The Prognostic Significance of Various 13q14 Deletions inChronic Lymphocytic Leukemia
Peter Ouillette2, RoxaneCollins2, Sajid Shakhan2, Jinghui Li2, Cheng Li1, Kerby Shedden3, and Sami N.Malek2
AbstractPurpose: To further our understanding of the biology and prognostic significance of various chromo-
somal 13q14 deletions in chronic lymphocytic leukemia (CLL).
Experimental Design: We analyzed data from SNP 6.0 arrays to define the anatomy of various 13q14
deletions in a cohort of 255CLLpatients andhave correlated two subsets of 13q14deletions (type I exclusive
of RB1 and type II inclusive of RB1) with patient survival. Furthermore, we measured the expression of the
13q14-resident microRNAs by quantitative PCR (Q-PCR) in 242 CLL patients and subsequently assessed
their prognostic significance. We sequenced all coding exons of RB1 in patients with monoallelic RB1
deletion and have sequenced the 13q14-resident miR locus in all patients.
Results: Large 13q14 (type II) deletions were detected in approximately 20%of all CLL patients andwere
associated with shortened survival. A strong association between 13q14 type II deletions and elevated
genomic complexity, as measured through CLL-FISH or SNP 6.0 array profiling, was identified, suggesting
that these lesions may contribute to CLL disease evolution through genomic destabilization. Sequence and
copy number analysis of the RB1 gene identified a small CLL subset that is RB1 null. Finally, neither the
expression levels of the 13q14-resident microRNAs nor the degree of 13q14 deletion, as measured through
SNP 6.0 array-based copy number analysis, had significant prognostic importance.
Conclusions:Our data suggest that the clinical course of CLL is accelerated in patients with large (type II)
13q14 deletions that span theRB1 gene, therefore justifying routine identification of 13q14 subtypes in CLL
management. Clin Cancer Res; 17(21); 6778–90. �2011 AACR.
Introduction
Chronic lymphocytic leukemia (CLL) has a variedclinical course, and genomic aberrations are recognizedas important to the diverse biological and clinical phe-notypes of CLL (1–4). The most frequent chromosomalabnormality in CLL is commonly referred to as del13q14,which is detectable by FISH or single-nucleotide poly-morphism (SNP) arrays in approximately 50% of all CLL(5–15). Although generally considered to be associatedwith favorable outcome, clinical experience and pub-lished data suggest that for a subset of CLL patients withthis deletion outcome is not favorable. The reasons for
this observation are not well understood and despiteapplication of currently available measurable CLL riskfactors, the reliable identification of the relatively unfa-vorable 13q14 patient subset is not possible.
Although the 13q14 probe used in the CLL-FISH panelidentifies all typical 13q14 deletions in CLL, regardless ofsize, recent analyses of CLL genomes using SNP array plat-forms have resulted in the identification of substantialanatomic heterogeneity of 13q14 deletions in CLL (16–19). This anatomic heterogeneity is reflected in large versussmall deletions, monoalleleic versus bialleleic deletions,and various deletion extensions into centromeric and telo-meric 13q regions. This heterogeneity provides clues tounderlying effects of various 13q14 deletions on CLL biol-ogy and clinical behavior andmakes it likely thatmore thanone pathobiological mechanism underlies the existence ofvarious 13q14 deletions in CLL.
To advance our knowledge of the effects of various CLL-associated 13q14 deletions on CLL patients, we have inves-tigated prognostic effects of various 13q14 types and asso-ciatedmolecular events, including 13q14-residentmiR15a/16-1 expression levels, in a prospectively collected cohort of255 CLL patients. In aggregate, our data provide solidsupport for a clinically meaningful separation of 13q14deletions into subtypes that are defined as good prognosistype I [exclusive of retinoblastoma gene (RB1);�30% of allCLL] and poor prognosis type II (inclusive of RB1;�20%of
Authors' Affiliations: 1Departments of Biostatistics and Biostatistics andComputational Biology, Harvard School of Public Health and the Dana-Farber Cancer Institute, Boston, Massachusetts; and 2Division of Hema-tology and Oncology, Department of Internal Medicine and 3Department ofStatistics, University of Michigan, Ann Arbor, Michigan
Note: Supplementary data for this article are available at Clinical CancerResearch Online (http://clincancerres.aacrjournals.org/).
Corresponding Author: Sami N. Malek, Division of Hematology andOncology, Department of Internal Medicine, University of Michigan,1500 E. Medical Center Drive, Ann Arbor, MI 48109. Phone: 734-763-2194; Fax: 734-647-9654; E-mail: [email protected]
all CLL) lesions and for routine measurements of thesesubtypes in CLL clinical management (20–23).
Materials and Methods
PatientsBetween January 2005 andSeptember 2009, a total of 266
patients evaluated at the University of Michigan Compre-hensive Cancer Center were enrolled onto this study. Thetrial was approved by the University of Michigan Institu-tional Review Board (IRBMED #2004-0962) and writteninformed consent was obtained from all patients prior toenrollment. Data from255 of 266 patients were included inthis report (5 patients enrolled on the study were excludedbecause of a diagnosis that was not CLL, and 6 patients hadinsufficient cryopreserved cells available for the analysesdescribed).Regardless of whether the subjects were originally diag-
nosed at our institution or another, we used the same CLLdiagnostic criteria, based on the National Cancer Institute-Working Group Guidelines for CLL (24). Eligible patientsneeded to have an absolute lymphocytosis (>5,000 maturelymphocytes per mL), and lymphocytes needed to expressCD19, CD23, sIg (weak), and CD5 in the absence of otherpan-T-cell markers.Time to first therapy (TTFT) and overall survival were
based on the date of trial enrollment (the date of spec-imen procurement) or alternatively the diagnosis date,respectively (25). CLL treatment was defined as cytotoxicchemotherapy and/or monoclonal antibody therapy forCLL. Clinical information, including Rai stage and alltreatments given, was collected for all patients. Patientsamples were characterized for selected CLL-associatedchromosomal aberrations as a routine clinical test at the
Mayo Clinic (Rochester, MN) by FISH. All biomarkerassessments were conducted on specimens procured attrial enrollment.
Cell isolationFlow cytometric sorting of CLL specimens. Cryopreserved
peripheral blood mononuclear cells from CLL blood speci-mens were prepared for fluorescence-activated cell sorting(FACS) intoCD19þ andCD3þ cells, as previously described(25).
Preparation of sample DNADNA used for SNP 6.0 profiling was extracted from
fluorescence-activated cell-sorted CD19þ and CD3þ cellsas described (25). For some cases (N ¼ 11), paired buccalDNA was used instead of CD3þ cell–derived DNA.
Array data analysisThe DNA was prepared for hybridization to SNP 6.0
arrays according to the manufacturers’ recommendations.Affymetrix CEL files for each cell sample were analyzed withGenotyping Console software for initial quality control,followed by the use of the Affymetrix "Birdseed" algorithmto generate tab-delimited SNP call files in text format. Callrates for the entire group of CD19þ samples included in thisreport were between 95.06% and 99.63%, with a mean callrate of 98.53%.Corresponding call rates for CD3þ or buccalDNA were between 93.14% and 99.66%, with a mean callrate of 98.49%.
