Accepted Manuscript Title: The Role of Reactive Oxygen Species and Metabolism on Cancer cells and their Microenvironment Author: Ana Costa Alix Scholer-Dahirel Fatima Mechta-Grigoriou PII: S1044-579X(14)00004-2 DOI: http://dx.doi.org/doi:10.1016/j.semcancer.2013.12.007 Reference: YSCBI 1097 To appear in: Seminars in Cancer Biology Received date: 30-7-2013 Revised date: 22-12-2013 Accepted date: 30-12-2013 Please cite this article as: Costa A, Scholer-Dahirel A, Mechta-Grigoriou F, The Role of Reactive Oxygen Species and Metabolism on Cancer cells and their Microenvironment, Seminars in Cancer Biology (2014), http://dx.doi.org/10.1016/j.semcancer.2013.12.007 This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Transcript
Accepted Manuscript
Title: The Role of Reactive Oxygen Species and Metabolismon Cancer cells and their Microenvironment
Author: Ana Costa Alix Scholer-Dahirel FatimaMechta-Grigoriou
Received date: 30-7-2013Revised date: 22-12-2013Accepted date: 30-12-2013
Please cite this article as: Costa A, Scholer-Dahirel A, Mechta-Grigoriou F, The Role ofReactive Oxygen Species and Metabolism on Cancer cells and their Microenvironment,Seminars in Cancer Biology (2014), http://dx.doi.org/10.1016/j.semcancer.2013.12.007
This is a PDF file of an unedited manuscript that has been accepted for publication.As a service to our customers we are providing this early version of the manuscript.The manuscript will undergo copyediting, typesetting, and review of the resulting proofbefore it is published in its final form. Please note that during the production processerrors may be discovered which could affect the content, and all legal disclaimers thatapply to the journal pertain.
Moreover, CAFs can indirectly enhance migration of tumor cells (Fig. 2). Indeed,
CAFs, similarly as fibroblasts exposed to chronic oxidative stress, express genes that
encode for proteases involved in ECM remodeling, including collagens, cell adhesion
molecules and MMPs [25, 72]. Interestingly, imaging of invasive co-cultures of
squamous cell carcinoma cells and fibroblasts showed that the leading cell is always
a fibroblast and that cancer cells move behind the fibroblast in the extracellular tracks
[98]. Interestingly, this effect is mediated through the Rho-dependent pathway, which
generates contractile forces in stromal fibroblasts in a ROS-dependent manner to
remodel ECM and create tracks for collective migration of tumor cells [99]. CAFs
contribute to increase the invasion and metastasis by promoting also EMT of
carcinoma cells. Likewise, MMP3 overexpression, which has been observed in early
stages of human breast cancers, leads to the expression of Rac1b and to the
stimulation of mitochondrial production of ROS. This increase of ROS levels
orchestrates the induction of the EMT program [75]. The involvement of ROS and
CAFs in EMT was also shown in prostate cancer, where CAFs secrete MMPs that
lead to the production of ROS by COX-2 in tumor cells, which is required for EMT,
stemness properties and dissemination of tumor cells [100-102]. Hence, the oxidative
tumor microenvironment not only modulates the conversion of fibroblasts into
myofibroblasts but also the production of paracrine signals and matrix remodeling
enzymes that impact on tumor epithelial cells, leading to invasion and metastasis. In
that context, the role of the redox-regulation of protein tyrosine phosphatases
(PTPases) is emerging as a key mechanism regulating signaling from cell surface
receptors, i.e. tyrosine kinase receptors and integrins [139]. Indeed, PTPase
inactivation by reversible oxidation of their active site has been shown to be
important regulator of growth factor signaling, especially for PDGFR-β-mediated
signaling [140]. This reciprocal crosstalk between redox regulators and signaling
Page 14 of 33
Accep
ted
Man
uscr
ipt
14
pathways control tumor cell proliferation and migration. The molecular mechanisms
controlled by ROS that impact on CAF function and crosstalk with epithelial cells is
described below.
Metabolic cross-talk between CAFs and tumor cells
One main feature characterizing tumor cells is the metabolic reprogramming that
allows cell growth and division. Growing evidence highlights the role of CAFs
supporting the metabolic reprogramming of tumor cells. As discussed above, most
cancer cells predominantly produce energy through a high rate of glycolysis and
lactic acid fermentation, even in the presence of normal levels of O2, a process
referred to as the “Warburg effect” [103]. Recently, it has been proposed that tumor
epithelial cells also induce aerobic glycolysis in neighboring fibroblasts [104]. These
CAFs then undergo a myofibroblastic differentiation and secrete lactate and
pyruvate, energy-rich metabolites that could be taken up by tumor cells. In this
model, tumor cells use CAFs for providing them high energetic sources and
transform the normal stroma into a wound healing-like stroma. This scenario is called
the “reverse Warburg effect”. In that sense, it has been proposed that cancer and
stroma co-evolve to give advantage to tumor cell growth [105, 106]. The lactate
uptake by cancer cells increases the surrounding pH, thus also protecting cells from
the extremely acid microenvironment. Consistently, oxidative stress in CAFs leads to
the up-regulation of the mono-carboxylate transporter MCT4, which shuttles lactate
to the epithelial cells [107]. Indeed, high expression levels of the glucose transporter
MCT4 in tumor stroma correlate with poor prognosis in triple negative breast cancer
patients [108].
