The role of endogenous scabies mite complement inhibitors in the development of Streptococcus pyogenes skin infections Lindsay Darryl Christian Bachelor of Applied Science (Biotechnology) Submitted in partial fulfilment of the requirements for the degree of Master of Applied Science (Research) School of Biomedical Sciences Institute of Health and Biomedical Innovation (IHBI) Faculty of Health Queensland University of Technology (QUT) & QIMR Berghofer Medical Research Institute August 2015
Transcript
The role of endogenous scabies mite
complement inhibitors in the development
of Streptococcus pyogenes skin infections
Lindsay Darryl Christian
Bachelor of Applied Science (Biotechnology)
Submitted in partial fulfilment of the requirements for the degree of
Master of Applied Science (Research)
School of Biomedical Sciences
Institute of Health and Biomedical Innovation (IHBI)
(A) was used as a negative control. The reaction was incubated aerobically at 37oC, 200 rpm; stopped after 40 min with
4% paraformaldehyde, and subsequently analysed with a FACS Canto A flow cytometer, measuring the fluorescence of
105 gated neutrophils at 530 nm. Results are displayed as Histograms reporting the trend percentage of FITC positive
and FITC negative cells from two independent assays.
A B
C
55
Optimising the isolation of human neutrophils using an immunomagnetic negative selection
To improve the purity a second isolation technique was trialled using the Easy Step Direct
Human Neutrophil Isolation kit (Stem Cell Technologies) per Materials and Methods section
2.5.3.
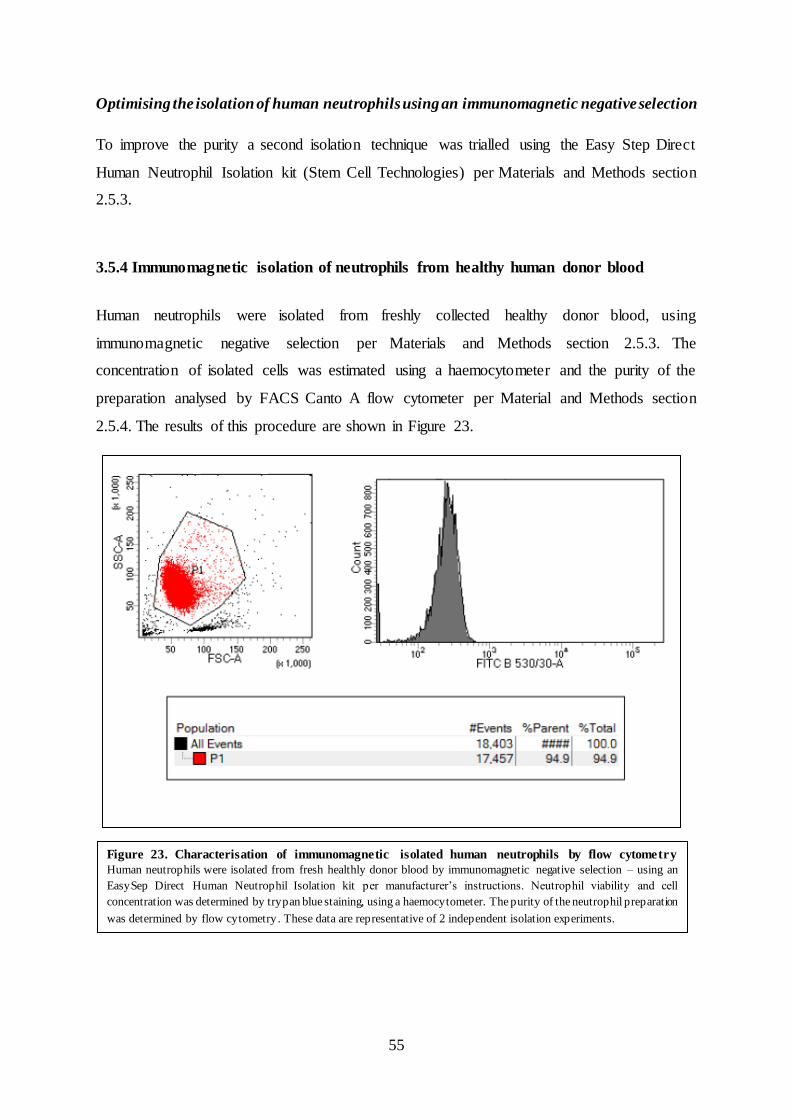
3.5.4 Immunomagnetic isolation of neutrophils from healthy human donor blood
Human neutrophils were isolated from freshly collected healthy donor blood, using
immunomagnetic negative selection per Materials and Methods section 2.5.3. The
concentration of isolated cells was estimated using a haemocytometer and the purity of the
preparation analysed by FACS Canto A flow cytometer per Material and Methods section
2.5.4. The results of this procedure are shown in Figure 23.
Figure 23. Characterisation of immunomagnetic isolated human neutrophils by flow cytometry
Human neutrophils were isolated from fresh healthly donor blood by immunomagnetic negative selection – using an
EasySep Direct Human Neutrophil Isolation kit per manufacturer’s instructions. Neutrophil viability and cell
concentration was determined by trypan blue staining, using a haemocytometer. The purity of the neutrophil preparation
was determined by flow cytometry . These data are representative of 2 independent isolation experiments.
56
The purity of the neutrophils as measured by FACS Canto A flow cytometer was found to be
approximately 95%. This data (Figure 23) is representative of two independent isolations. The
gated populations of cells (P1) represent the expected size of neutrophils (x-axis) and
intracellular complexity (y-axis) of neutrophils. The histogram displays the number of cells
and intensity of auto-fluorescence of isolated neutrophils corresponding to the FITC signal. To
verify that neutrophils were isolated, the CD66b marker expressed on peripheral neutrophils
was probed with a mouse anti-human CD66b antibody conjugated to FITC per Materials and
Methods section 2.5.5.
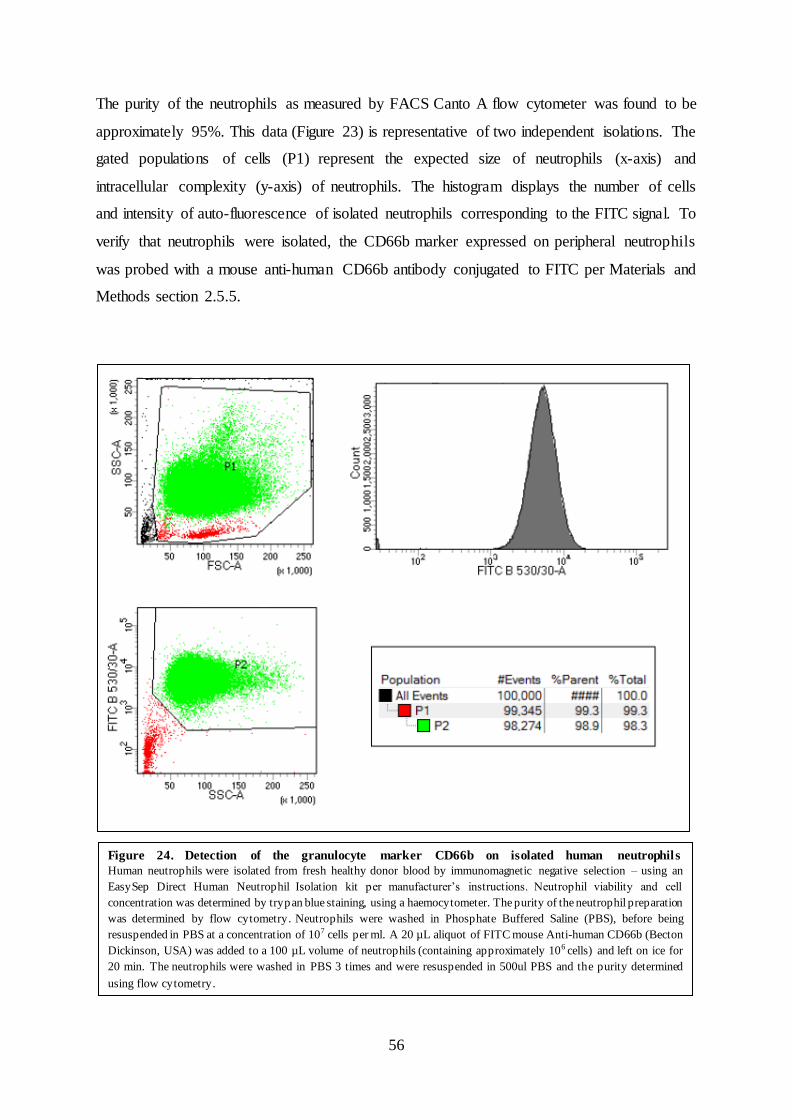
Figure 24. Detection of the granulocyte marker CD66b on isolated human neutrophils
Human neutrophils were isolated from fresh healthy donor blood by immunomagnetic negative selection – using an
EasySep Direct Human Neutrophil Isolation kit per manufacturer’s instructions. Neutrophil viability and cell
concentration was determined by trypan blue staining, using a haemocytometer. The purity of the neutrophil preparation
was determined by flow cytometry. Neutrophils were washed in Phosphate Buffered Saline (PBS), before being
resuspended in PBS at a concentration of 107 cells per ml. A 20 µL aliquot of FITC mouse Anti-human CD66b (Becton
Dickinson, USA) was added to a 100 µL volume of neutrophils (containing approximately 106 cells) and left on ice for
20 min. The neutrophils were washed in PBS 3 times and were resuspended in 500ul PBS and the purity determined
using flow cytometry.
