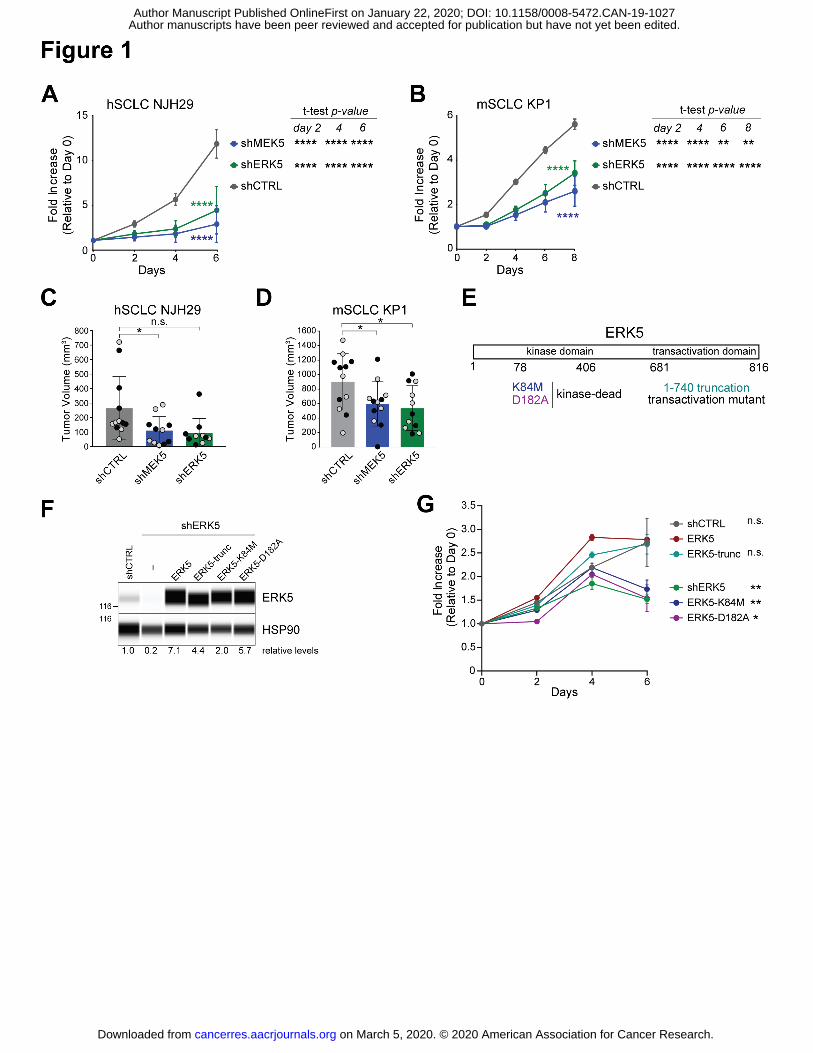
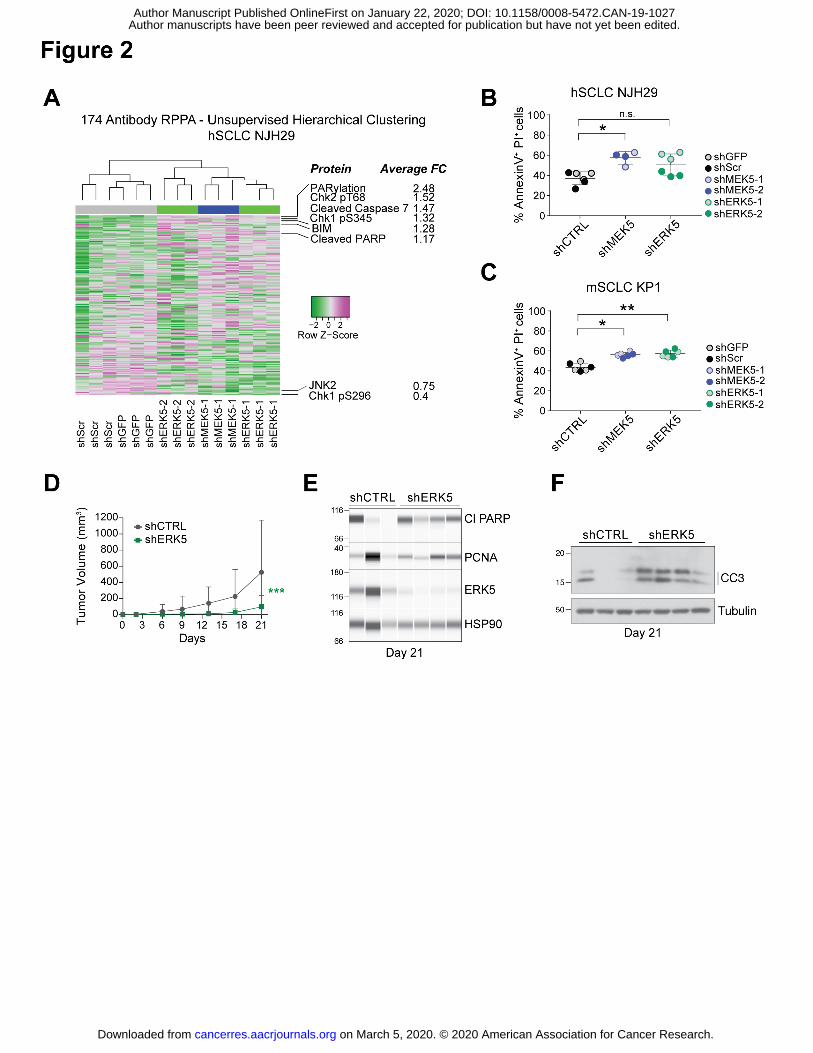
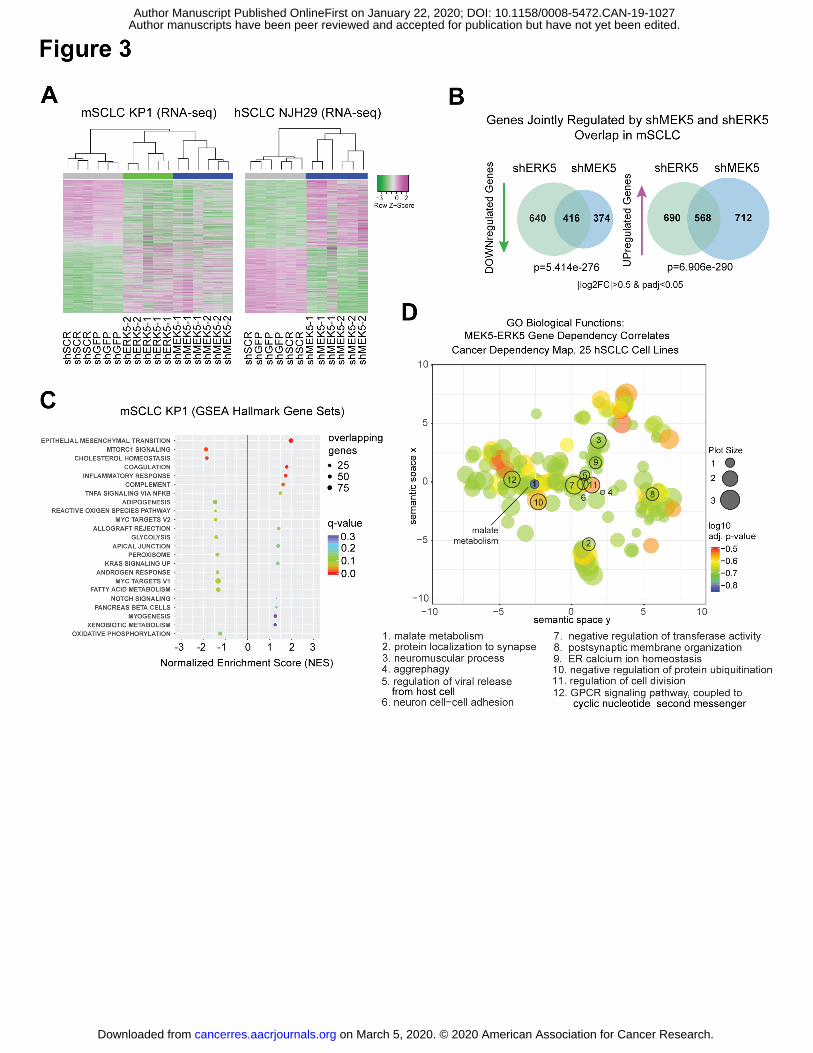
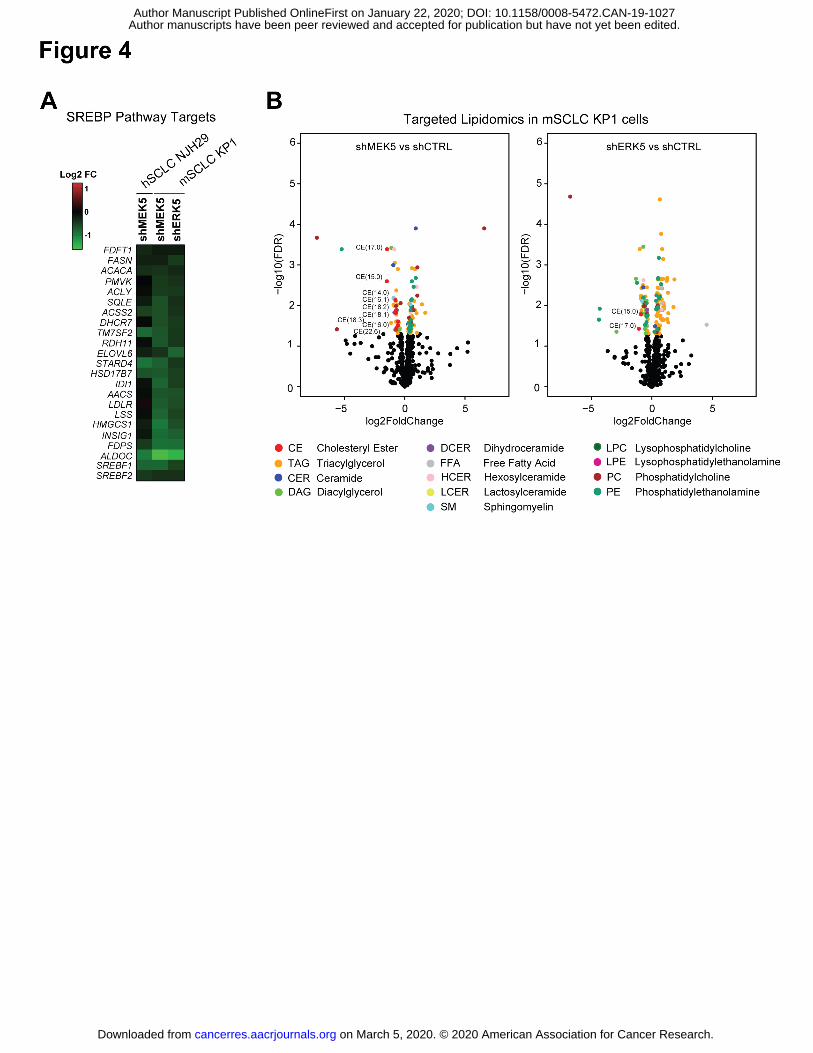
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
1. Bunn PA, Jr., Minna JD, Augustyn A, Gazdar AF, Ouadah Y, Krasnow MA, et al. Small Cell Lung Cancer: Can Recent Advances in Biology and Molecular Biology Be Translated into Improved Outcomes? J Thorac Oncol 2016;11:453-74
2. Sabari JK, Lok BH, Laird JH, Poirier JT, Rudin CM. Unravelling the biology of SCLC: implications for therapy. Nature reviews Clinical oncology 2017
3. Horn L, Mansfield AS, Szczesna A, Havel L, Krzakowski M, Hochmair MJ, et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N Engl J Med 2018;379:2220-9
4. Pietanza MC, Waqar SN, Krug LM, Dowlati A, Hann CL, Chiappori A, et al. Randomized, Double-Blind, Phase II Study of Temozolomide in Combination With Either Veliparib or Placebo in Patients With Relapsed-Sensitive or Refractory Small-Cell Lung Cancer. J Clin Oncol 2018:JCO2018777672
5. George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G, et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015;524:47-53
6. Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet 2012;44:1104-10
7. Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet 2012;44:1111-6
8. Mollaoglu G, Guthrie MR, Bohm S, Bragelmann J, Can I, Ballieu PM, et al. MYC Drives Progression of Small Cell Lung Cancer to a Variant Neuroendocrine Subtype with Vulnerability to Aurora Kinase Inhibition. Cancer Cell 2017;31:270-85
9. Jia D, Augert A, Kim DW, Eastwood E, Wu N, Ibrahim AH, et al. Crebbp Loss Drives Small Cell Lung Cancer and Increases Sensitivity to HDAC Inhibition. Cancer Discov 2018;8:1422-37
10. Knapp S, Sundstrom M. Recently targeted kinases and their inhibitors-the path to clinical trials. Curr Opin Pharmacol 2014;17C:58-63