Sample copy number heatmap displays were obtainedfrom CEL files through use of the freely available softwaredChip (26), adapted to run on a 64-bit computer environ-ment. For genomic copy number analysis, we visuallyinspected parallel heatmap copy number images of CD19þ
and paired CD3þ/buccal DNA samples generated throughdChipSNP and using the median smoothening functional-ity. Only those copy number changes detected in CD19þ
DNA that were not found at the same position in pairedCD3þ/buccal DNA were called somatic. This approachfollowed our previously externally referenced (FISH) SNP6.0 genomic lesion calling method in acute myelogenousleukemia that resulted in 100% concordance between SNP6.0 profiling and FISH results (for a total of 56 lesionsanalyzed; ref. 27). Furthermore, data on sensitivity andspecificity of acquired copy number (aCNA) calling calcu-lated on the basis of data for del17p, del11q, and del13q14derived from the clinical CLL-FISHpanel are summarized inthe Supplementary Methods and Results section. Using thisapproach, the 3 shortest identified aCNAs were 0.024,0.042, and 0.052 Mb in length and were defined by 18,27, and 19 consecutive SNP positions, respectively, and thevast majority of aCNAs were defined by more than 100consecutive SNP positions. In total, we detected 474 sub-chromosomal losses (size range: 0.024–108.73Mb) and 62subchromosomal gains (size range: 0.114–94.626Mb; ratioof losses to gains of 7.1:1).
The method used to nominate aCNAs by a statisticalalgorithm (27) is summarized in Supplementary Methods.
Translational Relevance
Genomic aberrations have important effects on chr-onic lymphocytic leukemia (CLL) biology and clinicaloutcome. Recently, a refined description of the substan-tial anatomic heterogeneity of the most commonCLL-associated chromosomal deletion, del13q14, hasemerged, and it is increasingly recognized that 13q14deletions cannot be reduced to a singular pathophysio-logic mechanism. Furthermore, knowledge is stillevolving about possible differential prognostic effectsimparted on CLL by various 13q14 subtypes. In thisreport, we provide evidence that large 13q14 deletionsthat are inclusive of the retinoblastoma gene (RB1) areprognostically adverse and that these deletions are fre-quently associated with unstable CLL genomes. Thisanalysis advances our understanding of the clinicalrelevance of the most common chromosomal deletiontype in CLL and informs future refinements in diagnosticapproaches to CLL management.
13q14 Deletion Subtypes and CLL Survival
www.aacrjournals.org Clin Cancer Res; 17(21) November 1, 2011 6779
Exon resequencing of p53, RB1, and the 13q14-residentmiR genomic locus
Primers to amplify and sequence exons 2 to 10 of humanp53 and all coding exons of RB1 and adjacent intronicsequences, including splice junctions, were designed by theprimer 3 program (http://frodo.wi.mit.edu/primer3/) andsequence information was generated as described (28).Mutations were confirmed using paired patient CD3þ/buc-cal DNA as templates. The primers to amplify and sequencethe 13q14-resident miR locus were as follows: F: TTC-TAAGCTCTGTTCAAAATGCT, R: TTGTGTTTCCTAACCTA-TAGCACTG, and SEQ: TAACAAGATTATCAATAATAC.
Determination of ZAP70 and IgVH statusDetermination of ZAP70 and IgVH status was carried out
as described (25).
Measurement of expression of 13q14-residentmicroRNAs 15a and 16-1 usingnormalized quantitativePCR
RNA was prepared from 2� 106 to 4� 106 fluorescence-activated cell-sorted CD19þ cells with the TRIzol reagentand resuspended in 100 mL diethyl pyrocarbonate (DEPC)-treated water. cDNA for microRNA reverse transcriptasequantitative PCR (Q-PCR) was made from approximately5 ng of RNA by the TaqMan MicroRNA Reverse Transcrip-tion Kit (Applied Biosystems) and RNA-specific primers.Primers and TaqMan-based probes were purchased fromApplied Biosystems (primers-on-demand). Primer/probemixtures included were as follows: miR16-1 (TM 391),miR15a (RT 389), RNU43 (TM 1095), and RNU49 (TM1005). Duplicate amplification reactions for microRNAsincluded primers/probes, TaqMan 2� Universal PCR Mas-terMix,NoAmpEraseUNG, and1.35mLof cDNA in a 20-mLreaction volume. Reactions were done on an ABI 7900HTmachine. Normalization of relative copy number estimatesfor RNA species of interest was done with the Ct values forthe RNU43 or RNU49 as reference (Ct mean gene of interest� Ct mean RNU43 or RNU49). Comparisons between CLLsubgroupswere carried out though subtractions ofmeans ofnormalized Ct values. Similar efficacies of amplification ofcDNA using primers for miR15, miR16-1, and RNU49 hadpreviously been verifiedwith 2-fold serial dilutions of cDNAmade from cell line–derived RNA over a range of eight 2-fold serial dilutions (18).
Statistical methodsTTFT or overall survival was defined as the time (in
months) between CLL diagnosis or CLL trial enrollmentand the date (in months) of first treatment or the patient’sdeath. For patients still untreated or alive, the month ofcensoring was November 2010. Univariate and bivariateanalyses were based on Kaplan–Meier estimates of survivorfunctions. Median survival times were estimated directlyfrom the survivor function estimates. Significance levels forgroupwise comparisons in the univariate and bivariateanalyses were based on the log-rank test. Assessment of therelationship between outcomes and various dichotomiza-
tions of a continuous risk factorwasmadeby considering allobserved values of the risk factor as potential dichotomi-zation thresholds and using univariate proportionalhazards regression to estimate the corresponding HR.
Results
Patient characteristicsCharacteristics of the 132CLLpatientswith typical 13q14
deletions as detected through SNP 6.0 profiling (identifiedwithin a prospectively enrolled and profiled cohort of 255CLL patients) analyzed in this study are summarizedin Table 1, stratified by 13q14 subtype and treatment status.Data for 123 patients with CLL without typical 13q14deletions are also summarized (Table 1). Of the 132patients with CLL with typical 13q14 deletions, 100(76%) were untreated (UT) and 32 (24%) relapsed (T) atthe time of study enrollment. Within the group of previ-ously untreated patients, the distribution of importantbiomarkers across type I or II 13q14 lesions was wellbalanced: Rai stage 0, 45%/41%; Rai stages 1 or 2, 51%/53%; IgVH unmutated, 32%/29%; ZAP70 positive, 30%/26%; p53 exons 2 to 10 mutated, 11%/6%; del17p present,7%/3%; and del11q present, 6%/12%. The median timefrom diagnosis to enrollment and from enrollment to dataanalysis for previously untreated patients is detailedin Table 1. All outcome analyses described later are basedon SNP 6.0 array analysis and biomarker measurementsthat were conducted on patient samples procured at studyenrollment, thus avoiding confounding effects of longitu-dinal biomarker instability. Outcome was calculated usingeither the CLL trial enrollment date or the diagnosis date asthe reference dates, as indicated, to minimize the effect oflead time biases.
The pathologic anatomy of acquired subchromosomalgenomic copy number changes spanning 13q14 in CLLas defined through SNP 6.0 array copy numberprofiling
We cataloged all aCNAs on chromosome 13 in our CLLcohort using visual inspection of simultaneous displays ofdChipSNP-based copy number estimates (heatmaps) forCD19þ cells andpairedCD3þ/buccalDNA(Fig. 1).Overall,51.7% (132 of 255) of CLL carried a classical 13q14 dele-tion as detected through SNP 6.0 arrays that included thegenomic region recognized in the clinically used CLL-FISHpanel (range of lesion sizes of 0.198–73.775 Mb; see Sup-plementary Table S1). In addition, rare atypical 13q dele-tions were identified. As previously described by lower-resolution SNP array platforms (Affymetrix SNP50k XbaIarrays), 13q14 deletions displayed substantial anatomicheterogeneity (18). Nonetheless, multiple distinct breakclusters located close to the 13q14-resident miR 15a/16-1loci and, alternatively, close to and inclusive of all or parts ofthe RB1 locus were identified. A telomeric cluster of breakswas located at approximately 50.2 to 50.7 Mb physicalposition, comprising the vastmajority of the breaks of short13q14 deletions. Of the short and relatively uniform 13q14
Ouillette et al.
