The spin-polarized extended BruecknerorbitalsCite as: J. Chem. Phys. 135, 094107 (2011); https://doi.org/10.1063/1.3629780Submitted: 15 June 2011 . Accepted: 08 August 2011 . Published Online: 06 September 2011
A. V. Luzanov, and O. V. Prezhdo
ARTICLES YOU MAY BE INTERESTED IN
Incremental full configuration interactionThe Journal of Chemical Physics 146, 104102 (2017); https://doi.org/10.1063/1.4977727
Projected Hartree–Fock theoryThe Journal of Chemical Physics 136, 164109 (2012); https://doi.org/10.1063/1.4705280
Hole-particle characterization of coupled-cluster singles and doubles and related modelsThe Journal of Chemical Physics 125, 154106 (2006); https://doi.org/10.1063/1.2360262
THE JOURNAL OF CHEMICAL PHYSICS 135, 094107 (2011)
The spin-polarized extended Brueckner orbitalsA. V. Luzanov1,a) and O. V. Prezhdo2,b)
1STC “Institute for Single Crystals,” National Academy of Sciences, Kharkov 61001, Ukraine2Department of Chemistry, University of Rochester, Rochester, New York 14627, USA
(Received 15 June 2011; accepted 8 August 2011; published online 6 September 2011)
One of the salient features of modern quantum chemistryis a wide use of non-canonical Hartree-Fock (HF) orbitals andeven non-HF orbitals for ab initio post-HF techniques. This isespecially true for the virtual space, where the canonical HForbitals are very inefficient.1 In this regard, the recent paper2
proposes an original and promising scheme for constructingstrongly localized virtual orbitals needed for electron cor-relation approaches. Among other non-standard orbital sets,the Brueckner orbitals (BOs) (Refs. 3–5) and closely relatedconstructs appear important for formulating optimized non-variational approaches such as the coupled cluster (CC) andrelated configuration interaction (CI) models.6–10 This fact isnot so unexpected if we recollect that BOs can be also de-fined as the best-overlap orbitals. BOs are even more prefer-able for interpretive purposes than the natural orbitals, whichare also extensively used in current electronic structure theory.(Refs. 11–22 are only a small part of an extremely rich litera-ture on this subject.)
We stress that although BOs are not frequently employedfor routine quantum-chemical calculations because they arerather demanding computationally, they remain an essentialpart of the current orbital methodology. For example, BOsturn out to be closely related to the Kohn-Sham orbitals ofdensity functional theory (DFT) and the corresponding gen-eralized Koopmans’ orbitals.23 Furthermore, in the recent
paper24 an effective one-body Hamiltonian was constructed,giving approximate BOs as its eigenvectors, and the negativeof ionization potentials and electron affinities as its eigenval-ues. Analogously, within DFT an appropriate computationaltechnique25 can be formulated by using local correlation po-tentials derived from a Brueckner double (BD) excitation cou-pled cluster wave-function (BDCC approximation). Some in-teresting applications of BDCC and related schemes can befound in the recent works.26–31 In particular, Brueckner or-bitals allow one to overcome the instability inherent in typi-cal simplified CC approaches, for low-symmetry diradicals tohave narrow HOMO-LUMO gaps.26, 27 All these facts demon-strate the importance of further development of the BO theoryfor modern quantum chemistry.
Introduction of BOs implies that they produce a singleSlater determinant that maximally overlaps with either a trueor a best approximate wave-function. However, a conven-tional spin-purity Slater determinant (even from BOs) cannotprovide a strong overlap with a highly correlated electronicstate, especially when quasidegenerate states appear. It wouldbe more advantageous if we could definitively interpret theexact state vector in terms of a generalized independent parti-cle model, even in the case of degenerate and quasidegeneratestates. As it turns out, it is indeed possible to do so directly forseveral simple systems. For instance, the two-electron prob-lem can be correctly treated by the Coulson-Fischer orbitals.32
In fact, the two-electron Coulson-Fischer wave functioncan be envisaged as a specific case of Löwdin’s extendedHartree-Fock (EHF) function.33 The latter is just the general-ized independent particle model that preserves physical clar-
094107-2 A. V. Luzanov and O. V. Prezhdo J. Chem. Phys. 135, 094107 (2011)
ity and, at the same time, includes correlation effects. In otherwords, the new different orbitals for different spins (DODS)should be constructed to ensure the maximal overlap betweenthe high-quality CI function under study and the appropri-ate spin-projected determinant. The orbitals so obtained willbe termed the spin-polarized extended (or simply, extended)BOs. Thus, the main goal of our paper is to present a newinterpretive scheme based on the extended. We would liketo stress that up to now the spin-polarized unrestricted MOsand related BOs remain a physically attractive basis for treat-ing electron correlation effects.34–41 Nevertheless, preservingspin-purity of electronic states is a principal point, and in ourstudy we eliminate spin contamination in a proper way.
The paper is organized as follows. In Sec. II the startingdefinitions and new computational equations concerning BOsare given. Section III contains the key equations for construct-ing the extended BOs from full CI (FCI) and related modelsof the complete active space self-consistent field (CASSCF)type. Section IV proposes a new interpretive tool for elec-tron correlation by spin-polarization diagrams derived fromBOs. Section V demonstrates how the given BOs and spin-polarization diagrams work for rather typical hydrogen clus-ters and carbon-containing systems, especially those havingopen-shell ground states (singlet diradicals) or those under-going bond breaking. Some results for excited states are alsopresented here. The last section concludes the paper, and Ap-pendices furnish the details on derivation of the main ex-pressions and some algorithmic formulas. Furthermore, inAppendix E we offer a plausible reasoning for the existenceof the extended BOs for any level of electron-correlation (incontrast to the unrestricted BOs).
II. DEFINITION OF BOs AND STARTING EXPRESSIONS
Here we briefly sketch the main concepts and derivenew computational expressions for BOs in the usual (non-extended) formulation. Let |�〉 be the given N-electron statevector with a total spin s. For practical purposes, |�〉 is un-derstood to be approximated from a “correlated” level of the-ory, i.e., CI or CASSCF. Moreover, a Slater determinant, |�〉,can be constructed from a set of (unknown) orthonormal spin-orbitals,
{|ϕi〉}1≤i≤N . (2.1)
The so-called Dirac-Fock density matrix, ρ, is associated withthis determinant as a projector onto the subspace of occupied(hole) spin-orbitals (2.1):
ρ = ρ2 =N∑
i=1
|ϕi〉〈ϕi |. (2.2)
Accordingly, I − ρ projects onto vacant (particle) orbitals{|ϕa〉}a≥N+1, that is I − ρ = ∑
a≥N+1 |ϕa〉〈ϕa|.By definition, BOs (2.1) should maximize the absolute
value of the overlap integral 〈� |�〉. In a more convenientform, the BOs furnish the maximal value of the non-negative-definite functional,
J = |〈� |�〉|2. (2.3)
The standard variational procedure gives the first varia-tion as
δJ = 2〈� |�〉〈δ� |�〉, (2.4)
where, for simplicity, these and subsequent quantities are as-sumed to be real. The prefactor 〈� |�〉 in Eq. (2.4) ensuresthe correct sign of δJ by accounting for the phase factors ofthe current orbitals. Therefore, the prefactor is retained de-spite its appearance as a common multiplier.
In order to obtain explicit equations for the determinationof BOs, the maximum of Eq. (2.4) is found by taking the firstvariation of the Slater determinant, |δ�〉, under the fixed |�〉.The variation |δ�〉 is of the conventional form
|δ�〉 =N∑
i=1
∑a≥N+1
�aia+a ai |�〉,
with ai and a+a being the annihilation and creation operators,
respectively, and �ai arbitrary parameters. The equivalent ex-pression (e.g., see Eq. (4.27) in Ref. 42 for a general rule) is
|δ �〉 =∑
1≤k≤N
�(k)|�〉, (2.5)
where the sum runs over all N electrons, and the one-electronoperator � is
� =∑i;a
�ai |ϕa〉〈ϕi |.
This � is in fact a particle-hole component of the first varia-tion δρ, that is � = (I − ρ)δρρ. Here and elsewhere we usethe customaty notation Z(k) to denote the given operator Zacting on electron k.
Then
〈δ� |�〉 = 〈�|∑
1≤k≤N
�+(k)|�〉, (2.6)
where �+ = ρδρ(I − ρ). Treating �+ in Eq. (2.6) as theusual one-electron matrix, we see that
〈δ� |�〉 = Tr �+ D�,�, (2.7)
where D�,� is the ordinary one-electron transition densitymatrix (for the general theory of the reduced density matri-ces see Refs. 11,43, and 44). As a result, requirement δJ = 0is tantamount to the matrix equation
(I − ρ)D�,�ρ = 0. (2.8)
It is enlightening to understand this equation in terms of theso-called Brillouin-Brueckner condition5 enforcing that sin-gle excitations do not enter |�〉 being expressed in the BOframe. For this purpose, we present the exact state vector |�〉in the standard CI expansion
|�〉 ={
C0 +∑
1≤k≤N
C1(k) +∑
1≤k<l≤N
C2(kl) + . . .
