J Biol Chem. 2011 Dec 23; 286(51): 43611–43621.Published online 2011 Oct 25. doi: 10.1074/jbc.M111.288928
PMCID: PMC3243568
The Thiazide-sensitive NaCl Cotransporter Is Targeted for Chaperone-dependent Endoplasmic Reticulum-associated DegradationPatrick G. Needham, Kasia Mikoluk, Pradeep Dhakarwal, Shaheen Khadem, Avin C. Snyder, Arohan R.Subramanya, and Jeffrey L. Brodsky
From the Department of Biological Sciences, University of Pittsburgh,the Department of Medicine, Renal-Electrolyte Division, University of Pittsburgh School of Medicine, andthe Veterans Affairs Pittsburgh Healthcare System, Pittsburgh, Pennsylvania 15261
To whom correspondence should be addressed: 3550 Terrace St., S832 Scaife Hall, Pittsburgh, PA 15261., Tel.: Phone: 412-648-9399;Fax: 412-647-6222; E-mail: [email protected].
The thiazide-sensitive NaCl cotransporter (NCC, SLC12A3) mediates salt reabsorption in the distalnephron of the kidney and is the target of thiazide diuretics, which are commonly prescribed to treathypertension. Mutations in NCC also give rise to Gitelman syndrome, a hereditary salt-wasting disorderthought in most cases to arise from impaired NCC biogenesis through enhanced endoplasmic reticulum-associated degradation (ERAD). Because the machinery that mediates NCC quality control is completelyundefined, we employed yeast as a model heterologous expression system to identify factors involved inNCC degradation. We confirmed that NCC was a bona fide ERAD substrate in yeast, as the majority ofNCC polypeptide was integrated into ER membranes, and its turnover rate was sensitive to proteasomeinhibition. NCC degradation was primarily dependent on the ER membrane-associated E3 ubiquitin ligaseHrd1. Whereas several ER luminal chaperones were dispensable for NCC ERAD, NCC ubiquitination anddegradation required the activity of Ssa1, a cytoplasmic Hsp70 chaperone. Compatible findings wereobserved when NCC was expressed in mammalian kidney cells, as the cotransporter was polyubiquitinatedand degraded by the proteasome, and mammalian cytoplasmic Hsp70 (Hsp72) coexpression stimulated thedegradation of newly synthesized NCC. Hsp70 also preferentially associated with the ER-localized NCCglycosylated species, indicating that cytoplasmic Hsp70 plays a critical role in selecting immature forms ofNCC for ERAD. Together, these results provide the first survey of components involved in the ERAD of amammalian SLC12 cation chloride cotransporter and provide a framework for future studies on NCC ERquality control.
The thiazide-sensitive NaCl cotransporter (NCC) is expressed at the apical surface of the distalconvoluted tubule of the kidney, where it mediates the reabsorption of 5–10% of filtered sodium chloride
(1). NCC plays an important role in determining the final sodium chloride content of urine entering thecollecting system and, hence, is critical for long term blood pressure regulation. Inhibition of NCC throughthe administration of thiazide diuretics has been associated with substantial reductions in hypertension-associated morbidity and mortality. Moreover, loss-of-function mutations of NCC cause Gitelmansyndrome, an autosomal recessive salt-wasting disorder (2, 3). Recently, it was shown that the carrier statefor these loss-of-function mutations confers protection from the development of hypertension, stronglysuggesting that factors governing the activity of NCC are key determinants of essential hypertension in thegeneral population (4–6).
NCC is a member of the SLC12 cation chloride cotransporter family of electroneutral transporters; othermembers include two Na K -2Cl cotransporters (NKCC1 and NKCC2) and four K -Cl cotransporters(KCC1–4) (7). These membrane proteins are important for the regulation of cell volume and ionhomeostasis in a variety of cell types, including neurons, erythrocytes, and diverse epithelia (7). The genesencoding these transporters are highly homologous and are predicted to share a complex topologyconsisting of 12 transmembrane domains, a sizable exofacial loop containing N-glycosylation sites, andlarge cytoplasmic amino and carboxyl termini flanking the transmembrane domains. Many of themutations that cause Gitelman syndrome are associated with reduced plasma membrane expression and ashift in the equilibrium of NCC expression toward its immature, glycosylated form (8–10). This findingimplies that many loss-of-function mutations that give rise to Gitelman syndrome cause the cotransporterto become misfolded within the early biosynthetic pathway, leading to the recognition of NCC by ERquality control mechanisms and degradation. Despite the relevance of NCC biogenesis to hypertension andthe pathogenesis of Gitelman syndrome, the molecular mechanisms involved in the quality control of NCCremain undefined.
Both wild type and mutant versions of membrane proteins with complex topologies, such as NCC, aresubject to ER quality control and degradation. For example, a significant portion of wild type cysticfibrosis transmembrane conductance regulator (CFTR) is selected for ER-associated degradation (ERAD)(11, 12), as is the epithelial sodium channel, ENaC (13–16). However, the deletion of a phenylalanine atposition 508 in CFTR is sufficient to divert nearly all of the protein to the ERAD pathway, which givesrise to cystic fibrosis (11, 12). In general, the selection of these and other proteins for ERAD requiressubstrate-specific interactions with molecular chaperones, such as members of the heat shock protein 70(Hsp70) and 40 (Hsp40) families (17). After chaperone-mediated selection in the ER lumen and/orcytoplasm, components of the ubiquitin-conjugation and ligation systems are recruited and append apolyubiquitin chain onto cytoplasmic domains of the substrate. Polyubiquitinated substrates are thenextracted from the ER by the Cdc48-p97 complex, which couples ATP hydrolysis to retro-translocation.Ultimately, the ERAD substrate is delivered to the proteasome and degraded. At present, it is impossible topredict a priori which of the many chaperones in mammalian cells might play a role in the selection of aspecific ERAD substrate, like NCC. This problem is compounded by the fact that distinct chaperones maybe involved in protein folding as well as degradation (18). Nevertheless, a thorough definition ofchaperone-substrate interactions for proteins such as NCC is critical; efforts to alter chaperone activity areunder way based on their proven ability to alter the conformations and promote the trafficking of disease-causing mutant proteins (19–21).
To begin to define the pathway by which NCC is subject to quality control, we employed yeast as a modelexpression system to compare mammalian NCC processing in wild type strains and in strains with targetedmutations of select components of the ERAD pathway. We verified that NCC is a bona fide ERADsubstrate in yeast, allowing us to co-opt the system to identify components of the ubiquitination machinery
and the chaperones that target NCC for degradation. Using genetic and biochemical tools, we find that oneof these chaperones, the cytoplasmic Hsp70, facilitates NCC polyubiquitination. We then utilize amammalian renal cell model for NCC and show that mammalian cytoplasmic Hsp70 selects NCC forERAD. Together, these data establish NCC as an ERAD substrate and identify Hsp70 as a critical mediatorof cation chloride cotransporter quality control.
EXPERIMENTAL PROCEDURES
A summary of the Saccharomyces cerevisiae strains used in this study isprovided in supplemental Table S1.