Clin Cancer Res; 17(21) November 1, 2011 Clinical Cancer Research6780
deletions, 54% (71 of 132) were between 0.678 and 1.944Mb in length and only 2 lesions were identified that wereshorter (CLL145 and CLL97, with lesion lengths of 0.198and 0.425 Mb, respectively; refs. 29–31).
Using a previously proposed classification schema fortypical 13q14 deletions into types I and II (exclusive andinclusive of RB1, respectively), we detected 85 type I lesionsand 54 type II lesions (including 7 CLL cases in which both
lesion types were identified, existing on separate chromo-somes) in this cohort. Furthermore, the frequency of 13q14type I and type II lesions in the entire CLL cohort (N¼ 255)was 33% and 21%, respectively.
Acquired uniparental disomy (aUPD) at 13q was iden-tified in 7CLL cases as previously described (18). In all thesecases, the LOH region was very large (Supplementary TableS2). Six of these 7 cases also contained a region of copy loss
Chromosome 13 overview Chromosome 13 zoom
47 Mb
47.954 Mb
47.776 Mb
RB1
miR15a,miR16-149.521 Mb
51 Mb82 Mb
30 Mb
Type IType II
CD3+ or buccal DNA CD3+ or buccal DNACD19+ DNA CD19+ DNA
Figure 1. Genomic copy number heatmap display of chromosome 13q of 255 CLL cases ranked by the position of centromeric 13q14 deletion break points:Copy number heatmap displays for paired DNA samples based on SNP 6.0 array profiling were generated with dChipSNP. Left, CD3þ or buccal DNA; right,CLL CD19þ DNA. Blue indicates copy loss, and red indicates copy gain. Each column represents one patient.
Table 1. Patient characteristics (Cont'd )
Patient characteristicsat enrollment
13q-Itreatmentnaïve, no. (%)
13q-Irelapsed,no. (%)
13q-IItreatmentnaïve, no. (%)
13q-IIrelapsed,no. (%)
Non-13qtreatmentnaïve, no. (%)
Non-13qrelapsed,no. (%)
Number of priortherapiesMedian NA 2 NA 1 NA 1Range NA 1–5 NA 1–5 NA 1–7
aSeven patients had both 13q-I and 13q-II lesions.bMultiple FISH abnormalities per patient were individually counted.
Ouillette et al.
Clin Cancer Res; 17(21) November 1, 2011 Clinical Cancer Research6782
at 13q14 (typical 13q14 deletions by FISH), whereas 1 casewas associated with a homozygous miR16-1-5p mutation(see later).
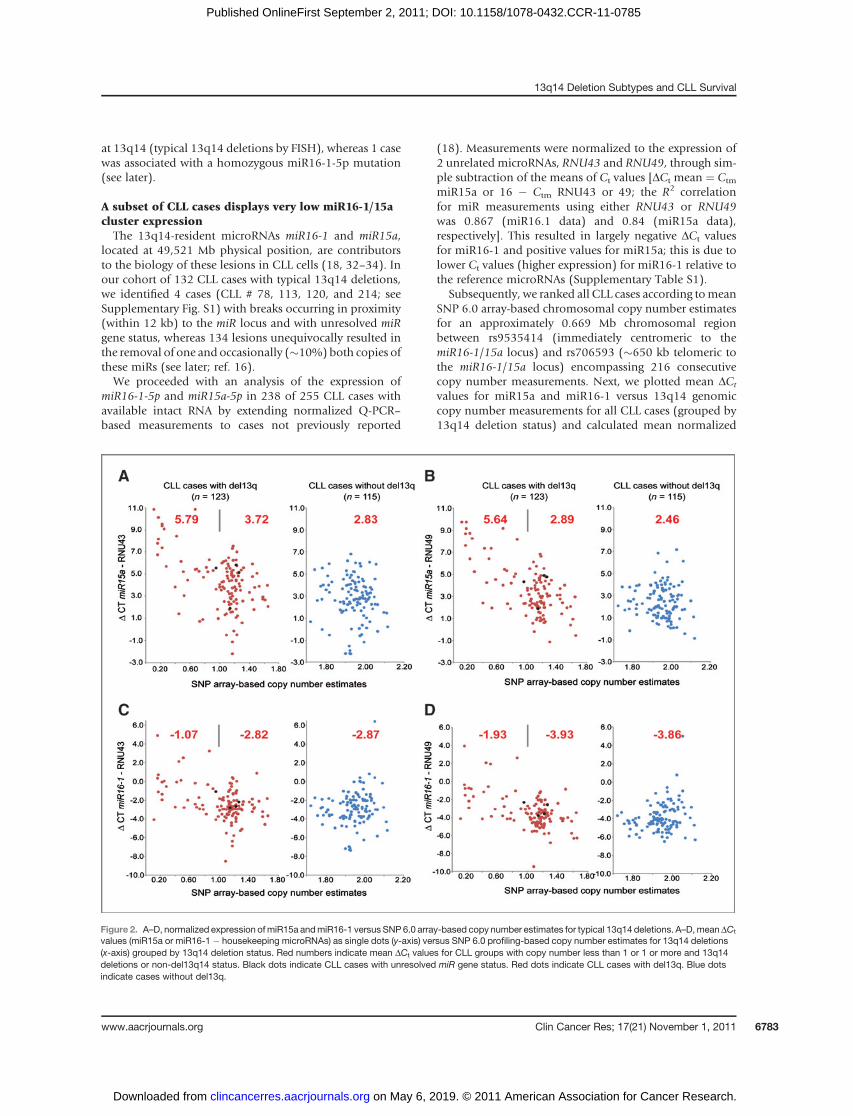
A subset of CLL cases displays very low miR16-1/15acluster expressionThe 13q14-resident microRNAs miR16-1 and miR15a,
located at 49,521 Mb physical position, are contributorsto the biology of these lesions in CLL cells (18, 32–34). Inour cohort of 132 CLL cases with typical 13q14 deletions,we identified 4 cases (CLL # 78, 113, 120, and 214; seeSupplementary Fig. S1) with breaks occurring in proximity(within 12 kb) to the miR locus and with unresolved miRgene status, whereas 134 lesions unequivocally resulted inthe removal of one and occasionally (�10%) both copies ofthese miRs (see later; ref. 16).We proceeded with an analysis of the expression of
miR16-1-5p and miR15a-5p in 238 of 255 CLL cases withavailable intact RNA by extending normalized Q-PCR–based measurements to cases not previously reported
(18). Measurements were normalized to the expression of2 unrelated microRNAs, RNU43 and RNU49, through sim-ple subtraction of the means of Ct values [DCt mean ¼ Ctm
miR15a or 16 � Ctm RNU43 or 49; the R2 correlationfor miR measurements using either RNU43 or RNU49was 0.867 (miR16.1 data) and 0.84 (miR15a data),respectively]. This resulted in largely negative DCt valuesfor miR16-1 and positive values for miR15a; this is due tolower Ct values (higher expression) for miR16-1 relative tothe reference microRNAs (Supplementary Table S1).
Subsequently, we ranked all CLL cases according tomeanSNP 6.0 array-based chromosomal copy number estimatesfor an approximately 0.669 Mb chromosomal regionbetween rs9535414 (immediately centromeric to themiR16-1/15a locus) and rs706593 (�650 kb telomeric tothe miR16-1/15a locus) encompassing 216 consecutivecopy number measurements. Next, we plotted mean DCt
values for miR15a and miR16-1 versus 13q14 genomiccopy number measurements for all CLL cases (grouped by13q14 deletion status) and calculated mean normalized
Figure 2. A–D, normalized expression ofmiR15a andmiR16-1 versus SNP6.0 array-based copy number estimates for typical 13q14 deletions. A–D,meanDCt
values (miR15a or miR16-1 � housekeeping microRNAs) as single dots (y-axis) versus SNP 6.0 profiling-based copy number estimates for 13q14 deletions(x-axis) grouped by 13q14 deletion status. Red numbers indicate mean DCt values for CLL groups with copy number less than 1 or 1 or more and 13q14deletions or non-del13q14 status. Black dots indicate CLL cases with unresolved miR gene status. Red dots indicate CLL cases with del13q. Blue dotsindicate cases without del13q.
