). Next day, cells were transfected with the different FABP1 promoter-reporter vectors and thenormalization plasmid pRL-SV40. Twenty-four hours post-transfection cells were exposed to the PPARα ligand GW7647 (1 μM) or vehicle (DMSO). Forty-eight hourspost-transfection, cells were lysed, and both firefly and Renilla reniformis luciferase activities were determined. Bars represent mean±SEM of 3–5 independent experiments,expressed as fold-change vs Ad-CONT. (A) ⁎⁎⁎pb0.001 vs. Ad-CONT (ANOVA and Tukey's test); ###pb0.001 vs. Ad-CONT (Student's t-test). (B) ⁎⁎pb0.01, ⁎⁎⁎pb0.001 vs. conditionwithout Ad-C/EBPα; #pb0.001 vs. Ad-FOXA1 alone; ¶pb0.001 vs. Ad-PPARα alone.
810 C. Guzmán et al. / Biochimica et Biophysica Acta 1831 (2013) 803–818
which overlaps with a new DR1 response element (Fig. 7A). Finally, inthe −96 bp proximal promoter we found a PPARα response element,which almost matches to the one previously described in the mousepromoter [24] (Fig. 7A).
We generated a series of reporter constructs with site-specificmutations to determine the relevance of these putative DR1-C/EBPand PPARα binding sites, as well as to assess the contribution ofperfect-match FOXA1 binding elements (Fig. 7A). Analysis of the basalreporter activity of the differentmutated constructs showed that muta-tion of the new composite DR1-C/EBP element caused a 95% decrease inbasal promoter activity (Fig. 7B), suggesting that a factor binding to thisDR1 response element (along with the already described, adjacentHNF1α) is involved in maintaining the high constitutive activity of theFABP1 gene. Mutation of the PPARα element also reduced substantiallythe basal FABP1 activity (84% decrease), whereas independent muta-tion of FOXA1 elements caused only partial inhibition (17–29% decreasevs. wild-type promoter activity; Fig. 7B).
Next, we transfected cells with the transcription factors to determinedifferences in activity between thewild-type and themutated promoters(Fig. 7C). Mutation of the PPARα element at−59 bp completely blockedthe induction by PPARα, whereas mutation of the DR1-C/EBP siteabolished repression by C/EBPα. This suggests that binding of C/EBPα tothe wild-type DR1-C/EBP element could displace an already bound factorwith higher activity and cause gene repression. Finally, mutation of theFOXA1 site 2 (mut 2) or double mutation of the sites 3+4 (mut 3) ledto a lower activation by FOXA1 (14–26% decrease), but none of themuta-tions completely suppressed the induction of reporter activity. Thisreinforces the idea that, in this region, there is a cluster with many alter-native FOXA1 binding sites and that mutation of only some of these sitescauses only partial inhibition.
3.5. FOXA1 and FOXA2 activate FABP1 through the same promoter regionand do not show cross-inhibition
FOXA1 and FOXA2 have high homology in the DNA binding domainand share recognition sequences on DNA [53]. Consequently, they cancompete for the same binding sites and may exhibit cross-inhibitoryeffects [54]. Moreover, transfection of FOXA1 in human hepatocytesand HepG2 cells causes FOXA2 down-regulation, and this could alsoindirectly affect FABP1 [34].
To investigate the potential relevance of the FOXA1/FOXA2 cross-talk on FABP1 we analyzed the response of the FABP1 promoter toFOXA1 and FOXA2 expression vectors, and found that both factorsonly transactivated the constructs containing the first −592 bp(Supplemental Fig. 4A). This confirms that the 363 bp region between−229 and −592 bp is the FABP1 region with the most significantresponse to FOXA factors. Next we analyzed if FOXA1 and FOXA2show cross-inhibitory effects on the −592 bp FABP1 construct andfound that co-transfection of FOXA1 and FOXA2 do not inhibit theresponse of the FABP1 promoter; rather, it had a statistically significantpositive effect (Supplemental Fig. 4B). These results demonstrate thatFOXA1 and FOXA2 have similar efficiency in the transactivation ofFABP1 and no competition and cross-inhibition is observed, in conso-nance with the abundant FOXA response elements in the FABP1 pro-moter region.
3.6. C/EBPα, FOXA1 and PPARα bind to the identified responsive regionsof the human FABP1 gene
Chromatin immunoprecipitation (ChIP) assays were performed toassess binding of the three transcription factors to the native FABP1
811C. Guzmán et al. / Biochimica et Biophysica Acta 1831 (2013) 803–818
5′-flanking region. Three DNA sequences responsive to FOXA1 (PCR-1),to C/EBPα (PCR-2) and to PPARα (PCR-3) were amplified afterimmunoprecipitation (Fig. 8A). Infection of HepG2 cells withAd-FOXA1 and Ad-PPARα caused a noticeable increase in the amountof immunoprecipitated FABP1 DNA by anti-FOXA1 and anti-PPARαantibodies, respectively (Fig. 8B upper panel), suggesting actual bindingof these transcription factors in vivo. Transfection of HepG2 cells withAd-C/EBPα caused also an increase in the amount of immunoprecipitatedDNA by anti-C/EBPα antibody but this increasewas lessmarked (Fig. 8Bupper panel). An exonic region in the RPLP0 (ribosomal protein, large,P0) gene devoid of FOXA1, C/EBPα and PPARα consensus bindingsites, and expected not to be enriched in the antibody-bound DNA frac-tions, was PCR-amplified as a negative control for ChIP assay (Fig. 8Blower panel).
3.7. HNF4α contributes to the high constitutive transcription of theFABP1 gen through the newly identified DR1-C/EBP element at −123 bp
One remaining question is: what transcription factor is replacedby C/EBPα in the composite DR1-C/EBP element? A nuclear receptorbinding to this DR1 site has to confer a high constitutive transcrip-tional activity to the FABP1 gene in the liver. We therefore thoughton HNF4α as a likely candidate.
To check this possibilitywe transfected thedifferent FABP1 promoterconstructs into the non-hepatic human cervical carcinoma HeLa cellline, which lacks HNF4α expression. We found that the basal reporteractivity of these constructs in HeLa cells was on average 104-foldlower than in HepG2 cells (Fig. 9A). More importantly, there were nosignificant differences among the activity of the different constructs inHeLa cells (Fig. 9A), which is in marked opposition with HepG2 cells,where an increase of 142-fold between the −96 and the −229 bpconstructs is observed (Fig. 5). The lack of response of this 133 bpfragment, containing the new DR1 element, in HeLa cells points toHNF4α as the likely factor competing with C/EBPα for the DR1-C/EBPelement.
To give support to this idea, we transfected the different FABP1constructs together withHNF4α in HeLa cells and found that the largestdifference in response to HNF4α was between the −96 (5-foldincrease) and the −229 bp fragment (70-fold increase), where thenew DR1 element is located (Fig. 9B). We also observed another increasein response to HNF4α in the −1205 bp fragment, which could suggestadditional HNF4α binding elements in the far distal FABP1 promoter.In addition, mutation of the DR1 element at −123 bp abolished most ofthe response associated to HNF4α (Fig. 9B).
Finally, we knocked-down HNF4α expression in HepG2 cells usinglentiviral shRNA and analyzed its effect on FABP1 expression. Cell trans-duction with four different HNF4α shRNAs caused a marked drop inHNF4α mRNA (42–82% decrease) and protein (Fig. 9C). HepG2 cellswith reduced HNF4α expression showed a concomitant reduction inFABP1 mRNA (47–81% decrease) (Fig. 9C). Altogether, our resultsdemonstrate that HNF4α is a key factor sustaining constitutive FABP1gene expression in the human liver.