All NCC clones used in these studies were derived from previously described and characterized cDNAs,including untagged mouse NCC (8), N-terminal hemagglutinin-tagged mouse NCC (HA-NCC) (22), and amouse NCC construct containing a double HA tag in the cotransporters second extracellular loop (2XHA-NCC) (22). To express NCC in yeast, the coding sequence was excised from pcDNA3.1 (see below) withEcoRI and ligated into the same site in the multicopy, uracil-selectable plasmid, pRS426GPD (23), inwhich expression is driven from a modest, constitutive promoter. Yeast were transformed with the plasmidusing a standard lithium acetate procedure, and colonies were selected on synthetic complete medialacking uracil (24). For experiments in mammalian cells, untagged NCC was subcloned from pgh19 (8)into the EcoRI site of pcDNA3.1. pRK5-HA-ubiquitin (25) was a gift of Paul Welling (University ofMaryland School of Medicine, Baltimore, MD), and human Hsp70 (Hsp72, HspA1A) in pcDNA3 was agift from Ron Rubenstein (University of Pennsylvania School of Medicine, Philadelphia, PA). An in-frameN-terminal myc epitope was added to the Hsp70 construct via PCR and subcloned into pMO-myc (22).Site-directed mutagenesis was performed using a PCR-based strategy with PfuTurbo DNA polymerase(QuikChange; Agilent).
The following commercial antibodies were used: mouse monoclonal anti-HA (HA-11, Covance),mouse monoclonal anti-myc (4A6, Millipore), rabbit polyclonal anti-c-myc (A-14, Santa Cruz), mousemonoclonal anti-human Hsp70 (Hsp72) (C92F3A-5, Stressgen/Enzo Life Sciences), rabbit polyclonal anti-tubulin (Sigma), horseradish peroxidase (HRP)-conjugated goat anti-mouse and goat anti-rabbit antibodies(Jackson ImmunoResearch), rabbit polyclonal anti-ubiquitin antibody (FL-76, Santa Cruz), and HRP-conjugated rat monoclonal anti-HA high affinity (3F10, Roche Applied Science). Polyclonal rabbit anti-mouse NCC antibody (26) was a gift from David Ellison (Oregon Health and Science University, PortlandOR), polyclonal rabbit anti-Pdi1 was provided by Vlad Denic (Harvard University, Cambridge, MA), andpolyclonal rabbit anti-Pma1 was a gift from Amy Chang (University of Michigan, Ann Arbor, MI).Polyclonal rabbit anti-Sec61 (27) and polyclonal rabbit anti-Kar2/BiP (28) were previously described.
Yeast cells transformed with the NCC expression plasmid were grown insynthetic complete media lacking uracil at 26 °C to an A of 0.8–1.0. These log-phase cultures were thentransferred to a water bath at either 30 °C or 37 °C (for assays using temperature-sensitive mutant strains).To stop protein translation, cycloheximide was added to a final concentration of 100 µg/ml, and aliquotswere removed at 0, 15, 30, and 60 min. Cells were isolated by centrifugation at 4 °C, quick-frozen, andstored at −80 °C, the cell pellets were thawed on ice, and total protein was TCA-precipitated as described(29). The protein samples were analyzed by SDS-PAGE and immunoblot analysis with HRP-conjugatedanti-HA antibody (Roche Applied Science). NCC was visualized using ECL (Pierce) and a Kodak 440CFimage station, and the data were quantified with Kodak 1D software. The amount of protein at the start ofthe chase (time 0) was set to 100%, and the amounts of protein at subsequent time points were expressedas a percent of the starting material.
Preparation of Mammalian Cell Lysates and Immunoblot Analysis
The integration of NCC into the yeast ER membrane was analyzed bycarbonate extraction (30). In brief, yeast microsomes were prepared as previously described (28) andincubated with ice-cold 100 mM Na CO , pH 13, or buffer 88 (20 mM HEPES, pH 6.8, 150 mM KOAc, 5mM MgOAc, 250 mM sorbitol) on ice for 30 min. After incubation, the membranes were centrifuged at~100,000 × g in a Sorvall RC M120EX centrifuge for 60 min at 4 °C. The supernatant was removed, andthe pellets were washed with buffer and centrifuged again for 15 min. The final pellets were solubilized inSDS sample buffer, and the pellet and supernatant fractions were subjected to SDS-PAGE and immunoblotanalysis with anti-HA antibody to detect NCC and anti-Sec61 and anti-Pdi1 antibodies to detect integralmembrane and soluble ER proteins, respectively.
The intracellular residence of NCC was determined by sedimentation in a sucrose gradient essentially asdescribed (31). A 30-ml culture was grown to an A of 0.8, and the cells were harvested bycentrifugation and then disrupted with agitation on a Vortex mixer with glass beads. The cell lysates werecleared of debris by low speed centrifugation, and the resulting lysate (300 µl) was layered on the top of an11-ml 30–70% sucrose gradient and centrifuged at 100,000 × g in a Beckman SW41 rotor for 14 h at 4 °C.Where indicated, the gradients either contained EDTA or Mg to release or maintain, respectively, theassociated ribosomes. Fractions were collected by pipeting from the top of the tube, and proteins wereanalyzed by SDS-PAGE and immunoblotting for NCC with anti-HA antibody. Specific antisera against theER (Kar2/BiP) and the plasma membrane (Pma1) resident proteins were also used (see above).
Yeast cells expressing NCC were grown to A of 0.8 at 26 °C. For experimentsemploying E3 ubiquitin ligase mutant strains, the cells were isolated at room temperature in a clinicalcentrifuge, washed 1 time in sterile water, and quick-frozen for storage at −80 °C. For experiments in theHsp70/Ssa1 mutant strains, the cells were shifted to 37 °C for 15 min and then immediately cooled in anice-water bath, and a final concentration of 10 mM NaN was added. The cells were then isolated in aclinical centrifuge and quick-frozen and stored at −80 °C. The preparation of cell lysates,immunoprecipitation of NCC, and analysis of conjugated ubiquitin were performed essentially asdescribed for CFTR (32). In brief, cells were disrupted by glass bead lysis, and membranes were pelletedby centrifugation. Membranes were treated with SDS buffer to liberate the integral membrane proteins,and the solution was diluted into a Triton X-100-containing buffer. Anti-HA antibody conjugated toagarose beads was added, and solutions were incubated overnight with rocking at 4 °C. The beads werewashed 3 times, and the bound proteins were liberated in SDS sample buffer at 37 °C for 30 min. Finally,the released proteins were subjected to SDS-PAGE and immunoblot analysis with anti-HA antibody todetect total NCC or anti-ubiquitin antibody to detect the ubiquitinated NCC fraction.
Madin Darby canine kidney (MDCK) cells were provided by Rebecca Hughey(University of Pittsburgh, Pittsburgh PA), and HEK-293T cells were donated by Kenneth Hallows(University of Pittsburgh, Pittsburgh, PA). Cells were cultured in high glucose Dulbecco's modified Eagle'smedium supplemented with 10% FBS, L-glutamine, and penicillin/streptomycin at 37 °C. Transienttransfections were performed using either FuGENE 6 (Roche Applied Science) or Lipofectamine 2000(Invitrogen) per the instructions of the manufacturer, and cells were subjected to analysis 24–48 h post-transfection.