57
The purity of the neutrophils as measured by the detection of CD66b by FACS Canto A flow
cytometer was found to be approximately 98% (Figure 24). This data is representative of two
independent isolations. The gated populations of cells (P1) represent the expected size of
neutrophils (x-axis) and intracellular complexity (y-axis) of neutrophils. Cell gate (P2)
displays the expected intracellular complexity (x-axis) over the neutrophils considered positive
for the FITC signal (y-axis). The histogram displays the intensity of isolated neutrophils
positive for the FITC signal (x-axis) over the number of cells corresponding to the intensity of
the FITC signal.
3.6 Statistical analysis
The statistical tests used to analyse the experimental data in this study included Two-way
ANOVA with Dunnett’s or Sidak’s multiple comparisons test. Two-way ANOVA is
appropriate for this study type as the experiments have been designed to measure the effect on
a dependent variable – in the case of the bactericidal assays in Results section 3.1, the bacterial
recovery by two independent factors; sample treatment and the concentration of the molecules
used in the treatment. Dunnett’s test is relevant for determining the statistical significance of
differences between the means of each individual sample treatments to the means of a control.
Here this has been used to analyse Figure 8, in which the means of treatment groups; BSA,
SMIPP-S I1 and SMS B4, were compared to the GVB control means. Sidak’s test was selected
for data sets to compare the means between all treatment groups in the assay, for example the
complement deposition assays in Results section 3.2 and 3.3, in which the mean measurements
of equal concentrations of BSA treatment was compared against the mean measurements of a
scabies complement inhibitor treatment. The significance threshold of all comparison tests
were set to 5% or a p-value of 0.05.
58
Chapter 4: Discussion
59
The ability of the individual to rapidly detect and destroy invading pathogens is critical to the
recovery of the organism. The complement component of the innate immune system is an
effective first line defence system, tagging pathogens for phagocytosis, and/or directly lysing
the cell. Pathogenic organisms have evolved sophisticated mechanisms to inhibit complement
mediated immune responses (Meri et al., 2013; Pearce et al., 1990; Tyson et al., 2007).
Streptococcus pyogenes (or group A Streptococcus (GAS)), a major bacterial pathogen in
humans, produces an assortment of virulence factors to disrupt various stages of the
complement cascade (Fernie-King et al., 2001; Lei et al., 2001; Podbielski et al., 1996; Thern
et al., 1995; von Pawel-Rammingen & Bjorck, 2003). Similarly, scabies mites have been
reported to produce a large range of secretory proteins, including multiple families of proteases
and protease inhibitors that disrupt the human complement system (Bergstrom et al., 2009;
Mika et al., 2012a; Mika et al., 2012b; Reynolds et al., 2014; Swe & Fischer, 2014).
Using immunohistochemistry approaches, previous research has shown that scabies mite
complement inhibitors are localised within the mite gut and are excreted into the epidermal
burrows with the mite faeces (Mika et al., 2012a; Willis et al., 2006). One of their likely
functions has been proposed to avert gut damage due to complement-mediated immune
processes; as the mite has been shown to feed on host serum (Rapp et al., 2006) and human
complement components have previously been localised in the mite gut (Mika et al., 2011).
Within epidermal burrows of the scabies mite, the release of multiple anti-complement proteins
may act synergistically and amount to levels that effectively inhibit complement activity
locally. As a consequence, this may provide a favourable environment for the establishment of
secondary bacterial infections in scabies burrows. GAS is an opportunistic pathogen that is
endemic in many regions of Northern Australia, a major cause of skin disease that often
coincides with or follows the onset of scabies (Clucas et al., 2008; Currie & Carapetis, 2000).
The association of scabies and bacterial pyoderma forms the basis of my hypothesis; that
scabies infections cause local complement inhibition in the epidermal burrows of its host,
promoting the establishment of opportunistic pathogens such as GAS.
The scabies mite complement inhibitors SMIPP-S I1 and SMS B4 have been reported to
promote the in vitro growth of a single GAS M1 strain (Mika et al., 2012a). More recently,
SMS B4 has also been shown to promote the growth of a range of Staphylococcus aureus
strains, isolated from patient skin infections in Queensland and the Northern Territory (Swe &
Fischer, 2014). The results presented here provide further evidence that SMIPP-S I1 and SMS
B4 both promote the growth of S. pyogenes. The data reported here has expanded from the
60
previous study by including various GAS clinical isolates from all three emm-pattern types.
Pattern A – C strains predominantly cause throat infections, pattern D predominately cause
skin infections and Pattern E strains are reported to cause both skin and throat infections. The
viability of all GAS strains was significantly increased in whole human blood treated with
SMIPP-S I1 and SMS B4 suggesting that this effect is not strain specific. As the complement
response is non-specific (Frank & Fries, 1991), this result was expected and these data show
the same trend as earlier studies into the effect of SMIPP-S I1 and SMS B4 on the viability of
different bacterial isolates to those examined here (Mika et al., 2012b; Swe & Fischer, 2014).
By focusing only on GAS 88-30, originally isolated from a scabies patient in the Northern
Territory (Brandt et al., 2000; CDC, 2014), it was shown that SMIPP-S I1 and SMS B4 both
reduce the blood mediated killing of GAS in a concentration dependent manner.
Bergstorm et al. and Mika et al. reported previously that SMIPP-S I1 and SMS B4 interfere
with the initiation and progression of all three complement pathways (Bergstrom et al., 2009;
Mika et al., 2012a). However, these studies assessed the human complement activation, using
homogenous artificial substrates such as zymosan, mannan or human IgG to activate the
Alternative, Lectin and Classical complement pathways respectively. To address the effect of
mite complement inhibitors on complement activation on the bacterial surface it was necessary
to accurately reflect the heterogeneous makeup of the bacterial cellular surface. Therefore
whole GAS cells were used to activate complement in the experiments presented here. Since
SMIPP-S I1 and SMS B4 belong to different classes of mite complement inhibitors, namely
serine proteases and serpins, it was postulated and shown that these molecules are likely to
impact at different steps of the complement cascade (Mika et al., 2012a; Reynolds et al., 2014).
This was also reflected in the results presented here – in the differences in the amount of
bacterial recovery induced by the two complement inhibitors as well as in the IC50 values in
the complement deposition assays. The IC50 of SMIPP-S I1 required for the reduction of
complement deposition on GAS was approximately 10-fold higher than that of SMS B4. In
these regards SMS B4 appeared to be a more potent inhibitor of complement components
required for neutralisation of GAS than SMIPP-S I1 and it is more likely that SMIPP-S I1 is
less advantageous to the bacteria. This complement inhibitor has been proposed to play a key
role in protecting the scabies mite itself; as MBL, the primary activator of the Lectin pathway
has been found to bind to the lining of the mite gut (Mika et al., 2011) and SMIPP-S I1 has
been shown to specifically interfere with the Lectin Pathway (Reynolds et al., 2014).