}|�〉,
(2.9)where C0 is the expansion coefficient of the reference deter-minant, and j-body excitation operators Cj (j ≥ 1) generate
j-fold excited configurations (with respect to |�〉). For in-stance, one-body (one-electron) operator C1 has the form
C1 =∑i;a
Cai |ϕa〉〈ϕi |, (2.10)
with Cai being appropriate configurational coefficients. Evi-dently, C1 = (I − ρ)C1ρ. This C1 generates a superpositionof singly excited configurations, |�i→a〉 = a+
a ai |�〉, that is∑1≤k≤N
C1(k) |�〉 =∑i;a
Cai |�i→a〉. (2.11)
(We recall again the above-mentioned correspondence rule inRef. 42.) In these terms, the transition density matrix D�,� ismerely
D�,� = C0ρ + C1. (2.12)
(See Appendix A for details.) Inserting the latter into Eq. (2.9)leads to
C1 = 0, (2.13)
thereby showing that indeed the singly excited configurationsmake no contribution to |�〉 when the reference determinantand excitation operators are built up from BOs. This prop-erty is usually taken as the definition of BOs. In practice, theiteration procedure with the Hermitian nonlinear eigenvalueproblem,
B|ϕi〉 = bi |ϕi〉, (2.14)
B = 〈� |�〉[D�,� + (D�,�)T ]/2, (2.15)
with superscript T denoting “transpose,” provides a reli-able algorithm for obtaining “self-consistent” BOs satisfyingEq. (2.8). For brevity, the operators determining BOs will betermed B-operators. Of course, there exist other general BOequations.45, 46
Now we need to derive working equations for the FCIcomputations of unrestricted BOs. To this end, the spin andspatial variables are separated, and the problem is reduced to aspin-free case. This can be achieved by a spin-separation tech-nique that is closely related to the Fock spin-free theory47, 48
(see Appendix B). Using this technique, the variationalspin-free formulation for conventional BOs can be obtained(Appendix C). The final equations are formulated as a spin-free eigenvalue problem for the common spin-up and spin-down (spin-free) orbitals{∣∣ϕα
i
⟩}1≤i≤n+s
,{∣∣ϕβ
i
⟩}1≤i≤n−s
. (2.16)
These orbitals specify the unrestricted determinant |�〉 for astate with the spin z-projection s. The variational equationsinvolve the usual spin-up and spin-down idempotent densitymatrices ραand ρβ (the projectors of type (2.2) for α- andβ- MOs (2.16), respectively). The coupled eigenproblem isproved to be
Bα∣∣ϕα
i
⟩ = bi
∣∣ϕαi
⟩, Bβ
∣∣ϕβ
i
⟩ = b′i
∣∣ϕβ
i
⟩, (2.17)
where the spin-free B-operators are defined by Eqs. (C9). Thematrices of these operators are computed in a spin-free orbital
basis taken from the set,{∣∣ϕ◦μ
⟩}1≤μ≤dim, (2.18)
with dim being the basis set size.
III. SPIN-EXTENDED BOs
At this stage, we can formulate our main problem in exactterms. The spin-projected determinant
|�ext〉 = Os |�〉/√
〈�|Os |�〉 (3.1)
is considered now instead of the Slater determinant |�〉. Inabove, Os is a projection operator onto a spin-pure N-electronstate with the spin z-projection s and the total spin value s (theso-called principal case). The initial N-electron state vector|�〉 is assumed to be a spin pure state with the same value ofs (in practice |�〉 is of the FCI or CASSCF type). Hence, therelation
|�〉 = Os |�〉 (3.2)
is true by construction. Furthermore, by defining the problemmore widely, one can invoke any reasonable type of projec-tions. For instance, one can exploit the complex-valued BOs,so the projection onto the real (or even imaginary) part of theN-electron state vector can be implied. To our knowledge, thismore general approach to BOs seems not duly recognized.
Equations for the spin-extended BOs, which are the keysubject of the paper, are obtained rather easily (the neededdetails are given in Appendix D). Utilizing spin-projection,the spin-extended BOs maximize the functional
J ext = 〈�ext |�〉2 = 〈� |�〉2/〈�|Os |�〉. (3.3)
We see that J ext differs from the previous functional (2.3) onlyby the denominator, which accounts for the renormalizationof the spin-projected determinant. Yet, this is an essential dif-ference. It noticeably modifies the variation in J, [Eq. (2.4)],as seen from Eq. (D1).
First, we briefly focus on the spin projector Os . The EHFtheory and related approaches33, 49–52 suggest that Löwdin’sspin-projection theory is especially pertinent here. In ourstudy, we follow Ref. 52 and use the elegant formula53 forOs in the form of an explicit polynomial of the ladder spin-operators
S+ =∑
1≤k≤N
|α(k)〉〈|β(k)〉|, S− = (S+)+. (3.4)
This formula (see also Ref. 48 (c) for more detail) can besuitably written as follows:
Os =n−s∑j=0
aj Sj− S
j+/(j !)2, aj = (−1)j
(2s + j + 1
j
)−1
.
(3.5)Consider in 〈�|Os |�〉, for instance, the first (j = 1) nontriv-ial term −〈�|S−S+|�〉/(2s + 2). The auxiliary ket S+|�〉 canbe viewed as a particular case of Eq. (2.11). Namely, by therule (A 1) applied for Z = S+, we get
S+|�〉 =∑
1≤k≤N
π (k)|�〉, (3.6)
094107-4 A. V. Luzanov and O. V. Prezhdo J. Chem. Phys. 135, 094107 (2011)
with π = (I − ρ)|α〉〈β|ρ; or explicitly π = π0|α〉〈β|, where
π0 = (I − ρα)ρβ. (3.7)
This matrix is not episodic. It occurred in the previous matrixformulation52 of the EHF theory. Notice that there exists an-other matrix-oriented EHF algorithm54 with many interestingapplications (see Refs. 49 and 55 for additional information).The π0-dependent expressions52 are preferred here, becauseπ0 vanishes in the restricted HF theory, suitably capturingelectron correlation. Furthermore, π0 conveniently describesthe spin-polarization effects considered in Sec. IV.
The resulting spin-extended BOs turn out to be eigenvec-tors of the coupled eigenvalue equations
Bαext
∣∣ϕαi
⟩ = bi
∣∣ϕαi
⟩, B
βext
∣∣ϕβ
i
⟩ = b′i
∣∣ϕβ
i
⟩, (3.8)
where Bαext and B
βext are specified by Eqs. (D7) and (D8). The
above system is reduced to Eqs. (2.17), if the one-determinantspin-purity condition,
ραρβ = ρβ, (3.9)
is satisfied.Applied to closed shells, the developed theory has fea-
tures resembling the conventional EHF theory. It is wellknown that the unrestricted Hartree-Fock (UHF) solutions,i.e., DODS without spin projecting, appear only under the so-called non-singlet (triplet) instability.56 The latter also servesas an indicator of the electron correlation strength. UnlikeUHF, however, there exist nontrivial EHF solutions for anynonzero electron correlation strength.57 In EHF solutions atleast one electron pair is spin-polarized. For the ith pair ofthis type, ∣∣ϕα
i
⟩ = ∣∣ϕβ
i
⟩. (3.10)
In fact, this was shown in Ref. 58 by taking as an example thesingle electron pair of H2.
The BO counterparts of UHF orbitals, i.e., unrestrictedBOs, and the corresponding conditions for their existencewere studied first in Ref. 59. It was determined that, similarlyto the usual UHF, unrestricted BOs also arise only when thecorresponding triplet instability condition is satisfied. Now,the question is under which conditions the spin-extended BOsappear. We cannot give a completely rigorous proof, but weare able to provide a plausible reasoning and show that non-trivial BO solutions such as in Eq. (3.10) are always possible(see Appendix E).
Before concluding this section, we need to discuss oneprincipal drawback of any approximation based on the spin-projected (or even half-projected) determinants. The draw-back stems from the fact that the Löwdin spin projector (3.5)is not multiplicatively separable (for detailed analysis seeRef. 60). Since the exact |�〉 is multiplicatively separable (un-der certain conditions), this lack of size-consistency is some-what alleviated, but does not vanish for functional (3.3). Asan example, consider the hydrogen molecule with the bondlength of 1 Å and treat this system at the FCI/6-31G level oftheory. The overlap integral, denoted by S[H2] ≡ 〈�ext |�〉,is equal to S[H2] = 0.9993 when the optimal, extended BOsare used. The minimal basis produces S[H2] = 1. Analo-gous calculations for the dimer H2 + H2 with the hydro-
gen molecules separated by 4 Å gives S[H2 + H2] = 0.9880.This is not equal to the squared quantity (S[H2])2 = 0.9986,even though the multiplicativity rule S[H2 + H2] = (S[H2])2
is expected to hold within a few thousandths at the distanceof 4 Å. In practice, however, this shortcoming does not un-dermine the extended BO theory, as will be demonstrated bythe illustrations of Sec. V. Nevertheless, the size-consistencydeficiency should not be forgotten. A possible remedy is tointroduce other BO determinants, for instance, those takenfrom the related electronic or geometric configurations. Insuch case, rather than Eq. (3.1), we consider a linear super-position of spin-projected BO determinants∣∣�ext
CIt
⟩ = cA
∣∣�extA
⟩ + cB
∣∣�extB
⟩ + . . . , (3.11)
where the individual kets {|�extA 〉} are obtained by the
procedure outlined above. The final result is simply ascalar product: J ext = U · V , with vectors U = ‖〈�ext
A |�〉‖and V = MU where M is the inverse metric matrix, M
= ‖〈�extA |�ext
B 〉‖−1.Alternative quasi-one-electron models, such as the gen-
eralized valence bond (GVB) approximation,61 may be con-sidered more appropriate than the EHF-like construction usedhere. However, it is difficult, if at all possible, to treat GVB ina pure matrix (more exactly, matrix-covariant) form; at leastthe authors could not succeed in formulating clear-cut matrixequations for the best-overlap GVB orbitals.
IV. INTERPRETING BOs IN TERMS OFSPIN-POLARIZATION INDICES
The BOs defined above are intended primarily for ana-lyzing molecular systems with essentially unpaired electrons,especially in the case of open-shell singlet states. For this rea-son we attempt to interpret the BOs in terms of the electronicindices that account for orbital spin-polarization (3.10) or therelated polarization of the idempotents ρα and ρβ that existswhen
ρα = ρβ. (4.1)
The simple technique proposed here can be indirectly con-nected with several important characteristics of effectivelyunpaired electrons developed in many works13, 62–65 (see alsoRef. 66).