3.8. FABP1 expression is repressed in NAFLD and correlates withdecreased levels of FOXA1 and PPARα
As FABP1 plays a key role in the physiological regulation of FAmetabolism in the liver it is conceivable that FABP1 expression couldbe altered in liver metabolic disorders such as NAFLD. To investigatethis possibility we studied FABP1 expression in two mouse modelsof NAFLD: mice fed a MCD diet and mice deficient in MAT1A.
MCD induces steatohepatitis rapidly. Histological analyses showhepatic inflammation after 3 days, severe macrovesicular steatosis by1 to 2 weeks and necroinflammation after 2 weeks, followed by fibrosis[55]. FABP1 mRNA and protein progressively decreased in mouse liveralong with the MCD diet up to a 25% of control level by 5 weeks
(Fig. 10A & B). We observed a similar behavior for FOXA1 and PPARα,the two FABP1 activators. Regarding the FABP1 repressor C/EBPα, wefound induced expression during the first 2 weeks of MCD diet, butthis trend reversed, and repressed C/EBPαmRNA levels were observedby 5 weeks (Fig. 10A). Data suggested that the repression of FABP1 inNAFLD could be due to the down-regulation of its transcription activators(FOXA1 and PPARα) and/or the up-regulation of its repressor C/EBPα(during the first 2 weeks).
To rule out that the observed effects of theMCDdietwere not due toa general repressive response caused by the severity of the disease, wedetermined the expression of other gene markers that should notbe down-regulated in this model: TNFα, iNOs, LXRα and fatty acidsynthase (FAS) ([56] & [S. Sanchez-Campos personal communication]).Results depicted in the Supplemental Fig. 5 demonstrate that thesegenes are induced as expected, thus indicating that in our experimentalconditions theMCDdiet led to the expected phenotypewithout causinggeneral non-specific gene repression.
To prove a causal link between FABP1 repression and transcriptionfactor deregulation in MCD-diet mice we attempted to rescue the re-pressed FABP1 by activating PPARα with its agonist Wy-14,643. Asshown in Fig. 10C, PPARα activation completely restored FABP1 mRNAto control level, indicating that PPARα repression contributes to FABP1down-regulation in NAFLD. Further experiments involving injection ofAd-FOXA1 and Ad-C/EBPα are needed to unequivocally prove thatFOXA1 repression and C/EBPα induction have a causal relationship withFABP1 down-regulation in NAFLD.
We also used MAT1A deficient mice as an alternative modelof NAFLD. These mice have reduced hepatic S-adenosylmethioninecontent and hyperplasia and spontaneously develop macrovesicularsteatosis and nonalcoholic steatohepatitis [31–33]. FABP1 mRNA andprotein were again significantly decreased in NAFL (Fig. 10D & E). How-ever, in these 3- and 9-month old MAT1A deficient mice only FOXA1followed a similar trend, whereas PPARα and C/EBPα did not demon-strate substantial alterations (Fig. 10D).
Finally, we investigated the regulation of FABP1 in human NAFLD.The expression of FABP1 was significantly lower in human NAFLsfrom a liver bank of non-obese liver donors (BMI 28.3±0.6,mean±SEM, n=43). FOXA1 and PPARα mRNAs were also signifi-cantly decreased, whereas C/EBPα showed trend toward inductionin steatotic livers (Fig. 11).
Overall, our results demonstrate that FABP1 is repressed in NAFLD,and suggest that the repression of FABP1 in NAFLD is associatedwith transcription factor deregulation, mainly FOXA1 and PPARα, butC/EBPα likely as well.
4. Discussion
We have accomplished a comprehensive study of FABP1 gene regula-tion in human liver cells. Most of the previous work on FABP1 regulationhas focused on the rat or the mouse FABP1 gene [26,27,29]. However,marked differences between rodents and humans in tissue-specificgene regulation [30,57] point to the need to investigate the regulationof human genes. Indeed, our results have revealed substantial differencesbetween the regulation of rodent and human FABP1 that are discussedbelow.
One of our most interesting findings is the repression of humanFABP1 by C/EBPα. However, previous work by Staloch et al. [28] dem-onstrated activation of rodent FABP1 by C/EBPα. Several experimentaldifferences could explain this apparent discrepancy. In the study byStaloch et al. [28] a C/EBPα binding site was identified at −78 bp ofthe FABP1 promoter. We have also observed activation by C/EBPαwhen our construct of −96 bp was analyzed, but this effect wascompletely reversed when longer FABP1 constructs were assayed(Fig. 6A). Secondly, the rat FABP1 gene was studied in intestinalCaCo-2 cells, and the−78 bp C/EBPα site was confirmed to be requiredfor FABP1 expression in murine small intestinal crypt cells but it was
812 C. Guzmán et al. / Biochimica et Biophysica Acta 1831 (2013) 803–818
Fig. 8. ChIP assays demonstrate binding of FOXA1, C/EBPα and PPARα to their responsive regions in the FABP1 promoter. A, Schematic diagram showing the positions of the threePCR primer sets designed to assess FOXA1, C/EBPα and PPARα binding to the native FABP1 5′-flanking region. PCR-1 encompasses the cluster of FOXA1 elements, PCR-2 covers thecomposite DR1-C/EBP element at −123 bp and PCR-3 the conserved PPARα DR1 element at −59 bp. B, formaldehyde cross-linked chromatin from adenovirus-infected HepG2cells was incubated with antibodies against FOXA1, C/EBPα and PPARα or with preimmune IgG; and quantitative PCR was performed on immunoprecipitated, purified DNA. Ali-quots (10 μl) of PCR amplifications from a representative experiment were subjected to electrophoresis on 2% agarose gel (upper panels). An exonic region of the RPLP0 genwas amplified as a negative control (lower panels). M, 100-bp DNA ladder.
813C. Guzmán et al. / Biochimica et Biophysica Acta 1831 (2013) 803–818
not functional in intestinal villus cells [28]. However, we have demon-strated the repressive effect of C/EBPα in human liver HepG2 cells andhepatocytes (Figs. 1 & 2). These results likely indicate that the FABP1gene is differentially regulated in different species, tissues and celltypes depending on the different repertoire of transcription factorsand their interactions.
Our data suggest that C/EBPα binds to an overlapped DR1-C/EBPcomposite element, where HNF4α is constitutively activating FABP1transcription. Activation of rat FABP1 by HNF4α has been previouslyreported [29]. In that study the identified HNF4α responsive element(mapped at −55 bp) 100% overlapped with the well characterizedPPARα element in the proximal FABP1 promoter [29].We have also ob-served a slight activation of the short −96 bp construct by HNF4α inHeLa cells (Fig. 9B), but we have identified a newDR1-C/EBP compositeelement at −123 bp which is required for the largest induction ofFABP1 by Ad-HNF4α in HeLa cells (Fig. 9B) and for the high constitutiveactivity of the promoter in HepG2 cells (Fig. 7B). The lack of response ofFABP1 to Ad-HNF4α in HepG2 cells (Fig. 1A) is likely due to the highexpression level of this transcription factor in this cell line and theconsequent saturation of response elements by endogenous HNF4α[42]. Indeed, HNF4α knockdown by shRNA in HepG2 cells resulted ina concomitant down-regulation of FABP1 (Fig. 9C).