11. Cristea S, Sage J. Is the Canonical RAF/MEK/ERK Signaling Pathway a Therapeutic Target in SCLC? J Thorac Oncol 2016
12. Sen T, Tong P, Stewart CA, Cristea S, Valliani A, Shames DS, et al. CHK1 inhibition in small cell lung cancer produces single-agent activity in biomarker-defined disease subsets and combination activity with cisplatin or olaparib. Cancer Res 2017
13. Sen T, Tong P, Diao L, Li L, Fan Y, Hoff J, et al. Targeting AXL and mTOR Pathway Overcomes Primary and Acquired Resistance to WEE1 Inhibition in Small-Cell Lung Cancer. Clin Cancer Res 2017
14. Lallo A, Frese KK, Morrow C, Szczepaniak Sloane R, Gulati S, Schenk MW, et al. The combination of the PARP inhibitor olaparib and the Wee1 inhibitor AZD1775 as a new therapeutic option for small cell lung cancer. Clin Cancer Res 2018
15. Wang X, Tournier C. Regulation of cellular functions by the ERK5 signalling pathway. Cell Signal 2006;18:753-60
16. Hoang VT, Yan TJ, Cavanaugh JE, Flaherty PT, Beckman BS, Burow ME. Oncogenic signaling of MEK5-ERK5. Cancer Lett 2017;392:51-9
17. Stecca B, Rovida E. Impact of ERK5 on the Hallmarks of Cancer. Int J Mol Sci 2019;20 18. Simoes AE, Rodrigues CM, Borralho PM. The MEK5/ERK5 signalling pathway in cancer:
a promising novel therapeutic target. Drug Discov Today 2016;21:1654-63 19. Liu F, Zhang H, Song H. Upregulation of MEK5 by Stat3 promotes breast cancer cell
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
20. Antoon JW, Martin EC, Lai R, Salvo VA, Tang Y, Nitzchke AM, et al. MEK5/ERK5 signaling suppresses estrogen receptor expression and promotes hormone-independent tumorigenesis. PloS one 2013;8:e69291
21. Lochhead PA, Gilley R, Cook SJ. ERK5 and its role in tumour development. Biochemical Society transactions 2012;40:251-6