61
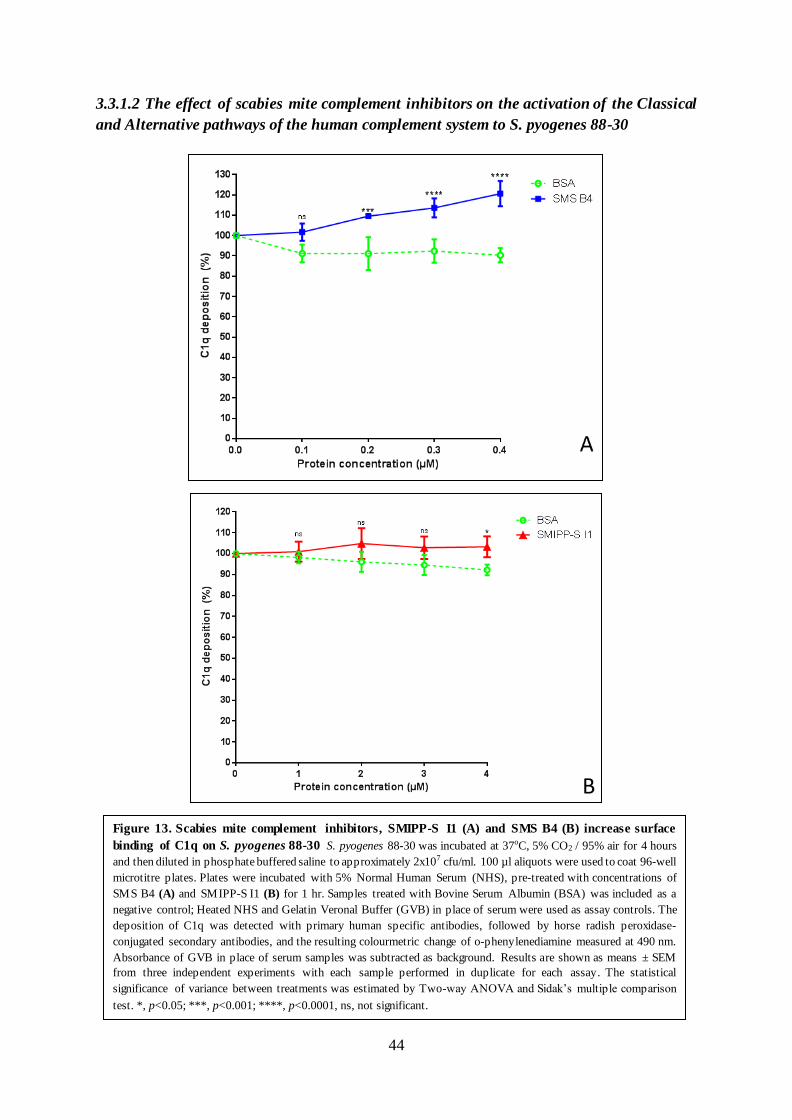
Both SMIPP-S I1 and SMS B4 increased the deposition of C1q, the pattern recognit ion
molecule that initiates the Classical pathway, on the surface of GAS. The effect I have observed
here appears to be marginal; with C1q deposition increased by approximately 5% at 4 µM of
SMIPP-S I1, and approximately 10 – 20% between 2 µM – 4 µM of SMS B4 respectively
(Figure 13, section 3.3.1). This requires further investigation, as previous findings have
demonstrated that SMS B4 decreased C1q deposition (Bergstrom et al., 2009; Mika et al.,
2012a). More recently, a similar study on SMS B4 reported a slight increase in C1q deposition
on Staphylococcus aureus similar to the levels observed here (Swe & Fischer, 2014). One
potential advantage in increasing C1q levels for the scabies mite and inadvertently for
associated bacterial pathogens, is highlighted in research by Potempa et al. (2009) where C1q
deposition was also found to be marginally increased during the initial stages of bacterial
infections. The authors have suggested this may lead to a low grade inflammation that is
beneficial to the pathogen by increasing the availability of nutrients and growth factors
produced during innate immune process and promoted bacterial colonisation (Potempa et al.,
2009). Both SMIPP-S I1 and SMS B4 reduced C4b deposition on the cellular surface of S.
pyogenes in this study, indicating that these proteins reduce with the amount of C3 convertase
(C4b2a) formed in the Classical and Lectin pathways. However, it is likely that only the C3
convertases of the Classical pathway is involved in this case, as I did not detect MBL on the
GAS surface. This finding suggests that the mechanism by which SMIPP-S I1 and SMS B4
reduce C4b deposition, is via disruption of the Classical pathway activation. Since C1q
deposition was found to be slightly increased, it is possible SMS B4 and SMIPP-S I1 are
affecting the function of C1 complex, namely cleavage of C4 to C4a and C4b, without affecting
the interaction of the C1 complex with the cell surface. I discuss a potential experiment to
investigate the effect of SMS B4 on C1q and the C1 complex in the “Conclusions and future
directions” section.
It has been reported previously that the Classical and Alternative pathways are the primary
innate immune response to S. pyogenes and other Streptococci (Brown et al., 2002; Wessels et
al., 1995; Yuste et al., 2006). Likewise, under our assay conditions, MBL did not bind to the
surface of S. pyogenes 88-30, suggesting that the MBL-dependent Lectin pathway is not
activated. It has previously been reported that L-ficolin, a pattern recognition molecule also
capable of activating the Lectin pathway, specifically detects Lipoteichoic acid; a cell wall
component present in all gram positive bacteria, including S. pyogenes (Lynch et al., 2004).
Thus the Lectin pathway may house additional roles in the complement response to S.
62
pyogenes. Both SMIPP-S I1 and SMS B4 were found to significantly reduce the binding of
properdin to S. pyogenes 88-30, as shown in Figure 14 of results section 3.3.1. The role of
properdin is to stabilise the C3 convertase (C3bBb) of the Alternative pathway, therefore a
reduction in properdin deposition may result in a decreased level of C3 convertase formation
in the Alternative pathway. C3 convertase formation in the Alternative pathway is initiated by
the deposition of C3b from spontaneous hydrolysis of basal C3 levels. If the scabies mite
complement inhibitors are directly interfering with the cleavage of C3 into C3a and C3b,
consequently the reduction in C3 convertase formed in the Alternative pathway will be
amplified. Properdin has also been found to act as a pattern recognition molecule to activate
the Alternative pathway independently (Bowen et al., 2014; Kobayashi & DeLeo, 2009;
Mounsey et al., 2010). This latter fact may also be relevant for complement activation to
scabies mite surface components, which may explain why SMIPP-S I1 and SMS B4 have
evolved to inhibit properdin binding.
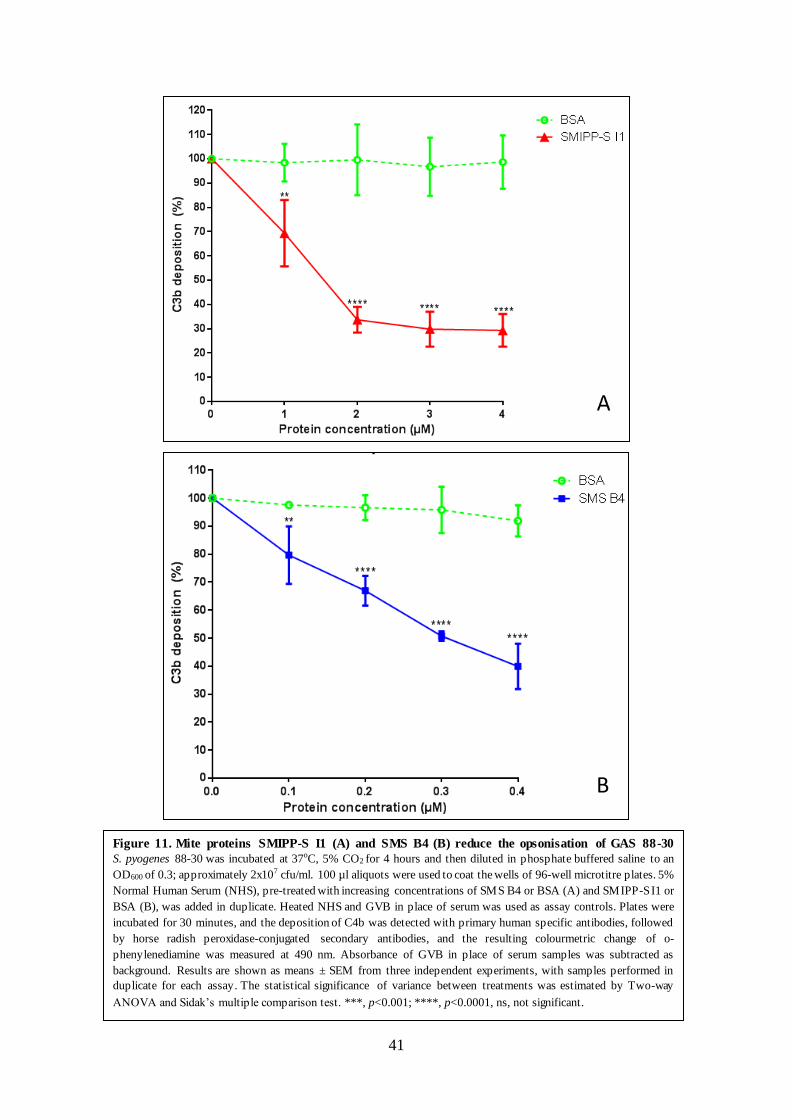
Reduced C3 convertase formation on the surface of GAS resulted in a reduction in the level of
C3b on GAS (Figure 11, Results section 3.2). C3b is the key opsonin which marks pathogens
for phagocytosis and once high concentrations are reached, initiates the formation of the C5
convertase. The inhibition of C3b deposition may in turn reduce the level of C5 convertase,
leading to a decreased production of the anaphylatoxin C5a, which plays a critical role in
attracting phagocytes to the site of infection. These results support previous findings with
SMIPP-S I1 and SMS B4 in which similar levels of disruption to the formation of the C3
convertase (Bergstrom et al., 2009; Mika et al., 2012a; Mika et al., 2012b; Swe & Fischer,
2014) and the C5 convertase (Swe & Fischer, 2014) were reported.