13q14 Deletion Subtypes and CLL Survival
www.aacrjournals.org Clin Cancer Res; 17(21) November 1, 2011 6783
expression values formiR16-1 andmiR15a for the CLL caseswith more extensive chromosomal loss (copy number esti-mates <1) and for the CLL cases with less extensive loss(copy number estimates �1) or no loss (cases without13q14 deletions). Data are summarized in Fig. 2A–D, withmean miR expression levels indicated with red numbers.
Mean normalized expression values of both miR16-1and miR15a were substantially lower (miR15a �8-foldand miR16-1 �4-fold, respectively) in the CLL subgroupwith more extensive del13q14 deletions (copy numberestimates <1) as opposed to the group with no del13q14.For CLL cases with monoallelic 13q14 deletions andassociated copy number estimates of 1 or more (themajority of such cases had copy number estimates of�1.2) as compared with cases without 13q14 deletions,there was evidence for a gene dosage effect for miR15aexpression (�1.3- to 1.9-fold lower) but no such effect formiR16-1 expression.
This combined data suggests that approximately 12% to15%ofCLL cases have very lowmiR16-1/15a levels and thatthe majority of CLL cases (�85%) display an overlappingrange of expression of these miRs, with mild relative reduc-tions inmiR15 expression inmonoallelically deleted 13q14cases.
Assessment of elevated HRs for short overall survivalbased on analysis of thresholded centromeric ortelomeric 13q14 lesion breakpoints
Given various published observations on CLL 13q14deletion sizes, associated gene deletions, and clinicalparameters, including our proposal for 13q14 subclassi-fication into types I and II based on RB1 gene status, wemodeled HRs for shortened survival for dichotomized13q14 groups defined by distinct physical chromosomalbreak positions. Specifically, various dichotomized13q14 groups were defined by sliding the separation/break points across all physical positions actually iden-tified in the 13q14 deletions in this cohort (based on thephysical positions of either telomeric or centromericbreaks; Supplementary Table S1). Using such anapproach, we determined that the location of centromericbreakpoints strongly influenced the risk for short overallsurvival in CLL, whereas the telomeric breakpoints hadlittle effect (Supplementary Fig. S2).
Specifically, the HR for short overall survival increased asthe centromeric separation break for the two 13q14 groupsmoved closer to the telomere, with the highest HRs iden-tified for breaks at approximately 48 to 48.5 Mb physicalposition, followed by a sharp drop in risk. The RB1, withwell-documented effects on cell-cycle control and chromo-somal instability in cancer cells, is located at 47.776 to47.954 Mb physical position and is very close to the cohortdichotomization breakpoints that are associated with peakHRs for short overall survival, thus providing justificationfor a 13q14 classification schema centered on RB1 (see alsoRB1 mutation and expression data later). Additional geneslocated telomeric to RB1 and in the area (48–48.5 Mbphysical position) demarcated by the highest HRs for short
overall survival included LPAR6, P2RY5, RCBTB2, andCYSLTR2, but none of these genes have well-establishedroles in biological processes that are known to affect cancercells. Finally, only 4 actual 13q14-associated chromosomalbreaks occurred between 48 and 48.5 Mb physical posi-tions, creatingmodest uncertainty about the actual physicalpositions associated with peak HRs as estimated by ourapproach.
Results of univariate outcome analyses of SNP 6.0array-detectable 13q14deletionsandoverall survival inCLL
We initially determined the prognostic value of SNP 6.0array-defined 13q14 type I and type II deletions on overallsurvival in the CLL cohort (UT þ T; N ¼ 132) usingunivariate analysis and either the CLL sample procure-ment/trial enrollment date or the CLL diagnosis date asthe reference date. For both analyses, overall survival wassignificantly shorter for CLL patients with 13q14 type IIlesions, as opposed to patients with 13q14 type I lesions(Kaplan–Meier plots for these analyses are displayed in Fig.3A andC). These findings were true in the analyses in whichCLL cases that carried both 13q14 lesion types (N¼ 7) wereincluded and hierarchically assigned to type II status(shown) or, alternatively, were excluded from analysis.Next, we analyzed the prognostic value of SNP 6.0 array-defined 13q14 type I and type II deletions on overallsurvival in the untreated subset of patients (UT; N ¼100) and again noted shortened survival of CLL patientswith 13q14 type II, as opposed to type I lesions (Kaplan–Meier plots for these analyses are displayed in Fig. 3Band D).
Finally, given that the nature of 13q14 deletion mayalso influence initial CLL disease progression, we ana-lyzed TTFT estimates for the 2 13q14 types but found nostrong effects.
Deletion 13q14 type II lesions are associated with ahigher incidence of coexisting CLL-FISH–detectablechromosomal abnormalities than 13q14 type Ilesions
Next, we determined the frequency of 13q14 deletions oftype I and II lesions that are associated with coexistinggenomic lesions as detected through (i) CLL-FISH-25 or(ii) SNP 6.0 array profiling (see later). We defined 13q14deletions of either type I or II lesions as "FISH-complex" ifthey were associated with one or more additional abnor-malities in the clinically used CLL-FISH panel [and if theseabnormalities were detected in �25% of nuclei analyzed(FISH-25); this is to remove low percentage lesions ofunclear clinical or biological significance]. Using thesecriteria, the following frequencies were measured: 13q14-I (N ¼ 85): sole abnormality (74 of 85 ¼ 87%), FISH-complex (11 of 85 ¼ 13%); 13q14-II (N ¼ 54): soleabnormality (38 of 54 ¼ 70%), FISH-complex [16 of54 ¼ 30%; (P ¼ 0.03, the Fisher exact test); CLL cases withboth type I and II lesions (N ¼ 7) were counted as 13q14-Iand 13q14-II].
Ouillette et al.
Clin Cancer Res; 17(21) November 1, 2011 Clinical Cancer Research6784
Outcome analysis for del13q14 subtypes andassociated CLL-FISH lesionsWe proceeded with an outcome analysis of the relative
contributions of 13q14 subtypes by category (I vs. II)versus associated CLL-FISH-25–based genomic complex-ity. Initially, we focused on bivariate analyses in which13q14 lesion types were further separated into solelesions or lesions coexisting with any other CLL-FISH-25 lesion (CLL cases that carried both 13q14 lesion typeswere included and hierarchically assigned to type IIstatus). Data are summarized in Fig. 3E–H. From theseanalyses, the following conclusions can be supported: (i)sole 13q14 lesions of either type were associated with arelatively better prognosis than either type existing in thepresence of additional FISH findings; (ii) trends forshorter overall survival for 13q14 type II deletions withCLL-FISH complexity versus 13q14 type I deletions withCLL-FISH complexity were observed; (iii) trends forshorter overall survival for sole 13q14 type II versus sole13q14 type I lesions were observed, albeit more pro-nounced in CLL cases that were relapsed at the time ofanalysis (UT þ T plots); and (iv) 13q14 type II deletionswith CLL-FISH complexity were substantially enriched inrelapsed CLL patients (frequency 45%).
Deletion of 13q14 type II lesions is associated with ahigher incidence of coexisting SNP 6.0 array-detectableacquired subchromosomal copy number aberrationsthan 13q14 type I lesions
Next, we determined the frequencies of either 13q14lesion type that were associated with 2 or more additionalSNP 6.0 array-based subchromosomal aCNAs (total aCNAcomplexity �3). The following frequencies were measured:(i) 13q14-I (N ¼ 85), sole abnormality (45 of 85 ¼ 53%)and 13q14-I plus 2 or more aCNA (16 of 85 ¼ 19%); (ii)13q14-II (N¼ 54), sole abnormality (20 of 54¼ 37%) and13q14-II plus 2 or more aCNAs (18 of 54¼ 33%; P¼ 0.02,the Fisher exact test; CLL cases with both type I and II lesionswere counted as 13q14-I and 13q14-II). For comparison,the frequency of 3 or more SNP 6.0 array-based subchro-mosomal aCNAs in our entire CLL cohort of 255 profiledpatients was 20% (35, 36).
