Vol. 187, No. 2, 1992 September 16, 1992 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS Pages 657-663 THE STRUCTURE AND BIOLOGICAL ACTIVITIES OF THE WIDELY USED PROTEIN KINASE INHIBITOR, H7, DIFFER DEPENDING ON THE COMMERCIAL SOURCE James Quick, J. Anthony Wares and Paul E. Driedger* Alder Research Center, 165 New Boston Street, Woburn, MA 01801 3 Harvard-Thorndike Laboratories of the Department of Medicine (Cardiovascular Division), Beth Israel Hospital, Harvard Medical School, Boston, MA 02215 Received July 10, 1992 SUMMARY: The protein kinase inhibitor I-(5’-isoquinolinesulfonyl)-2-methylpiperazine (H7) has been widely used because of its ability to inhibit cyclic AMP- and cyclic GMP- dependent protein kinases (PKA and PKG) and protein kinase C (PKC) at roughly equal concentrations; it is much less potent on other kinases. Previous studies in other laboratories have found that H7 samples from different commercial sources have different properties in cellular studies and protein kinase C inhibition assays. We now report the results of chemical and biological tests which show that H7 samples also differ in chemical structure, again depending on their commercial source. Chemical synthesis and NMR spectroscopy indicate that H7 from most suppliers has the structure originally proposed for H7, while “H7” from another supplier is in fact its 3-methylpiperazine positional isomer. 0 1992 A‘ademlc Press, 1°C. In 1984 Hidaka et al. reported the protein kinase inhibitory activities of a series of isoquinolinesulfonamides (1). One of these compounds, 117, showed I(i values of 3.0 PM, 5.8 PM and 6.0 PM for PKA, PKG and PKC, respectively. H7 was a much weaker inhibitor of four other protein kinases, and as a result this compound has been widely used for studying the results of general PKA, PKG and PKC inhibition. When used in conjunction with a negative control, such as N-(2-guanidinoethyl)-5- isoquinolinesulfonamide (HA-1004), a potent inhibitor of PKA and PKG which is much less active on PKC, H7 can yield specific information on the effects of inhibiting PKC. Recently Kischel et al. reported (2) that H7 samples from two manufacturers, Seikagaku and Sigma, differed in their PKC inhibitory potencies by a factor of 4, with the Seikagaku H7 being the more potent. This difference was detected in a functional *To whom correspondence should be addressed. Abbreviations: CBZ, N-carbobenzyloxycarbonyl; H7, 1-(5’-isoquinolinesulfonyl)-2- methylpiperazine; HA-1004, N-(2-guanidinoethyl)-5-isoquinolinesulfonamide; Iso-H7, l- (5’-isoquinolinesulfonyl)-3-methylpiperazine; PKA, cyclic AMP-dependent protein kinase; PKC, calcium/phospholipid-dependent protein kinase; PKG, cyclic GMP-dependent protein kinase; PMA, phorbol 12-myristate 13-acetate. 0006-291X192 $4.00 657 Copyright 0 1992 by Academic Press, Inc. All righrs oj reproduction in atzy form reserved.
Transcript
Vol. 187, No. 2, 1992
September 16, 1992
BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS
Pages 657-663
THE STRUCTURE AND BIOLOGICAL ACTIVITIES OF THE WIDELY USED PROTEIN KINASE INHIBITOR, H7, DIFFER
DEPENDING ON THE COMMERCIAL SOURCE
James Quick, J. Anthony Wares and Paul E. Driedger*
Alder Research Center, 165 New Boston Street, Woburn, MA 01801
3 Harvard-Thorndike Laboratories of the Department of Medicine (Cardiovascular Division), Beth Israel Hospital, Harvard Medical School, Boston, MA 02215
Received July 10, 1992
SUMMARY: The protein kinase inhibitor I-(5’-isoquinolinesulfonyl)-2-methylpiperazine (H7) has been widely used because of its ability to inhibit cyclic AMP- and cyclic GMP- dependent protein kinases (PKA and PKG) and protein kinase C (PKC) at roughly equal concentrations; it is much less potent on other kinases. Previous studies in other laboratories have found that H7 samples from different commercial sources have different properties in cellular studies and protein kinase C inhibition assays. We now report the results of chemical and biological tests which show that H7 samples also differ in chemical structure, again depending on their commercial source. Chemical synthesis and NMR spectroscopy indicate that H7 from most suppliers has the structure originally proposed for H7, while “H7” from another supplier is in fact its 3-methylpiperazine positional isomer. 0 1992 A‘ademlc Press, 1°C.