ROS, HIF and metabolism
It is recognized that metabolic reprogramming is under control of oxidative stress and
hypoxia. The transcription factors HIF-1 and HIF-2 play a crucial role in the metabolic
reprogramming of both CAFs and epithelial cells. Although their role remains
controversial under hypoxia, clear evidence now exist indicating that ROS stabilize
the HIF proteins under normoxic conditions through modulation of PHD enzyme
activity [5, 109-111]. Chiarugi and collaborators have shown that accumulation of
Page 15 of 33
Accep
ted
Man
uscr
ipt
15
ROS and HIF stabilization in CAFs result also from the down-regulation of SIRT3, a
mitochondrial NAD-dependent deacetylase [112]. HIF through SIRT3 can regulate
CAFs metabolism driving the Warburg phenotype. In addition, HIF-1 regulates
several genes involved in glucose metabolism, as glucose transporters (GLUT1 and
GLUT3) in order to increase the glucose uptake in the cell. Moreover, HIF targets
MCT4, responsible for the export of lactate [113]. Finally, HIF also activates pyruvate
kinase M2 (PKM2) transcription, contributing to reprogram the glucose metabolism of
epithelial cells [114]. In summary, oxidative stress induced by ROS production has
severe implications in CAFs metabolism, driving aerobic glycolysis that in turn has
profound effects on tumor microenvironment. In a simplistic view, CAFs feed the
cancers with high-rich compounds and induce anti-oxidant defense in cancer cells
allowing cancer cells to proliferate. Although the main focus of research has been
attributed until now to the interactions between CAFs and cancer cells, the effects of
oxidative stress on other components of the tumor microenvironment and the
interaction of “stressed”-CAFs with other cell types in the microenvironment remain
to be investigated, as well as further implications in tumor aggressiveness and
resistance to treatment.
ROS impact on CAF interaction with tumor epithelial cells
The high levels of ROS generated by mitochondrial dysfunction, NOX
overexpression or any other mechanisms, promote tumor cell proliferation and
motility in breast cancers [115]. In this regard, there are increasing reports in
literature demonstrating the role of ROS in several signaling pathways that control
properties of tumor cells. Here, we focus on two well-known examples of how ROS
modulate the cross-talk between cancer cells and CAFs that ultimately leads to
enhanced tumor cell motility and invasion. Cancer migration and metastasis are
mediated by chemokines receptors [116]. Chemokines can act as paracrine factors,
establishing the communication between tumor epithelial cells and the cellular
components of the tumor microenvironment to promote tumorigenesis, tumor growth
and metastasis. The two examples considered below, namely CXCL12(SDF-
1)/CXCR4 and HGF/c-met, are regulated by the HIF transcription factors under
normoxia in a ROS-dependent manner [25, 117-121].
Page 16 of 33
Accep
ted
Man
uscr
ipt
16
The activation of CXCL12/CXCR4 signaling pathway is one striking example of
cross-talk between tumor epithelial cells and CAFs that can be affected by ROS [90].
The cytokine CXCL12 is highly secreted by CAFs, while its receptor CXCR4 is found
mainly at the surface of tumor epithelial cells. CXCL12 signaling via its receptor
CXCR4 drives proliferation of tumor cells and promotes neo-angiogenesis by the
recruitment of endothelial progenitor cells into the tumor stroma [90].
CXCL12/CXCR4 signaling pathway has been found to be up-regulated in various
tumor subtypes, such as HER2 breast adenocarcinoma subtype where both CXCR4
and CXCL12 accumulate and play key roles in metastases. Indeed, up-regulation of
CXCR4 in HER2 cancer cells is essential to HER2-mediated tumor metastases [122].
In addition, accumulation of CXCL12 in the stroma of this breast cancer subtype
correlates with stromal alterations, including myofibroblast proportion and neo-
angiogenesis, NOX4 accumulation and enhanced rate of metastases [25, 123, 124].
Another example of a cross-talk between tumor epithelial cells and CAFs modulated
by ROS is the HGF/c-Met signaling pathway. Activation of HGF/c-Met pathway is
involved in the acquisition of an aggressive phenotype and resistance to therapy, as
shown in breast cancer. HGF is mainly expressed and secreted by tumor-associated
fibroblasts, while the proto-oncogene product c-Met is overexpressed at the surface
of tumor epithelial cells [125]. HGF binding to its cognate receptor c-Met increases
ROS levels in tumor cells, subsequently leading to c-Met phosphorylation and
activation of downstream signaling pathway. Activation of HGF/c-met signaling
promotes ultimately tumor cell motility and invasion [126]. Additionally, ROS levels in
the tumor bed can influence HGF bioavailability. Indeed, HGF can be sequestered in
the ECM by thrombospondin-1 [127] while high ROS levels inhibit thrombospondin-1
scavenger activity [128]. As a result, high ROS levels in the tumor bed lead to
increased HGF bioavailability and activation of the HGF/c-Met pathway.
Taken together, those observations indicate that oxidative stress in the tumor bed
can modulate the interactions between tumor epithelial cells and CAFs, contributing
to tumor progression and metastasis.
4. ROS and CAFs: similarities in cancer and wound healing
Page 17 of 33
Accep
ted
Man
uscr
ipt
17
The tumor microenvironment and the granulation tissue formed during wound healing
share remarkable histological resemblances [129]. Indeed, both tissues are
characterized by a reactive stroma composed of activated fibroblasts and specific
ECM components, immune cells and newly formed blood and lymph vessels.
However, in wounds, the stromal reaction is only a transient state while it persists in
tumors leading to Dvorak tumor definition as “wounds that do not heal”. Interestingly,
ROS have been involved in the initiation of the wound healing process, in particular
in cell attraction, migration and adhesion [130, 131]. Thus ROS-mediated signaling is
a key event share by both cancer and wound healing (Fig. 3).
ROS and inflammation: impact on CAFs
ROS, mainly H2O2, are generated immediately after injury and can act as
chemoattractant for immune cells [74, 132]. In zebrafish, tissue-scale gradient of
H2O2 allows leukocyte recruitment to the site of injury across distances of hundreds
of micrometers within minutes of wounding. Indeed, leukocyte recruitment coincides
with the presence of H2O2 in the blood vessels while H2O2 dissipation leads to a
decreased recruitment of immune cells [74]. Moreover, Feng et al. [133] showed,
using zebrafish as a model, that H2O2 is a key signal involved in early recruitment of
leukocytes to transformed cell burden. Interestingly, the authors provide evidence
that both transformed cells and normal surrounding cells contribute to generate H2O2.
To what extent H2O2 may play a similar role in higher organisms remains to be
established. In wound healing, a major consequence of leukocyte recruitment to the
wound is the secretion of inflammatory molecules. Thus, fibroblast activation and
inflammatory response are co-occurring in wound healing and cancer. Similarly,
chronic inflammation is also associated to fibroblast conversion into myofibroblasts
suggesting that inflammatory cells could induce CAF activation. Consistently,
inflammatory cytokines such as IL1β, TNF-α or TNF-β contribute to fibroblast
activation in chronic inflammatory disease. One might thus propose that H2O2-
recruited inflammatory cells could impact CAF heterogeneity and functions.