Given the known strong association of p53 mutationswith elevated genomic complexity in CLL, we determinedp53 exon 2 to 10mutation frequencies in the CLL cases with13q14 type I or II deletions and 2 or more additional SNP6.0-based aCNAs: There were 16 cases with 13q14-I plus 2ormore aCNA,ofwhich7 (44%)weremutated in p53 exons2 to10, and therewere 18 caseswith13q14-II plus 2ormore
UT+TA B E F
C D G H
Pro
port
ion s
urv
ivin
g
1.0
0.8
0.6
0.4
0.2
0.0
Pro
port
ion s
urv
ivin
g
1.0
0.8
0.6
0.4
0.2
0.0
Pro
port
ion s
urv
ivin
g
1.0
0.8
0.6
0.4
0.2
0.0
Pro
port
ion s
urv
ivin
g
Pro
port
ion s
urv
ivin
g
1.0
0.8
0.6
0.4
0.2
0.0
Pro
port
ion s
urv
ivin
g
1.0
0.8
0.6
0.4
0.2
0.0
Pro
port
ion s
urv
ivin
g
1.0
0.8
0.6
0.4
0.2
0.0
1.0
0.8
0.6
0.4
0.2
0.0
Pro
port
ion s
urv
ivin
g
1.0
0.8
0.6
0.4
0.2
0.0
0 10 20 30 40
13q-I (n = 78)
Time from enrollment (mo)
0 50 100 150 200 250 300 350
Time from diagnosis (mo)
0 50 100 150 200 250 300 350
Time from diagnosis (mo)
0 50 100 150 200 250 300 350
Time from diagnosis (mo)
0 50 100 150 200 250 300 350
Time from diagnosis (mo)
P < 0.00113q-II (n = 54)
13q-I (n = 78)
P = 0.0113q-II (n = 54)
13q-I (n = 66)
13q-I single (n = 67)
13q-II single (n = 38)
13q-I complex (n = 11)
13q-II complex (n = 16)
13q-I single (n = 67)
13q-II single (n = 38)
13q-I complex (n = 11)
13q-II complex (n = 16)
13q-I single (n = 58)
13q-II single (n = 27)
13q-I complex (n = 8)
13q-II complex (n = 7)
13q-I single (n = 58)
13q-II single (n = 27)
13q-I complex (n = 8)
13q-II complex (n = 7)
P = 0.04
P = 0.03
P = 0.04
P = 0.15
P < 0.001
P < 0.001
P = 0.03
P = 0.13
P = 0.13
P = 0.04
P = 0.07
P < 0.001
P < 0.001
13q-II (n = 34)
13q-I (n = 66)
P = 0.0713q-II (n = 34)
50 60 70 0 10 20 30 40
Time from enrollment (mo)
50 60 70 0 10 20 30 40
Time from enrollment (mo)
50 60 70 0 10 20 30 40
Time from enrollment (mo)
50 60 70
UT only UT+T UT only
UT+T UT only UT+T UT only
Figure 3. A–H, deletions 13q14 types I or II and overall survival in CLL (Kaplan–Meier plots). A–D, 13q14 type I or II deletions and overall survival in CLL. UT,untreated at enrollment; UT þ T, untreated or relapsed at enrollment. A and B, overall survival from date of enrollment. C and D, overall survival from date ofdiagnosis. E–H, pairwise groupings by 13q14 status (I or II) and any associated FISH lesions. The suffix "complex" indicates a 13q14 deletion with any (�1)coexisting FISH-25 finding.
13q14 Deletion Subtypes and CLL Survival
www.aacrjournals.org Clin Cancer Res; 17(21) November 1, 2011 6785
aCNAs, of which 6 (34%) were mutated in p53 exons 2to 10.
Resultsofoutcomeanalysesof 13q14deletionsof type Ior II lesions stratified by associated SNP 6.0 arrayprofiling–detectable subchromosomal aCNA loadsversus overall survival in CLL
We proceeded with bivariate outcome analysis of 13q14deletions of type I and II lesions grouped by associated SNP6.0 array-based subchromosomal aCNA status of 2 or moreand 3 or more, respectively (CLL cases that carried both13q14 lesion types were included and hierarchicallyassigned to type II status). Kaplan–Meier plots for theseanalyses are displayed in Fig. 4A–H. From these analyses,the following conclusions are supported: (i) 13q14 lesionsof either type with elevated associated aCNA counts wereassociated with a significantly worse prognosis than eithertype with no or additional aCNAs below indicated thresh-olds; and (ii) 13q14 type II deletions were prognosticallymore adverse than 13q14 type I deletions, as evidenced byan accelerated disease course in cases with low associatedcomplexity (aCNA �1 or �2, respectively), a finding thatwas particularly evident in CLL cases that were relapsed atthe time of analysis (UT þ T plots).
Next, we conducted bivariate outcome analysis of13q14 deletions of type I and II lesions stratified by
associated SNP 6.0 array-based subchromosomal aCNAstatus of 2 or more and 3 or more, respectively, basedon aCNA nominations that had been made by algorith-mic aCNA calling methods (see Supplementary Meth-ods). Results were similar to results based on aCNAnominations made through visual heatmap inspection.Kaplan–Meier plots are displayed in Supplementary Fig.S3A–H.
Degree of 13q14 deletions and outcome in CLLThe degree of 13q14 deletions was quantified using the
mean of 216 consecutive copy number estimates based onprobes located within 13q14 deletions and correlatedwith the outcome measures TTFT and overall survival forthe group of 132 CLL patients with 13q14 deletions. TheCLL cohort was dichotomized at every actual copy num-ber measurement and HRs for short TTFT or overallsurvival were computed. As can be seen in SupplementaryFig. S4A–H, a copy number estimate of approximately 1(N ¼ 32 for copy number <1 and N ¼ 100 for copynumber �1) and high copy number cutoffs (the latterbased on small N) optimally separated the cohort, sug-gesting that the degree of 13q14 deletion could havenegative effects on CLL outcome once larger cohorts arestudied (for this cohort, the 95% confidence bands in grayoverlapped an HR of 1; ref. 37).