In 1984 Hidaka et al. reported the protein kinase inhibitory activities of a series
of isoquinolinesulfonamides (1). One of these compounds, 117, showed I(i values of 3.0
PM, 5.8 PM and 6.0 PM for PKA, PKG and PKC, respectively. H7 was a much weaker
inhibitor of four other protein kinases, and as a result this compound has been widely
used for studying the results of general PKA, PKG and PKC inhibition. When used in
conjunction with a negative control, such as N-(2-guanidinoethyl)-5-
isoquinolinesulfonamide (HA-1004), a potent inhibitor of PKA and PKG which is much
less active on PKC, H7 can yield specific information on the effects of inhibiting PKC.
Recently Kischel et al. reported (2) that H7 samples from two manufacturers,
Seikagaku and Sigma, differed in their PKC inhibitory potencies by a factor of 4, with the
Seikagaku H7 being the more potent. This difference was detected in a functional
*To whom correspondence should be addressed.
Abbreviations: CBZ, N-carbobenzyloxycarbonyl; H7, 1-(5’-isoquinolinesulfonyl)-2- methylpiperazine; HA-1004, N-(2-guanidinoethyl)-5-isoquinolinesulfonamide; Iso-H7, l- (5’-isoquinolinesulfonyl)-3-methylpiperazine; PKA, cyclic AMP-dependent protein kinase; PKC, calcium/phospholipid-dependent protein kinase; PKG, cyclic GMP-dependent protein kinase; PMA, phorbol 12-myristate 13-acetate.
0006-291X192 $4.00
657 Copyright 0 1992 by Academic Press, Inc.
All righrs oj reproduction in atzy form reserved.
Vol. 187, No. 2, 1992 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS
bioassay as well. The authors’ attempts to find a chemical or spectroscopic difference
between H7 samples from the two sources were not successful. A subsequent report (3)
found H7 from Seikagaku to be active in a series of bioassays, with IC5O’s of IO-32 PM,
while H7 from Sigma was inactive at 70 PM.
There are inconsistencies in the synthetic routes given for H7 in Ref. 1 and an
associated U.S. patent (4) and European patent application (5). These discrepancies,
along with the reported discrepancies in biological effects using H7 from different
commercial sources (2,3) prompted the present investigation. We now present chemical
evidence that H7 samples obtained from various commercial sources differ in chemical
structure, as suggested by their differing biological activity. Based on the available
evidence the chemical stucture of Seikagaku H7 is assigned as 1-(5-isoquinolinesulfonyl)-
2-methylpiperazine, which is the same as its original designation (1). The structure of
Sigma “H7”, however, is 1-(5-isoquinolinesulfonyl)-3-methylpiperazine.
MATERIALS AND METHODS
Chemicals. Samples labeled “H7” were obtained from Sigma (free base, lot No. 116F-5860, and di-HCl salt, lot No. 120H-5816); Seikagaku, di-HCI salt, lot No. H91402; Calbiochem, di-HCl salt, lot No. 91X009; Biomol, di-HCl salt, lot No. F379; Cookson Chemicals, di-HCl salt, lot No. 5386; Toronto Biochemicals, di-HCl salt, lot No. 3-FW-79 and Research Biochemicals Inc., mono-HCl salt, lot No. CT-890B. A sample of 1-(5- isoquinolinesulfonyl)-3-methylpiperazine was obtained as the di-HCl salt from Toronto Biochemicals, lot No. l-HW-143. See Table 1 for the supplier product numbers for each of these compounds. Samples of the two isomeric 1-(5-isoquinolinesulfonyl)-2- and -3- methylpiperazines, prepared by the following routes, were obtained from LC Services Corporation. The compound identified in this paper as H7 was prepared as shown in Scheme 1, by reaction of 5-isoquinolinesulfonyl chloride with the product resulting from introducing a single N-carbobenzyloxycarbonyl (CBZ) blocking group onto 2- methylpiperazine, followed by removal of the CBZ group with HBr in acetic acid. The compound identified herein as Iso-H7 was prepared as shown in Scheme 2, by direct condensation of 5-isoquinolinesulfonyl chloride and 2-methylpipgpine; this follows the route gtven as tllustrattve of the syntheses of H7 by Htdaka (1). [ P]ATP was purchased from New England Nuclear and other reagents used were the best available grade from standard commercial sources.