Ca2+ and ATP: primary signals inducing ROS in wound healing
Knowledge about the signals that mediate wound healing is emerging. The wound
healing response is a rapid process and the earliest molecular events identified are
Page 18 of 33
Accep
ted
Man
uscr
ipt
18
diffusible damage signals that include Ca2+ flashes, ATP release and H2O2 gradient.
These events induce short-term transcription-independent cellular effects while gene
expression is being adjusted [134]. Wounding triggers an instantaneous Ca2+ flash in
a variety of species including zebrafish, C.elegans and X.laevis [135-137]. Blocking
this Ca2+ flash inhibits H2O2 release at the wound site and leads to a reduction in the
number of immune cells migrating to the wound. The wound-induced Ca2+ flash was
shown to activate DUOX and thus to trigger the production of the attractant damage
cue H2O2 [132]. Consistently, in zebrafish and drosophila, inhibition of DUOX activity
abrogates the formation of H2O2 gradients [74, 132]. Accordingly, DUOX knockdown
results in decreased immune cell recruitment and reduces the number of transformed
cells, showing the clear impact of this signaling pathway in tumorigenesis [133]. ATP
release is also an early event triggered by wounding, and recent studies have shown
that ATP release can lead to an increase of Ca2+ and H2O2, serving as messenger
molecule to activate a cascade of signaling events in surrounding cells. This
signaling cascade is in part mediated by Ca2+ increase, subsequently leading to
activation of signaling pathways, such as EGF-dependent pathway, that ultimately
contributes to the healing of the wound [138]. As both Ca2+ and ATP have been
shown to modulate DUOX activity and subsequent H2O2 production, it is possible that
those diffusible signals might impact stromal components, such as CAFs. Consistent
with that hypothesis, H2O2, Ca2+ and ATP affect molecules, such as Rho GTPases,
that regulate the cytoskeleton and migratory properties similarly to phenotype of
tumor-associated myofibroblasts [25]. Further studies will be required to investigate
the role of Ca2+ flashes, ATP release, DUOX activity and H2O2 gradient in CAF
heterogeneity and functions and more generally in cancer biology in mammals. Being
the earliest events in wound signaling, and if homology is shared cancer, the control
of these signals could uncover new strategies in cancer therapy.
5. Conclusion Chronic oxidative stress has severe implications in tumor initiation, growth and
metastasis. ROS can be produced by tumor epithelial cells or various stromal cell
types. They act as crucial signaling molecules not only in cancer cells but also in
surrounding stromal components, through diffusion of the most stable or diffusible
ROS compounds, such as H2O2. ROS mediate cell motility and invasive properties of
Page 19 of 33
Accep
ted
Man
uscr
ipt
19
tumor cells, contribute to ECM remodeling, increase neo-angiogenesis and are
involved in the metabolic reprogramming of both tumor cells and CAFs. As described
in this review, one of the major effects of ROS is the conversion of fibroblasts into
myofibroblasts, thus shaping the tumor microenvironment. As CAFs have been newly
defined as heterogeneous populations, the effect of ROS in the genesis of these
different sub-populations and their biological functions needs to be investigated.
Moreover, ROS sustain the reactive stroma, which characterizes both wound healing
and cancers, not only by affecting CAFs but also by mediating the recruitment of
inflammatory cells. To what extent ROS-recruited inflammatory cells have an impact
on fibroblast heterogeneity is not yet known and deserves further investigation. In
summary, ROS are unarguably main drivers, which alter major stromal components
and affect the shape and the tension of the tumor microenvironment. Better
understanding of their functions in cancer prognosis and chemo-sensitivity could
therefore contribute to pave the way for new concepts of therapy.
Page 20 of 33
Accep
ted
Man
uscr
ipt
20
FIGURE LEGENDS Figure 1: Main sources of reactive oxygen species (ROS) in epithelial cancers
ROS generated from epithelial cells can result from increased metabolism associated
with dysfunctional mitochondria, oncogene activation or cytokine/chemokine
signalling that activate ROS-producing enzymes: NADPH oxidases (NOX),
clycloxygenases (COX, that mediates the production of prostanoids) and
lipoxygenases (LOX, that catalyze the dioxygenation of polyunsaturated fatty acids,
FA). In solid tumors, hypoxia could also contribute to maintain a certain rate of
oxidative stress. Additionally, levels of antioxidant defenses are often reduced. In the
tumor microenvironment, CAFs and inflammatory cells are also important producers