Fig. 7. Map of predicted C/EBPα, FOXA1 and PPARα elements in the FABP1 promoter and funto C/EBPα, FOXA1 and PPARα were analyzed by using the MatInspector Software and PAZsequences. In the composite DR1-C/EBP element the predicted imperfect C/EBP site is shaB, Basal reporter activity of different FABP1 5′-flanking constructs with mutations in thethe mutated FABP1 reporter constructs to C/EBPα, FOXA1 and PPARα+1 μM GW7647. ⁎
mean±SEM from 4 to 5 independent experiments, expressed as fold-change vs Ad-CONT.
Our data also suggest that an increase in C/EBPα levels leads to com-petition between C/EBPα and HNF4α for binding to the overlappedDR1-C/EBP composite element. Displacement of HNF4α would cause adrop in constitutive FABP1 expression. This could be due to reducedtranscription activation by C/EBPα or because the newly bound C/EBPαrecruits corepressors such as CtBP1 and CtBP2 [58] or HDAC1 [59].
Another important finding is the activation of human FABP1 byFOXA1, which is in agreement with the previously reported activationof the rat FABP1 promoter by endodermal FOXA factors [27]. In thatstudy a −596 bp construct of the rat FABP1 responded to FOXA2 inCaCo and HepG2 cells. The FOXA response elements were predictedat −169, −155 and −94 bp, and mutation of all these sites reducedbasal and FOXA2-induced reporter activity [27]. These data do notagree with our results supporting that FOXA1 response lies in a highdensity cluster of FOXA binding sites between −229 and −592 bp(Fig. 6A & Supplemental Fig. 3). One possible explanation for thisdiscrepancy is that in the study by Divine et al. [27] simultaneousmutation of the putative FOXA sites also destroys the HNF1α bindingsite at −95 bp, which has been reported to be essential for FABP1activity [26].
While preparing this manuscript, Wu et al. [60] published a studyon human FABP1, but with substantially different conclusions. The
ctional analysis by site-specific mutagenesis. A, FABP1 promoter sequences responsiveAR database matrices. Underlined sequences denote predicted elements and mutatedded in gray. TATA box and coding sequences are written in italicized capital letters.predicted sites. ⁎pb0.05, ⁎⁎pb0.01 and ⁎⁎⁎pb0.001 vs. wt-promoter. C. Response ofpb0.05 and ⁎⁎⁎pb0.001 vs. Ad-CONT. EEEpb0.001 vs. wt promoter. Bars represent
Fig. 9. Transcriptional regulation of FABP1 byHNF4α. A, HeLa cells, which lackHNF4α, weretransfected with the FABP1 reporter constructs. Forty-eight hours post-transfection cellswere lysed and luciferase activities determined. Bars represent mean±SEM of 4 indepen-dent experiments. ⁎⁎pb0.01 vs. pGL3B; ns: non-significant vs. −96 bp FABP1-LUC. B, HeLacells were infected with Ad-HNF4α (30 MOI) or Ad-CONT (30 MOI). Next day, cellswere transfected with the FABP1 reporter constructs. Forty-eight hours post-transfection,cells were lysed, and luciferase activities determined. Bars represent mean±SEM from 4independent experiments, expressed as fold-change vs Ad-CONT. ⁎⁎pb0.01, ⁎⁎⁎pb0.001vs. −96 bp FABP1-LUC. C, HepG2 cells were infected with the control lentivirus pLKO.1-shRNA-LUC (sh-control) or with lentivirus encoding four HNF4α-targeting shRNAs(sh1-sh4). Seventy-two hours after infection, mRNA level of HNF4α and FABP1 was deter-mined by quantitative RT-PCR and total protein extracts were immunoblotted with anti-HNF4α (lower panel).
814 C. Guzmán et al. / Biochimica et Biophysica Acta 1831 (2013) 803–818
study by Wu et al. [60] showed that both C/EBPα and FOXA2 areactivators of FABP1 through an overlapped element at−175 bp. How-ever, our data support that C/EBPα is an important repressor by binding
to a composite DR1-C/EBP element at−123 bp, whereas both FOXA1and FOXA2 achieve activation through the region between −229 and−592 bp (Supplemental Fig. 4). These discrepancies are difficult to ex-plain by different cell models/species or technical conditions, but it isimportant to note that in the study byWu et al. [60] the different dele-tion constructs were not co-transfected with transcription factors toprove which region displays the primary response. Similarly, thesite-mutated reporter plasmids were not assayed with transcriptionfactors to demonstrate the relevance of the predicted sites. Moreover,Wu et al. [60] do not prove activation of the native human FABP1gene by transfected C/EBPα or FOXA. In our study, we demonstratethe activating/repressing effect of each factor on the native FABP1gene and confirmed results in cultured human hepatocytes. We havetested all the wild-type and mutated FABP1 constructs with transcrip-tion factors to undoubtedly identify the responsive region and therelevance of the putative binding elements for repression and activa-tion. We have combined factors to demonstrate dominant negative oradditive responses. Finally, we have studied the mechanism of FABP1repression by C/EBPα and identified competition with HNF4α as thelikely mechanism.
We have also used immunoblotting analysis and immunofluores-cence microscopy to demonstrate that the effects of C/EBPα, FOXA1 andPPARα on FABP1 mRNA are translated to the protein level (Figs. 3 & 4).Interestingly, we also found that FABP1 protein seems to be localized inthe nuclear/perinuclear region in HepG2 cells, and that FOXA1 andPPARα favor the induction of FABP1 in the cytoplasm (Fig. 4 and Supple-mental Fig. 2). It has been shown that FABP1 colocalizes with PPARα andenhances ligand distribution to the nuclei of living cells [61]. This wouldexplain the presence of FABP1 in nuclei. Our data also supports thatthe induction of FABP1 by PPARα causes an increase of FABP1 in thecytoplasmic compartment, where this protein has to accomplish itsmajor role in FA trafficking and distribution [4].
The regulation of FABP1 could have clinical significance as far asstudies with FABP1 deficient mice have proven that FABP1 is involvedin the control of body weight, obesity and fatty liver [8,12,14–16].Moreover, genetic variations within FABP1 influence susceptibilityto NAFLD in humans [22]. These facts led us to investigate if FABP1expression is altered in metabolic conditions and found that FABP1is significantly repressed in human NAFL and in two different animalmodels of NAFLD (Figs. 10 & 11). Our findings are in agreement witha clinical study showing that FABP1 is underexpressed in obese pa-tients with nonalcoholic steatohepatitis [20]. FABP1 levels were alsodecreased in a steatotic rat model established by administration of17α-ethynylestradiol [21].
In the present study we have also determined if the down-regulation of FABP1 in NAFLD correlates with alterations in the factorscontrolling FABP1 transcription. All of our results demonstrated thatthe repression of FABP1 in NAFLD correlates more closely with thedown-regulation of FOXA1 and PPARα, but increased C/EBPα expres-sion could also contribute to this effect (Figs. 10 & 11). The repressionof FOXA1 in NAFLD has been recently reported by us [34] and therepression of PPARα in human NAFL and animal models of NAFLD isin agreement with PPARα down-regulation in the liver of OtsukaLong-Evans Tokushima fatty rats [62]. All these resultsmake it temptingto suggest that NAFLD triggers down-regulation of FOXA1 and PPARαand that this in turns negatively affects the expression of target genessuch as FABP1. The rescue of the repressed FABP1 in MCD mice byactivating endogenous PPARα (Fig. 10C) represents a proof-of-conceptsupporting this mechanism.