22. Denny SK, Yang D, Chuang CH, Brady JJ, Lim JS, Gruner BM, et al. Nfib Promotes Metastasis through a Widespread Increase in Chromatin Accessibility. Cell 2016;166:328-42
23. Byers LA, Wang J, Nilsson MB, Fujimoto J, Saintigny P, Yordy J, et al. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov 2012;2:798-811
24. Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, et al. Defining a Cancer Dependency Map. Cell 2017;170:564-76 e16
25. Rudin CM, Poirier JT, Byers LA, Dive C, Dowlati A, George J, et al. Molecular subtypes of small cell lung cancer: a synthesis of human and mouse model data. Nat Rev Cancer 2019
26. Kasler HG, Victoria J, Duramad O, Winoto A. ERK5 is a novel type of mitogen-activated protein kinase containing a transcriptional activation domain. Mol Cell Biol 2000;20:8382-9
27. Nithianandarajah-Jones GN, Wilm B, Goldring CE, Muller J, Cross MJ. ERK5: structure, regulation and function. Cell Signal 2012;24:2187-96
28. Tibes R, Qiu Y, Lu Y, Hennessy B, Andreeff M, Mills GB, et al. Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Molecular cancer therapeutics 2006;5:2512-21
29. Khan AUH, Allende-Vega N, Gitenay D, Gerbal-Chaloin S, Gondeau C, Vo DN, et al. The PDK1 Inhibitor Dichloroacetate Controls Cholesterol Homeostasis Through the ERK5/MEF2 Pathway. Sci Rep 2017;7:10654
30. Belkahla S, Haq Khan AU, Gitenay D, Alexia C, Gondeau C, Vo DN, et al. Changes in metabolism affect expression of ABC transporters through ERK5 and depending on p53 status. Oncotarget 2018;9:1114-29
31. Horton JD, Shah NA, Warrington JA, Anderson NN, Park SW, Brown MS, et al. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proceedings of the National Academy of Sciences of the United States of America 2003;100:12027-32
32. Guo D, Bell EH, Mischel P, Chakravarti A. Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr Pharm Des 2014;20:2619-26