In Results section 3.4 I aimed to determine which pathway(s) of the human complement is
most critical for the host response to GAS. The data overall indicate that a deficiency in the
C1q component of the Classical pathway led to the greatest reduction in opsonisation of GAS.
Figure 15 (section 3.4) suggested that a deficiency in MBL resulted in the highest reduction of
C3b deposition, however the addition of heated human serum with high levels of GAS
antibodies recovered the level of C3b deposition to a similar level as seen when NHS was tested
in a control assay. The MBL deficient serum was then found to contain an excessive C1q
concentration of 160% compared with the NHS, as reported in Figure 16, suggesting antibody
dependent activation of complement is most important in the activation of complement to S.
pyogenes. Interestingly, the level of C3b deposition in the C1q deficient serum sample was also
recovered following addition of heated human serum with high levels of S. pyogenes antibodies
63
in Figure 16. This result is unexpected, though there is a potential explanation for this result.
While it is well-established that antibody-dependent activation of complement occurs by the
Classical pathway, antibody-dependent activation of the Alternative pathway has been reported
for several disease processes in humans and animals with C1q deficiencies (Banda et al., 2007;
Cutler et al., 1991; Zhou et al., 2012). The Alternative pathway is known to amplify both the
Classical and the Lectin pathways, thus it is possible that the antibody-directed properdin-
activation of complement occurs when the Classical pathway is absent or non-functional.
Complement-mediated immune responses facilitate the killing of pathogens via phagocytos is,
or via direct cell destruction by the formation of the membrane attack complex. The latter
process is ineffective against gram positive bacteria, due to the structural composition of the
cell wall (Berends et al., 2013; Joiner et al., 1983). Therefore, the recovery of the GAS clinica l
isolates observed in this study is most likely due to interference with phagocytosis. Neutrophils
are the most abundant human leucocyte, and play an essential role in complement mediated
phagocytosis (Kobayashi & DeLeo, 2009). An additional aim of this study was to determine if
SMIPP-S I1 and SMS B4 interfere with the phagocytosis of S. pyogenes by neutrophils. The
methods used to generate the preliminary data (section 3.5) are based on a similar study
investigating the effect of SMS B4 on the phagocytosis of S. aureus by neutrophils (Swe &
Fischer, 2014). The method for GAS phagocytosis assay is currently being optimised. As the
negative control experiment contains heat inactivated serum, very little phagocytosis should be
observed, however a FITC signal indicating phagocytosis was observed, suggesting that this
control experiment requires optimisation, most likely with regards to the concentrat ion of FITC
labelled bacteria. An additional consideration is that the purity of neutrophils obtained by
gradient centrifugation (60%; as shown in Figure 20, Results section 3.5.2) was lower than that
used in the study by Swe and Fischer (2014), in which a minimum neutrophil purity of 70%
was considered necessary. This method required a significant amount of time and neutrophils
are short lived once isolated from the host, with an estimated viability of up to 4 hours (Oh et
al., 2008). The time taken and the process involved in gradient centrifugation may have
activated or damaged the neutrophils, reducing viability and functionality. The purity of the
neutrophils obtained using immunomagnetic negative selection was considerably higher, at
95%, as reported in results section 3.5.4. Due to time limitations, I was not able to repeat the
assays with neutrophils isolated using this method. In the future it would be beneficial to ensure
that the viability and functionality of the neutrophils is maintained following immunomagnetic
isolation, using techniques such as the oxidative burst test (Hashiguchi et al., 2005). There may
64
also be differences in the time required for the neutrophils to phagocytose S. pyogenes,
compared to S. aureus; 40 minutes was selected here based on the conditions in the Swe and
Fischer (2014) study, which investigated phagocytosis of S. aureus. It will be beneficial to
measure the level of FITC-GAS phagocytosis over a time course, to ensure the optimal time is
selected.
The findings reported in this study provides supportive data for the hypothesis that disruption
of complement mediated immune responses to scabies provide favourable conditions not only
for scabies mites themselves to survive and proliferate in the host, but also for bacteria that
colonise the mite infected epidermis. Consequently GAS pyoderma may be promoted by the
presence of mites and may in unfortunate circumstances progress to an invasive state leading
to systemic bacteraemia. The ability of S. pyogenes infection to become systemic may induce
an adaptive immune response, priming the host immune system for secondary sequelae such
as rheumatic heart disease in susceptible individuals. A recent study by Ellis et al. (2010) has
suggested that individuals susceptible to rheumatic heart disease may possess similar cardiac
myosin epitopes that share structural similarities with group A streptococcal surface
components found across all emm types (Ellis et al., 2010). According to their findings, any S.
pyogenes infection that is persistent enough to induce human antibody responses that cross
react with host tissue epitopes may have the potential to lead to rheumatic fever and rheumatic
heart disease. Instances where this scenario has been identified have been published. For
example, a recent study in Hawaii, where rheumatic heart disease occurs at one of the highest
rates worldwide, reported that the GAS emm types implicated in the development of ARF in
Hawaii were not among the so-called rheumatogenic emm types found in temperate climates
(Erdem et al., 2007). This suggests that different GAS emm types may be capable of inducing
rheumatic fever and rheumatic heart disease between temperate and tropical climates. This
notion has been suggested by several research groups (McDonald et al., 2007; McDonald et
al., 2004; Parks et al., 2012). The current hypothesis in the literature is that GAS pyoderma
may be responsible for the high rates of rheumatic heart disease observed in tropical climates,
in accord with the low reported rates of GAS pharyngitis. In tropical regions of Australia and
the Pacific, GAS pyoderma is implicated as the primary cause of invasive GAS disease
(Carapetis et al., 1999; Le Hello et al., 2010; Romani et al., 2015). Scabies is recognised as a
major risk factor for pyoderma in Aboriginal communities of tropical northern Australia
(Clucas et al., 2008; Currie & Carapetis, 2000; Kearns et al., 2013). The finding that SMIPP-
S I1 and SMS B4 promote the recovery of GAS and inhibit complement mediated responses
65
highlights the link between scabies and associated bacterial infections, emphasizing that urgent
attention is required towards scabies infections in these communities. Prevention and
eradication of scabies may in turn reduce the heavy burden of pyoderma observed and
potentially prevent the development of rheumatic fever and rheumatic heart disease.
66
Chapter 5: Conclusion
and future directions
67
This study provides further evidence that SMIPP-S I1 and SMS B4 interfere with the human
complement defence. SMIPP-S I1 and SMS B4 reduced the amount of C3 convertase formed
on the surface of the bacteria and consequently reduced deposition of C3b, a potent opsonin on
the surface of S. pyogenes. This in turn interfered with the complement-mediated neutralisa t ion
of GAS. This study suggests that the function of the Classical and Alternative pathways are
predominately effected by SMS B4 and SMIPP-S I1 in response to GAS.
Limitations of study
The above findings may not be the sole mechanisms of these scabies mite complement
inhibitors in the pathogenicity of scabies associated bacterial infections. The potential effects
of SMIPP-S I1 and SMS B4 on the ficolins and collectins in complement activation to GAS
have not been addressed in this study, leaving the Lectin pathway for future investigat ion.
There are additional controls that may be suitable for future repeats of the assays performed in
this study, these include: heat inactivated serum as a positive control for anti-complement
activity in bactericidal assays and heat inactivated scabies mite complement inhibitors to
demonstrate the specificity of their activity. The anti-complement protein cobra venom factor
has been used as a positive control in similar bactericidal assays to those performed in this
study (Swe & Fischer, 2014), however the stock batches obtained recently have yielded
inconsistent results and was left out of the study due to time restrictions. Cobra venom factor
is also not suitable for use in the ELISA assays to measure the deposition of C3b as it is
structurally homologous to C3b and has been found to cross-react with C3b antibodies (Vogel
et al., 1984). Additionally, the number of samples and time available to perform all assays in
this study is limited due to the use of human blood and blood products that expire within a
relatively short time frame. Therefore it was not feasible to include these additional controls in
this study.
This study has investigated the activity of two recombinant scabies mite complement inhibito rs
within an in-vitro system. The artificial assay systems used here will not entirely represent the
complex in vivo conditions during scabies infestation and subsequent secondary bacterial
infections in the skin. Likely the percentage of biologically active molecules is not 100% in the
preparations of recombinantly produced proteins. In addition, during an active scabies
infection, it is likely that mite complement inhibitors are not produced in isolation. SMIPPS-
I1 and SMS B4 are members of two multi-copy families of predicted protein homologs
68
discovered previously in a gene discovery project analysing scabies mite mRNA transcripts
(Fischer et al., 2003). It is very likely that multiple homologous proteins of each family are
expressed simultaneously by multiple mites present within the skin burrows. While the in vivo
concentrations of these molecules in the mite gut is unknown, a cumulative effect of many
anti-complement activities coming from the large family of SMIPP-Ss and from at least two if
not more serine protease inhibitors is most likely. As many of these molecules may act on
several levels in the complement system.