A lower expression of FABP1 in NAFLD could be an adaptive re-sponse against accumulated fat in the liver. Indeed, lower triglycer-ides have been found in FABP1 deficient mice under specific dietsand conditions [6,8,15–17]. Lipid analysis of FABP1 gene-ablated he-patocytes has also revealed decreased triglyceride levels [18]. There-fore, lower levels of FABP1 (via lower FOXA1/PPARα) could stop orreduce fat accumulation in the liver. This scenario is in consonance
Fig. 10. Changes in FABP1, C/EBPα, FOXA1 and PPARα expressions in liver of two animal models of NAFLD. Mice were fed a MCD diet or a MCD diet supplemented with methionineand choline (control mice). At the indicated times livers were collected and analyzed (A–B). Other groups of mice fed a MCD diet for 5 weeks received a daily gavage dose of eitherthe PPARα agonist Wy-14,643 or the carrier methylcellulose for 4 days. Twenty-four hours after the last dose mice were sacrificed (C). Mice deficient in MAT1A, which spontane-ously develop nonalcoholic steatohepatitis, and their wild-type C57BL/6 littermates were allowed food and water ad libitum. After 3 or 9-month livers were excised and analyzed(D–E). FABP1, C/EBPα, FOXA1 and PPARαmRNA were determined by Q-RT-PCR (A, C, D) and FABP1 protein analyzed by immunoblotting (B–E). Data represent mean±SEM of 4–7mice, expressed as % of controls (dashed line or CONT). ⁎pb0.05, ⁎⁎pb0.01 and ⁎⁎⁎pb0.001 vs. control paired mice.
815C. Guzmán et al. / Biochimica et Biophysica Acta 1831 (2013) 803–818
with the well-known decrease in hepatic triglycerides during theprogression of NAFLD [63,64]. However, a lower FABP1 expressioncould also be a double-edge sword as the capacity to attenuate the
cytotoxic detergent effect of free FAs could be decreased andlipotoxicity enhanced, thus contributing to the development of in-flammation, steatohepatitis and NAFLD progression [65].
Fig. 11. Expression analysis of FABP1 and its transcription factors in human NAFL. Total RNA was purified from 18 NAFL samples (total lipids>600 μg/mg prot and TG>500 μg/mg prot)and from 25 nonsteatotic livers (total lipids b400 μg/mg prot and TAGb300 μg/mg prot) (Human Liver Bank HLaFe-CIBERehd). The mRNA concentration of FABP1, FOXA1,PPARα, C/EBPα and HNF4α was determined by real-time Q-RT-PCR. GAPDH mRNA was used for normalization. Box plots represent the 25th, the median and the 75th percen-tiles, a dashed line depicts the mean, whiskers indicate the 95th and 5th percentiles, and dots represent outliers. Statistical significance between groups was determined by the nonpara-metric Mann–Whitney test.
816 C. Guzmán et al. / Biochimica et Biophysica Acta 1831 (2013) 803–818
Fig. 12 summarizes our observations and provides a picture of theintricate regulation of human FABP1 transcription. We conclude that inthe human liver, FABP1 is subjected to a complex transcriptional regula-tion that includes activators (FOXA1, HNF4α and PPARα) and repressors(C/EBPα) that modulate the levels of expression of FABP1 in differentphysiological and pathological conditions such as NAFLD. The study ofFABP1 regulation allows a better comprehension of the mechanismscontrolling lipid homeostasis and its related metabolic disorders.
Acknowledgments
This work has been supported by grants PI 10/00194 from Fondo deInvestigación Sanitaria (FIS, Instituto de Salud Carlos III, Ministerio de
Fig. 12. A model for transcription regulation of the human FABP1 gene. Gene activation byinduces FABP1 in response to fatty acids and their derivatives, and to synthetic fibrate druFABP1 transcription in the human liver. NAFLD alters expression of transcription factors, wfree unbound FA, and potentiate lipotoxicity and NAFLD progression.
Economía y Competitividad), BFU2010-15784 from Ministerio deEducación y Ciencia and GRS 482/A/10 from Junta de Castilla y León.C.G. was a recipient of a contract (CA 09/00122) from the Instituto deSalud Carlos III. M.B. (EHD‐10‐DOC2) and M.V. G.‐M (EHD-24-DOC)were recipients of CIBERehd contracts. The authors also wish to thankto Dr. David Hervás (Bioinformatics Unit, IIS La Fe) for his help andprofessional advice.
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.bbalip.2012.12.014.
HNF4α and FOXA1 contributes to maintain high FABP1 expression in the liver. PPARαgs. C/EBPα competes with HNF4α for binding and reduces constitutive and induciblehich leads in turn to FABP1 repression. This could favor lower triglycerides and more
817C. Guzmán et al. / Biochimica et Biophysica Acta 1831 (2013) 803–818
References
[1] M.R. Teli, O.F. James, A.D. Burt, M.K. Bennett, C.P. Day, The natural history ofnonalcoholic fatty liver: a follow-up study, Hepatology 22 (1995) 1714–1719.
[2] M.J. McArthur, B.P. Atshaves, A. Frolov, W.D. Foxworth, A.B. Kier, F. Schroeder,Cellular uptake and intracellular trafficking of long chain fatty acids, J. Lipid Res.40 (1999) 1371–1383.
[3] J. Storch, B. Corsico, The emerging functions and mechanisms of mammalian fattyacid-binding proteins, Annu. Rev. Nutr. 28 (2008) 73–95.
[4] B.P. Atshaves, G.G. Martin, H.A. Hostetler, A.L. McIntosh, A.B. Kier, F. Schroeder, Liverfatty acid-binding protein and obesity, J. Nutr. Biochem. 21 (2010) 1015–1032.
[5] F. Schroeder, A.D. Petrescu, H. Huang, B.P. Atshaves, A.L. McIntosh, G.G. Martin, H.A.Hostetler, A. Vespa, D. Landrock, K.K. Landrock, H.R. Payne, A.B. Kier, Role of fatty acidbinding proteins and long chain fatty acids in modulating nuclear receptors andgene transcription, Lipids 43 (2008) 1–17.
[6] G.G. Martin, B.P. Atshaves, H. Huang, A.L. McIntosh, B.J. Williams, P.J. Pai, D.H.Russell, A.B. Kier, F. Schroeder, Hepatic phenotype of liver fatty acid bindingprotein gene-ablated mice, Am. J. Physiol. Gastrointest. Liver Physiol. 297(2009) G1053–G1065.
[7] Y. Xie, E.P. Newberry, S.M. Kennedy, J. Luo, N.O. Davidson, Increased susceptibility todiet-induced gallstones in liver fatty acid binding protein knockoutmice, J. Lipid Res.50 (2009) 977–987.
[8] E.P. Newberry, Y. Xie, S.M. Kennedy, J. Luo, N.O. Davidson, Protection againstWestern diet-induced obesity and hepatic steatosis in liver fatty acid-bindingprotein knockout mice, Hepatology 44 (2006) 1191–1205.