Cells were washed twice with phosphate-buffered saline (PBS), scraped, collected, and isolated by centrifugation at 1000g for 5 min. Post-nuclearlysate supernatants were obtained by passing the pellets 25 times through a 20–200-µl pipette tip in 1 of 3lysis buffers depending on the experiment: cell lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM
Na EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1
Metabolic Labeling and Pulse-Chase Studies in Mammalian Cell Lines
Deglycosylation Studies
NCC Co-immunoprecipitation Analysis
mM Na VO , and 1 µg/ml leupeptin, 1 mM PMSF), radioimmune precipitation assay buffer (identical tocell lysis buffer except Triton X-100 was replaced with 1% Nonidet P-40 and 1% sodium deoxycholate),or detergent solution (50 mM Tris-HCl, pH 8.5, 1% Nonidet P-40, 0.4% sodium deoxycholate, and 62.5 mM
EDTA supplemented with 1 tablet Roche Complete Protease Inhibitor Mixture and 1 mM PMSF). Thesamples were incubated on ice for 15 min, and insoluble material was removed by centrifugation at 16,000× g for 5 min. Protein concentrations were determined by the Bradford method (Bio-Rad protein assay kit).Lysates were diluted according to the instructions of the manufacturer, so that the detergents in the lysisbuffers would not interfere with the protein assay. For samples subjected to SDS-PAGE, lysates weredenatured in Laemmli buffer, maintained at room temperature for 30 min, and loaded in a 4 °C cold roomonto 10% polyacrylamide gels preincubated with chilled SDS buffer, and the proteins were resolved at 4°C on the polyacrylamide gels. Immunoblot analysis was performed as described previously (22).
HEK293T or MDCK cells expressing N-terminal HA-tagged NCC in 6-well plates were assayed 24 h post-transfection. Cells were washed twicewith prewarmed starving medium (cysteine/methionine-free high-glucose DMEM supplemented with 10%dialyzed FBS, penicillin/streptomycin, and glutamine) and incubated in fresh starving medium in a 5%CO incubator at 37 °C for 30 min. The cells were then washed twice on ice with ice-cold PBS andmetabolically labeled at 37 °C in the CO incubator in starving medium containing 150 µCi/ml Translabel(MP Biomedicals) for 30 min. The radiolabel was aspirated from the samples, and the cells were washedtwice in prewarmed chasing medium (complete high glucose DMEM supplemented with 10 mg/ml each ofunlabeled methionine and cysteine). The cells were then “chased” in the incubator at 37 °C for theindicated times and washed on ice 3 times with ice-cold PBS. The cells were then lysed in 400 µl ofradioimmune precipitation assay buffer/well by adding the lysis buffer directly to the plates and rockingthe cells at room temperature for 1 h. Lysates were collected into microcentrifuge tubes and clarified bycentrifugation at 1000 − g for 5 min. HA-NCC was then immunoprecipitated from the cleared lysates byadding 1 µg of HA-11 antibody and incubating the solution overnight at 4 °C with end-over-end rotation.NCC-antibody complexes were subsequently precipitated from the sample by adding 30 µg of protein A/Gagarose slurry and rotating the mixture at room temperature for 1 h. The beads were washed five times in500 µl of PBS, and the immunoprecipitates were eluted from the beads in 5× Laemmli buffer andfractionated by SDS-PAGE. The gels were dried (Dry-Ease gel drying kit, Invitrogen), and radiolabeledNCC was detected with a Personal Molecular Imager system (Bio-Rad) using a TR phosphorimagingscreen.
Endoglycosidase H and peptide N-glycosidase F were obtained from New EnglandBiolabs. HEK293 cells expressing HA-NCC were metabolically labeled for 2 h in starving mediumcontaining 150 µCi/ml Translabel, cell lysates were prepared, and NCC was immunoprecipitated asdescribed above. For both reactions, immunoprecipitates were eluted from the anti-HA immunoaffinityresin by adding 10 µl of 1× glycoprotein denaturing buffer (New England Biolabs, included with theendoglycosidase H and peptide N-glycosidase F enzymes) and heating the samples at 90 °C for 2 min. Theeluates were cooled to room temperature and adjusted to a final volume of 20 µl containing either 2500units of endoglycosidase H or 1000 units of peptide N-glycosidase F and their requisite buffers per theinstructions of the manufacturer. Samples were incubated at 37 °C with endoglycosidase H for 3 h orpeptide N-glycosidase F for 1 h before SDS-PAGE and autoradiography as described above.
Cells transiently expressing the indicated constructs were lysed in celllysis buffer as described above. A total of 300 µg of the lysate was diluted to a total of 300 µl andprecleared with 30 µl of protein A/G-agarose slurry (Calbiochem) by end-over-end rotation at 4 °C for 2 h.
NCC Can Be Expressed in the Yeast, S. cerevisiae, and Resides Predominantly in the ER Membrane
Next, the indicated amount of antibody was added to the precleared lysates, and the samples were rotatedin fresh 30-µl aliquots of protein A/G beads overnight at 4 °C. The samples were then centrifuged at lowspeed, and the beads were washed once in 500 µl of PBS, twice in 500 µl of PBS containing 0.5% TritonX-100, and once more in 500 µl of PBS. Immunoprecipitated proteins were eluted by incubating the beadsat room temperature for 30 min in 5× Laemmli buffer, separated by SDS-PAGE, and analyzed byimmunoblotting as described above.
For Western blot quantification, densitometry was carried out with NIH ImageJ software. Bio-Rad QuantityOne was used for analysis of phosphorimaging data. GraphPad Prism software was used forstatistical analyses. Comparisons between two groups were determined by a Student's t test.
RESULTS
We previouslydeveloped yeast expression systems to define and characterize the ERAD requirements for severalclinically relevant mammalian ion channels, including the chloride channel CFTR (29, 32–34) and thetrimeric sodium channel, ENaC (13, 14). The yeast system can then be exploited to identify the uniquedegradation requirements for each substrate by examining the protein fate in cells mutated for specificgenes, such as those required for ERAD (35). These include both cytosolic and ER luminal chaperones, E3ubiquitin ligases, and components required to retrotranslocate substrates from the ER to the cytoplasm fordelivery to the proteasome. Results from the yeast system can subsequently be confirmed in mammaliancells, thus establishing this model organism as a tool to expedite gene discovery. For example, weidentified the contributions of small heat shock proteins (Hsps) during the ERAD of CFTR and ENaC inboth yeast and vertebrate/mammalian cell systems (13, 32). Recently, we identified a role for two ERluminal Hsp40s, Scj1 and Jem1, during the selection and ERAD of ENaC in yeast; the homologous humanluminal Hsp40s were similarly required to facilitate the destruction of the ENaC subunits, as assessed in anXenopus oocyte expression system (14).