Future directions
The finding that SMS B4 slightly increased the deposition of C1q on the surface of
Staphylococcus aureus (Swe & Fischer, 2014) and now GAS (reported in Figure 13, Results
section 3.3.1) are conflicting observations with previous studies which have demonstrated that
SMS B4 decreased C1q deposition (Bergstrom et al., 2009; Mika et al., 2012a). A potential
experiment to determine the functional effect(s) of SMS B4 on C1q deposition, is to investiga te
if formation of the activated C1 complex is disrupted, by performing a C1 reconstitution assay
as described by (Arlaud & Thielens, 1993) in the presence of SMS B4 or controls. By treating
assay samples with SMS B4 or controls and performing SDS-PAGE on the resulting reaction
mixtures, it is possible to determine if SMS B4 prevents the conversion of the C1 complex
proenzymes C1r and C1s to their active forms. The active forms of C1r and C1s are functiona l
serine proteases resulting in the downstream progression of the Classical pathway, by
catalysing the cleavage of C4 to C4a and C4b. Activated forms of these complement factors
will produce two distinct protein bands each of 57 and 35 kDa (C1r) and 55 and 30 kDa (C1s),
while the inactivated proenzymes will be visible as single 85 kDa (C1r) and 84 kDa (C1s)
bands by SDS PAGE respectively. A western blot could also be performed to be more confident
in the differential identification of these molecules.
The functional analysis of the two recombinant scabies mite protein reported here may very
likely not completely reflect the natural pathogenesis. Further research is required to elucidate
the collective roles of complement inhibitors released during the scabies infection. It has been
shown that secondary bacterial infections of the mite burrows often involve co-infections of
both S. pyogenes and S. aureus (Bowen et al., 2014; Steer et al., 2009; Whitehall et al., 2013).
It would be ideal to extend the work from earlier studies and here, to include combinations of
mite complement inhibitors in the same assay at lower concentrations. Human scabies samples
69
are very difficult to obtain. The porcine integumentary system closely resembles that of human,
and since 2010 a porcine model of scabies has allowed for complete study of the mite lifecyc le
and disease process (Mounsey et al., 2010). Using this model it may be possible to both extend
the bactericidal assays to an in vivo platform, and also to utilise transcriptome based methods
to identify the presence and population of scabies mite complement inhibitors released during
the disease process. As recently shown in this porcine model (Swe et al., 2014) scabies
infections have a significant effect on the composition of the cutaneous microbiome.
Investigating the potential of scabies induced microbiome changes in humans should be high
on the future research agenda, despite the challenges of limited access to scabies patients and
ethical limitations with regards to longitudinal studies. Understanding whether the relationship
between scabies mites and pathogens interferes with the dynamics of the host-benefic ia l
microflora may provide further insights into the pathogenesis of scabies and associated
bacterial disease. Investigation of the skin microbiome in scabies patients may reveal novel
information including a novel diagnostic marker, as scabies infections are notoriously difficult
to reliably diagnose by current methods (Walter et al., 2011).
This study demonstrates that the scabies mite complement inhibitors SMIPP-S I1 and SMS B4
interfere with the complement cascade, disrupting a critical component of the early stages of
the host immune response, in turn providing an optimal environment for the establishment and
proliferation of opportunistic pathogens such as Group A Streptococcus. The practical
outcomes of this finding are that this knowledge invites furthers research as well as highlights
the need for early intervention into scabies infections. Changes in policies and practices in this
regard may ultimately prevent the onset of secondary bacterial infections and the potentially
fatal downstream diseases that may result.
70
Bibliography
71
AIHW. (2004). Rheumatic heart disease: all but forgotten in Australia except among Aboriginal and Torres Strait Islander peoples. Canberra: Australian Institute of Health
and Welfare. Retrieved March 26, 2014, from http://www.aihw.gov.au/publication-detail/?id=6442467621
Andrews, R. M., Kearns, T., Connors, C., Parker, C., Carville, K., Currie, B. J., & Carapetis, J. R. (2009). A regional initiative to reduce skin infections amongst aboriginal children living in remote communities of the Northern Territory, Australia. PLoS Negl Trop Dis,
3(11), e554. Arlaud, G. J., & Thielens, N. M. (1993). Human complement serine proteases C1r and C1s and
their proenzymes. Methods Enzymol, 223, 61-82. Arlian, L. G., Runyan, R. A., & Estes, S. A. (1984). Cross infestivity of Sarcoptes scabiei. J
Am Acad Dermatol, 10(6), 979-986.
Azevedo, P. M., Pereira, R. R., & Guilherme, L. (2012). Understanding rheumatic fever. Rheumatol Int, 32(5), 1113-1120.
Banda, N. K., Takahashi, K., Wood, A. K., Holers, V. M., & Arend, W. P. (2007). Pathogenic complement activation in collagen antibody-induced arthritis in mice requires amplification by the alternative pathway. J Immunol, 179(6), 4101-4109.
Berends, E. T. M., Dekkers, J. F., Nijland, R., Kuipers, A., Soppe, J. A., van Strijp, J. A. G., & Rooijakkers, S. H. M. (2013). Distinct localization of the complement C5b-9 complex
on Gram-positive bacteria. Cell Microbiol, 15(12), 1955-1968. Bergstrom, F. C., Reynolds, S., Johnstone, M., Pike, R. N., Buckle, A. M., Kemp, D. J., Fischer,
K., & Blom, A. M. (2009). Scabies mite inactivated serine protease paralogs inhibit the
human complement system. J Immunol, 182(12), 7809-7817. Bessen, D. E., Carapetis, J. R., Beall, B., Katz, R., Hibble, M., Currie, B. J., Collingridge, T.,
Izzo, M. W., Scaramuzzino, D. A., & Sriprakash, K. S. (2000). Contrasting molecular epidemiology of group A streptococci causing tropical and nontropical infections of the skin and throat. J Infect Dis, 182(4), 1109-1116.
Bessen, D. E., Fiorentino, T. R., & Hollingshead, S. K. (1997). Molecular markers for throat and skin isolates of group A streptococci. Adv Exp Med Biol, 418, 537-543.
Bisno, A. L., Pearce, I. A., Wall, H. P., Moody, M. D., & Stollerman, G. H. (1970). Contrasting epidemiology of acute rheumatic fever and acute glomerulonephritis. N Engl J Med, 283(11), 561-565.
Blom, A. M., Hallstrom, T., & Riesbeck, K. (2009). Complement evasion strategies of pathogens-acquisition of inhibitors and beyond. Mol Immunol, 46(14), 2808-2817.
Blyth, C. C., Robertson, P. W., & Rosenberg, A. R. (2007). Post-streptococcal glomerulonephritis in Sydney: A 16-year retrospective review. J Paediatr Child Health, 43(6), 446-450.
Bouvresse, S., & Chosidow, O. (2010). Scabies in healthcare settings. Curr Opin Infect Dis, 23(2), 111-118.
Bowen, A. C., Tong, S., Chatfield, M. D., & Carapetis, J. R. (2014). The microbiology of impetigo in Indigenous children: associations between Streptococcus pyogenes , Staphylococcus aureus, scabies, and nasal carriage. BMC Infect Dis, 14(1), 3854.
Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem, 72,
248-254. Brandt, E. R., Sriprakash, K. S., Hobb, R. I., Hayman, W. A., Zeng, W., Batzloff, M. R.,
Jackson, D. C., & Good, M. F. (2000). New multi-determinant strategy for a group A
streptococcal vaccine designed for the Australian Aboriginal population. Nat Med, 6(4), 455-459.
Brown, J. S., Hussell, T., Gilliland, S. M., Holden, D. W., Paton, J. C., Ehrenstein, M. R., Walport, M. J., & Botto, M. (2002). The classical pathway is the dominant complement
pathway required for innate immunity to Streptococcus pneumoniae infection in mice. Proceedings of the National Academy of Sciences, 99(26), 16969-16974.
Burgess, I. (1994). Sarcoptes scabiei and Scabies. In J. R. Baker & R. Muller (Eds.), Adv Parasitol (Vol. 33, pp. 235-292): Academic Press.
Carapetis, J. R., Connors, C., Yarmirr, D., Krause, V., & Currie, B. J. (1997). Success of a
scabies control program in an Australian aboriginal community. Pediatr Infect Dis J, 16(5), 494-499.