[9] M.O. Weickert, C.V. Loeffelholz, M. Roden, V. Chandramouli, A. Brehm, P. Nowotny,M.A. Osterhoff, F. Isken, J. Spranger, B.R. Landau, A.F. Pfeiffer, M. Mohlig, A Thr94Alamutation in human liver fatty acid-binding protein contributes to reduced hepaticglycogenolysis and blunted elevation of plasma glucose levels in lipid-exposed sub-jects, Am. J. Physiol. Endocrinol. Metab. 293 (2007) E1078–E1084.
[10] H.A. Hostetler, M. Balanarasimha, H. Huang, M.S. Kelzer, A. Kaliappan, A.B. Kier, F.Schroeder, Glucose regulates fatty acid binding protein interaction with lipidsand peroxisome proliferator-activated receptor alpha, J. Lipid Res. 51 (2010)3103–3116.
[11] G.G. Martin, B.P. Atshaves, A.L. McIntosh, H.R. Payne, J.T. Mackie, A.B. Kier, F.Schroeder, Liver fatty acid binding protein gene ablation enhances age-dependentweight gain in male mice, Mol. Cell. Biochem. 324 (2009) 101–115.
[12] G.G. Martin, B.P. Atshaves, A.L. McIntosh, J.T. Mackie, A.B. Kier, F. Schroeder, Liver fattyacid-binding protein gene-ablated female mice exhibit increased age-dependent obe-sity, J. Nutr. 138 (2008) 1859–1865.
[13] G.G. Martin, B.P. Atshaves, A.L. McIntosh, J.T. Mackie, A.B. Kier, F. Schroeder, Liverfatty acid binding protein gene ablation potentiates hepatic cholesterol accumu-lation in cholesterol-fed female mice, Am. J. Physiol. Gastrointest. Liver Physiol.290 (2006) G36–G48.
[14] B.P. Atshaves, A.L. McIntosh, S.M. Storey, K.K. Landrock, A.B. Kier, F. Schroeder,High dietary fat exacerbates weight gain and obesity in female liver fatty acidbinding protein gene-ablated mice, Lipids 45 (2010) 97–110.
[15] E.P. Newberry, S.M. Kennedy, Y. Xie, B.T. Sternard, J. Luo, N.O. Davidson, Diet-inducedobesity and hepatic steatosis in L-Fabp/mice is abrogated with SF, but not PUFA,feeding and attenuated after cholesterol supplementation, Am. J. Physiol.Gastrointest. Liver Physiol. 294 (2008) G307–G314.
[16] E.P. Newberry, S.M. Kennedy, Y. Xie, J. Luo, R.M. Crooke, M.J. Graham, J. Fu, D.Piomelli, N.O. Davidson, Decreased body weight and hepatic steatosis with alteredfatty acid ethanolamide metabolism in aged L-Fabp −/− mice, J. Lipid Res. 53(2012) 744–754.
[17] E.P. Newberry, Y. Xie, S. Kennedy, X. Han, K.K. Buhman, J. Luo, R.W. Gross, N.O.Davidson, Decreased hepatic triglyceride accumulation and altered fatty acid uptakein mice with deletion of the liver fatty acid-binding protein gene, J. Biol. Chem. 278(2003) 51664–51672.
[18] B.P. Atshaves, A.M. McIntosh, O.I. Lyuksyutova, W. Zipfel, W.W. Webb, F.Schroeder, Liver fatty acid-binding protein gene ablation inhibits branched-chain fatty acid metabolism in cultured primary hepatocytes, J. Biol. Chem. 279(2004) 30954–30965.
[19] N. Gao, X. Qu, J. Yan, Q. Huang, H.Y. Yuan, D.S. Ouyang, L-FABP T94A decreasedfatty acid uptake and altered hepatic triglyceride and cholesterol accumulationin Chang liver cells stably transfected with L-FABP, Mol. Cell. Biochem. 345(2010) 207–214.
[20] M. Charlton, K. Viker, A. Krishnan, S. Sanderson, B. Veldt, A.J. Kaalsbeek, M. Kendrick, G.Thompson, F. Que, J. Swain, M. Sarr, Differential expression of lumican and fatty acidbinding protein-1: new insights into the histologic spectrum of nonalcoholic fattyliver disease, Hepatology 49 (2009) 1375–1384.
[21] D.Y. Hung, G.A. Siebert, P. Chang, F.J. Burczynski, M.S. Roberts, Reduced hepaticextraction of palmitate in steatosis correlated to lower level of liver fatty acidbinding protein, Am. J. Physiol. Gastrointest. Liver Physiol. 288 (2005) G93–G100.
[22] X.E. Peng, Y.L. Wu, Q.Q. Lu, Z.J. Hu, X. Lin, Two genetic variants in FABP1 andsusceptibility to non-alcohol fatty liver disease in a Chinese population,Gene 500 (2012) 54–58.
[23] C. Schachtrup, T. Emmler, B. Bleck, A. Sandqvist, F. Spener, Functional analysis ofperoxisome-proliferator-responsive element motifs in genes of fatty acid-bindingproteins, Biochem. J. 382 (2004) 239–245.
[24] C. Wolfrum, P. Ellinghaus, M. Fobker, U. Seedorf, G. Assmann, T. Borchers, F.Spener, Phytanic acid is ligand and transcriptional activator of murine liverfatty acid binding protein, J. Lipid Res. 40 (1999) 708–714.
[25] J.F. Landrier, C. Thomas, J. Grober, H. Duez, F. Percevault, M. Souidi, C. Linard, B.Staels, P. Besnard, Statin induction of liver fatty acid-binding protein (L-FABP)
[28] L.J. Staloch, J.K. Divine, J.T. Witten, T.C. Simon, C/EBP and Cdx family factors regulateliver fatty acid binding protein transgene expression in the small intestinal epithelium,Biochim. Biophys. Acta 1731 (2005) 168–178.
[29] C.W. Rowley, L.J. Staloch, J.K. Divine, S.P. McCaul, T.C. Simon, Mechanisms of mutualfunctional interactions betweenHNF-4alpha andHNF-1alpha revealed bymutationsthat cause maturity onset diabetes of the young, Am. J. Physiol. Gastrointest. LiverPhysiol. 290 (2006) G466–G475.
[30] D.T. Odom, R.D. Dowell, E.S. Jacobsen, W. Gordon, T.W. Danford, K.D. MacIsaac,P.A. Rolfe, C.M. Conboy, D.K. Gifford, E. Fraenkel, Tissue-specific transcriptionalregulation has diverged significantly between human and mouse, Nat. Genet.39 (2007) 730–732.
[31] S.C. Lu, L. Alvarez, Z.Z. Huang, L. Chen, W. An, F.J. Corrales, M.A. Avila, G. Kanel, J.M.Mato, Methionine adenosyltransferase 1A knockout mice are predisposed to liverinjury and exhibit increased expression of genes involved in proliferation, Proc.Natl. Acad. Sci. U. S. A. 98 (2001) 5560–5565.
[32] A. Cano, X. Buque,M.Martinez-Una, I. Aurrekoetxea, A.Menor, J.L. Garcia-Rodriguez,S.C. Lu, M.L. Martinez-Chantar, J.M. Mato, B. Ochoa, P. Aspichueta, Methionineadenosyltransferase 1A gene deletion disrupts hepatic very low-density lipoproteinassembly in mice, Hepatology 54 (2011) 1975–1986.