Previous observations of NCC biogenesis in heterologous expression systems, including Xenopus laevisoocytes and mammalian cells, indicate that the cotransporter exists in two glycosylated species at steadystate: a core (high mannose) endoglycosidase H-sensitive band that migrates at 110 kDa and a matureendoglycosidase H-resistant band that resolves more broadly between 110–150 kDa (8, 36, 37). Both ofthese species have also been observed in the mammalian kidney (8). Because high mannose glycospeciesreside within the biosynthetic pathway proximal to the pre-medial Golgi (38), these observations suggestthat NCC is substantially ER-localized and subject to ER quality control mechanisms that monitor andfacilitate its folding or degradation. To test this hypothesis and to identify and characterize the factorsunderlying NCC ERAD, we developed a yeast expression system to study NCC processing. To facilitateprotein detection, an NCC construct with a double HA tag incorporated into the second extracellular loop(2XHA-NCC) was employed. Notably, the tagged and untagged versions of NCC are equally efficient atfacilitating Na uptake, demonstrating that the inclusion of the epitope does not alter NCC transportcharacteristics (22). Therefore, this 2XHA-NCC cDNA was subcloned into a yeast expression vector, andNCC expression was verified in various yeast strains (see “Experimental Procedures”; data not shown).
To determine the subcellular localization of NCC in yeast, we subjected whole cell extracts to sucrosegradient centrifugation (see “Experimental Procedures”). In the absence of Mg and presence of EDTA,this technique separates ER-derived membranes from plasma membranes. As seen in Fig. 1A, the ERresident chaperone Kar2/BiP resides in less dense fractions, whereas the plasma membrane resident, Pma1,migrates to denser fractions at the bottom of the gradient. We observed that essentially all of the NCC in
the gradient mirrored Kar2/BiP residence, strongly suggesting that NCC is predominately in the yeast ER.To confirm ER localization, we then performed the sucrose gradient centrifugation in the presence ofMg and absence of EDTA, which retains ribosomes on the ER membrane and leads to a characteristicshift of this organelle to denser regions of the gradient. As displayed in Fig. 1B, both Kar2/BiP and NCCexhibited a Mg -dependent shift, further supporting ER residence.
To confirm that NCC is integrated into the ER membrane and is not peripherally associated with the lipidbilayer, we isolated ER-enriched membranes and subjected them to a high pH, Na CO wash (see“Experimental Procedures:). Under these conditions, peripherally associated proteins are liberated from themembrane and reside in the supernatant after centrifugation, whereas integral membrane proteins pelletwith the membranes (30). After Na CO treatment, NCC remained in the pellet, as did an integralmembrane component of the translocation channel, Sec61 (Fig. 1C). In contrast, a soluble protein in theER, Pdi1, was released into the supernatant. Together, these data indicate that NCC integrates into the ERmembrane when expressed in yeast.
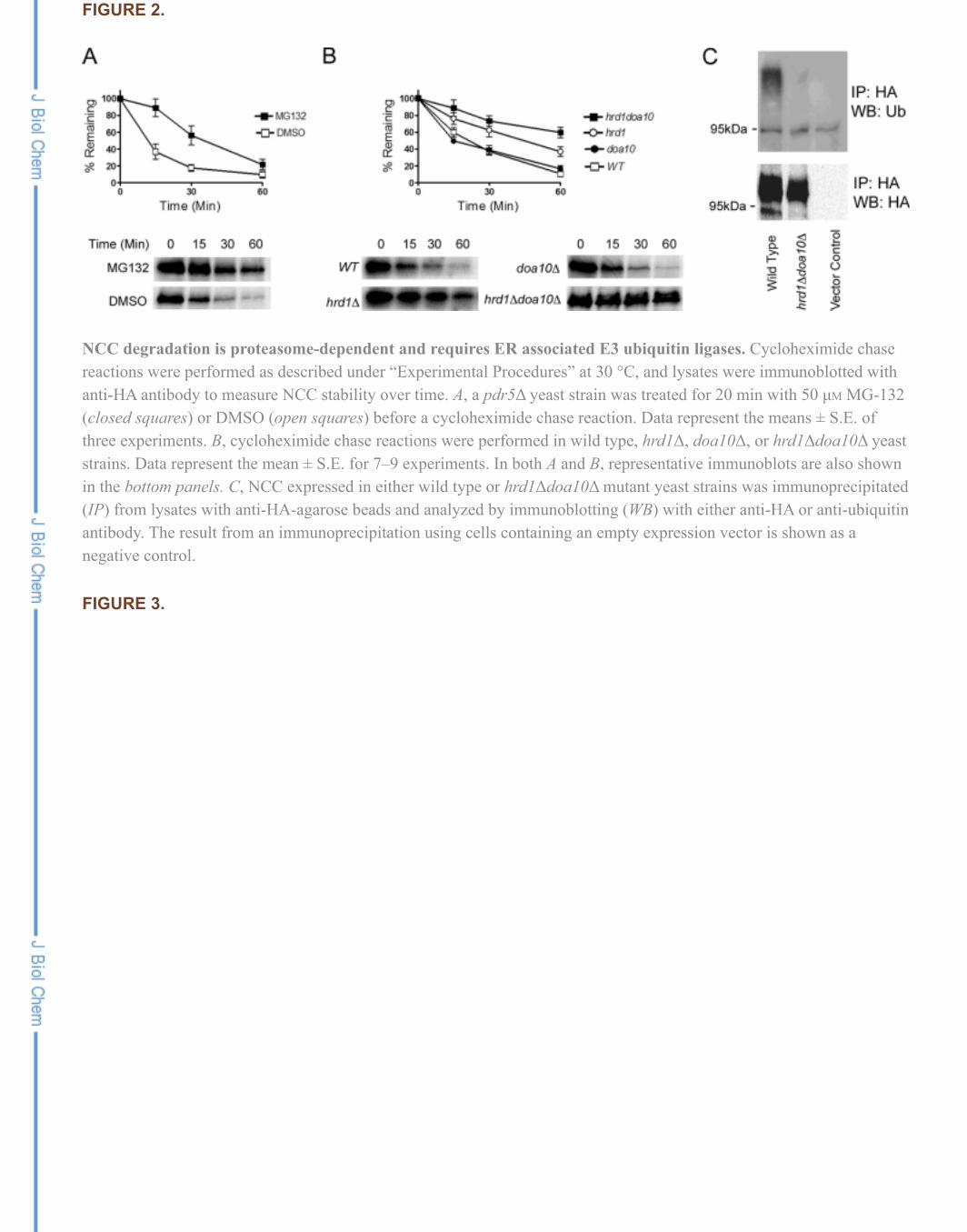
To determine whether NCC is targeted for ERAD, we performedcycloheximide chase assays to measure the rate of protein degradation after protein synthesis was arrested.We found that NCC was quite unstable in yeast, such that <10% of the starting material remained after 60min. To determine if the degradation of NCC is proteasome-dependent, which is a hallmark of ERAD, wefirst introduced the NCC expression vector into a pdr5Δ strain, which lacks a multidrug pump andfacilitates the uptake of the proteasome-specific inhibitor, MG-132 (39). As seen in Fig. 2A, NCC wassignificantly stabilized after cycloheximide addition in cells treated with MG-132 when compared withcells treated with the vehicle, DMSO. Interestingly, stabilization was most dramatic at early time points.Because MG-132 inhibits only one of the three protease activities of the proteasome (40), it is possible thatthe two uninhibited proteases act secondarily during degradation but eventually can compensate for theinhibited activity. It is also possible that NCC is shunted to the vacuole for degradation over time. To testthis hypothesis, we examined whether there was vacuole-dependent degradation by measuring NCCstability in a pep4Δ strain, which lacks nearly all vacuole-associated protease activity (41). However, NCCdegradation was robust in this strain (supplemental Fig. S1).