Carapetis, J. R., Walker, A. M., Hibble, M., Sriprakash, K. S., & Currie, B. J. (1999). Clinica l and epidemiological features of group A streptococcal bacteraemia in a region with hyperendemic superficial streptococcal infection. Epidemiol Infect, 122(1), 59-65.
Cavalcante, R. R., Pereira, M. H., & Gontijo, N. F. (2003). Anti-complement activity in the saliva of phlebotomine sand flies and other haematophagous insects. Parasitology,
127(Pt 1), 87-93. CDC. (2010). Parasites; Scabies. Biology; Life cycle. Retrieved 30 March, 2015, from
http://www.cdc.gov/parasites/scabies/biology.html
CDC. (2014). emm97 - Isolated from scabetic lesions on a child in Darwin. emm types and sequence types. Retrieved 27 March, 2014, from http://www.cdc.gov/streplab/types-
emm103-124.html Chakravarty, S. D., Zabriskie, J. B., & Gibofsky, A. (2014). Acute rheumatic fever and
streptococci: the quintessential pathogenic trigger of autoimmunity. Clin Rheumatol,
33(7), 893-901. Chalaire, K. C., Kim, T. K., Garcia-Rodriguez, H., & Mulenga, A. (2011). Amblyomma
americanum (L.) (Acari: Ixodidae) tick salivary gland serine protease inhibitor (serpin) 6 is secreted into tick saliva during tick feeding. J Exp Biol, 214(Pt 4), 665-673.
Chehoud, C., Rafail, S., Tyldsley, A. S., Seykora, J. T., Lambris, J. D., & & Grice, E. A. (2013).
Complement modulates the cutaneous microbiome and inflammatory milieu. Proc Natl Acad Sci U S A, 110(37), 15061-15066.
Chiller, K., Selkin, B. A., & Murakawa, G. J. (2001). Skin microflora and bacterial infect ions of the skin. J Investig Dermatol Symp Proc, 6(3), 170-174.
Clucas, D. B., Carville, K. S., Connors, C., Currie, B. J., Carapetis, J. R., & Andrews, R. M.
(2008). Disease burden and health-care clinic attendances for young children in remote aboriginal communities of northern Australia. Bull World Health Organ, 86(4), 275-
281. Cook, A. M., & Romanelli, F. (2003). Ivermectin for the treatment of resistant scabies. Ann
Pharmacother, 37(2), 279-281.
Cunningham, M. W. (2008). Pathogenesis of group A streptococcal infections and their sequelae. Adv Exp Med Biol, 609, 29-42.
Currie, B. J., & Carapetis, J. R. (2000). Skin infections and infestations in Aborigina l communities in northern Australia. Australas J Dermatol, 41(3), 139-143; quiz 144-135.
Currie, B. J., Maguire, G. P., & Wood, Y. K. (1995). Ivermectin and crusted (Norwegian) scabies. Med J Aust, 163(10), 559-560.
Currie, B. J., & McCarthy, J. S. (2010). Permethrin and ivermectin for scabies. N Engl J Med, 362(8), 717-725.
Cutler, C. W., Kalmar, J. R., & Arnold, R. R. (1991). Antibody-dependent alternate pathway
of complement activation in opsonophagocytosis of Porphyromonas gingivalis. Infect Immun, 59(6), 2105-2109.
Daix, V., Schroeder, H., Praet, N., Georgin, J. P., Chiappino, I., Gillet, L., de Fays, K., Decrem, Y., Leboulle, G., Godfroid, E., Bollen, A., Pastoret, P. P., Gern, L., Sharp, P. M., &
Vanderplasschen, A. (2007). Ixodes ticks belonging to the Ixodes ricinus complex encode a family of anticomplement proteins. Insect Mol Biol, 16(2), 155-166.
Degn, S. E., & Thiel, S. (2013). Humoral pattern recognition and the complement system. Scand J Immunol, 78(2), 181-193.
Elgart, M. L. (1996). A risk-benefit assessment of agents used in the treatment of scabies. Drug
Saf, 14(6), 386-393. Ellis, N. M., Kurahara, D. K., Vohra, H., Mascaro-Blanco, A., Erdem, G., Adderson, E. E.,
Veasy, L. G., Stoner, J. A., Tam, E., Hill, H. R., Yamaga, K., & Cunningham, M. W. (2010). Priming the immune system for heart disease: a perspective on group A streptococci. J Infect Dis, 202(7), 1059-1067.
Engelman, D., Kiang, K., Chosidow, O., McCarthy, J., Fuller, C., Lammie, P., Hay, R., Steer, A., & Members Of The International Alliance For The Control Of, S. (2013). Toward
the global control of human scabies: introducing the International Alliance for the Control of Scabies. PLoS Negl Trop Dis, 7(8), e2167.
Erdem, G., Mizumoto, C., Esaki, D., Reddy, V., Kurahara, D., Yamaga, K., Abe, L., Johnson,
D., Yamamoto, K., & Kaplan, E. L. (2007). Group A Streptococcal Isolates Temporally Associated with Acute Rheumatic Fever in Hawaii: Differences from the Continenta l
United States. Clin Infect Dis, 45(3), e20-e24. Fernie-King, B. A., Seilly, D. J., Willers, C., Wurzner, R., Davies, A., & Lachmann, P. J.
(2001). Streptococcal inhibitor of complement (SIC) inhibits the membrane attack
complex by preventing uptake of C567 onto cell membranes. Immunology, 103(3), 390-398.
Fischer, K., Holt, D., Currie, B., & Kemp, D. (2012). Scabies: Important Clinical Consequences Explained by New Molecular Studies. In D. Rollinson & S. I. Hay (Eds.), Adv Parasitol (Vol. 79, pp. 339-373): Academic Press.
Fischer, K., Holt, D. C., Harumal, P., Currie, B. J., Walton, S. F., & Kemp, D. J. (2003). Generation and characterization of cDNA clones from Sarcoptes scabiei var. hominis
for an expressed sequence tag library: identification of homologues of house dust mite allergens. Am J Trop Med Hyg, 68(1), 61-64.
Frank, M. M., & Fries, L. F. (1991). The role of complement in inflammation and phagocytos is.
Immunol Today, 12(9), 322-326. Guilherme, L., & Kalil, J. (2010). Rheumatic fever and rheumatic heart disease: cellular
mechanisms leading autoimmune reactivity and disease. J Clin Immunol, 30(1), 17-23. Haidan, A., Talay, S. R., Rohde, M., Sriprakash, K. S., Currie, B. J., & Chhatwal, G. S. (2000).
Pharyngeal carriage of group C and group G streptococci and acute rheumatic fever in
an Aboriginal population. Lancet, 356(9236), 1167-1169. Hanna, J. N., & Heazlewood, R. J. (2005). The epidemiology of acute rheumatic fever in
Indigenous people in north Queensland. Aust N Z J Public Health, 29(4), 313-317. Hashiguchi, N., Chen, Y., Rusu, C., Hoyt, D., & Junger, W. (2005). Whole-Blood Assay to
Measure Oxidative Burst and Degranulation of Neutrophils for Monitoring Trauma
Patients. Eur J Trauma, 31(4), 379-388. Hengge, U. R., Currie, B. J., Jäger, G., Lupi, O., & Schwartz, R. A. (2006). Scabies: a
ubiquitous neglected skin disease. Lancet Infect Dis, 6(12), 769-779. Holt, D. C., Fischer, K., Allen, G. E., Wilson, D., Wilson, P., Slade, R., Currie, B. J., Walton,
S. F., & Kemp, D. J. (2003). Mechanisms for a novel immune evasion strategy in the
scabies mite sarcoptes scabiei: a multigene family of inactivated serine proteases. J Invest Dermatol, 121(6), 1419-1424.
74
Hourcade, D. E. (2006). The role of properdin in the assembly of the alternative pathway C3 convertases of complement. J Biol Chem, 281(4), 2128-2132.
Jackson, A., Heukelbach, J., Filho, A. F. d. S., Campelo Júnior, E. d. B., & Feldmeier, H. (2007). Clinical features and associated morbidity of scabies in a rural community in
Alagoas, Brazil. TM & IH, 12(4), 493-502. Joiner, K., Brown, E., Hammer, C., Warren, K., & Frank, M. (1983). Studies on the mechanism
of bacterial resistance to complement-mediated killing. III. C5b-9 deposits stably on
rough and type 7 S. pneumoniae without causing bacterial killing. J Immunol, 130(2), 845-849.
Kaplan, E. L., Anthony, B. F., Chapman, S. S., Ayoub, E. M., & Wannamaker, L. W. (1970). The influence of the site of infection on the immune response to group A streptococci. J Clin Invest, 49(7), 1405-1414.
Kaplan, E. L., & Bisno, A. L. (2006). Antecedent streptococcal infection in acute rheumatic fever. Clin Infect Dis, 43(6), 690-692.