[33] M.L. Martinez-Chantar, F.J. Corrales, L.A. Martinez-Cruz, E.R. Garcia-Trevijano, Z.Z.Huang, L. Chen, G. Kanel, M.A. Avila, J.M. Mato, S.C. Lu, Spontaneous oxidativestress and liver tumors in mice lacking methionine adenosyltransferase 1A,FASEB J. 16 (2002) 1292–1294.
[34] M. Moya, M. Benet, C. Guzman, L. Tolosa, C. Garcia-Monzon, E. Pareja, J.V. Castell,R. Jover, Foxa1 reduces lipid accumulation in human hepatocytes and isdown-regulated in nonalcoholic fatty liver, PLoS One 7 (2012) e30014.
[35] M. Moya, M. Jose Gomez-Lechon, J.V. Castell, R. Jover, Enhanced steatosis by nuclearreceptor ligands: a study in cultured human hepatocytes and hepatoma cells with acharacterized nuclear receptor expression profile, Chem. Biol. Interact. 184 (2010)376–387.
[36] J.B. Lowe,M.S. Boguski, D.A. Sweetser, N.A. Elshourbagy, J.M. Taylor, J.I. Gordon, Humanliver fatty acid binding protein. Isolation of a full length cDNA and comparativesequence analyses of orthologous and paralogous proteins, J. Biol. Chem. 260 (1985)3413–3417.
[37] M. Pascual, M.J. Gomez-Lechon, J.V. Castell, R. Jover, ATF5 is a highly abundantliver-enriched transcription factor that cooperates with constitutive androstanereceptor in the transactivation of CYP2B6: implications in hepatic stress re-sponses, Drug Metab. Dispos. 36 (2008) 1063–1072.
[38] M. Benet, A. Lahoz, C. Guzman, J.V. Castell, R. Jover, CCAAT/enhancer-bindingprotein alpha (C/EBPalpha) and hepatocyte nuclear factor 4alpha (HNF4alpha)synergistically cooperate with constitutive androstane receptor to transactivatethe human cytochrome P450 2B6 (CYP2B6) gene: application to the developmentof a metabolically competent human hepatic cell model, J. Biol. Chem. 285 (2010)28457–28471.
[39] C. Rodriguez-Antona, R. Bort, R. Jover, N. Tindberg, M. Ingelman-Sundberg, M.J.Gomez-Lechon, J.V. Castell, Transcriptional regulation of human CYP3A4 basal ex-pression by CCAAT enhancer-binding protein alpha and hepatocyte nuclearfactor-3 gamma, Mol. Pharmacol. 63 (2003) 1180–1189.
[40] C.P. Martinez-Jimenez, M.J. Gomez-Lechon, J.V. Castell, R. Jover, Transcriptionalregulation of the human hepatic CYP3A4: identification of a new distal enhancerregion responsive to CCAAT/enhancer-binding protein beta isoforms (liver activat-ing protein and liver inhibitory protein), Mol. Pharmacol. 67 (2005) 2088–2101.
[41] R. Jover, R. Bort, M.J. Gomez-Lechon, J.V. Castell, Down-regulation of human CYP3A4by the inflammatory signal interleukin-6: molecular mechanism and transcriptionfactors involved, FASEB J. 16 (2002) 1799–1801.
[42] C.P. Martinez-Jimenez, M.J. Gomez-Lechon, J.V. Castell, R. Jover, Underexpressedcoactivators PGC1alpha and SRC1 impair hepatocyte nuclear factor 4 alpha func-tion and promote dedifferentiation in human hepatoma cells, J. Biol. Chem. 281(2006) 29840–29849.
[43] A. Gomez-Foix, W. Coats, S. Baque, T. Alam, R. Gerard, C. Newgard, Adenovirus-mediated transfer of the muscle glycogen phosphorylase gene into hepatocytesconfers altered regulation of glycogen metabolism, J. Biol. Chem. 267 (1992)25129–25134.
[44] J. Castell, D. Hernandez, F.A.M. Gomez-Foix, I. Guillen, T. Donato, L.M.J. Gomez-Lechon,Adenovirus-mediated gene transfer into human hepatocytes: analysis of the biochemicalfunctionality of transduced cells, Gene Ther. 4 (1997) 455–464.
[45] E. Lima-Cabello, M.V. Garcia-Mediavilla, M.E. Miquilena-Colina, J. Vargas-Castrillon,T. Lozano-Rodriguez, M. Fernandez-Bermejo, J.L. Olcoz, J. Gonzalez-Gallego, C.Garcia-Monzon, S. Sanchez-Campos, Enhanced expression of pro-inflammatoryme-diators and liver X-receptor-regulated lipogenic genes in non-alcoholic fatty liverdisease and hepatitis C, Clin. Sci. (Lond.) 120 (2011) 239–250.
[46] N. Dionisio, M.V. Garcia-Mediavilla, S. Sanchez-Campos, P.L. Majano, I. Benedicto, J.A.Rosado, G.M. Salido, J. Gonzalez-Gallego, Hepatitis C virus NS5A and core proteinsinduce oxidative stress-mediated calcium signalling alterations in hepatocytes,J. Hepatol. 50 (2009) 872–882.
[47] M.V. Garcia-Mediavilla, S. Pisonero-Vaquero, E. Lima-Cabello, I. Benedicto, P.L. Majano,F. Jorquera, J. Gonzalez-Gallego, S. Sanchez-Campos, Liver X receptor alpha-mediated
818 C. Guzmán et al. / Biochimica et Biophysica Acta 1831 (2013) 803–818
regulation of lipogenesis by core and NS5A proteins contributes to HCV-induced liversteatosis and HCV replication, Lab. Invest. 92 (2012) 1191–1202.
[48] B. Desvergne, L. Michalik, W. Wahli, Transcriptional regulation of metabolism,Physiol. Rev. 86 (2006) 465–514.
[49] H. Schrem, J. Klempnauer, J. Borlak, Liver-enriched transcription factors in liverfunction and development. Part I: the hepatocyte nuclear factor network andliver-specific gene expression, Pharmacol. Rev. 54 (2002) 129–158.
[50] H. Schrem, J. Klempnauer, J. Borlak, Liver-enriched transcription factors in liverfunction and development. Part II: the C/EBPs and D site-binding protein in cellcycle control, carcinogenesis, circadian gene regulation, liver regeneration, apo-ptosis, and liver-specific gene regulation, Pharmacol. Rev. 56 (2004) 291–330.
[51] K. Cartharius, K. Frech, K. Grote, B. Klocke, M. Haltmeier, A. Klingenhoff, M. Frisch,M. Bayerlein, T. Werner, MatInspector and beyond: promoter analysis based ontranscription factor binding sites, Bioinformatics 21 (2005) 2933–2942.
[52] E. Portales-Casamar, S. Kirov, J. Lim, S. Lithwick, M.I. Swanson, A. Ticoll, J. Snoddy,W.W. Wasserman, PAZAR: a framework for collection and dissemination ofcis-regulatory sequence annotation, Genome Biol. 8 (2007) R207.
[53] J.R. Friedman, K.H. Kaestner, The Foxa family of transcription factors in developmentand metabolism, Cell Mol. Life Sci. 63 (2006) 2317–2328.
[54] S.A. Duncan, M.A. Navas, D. Dufort, J. Rossant, M. Stoffel, Regulation of a tran-scription factor network required for differentiation and metabolism, Science281 (1998) 692–695.
[55] C.C. Almonacid-Urrego, S. Sanchez-Campos, M.J. Tunon, J. Gonzalez-Gallego,Non-alcoholic steatohepatitis: what can we learn from animal models? Curr. Med.Chem. 19 (2012) 1389–1404.