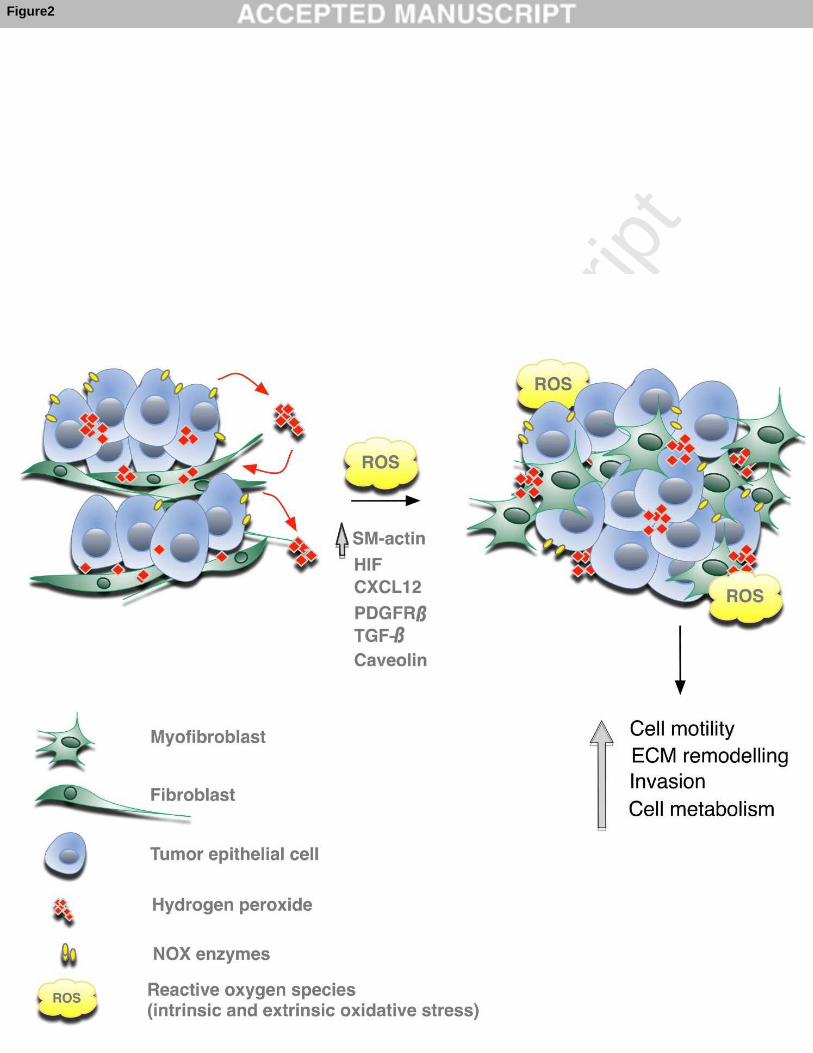
of ROS and thus contribute to persistent oxidative stress in tumors. Figure 2: Role of ROS in myofibroblast differentiation and crosstalk with tumor epithelial cells Chronic oxidative stress in tumor microenvironment, mainly represented by hydrogen
peroxide due to its highly diffusible properties, originate mostly from tumor epithelial
cells (e.g. by up-regulation of the NADPH oxidase (NOX) enzymes) and to a lower
extent from other cell types, such as myeloid cells or fibroblasts themselves. ROS
modulate several factors and signaling pathways (including HIF, CXCL12, PDGFR-β,
TGF-β, caveolin), which have been shown to play key roles in the conversion of
fibroblasts into myofibroblasts (α-smooth muscle actin-positive fibroblasts with high
migration propensity). In turn, ROS levels in activated fibroblasts stimulate paracrine
signals, which promote tumor epithelial cell motility and invasion, increase
extracellular matrix (ECM) remodeling, stimulate neo-angiogenesis and modulate
tumor cell metabolism, altogether contributing to tumor progression and metastasis.
Figure 3: Similarities between cancer and wound healing driven by ROS and CAFs
ROS, through hydrogen peroxide, modulate fibroblast conversion into myofibroblasts
and participate in the recruitment of inflammatory cells both in wound healing and
cancer. When healing process is achieved, ROS levels decrease and myofibroblasts
undergo nemesis. In contrast, in tumors, the chronic oxidative stress sustains a
persistent myofibroblastic and inflammatory environment. Moreover, in wound
healing, calcium waves and ATP release stimulate hydrogen peroxide release
Page 21 of 33
Accep
ted
Man
uscr
ipt
21
(through DUOX activation), while in tumors, these mechanisms are still poorly
characterized. Specifically, how ROS, calcium or ATP modulates fibroblast
heterogeneity and impact on their functions remains to be investigated.
Page 22 of 33
Accep
ted
Man
uscr
ipt
22
BIBLIOGRAPHY [1] Gueraud F, Atalay M, Bresgen N, Cipak A, Eckl PM, Huc L, et al. Chemistry and biochemistry of lipid peroxidation products. Free Radic Res. 2010;44:1098-124. [2] Oakley FD, Abbott D, Li Q, Engelhardt JF. Signaling components of redox active endosomes: the redoxosomes. Antioxid Redox Signal. 2009;11:1313-33. [3] Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 2006;97:1634-58. [4] Dikalov S, Griendling KK, Harrison DG. Measurement of reactive oxygen species in cardiovascular studies. Hypertension. 2007;49:717-27. [5] Gerald D, Berra E, Frapart YM, Chan DA, Giaccia AJ, Mansuy D, et al. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell. 2004;118:781-94. [6] Laurent G, Solari F, Mateescu B, Karaca M, Castel J, Bourachot B, et al. Oxidative stress contributes to aging by enhancing pancreatic angiogenesis and insulin signaling. Cell Metab. 2008;7:113-24. [7] Dolado I, Swat A, Ajenjo N, De Vita G, Cuadrado A, Nebreda AR. p38alpha MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer cell. 2007;11:191-205. [8] Gommeaux J, Cano C, Garcia S, Gironella M, Pietri S, Culcasi M, et al. Colitis and colitis-associated cancer are exacerbated in mice deficient for tumor protein 53-induced nuclear protein 1. Molecular and cellular biology. 2007;27:2215-28. [9] Dudley SC, Jr., Hoch NE, McCann LA, Honeycutt C, Diamandopoulos L, Fukai T, et al. Atrial fibrillation increases production of superoxide by the left atrium and left atrial appendage: role of the NADPH and xanthine oxidases. Circulation. 2005;112:1266-73. [10] Brand MD. The sites and topology of mitochondrial superoxide production. Exp Gerontol. 2010;45:466-72. [11] Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1-13. [12] Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell. 2012;48:158-67. [13] Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245-313. [14] Storz P. Reactive oxygen species in tumor progression. Front Biosci. 2005;10:1881-96. [15] Lu W, Hu Y, Chen G, Chen Z, Zhang H, Wang F, et al. Novel role of NOX in supporting aerobic glycolysis in cancer cells with mitochondrial dysfunction and as a potential target for cancer therapy. PLoS Biol. 2012;10:e1001326. [16] Puntel RL, Roos DH, Grotto D, Garcia SC, Nogueira CW, Rocha JB. Antioxidant properties of Krebs cycle intermediates against malonate pro-oxidant activity in vitro: a comparative study using the colorimetric method and HPLC analysis to determine malondialdehyde in rat brain homogenates. Life Sci. 2007;81:51-62. [17] Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A. 1998;95:11715-20. [18] Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721-32. [19] Pouyssegur J, Mechta-Grigoriou F. Redox regulation of the hypoxia-inducible factor. Biol Chem. 2006;387:1337-46.