UT+T UT+TA B E F
C D G H
Pro
port
ion s
urv
ivin
g
1.0
0.8
0.6
0.4
0.2
0.0
Pro
port
ion s
urv
ivin
g
1.0
0.8
0.6
0.4
0.2
0.0
Pro
port
ion s
urv
ivin
g
1.0
0.8
0.6
0.4
0.2
0.0
Pro
port
ion s
urv
ivin
g
1.0
0.8
0.6
0.4
0.2
0.0
Pro
port
ion s
urv
ivin
g
1.0
0.8
0.6
0.4
0.2
0.0
Pro
port
ion s
urv
ivin
g
1.0
0.8
0.6
0.4
0.2
0.0
Pro
port
ion s
urv
ivin
g
1.0
0.8
0.6
0.4
0.2
0.0
Pro
port
ion s
urv
ivin
g
1.0
0.8
0.6
0.4
0.2
0.0
0 10 20 30 40
Time from enrollment (mo)
0 50 100 150 200 250 300 350
Time from diagnosis (mo)
0 50 100 150 200 250 300 350
Time from diagnosis (mo)
13q-I ≤1 sc (n = 66)
13q-II ≤1 sc (n = 36)
13q-I ≥2 sc (n = 12)
13q-II ≥2 sc (n = 18)
13q-I ≤1 sc (n = 57)
13q-II ≤1 sc (n = 28)
13q-I ≥2 sc (n = 9)
13q-II ≥2 sc (n = 6)
13q-I ≤1 sc (n = 57)
13q-II ≤1 sc (n = 28)
13q-I ≥2 sc (n = 9)
13q-II ≥2 sc (n = 6)
13q-I ≤1 sc (n = 66)
13q-II ≤1 sc (n = 36)
13q-I ≥2 sc (n = 12)
13q-II ≥2 sc (n = 18)
13q-I ≤2 sc (n = 72)
13q-II ≤2 sc (n = 43)
13q-I ≥3 sc (n = 6)
13q-II ≥3 sc (n = 11)
13q-I ≤2 sc (n = 62)
13q-II ≤2 sc (n = 31)
13q-I ≥3 sc (n = 4)
13q-II ≥3 sc (n = 3)
13q-I ≤2 sc (n = 62)
13q-II ≤2 sc (n = 31)
13q-I ≥3 sc (n = 4)
13q-II ≥3 sc (n = 3)
13q-I ≤2 sc (n = 72)
13q-II ≤2 sc (n = 43)
13q-I ≥3 sc (n = 6)
13q-II ≥3 sc (n = 11)
P < 0.001P < 0.001
P < 0.001
P = 0.01 P = 0.01
P < 0.001
P < 0.001
P < 0.001
P < 0.001
P < 0.001
P < 0.001 P < 0.001
P = 0.03
P < 0.001
P = 0.01
P = 0.06
P = 0.05
P = 0.05 P = 0.01 P = 0.07P = 0.08
50 60 70 0 10 20 30 40
Time from enrollment (mo)
50 60 70
0 10 20 30 40
Time from enrollment (mo)
0 50 100 150 200 250 300 350
Time from diagnosis (mo)
0 50 100 150 200 250 300 350
Time from diagnosis (mo)
50 60 70 0 10 20 30 40
Time from enrollment (mo)
50 60 70
UT+T UT+T
UT only UT only UT only UT only
Figure 4. A–H, deletion 13q14 types I or II and associated SNP 6.0 array profiling–based aCNA and overall survival in CLL (Kaplan–Meier plots). A–D, overallsurvival from date of enrollment. E–H, overall survival from date of diagnosis. UT, untreated at enrollment; UT þ T, untreated or relapsed at enrollment. Thesuffix "sc" indicates subchromosomal aCNA present at indicated thresholds.
Ouillette et al.
Clin Cancer Res; 17(21) November 1, 2011 Clinical Cancer Research6786
13q14-resident miR15a/16-1 expression and outcomein CLLThe relative expression levels of the 13q14-resident miRs
as measured by Q-PCR (see earlier) were tested as a prog-nostic factor. The CLL cohort was dichotomized at everyactual normalizedmiR expressionmeasurement (actualDCt
values) andHRs computed.No clear prognostic effects wereidentified (Supplementary Fig. S5A–H and S6A–H). Thiswas equally true for 13q14-resident miR levels tested asprognostic factors in the entire CLL cohort (N ¼ 255; datanot shown).
CLL with 13q14 type II are associated with lower RB1mRNA expression levels and RB1 frameshiftmutations or bialleleic RB1 deletions in a minoritysubset of casesThe RB1 gene coding exons were resequenced in 53 of the
CLL with 13q14 type II deletions, resulting in the identifi-cation of 2 cases (CLL# 158 and 173) with somaticallyacquired truncating frameshift mutations. Together withone case of CLL with biallelic RB1 deletion (CLL# 13), thisindicates that approximately 5% of CLL with 13q14 type IIdeletions are RB1 null.
Next, wemeasured RB1mRNA expression by normalizedQ-PCR in the first 160 consecutively enrolled cases withintact RNA and identified significantly lower RB1 expres-sion in CLL with 13q14 type II deletions as opposed to CLLwith 13q14 type I deletions or no 13q14 deletions. Data aresummarized in Fig. 5.
Identification of rare somatically acquired mutationsin 13q14-resident miR16-3p and miR16-5p in CLL
Sequence analysis of the genomic locus for miR16 and15a and flanking sequences in the entire CLL cohort iden-tified 2 somatically mutated miR16 cases and no miR15amutations. CLL # 61 harbored a heterozygous 6 bp deletionspanning miR16-3p, whereas CLL # 70 carried a homozy-gous 1 nucleotide deletion in miR16-5p in the setting ofchromosome 13 aUPD. The vast majority of CLL cases(99%) carried wild-type miR 16 and 15a genes. Data aresummarized in Fig. 6.
Discussion
In this study, we used SNP 6.0 arrays to interrogategenomic DNA isolated from fluorescence-activated cell-
Normalized Rb1 mRNA levels
*
A B Rb1 mutation data
12.0
10.0
8.0
ΔCT V
alu
es (
Rb
-GA
PD
)
6.0
4.0
100 100
100
CLL-158 CD19+ DNA
deletion 757A
truncated Rb1 Δ 200-928
T T T T T T TC CCCC CG GA A AA AAAAAG
T T T T T T TC CCCC CG GA A A AA AAAAAG
CLL-173 CD19+ DNA
1238C>C/T
truncated Rb1 Δ 358-928
CLL-158 CD3+ DNA
wild-type
Nucleotides are numbered according toNCBI sequence NM_000321.2
CLL-173 CD3+ DNA
wild-type
100
2.0
= P < 10–6
0.0
del13q
Type I
del13q
Type IInon-del 13q
Figure 5. A and B, identification of somatically acquired RB1mutations and results of Q-PCR–based RB1 expression analysis. A, DCt values [Ctm RB1 � Ctm
glyceraldehyde 3-phosphate dehydrogenase (GAPDH)] with each dot representing the mean of duplicate measurements in individual patients (N ¼ 160).Groupings are by 13q14 status. B, RB1 mutation results in CD19þ-derived DNA versus CD3þ-derived DNA.
13q14 Deletion Subtypes and CLL Survival
www.aacrjournals.org Clin Cancer Res; 17(21) November 1, 2011 6787
sorted CD19þ cells compared with either paired sortedCD3þ cells (96%) or paired buccal (4%) DNA from 255CLL patients for acquired chromosomal copy numberchanges (aCNA) and LOH with a detailed focus on13q14 deletions. The novel findings from this study are(i) that 13q14 deletions have varying effects on CLL clinicalbehavior, with large 13q14 deletions (type II: inclusive ofRB1) associated with an inferior prognosis; (ii) that 13q14type II deletions are frequently associated with additionalaCNA (a total aCNA count of �3 was detected in �35% ofcases; ref. 35); (iii) that approximately 10% to 15% of CLLwith bialleleic 13q14 deletions display markedly reducedmiR15a/16-1 levels but that the effect of monoallelic miRlossonmiRexpression is subtle andonly shown formiR15a;(iv) that the relative expression levels of the 13q14-residentmiRsdonot substantially affectCLLprognosis; (v) that a fewCLL cases with 13q14 type II deletions carry RB1 frameshiftmutations and therefore are RB1 null; and (vi) that rare CLLcases harbor acquired mutations in miR16.1.
This study is characterized by several methodologicstrengths including paired (CD19þ vs. CD3þ) array anal-ysis for all CLL samples, ultrahigh-purity (sorted) cells asa source of DNA and RNA (for miR expression analysis),and a rigorous and conservative genomic copy numberdata analysis schema that had previously been externallyvalidated by FISH (27). The analysis of paired samples isof particular importance for the accurate determination ofgenomic complexity in cancer cell genomes in the settingof ultrahigh-resolution SNP arrays, given that the vastmajority of copy number variations (CNV) are less than 1Mb in length. In all, our approach results in very high
specificity and sensitivity for aCNA, thus avoiding pro-blems for the precision of genomic clinical correlativeanalysis resulting from under- or overcalling of genomiclesions.