[56] Y. Yamazaki, S. Kakizaki, N. Horiguchi, N. Sohara, K. Sato, H. Takagi, M. Mori, M.Negishi, The role of the nuclear receptor constitutive androstane receptor in thepathogenesis of non-alcoholic steatohepatitis, Gut 56 (2007) 565–574.
[57] M.D. Wilson, D.T. Odom, Evolution of transcriptional control in mammals, Curr.Opin. Genet. Dev. 19 (2009) 579–585.
[58] C. Vernochet, S.B. Peres, K.E. Davis, M.E. McDonald, L. Qiang, H. Wang, P.E. Scherer,S.R. Farmer, C/EBPalpha and the corepressors CtBP1 and CtBP2 regulate repressionof select visceral white adipose genes during induction of the brown phenotype inwhite adipocytes by peroxisome proliferator-activated receptor gamma agonists,Mol. Cell. Biol. 29 (2009) 4714–4728.
[59] G.L. Wang, E. Salisbury, X. Shi, L. Timchenko, E.E. Medrano, N.A. Timchenko,HDAC1 cooperates with C/EBPalpha in the inhibition of liver proliferation in oldmice, J. Biol. Chem. 283 (2008) 26169–26178.
[60] Y.L. Wu, X.E. Peng, D. Wang, W.N. Chen, X. Lin, Human liver fatty acid bindingprotein (hFABP1) gene is regulated by liver-enriched transcription factorsHNF3beta and C/EBPalpha, Biochimie 94 (2012) 384–392.
[61] H. Huang, O. Starodub, A. McIntosh, B.P. Atshaves, G. Woldegiorgis, A.B. Kier, F.Schroeder, Liver fatty acid-binding protein colocalizes with peroxisome proliferatoractivated receptor alpha and enhances ligand distribution to nuclei of living cells,Biochemistry 43 (2004) 2484–2500.