Page 23 of 33
Accep
ted
Man
uscr
ipt
23
[20] Adachi Y, Shibai Y, Mitsushita J, Shang WH, Hirose K, Kamata T. Oncogenic Ras upregulates NADPH oxidase 1 gene expression through MEK-ERK-dependent phosphorylation of GATA-6. Oncogene. 2008;27:4921-32. [21] Kodama R, Kato M, Furuta S, Ueno S, Zhang Y, Matsuno K, et al. ROS-generating oxidases Nox1 and Nox4 contribute to oncogenic Ras-induced premature senescence. Genes Cells. 2013;18:32-41. [22] Mitsushita J, Lambeth JD, Kamata T. The superoxide-generating oxidase Nox1 is functionally required for Ras oncogene transformation. Cancer Res. 2004;64:3580-5. [23] Seru R, Mondola P, Damiano S, Svegliati S, Agnese S, Avvedimento EV, et al. HaRas activates the NADPH oxidase complex in human neuroblastoma cells via extracellular signal-regulated kinase 1/2 pathway. J Neurochem. 2004;91:613-22. [24] Shinohara M, Shang WH, Kubodera M, Harada S, Mitsushita J, Kato M, et al. Nox1 redox signaling mediates oncogenic Ras-induced disruption of stress fibers and focal adhesions by down-regulating Rho. J Biol Chem. 2007;282:17640-8. [25] Toullec A, Gerald D, Despouy G, Bourachot B, Cardon M, Lefort S, et al. Oxidative stress promotes myofibroblast differentiation and tumour spreading. EMBO Mol Med. 2010;2:211-30. [26] Lim SD, Sun C, Lambeth JD, Marshall F, Amin M, Chung L, et al. Increased Nox1 and hydrogen peroxide in prostate cancer. Prostate. 2005;62:200-7. [27] Szanto I, Rubbia-Brandt L, Kiss P, Steger K, Banfi B, Kovari E, et al. Expression of NOX1, a superoxide-generating NADPH oxidase, in colon cancer and inflammatory bowel disease. J Pathol. 2005;207:164-76. [28] DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106-9. [29] Mateescu B, Batista L, Cardon M, Gruosso T, de Feraudy Y, Mariani O, et al. miR-141 and miR-200a act on ovarian tumorigenesis by controlling oxidative stress response. Nat Med. 2011;17:1627-35. [30] Sporn MB, Liby KT. NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer. 2012;12:564-71. [31] Batista L, Gruosso T, Mechta-Grigoriou F. Ovarian cancer emerging subtypes: role of oxidative stress and fibrosis in tumour development and response to treatment. Int J Biochem Cell Biol. 2013;45:1092-8. [32] Hu Y, Rosen DG, Zhou Y, Feng L, Yang G, Liu J, et al. Mitochondrial manganese-superoxide dismutase expression in ovarian cancer: role in cell proliferation and response to oxidative stress. J Biol Chem. 2005;280:39485-92. [33] Min JY, Lim SO, Jung G. Downregulation of catalase by reactive oxygen species via hypermethylation of CpG island II on the catalase promoter. FEBS Lett. 2010;584:2427-32. [34] Oberley TD, Oberley LW. Antioxidant enzyme levels in cancer. Histol Histopathol. 1997;12:525-35. [35] Teoh-Fitzgerald ML, Fitzgerald MP, Jensen TJ, Futscher BW, Domann FE. Genetic and epigenetic inactivation of extracellular superoxide dismutase promotes an invasive phenotype in human lung cancer by disrupting ECM homeostasis. Mol Cancer Res. 2012;10:40-51. [36] Brandon M, Baldi P, Wallace DC. Mitochondrial mutations in cancer. Oncogene. 2006;25:4647-62.
Page 24 of 33
Accep
ted
Man
uscr
ipt
24
[37] Petros JA, Baumann AK, Ruiz-Pesini E, Amin MB, Sun CQ, Hall J, et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc Natl Acad Sci U S A. 2005;102:719-24. [38] Ohsawa S, Sato Y, Enomoto M, Nakamura M, Betsumiya A, Igaki T. Mitochondrial defect drives non-autonomous tumour progression through Hippo signalling in Drosophila. Nature. 2012;490:547-51. [39] Allinen M, Beroukhim R, Cai L, Brennan C, Lahti-Domenici J, Huang H, et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer cell. 2004;6:17-32. [40] Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9:239-52. [41] Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557-63. [42] De Wever O, Nguyen QD, Van Hoorde L, Bracke M, Bruyneel E, Gespach C, et al. Tenascin-C and SF/HGF produced by myofibroblasts in vitro provide convergent pro-invasive signals to human colon cancer cells through RhoA and Rac. FASEB J. 2004;18:1016-8. [43] Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392-401. [44] Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer cell. 2010;17:135-47. [45] Pietras K, Sjoblom T, Rubin K, Heldin CH, Ostman A. PDGF receptors as cancer drug targets. Cancer cell. 2003;3:439-43. [46] Goetz JG, Minguet S, Navarro-Lerida I, Lazcano JJ, Samaniego R, Calvo E, et al. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell. 2011;146:148-63. [47] Strutz F, Okada H, Lo CW, Danoff T, Carone RL, Tomaszewski JE, et al. Identification and characterization of a fibroblast marker: FSP1. J Cell Biol. 1995;130:393-405. [48] Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science. 2010;330:827-30. [49] Goretzki L, Burg MA, Grako KA, Stallcup WB. High-affinity binding of basic fibroblast growth factor and platelet-derived growth factor-AA to the core protein of the NG2 proteoglycan. J Biol Chem. 1999;274:16831-7. [50] Sugimoto H, Mundel TM, Kieran MW, Kalluri R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol Ther. 2006;5:1640-6. [51] Pula B, Jethon A, Piotrowska A, Gomulkiewicz A, Owczarek T, Calik J, et al. Podoplanin expression by cancer-associated fibroblasts predicts poor outcome in invasive ductal breast carcinoma. Histopathology. 2011;59:1249-60. [52] Schoppmann SF, Berghoff A, Dinhof C, Jakesz R, Gnant M, Dubsky P, et al. Podoplanin-expressing cancer-associated fibroblasts are associated with poor prognosis in invasive breast cancer. Breast Cancer Res Treat. 2012;134:237-44. [53] Neilson EG. Mechanisms of disease: Fibroblasts--a new look at an old problem. Nat Clin Pract Nephrol. 2006;2:101-8. [54] Radisky ES, Radisky DC. Stromal induction of breast cancer: inflammation and invasion. Rev Endocr Metab Disord. 2007;8:279-87. [55] Zavadil J, Haley J, Kalluri R, Muthuswamy SK, Thompson E. Epithelial-mesenchymal transition. Cancer Res. 2008;68:9574-7.