Our objective is to describe the behavior of “odd”62 elec-trons in terms of proper polarization indices using ρα and ρβ
only. The matrix π0 in Eqs. (3.6) and (3.7) may be duly uti-lized to this end. Indeed, S+|�〉 is non-vanishing as a directconsequence of unpaired electrons (4.1). Thus,
‖S+|�〉‖2 = TrπT π = TrπT0 π0 = Trπ0. (4.2)
S−|�〉 proves also important for our purpose, along withEq. (3.6). This vector must vanish only for spin-pure singletstates. In the case of non-zero spin states, we have S−|�〉 = 0(the principal case is implied), even though the state is free ofspin-contamination. Explicitly,
(The rule (A1) is used for Z = S−.) If s > 0, this matrixis never null, even when a spin-restricted determinant isemployed, such that υ0 = ρα − ρβ . Hence, the squared normtakes the form,
‖S−|�〉‖2 = Tr υT υ = Tr υT0 υ0 = Trυ0. (4.5)
It gives a possible count of the unpaired electrons for non-zerospin states. Together, Eqs. (4.2) and (4.5) give Tr(π0 + υ0)= Tr(ρα − ρβ)2, so that
‖S+|�〉‖2 + ‖S−|�〉‖2 = ‖ρα − ρβ‖2. (4.6)
The measure displayed in Eq. (4.6) gives the number of “odd”electrons as defined in Ref. 64 [in this paper the proper term“effectively unpaired electrons” was coined]. For an unre-stricted Slater determinant, Eq. (4.6), otherwise expressed asNU
odd, can be also rewritten as
NUodd = N − 2Tr ραρβ. (4.7)
Note that Eq. (4.7) is a particular case of the more gen-eral number, Nodd, of effectively unpaired electrons. Bydefinition62–64 the general index is
Nodd = 2N − Tr(o
D1�)2, (4.8)
witho
D1�being the spin-free density matrix of a multi-
configurational wave function. This popular approach, incor-porated in the MOLPRO package,67 was critiqued in Ref. 13.While the criticism was partially rebutted63 the problem ofpossible overestimations of effectively unpaired electrons re-main real. In many cases, the modified index from Ref. 13appears more suitable. We will signify this index by Nunpair. Itcan be explicitly calculated from the natural occupation num-bers, λk , as follows:
Nunpair =∑
i
min[λi, 2 − λi]. (4.9)
Notice that Nunpair can be directly connected with the hole-particle interpretation of CI wave-functions.66 This factprompts us to modify the polarization index (4.7) as well.
Before introducing such adapted polarization indices, weremark that in the hole-particle interpretation,66 the numberof unpaired electrons from Ref. 13 can be understood in sim-ple CI terminology. This number is mainly determined bythe squared CI coefficients of doubly excited configurations.Turning to the analysis given in Appendix E [see Eq. (E4)],we suggest that these squared coefficients can be estimatedas t2 ≈ z4, where z is the orbital splitting parameter as de-fined in Eqs. (E2). The orbitals (E2) lead to the rank-1 ma-trix π0, so that TrπT
0 π0 = Trπ0 = 4z2(1 + z2)−2 ≈ z2 ≈ |t |.Hence, quantities such as t2 are related to matrix (πT
0 π0)2,and the number of unpaired electrons can be identified withthe invariant
Nβ
pol = TrDβ
pol, (4.10)
where
Dβ
pol = (πT
0 π0)2
. (4.11)
Similarly to Eqs. (4.10) and (4.11), we define
Nαpol = TrDα
pol, (4.12)
Dαpol = (
υT0 υ0
)2. (4.13)
Taken together, the two invariants, (4.10) and (4.12), lead tothe total spin-polarization index,
Npol = Nαpol + N
β
pol = 2(s + N
β
pol
), (4.14)
producing an adequate estimate of the effective number of un-paired electrons in spin polarization terminology. The identity
Nαpol = N
β
pol + 2s, (4.15)
used in Eq. (4.14) is deduced by simple matrix manipula-tions on Eqs. (4.10) and (4.12), and by taking into accountEqs. (3.7) and (4.4). From Eqs. (4.7) and (4.14), it is easy toprove that Npol ≤ Nodd for any unrestricted determinant state.
A few comments about distributions of the odd, i.e., ef-fectively unpaired electrons over atoms in a molecule are inorder. The odd-electron atomic indices are expressed sim-ply via atomic diagonal elements of DU
odd = (ρα − ρβ )2.64 Amore detailed picture can be provided within the above spin-polarization analysis. Two kinds of atomic distributions arecomputed, based on the special matrices (4.11) and (4.13). Weintroduce the following α− and β− polarization distributionscomposed of the atomic indices,
αA =
∑μ∈A
(Dα
pol
)μμ
, (4.16)
β
A =∑μ∈A
(D
β
pol
)μμ
, (4.17)
respectively. Here, A is a selected atom in a molecule. Thesubscript μμ indicates a diagonal element of the matrix in theorthonormal Löwdin-type AO basis. The total spin polariza-tion index assigned to atom A is
A = αA +
β
A, (4.18)
such that the sum of A over all atoms reproduces Npol (4.14).Closing this section, we list the main indices that
will be used for describing effectively unpaired electrons.These are the known, general-type indices, Nunpair and Nodd
[see Eqs. (4.8) and (4.9)], and the new spin polarization in-dex (4.14). The latter specifies the unpairing for DODS-typemodels only. The corresponding atomic polarization indicesare determined by Eqs. (4.16) and (4.17). We stress that inour approach the DODS orbitals are taken as spin-extendedBOs, so that the polarization indices (4.14) and (4.16)–(4.18)give reasonable results when the squared overlap (3.3) is large(near to 1). Other specialized spin-polarization indices are de-signed within the DFT methodology.35 However, they are dif-ficult to extend beyond DFT.
V. APPLICATIONS
We illustrate the theoretical results described above firstby considering simple systems, such as hydrogen clustersand small molecules, for which FCI computations are easily
094107-6 A. V. Luzanov and O. V. Prezhdo J. Chem. Phys. 135, 094107 (2011)
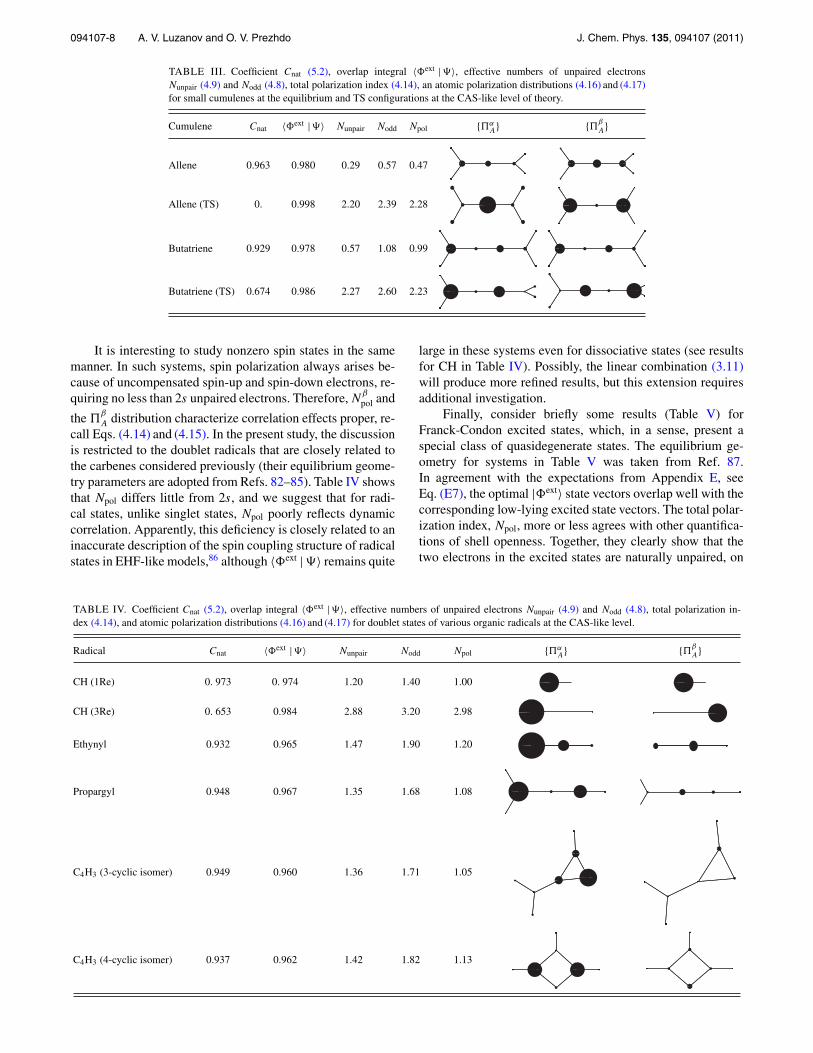
TABLE I. Coefficient Cnat (5.2), overlap integral 〈�ext |�〉, effective numbers of unpaired electronsNunpair (4.9) and Nodd (4.8), total polarization index (4.14), and atomic polarization distributions (4.16) and (4.17)for cyclic hydrogen clusters at the FCI/6-31G level.
feasible with small basis sets. The wave-function opera-tor technique68 is implemented to obtain the one-electrontransition matrices, needed for solving the extended BO equa-tions (3.8) at the FCI level (see Eqs. (C3)–(C13) and (D7)–(D9), and the overall description of the algorithm in the lastparagraph of Appendix D). For more complex systems, suchas carbenes, we make use of a simplified CAS-like approach.Namely, to update the active orbital space, we compute thecorresponding projectors ρα and ρβ for all electrons,
ρα = ρ0core + ρα
act, ρβ = ρ0core + ρ
βact (5.1)
The operator, ρ0core, is a fixed projector on frozen-core
orbitals, and ραact, ρ
βact are obtained from the extended BOs of
the current FCI solution for the chosen active orbital space.The full conventional Fockians Fα and Fβ are constructedfrom the above ρα , ρβ thus producing, via the Fockianseigenkets, the full orbital spaces for spin-up and spin-downelectrons. Then, the updated ραand ρβ are commonlycomputed from the new orbitals. The natural orbitals areconstructed from ρα + ρβ in the usual way and are used togenerate the updated active space, as in Ref. 12 (a). Thisvery simple procedure does not involve second order densitymatrices and provides a good approximation to the trueCASSCF solution. Approximately, 80–90% of the CASSCFcorrelation energy is captured, and such accuracy is sufficientfor our demonstration purposes. There exist other approachesto mimic the CASSCF results.16 This particular procedure ischosen, since it allows us to incorporate the extended BOsinto the above specified computational scheme.