[62] J.E. Yeon, K.M. Choi, S.H. Baik, K.O. Kim, H.J. Lim, K.H. Park, J.Y. Kim, J.J. Park, J.S.Kim, Y.T. Bak, K.S. Byun, C.H. Lee, Reduced expression of peroxisome proliferator-activated receptor-alpha may have an important role in the development ofnon-alcoholic fatty liver disease, J. Gastroenterol. Hepatol. 19 (2004) 799–804.
[63] T. Nagaya, N. Tanaka,M. Komatsu, T. Ichijo, K. Sano, A. Horiuchi, S. Joshita, T. Umemura,A. Matsumoto, K. Yoshizawa, T. Aoyama, K. Kiyosawa, E. Tanaka, Development fromsimple steatosis to liver cirrhosis and hepatocellular carcinoma: a 27-year follow-upcase, Clin. J. Gastroenterol. 1 (2008) 116–121.
[64] T. Nagaya, N. Tanaka, T. Suzuki, K. Sano, A. Horiuchi, M. Komatsu, T. Nakajima,T. Nishizawa, S. Joshita, T. Umemura, T. Ichijo, A. Matsumoto, K. Yoshizawa, J.Nakayama, E. Tanaka, T. Aoyama, Down-regulation of SREBP-1c is associatedwith the development of burned-out NASH, J. Hepatol. 53 (2010) 724–731.
[65] B.A. Neuschwander-Tetri, Hepatic lipotoxicity and the pathogenesis of nonalcoholicsteatohepatitis: the central role of nontriglyceride fatty acidmetabolites, Hepatology52 (2010) 774–788.




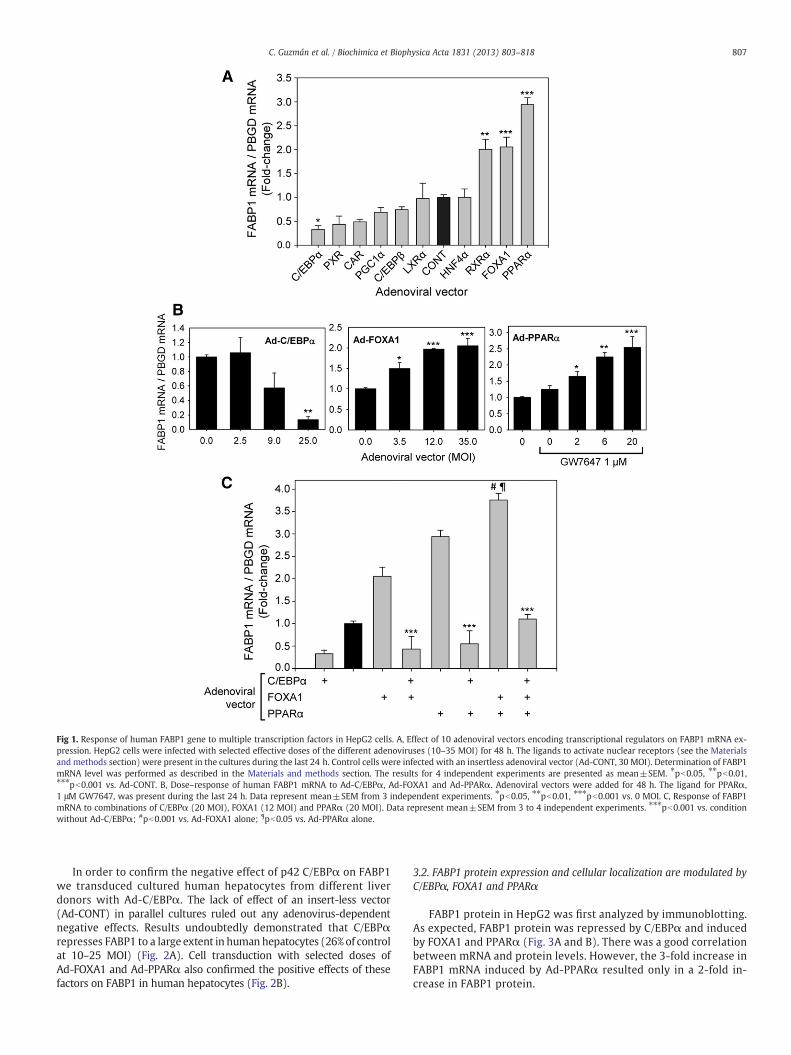
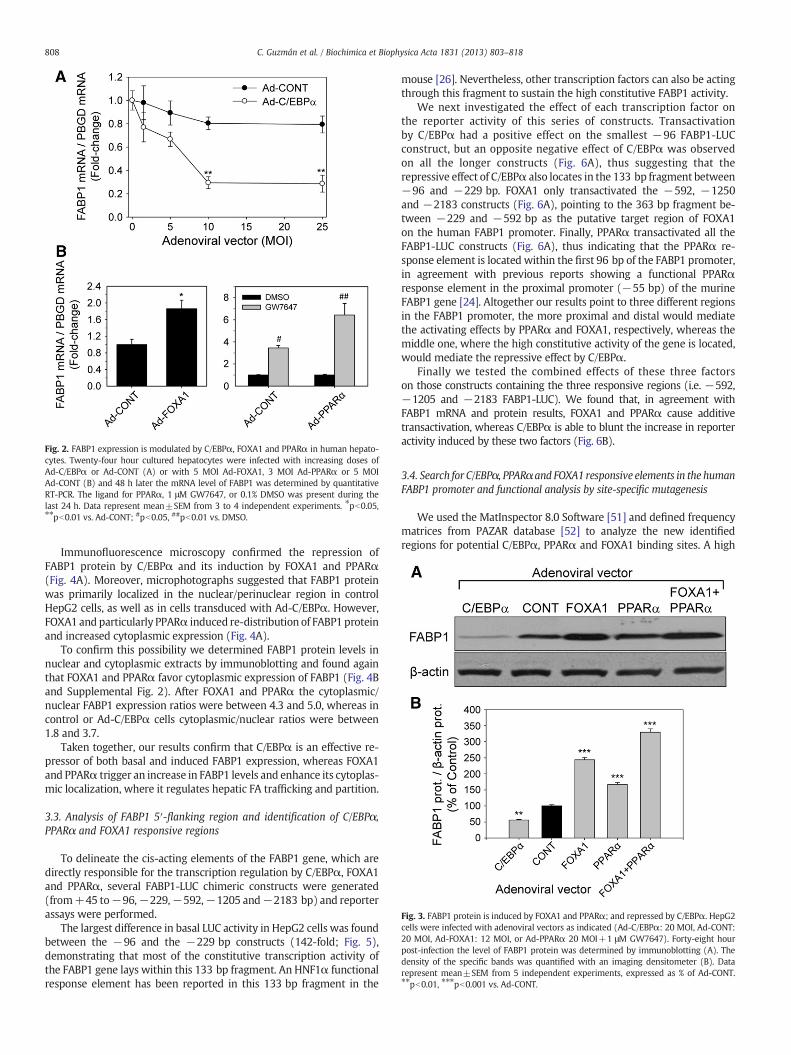



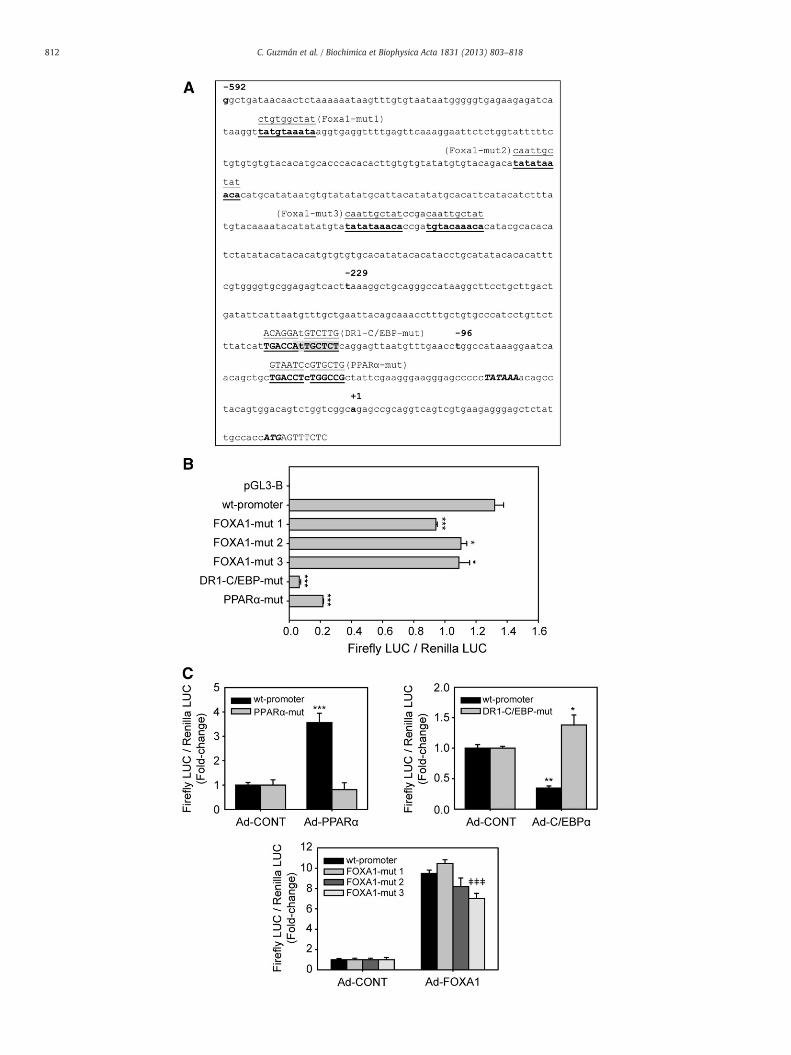

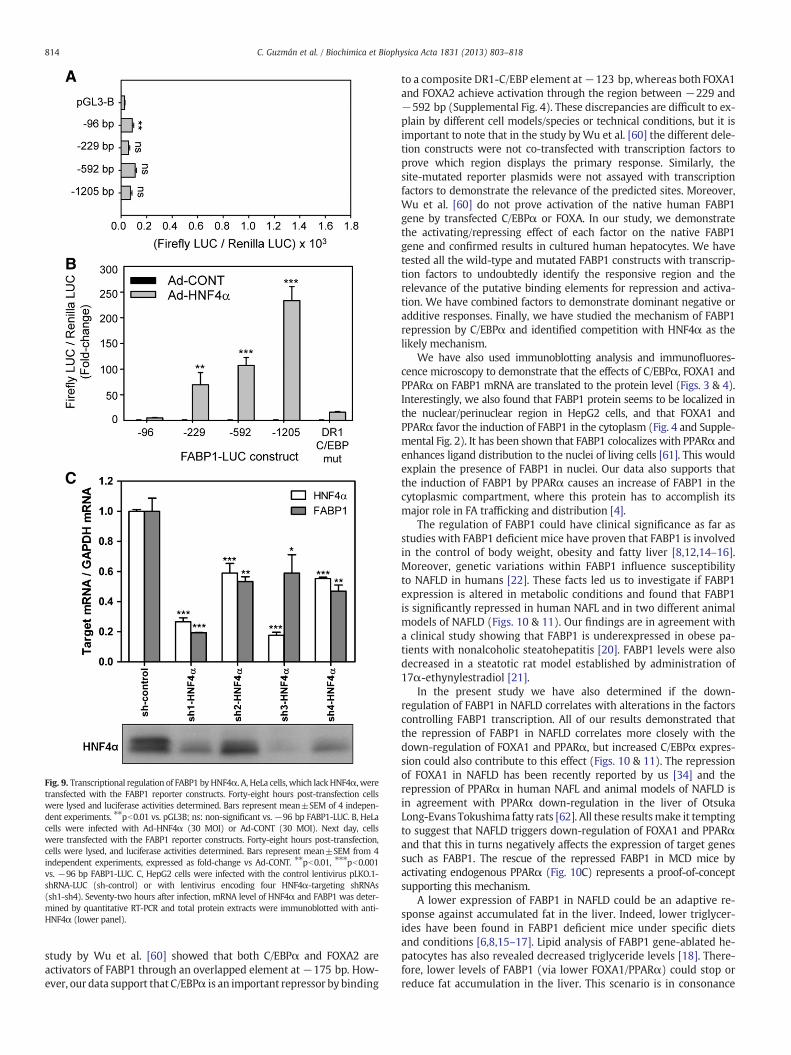














![Fatty liver disease with Diabetes Mellitus [BANGLADESH]](https://static.documents.pub/doc/80x56/587eb3901a28abbb688b57dd/fatty-liver-disease-with-diabetes-mellitus-bangladesh.jpg)