Page 25 of 33
Accep
ted
Man
uscr
ipt
25
[56] Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007;67:10123-8. [57] Qiu W, Hu M, Sridhar A, Opeskin K, Fox S, Shipitsin M, et al. No evidence of clonal somatic genetic alterations in cancer-associated fibroblasts from human breast and ovarian carcinomas. Nat Genet. 2008;40:650-5. [58] Direkze NC, Hodivala-Dilke K, Jeffery R, Hunt T, Poulsom R, Oukrif D, et al. Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Res. 2004;64:8492-5. [59] Ishii G, Sangai T, Oda T, Aoyagi Y, Hasebe T, Kanomata N, et al. Bone-marrow-derived myofibroblasts contribute to the cancer-induced stromal reaction. Biochem Biophys Res Commun. 2003;309:232-40. [60] Jeon ES, Moon HJ, Lee MJ, Song HY, Kim YM, Cho M, et al. Cancer-derived lysophosphatidic acid stimulates differentiation of human mesenchymal stem cells to myofibroblast-like cells. Stem Cells. 2008;26:789-97. [61] Paunescu V, Bojin FM, Tatu CA, Gavriliuc OI, Rosca A, Gruia AT, et al. Tumour-associated fibroblasts and mesenchymal stem cells: more similarities than differences. J Cell Mol Med. 2011;15:635-46. [62] Quante M, Tu SP, Tomita H, Gonda T, Wang SS, Takashi S, et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer cell. 2011;19:257-72. [63] Spaeth EL, Dembinski JL, Sasser AK, Watson K, Klopp A, Hall B, et al. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS One. 2009;4:e4992. [64] Dirat B, Bochet L, Dabek M, Daviaud D, Dauvillier S, Majed B, et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011;71:2455-65. [65] Kidd S, Spaeth E, Watson K, Burks J, Lu H, Klopp A, et al. Origins of the tumor microenvironment: quantitative assessment of adipose-derived and bone marrow-derived stroma. PLoS One. 2012;7:e30563. [66] Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170:1807-16. [67] Kojima Y, Acar A, Eaton EN, Mellody KT, Scheel C, Ben-Porath I, et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci U S A. 2010;107:20009-14. [68] Ronnov-Jessen L, Petersen OW. Induction of alpha-smooth muscle actin by transforming growth factor-beta 1 in quiescent human breast gland fibroblasts. Implications for myofibroblast generation in breast neoplasia. Lab Invest. 1993;68:696-707. [69] Mueller L, Goumas FA, Affeldt M, Sandtner S, Gehling UM, Brilloff S, et al. Stromal fibroblasts in colorectal liver metastases originate from resident fibroblasts and generate an inflammatory microenvironment. Am J Pathol. 2007;171:1608-18. [70] Ostman A, Augsten M. Cancer-associated fibroblasts and tumor growth--bystanders turning into key players. Curr Opin Genet Dev. 2009;19:67-73. [71] Jain M, Rivera S, Monclus EA, Synenki L, Zirk A, Eisenbart J, et al. Mitochondrial reactive oxygen species regulate transforming growth factor-beta signaling. J Biol Chem. 2013;288:770-7. [72] Artaud-Macari E, Goven D, Brayer S, Hamimi A, Besnard V, Marchal-Somme J, et al. Nuclear factor erythroid 2-related factor 2 nuclear translocation induces
Page 26 of 33
Accep
ted
Man
uscr
ipt
26
myofibroblastic dedifferentiation in idiopathic pulmonary fibrosis. Antioxid Redox Signal. 2013;18:66-79. [73] Lee JM, Calkins MJ, Chan K, Kan YW, Johnson JA. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J Biol Chem. 2003;278:12029-38. [74] Niethammer P, Grabher C, Look AT, Mitchison TJ. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature. 2009;459:996-9. [75] Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005;436:123-7. [76] Bae YS, Sung JY, Kim OS, Kim YJ, Hur KC, Kazlauskas A, et al. Platelet-derived growth factor-induced H(2)O(2) production requires the activation of phosphatidylinositol 3-kinase. J Biol Chem. 2000;275:10527-31. [77] Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell. 2002;9:387-99. [78] Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296-9. [79] Xu D, Rovira, II, Finkel T. Oxidants painting the cysteine chapel: redox regulation of PTPs. Dev Cell. 2002;2:251-2. [80] Thannickal VJ, Day RM, Klinz SG, Bastien MC, Larios JM, Fanburg BL. Ras-dependent and -independent regulation of reactive oxygen species by mitogenic growth factors and TGF-beta1. FASEB J. 2000;14:1741-8. [81] Salmeen A, Park BO, Meyer T. The NADPH oxidases NOX4 and DUOX2 regulate cell cycle entry via a p53-dependent pathway. Oncogene. 2010;29:4473-84. [82] Sotgia F, Martinez-Outschoorn UE, Howell A, Pestell RG, Pavlides S, Lisanti MP. Caveolin-1 and cancer metabolism in the tumor microenvironment: markers, models, and mechanisms. Annu Rev Pathol. 2012;7:423-67. [83] Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Daumer KM, Milliman JN, Chiavarina B, et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle. 2010;9:2423-33. [84] Martinez-Outschoorn UE, Whitaker-Menezes D, Lin Z, Flomenberg N, Howell A, Pestell RG, et al. Cytokine production and inflammation drive autophagy in the tumor microenvironment: role of stromal caveolin-1 as a key regulator. Cell Cycle. 2011;10:1784-93. [85] Luanpitpong S, Talbott SJ, Rojanasakul Y, Nimmannit U, Pongrakhananon V, Wang L, et al. Regulation of lung cancer cell migration and invasion by reactive oxygen species and caveolin-1. J Biol Chem. 2010;285:38832-40. [86] Rosc-Schluter BI, Hauselmann SP, Lorenz V, Mochizuki M, Facciotti F, Pfister O, et al. NOX2-derived reactive oxygen species are crucial for CD29-induced pro-survival signalling in cardiomyocytes. Cardiovasc Res. 2012;93:454-62. [87] Hu M, Peluffo G, Chen H, Gelman R, Schnitt S, Polyak K. Role of COX-2 in epithelial-stromal cell interactions and progression of ductal carcinoma in situ of the breast. Proc Natl Acad Sci U S A. 2009;106:3372-7. [88] Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A, et al. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008;68:918-26.