The 6-31G basis set is employed for all systems. TheCAS-like model calculations (12 active MOs and 12 or soactive electrons) are performed using an extension of our ear-lier code, described in Ref. 68 (b). This code is written forthe Mathematica environment69 and has fairly restricted ca-pabilities, limited to simple basis sets for few-atom systems.Yet, it is sufficient to test the main ideas of this paper. In or-der to visualize spin-polarization, we draw the atomic spin-polarization distributions α
A and β
A using atom-centeredblack circles with radii proportional to the square root of thecorresponding quantity. Other details will be specified as nec-essary.
First, consider small clusters of hydrogen atoms. Theywere explored within the valence bond method in book,70
from which we take the Dnh structures of the transition statesof cyclic H4, H6, and H8 (no local minima exist for cyclicH-clusters). We treat these systems at the FCI/6-31G level oftheory, except in the case of H8, where the CAS-like tech-nique is employed. These systems resemble iso-π−electroniccyclic polyenes.71 Therefore, it not surprising that, in agree-ment with the counterpart of Hückel’s rule, the singlet groundstates in H4 and H8 are of an open-shell nature and re-quire at least a two-determinant description. The ground stateof H6 is a closed-shell singlet. The unpaired electron in-dices (4.8) and (4.9) [“shell openness indices” termed so inRef. 68 (c)] should be close to 2 in H4 and H8, in good agree-ment with the data in Table I. We see that the total polar-ization index, Npol, is quite consistent with the characteris-tics of electron unpairing directly obtained from Nunpair andNodd. Furthermore, the polarization distributions α
A and β
A
are rather expressive. By their nature, they correspond to theorbital pictures known for the alternant MOs (e.g., see Fig. 3in Ref. 33). The related GVB orbital pictures for cyclobuta-diene can be found in Ref. 72. At the same time, the polar-ization diagrams shown here represent the full unpaired elec-tron densities rather than the individual orbital contributions.It would be natural in our approach to display also the singlevalue decomposition orbitals from the basic matrices definedin Eqs. (3.7) and (4.4). To save space we present here andbelow only the cumulative polarization picture which is pro-vided by the polarization diagrams α
A and β
A.The proximity of |�ext〉 to the “exact” |�〉 is reflected by
the overlap 〈�ext |�〉. This quantity is compared in Table I tothe coefficients
Cnat = 〈�nat |�〉, (5.2)
where |�nat〉 is the closed-shell natural orbital determinant.From Table I, we see that Cnat
∼= 〈�ext |�〉 is only for theclosed-shell ground state of H6. At the same time, the open-shell singlet states in cyclic H4 and H8 are described verywell only by the spin-extended BO model. The spin polariza-tion diagrams in Table I and in other tables below describeunpaired electron distributions resulting mainly from static
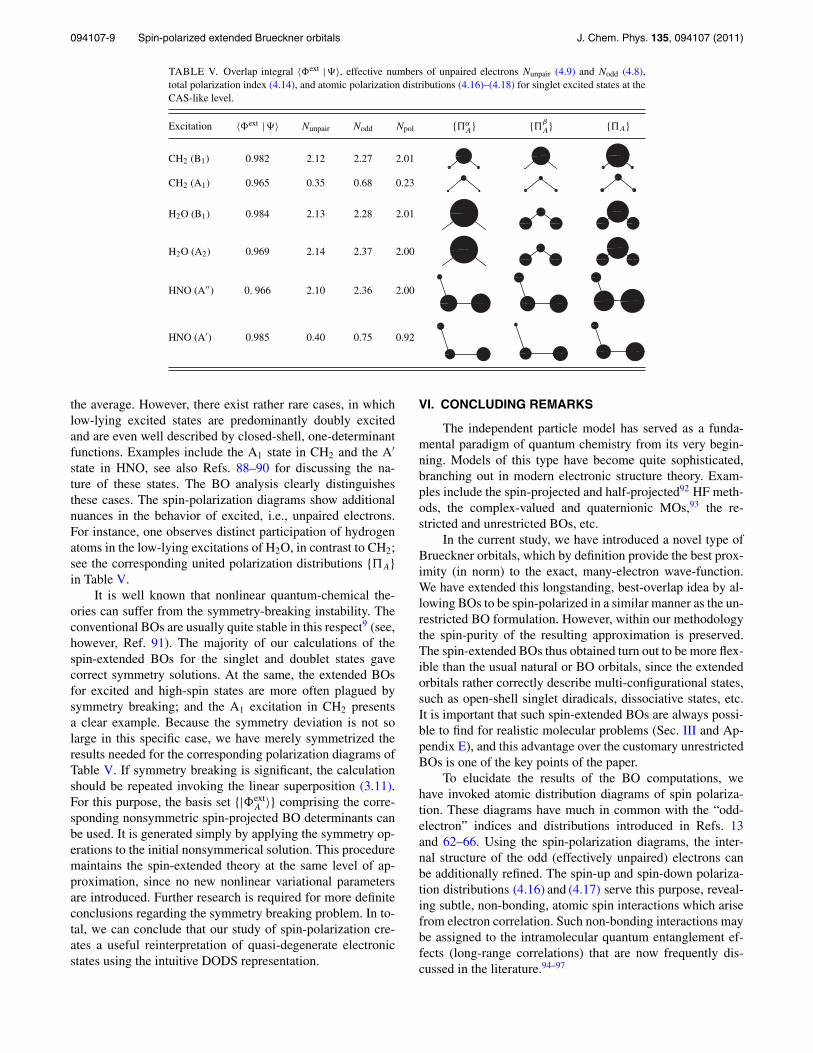
TABLE II. Coefficient Cnat (5.2), overlap integral 〈�ext |�〉, effective numbers of unpaired electrons Nunpair (4.9) and Nodd (4.8), total polarizationindex (4.14), and atomic polarization distributions (4.16) and (4.17) for carbenes at the FCI and CAS-like levels.
electron correlation. Hence, the coupling that is invoked bythis spin polarization is not of the same physical nature as thespin coupling leading to the common chemical bonding forequilibrium configurations. In other words, the singlet-typecoupling of unpaired electrons is maintained, owing to theconservation of the total spin, but this coupling is nonbond-ing.
Similar results are obtained for more complex systems,as demonstrated by the typical carbene molecules (Table II),including methylene and the simplest “dicarbene,” C2. Thesesystems are considered at both the equilibrium geometry,taken from Refs. 73 and 74, respectively, and the “dissocia-tive” geometry in which all bonds are triply stretched. Theequilibrium structures for polyatomic carbenes are taken fromRefs. 75–77. The methylene molecule is treated at the FCIlevel of theory with a frozen 1s-core on the carbon atom,while the other carbenes are described at the CAS-like levelof theory. It follows from Table II that |�ext〉 gives a goodrepresentation of the multi-configurational states even for thestretched geometry of the simplest carbenes. Comparable re-sults are obtained for open-shell singlet states such as that in1,2-cyclobutadiene, in which again the natural orbital deter-minant fails to describe this unusual, singlet diradical.
As further pertinent examples, we consider two cumu-lenes, namely, allene and butatriene, undergoing internal ro-tation. Previously, we treated this internal rotation problemat the π−electron FCI level of theory,78 which generatedreasonable energy barriers (compare the data from Refs. 78and 79). A detailed analysis was not given at that time.Now, these molecules are briefly revisited within the CAS-like approach supplemented by the BO analysis. The results,based on molecular geometry from Refs. 79 and 80, are il-lustrated in Table III. We again remark that the BO repre-sentation adequately reflects the multi-configurational nature
of both the equilibrium state and the twisted state (TS). TheTS in these and similar conjugated systems are predominantlytwo-configuration in its nature, which is rather typical of di-radicaloid systems.72, 81 Moreover, the spin-extended state ismarkedly more similar to the initial CAS-like state in thetwisted geometry rather than the equilibrium (non-twisted)configuration. For both configurations of the cumulene sys-tems, the terminal CH2 groups are more spin-polarized thanthe inner atoms.
When correlated spin-up and spin-down electrons arelocalized in well separated regions, as is the case for1,2-cyclobutadiene and twisted allene, the open-shell struc-ture of the diradicals can be put in a more graphical way.For the systems just mentioned, spin-polarization can be ex-pressed as follows:
(5.3)
The hydrogen atoms are ignored in this representation, sincethey contribute little to the spin polarization. Often, both cor-related spin-up and spin-down electrons may occupy identicalsites simultaneously. Then, the above schemes lose their ap-peal, and each distribution α
A and β
A should be displayedseparately. In the case of singlet states the time-reversal sym-metry requires that the spin-up and spin-down electrons enjoyequal rights. Therefore, there exist no real spin densities fors = 0, and one should keep in mind that all the displayed spin-polarization pictures imply equivalent pictures for the oppo-site spin orientation.
094107-8 A. V. Luzanov and O. V. Prezhdo J. Chem. Phys. 135, 094107 (2011)
TABLE III. Coefficient Cnat (5.2), overlap integral 〈�ext |�〉, effective numbers of unpaired electronsNunpair (4.9) and Nodd (4.8), total polarization index (4.14), an atomic polarization distributions (4.16) and (4.17)for small cumulenes at the equilibrium and TS configurations at the CAS-like level of theory.