NMR Suectrosconv. Proton NMR spectra were recorded at room temperature on a Varian XL-400 fourier transform NMR spectrometer at 400 MHz using D 0 as solvent. Samples of H7 in the free base form were converted to the di-hydroch f . ortde salt form prior to NMR analysis.
PKC Catalvtic Assavs. Purified rat brain PKC (a mixture of isotypes) was purchased from Lipidex. PKC catalytic activity was quantitated by the ability of the enzyme to phosphorylate histone III-S, as previously described (6). The final reaction mixture (100 ~1) co III-S, 0.33 PM [r- 3?
tained 50 mM Tris-HCl pH 7.0, 10 mM MgC12, 200 pg/ml histone P]ATP, appropriate amounts of the PKC isotype mixture, 50 pg/ml
phosphatidylserine and 1 mM CaC12. Separate control and comparison experiments were performed with 1 mM EGTA substituted for CaC12, in the absence of PS, and following the addition of phorbol esters. The inhibitors H7 and Iso-H7 were added at a concentration range of 0.1-300 PM. The reactions were maintained at 30° C for 5 minutes and then were terminated by addition of 35 ~1 of 25% TCA and 200 ~1 of 0.1% BSA. The samples were spun in an Eppendorf centrifuge and pellets were washed with 10% TCA. Pellets were dissolved in 30 ~1 NaOH, scintillation fluid was added, and the radioactivity was measured. Results are expressed as means +/- S.D. for 3-6 separate determinations.
658
Vol. 187, No. 2, 1992 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS
RESULTS
Schemes 1 and 2 depict the synthetic routes for the H7 and Iso-H7, respectively,
used in the experiments below. The structural assignments are based on the well-
accepted and precedented (7) presumption that the steric hindrance of the methyl group
in 2-methylpiperazine makes the l-nitrogen atom less reactive than the 4-nitrogen in the
present chemical reactions. Note that the numbering of the methyl group in the starting
2-methylpiperazine changes at several points in these routes. After the present work was
completed, similar syntheses were published by another laboratory (8).
The samples of H7 and Iso-H7 thus synthesized were then characterized by 400
MHz proton NMR. Fig. 1 shows expansions of the NMR region that differs most
noticeably between H7 and Iso-H7, namely the resonances for the 2-methylpiperazine
protons. The differences in chemical shift and splitting for H7 and Iso-H7 are slight but
nonetheless quite distinct under these high resolution NMR conditions.
After these standards were established, samples of H7 from all commercial
sources known to us were analyzed under the same NMR conditions. In addition, a
commercial sample of Iso-H7 from Toronto Research Chemicals was also studied. Table
1 summarizes the comparisons of NMR spectra from each of these samples. The NMR
spectrum of the compound synthesized in Scheme 1 exactly matched the spectra of
commercial “H7” samples from five suppliers, including Seikagaku. In contrast, the
spectrum of the compound synthesized in Scheme 2 exactly matched the spectrum of
“H7” from a sixth supplier, Sigma, and of a commercial sample labeled “l-(5-
isoquinolinesulfonyl)-3-methylpiperazine”, i.e. Iso-H7, from Toronto Research
Chemicals.
Based on the fact that Seikagaku H7 was recently found to be as potent (2) as
originally reported (1) for H7, we have designated the product of Scheme 1 as H7.