Page 27 of 33
Accep
ted
Man
uscr
ipt
27
[89] Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999;59:5002-11. [90] Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335-48. [91] Vicent S, Sayles LC, Vaka D, Khatri P, Gevaert O, Chen R, et al. Cross-species functional analysis of cancer-associated fibroblasts identifies a critical role for CLCF1 and IL-6 in non-small cell lung cancer in vivo. Cancer Res. 2012;72:5744-56. [92] Yang G, Rosen DG, Zhang Z, Bast RC, Jr., Mills GB, Colacino JA, et al. The chemokine growth-regulated oncogene 1 (Gro-1) links RAS signaling to the senescence of stromal fibroblasts and ovarian tumorigenesis. Proc Natl Acad Sci U S A. 2006;103:16472-7. [93] Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer cell. 2012;21:309-22. [94] Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74. [95] Polyak K, Haviv I, Campbell IG. Co-evolution of tumor cells and their microenvironment. Trends Genet. 2009;25:30-8. [96] Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, et al. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004;303:848-51. [97] Guo X, Oshima H, Kitmura T, Taketo MM, Oshima M. Stromal fibroblasts activated by tumor cells promote angiogenesis in mouse gastric cancer. J Biol Chem. 2008;283:19864-71. [98] Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, Harrington K, et al. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol. 2007;9:1392-400. [99] Sanz-Moreno V, Gaggioli C, Yeo M, Albrengues J, Wallberg F, Viros A, et al. ROCK and JAK1 signaling cooperate to control actomyosin contractility in tumor cells and stroma. Cancer cell. 2011;20:229-45. [100] Giannoni E, Bianchini F, Calorini L, Chiarugi P. Cancer associated fibroblasts exploit reactive oxygen species through a proinflammatory signature leading to epithelial mesenchymal transition and stemness. Antioxid Redox Signal. 2011;14:2361-71. [101] Giannoni E, Bianchini F, Masieri L, Serni S, Torre E, Calorini L, et al. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res. 2010;70:6945-56. [102] Giannoni E, Parri M, Chiarugi P. EMT and oxidative stress: a bidirectional interplay affecting tumor malignancy. Antioxid Redox Signal. 2012;16:1248-63. [103] Warburg O, Wind F, Negelein E. The Metabolism of Tumors in the Body. J Gen Physiol. 1927;8:519-30. [104] Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8:3984-4001. [105] Fiaschi T, Marini A, Giannoni E, Taddei ML, Gandellini P, De Donatis A, et al. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012;72:5130-40.
Page 28 of 33
Accep
ted
Man
uscr
ipt
28
[106] Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle. 2010;9:3256-76. [107] Whitaker-Menezes D, Martinez-Outschoorn UE, Lin Z, Ertel A, Flomenberg N, Witkiewicz AK, et al. Evidence for a stromal-epithelial "lactate shuttle" in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle. 2011;10:1772-83. [108] Witkiewicz AK, Whitaker-Menezes D, Dasgupta A, Philp NJ, Lin Z, Gandara R, et al. Using the "reverse Warburg effect" to identify high-risk breast cancer patients: stromal MCT4 predicts poor clinical outcome in triple-negative breast cancers. Cell Cycle. 2012;11:1108-17. [109] Park JH, Kim TY, Jong HS, Kim TY, Chun YS, Park JW, et al. Gastric epithelial reactive oxygen species prevent normoxic degradation of hypoxia-inducible factor-1alpha in gastric cancer cells. Clin Cancer Res. 2003;9:433-40. [110] Ryu JH, Li SH, Park HS, Park JW, Lee B, Chun YS. Hypoxia-inducible factor alpha subunit stabilization by NEDD8 conjugation is reactive oxygen species-dependent. J Biol Chem. 2011;286:6963-70. [111] Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer cell. 2005;7:77-85. [112] Fiaschi T, Chiarugi P. Oxidative stress, tumor microenvironment, and metabolic reprogramming: a diabolic liaison. Int J Cell Biol. 2012;2012:762825. [113] Ullah MS, Davies AJ, Halestrap AP. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J Biol Chem. 2006;281:9030-7. [114] Luo W, Hu H, Chang R, Zhong J, Knabel M, O'Meally R, et al. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011;145:732-44. [115] Pelicano H, Lu W, Zhou Y, Zhang W, Chen Z, Hu Y, et al. Mitochondrial dysfunction and reactive oxygen species imbalance promote breast cancer cell motility through a CXCL14-mediated mechanism. Cancer Res. 2009;69:2375-83. [116] Muller G, Lipp M. Signal transduction by the chemokine receptor CXCR5: structural requirements for G protein activation analyzed by chimeric CXCR1/CXCR5 molecules. Biol Chem. 2001;382:1387-97. [117] Arora S, Bhardwaj A, Singh S, Srivastava SK, McClellan S, Nirodi CS, et al. An undesired effect of chemotherapy: gemcitabine promotes pancreatic cancer cell invasiveness through ROS-dependent, NF-kappaB- and HIF-1alpha-mediated upregulation of CXCR4. J Biol Chem. 2013. [118] Chen HH, Su WC, Lin PW, Guo HR, Lee WY. Hypoxia-inducible factor-1alpha correlates with MET and metastasis in node-negative breast cancer. Breast Cancer Res Treat. 2007;103:167-75. [119] Chetram MA, Hinton CV. ROS-mediated regulation of CXCR4 in cancer. Frontiers in Biology. 2013;8:273-8. [120] Comito G, Calvani M, Giannoni E, Bianchini F, Calorini L, Torre E, et al. HIF-1alpha stabilization by mitochondrial ROS promotes Met-dependent invasive growth and vasculogenic mimicry in melanoma cells. Free Radic Biol Med. 2011;51:893-904. [121] Scarpino S, Cancellario d'Alena F, Di Napoli A, Pasquini A, Marzullo A, Ruco LP. Increased expression of Met protein is associated with up-regulation of hypoxia