It is interesting to study nonzero spin states in the samemanner. In such systems, spin polarization always arises be-cause of uncompensated spin-up and spin-down electrons, re-quiring no less than 2s unpaired electrons. Therefore, Nβ
pol and
the β
A distribution characterize correlation effects proper, re-call Eqs. (4.14) and (4.15). In the present study, the discussionis restricted to the doublet radicals that are closely related tothe carbenes considered previously (their equilibrium geome-try parameters are adopted from Refs. 82–85). Table IV showsthat Npol differs little from 2s, and we suggest that for radi-cal states, unlike singlet states, Npol poorly reflects dynamiccorrelation. Apparently, this deficiency is closely related to aninaccurate description of the spin coupling structure of radicalstates in EHF-like models,86 although 〈�ext | �〉 remains quite
large in these systems even for dissociative states (see resultsfor CH in Table IV). Possibly, the linear combination (3.11)will produce more refined results, but this extension requiresadditional investigation.
Finally, consider briefly some results (Table V) forFranck-Condon excited states, which, in a sense, present aspecial class of quasidegenerate states. The equilibrium ge-ometry for systems in Table V was taken from Ref. 87.In agreement with the expectations from Appendix E, seeEq. (E7), the optimal |�ext〉 state vectors overlap well with thecorresponding low-lying excited state vectors. The total polar-ization index, Npol, more or less agrees with other quantifica-tions of shell openness. Together, they clearly show that thetwo electrons in the excited states are naturally unpaired, on
TABLE IV. Coefficient Cnat (5.2), overlap integral 〈�ext |�〉, effective numbers of unpaired electrons Nunpair (4.9) and Nodd (4.8), total polarization in-dex (4.14), and atomic polarization distributions (4.16) and (4.17) for doublet states of various organic radicals at the CAS-like level.
TABLE V. Overlap integral 〈�ext |�〉, effective numbers of unpaired electrons Nunpair (4.9) and Nodd (4.8),total polarization index (4.14), and atomic polarization distributions (4.16)–(4.18) for singlet excited states at theCAS-like level.
Excitation 〈�ext |�〉 Nunpair Nodd Npol { αA} { β
A} { A}
CH2 (B1) 0.982 2.12 2.27 2.01
CH2 (A1) 0.965 0.35 0.68 0.23
H2O (B1) 0.984 2.13 2.28 2.01
H2O (A2) 0.969 2.14 2.37 2.00
HNO (A′′) 0. 966 2.10 2.36 2.00
HNO (A′) 0.985 0.40 0.75 0.92
the average. However, there exist rather rare cases, in whichlow-lying excited states are predominantly doubly excitedand are even well described by closed-shell, one-determinantfunctions. Examples include the A1 state in CH2 and the A′
state in HNO, see also Refs. 88–90 for discussing the na-ture of these states. The BO analysis clearly distinguishesthese cases. The spin-polarization diagrams show additionalnuances in the behavior of excited, i.e., unpaired electrons.For instance, one observes distinct participation of hydrogenatoms in the low-lying excitations of H2O, in contrast to CH2;see the corresponding united polarization distributions { A}in Table V.
It is well known that nonlinear quantum-chemical the-ories can suffer from the symmetry-breaking instability. Theconventional BOs are usually quite stable in this respect9 (see,however, Ref. 91). The majority of our calculations of thespin-extended BOs for the singlet and doublet states gavecorrect symmetry solutions. At the same, the extended BOsfor excited and high-spin states are more often plagued bysymmetry breaking; and the A1 excitation in CH2 presentsa clear example. Because the symmetry deviation is not solarge in this specific case, we have merely symmetrized theresults needed for the corresponding polarization diagrams ofTable V. If symmetry breaking is significant, the calculationshould be repeated invoking the linear superposition (3.11).For this purpose, the basis set {|�ext
A 〉} comprising the corre-sponding nonsymmetric spin-projected BO determinants canbe used. It is generated simply by applying the symmetry op-erations to the initial nonsymmerical solution. This proceduremaintains the spin-extended theory at the same level of ap-proximation, since no new nonlinear variational parametersare introduced. Further research is required for more definiteconclusions regarding the symmetry breaking problem. In to-tal, we can conclude that our study of spin-polarization cre-ates a useful reinterpretation of quasi-degenerate electronicstates using the intuitive DODS representation.
VI. CONCLUDING REMARKS
The independent particle model has served as a funda-mental paradigm of quantum chemistry from its very begin-ning. Models of this type have become quite sophisticated,branching out in modern electronic structure theory. Exam-ples include the spin-projected and half-projected92 HF meth-ods, the complex-valued and quaternionic MOs,93 the re-stricted and unrestricted BOs, etc.
In the current study, we have introduced a novel type ofBrueckner orbitals, which by definition provide the best prox-imity (in norm) to the exact, many-electron wave-function.We have extended this longstanding, best-overlap idea by al-lowing BOs to be spin-polarized in a similar manner as the un-restricted BO formulation. However, within our methodologythe spin-purity of the resulting approximation is preserved.The spin-extended BOs thus obtained turn out to be more flex-ible than the usual natural or BO orbitals, since the extendedorbitals rather correctly describe multi-configurational states,such as open-shell singlet diradicals, dissociative states, etc.It is important that such spin-extended BOs are always possi-ble to find for realistic molecular problems (Sec. III and Ap-pendix E), and this advantage over the customary unrestrictedBOs is one of the key points of the paper.
To elucidate the results of the BO computations, wehave invoked atomic distribution diagrams of spin polariza-tion. These diagrams have much in common with the “odd-electron” indices and distributions introduced in Refs. 13and 62–66. Using the spin-polarization diagrams, the inter-nal structure of the odd (effectively unpaired) electrons canbe additionally refined. The spin-up and spin-down polariza-tion distributions (4.16) and (4.17) serve this purpose, reveal-ing subtle, non-bonding, atomic spin interactions which arisefrom electron correlation. Such non-bonding interactions maybe assigned to the intramolecular quantum entanglement ef-fects (long-range correlations) that are now frequently dis-cussed in the literature.94–97
094107-10 A. V. Luzanov and O. V. Prezhdo J. Chem. Phys. 135, 094107 (2011)
Specific examples given in Sec. V indicate that in the caseof singlet states, the multi-configuration state vector can bewell condensed to a single-particle, extended BO represen-tation, providing a suitable visualization tool by way of thespin-polarization diagrams. For nonzero spin states, the pro-jected determinant formed with extended BOs also provides agood representation of the multi-configurational state, but, inthis case, spin-polarization effects are less pronounced.
Several points remain to be discussed in a critical manner.At the end of Sec. III we analyzed the extent to which the ap-proach based on the spin projection technique can violate thesize-consistency requirement. Based on the specific examplesand the fact that the correct state vector is size-consistent, weargued that size-consistency does not aggravate BO problemsin practice. If needed, the more sophisticated representation|�ext
CIt〉 (3.11) can be used. The spin polarization diagrams areeasy to obtain for |�ext
CI 〉 as well.At present, we could employ the extended BOs pri-
marily for interpretive purposes. This is in contrast to theconventional BOs, which form the basis for many effe-ctive electron-correlation methods, such as BDCC.6 Here,the use of extended BOs is limited to our iterative CAS-likeprocedure (Sec. V), avoiding the standard CASSCF schemethat demands repeated computations of the second orderdensity matrix. This CAS-like scheme is a simple and roughapproximation to the true CASSCF. Nevertheless, it providesa reasonable characterization of the electronic states understudy. It remains to be seen whether it will be possible toinclude the spin-extended BOs in other iterative electron-correlation procedures, for instance, to improve convergenceof CC methods in restricted active spaces. We can supposethat generalizing the proposed procedures to the CC method-ology will be useful for interpretative goals at the very least.
We stress again that in the present paper, the extendedBO technique has only been developed within the FCI andCAS approaches. Since CASSCF can be applied to a varietyof systems and processes, especially to chemical and photo-chemical reactions,98, 99 we hope that the spin-extended BOmethod will be useful as an additional interpretive tool formany practical problems.
ACKNOWLEDGMENTS
The authors are indebted to Dr. Heather M. Jaeger forvaluable comments and to Dr. Oleg A. Zhikol for providingtest calculations on the CASSCF models. The research wassupported in part by the National Science Foundation (NSF)(Grant No. CHE-1050405).
APPENDIX A: DERIVATION OF EQ. (2.12)
Here we compute the transition density matrix D�,�
where |�〉 is given in Eq. (2.9), or in equivalent form,
|�〉 = C0|�〉 +∑i;a
Cai |�i→a〉 + . . . .
The terms from high-order excited configurations |�i→aj→b
〉etc., are not given explicitly, since they do not contributeto D�,�. The transition density between |�i→a〉 and |�〉 is
simply |ϕa〉〈ϕi |, and the total contribution to D�,� from thesingly excited configurations is just C1∑
i;a
Cai |ϕa〉〈ϕi |.
Moreover, the “transition” density between |�〉 and |�〉is the projector (2.2), leading, as a result, to Eq. (2.12). We re-fer the reader to Ref. 100 for more general expressions. Fromthe general rules given there one deduces the following for-mula used in the current work,∑
1≤k≤N
Z(k)|�〉 = (TrZρ)|�〉 +∑
1≤k≤N
Z−(k)|�〉, (A1)
where Z is an arbitrary one-electron operator, and Z− = (I− ρ)Zρ.
APPENDIX B: MATRIX REPESENTATION OF THE FULLCI STATE VECTOR
In this appendix we provide preliminary details for thespin separation procedure based on the Fock theory. In whatfollows, a half-integer quantity, n ≡ N/2, will be used. In theFock spin-free theory we start with the total wave-function48
|�〉 =(
N
n + s
)1/2
AN |�o〉|χσ 〉, (B1)
where |�o〉 is a spin-free N-electron state vector, and AN
is the conventional N-electron antisymmetrizer. The spincomponent,
with |α〉 and |β〉 being the usual spin eigenkets of a singleelectron, guarantees that |�〉 is an eigenvector of the totalz-component spin operator with the eigenvalue s. In this case,commonly known as the principal case, the eigenvalue of thez-projection is the same as the total spin s. We assume that allspin-purity conditions are satisfied by construction or by anappropriate variational procedure. In particular, |�o〉 allowsthe bilinear expansion
|�o〉 =∑I ;P
XIP
∣∣�oI
⟩ ⊗ ∣∣�oP
⟩(B3)
where |�oI 〉 and |�o
P 〉 are spinless (n ± s)−electron deter-minants, and {XIP } is a set of linear variational parameters(configuration coefficients). A set of {|�o
I 〉} can be associatedwith a subsystem of spin-up (n + s)-electron states, whereas{|�o
P 〉} corresponds to a subsystem of spin-down (n − s)-electron states. Furthermore, we can identify subscripts I in|�o
This determinantal basis is generated by the spin-free orbitals,taken from the set (2.18).