Consequently the product of Scheme 2 and, by NMR comparison, Sigma “H7”, are
designated as Iso-H7.
CBZ I
+ CBZ-Cl A
Scheme 1. Route used to synthesize H7.
659
Vol. 187, No. 2, 1992 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS
~O*Cl
Scheme 2. Route used to synthesize Iso-H7.
H N
5 4 3
(7
CH3
6 2
IT
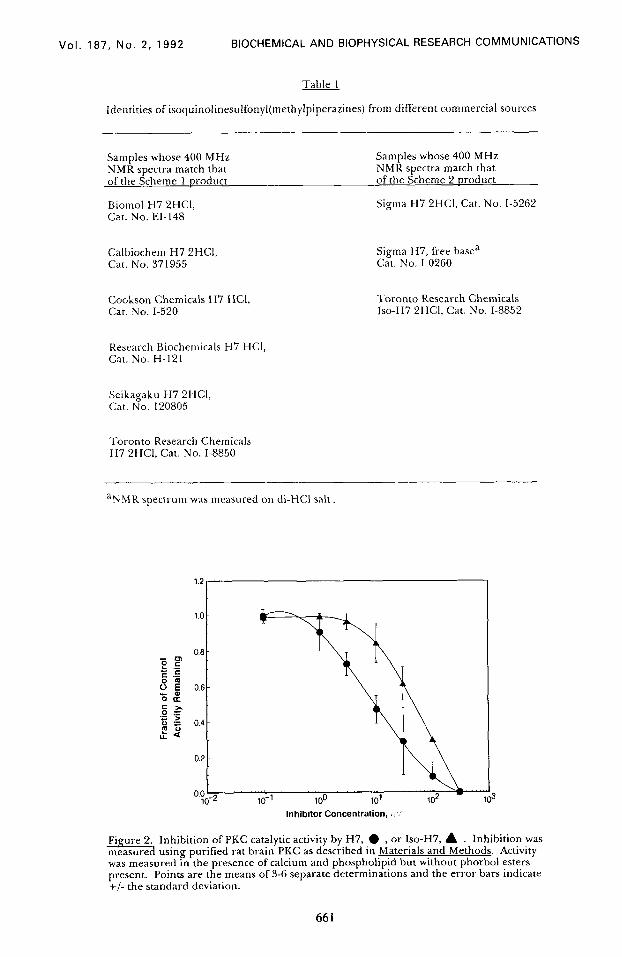
To verify the reported biological data, the products of Schemes 1 and 2 were
tested for their ability to inhibit purified rat brain PKC. Fig. 2 shows the concentration-
response curves for our samples of H7 and ISO-H7. As expected from previous reports
(2,3) in which Seikagaku’s H7 was compared to Sigma’s “H7”, identified here as Iso-H7,
authentic H7 (from Scheme 1) was distinctly more potent as a PKC inhibitor than was
Iso-H7 from Scheme 2. The IC59 values, 9 PM for H7 and 49 PM for Iso-H7, differed by
a factor of 5.4-fold, in good agreement with the 4-fold difference between Seikagaku and
Sigma “H7” products reported previously (2).
DISCUSSION
Selective inhibitors of PKC are currently much sought-after as tools for
elucidating the functions of this complex family of isotypes. H7, the properties of which
were first reported (1) in 1984, is one of the most widely used protein kinase inhibitors,
and a detailed knowledge of its biological properties is thus of considerable importance.
Fi me 1 7?ikc 400 MHz NMR spectra of (a) H7 and (b) Iso-H7. The traces show the region to 4.5 ppm relative to tetramethylsilane.