Kasper, G., Brown, A., Eberl, M., Vallar, L., Kieffer, N., Berry, C., Girdwood, K., Eggleton, P., Quinnell, R., & Pritchard, D. I. (2001). A calreticulin-like molecule from the human hookworm Necator americanus interacts with C1q and the cytoplasmic signall ing
domains of some integrins. Parasite Immunol, 23(3), 141-152. Kearns, T., Clucas, D., Connors, C., Currie, B. J., Carapetis, J. R., & Andrews, R. M. (2013).
Clinic attendances during the first 12 months of life for Aboriginal children in five remote communities of northern Australia. PLoS One, 8(3), e58231.
Kobayashi, S. D., & DeLeo, F. R. (2009). Role of neutrophils in innate immunity: a systems
biology- level approach. Wiley Interdiscip Rev Syst Biol Med, 1(3), 309-333. Lassa, S., Campbell, M. J., & Bennett, C. E. (2011). Epidemiology of scabies prevalence in the
U.K. from general practice records. Br J Dermatol, 164(6), 1329-1334. Le Hello, S., Doloy, A., Baumann, F., Roques, N., Coudene, P., Rouchon, B., Lacassin, F., &
Bouvet, A. (2010). Clinical and Microbial Characteristics of Invasive Streptococcus
pyogenes Disease in New Caledonia, a Region in Oceania with a High Incidence of Acute Rheumatic Fever. J Clin Microbiol, 48(2), 526-530.
Lei, B., DeLeo, F. R., Hoe, N. P., Graham, M. R., Mackie, S. M., Cole, R. L., Liu, M., Hill, H. R., Low, D. E., Federle, M. J., Scott, J. R., & Musser, J. M. (2001). Evasion of human innate and acquired immunity by a bacterial homolog of CD11b that inhib its
opsonophagocytosis. Nat Med, 7(12), 1298-1305. Lennon, D. (2000). Rheumatic fever, a preventable disease? The New Zealand experience.
Streptococci and streptococcal diseases: entering the new millennium. Porirua: Institute of Environmental Science and Research, 503-512.
Lynch, N. J., Roscher, S., Hartung, T., Morath, S., Matsushita, M., Maennel, D. N., Kuraya,
M., Fujita, T., & Schwaeble, W. J. (2004). L-ficolin specifically binds to lipoteicho ic acid, a cell wall constituent of Gram-positive bacteria, and activates the lectin pathway
of complement. J Immunol, 172(2), 1198-1202. McDonald, M., Brown, A., Edwards, T., Hope, A., Amu, M., Morey, F., Currie, B. J., &
Carapetis, J. R. (2007). Apparent contrasting rates of pharyngitis and pyoderma in
regions where rheumatic heart disease is highly prevalent. Heart Lung Circ, 16(4), 254-259.
McDonald, M., Currie, B. J., & Carapetis, J. R. (2004). Acute rheumatic fever: a chink in the chain that links the heart to the throat? Lancet Infect Dis, 4(4), 240-245.
McDonald, M. I., Towers, R. J., Andrews, R., Benger, N., Fagan, P., Currie, B. J., & Carapetis,
J. R. (2008). The dynamic nature of group A streptococcal epidemiology in tropical communities with high rates of rheumatic heart disease. Epidemiol Infect, 136(4), 529-
539.
75
McMillan, D. J., Dreze, P. A., Vu, T., Bessen, D. E., Guglielmini, J., Steer, A. C., Carapetis, J. R., Van Melderen, L., Sriprakash, K. S., & Smeesters, P. R. (2013). Updated model of
group A Streptococcus M proteins based on a comprehensive worldwide study. Clin Microbiol Infect, 19(5), E222-229.
Mellanby, K. (1944). The development of symptoms, parasitic infection and immunity in human scabies. Parasitology, 35(04), 197-206.
Meri, T., Amdahl, H., Lehtinen, M. J., Hyvarinen, S., McDowell, J. V., Bhattacharjee, A., Meri,
S., Marconi, R., Goldman, A., & Jokiranta, T. S. (2013). Microbes bind complement inhibitor factor H via a common site. PLoS Pathog, 9(4), e1003308.
Mika, A., Goh, P., Holt, D. C., Kemp, D. J., & Fischer, K. (2011). Scabies mite peritrophins are potential targets of human host innate immunity. PLoS Negl Trop Dis, 5(9), e1331.
Mika, A., Reynolds, S. L., Mohlin, F. C., Willis, C., Swe, P. M., Pickering, D. A., Halilovic,
V., Wijeyewickrema, L. C., Pike, R. N., Blom, A. M., Kemp, D. J., & Fischer, K. (2012a). Novel scabies mite serpins inhibit the three pathways of the human
complement system. PLoS One, 7(7), e40489. Mika, A., Reynolds, S. L., Pickering, D., McMillan, D., Sriprakash, K. S., Kemp, D. J., &
Fischer, K. (2012b). Complement inhibitors from scabies mites promote streptococcal
growth--a novel mechanism in infected epidermis? PLoS Negl Trop Dis, 6(7), e1563. Mounsey, K., Ho, M. F., Kelly, A., Willis, C., Pasay, C., Kemp, D. J., McCarthy, J. S., &
Fischer, K. (2010). A tractable experimental model for study of human and animal scabies. PLoS Negl Trop Dis, 4(7), e756.
Mounsey, K. E., Holt, D. C., McCarthy, J., Currie, B. J., & Walton, S. F. (2008). Scabies:
molecular perspectives and therapeutic implications in the face of emerging drug resistance. Future Microbiol, 3(1), 57-66.
Mounsey, K. E., Holt, D. C., McCarthy, J. S., Currie, B. J., & Walton, S. F. (2009). Longitudinal evidence of increasing in vitro tolerance of scabies mites to ivermectin in scabies-endemic communities. Arch Dermatol, 145(7), 840-841.
Mulenga, A., Tsuda, A., Onuma, M., & Sugimoto, C. (2003). Four serine proteinase inhibito rs (serpin) from the brown ear tick, Rhiphicephalus appendiculatus; cDNA cloning and
preliminary characterization. Insect Biochem Mol Biol, 33(2), 267-276. Nissenson, A. R., Baraff, L. J., Fine, R. N., & Knutson, D. W. (1979). Poststreptococcal acute
glomerulonephritis: fact and controversy. Ann Intern Med, 91(1), 76-86.
Nunn, M. A., Sharma, A., Paesen, G. C., Adamson, S., Lissina, O., Willis, A. C., & Nuttall, P. A. (2005). Complement inhibitor of C5 activation from the soft tick Ornithodoros
moubata. J Immunol, 174(4), 2084-2091. Oh, H., Siano, B., & Diamond, S. (2008). Neutrophil Isolation Protocol. J Vis Exp(17). Oh, J., Freeman, A. F., Program, N. C. S., Park, M., Sokolic, R., Candotti, F., Holland, S. M.,
Segre, J. A., & Kong, H. H. (2013). The altered landscape of the human skin microbiome in patients with primary immunodeficiencies. Genome Res, 23(12), 2103-
2114. Parks, T., Smeesters, P. R., & Steer, A. C. (2012). Streptococcal skin infection and rheumatic
Pearce, E. J., Hall, B. F., & Sher, A. (1990). Host-specific evasion of the alternat ive complement pathway by schistosomes correlates with the presence of a phospholipase
Pils, B., & Schultz, J. (2004). Inactive enzyme-homologues find new function in regulatory
processes. J Mol Biol, 340(3), 399-404.
76
Podbielski, A., Schnitzler, N., Beyhs, P., & Boyle, M. D. (1996). M-related protein (Mrp) contributes to group A streptococcal resistance to phagocytosis by human granulocytes.
Mol Microbiol, 19(3), 429-441. Potempa, M., Potempa, J., Kantyka, T., Nguyen, K. A., Wawrzonek, K., Manandhar, S. P.,
Popadiak, K., Riesbeck, K., Eick, S., & Blom, A. M. (2009). Interpain A, a cysteine proteinase from Prevotella intermedia, inhibits complement by degrading complement factor C3. PLoS Pathog, 5(2), e1000316.
Ralph, A. P., & Carapetis, J. R. (2013). Group a streptococcal diseases and their global burden. Curr Top Microbiol Immunol, 368, 1-27.
Rapp, C. M., Morgan, M. S., & Arlian, L. G. (2006). Presence of host immunoglobulin in the gut of Sarcoptes scabiei (Acari: Sarcoptidae). J Med Entomol, 43(3), 539-542.
Rawlings, N. D., & Barrett, A. J. (1994). Families of serine peptidases. Methods Enzymol, 244,
19-61. Reynolds, S. L., Pike, R. N., Mika, A., Blom, A. M., Hofmann, A., Wijeyewickrema, L. C.,
Kemp, D., & Fischer, K. (2014). Scabies mite inactive serine proteases are potent inhibitors of the human complement lectin pathway. PLoS Negl Trop Dis, 8(5), e2872.