For formal manipulations a wave-function matrix,
X = ‖XIP ‖, (B7)
can be introduced, but we will not deeply pursue this issue.We also notice that Eq. (B3) can be viewed as the Hartree-Waller double determinant expansion considered in manyworks (e.g., see Ref. 101). At the same time, the above for-malism is inherently connected with the known determinantalFCI method102 due to the fact that each Slater determinantis in one-to-one correspondence with a spin-free doubledeterminant. The multi-indices (B4) correspond to alpha-and beta-strings of the determinantal CI method (compareEq. (B3) with Eq. (11.8.64) in book.102 This correspondenceshould be taken into account in practical computations basedon CASSCF model within determinantal CI algorithm.
Here we derive a spin-free form of functional (2.3) andthe corresponding variational equations. Formal manipula-tions on Eq. (2.3) are straightforward, owing to the fact thatMcWeeny’s generalized group theory43 can be applied toEq. (B1) because of the strong orthogonality between thespin-up and spin-down subsystems in state (B1). In doing sothe spin-unrestricted Slater determinant |�〉 is replaced with aspin-free vector of the form |�α ⊗ �β〉, where spin-free |�α〉and |�β〉 are spinless determinants built up from the spin-upand spin-down spin-free orbital sets |ϕα
i 〉, |ϕβ
i 〉, respectively.Then, elementary manipulations lead to the spin-free repre-sentation,
〈� |�〉 = 〈�α ⊗ �β |�o〉 ≡ Xαβ, (C1)
and, accordingly,
J = (Xαβ)2. (C2)
We can rewrite |�α〉 = ∑I |�o
I 〉〈�oI |�α〉 and |�β〉
= ∑P |�o
P 〉〈�oP |�β〉, and this transforms the total overlap
integral (C1) to
Xαβ =∑I ;P
s(α)I XIP s
(β)P , (C3)
where elementary overlaps s(α)I and s
(β)P are introduced as fol-
lows:
s(α)I = ⟨
�oI |�α〉, s
(β)P = ⟨
�oP |�β
⟩. (C4)
[See also Eqs. (C10) and (C11).] Notice that Xαβ can bethought as a special off-diagonal matrix element of wave-function matrix (B7).
For computing Eq. (C3) and the following expressions itis suitable to define the sets
X(β)I =
∑P
s(β)P XIP , X
(α)P =
∑I
s(α)I XIP , (C5)
soXαβ =
∑P
s(β)P X
(α)P . (C6)
Computations of the first variations 〈δ�α| and |δ�β〉 inEq. (C1) are similar to those in Eqs. (2.5) and (2.6). The aux-iliary sets of one-electron transitional matrices
dα;I =∑
J
s(α)J dI→J , dβ;P =
∑Q
s(β)Q dP→Q, (C7)
appear naturally. Here, for instance, dI→J is elementary tran-sition density matrix between |�o
I 〉 and |�oJ 〉 (see below).
Then instead of D�,� in Eq. (2.15), two main spin-free ma-trices dα and dβ arise
dα =∑
I
X(β)I dα;I , dβ =
∑P
X(α)P dβ;P . (C8)
As a result, the maximum of the functional (C2) is at-tained by solutions of Fock-type coupled eigenvalue prob-lems (2.17), where the spin-free B-operators are defined asfollows:
Now we present the working expressions for the auxil-iary overlap integrals (C4) and the transition matrices dI ;α
and dβ;P (C8) needed for Eq. (C9). Elementary overlaps aredirectly computed as ordinary determinants,
s(α)I ≡ s
(α)i1i2...in+s
= Det
∥∥∥∥∥∥∥U
(α)1,i1
U(α)1,i2
. . . U(α)1,in+s
· · . . .
U(α)n+s,i1
U(α)n+s,i2
. . . U(α)n+s,in+s
∥∥∥∥∥∥∥ ,
(C10)
s(β)P ≡ s(β)
p1p2...pn−s= Det
∥∥∥∥∥∥U
(β)1,p1
U(β)1,p2
. . . U(β)1,pn−s
. . . . .
U(β)n−s,p1
U(β)n−s,p2
. . . U(β)n−s,pn−s
∥∥∥∥∥∥ ,
(C11)
where matrix U (α) = ‖〈ϕαi |ϕo
μ〉‖ is composed from the ex-pansion coefficients of α−MOs [Eq. (2.16)] in the ba-sis (2.18). Analogously, U (β) = ‖〈ϕβ
i |ϕoμ〉‖.
Auxiliary matrices dI ;α in Eq. (C8) computed in the form
dI ;αμν =
∑J
s(α)J dI→J
μν , (C12)
where dI→Jμν = 〈ϕo
μ|dI→J |ϕoν 〉. The latter matrices are easily
evaluated by the Slater rules for determinants. Namely, ifI = J then dI→J
μν = δμνθ [I, μ] where θ [I, μ] is equal to 1 ifmulti-index I contains small index μ, otherwise θ [I, μ] = 0.If I = J , the nonzero result arises only when the multi-indices I and J differ in one small index, or symbolically, I
= i1 . . . a . . . in+s , J = j1 . . . b . . . jn+s , and I/a = J/b. ThendI→J
μν = (−1)ηa+ηbδμbδνa,
with ηa being position number of a in the multi-index I.Similarly, we compute
dP ;βμν =
∑Q
s(β)Q dP→Q
μν , (C13)
where dP→Qμν are obtained by the rule above.
094107-12 A. V. Luzanov and O. V. Prezhdo J. Chem. Phys. 135, 094107 (2011)
APPENDIX D: VARIATIONAL EQUATIONSFOR EXTENDED BOs
The equations for the spin-extended BOs are obtained inthe usual way. The first variation of J ext in Eq. (3.3) can becast in the form
Owing to Eq. (3.5), the normalization factor 〈�|Os |�〉 can bereduced to the form
Ns ≡ 〈�|Os |�〉 =n−s∑j=0
aj ξ(j ), (D2)
where
ξ (j ) = 〈�|Sj−S
j+|�〉/(j !)2. (D3)
In turn, these quantities are transformed to the special matrixinvariants of the spin-free matrix π0 defined by Eq. (3.7) (forderivation see Ref. 52). Namely, these ξ (j ) turn out to be thecharacteristic coefficients of the matrix π0. They can be read-ily computed by the known Leverrier-Faddeev method (forsome properties and recent applications of similar invariantssee Ref. 103),
ξ (j ) = j−1Trπ0σ(j ), (D4)
where ξ (0) = 1. Matrices σ (j ) can be obtained recurrentlyfrom the equation,
σ (j ) = ξ (j−1)I − σ (j−1)π0, (D5)
starting with the marix σ (0) = 0. The variational property,
δξ (j ) = Trδπ0σ(j ), (D6)
is useful for formal manipulations as well. Note that our ex-pression for Ns is closely related to Eq. (24) in Ref. 104, but aclear and direct matrix representation was not provided there.
After simple algebraic manipulations involvingEqs. (3.7) and (D2) and (D6), we obtain the workingequations (3.8), which follow from the requirement δJ ext = 0with δJ ext of the form (D1). The extended B-operators inEqs. (3.8) take the form
Bαext = NsB
α + Xαβ{ρβσ aver + σ averρβ}/2, (D7)
Bβext = NsB
β − Xαβ{σ aver(I − ρα) + (I − ρα)σ aver}/2,
(D8)with the matrix σ aver equal to
σ aver =n−s∑j=0
ajσ(j ). (D9)
Thus, we can succinctly describe the algorithm for BOs.We assume that the set of expansion coefficients {XIP } is ob-tained from the determinantal FCI or CASSCF method in thefixed basis set (2.18). Then, we perform the following steps:
(i) precalculate integrals s(α)I and s
(β)P [Eqs. (C10) and
(C11)], form X(β)I and X
(α)P by Eq. (C5), and evaluate
Xαβ by Eq. (C6);
(ii) find dI ;αμν and dP ;β
μν from Eqs. (C12) and (C13), and com-pute matrix elements for Eq. (C8)
dαμν =
∑I
X(β)I dI ;α
μν , dβμν =
∑P
X(α)P dβ;P
μν ;
(iii) calculate the “unrestricted” B-matrices (C9):
μν ‖(0 ≤ j ≤ n − s) by Eqs. (3.7),(D4) and (D5), and compute σ aver
μν = ∑n−sj=0 ajσ
(j )μν ;
(v) compute the “extended” B-matrices by Eqs. (D7) and(D8) and solve eigenproblem (3.8).
APPENDIX E: EXISTENCE OF SPIN-EXTENDED BOsFOR CLOSED SHELLS
The proof of existence of spin-polarized solutionbegins as in Ref. 105 for the similar problem within thehalf-projected HF theory. We start with the closed shelldeterminant, |�0〉, in which all n spinless orbitals |ϕ◦
i 〉 are
doubly occupied, that is |�0〉 = |+ϕ◦1
−ϕ◦
1 . . .+ϕ◦
i
−ϕ◦
1 . . .+ϕ◦
1
−ϕ◦
1〉where |+ϕ◦
1〉 = |ϕ◦i 〉|α〉, |+ϕ◦
1〉 = |ϕ◦i 〉|β〉. We replace the given
ith electron pair by a spin polarized pair,∣∣+ϕ◦
1
−ϕ◦
1
⟩ → ∣∣+ϕ◦
1
−ϕ◦
1
⟩, (E1)
where ∣∣ϕαi
⟩ = (1 + z2)−1/2{∣∣ϕ◦
i
⟩ + z∣∣ϕ◦
a
⟩},∣∣ϕβ
i
⟩ = (1 + z2)−1/2{∣∣ϕ◦
i
⟩ − z∣∣ϕ◦
a
⟩}. (E2)
Here, |ϕ◦a〉 is a vacant orbital, and z is an arbitrary mixing
parameter. In the conventional notation the correspondingspin-polarized determinant, |�〉, takes the following form:
|�〉 = {|�0〉 +√
2z∣∣�S=1
i→a
⟩ − z2|�ii→aa〉}/(1 + z2).