33. Buhaescu I, Izzedine H. Mevalonate pathway: a review of clinical and therapeutical implications. Clin Biochem 2007;40:575-84
34. Jones P, Kafonek S, Laurora I, Hunninghake D. Comparative dose efficacy study of atorvastatin versus simvastatin, pravastatin, lovastatin, and fluvastatin in patients with hypercholesterolemia (the CURVES study). Am J Cardiol 1998;81:582-7
35. Polley E, Kunkel M, Evans D, Silvers T, Delosh R, Laudeman J, et al. Small Cell Lung Cancer Screen of Oncology Drugs, Investigational Agents, and Gene and microRNA Expression. J Natl Cancer Inst 2016;108
36. Hayashi M, Kim SW, Imanaka-Yoshida K, Yoshida T, Abel ED, Eliceiri B, et al. Targeted deletion of BMK1/ERK5 in adult mice perturbs vascular integrity and leads to endothelial failure. The Journal of clinical investigation 2004;113:1138-48
37. Watson FL, Heerssen HM, Bhattacharyya A, Klesse L, Lin MZ, Segal RA. Neurotrophins use the Erk5 pathway to mediate a retrograde survival response. Nat Neurosci 2001;4:981-8
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
38. Vaseva AV, Blake DR, Gilbert TSK, Ng S, Hostetter G, Azam SH, et al. KRAS Suppression-Induced Degradation of MYC Is Antagonized by a MEK5-ERK5 Compensatory Mechanism. Cancer Cell 2018;34:807-22 e7
39. Charni S, de Bettignies G, Rathore MG, Aguilo JI, van den Elsen PJ, Haouzi D, et al. Oxidative phosphorylation induces de novo expression of the MHC class I in tumor cells through the ERK5 pathway. Journal of immunology 2010;185:3498-503
40. Morita M, Sato T, Nomura M, Sakamoto Y, Inoue Y, Tanaka R, et al. PKM1 Confers Metabolic Advantages and Promotes Cell-Autonomous Tumor Cell Growth. Cancer Cell 2018;33:355-67 e7
41. Nomura M, Morita M, Tanuma N. A metabolic vulnerability of small-cell lung cancer. Oncotarget 2018;9:32278-9
42. Huang F, Ni M, Chalishazar MD, Huffman KE, Kim J, Cai L, et al. Inosine Monophosphate Dehydrogenase Dependence in a Subset of Small Cell Lung Cancers. Cell Metab 2018;28:369-82 e5
43. Yu L, Li K, Xu Z, Cui G, Zhang X. Integrated omics and gene expression analysis identifies the loss of metabolite-metabolite correlations in small cell lung cancer. Onco Targets Ther 2018;11:3919-29
44. Zhou T, Zhan J, Fang W, Zhao Y, Yang Y, Hou X, et al. Serum low-density lipoprotein and low-density lipoprotein expression level at diagnosis are favorable prognostic factors in patients with small-cell lung cancer (SCLC). BMC Cancer 2017;17:269
45. Khanzada UK, Pardo OE, Meier C, Downward J, Seckl MJ, Arcaro A. Potent inhibition of small-cell lung cancer cell growth by simvastatin reveals selective functions of Ras isoforms in growth factor signalling. Oncogene 2006;25:877-87
46. Arcaro A, Aubert M, Espinosa del Hierro ME, Khanzada UK, Angelidou S, Tetley TD, et al. Critical role for lipid raft-associated Src kinases in activation of PI3K-Akt signalling. Cell Signal 2007;19:1081-92
47. Ahmadi Y, Ghorbanihaghjo A, Argani H. The balance between induction and inhibition of mevalonate pathway regulates cancer suppression by statins: A review of molecular mechanisms. Chem Biol Interact 2017;273:273-85
48. Park KS, Martelotto LG, Peifer M, Sos ML, Karnezis AN, Mahjoub MR, et al. A crucial requirement for Hedgehog signaling in small cell lung cancer. Nat Med 2011;17:1504-8
49. Chen C, Breslin MB, Lan MS. Sonic hedgehog signaling pathway promotes INSM1 transcription factor in neuroendocrine lung cancer. Cell Signal 2018;46:83-91
50. Clendening JW, Penn LZ. Targeting tumor cell metabolism with statins. Oncogene 2012;31:4967-78
51. Seckl MJ, Ottensmeier CH, Cullen M, Schmid P, Ngai Y, Muthukumar D, et al. Multicenter, Phase III, Randomized, Double-Blind, Placebo-Controlled Trial of Pravastatin Added to First-Line Standard Chemotherapy in Small-Cell Lung Cancer (LUNGSTAR). J Clin Oncol 2017;35:1506-14
52. Ung MH, MacKenzie TA, Onega TL, Amos CI, Cheng C. Statins associate with improved mortality among patients with certain histological subtypes of lung cancer. Lung Cancer 2018;126:89-96
53. Mahoney CE, Pirman D, Chubukov V, Sleger T, Hayes S, Fan ZP, et al. A chemical biology screen identifies a vulnerability of neuroendocrine cancer cells to SQLE inhibition. Nat Commun 2019;10:96
54. Balanis NG, Sheu KM, Esedebe FN, Patel SJ, Smith BA, Park JW, et al. Pan-cancer Convergence to a Small-Cell Neuroendocrine Phenotype that Shares Susceptibilities with Hematological Malignancies. Cancer Cell 2019;36:17-34 e7
55. Nguyen D, Lemos C, Wortmann L, Eis K, Holton SJ, Boemer U, et al. Discovery and Characterization of the Potent and Highly Selective (Piperidin-4-yl)pyrido[3,2-
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
d]pyrimidine Based in Vitro Probe BAY-885 for the Kinase ERK5. J Med Chem 2019;62:928-40
56. Xia Y, Xie Y, Yu Z, Xiao H, Jiang G, Zhou X, et al. The Mevalonate Pathway Is a Druggable Target for Vaccine Adjuvant Discovery. Cell 2018;175:1059-73 e21
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Published OnlineFirst January 22, 2020.Cancer Res Sandra Cristea, Garry L. Coles, Daniel Hornburg, et al. cell lung cancerThe MEK5-ERK5 kinase axis controls lipid metabolism in small
Updated version
10.1158/0008-5472.CAN-19-1027doi:
Access the most recent version of this article at:
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/early/2020/01/22/0008-5472.CAN-19-1027To request permission to re-use all or part of this article, use this link
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027