As part of the work presented here, we provide refinedestimates for optimal physical separation cutoffs for 13q14deletions into prognostically significant subtypes. From apractical perspective, routine inclusion of a probe forRB1orgenes located within 300 to 500 kb telomeric region to RB1should reliably identify CLL patients carrying more adverse13q14 subtypes (17).
Our novel identification of somatically acquired RB1null states, including somatically acquired RB1 muta-tions, provides evidence that for a subset of 13q14 dele-tions, RB1 is a relevant target gene. Furthermore, thisconclusion is supported by the well-studied effects of RB1on cell-cycle control and genomic stability (38). Howev-er, given the identification of multiple 13q14 deletiontypes, it is likely that additional 13q14-resident genesactively influence CLL biology and clinical phenotypesand that the identification of such genes remains animportant research objective.
The identification of rare CLL with miR16.1 mutationsprovides additional evidence for a functional role of thismicroRNA in CLL pathophysiology. Interestingly, 1 of the 2miR16.1mutations occurred inmiR16.1-3p, indicating thatfurther studies of this gene product are warranted. Of note,however, monoallelic miR16.1 loss is insufficient to affectmiR16.1 expression compared with nondeleted CLL cases,thus providing constraints on models relating to the exactrole of this microRNA in 13q14 biology.
T T T T
T T
C C N G
G G G G
G80 8070
130 130
70C
C C C
C C C CA A
A A A T TTG G G GC C CA A A
A A T T TTC C GGN C C C C CA AA A
CD19+, CLL-61A
B
hsa-miR-16-1* hsa-miR-16
hsa-miR-16-1* hsa-miR-16
CD19+, CLL-70 CD3+, CLL-70
CD3+, CLL-61
Figure 6. A and B, identification ofsomatically acquired miR16.1mutations: miR16.1 mutationresults in CD19þ-derived DNAversus CD3þ-derived DNA. Thenucleotide sequenceof themiR16.1gene located within 13q14 isindicated and miR16.1-3p andmiR16.1-5p are highlighted inyellow.
Ouillette et al.
Clin Cancer Res; 17(21) November 1, 2011 Clinical Cancer Research6788
This report is based on CLL patients enrolled prospec-tively at a single center with an outcome focus on overallsurvival. To minimize the effect of biases introducedthrough sample collections relative to prior treatments oractual dates of diagnosis, we have analyzed and presenteddata for the untreated and combined (untreated andrelapsed) CLL cohort separately, using either the diagnosisor trial enrollment date as the reference dates.Such an analysis approach seems particularly relevant to
biomarkers that change over the course of a patient’s illnessor for markers for which such a change is likely but not yetproven through longitudinal analysis (inclusive of thehypothesis that 13q14 type II deletions arise during CLLdisease evolution). About the conclusion that 13q14 type IIdeletions constitute a factor associated with and contribu-tory tomore aggressive CLL, wewere able to show that theselesions are disproportionally associated with elevated geno-mic complexity inCLL: a trait associatedwith aggressiveCLL(25). It is, however, important to note that additionalstudieswith substantially larger patient numbers are neededto be able to measure the relative contribution of 13q14deletion subtypes versus other factors known to affect CLLclinical behavior.One of the current, still speculative, interpretations of the
combined available data on 13q14 deletions in CLL is thatshort 13q14 deletions affect genes that confer a growth orsurvival/antiapoptotic advantage on CLL cells, whereaslonger 13q14 deletions are part of CLL disease evolutionand degeneration into more aggressive subtypes; the latterwould be due to increased genomic instability (as also seenwith 17p and 11q deletions) and an ensuing potential foroutgrowth of more aggressive CLL subclones (35, 39) andlikely other as-yet unidentified mechanisms.
In summary, our data show that 13q14 deletions inCLL are biologically and prognostically distinct entitiesand that routine clinical identification of 13q14 type Iand II deletions would allow for refined CLL patient riskstratification.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Authors' Contributions
P. Ouillette, J. Li, and S. Malek carried out the laboratory research. R. Collinsand S. Malek analyzed clinical data. K. Shedden, S. Shakhan, and C. Liassisted with statistical analysis and software development for data analysis.S. Malek conceived the study and supervised the work. P. Ouillette, K.Shedden, and S. Malek wrote the manuscript.
Acknowledgments
The authors thank the microarray core of the University of MichiganComprehensive Cancer Center for the services provided.
Grant Support
This work was supported by the NIH through grant 1R01 CA136537-01 (to S.N. Malek), the Translational Research Program of the Leukemiaand Lymphoma Society of America (to S.N. Malek), and a Scholar inClinical Research Award from the Leukemia and Lymphoma Society ofAmerica (to S.N. Malek). This research is supported (in part) by the NIHthrough the University of Michigan’s Cancer Center Support grant (5 P30CA46592).
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicatethis fact.
ReceivedMarch 23, 2011; revised July 14, 2011; accepted August 19, 2011;published OnlineFirst September 2, 2011.
References1. Dohner H, Stilgenbauer S, Benner A, Leupolt E, Krober A, Bullinger L,
et al. Genomic aberrations and survival in chronic lymphocytic leuke-mia. N Engl J Med 2000;343:1910–6.
2. Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. N EnglJ Med 2005;352:804–15.
3. Shanafelt TD, Hanson C, Dewald GW, Witzig TE, LaPlant B, Abra-hamzonJ, et al. Karyotype evolutionon fluorescent in situhybridizationanalysis is associated with short survival in patients with chroniclymphocytic leukemia and is related toCD49d expression. JClinOncol2008;26:e5–6.
4. Juliusson G, Oscier DG, Fitchett M, Ross FM, Stockdill G, Mackie MJ,et al. Prognostic subgroups in B-cell chronic lymphocytic leukemiadefined by specific chromosomal abnormalities. N Engl J Med1990;323:720–4.
5. Peterson LC, Lindquist LL, Church S, Kay NE. Frequent clonalabnormalities of chromosome band 13q14 in B-cell chronic lym-phocytic leukemia: multiple clones, subclones, and nonclonalalterations in 82 midwestern patients. Genes Chromosomes Cancer1992;4:273–80.
6. Bullrich F, Veronese ML, Kitada S, Jurlander J, Caligiuri MA, Reed JC,et al. Minimal region of loss at 13q14 in B-cell chronic lymphocyticleukemia. Blood 1996;88:3109–15.
7. Liu Y, HermansonM,Grander D,MerupM,WuX, HeymanM, et al. 13qdeletions in lymphoid malignancies. Blood 1995;86:1911–5.
8. RowntreeC, Duke V, Panayiotidis P, Kotsi P, PalmisanoGL, HoffbrandAV, et al. Deletion analysis of chromosome 13q14.3 and characteri-
sation of an alternative splice form of LEU1 in B cell chronic lympho-cytic leukemia. Leukemia 2002;16:1267–75.
9. Wolf S,MertensD,SchaffnerC,KorzC,DohnerH,StilgenbauerS, et al.B-cell neoplasia associated gene with multiple splicing (BCMS): thecandidateB-CLLgeneon13q14comprisesmore than560 kbcoveringall critical regions. Hum Mol Genet 2001;10:1275–85.
10. Rondeau G, Moreau I, Bezieau S, Petit JL, Heilig R, Fernandez S, et al.Comprehensive analysis of a large genomic sequence at the putativeB-cell chronic lymphocytic leukaemia (B-CLL) tumour suppressergene locus. Mutat Res 2001;458:55–70.
11. Migliazza A, Bosch F, Komatsu H, Cayanis E, Martinotti S, Toniato E,et al.Nucleotidesequence, transcriptionmap, andmutation analysis ofthe 13q14 chromosomal region deleted in B-cell chronic lymphocyticleukemia. Blood 2001;97:2098–104.