(E3)Here the triplet |�S=1
i→a〉 component causes spin-contamination. The extended solution (3.1) removesthis component, so
|�ext〉 = {|�0〉 − z2|�ii→aa〉}/√
1 + z4. (E4)
In order to calculate J ext or the related quantity 〈�ext |�〉,we need a proper representation of the exact |�〉. The stateof interest is a ground state for which the overlap integral c0
= 〈�0 |�〉 is assumed to be nonzero. Hence, up to a factor c0,the correlated state is taken as |�〉 = |�0〉 + t |�ii→aa〉 + . . .,where t is a diagonal 2-electron particle-hole amplitude. Theomitted terms do not influence the discussion below. Forthe two-electron ground state problem, e.g., the hydrogenmolecule, the amplitude t is strictly negative, as proved bysolving directly the associated secular problem in a minimalbasis set. The same holds true for many-electron systems inthe second-order Møller-Plesset perturbation theory (MP2),as shown by the explicit formulas of MP2. At least, therealways exist essentially non-negative “diagonal” amplitudesin MP2 and related coupled cluster approaches. Thus, inrealistic molecular systems, the inequality t < 0 appears
valid without exception, at least for the one “diagonal” doubleexcitation ii → aa. Having Eqs. (E3) and (E4) in mind, theoverlap integral is computed according to
〈�ext |�〉 = {1 − tz2}/√
1 + z4. (E5)
Two extremal points: z = 0 (no spin polarization) and z2
= −t occur. The latter extremum gives the spin-polarizedsolution that exists always owing to the negativity of t .This completes the informal argument supporting the mainassertion of this appendix. In practice, we never encounteredmolecules for which the spin-extended BOs could not befound for the ground state.
A similar BO approach is possible for low-lying excitedstates in molecules. Typically, the lowest lying singlet excitedstate wave-function is dominated by a singly excited config-uration |�i→a〉. This state is formed by the standard replace-ment,
∣∣+ϕ◦
i
−ϕ◦
i
⟩ → 1√2
∣∣+ϕ◦
i
−ϕ◦
a + +ϕ◦
a
−ϕ◦
i
⟩, (E6)
and corresponds to the specific solution for |�ext〉 with |ϕαi 〉
= |ϕ◦i 〉, |ϕβ
i 〉 = |ϕ◦a〉. The remaining orbitals of |�0〉 are un-
changed, leading to
〈�ext |�i→a〉 = 1 (E7)
and |�ext〉 = |�i→a〉. For typical molecular systems, spin-extended BOs can provide a good description of excitedstates as well. In other context, specific examples of excitedstates were given previously55 using the conventional EHF ap-proach.
The above BO reconstruction of |�i→a〉 can be also re-formulated in terms of the complex-valued BOs. In this case,another BO approach can be designed by extending the ear-lier BO approach into the complex plane (see the first para-graph of Sec. III). To obtain an excited state model, one musttake the imaginary part of the corresponding complex-valuedSlater determinant.105 This problem deserves a separate study,which is beyond scope of the current paper.
1S. Saebø and P. Pulay, Annu. Rev. Phys. Chem. 44, 213 (1993).2B. Jansík, S. Høst, K. Kristensen, and P. Jørgensen, J. Chem. Phys. 134,194104 (2011).
3K. A. Brueckner and W. Wada, Phys. Rev. 103, 1008 (1956).4R. K. Nesbet, Phys. Rev. 109, 1632 (1958); R. K. Nesbet, VariationalPrinciples and Methods in Theoretical Physics and Chemistry (CambridgeUniversity Press, New York, 2003).
5P.-Q. Löwdin, J. Math. Phys. 3, 1171 (1962).6C. E. Dykstra, Chem. Phys. Lett. 45, 466 (1977); L. Z. Stolarczyk andH. J. Monkhorst, Int. J. Quantum Chem., Quantum Chem. Symp. S18,267 (1984); N. C. Handy, J. A. Pople, M. Head-Gordon, K. Raghavachari,and G. W. Trucks, Chem. Phys. Lett. 164, 185 (1989).
7D. Crawford and H. F. Schaefer, Rev. Comput. Chem. 14, 33 (2000).8J. F. Stanton, J. Gauss, and R. J. Bartlett, J. Chem. Phys. 97, 5554 (1992);R. J. Bartlett and J. F. Stanton, Rev. Comput. Chem. 5, 65 (1994).
9G. E. Scuseria and H. F. Schaefer III, Chem. Phys. Lett. 142, 354 (1987);C. D. Sherrill, A. I. Krylov, E. F. C. Byrd, and M. Head-Gordon, J. Chem.Phys. 109, 4171 (1998).
10K. Jankowski and K. Rubiniec, Mol. Phys. 100, 1741 (2002).11E. R. Davidson, Reduced Density Matrices in Quantum Chemistry (Aca-
demic, New York, 1976).12J. M. Boffil and P. Pulay, J. Chem. Phys. 90, 3637 (1989); R. G. A. Bone
and P. Pulay, Int. J. Quantum Chem. 45, 133 (1993).13M. Head-Gordon, Chem. Phys. Lett. 372, 508 (2003).14S. Goedecker and C. J. Umrigar, Phys. Rev. Lett. 81, 866 (1998).15M. S. Gordon, M. W. Schmidt, G. M. Chaban, K. R. Glaesemann,
W. J. Stevens, and C. Gonzalez, J. Chem. Phys. 110, 4199 (1999).16M. L. Abrams and C. D. Sherrill, Chem. Phys. Lett. 395, 227 (2004).17G. Fagas, P. Delaney, and J. C. Greer, Phys. Rev. B 73, 241314(R) (2006);
J. C. Greer, Mol. Phys. 106, 1363 (2008); I. Yeriskin, S. McDermott,R. J. Bartlett, G. Fagas, and J. C. Greer, J Phys. Chem C 114, 20564(2010).
18A. Köhn and J. Olsen, J. Chem. Phys. 125, 174110 (2006).19F. Weinhold and C. R. Landis Valency and Bond-
ing: A Natural Bond Orbital Donor-Acceptor Perspective(Cambridge University Press, Cambridge, England, 2005).
20F. Aquilante, T. K. Todorova, L. Gagliardi, T. B. Pedersen, and B. O. Roos,J. Chem. Phys. 131, 034113 (2009).
21A. G Taube and R. J. Bartlett, J. Chem. Phys. 128, 164101 (2008);A. Landau, K. Khistyaev, S. Dolgikh, and A. I. Krylov, J. Chem. Phys.132, 014109 (2010).
22A. V. Luzanov and O. A. Zhikol, Int. J. Quantum Chem. 110, 902 (2010).23I. Lindgren and S. Salomonson, Int. J. Quantum Chem. 90, 294 (2002).24A. Beste and R. J. Bartlett, J. Chem. Phys. 123, 154103 (2005).25A. Heßelmann, J. Chem. Phys., 122, 244108 (2005); Phys. Chem. Chem.
Phys. 8, 563 (2006).26C. J. Cramer, J. Am. Chem. Soc. 120, 6261 (1998).27M. Prall, A. Wittkopp, and P. R. Schreiner, J. Phys. Chem. A 105, 9265
(2001).28S. Tschumper, M. L. Leininger, B. C. Hoffman, E. F. Valeev, H. F.
Schaefer, and M. Quack, J. Chem. Phys. 116, 690 (2002).29I. M. B. Nielsen, C. L. Janssen, and M. D. Allendorf, J. Phys. Chem. A
107, 5122 (2003).30T. Tuttle, E. Kraka, and D. Cremer, J. Am. Chem. Soc. 127, 9469
(2005).31M. Caricato, G. Scalmani, and M. J. Frisch, J. Chem. Phys. 134, 244113
(2011).32C. A. Coulson and I. Fischer, Philos. Mag. 40, 386 (1949).33P.-O. Löwdin, Phys. Rev. 97, 1509 (1955).34C. J. Cramer, F. J. Dulles, D. J. Giesen, and J. Almlöf, Chem. Phys. Lett.
245, 165 (1995).35P. Pérez, J. Andrés, V. S. Safont, O. Tapia, and R. Contreras, J. Phys.
Chem. A 106, 5353 (2002); E. Chamorro, P. Perez, F. De Proft, andP. Geerlings, J. Chem. Phys. 124, 044105 (2006); E. Chamorro, J. C.Santos, C. A. Escobar, and P. Perez, Chem. Phys. Lett. 431, 210 (2006).
36J. Grafenstein, E. Kraka, M. Filatov, and D. Cremer, Int. J. Mol. Sci. 2,360 (2002).
37P. G. Szalay, J. Vázquez, C. Simmons, and J. F. Stanton, J. Chem. Phys.121, 7624 (2004).
38A. G. Taube and R. J. Bartlett J. Chem. Phys. 128, 044110 (2008).39U. De Giovannini, F. Cavaliere, R. Cenni, M. Sassetti, and B. Kramer,
Phys. Rev. B 77, 035325 (2008).40W. Purwanto, W. A. Al-Saidi, H. Krakauer, and S. Zhang, J. Chem. Phys.
128, 114309 (2008).41E. C. Barnes, G. A. Petersson, J. A. Montgomery, Jr., M. J. Frisch, and
J. M. L. Martin, J. Chem. Theory Comput. 5, 2687 (2009); T. Tsuchimochiand G. E. Scuseria, J. Chem. Phys. 134, 064101 (2011).