To provide further evidence that NCC is an ERAD substrate in yeast, cycloheximide chase assays wereperformed in strains lacking one or both of the two ERAD-requiring ubiquitin ligases, Hrd1 and Doa10. Asshown in Fig. 2B, NCC turn-over was identical in the wild type and doa10Δ strains but was significantlyslowed in the hrd1Δ strain. Moreover, NCC was stabilized further in the hrd1Δdoa10Δ mutant, suggestingthat Doa10 plays a secondary role during NCC degradation, possibly due to greater misfolding of thecytoplasmic domains of NCC, as it accumulates in the ER. Of note, a reciprocal effect was observed whenSte6* degradation was measured; here, ERAD was primarily Doa10-dependent and largely Hrd1-independent, but more pronounced stabilization was observed when both E3s were absent (42). To confirmthat the E3-deficient strains exhibited the expected phenotypes, we repeated these experiments using adifferent protein, a mutated form of carboxypeptidase Y known as CPY* (supplemental Fig. S2), a wellcharacterized ERAD substrate that shows Hrd1-dependent degradation (43). As anticipated, CPY*carboxypeptidase Y was exclusively Hrd1-dependent. We also established that the presence of the epitopetag did not lead to an artificial degradation signal by comparing the degradation rate of an HA-tagged to -untagged NCC in cycloheximide chase reactions. Notably, the degradation rates of both forms of NCCwere identical, and both NCC species exhibited E3-dependent degradation (supplemental Fig. S3).
Formally, the deletion of the E3s may have led to secondary effects, which might grossly alter ER and/orcellular homeostasis and slow the degradation rate of NCC. To exclude this possibility, we measured NCC
The Cytoplasmic Hsp70, Ssa1, Facilitates NCC Degradation
Ssa1 Function Contributes to NCC Ubiquitination in Yeast
ubiquitination after its immunoprecipitation from wild type and hrd1Δdoa10Δ yeast strains. As shown in Fig. 2C, NCC ubiquitination was essentially absent in the hrd1Δdoa10Δ mutant, whereas a strong signalcorresponding to NCC was observed in wild type cells. Based on the fact that NCC is ER-retained, isubiquitinated, and is degraded in a ubiquitin-proteasome-dependent manner, we can conclude that thisprotein is an ERAD substrate in yeast. In further support of this conclusion, we also found that NCCdegradation required the function of Cdc48, the AAA ATPase that extracts ubiquitinated substrates fromthe ER before their proteasome-dependent degradation (17) (supplemental Fig. S4).
Because the selection of ERAD substrates usuallyrequires specific chaperones, we next sought to identify the Hsp70s and/or Hsp40s that facilitate NCCdegradation. Both cytoplasmic and ER luminal chaperones have been identified as being important for thedegradation of various ERAD substrates in yeast; however, the exact make-up of the required chaperonesis substrate-specific and is often influenced by a substrate membrane topology (17). NCC contains 12transmembrane segments, 2 large cytoplasmic domains, and a large ER-exposed exofacial loop. Given thefact that the cotransporter contains domains in the ER and cytoplasm, NCC could potentially interact withchaperones on either side or on both sides of the ER membrane.
By utilizing temperature sensitive and deletion mutants, we analyzed NCC degradation when the functionsof the major Hsp70 and Hsp40 chaperones in the cytoplasm and in the ER were ablated. The outcome ofthese experiments is presented in Fig. 3. Overall, we found that the only chaperone that affected NCCdegradation was the cytoplasmic Hsp70, Ssa1. Specifically, after temperature shift to 37 °C, NCC wasmoderately stabilized in the ssa1–45 mutant compared with the wild type SSA1 strain (Fig. 3A). Somewhatsurprisingly, two functionally redundant cytoplasmic Hsp40s, Hlj1 and Ydj1 (33), were dispensable forNCC degradation (Fig. 3C). These results suggest that Ssa1 action is Hsp40-independent or may requiremembers of another chaperone class to promote maximal rates of NCC degradation. As an initial test ofthis hypothesis, we examined whether the deletion of the genes encoding the small Hsps, Hsp26 andHsp42, which contributed to CFTR and ENaC ERAD (13, 32), had an effect on NCC turnover; however,NCC degradation was unchanged between the wild type and hsp26Δhsp42Δ strains (supplemental Fig. S5).Finally, in contrast to the ability of a cytoplasmic chaperone to facilitate NCC turnover, neither the ERluminal Hsp70 Kar2/BiP nor the functionally redundant luminal Hsp40s, Scj1 and Jem1, contributed toNCC degradation (Fig. 3, B and D). As a control and consistent with published results (44), we confirmedthat carboxypeptidase Y was significantly stabilized in these strains (data not shown).
The stabilizing effect of compromised Ssa1 functionon NCC degradation could be due to a direct effect during substrate ubiquitination. Alternatively or inaddition, Ssa1 might act at a post-ubiquitination step, perhaps retaining NCC in a soluble conformationduring proteasome delivery (45–47). To determine if Ssa1 acts at a pre- and/or post-ubiquitination step, weimmunoprecipitated NCC from wild type SSA1 yeast and the temperature sensitive ssa1–45 mutant strainafter a brief temperature shift to inactivate the ssa1–45 protein (48). After only 15 min at 37 °C, theamount of HA-tagged NCC declined in the wild type cells but accumulated in the ssa1–45 mutant,consistent with the stabilization seen in the cycloheximide chase (compare Fig. 4A, IP: HA, WB: HA, to Fig. 3A, 15 min). Even though the amount of ubiquitination is comparable in the wild type and ssa1–45strains (Fig. 4A, IP: HA, WB: Ub), when this signal is normalized to the total protein (HA) signal, therelative amount of ubiquitinated NCC is 2.3-fold higher in wild type than in ssa1–45 mutant cells (p =0.004; Fig. 4B). The ssa1-dependent reduction in ubiquitinated NCC is absent if cells are grownexclusively at 26 °C (data not shown), demonstrating the specificity of the ssa1 mutant effect. Thereduction in ubiquitination under the examined conditions is consistent with Ssa1 acting primarily at a
NCC Is a Rapidly Degraded ERAD Substrate in Mammalian Cells
recognition step during NCC ubiquitination. These results are in accordance with other studies in whichthe level of ubiquitination of an integral membrane ERAD substrate was shown to be decreased in yeastcontaining mutant forms of Ssa1 (46, 49).
To verify the relevance of the observationsobtained in the yeast model, we analyzed NCC expression, proteasomal degradation, and ubiquitination inmammalian cells. First, a metabolic labeling pulse-chase assay was employed to specifically examinenascent cotransporters that undergo processing in the ER immediately after biosynthesis. To monitor thisnewly produced pool, HEK293 cells expressing N-terminal HA-tagged NCC were pulse-labeled with[ S]cysteine and [ S]methionine. The radiolabeled cotransporter was then immunoprecipitated fromwhole cell extracts with anti-HA antibodies, fractionated by SDS-PAGE, and detected by autoradiography.Using this approach, we identified a radiolabeled band that migrated at 110 kDa (supplemental Fig. S6),consistent with the core-glycosylated, ER-localized immature form of NCC (8, 36, 37). To verify that thisband corresponded to core NCC, we performed deglycosylation reactions of radiolabeled NCC in HEK293cells (supplemental Fig. S6). Cells were pulse-labeled with S, and anti-HA-NCC immunoprecipitateswere subjected to treatment with the N-glycosidases endoglycosidase H (which selectively cleaves highmannose core N-glycans), or peptide N-glycosidase F, which cleaves both mature and core N-glycans). Aspredicted, the 110-kDa band shifted slightly in response to treatment with both enzymes, confirming thatthis band corresponds to core NCC (supplemental Fig. S6, left panel). Of note, we observed that themature endoglycosidase H-resistant glycosylated species tended to migrate closely to the immature form asa low abundance smear of about 110–130 kDa. This species was visible in both in immunoblots ofHEK293 cell lysates (Fig. 5B, right panel) and in metabolic labeling studies after longer exposures of theimaging screen (supplemental Fig. S6, right panel). These findings are compatible with previous reportsby other groups that have employed HEK293 cells as an expression system to study NCC expression andregulation (50, 51). These results indicate that when expressed in mammalian cells, NCC predominantlyexists in its core glycosylated state, suggesting that the cotransporter undergoes extensive biosyntheticprocessing in the ER and is strongly sensitive to chaperone-dependent ERAD.
Despite the inefficient maturation of NCC in HEK293 cells, we found that the wild type cotransporter wasstill processed more efficiently than several NCC mutants harboring missense or nonsense mutationsknown to cause Gitelman syndrome. These NCC mutants (R948X, G738R, R989X, and T997I) werepreviously characterized in an amphibian expression system and exhibit reduced sodium transport activitydue to impaired biosynthetic processing (8–10). We confirmed that these mutant proteins behavedsimilarly in HEK293 cells cultured at physiologic temperature, as they exhibited a substantial reduction inthe steady state abundance of mature NCC as assessed by immunoblot analysis (supplemental Fig. S7). Forexample, the R989X mutant, which manifested the most mature glycosylation of the four Gitelmanmutants tested, exhibited an apparent maturation efficiency of only 12% compared with 31% for the wildtype protein. These data indicate that although wild type NCC is inefficiently processed in mammalian cellculture systems, it still undergoes productive folding and maturation to a greater extent than defective loss-of-function mutants known to cause human disease through impaired cotransporter biogenesis.
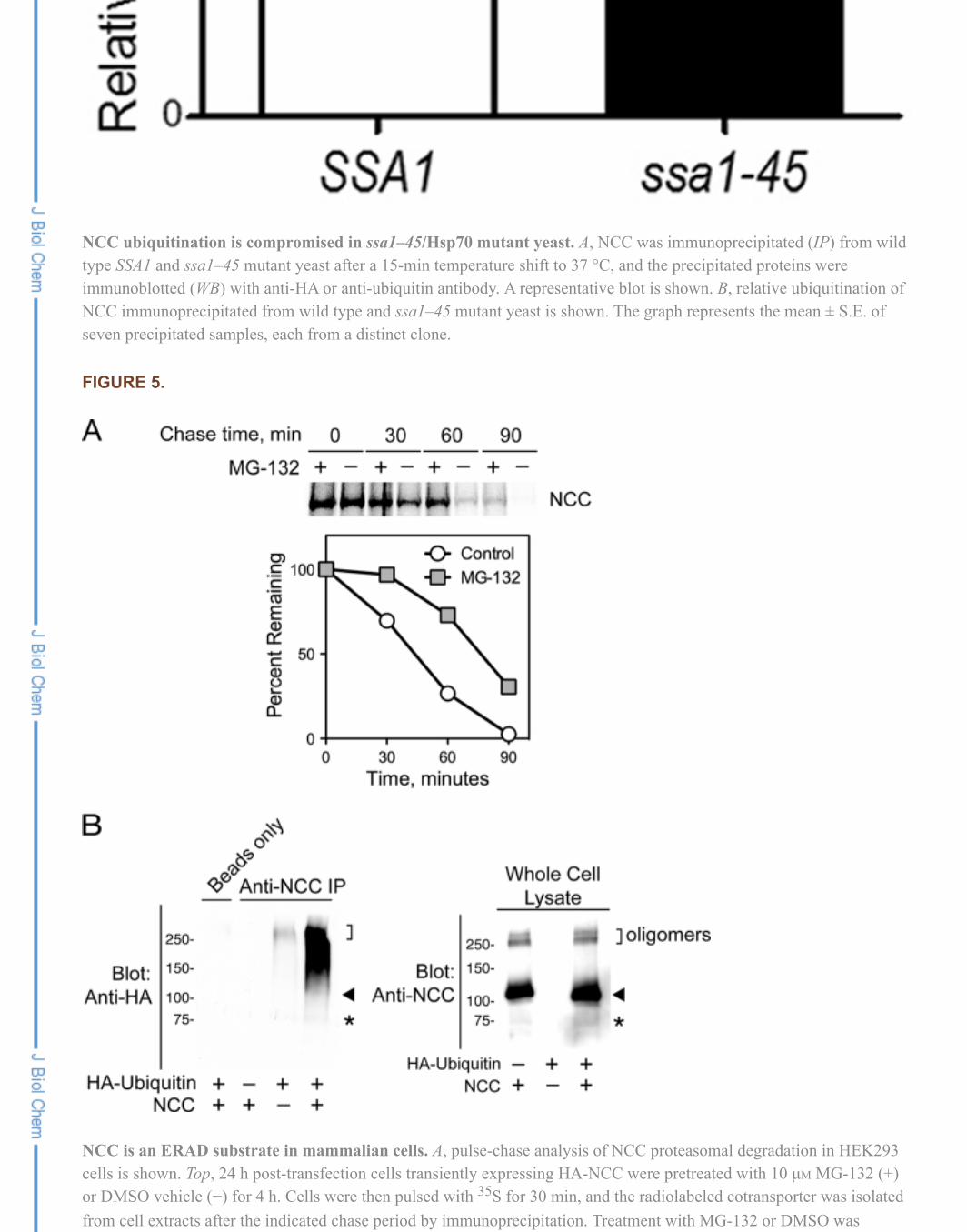
To determine the effect of proteasome inhibition on the degradation of newly synthesized NCC, HEK293cells expressing the cotransporter were pulse-labeled in the absence or presence of MG-132 and weresubsequently chased with excess unlabeled cysteine and methionine for the times indicated in Fig. 5A. Incontrol (DMSO vehicle-treated) cells transfected with NCC, the cotransporter was rapidly degraded over90 min. In contrast, the NCC degradation rate was attenuated substantially in cells that were pretreated,pulsed, and chased in the presence of 10 µM MG-132. Notably, the effect of MG-132 on the NCC decay
Cytoplasmic Hsp70 Promotes NCC ERAD in MDCK Epithelia
rate was analogous to our observations of NCC degradation in yeast (Fig. 2A), with higher MG-132effectiveness occurring at earlier points in the time course. These data confirm that the cotransporter isdegraded rapidly by the proteasome in mammalian cells, with most newly synthesized NCC undergoingERAD before export to the Golgi.
To confirm that NCC is polyubiquitinated in mammalian cells, we performed a coimmunoprecipitationexperiment in HEK293 cells containing either an empty vector control or expressing an untagged versionof NCC with or without a second plasmid encoding an HA epitope-tagged ubiquitin cDNA. Whole celllysates from these cells were immunoprecipitated with a previously characterized rabbit anti-mouse NCCpolyclonal antibody (see “Experimental Procedures”). The samples were subsequently fractionated bySDS-PAGE and subjected to anti-HA immunoblotting. In cells transfected with HA-ubiquitin alone, a fainthigh molecular weight signal was seen, consistent with the nonspecific precipitation of HA-ubiquitinatedproteins by the polyclonal NCC antibody. By contrast, in cells cotransfected with both NCC and HA-ubiquitin, immunoprecipitation with the anti-NCC antibody yielded a robust immunoreactive smear thatwas strongest at a molecular mass of 110 kDa and higher (Fig. 5B, left panel, arrow). The signal isconsistent with the specific covalent attachment of multiple HA-ubiquitin chains to the cotransporter. Wealso noted a lower-intensity smear from ~75 kDa, extending up to 110 kDa. Based on an analysis of theNCC migration pattern in the whole cell lysates (Fig. 5B, right panel), this signal corresponded to theubiquitination of a low abundance proteolytic fragment of the cotransporter (Fig. 5B, asterisk).Collectively, this pattern of immunoreactivity indicates that a substantial fraction of the total steady stateNCC pool is polyubiquitinated when expressed in mammalian cells.
To further evaluate the relevance of theobservations made in yeast, we analyzed the effect of human cytoplasmic Hsp70 (Hsp72) on NCCprocessing in MDCK epithelial cells. In these cells transient coexpression of Hsp70 with NCC resulted in a>90% reduction in the mature, glycosylated form of NCC (Fig. 6A). These results demonstrate thatcytoplasmic Hsp70 overexpression suppresses NCC maturation in the biosynthetic pathway. Closerinspection of the NCC expression pattern in the whole cell lysates revealed that Hsp70 coexpression didnot increase the total amount of immature core-glycosylated NCC. Based on this finding, we reasoned thatcytoplasmic Hsp70 may not simply act as a holding factor that promotes NCC ER retention in MDCKcells but that it may also interact in a protein complex with NCC to promote its degradation. Indeed, Smetabolic labeling and pulse-chase studies in MDCK cells confirmed that Hsp70 overexpressionaccelerated the rate of NCC degradation by ~50% (Fig. 6B). Because Hsp70 dramatically reduced theabundance of mature NCC without affecting core NCC abundance at steady state, these results suggest thatHsp70 accelerates NCC ERAD to a degree that approximates the rate of NCC biosynthetic maturation inMDCK cells under control conditions. Because these rates appear to match one another, this explains thesimilarity in the steady state abundance of the core-glycosylated band in the control and experimentalgroups.
To test the hypothesis that Hsp70 resides in a chaperone complex that physically interacts with NCC topromote its degradation, we performed co-immunoprecipitation experiments in MDCK cells expressingtagged versions of NCC and Hsp70. As shown in Fig. 6C, left, an antibody with high specificity forcytoplasmic Hsp70 immunoprecipitated N-terminal HA-tagged NCC only in cell lysates when both NCCand myc-tagged Hsp70 were co-expressed. The immunoprecipitation was specific for the 110-kDa NCCspecies, indicating that Hsp70 interacted preferentially with the immature ER-localized form of NCC. Wealso observed co-immunoprecipitation of Hsp70 and NCC in the reciprocal direction. In these experimentslow grade Hsp70 background binding to the protein A- and G-Sepharose resin was noted; however, we
observed a substantial increase in the Hsp70 signal only in the anti-NCC immunoprecipitates from celllysates when the chaperone was coexpressed with NCC (Fig. 6C, right). Together these data providestrong evidence that NCC associates with Hsp70 in a protein complex in mammalian cells and that thiscomplex selects immature forms of NCC for ERAD.
DISCUSSION
In this study we used yeast and mammalian expression systems to begin to define the mechanismsunderlying NCC ERAD. Yeast were initially employed as a genetically tractable system to identify specificevolutionarily conserved elements involved in NCC degradation. After confirming that the cotransporter isa bona fide ERAD substrate in yeast, we found that NCC is degraded via a pathway that exhibits definedrequirements for chaperone-dependent recognition, ubiquitination, membrane extraction, and disposal.Specifically, our data indicate that NCC is recognized by the cytoplasmic yeast Hsp70 Ssa1, marked fordegradation primarily by the ER-associated E3 ubiquitin ligase Hrd1, extracted from the membrane by theAAA+ ATPase Cdc48, and degraded via the proteasome. We then verified the relevance of these findingsin parallel studies of NCC degradation, ubiquitination, and chaperone interaction in mammalian cells.
Our data represent the first survey of molecular candidates involved in the ER quality control of an SLC12cation chloride cotransporter. Accordingly, these experiments have delineated some key differencesbetween the mechanisms underlying the ER quality control of cation chloride cotransporters and otherintegral membrane proteins. For example, the chaperones that participate in the recognition of ENaC forERAD are different from those involved in NCC recognition. Whereas all ER luminal chaperones tested inthis study were found to be dispensable for NCC ERAD, ENaC requires Scj1 and Jem1 to be targeted forproteasomal degradation (14). In contrast, yeast cytoplasmic Hsp70 appears to have a more robust effecton NCC turnover than ENaC (Fig. 3 and Ref. 14). Collectively, these observations are compatible with thetransmembrane topologies of the two proteins, as two-thirds of ENaC resides in the ER lumen (14),whereas over half of the NCC polypeptide is positioned in the cytoplasm, with the large NCC C terminuscomprising a structured domain that consumes ~40% of the cotransporter sequence (7).
In our evaluation of components involved in NCC ERAD, we found that the cytoplasmic Hsp70 homolog,Ssa1, was the only chaperone that influenced the cotransporter turnover rate. NCC degradation, however,was not completely suppressed in the ssa1–45 mutant strain (Fig. 4). We envision three explanations thatcould account for this finding. First, under the conditions of the experiment, only a partial loss of Ssa1activity may have occurred as ssa1–45 is not a complete loss-of-function allele; thus, the cotransportercould still be recognized, albeit inefficiently, for ERAD by residual Ssa1 function. Second, otherchaperones with functions that partially overlap with Ssa1 could participate in NCC ER quality control.For example, the Ssb and Ssz cytoplasmic 70-kDa chaperones could potentially take part in acotranslational NCC ERAD checkpoint in yeast, as previously work has established their presence onribosome-bound nascent polypeptide chains (52). The yeast Hsp90s have also been reported to possesssome functional overlap with Ssa1 (53), and Hsp90s in general may be biased toward promoting substratedegradation, depending on the cochaperones with which they interact (54). Third, Ssa1 may interact withthe cotransporter both during and after ubiquitination. Thus, separate from its role in ERAD substraterecognition, cytoplasmic Hsp70 may help maintain NCC solubility after ubiquitination and duringsubstrate retro-translocation and proteasomal delivery.