660
Vol. 187, No. 2, 1992 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS
Table 1
Identities of isoquinolinesulfonyl(methylpiperazines) from different commercial sources
Samples whose 400 MHz NMR spectra match that of the Scheme 1 oroduct
Biomol H7 2HCl, Cat. No. El-148
Samples whose 400 MHz NMR spectra match that of the Scheme 2 oroduct
Sigma H7 2HCl, Cat. No. I-5262
Calbiochem H7 2HCI, Cat. No. 371955
Sigma H7, free basea Cat. No. I-0260
Cookson Chemicals H7 HCI, Cat. No. I-520
Toronto Research Chemicals Iso-H7 2HCI, Cat. No. l-8852
Research Biochemicals H7 HCI, Cat. No. H-121
Seikagaku H7 2HCI, Cat. No. 120805
.I*oronto Research Chemicals H7 2HC1, Cat. No. I-8850
a,NMR spectrum was measured 011 di-HCI salt,
Inhibitor Concentration, ,A’
Fi ure 2. Inhibition of PKC catalytic activity by H7, l , or Iso-H7, A . Inhibition was %. measure usmg purified rat brain PKC as described in Materials and Methods. Activity was measured in the presence of calcium and phospholipid but without phorbol esters present. Points are the means of 3-6 separate determinations and the error bars indicate +/- the standard deviation.
Vol. 187, No. 2, 1992 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS
The present work clearly demonstrates that two different chemical compounds
have been designated as H7. Furthermore, this and previous studies (2,3) have shown
that these two compounds differ in their ability to inhibit PKC.
Interpretation of the results in at least five reports (9-13) may have been
adversely affected by the unrecognized substitution of Iso-H7 for H7. In each of these,
Iso-H7 (Sigma’s H7) did not inhibit the effects of known PKC activators such as phorbol
esters or teleocidin at concentrations consistent with H7’s reported potency as a PKC
inhibitor. Based on these observations, the studies concluded that some effects of the
phorbol esters and teleocidin may be indenendent of PKC. It now appears that these
conclusions should be reevaluated; those effects designated as “PKC-independent” effects
may instead reflect the low potency of Iso-H7 as a PKC inhibitor. It is also possible that
Iso-H7 fails to inhibit one or more isotypes of PKC, making it inactive as an inhibitor of
biological effects putatively mediated solely by such isotypes.
Moreover, to our knowledge the effects of Iso-H7 on non-PKC kinases have not
been reported. This is an important question because the original report (1) on the
kinase inhibitory properties of the isoquinolinesulfonamide series showed that very small
structural changes in the amide portion of the inhibitors led to large changes in
inhibitory profiles against a variety of kinases. In the absence of kinase specificty data, it
is difftcult to interpret the results obtained for Iso-H7 in various experimental systems.
In addition to the problems associated with the unrecognized substitution of Iso-
H7 for H7 in many published experiments, numerous reports indicate that the
pharmacological properties of both H7 and Iso-H7 are potentially quite complex. On
one hand, as would be expected, several studies (14-16) confirm that Seikagaku H7 has
an IC50 of about 6 PM in blocking functions of intact cells thought to be mediated by
PKC, and in each of these studies the negative control compound HA-1004 was inactive
or nearly so. The potency of H7 in these reports was the same as that originally reported
for H7 as a direct PKC inhibitor (1).
On the other hand, authentic H7 has failed to block several biological responses
induced by phorbol esters, such as PMA-enhanced potentiation of prostaglandin E2
release (17) and PMA-stimulated phosphatidylethanol production (18). These results
suggest that either authentic H7 does not inhibit one or more PKC isotypes or that PMA
can induce biological effects via pathways that do not require PKC catalytic activity.
Furthermore, other laboratories have found several instances of unexpectedly
good potency for Iso-H7. In one (19), Iso-H7 (i.e. Sigma H7) showed a potency of5 $kf
in inhibiting human natural killer cell activity, an assay in which HA-1004 was active
only at an extremely high concentration. In another study (20), 5 PM Iso-H7 inhibited
poly(rI).poly(rC)-induced interferon-/? production. The latter study did not include a
negative control. These results indicate that Iso-H7 may potently inhibit a non-PKC
target, or that it may specifically inhibit a PKC isotype with a potency mu& greater than
that seen for Iso-H7 inhibition of other PKC isotypes.