Page 29 of 33
Accep
ted
Man
uscr
ipt
29
inducible factor-1 (HIF-1) in tumour cells in papillary carcinoma of the thyroid. J Pathol. 2004;202:352-8. [122] Li YM, Pan Y, Wei Y, Cheng X, Zhou BP, Tan M, et al. Upregulation of CXCR4 is essential for HER2-mediated tumor metastasis. Cancer cell. 2004;6:459-69. [123] Boimel PJ, Smirnova T, Zhou ZN, Wyckoff J, Park H, Coniglio SJ, et al. Contribution of CXCL12 secretion to invasion of breast cancer cells. Breast Cancer Res. 2012;14:R23. [124] Hinton CV, Avraham S, Avraham HK. Role of the CXCR4/CXCL12 signaling axis in breast cancer metastasis to the brain. Clin Exp Metastasis. 2010;27:97-105. [125] Matsumoto K, Nakamura T. Hepatocyte growth factor and the Met system as a mediator of tumor-stromal interactions. Int J Cancer. 2006;119:477-83. [126] Jagadeeswaran R, Jagadeeswaran S, Bindokas VP, Salgia R. Activation of HGF/c-Met pathway contributes to the reactive oxygen species generation and motility of small cell lung cancer cells. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1488-94. [127] Margosio B, Marchetti D, Vergani V, Giavazzi R, Rusnati M, Presta M, et al. Thrombospondin 1 as a scavenger for matrix-associated fibroblast growth factor 2. Blood. 2003;102:4399-406. [128] Chen JK, Zhan YJ, Yang CS, Tzeng SF. Oxidative stress-induced attenuation of thrombospondin-1 expression in primary rat astrocytes. J Cell Biochem. 2011;112:59-70. [129] Dvorak HF, Galli SJ, Dvorak AM. Cellular and vascular manifestations of cell-mediated immunity. Hum Pathol. 1986;17:122-37. [130] Hurd TR, DeGennaro M, Lehmann R. Redox regulation of cell migration and adhesion. Trends Cell Biol. 2012;22:107-15. [131] Reth M. Hydrogen peroxide as second messenger in lymphocyte activation. Nat Immunol. 2002;3:1129-34. [132] Razzell W, Evans IR, Martin P, Wood W. Calcium flashes orchestrate the wound inflammatory response through DUOX activation and hydrogen peroxide release. Curr Biol. 2013;23:424-9. [133] Feng Y, Santoriello C, Mione M, Hurlstone A, Martin P. Live imaging of innate immune cell sensing of transformed cells in zebrafish larvae: parallels between tumor initiation and wound inflammation. PLoS Biol. 2010;8:e1000562. [134] Cordeiro JV, Jacinto A. The role of transcription-independent damage signals in the initiation of epithelial wound healing. Nat Rev Mol Cell Biol. 2013;14:249-62. [135] Benink HA, Bement WM. Concentric zones of active RhoA and Cdc42 around single cell wounds. J Cell Biol. 2005;168:429-39. [136] Xu S, Chisholm AD. A Galphaq-Ca(2)(+) signaling pathway promotes actin-mediated epidermal wound closure in C. elegans. Curr Biol. 2011;21:1960-7. [137] Yoo SK, Freisinger CM, LeBert DC, Huttenlocher A. Early redox, Src family kinase, and calcium signaling integrate wound responses and tissue regeneration in zebrafish. J Cell Biol. 2012;199:225-34. [138] Yin J, Xu K, Zhang J, Kumar A, Yu FS. Wound-induced ATP release and EGF receptor activation in epithelial cells. J Cell Sci. 2007;120:815-25. [139] Frijhoff J, Dagnell M, Godfreay R, Ostman A. Regulation of Protein Tyrosine Phosphatase Oxidation in cell adhesion and Migration. Antioxid Redox Signal. 2013 Oct10. In press. [140] Dagnell M, Frijhoff J, Pader I, Augsten M, Boivin B, Xu J, Mandal PK, Tonks NK, Hellberg C, Conrad M, Arnér ES, Ostman A. Selective actvation of oxidized
Page 30 of 33
Accep
ted
Man
uscr
ipt
30
PTP1B by the thioredoxin system modulates PDGF-b receptor tyrosine kinase signaling.PNAS. 2013;110(33):13398-403. [141] Calabrese C, Iommarini L, Kurelac I, Calvaruso MA, Capristo M, Lollini PL, Nanni P, Bergamini C, Nicoletti G, Giovanni CD, Ghelli A, Giorgio V, Caratozzolo MF, Marzano F, Manzari C, Betts CM, Carelli V, Ceccarelli C, Attimonelli M, Romeo G, Fato R, Rugolo M, Tullo A, Gasparre G, Porcelli AM. Respiratory complex I is essential to induce a Warburg profile in mitochondria-defective tumor cells. Cancer Metab. 2013 Mar 18;1(1):11. [142] Caro P, Kishan AU, Norberg E, Stanley IA, Chapuy B, Ficarro SB, Polak K, Tondera D, Gounarides J, Yin H, Zhou F, Green MR, Chen L, Monti S, Marto JA, Shipp MA, Danial NN. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell. 2012;22(4):547-60. [143] Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D, Körbel C, Laschke MW, Gimotty PA, Philipp SE, Krause E, Pätzold S, Villanueva J, Krepler C, Fukunaga-Kalabis M, Hoth M, Bastian BC, Vogt T, Herlyn M. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell. 2013;23(6):811-25.