Rodriguez-Iturbe, B., & Batsford, S. (2007). Pathogenesis of poststreptococcal
glomerulonephritis a century after Clemens von Pirquet. Kidney Int, 71(11), 1094-1104. Romani, L., Koroivueta, J., Steer, A. C., Kama, M., Kaldor, J. M., Wand, H., Hamid, M., &
Whitfeld, M. J. (2015). Scabies and Impetigo Prevalence and Risk Factors in Fiji: A National Survey. PLoS Negl Trop Dis, 9(3).
Ross, J., Jiang, H., Kanost, M. R., & Wang, Y. (2003). Serine proteases and their homologs in
the Drosophila melanogaster genome: an initial analysis of sequence conservation and phylogenetic relationships. Gene, 304, 117-131.
Sanford, J. A., & Gallo, R. L. (2013). Functions of the skin microbiota in health and disease. Semin Immunol, 25(5), 370-377.
Sigma-Aldrich. (2011). Density Gradient Centrifugation using Histopaque®-1119.
Centrifugation Separations. Retrieved 30 March, 2015, from http://www.sigmaaldrich.com/content/dam/sigma-aldrich/docs/Sigma-
Aldrich/Brochure/1/biofiles_v6_n5.pdf Simser, J. A., Mulenga, A., Macaluso, K. R., & Azad, A. F. (2004). An immune responsive
factor D-like serine proteinase homologue identified from the American dog tick,
Dermacentor variabilis. Insect Mol Biol, 13(1), 25-35. Sriprakash, K. S., & Hartas, J. (1996). Lateral genetic transfers between group A and G
streptococci for M-like genes are ongoing. Microb Pathog, 20(5), 275-285. Steer, A. (2014). Scabies joins the list of WHO neglected tropical diseases. The Lancet, Global
Steer, A. C., Jenney, A. W., Kado, J., Batzloff, M. R., La Vincente, S., Waqatakirewa, L., Mulholland, E. K., & Carapetis, J. R. (2009). High burden of impetigo and scabies in a tropical country. PLoS Negl Trop Dis, 3(6), e467.
Stoevesandt, J., Carle, L., Leverkus, M., & Hamm, H. (2012). Control of large institutiona l scabies outbreaks. J Dtsch Dermatol Ges, 10(9), 637-647.
Suchitra, S., & Joshi, P. (2005). Characterization of Haemonchus contortus calreticul in suggests its role in feeding and immune evasion by the parasite. Biochim Biophys Acta, 1722(3), 293-303.
Swe, P. M., & Fischer, K. (2014). A scabies mite serpin interferes with complement-media ted neutrophil functions and promotes staphylococcal growth. PLoS Negl Trop Dis, 8(6),
Swe, P. M., Zakrzewski, M., Kelly, A., Krause, L., & Fischer, K. (2014). Scabies mites alter the skin microbiome and promote growth of opportunistic pathogens in a porcine
model. PLoS Negl Trop Dis, 8(5), e2897. Taplin, D., Meinking, T. L., Chen, J. A., & Sanchez, R. (1990). Comparison of crotamiton 10%
cream (Eurax) and permethrin 5% cream (Elimite) for the treatment of scabies in children. Pediatr Dermatol, 7(1), 67-73.
Taplin, D., Porcelain, S. L., Meinking, T. L., Athey, R. L., Chen, J. A., Castillero, P. M., &
Sanchez, R. (1991). Community control of scabies: a model based on use of permethrin cream. Lancet, 337(8748), 1016-1018.
Thern, A., Stenberg, L., Dahlback, B., & Lindahl, G. (1995). Ig-binding surface proteins of Streptococcus pyogenes also bind human C4b-binding protein (C4BP), a regulatory component of the complement system. J Immunol, 154(1), 375-386.
Towers, R. J., Gal, D., McMillan, D., Sriprakash, K. S., Currie, B. J., Walker, M. J., Chhatwal, G. S., & Fagan, P. K. (2004). Fibronectin-binding protein gene recombination and
horizontal transfer between group A and G streptococci. J Clin Microbiol, 42(11), 5357-5361.
Tyson, K., Elkins, C., Patterson, H., Fikrig, E., & de Silva, A. (2007). Biochemical and
functional characterization of Salp20, an Ixodes scapularis tick salivary protein that inhibits the complement pathway. Insect Mol Biol, 16(4), 469-479.
Tyson, K. R., Elkins, C., & de Silva, A. M. (2008). A novel mechanism of complement inhibition unmasked by a tick salivary protein that binds to properdin. J Immunol, 180(6), 3964-3968.
Vogel, C. W., Smith, C. A., & Muller-Eberhard, H. J. (1984). Cobra venom factor: structural homology with the third component of human complement. J Immunol, 133(6), 3235-
3241. von Pawel-Rammingen, U., & Bjorck, L. (2003). IdeS and SpeB: immunoglobulin-degrad ing
cysteine proteinases of Streptococcus pyogenes. Curr Opin Microbiol, 6(1), 50-55.
Walport, M. J. (2001). Complement. First of two parts. N Engl J Med, 344(14), 1058-1066. Walter, B., Heukelbach, J., Fengler, G., Worth, C., Hengge, U., & Feldmeier, H. (2011).
Comparison of dermoscopy, skin scraping, and the adhesive tape test for the diagnosis of scabies in a resource-poor setting. Arch Dermatol, 147(4), 468-473.
Walton, S. F., Beroukas, D., Roberts-Thomson, P., & Currie, B. J. (2008). New insights into
disease pathogenesis in crusted (Norwegian) scabies: the skin immune response in crusted scabies. Br J Dermatol, 158(6), 1247-1255.
Walton, S. F., & Currie, B. J. (2007). Problems in diagnosing scabies, a global disease in human and animal populations. Clin Microbiol Rev, 20(2), 268-279.
Walton, S. F., McBroom, J., Mathews, J. D., Kemp, D. J., & Currie, B. J. (1999). Crusted
Scabies: A Molecular Analysis of Sarcoptes scabiei var. hominis Populations from Patients with Repeated Infestations. Clin Infect Dis, 29(5), 1226-1230.
Wannamaker, L. W. (1973). The chain that links the heart to the throat. Circulation, 48(1), 9-18.
Wessels, M. R., Butko, P., Ma, M., Warren, H. B., Lage, A. L., & Carroll, M. C. (1995). Studies
of group B streptococcal infection in mice deficient in complement component C3 or C4 demonstrate an essential role for complement in both innate and acquired immunity.
Proc Natl Acad Sci U S A, 92(25), 11490-11494. Whitehall, J., Kuzulugil, D., Sheldrick, K., & Wood, A. (2013). Burden of paediatric pyoderma
and scabies in North West Queensland. J Paediatr Child Health, 49(2), 141-143.
Willis, C., Fischer, K., Walton, S. F., Currie, B. J., & Kemp, D. J. (2006). Scabies mite inactivated serine protease paralogues are present both internally in the mite gut and
externally in feces. Am J Trop Med Hyg, 75(4), 683-687.
78
Wong, L. C., Amega, B., Connors, C., Barker, R., Dulla, M. E., Ninnal, A., Kolumboort, L., Cumaiyi, M. M., & Currie, B. J. (2001). Outcome of an interventiona l program for
scabies in an Indigenous community. Med J Aust, 175(7), 367-370. Yuste, J., Ali, S., Sriskandan, S., Hyams, C., Botto, M., & Brown, J. S. (2006). Roles of the
alternative complement pathway and C1q during innate immunity to Streptococcus pyogenes. J Immunol, 176(10), 6112-6120.
Zeeuwen, P. L., Boekhorst, J., van den Bogaard, E. H., de Koning, H. D., van de Kerkhof, P.
M., Saulnier, D. M., van, S., II, van Hijum, S. A., Kleerebezem, M., Schalkwijk, J., & Timmerman, H. M. (2012). Microbiome dynamics of human epidermis following skin
barrier disruption. Genome Biol, 13(11), R101. Zhou, H. F., Yan, H., Stover, C. M., Fernandez, T. M., Rodriguez de Cordoba, S., Song, W.
C., Wu, X., Thompson, R. W., Schwaeble, W. J., Atkinson, J. P., Hourcade, D. E., &
Pham, C. T. (2012). Antibody directs properdin-dependent activation of the complement alternative pathway in a mouse model of abdominal aortic aneurysm. Proc
Natl Acad Sci U S A, 109(7), E415-422.
79
Appendix
80
Growth curve results – for estimation of GAS cfu challenge dose.
Time Dilution Counts per 10uL (GAS 88/30) Total Average Cfu/ml