12. Mabuchi H, Fujii H, Calin G, Alder H, Negrini M, Rassenti L, et al.Cloning and characterization of CLLD6, CLLD7, and CLLD8, novelcandidate genes for leukemogenesis at chromosome 13q14, a regioncommonly deleted inB-cell chronic lymphocytic leukemia. Cancer Res2001;61:2870–7.
13. Kitamura E, Su G, Sossey-Alaoui K, Malaj E, Lewis J, Pan HQ, et al. Atranscription map of the minimally deleted region from 13q14 in B-cellchronic lymphocytic leukemia as defined by large scale sequencing ofthe 650 kb critical region. Oncogene 2000;19:5772–80.
14. Kapanadze B, Makeeva N, Corcoran M, Jareborg N, Hammarsund M,Baranova A, et al. Comparative sequence analysis of a region onhuman chromosome 13q14, frequently deleted in B-cell chronic
13q14 Deletion Subtypes and CLL Survival
www.aacrjournals.org Clin Cancer Res; 17(21) November 1, 2011 6789
lymphocytic leukemia, and its homologous region on mouse chromo-some 14. Genomics 2000;70:327–34.
15. Grubor V, Krasnitz A, Troge JE, Meth JL, Lakshmi B, Kendall JT, et al.Novel genomic alterations and clonal evolution in chronic lymphocyticleukemia revealed by representational oligonucleotide microarrayanalysis (ROMA). Blood 2009;113:1294–303.
16. Mosca L, Fabris S, Lionetti M, Todoerti K, Agnelli L, Morabito F, et al.Integrative genomics analyses reveal molecularly distinct subgroupsof B-cell chronic lymphocytic leukemia patients with 13q14 deletion.Clin Cancer Res 2010;16:5641–53.
17. Parker H, Rose-Zerilli MJ, Parker A, Chaplin T, Wade R, Gardiner A,et al. 13q deletion anatomy and disease progression in patients withchronic lymphocytic leukemia. Leukemia 2011;25:489–97.
18. Ouillette P, Erba H, Kujawski L, Kaminski M, Shedden K, Malek SN.Integrated genomic profiling of chronic lymphocytic leukemiaidentifies subtypes of deletion 13q14. Cancer Res 2008;68:1012–21.
19. Pfeifer D, Pantic M, Skatulla I, Rawluk J, Kreutz C, Martens UM,et al. Genome-wide analysis of DNA copy number changes andLOH in CLL using high-density SNP arrays. Blood 2007;109:1202–10.
20. Liu Y, Grander D, Soderhall S, Juliusson G, Gahrton G, Einhorn S.Retinoblastoma gene deletions in B-cell chronic lymphocytic leuke-mia. Genes Chromosomes Cancer 1992;4:250–6.
21. Liu Y, Szekely L, Grander D, Soderhall S, Juliusson G, Gahrton G, et al.Chronic lymphocytic leukemia cells with allelic deletions at 13q14commonly have one intact RB1gene: evidence for a role of an adjacentlocus. Proc Natl Acad Sci U S A 1993;90:8697–701.
22. Dohner H, Pilz T, Fischer K, Cabot G, Diehl D, Fink T, et al. Molecularcytogenetic analysis of RB-1 deletions in chronic B-cell leukemias.Leuk Lymphoma 1994;16:97–103.
23. Stilgenbauer S, Dohner H, Bulgay-Morschel M, Weitz S, Bentz M,Lichter P. High frequency of monoallelic retinoblastoma gene deletionin B-cell chronic lymphoid leukemia shown by interphase cytogenet-ics. Blood 1993;81:2118–24.
24. ChesonBD, Bennett JM,GreverM, KayN, KeatingMJ,O'Brien S, et al.National Cancer Institute-sponsored Working Group guidelines forchronic lymphocytic leukemia: revised guidelines for diagnosis andtreatment. Blood 1996;87:4990–7.
26. Lin M, Wei LJ, Sellers WR, Lieberfarb M, Wong WH, Li C. dChipSNP:significance curve and clustering of SNP-array-based loss-of-hetero-zygosity data. Bioinformatics 2004;20:1233–40.
27. Parkin B, Erba H, Ouillette P, Roulston D, Purkayastha A, Karp J, et al.Acquired genomic copy number aberrations and survival in adult acutemyelogenous leukemia. Blood 2010;116:4958–67.
28. Long J, ParkinB,Ouillette P,BixbyD, SheddenK, ErbaH, et al.Multipledistinct molecular mechanisms influence sensitivity and resistance toMDM2 inhibitors in adult acute myelogenous leukemia. Blood2010;116:71–80.
29. Bouyge-Moreau I, Rondeau G, Avet-Loiseau H, Andre MT, Bezieau S,CherelM, et al. Construction of a 780-kbPAC,BAC, and cosmid contigencompassing the minimal critical deletion involved in B cell chroniclymphocytic leukemia at 13q14.3. Genomics 1997;46:183–90.
30. Kalachikov S, Migliazza A, Cayanis E, Fracchiolla NS, Bonaldo MF,Lawton L, et al. Cloning and gene mapping of the chromosome 13q14region deleted in chronic lymphocytic leukemia. Genomics1997;42:369–77.
31. Palamarchuk A, Efanov A, Nazaryan N, SantanamU, Alder H, RassentiL, et al. 13q14 deletions in CLL involve cooperating tumor suppres-sors. Blood 2010;115:3916–22.
32. Fulci V, Chiaretti S, Goldoni M, Azzalin G, Carucci N, Tavolaro S, et al.Quantitative technologies establish a novel microRNA profile of chron-ic lymphocytic leukemia. Blood 2007;109:4944–51.
33. Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, et al.Frequent deletions and down-regulation of micro-RNA genes miR15and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl AcadSci U S A 2002;99:15524–9.
34. Raveche ES, Salerno E, Scaglione BJ, Manohar V, Abbasi F, Lin YC,et al. Abnormal microRNA-16 locus with synteny to human 13q14linked to CLL in NZB mice. Blood 2007;109:5079–86.
35. Ouillette P, Fossum S, Parkin B, Ding L, Bockenstedt P, Al-Zoubi A,et al. Aggressive chronic lymphocytic leukemia with elevated genomiccomplexity is associated with multiple gene defects in the response toDNA double-strand breaks. Clin Cancer Res 2010;16:835–47.
36. Ouillette P, Collins R, Shakhan S, Li J, Peres E, Kujawski L, Talpaz M,et al. Acquired copy number aberrations and survival in chroniclymphocytic leukemia. Blood 2011;118:3051–61.
37. Van Dyke DL, Shanafelt TD, Call TG, Zent CS, Smoley SA, Rabe KG,et al. A comprehensive evaluation of the prognostic significance of 13qdeletions in patients with B-chronic lymphocytic leukaemia. Br JHaematol 2010;148:544–50.
38. Hernando E, Nahle Z, JuanG, Diaz-Rodriguez E, AlaminosM, HemannM, et al. Rb inactivation promotes genomic instability by uncouplingcell cycle progression frommitotic control. Nature 2004;430:797–802.
39. HammarsundM,CorcoranMM,WilsonW,ZhuC,EinhornS,SangfeltO,et al. Characterizationof a novel B-CLLcandidate gene–DLEU7–locatedin the 13q14 tumor suppressor locus. FEBS Lett 2004;556:75–80.
Ouillette et al.
Clin Cancer Res; 17(21) November 1, 2011 Clinical Cancer Research6790
2011;17:6778-6790. Published OnlineFirst September 2, 2011.Clin Cancer Res Peter Ouillette, Roxane Collins, Sajid Shakhan, et al. Lymphocytic LeukemiaThe Prognostic Significance of Various 13q14 Deletions in Chronic
Updated version
10.1158/1078-0432.CCR-11-0785doi:
Access the most recent version of this article at: