The Role of platelets aggregation tests in the diagnosis of clopidogril resistance rate in patients undergoing pecutaneous coronary intervension Introduction: Platelets play a key role in the pathophysiology of thrombosis after plaque rupture (1) .Plaque rupture occurs spontaneously in patients with acute coronary syndromes (ACS), or may be iatrogenically induced in patients undergoing percutaneous coronary interventions (PCI).significant reduction in such vascular events in high risk patients with coronary artery diseases or strokes has been achieved using antiplatelet therapy (2) . Clopidogrel is a potent antiplatelet drug that is effective in reducing the risk of vascular events in patients with established vascular disease (3-5) .In addition, it potentiates the effect of aspirin in reducing ischemic and vascular events in the setting of 1
Transcript
The Role of platelets aggregation tests in the diagnosis of clopidogril
resistance rate in patients undergoing pecutaneous coronary
intervension
Introduction:
Platelets play a key role in the pathophysiology of thrombosis after plaque
rupture (1) .Plaque rupture occurs spontaneously in patients with acute coronary
syndromes (ACS), or may be iatrogenically induced in patients undergoing
percutaneous coronary interventions (PCI).significant reduction in such vascular
events in high risk patients with coronary artery diseases or strokes has been
achieved using antiplatelet therapy (2) .
Clopidogrel is a potent antiplatelet drug that is effective in reducing the risk
of vascular events in patients with established vascular disease (3-5) .In addition, it
potentiates the effect of aspirin in reducing ischemic and vascular events in the
setting of cardiovascular diseases (6-8). However, a substantial percentage of
patients with Cardiovascular Disease (CAD) showed low or no response to
clopidogrel therapy (9-11) .
The term ‘Clopidogrel Resistance’ has been used to denote non-
responsiveness of Adenosine Di-Phophatase “ADP” induced platelet aggregation
following standard clopidogrel therapy(12). Several factors were suggested to
explain low response to clopidogrel; including genetic polymorphisms, cellular
factors “e.g. accelerated platelet turnover” and drug-drug interaction” (13) .
Platelet Aggregation:
1
Adenosine diphosphate plays an important role in platelet activation and
aggregation (14,15). Upon damaged or disrupted endothelium, circulating platelets
adhere to the vessel wall through interactions with the subendothelium constituents
(collagen, von Willebrand factor, and other adhesive proteins such as fibronectin,
laminin, and vitronectin) (15,16). After adhesion, these anchored platelets undergo
conformational changes through the action of several extrinsic activators such as
collagen, thrombin, and epinephrine.Once activated, platelets release the contents
of their dense (platelet agonists such as ADP and serotonin) and alpha-granules
(fibrinogen, von Willebrand factor, other adhesive proteins, proinflammatory
factors, and prothrombotic factors), which trigger platelet-activating intracellular
signals in surrounding platelets. Activated platelets also synthesize and release
thromboxane A2 in circulation. Activated and degranulated platelets expose
glycoprotein (GP) IIb/IIIa receptors at their surface allowing fibrinogen binding,
which forms bridges between adjacent activated platelets causing platelet
aggregation. In addition, the release of granule contents amplifies the coagulation
and inflammatory processes.
ADP and its receptor. Adenosine diphosphate binds to neighboring
platelets through two G-protein coupled receptors (P2Y1 and P2Y12) and the
surface of the platelets (GPIa/IIa, GPIb/V/IX, GPVI, etc.) bind the former
molecules, causing the platelet to adhere to the site of the endothelial injury (23,24).
Platelet adhesion leads to activation of the cell through intracellular
metabolic cascades. As a result,platelets aggregate through fibrinogen bridges,
which bind to the activated platelet receptor-integrin αIIbβ3 (GPIIb/IIIa). Activated
platelets release biologically active substances, stored inside the cell or newly
synthesised,among them adenosine diphosphate (ADP), arachidonic acid, platelet-
activating factor (PAF) and serotonin, which induce and preserve platelet
activation and aggregation through positive feedback mechanisms(24,25).
The secretion of pre-coagulant factors from the platelets (e.g. Factor V) and
the interaction with the negatively charged phospholipids of the platelet membrane
maximise the reaction of thrombin synthesis,which has been initiated by the
intravascular exposure of tissue factor and is one of the most powerful platelet
activators. These mechanisms may partly explain the recurrence of thrombotic
episodes in patients already on antiplatelet medication, and justify the need for this
type of drug in cases of acute ischaemic events(26,27) .
Apart from the established importance of platelet actions in the thrombotic
procedure, they play a significant role in the formation of the atheromatous plaque.
According to recent reports, they adhere to the endothelium under mild
inflammatory conditions and attract monocytes, which penetrate the sub-
endothelium and are transformed into macrophages and foam cells.
Several adhesion molecules (P-selectin, ICAM-1) and chemokines (MCP-1,
SDF-1, IL1β, IL- 8, CD40L, RANTES, ENA-78, etc.) participate in these
intercellular interactions and enhance the inflammation in the arterial wall(28).
5
Apart from monocytes,endothelial progenitor cells (EPCs) are also recruited
by the activated platelets and have the potential to transform into either foam cells,
promoting atherogenesis, or into endothelial cells, leading to endothelial
regeneration (29).
Figure2: Central role of ADP-P2Y12 Receptor Interaction In Platelet Activation And
Aggregation.
Mechanism of action and metabolism of Clopidogrel:
Clopidogrel is a prodrug that requires hepatic conversion into an active
metabolite to exert its antiplatelet response .Most of absorbed clopidogrel (85% to
90%) is hydrolyzed by carboxylase to an inactive carboxylic acid
metabolite,SR26334, whereas the remaining 10% to 15% is rapidly metabolized by
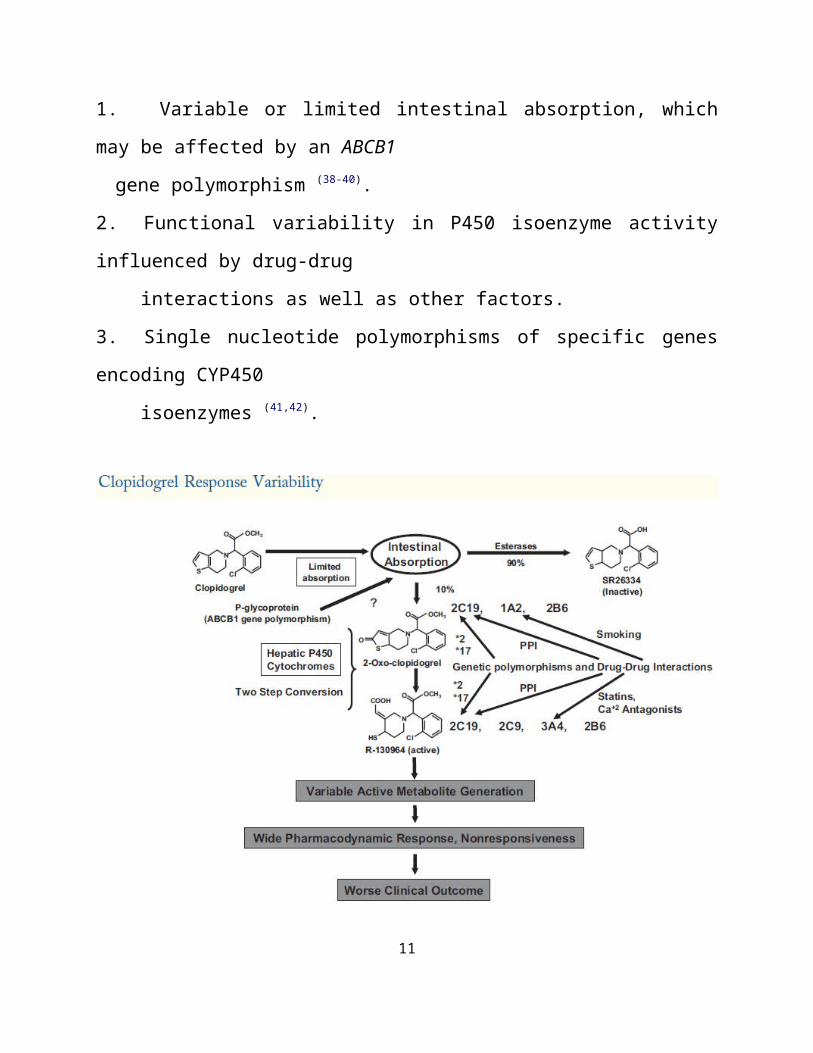
hepatic cytochrome (CYP) P450 isoenzymes in a 2-step process ( Fig. 3).