42P. R. Surján, Second Quantization Approach to Quantum Chemistry(Springer-Verlag, Berlin, 1989).
44J. A. Coleman and V. J. Yukalov, Reduced Density Matrices: CoulsonChallenge (Springer, New York, 2000).
45W. Kutzelnigg and V. H. Smith, Jr., J. Chem. Phys. 41, 896 (1964);A. Kozlov and V. I. Pupyshev, Chem. Phys. Lett. 206, 151 (1993).
46J. V. Ortiz, Int. J. Quantum Chem. 100, 1131 (2004).47V. A. Fock, Zh. Eksp. Teor. Fiz. 10, 961 (1940); Selected Works: Quantum
Mechanics and Quantum Field Theory (Chapman and Hall/CRC, London,2004).
48G. E. Vaiman, A. V. Luzanov, and M. M. Mestechkin, Theor. Math. Phys.28, 634 (1976); G. T. Klimko and M. M. Mestechkin, Int. J. QuantumChem. 17, 415 (1980); A. V. Luzanov, “Spin-free quantum chemistry:
094107-14 A. V. Luzanov and O. V. Prezhdo J. Chem. Phys. 135, 094107 (2011)
What one can gain from Fock’s cyclic symmetry,” Int. J. Quantum Chem.(in press).
49I. Mayer, Adv. Quantum Chem. 12, 189 (1980).50H. Yuan and D. Cremer, Chem. Phys. Lett. 324, 389 (2000).51J. L. Sonnenberg, H. P. Hratchian, and H. B. Schlegel, in Encyclopedia of
Inorganic Chemistry, 3rd ed. (Wiley, New York, 2009), p. 173.52A. V. Luzanov and V. V. Ivanov, Teor. Eksp. Khim. 26, 385 (1989); Theor.
Exp. Chem. 26, 363 (1991).53P.-O. Löwdin, Rev. Mod. Phys. 36, 966 (1964).54M. M. Mestechkin and G. E. Whyman, Int. J. Quantum Chem. 8, 45
(1974); M. M. Mestechkin, A. G. Gershikov, and G. E. Whyman, Chem.Phys. Lett. 91, 443 (1982).
55S. I. Smirnov, M. M. Mestechkin, and G. E. Vaiman, Theor. Exp. Chem.24, 202 (1988); M. M. Mestechkin and G. E. Whyman, Mol. Phys. 69, 775(1990).
56J. Cížek and J. Paldus, J. Chem. Phys. 47, 3976 (1967).57M. M. Mestechkin, Int. J. Quantum Chem. 13, 469 (1978).58A. Hibbert and C. A. Coulson, Proc. Phys. Soc. Jpn. 92, 17 (1967).59J. Paldus, J. Cížek, and B. A. Keating, Phys. Rev. A 8, 640 (1973).60E. R. Davidson and A. E. Clark, Int. J. Quantum Chem. 103, 1 (2005).61F. W. Bobrowicz and W. A. Goddard III, in Modern Theoretical Chem-
istry: Methods of Electronic Structure Theory, edited by H. F. Schaefer III(Plenum, New York, 1977), Chap. 4, p. 71.
62K. Takatsuka, T. Fueno, and K. Yamaguchi, Theor. Chim. Acta 48, 175(1978).
63R. C. Bochicchio, J. Mol. Struct.: THEOCHEM 429, 229 (1998);R. C. Bochicchio, A. Torre, and L. Lain, Chem. Phys. Lett. 380, 486(2003); L. Lain, A. Torre, D. R. Alcoba, and R. C. Bochicchio, Chem.Phys. Lett. 476, 101 (2009).
64V. N. Staroverov and E. R. Davidson, Chem. Phys. Lett. 330, 161 (2000);E. R. Davidson and A. E. Clark, Phys. Chem. Chem. Phys. 9, 1881 (2007).
65A. D. Dutoi, Y. Jung, and M. Head-Gordon, J. Phys. Chem. A 108, 10270(2004).
66A. V. Luzanov and O. V. Prezhdo, J. Chem. Phys. 124, 224109 (2006).67MOLPRO, a package of ab initio programs designed by H.-J. Werner and
P. J. Knowles, version 2009.1, R. D. Amos, A. Bernhardsson, A. Berninget al.
68A. V. Luzanov, A. L. Wulfov, V. O. Krouglov, Chem. Phys. Lett. 197, 614(1992); A. V. Luzanov and O. V. Prezhdo, Int. J. Quantum Chem. 102, 582(2005); A. V. Luzanov and O. A. Zhikol, Int. J. Quantum Chem. 104, 167(2005).
70G. A. Gallup, Valence Bond Method (Cambridge University Press,Cambridge, England, 2002).
71B. S. Jursic, Int. J. Quantum Chem. 69, 679 (1998).72W. T. Borden, In Encyclopedia of Computational Chemistry, Vol. 1, edited
by P. v. R. Schleyer (Wiley, Chichester, 1998), p. 708.73C. D. Sherrill, M. L. Leininger, T. J. Van Huis, and H. F. Schaefer, J.
Chem. Phys. 108, 1040 (1998).74K. P. Huber and G. Herzberg, Constants of Diatomic Molecules, Molec-
ular Spectra and Molecular Structure (Van Nostrand, New York, 1979),Vol. IV.
75C. D. Sherrill, E. F. C. Byrd, and M. Head-Gordon, J. Chem. Phys. 113,1447 (2000).
76Q. Wu, Q. Cheng, Y. Yamaguchi, Q. Li, and H. F. Schaefer, J. Chem. Phys.132, 044308 (2010).
77K. J. Daoust, S. M. Hernandez, K. M. Konrad, I. D. Mackie, J. Winstanley,and R. P. Johnson, J. Org. Chem. 71, 5708 (2006).
78Y. F. Pedash, V. V. Ivanov, and A. V. Luzanov, Theor. Exp. Chem. 28, 19(1992).
79P. D. Jarowski, F. Diederich, and K. N. Houk, J. Phys. Chem. A 110, 7237(2006).
80H. Bettinger, P. Schreiner, P. v. R. Schleyer, and H. Schaefer, J. Phys.Chem. 100, 16147 (1996).
81L. Salem, Pure Appl. Chem. 33, 317 (1973).82F Stahl, P. V. Schleyer, H. F. Bettinger, R. I. Kaiser, Y. T. Lee, and
H. F. Schaefer, J. Chem. Phys. 114, 3476 (2001).83S. E. Wheeler, K. A. Robertson, W. D. Allen, and H. F. Schaefer, J. Phys.
Chem. A 111, 3819 (2007).84T. N. Le, A. M. Mebel, and R. I. Kaiser, J. Comput. Chem. 22, 1522
(2001).85B. Ceursters, H. M. T. Nguyen, J. Peeters, and M. T. Nguyen, Chem. Phys.
262, 243 (2000).86C. P. Byrman, J. H. van Lenthe, and J. Verbeek, Theor. Chim. Acta 86, 129
(1993).87G. Herzberg, Molecular Spectra and Molecular Structure III (Van
Nostrand, New York, 1966).88A. I. Krylov, C. D. Sherrill, and M. Head-Gordon, J. Chem. Phys. 113,
6509 (2000); A. Kalemos, T. Dunning, Jr., A. Mavridis, and J. F. Harrison,Can. J. Chem. 82, 684 (2004).
89A. A. Wu, S. D. Peyerimhoff, and R. J. Buenker, Chem. Phys. Lett. 35,316 (1975).
90E. R. Davidson and L. E. McMurchie, in Excited States, edited byE. C. Lim (Academic, New York, 1982), Vol. 5, p. 1.
91T. D. Crawford and J. F. Stanton, J. Chem. Phys. 112, 7873 (2000).92Y. G. Smeyers and L. Doreste, Int. J. Quantum Chem. 7, 687 (1973);
Y. G. Smeyers, P. Fernandez-Serra, and M. B. Ruiz, in Strategies andApplications in Quantum Chemistry, edited by M. Defranceschi andY. Ellinger (Kluwer, Dordrecht, 1996), p. 175.
93J. Hendekovic, Int. J. Quantum Chem. 8, 799 (1974); M. Seel and J. Ladik,Chem. Phys. 39, 65 (1979).
94S. Kais, Adv. Chem. Phys. 121, 537 (2007).95T. Juhász and D. A. Mazziotti, J. Chem. Phys. 125, 174105 (2006).96A. V. Luzanov and O. V. Prezhdo, Mol. Phys. 105, 2879 (2007).97D. R. Alcoba, R. C. Bochicchio, L. Lain, and A. Torre, J. Chem. Phys.
133, 144104 (2010).98M. Garavelli, F. Bernardi, and A. Cembran, in Computational Photochem-
istry, Theoretical and Computational Chemistry, edited by M. Olivucci(Elsevier, Amsterdam, 2005), Vol. 16, p. 191.
99M. J. Bearpark, F. Ogliaro, T. Vreven, M. Boggio-Pasqua, M. J. Frisch,S. M. Larkin, M. Morrison, and M. A. Robb, J. Photochem. Photobiol., A190, 207 (2007).
100A. V. Luzanov, Theor. Math. Phys. 30, 232 (1977); Int. J. Quantum Chem.108, 671 (2008).
101T. K. Lim and C. G. Jesudason, Int. J. Quantum Chem. S16, 259(1982); R. Pauncz, ibid. 35, 717 (1989); D. J. Klein, ibid. 111, 76(2011).
102T. Helgaker, P. Jørgensen, and J. Olsen, Molecular Electronic StructureTheory (Wiley, New York, 2000).
103A. V. Luzanov, Int. J. Quantum Chem. 111, 2196 (2011).104J. E. Harriman, Colloq. Int. C. N. R. S. 164, 139 (1967).105A. V. Luzanov, Zh. Strukt. Khim. 25 (6), 3 (1984); J. Struct. Chem. 25,