Obviously, many of the questions raised by the disparate results observed for
both H7 and ISO-H7 in numerous experimental systems could be clarified by studying
662
Vol. 187, No. 2, 1992 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS
their inhibitory properties on the known PKC isotypes in pure form. For H7, at least
one PKC isotype has been tested: PKC-/I1 was inhibited by Seikagaku H7 with an IC50
of 10 PM (2 1). Iso-H7 was found to inhibit three chromatographically purified rat brain
PKC isotypes with roughly equal IC5O’s of 60 PM (22).
ACKNOWLEDGMENTS
We thank LC Services Corporation for providing H7, Iso-H7 and the details of
their synthesis and characterization. The assistance of Ms. Cai Ping of Boston University
in running 400 MHz proton NMR spectra and the technical assistance of Ms. Ellen
Johnson are gratefully acknowledged. This work was supported in part by grant No.
HL38820 from the NIH and a Research Career Development Award (HL02271), both to
JAW.
REFERENCES
1. Hidaka, H., Inagaki, M., Kawamoto, S., and Sasaki, Y. (1984) Biochemistry - USA 23, 5036-5041. 2. Kischel, T., Harbers, M., Stabel, S., Borowski, P., Mullet-, K. and Hilz, H. (1989) Biochem. Biophys. Res. Comm. 165,981-987. 3. Lacombe, D., Darcy, K. and Ip, M. (1991) Proc. AACR 32,208. 4. Hidaka, H., Sone, T., Sasaki, Y. and Sugihara, T. (1985) U.S. Patent No. 4,560,755. 5. Hidaka, H., Sone, T., Sasaki, Y. and Sugihara, T. (1982) European Patent Application 82 10229.0, Publication No. 0,06 1,673. 6. Grabarek, J., Raychowdhury, M., Ravid, K., Kent, K.C., Newman, P.J. and Ware, J.A. (1992) J. Biol. Chem. 267, 1001 l-10017. 7. Beck, K., Hamlin, K. and Weston, A. (1952) J. Am. Chem. Sot. 74, 605-608. 8. Morikawa, A, Sone, T. and Asano, T. (1992) Chem. Pharm. Bull. 40, 770-773. 9. Kaneko, Y., Toda, G. and Oka, H. (1987) Biochem. Biophys. Res. Commun. 145,549- 555. 10. Villalobos-Molina, R., Ransanz, V., Torres-Marquez, M. Hong, E. and Garcia-Sainz, J. (1990) Biochem. Biophys. Res. Commun. 171, 618-624. 11. Murphy, J., Yaxley, J. and Norton, J. (1991) Biochim. Biophys. Acta 1092, 110-l 18. 12. Keller, H., Niggli, V., Zimmerman, A. and Portmann, R. (1990) J. Cell. Sci. 96, 99- 106. 13. Keller, H. (1990) J. Cell. Physiol. 145,465-471. 14. Clark, R., Love, T., Sgroi, D., Lingenheld, E. and Sha’afi, R. (1987) Biochem. Biophys. Res. Commun. 145, 666-672. 15. Ito, M., Tanabe, F., Sato, A., Takami, Y. and Shigeta, S. (1988) Int. J. Immunopharmacol. 10,211-216. 16. Wepsic, H., DufIie, G. and Ellis, N. (1989) Immunopharmacol. Immunotoxicol. 11, 81-99. 17. Donati, D., Baldari, C., Macchia, G., Massone, A., Telford J. and Parente, L. (1990) J. Immunol. 145,4115-4120. 18. Cao, Y., Reddy, C. and Mastro, A. (1990) Bioch. Biophys. Res. Commun. 171, 955- 962. 19. Steel, T. and Brahme, Z. (1988) J. Immunol. 141, 3164-3169. 20. Thacore, H., Lin, H.-Y., Davis, P. and Schoenl, M. (1990) J. Gen. Virol. 71, 2833- 2839. 21. Rotenberg, S., Krauss, R., Borner, C., and Weinstein, I. (1990) Biochem. J. 266, 173- 178. 22. Pelosin, J.-M., Keramidas, M., Souvignet, C. and Chambaz, E. (1990) Biochem. Biophys. Res. Commun. 169, 1040-1048.