6
In the first step, the thiophene ring of clopidogrel is oxidized to 2-oxo-
clopidogrel, which is then hydrolyzed to a highly labile active metabolite, R-
130964, which has both carboxylic acid and thiol groups (30-32).
Recent studies indicate that CYP2C19, CYP1A2, and CYP2B6 participate in
the first metabolic step, whereas CYP2C19, CYP2C9, CYP2B6, and CYP3A are
responsible for the second step (30,31) (Fig. 3).
The highly unstable active metabolite, R-130964, covalently binds to
platelet P2Y12 receptor specifically and irreversibly during passage through the
hepatic circulation resulting in inhibition of ADP-induced platelet activation-
aggregation for the life span of the platelet (33) (Fig 1).
This metabolic activation scheme is consistent with the time-dependent
cumulative inhibition of ADP-induced platelet aggregation as observed with
repeated daily dosing of clopidogrel and is further highlighted by slow recovery of
platelet function following drug withdrawal (34-36).
Multiple lines of evidence strongly suggest that variable and insufficient
active metabolite generation are the primary explanations for clopidogrel response
variability and nonresponsiveness, respectively (37).
Variable levels of active metabolite generation following clopidogrel
administration could be explained by:
1. Variable or limited intestinal absorption, which may be affected by an ABCB1
gene polymorphism (38-40).
2. Functional variability in P450 isoenzyme activity influenced by drug-drug
interactions as well as other factors.
3. Single nucleotide polymorphisms of specific genes encoding CYP450
isoenzymes (41,42).
7
Figure 3:Clopidogrel response variability is a pharmacokinetic problem primarily influenced by the activity of cytochrome P450 isoenzymes in the generation of the active metabolite.Absorption may be affected by polymorphism of the ABCB1 gene.The activity of hepatic cytochrome isoenzymes are influenced by drug-drug interactions, single nucleotide polymorphisms, and environmental influences (smoking).
Pharmacokinetic and Pharmacodynamic properties of Clopidegrel:
Clopidogrel is a member of the thienopyridine family, along with
ticlopidyne and prasugrel, and is a powerful antiplatelet agent (43). It is a product,
which is absorbed in the gut with the aid of the ABCB1/MDR1 protein transporter.
Subsequently, it is converted to the active metabolite by several isoforms of
cytochrome P450 in the liver, mainly CYP2C19. CYP3A4, CYP3A5,CYP1A2,
CYP2B6 and CYP2C9 also participate in the procedure.
8
The maximum concentration of the active metabolite in blood is reached
within 1 hour after administration of 600 mg clopidogrel (44). interestingly, 85% of
the absorbed drug is hydrolysed by plasma and intestinal mucosa esterases to form
inactive product. The half-life of the active metabolite after a single or multiple
doses is about 8 hours (45). This active metabolite is a potent selective inhibitor of
the P2Y12ADP receptor, which exerts its action by forming disulphuric bonds with
2 serine residues (ser-17, ser-270) of the receptor molecule.
The maximum inhibitory activity of clopidogrel is reached in 24 hours after
administration of 75 mg, in six hours after 300 mg, and in two hours after 600 mg.
as the P2Y12 receptor blockade is irreversible, and 10% of platelets are renewed
daily, at 5 days after treatment cessation 50% of the circulating platelets will be
completely functional and capable of producing adequate hemostasis(46).
Clopidogrel resistance — definition
No single receptor signaling pathway mediating platelet activation is
responsible for all thrombotic complications. Therefore, a single treatment strategy
directed against a specific receptor cannot overcome all thrombotic complications.
With this in mind, it is our opinion that the optimal definition of resistance or non
responsiveness to an antiplatelet agent is the failure of the antiplatelet agent to
inhibit the target of its action. The identification of resistance would therefore
9
utilize a laboratory technique that detects residual activity of the target. Therefore,
clopidogrel resistance is best demonstrated by evidence of residual post-treatment
P2Y12 activity by measuring ADP-induced platelet aggregation before and after
treatment. Since thrombosis involves multiple signaling pathways, treatment
failure is not synonymous with drug resistance (47).
Aim of the study:
To estimate the incidence of clopidogrel resistance among Kurdish
population and to find the predictors of its resistance.
Materials and methods:
This is a prospective study, 100 patients planned to undergo