The recognition steps involved in NCC ERAD are comparable with those previously described for CFTR,which is also Ssa1-dependent in yeast (29). Interestingly, however, the specific cytoplasmic Hsp70sinvolved in CFTR ERAD in mammalian cells appear to be different from those involved in NCC
degradation. Rubenstein and Zeitlin (55) previously showed that the histone deacetylase inhibitor 4-phenylbutryate suppresses CFTR ERAD by down-regulating Hsc70 (HspA8), the constitutively expressedcytoplasmic 70-kDa chaperone in mammals. Under the same conditions, 4-phenylbutryate promotesphysical interactions between the stress-inducible chaperone Hsp70 (Hsp72) and CFTR (56), suggestingthat Hsc70 stimulates CFTR ERAD, whereas Hsp70 promotes CFTR maturation. We have found that thesesame cytoplasmic Hsp70 homologs have opposite effects on NCC; Hsp70 is a potent stimulator of NCCERAD (Fig. 6), and preliminary analyses of the effect of Hsc70 on NCC in MDCK cells indicate that it isless efficient than Hsp70 at promoting NCC degradation (data not shown). These observations aretempered by evidence that 4-phenylbutryate changes the levels of many factors, including severalchaperones that may also affect CFTR biogenesis (57). Nevertheless, they reinforce the view thatmammalian cytoplasmic Hsp70s are not functionally redundant (58). Our results also support a model inwhich the relative tendencies of Hsp70 and Hsc70 to stimulate ERAD are not fixed; rather, the abilities ofthese chaperones to select substrates for folding or degradation vary depending upon the client with whichthey interact.
In this study we also identified Hrd1 as an important ER-associated E3 ligase that marks NCC for ERADthrough ubiquitination. In yeast, Hrd1 is a key participant in the ERAD pathways responsible for thedegradation of substrates with folding defects in either the ER luminal (ERAD-L) or transmembrane(ERAD-M) domains (43, 59). Because ER luminal chaperones are dispensable for NCC ERAD in yeast,our observations imply that NCC is at least partially processed for degradation through the ERAD-Mpathway. This hypothesis seems reasonable as all cation chloride cotransporters possess 12 transmembranedomains that must pack together into a stable tertiary structure during biogenesis. To a limited extent, NCCturnover was also Doa10-dependent, as a doa10Δhrd1Δ-deficient strain stabilized NCC ERAD to a greaterdegree than strains in which Hrd1 alone was ablated. Doa10 participates in the degradation of cytoplasmic(ERAD-C) substrates (59, 60). The observation that the Doa10 effect was only seen after ablating Hrd1suggests that in yeast, ERAD-M processing of NCC may overshadow its quality control by ERAD-C-dependent mechanisms. The mammalian versions of the yeast ER-associated E3 ligases specificallyinvolved in NCC ERAD are currently unknown, but potential candidates for future study include theDoa10 homolog TEB4 (MARCH-VI) (61), the HRD1-SEL1L complex (a homolog of the yeast Hrd1-Hrd3complex) (62), and the Hrd1-like E3 gp78 (63).
In sum, the work presented in this report outlines a framework for exploring the mechanistic basis ofGitelman syndrome in future studies. In Gitelman syndrome, a defect in NCC function impairs sodiumchloride reabsorption in the distal convoluted tubule of the kidney, resulting in renal salt wasting,hypokalemia, and metabolic alkalosis. Nearly all cases of Gitelman syndrome are caused by mutations thatreduce NCC plasma membrane expression (64). For many of these mutants, the reduced surface expressioncorrelates with changes in glycosylation, consistent with a reduction in anterograde ER export due to NCCmisfolding during biogenesis (Refs. 8–10 and 64 and see supplemental Fig. S7). Our data collectivelysuggest that chaperone complexes responsible for shunting their clients into ERAD-M or ERAD-Cdegradation pathways influence NCC turnover. Consequently, we propose that these same pathways sensemisfolded conformational intermediates of NCC, targeting the cotransporter for accelerated degradation.We anticipate that the yeast and mammalian expression systems developed here will provide a novelapproach to analyze further the processing and ER quality control of wild type and mutant NCC isoforms.
We thank Karina Pena and Joseph Tran for technical assistance, Ronald Rubenstein, Rebecca Hughey, KenHallows, Paul Welling, and Ora Weisz for reagents and helpful technical advice, and Thomas Kleyman forvaluable insights during the course of these studies.
This work was supported, in whole or in part, by National Institutes of Health Grants DK79307 (to the PittsburghCenter for Kidney Research-Model Organisms Core), GM75061 (to J. L. B.), and DK84566 (to A. R. S.). This work wasalso supported by a Mid-Level Career Development Award from the United States Department of Veterans Affairs (to A.R. S.) and James A. Shaver Fund of the American Heart Association Grant 10BGIA3890010 (to A. R. S.).
The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1 and Figs. S1–S7.
1. Ellison D. H., Velázquez H., Wright F. S. (1987) Am. J. Physiol. 253, F546–F554 [PubMed: 3631283]
2. Simon D. B., Nelson-Williams C., Bia M. J., Ellison D., Karet F. E., Molina A. M., Vaara I., Iwata F.,Cushner H. M., Koolen M., Gainza F. J., Gitleman H. J., Lifton R. P. (1996) Nat. Genet. 12, 24–30[PubMed: 8528245]
3. Vargas-Poussou R., Dahan K., Kahila D., Venisse A., Riveira-Munoz E., Debaix H., Grisart B., BridouxF., Unwin R., Moulin B., Haymann J. P., Vantyghem M. C., Rigothier C., Dussol B., Godin M., Nivet H.,Dubourg L., Tack I., Gimenez-Roqueplo A. P., Houillier P., Blanchard A., Devuyst O., Jeunemaitre X.(2011) J. Am. Soc. Nephrol. 22, 693–703 [PMCID: PMC3065225] [PubMed: 21415153]
4. Ji W., Foo J. N., O'Roak B. J., Zhao H., Larson M. G., Simon D. B., Newton-Cheh C., State M. W.,Levy D., Lifton R. P. (2008) Nat. Genet. 40, 592–599 [PMCID: PMC3766631] [PubMed: 18391953]
5. Subramanya A. R., Welling P. A. (2011) Am. J. Physiol. Renal Physiol. 300, F838–F839[PMCID: PMC3075001] [PubMed: 21325498]
6. Devuyst O. (2008) Nat. Genet. 40, 495–496 [PubMed: 18443583]
7. Gamba G. (2005) Physiol. Rev. 85, 423–493 [PubMed: 15788703]
8. Kunchaparty S., Palcso M., Berkman J., Velázquez H., Desir G. V., Bernstein P., Reilly R. F., Ellison D.H. (1999) Am. J. Physiol. 277, F643–F649 [PubMed: 10516289]
9. De Jong J. C., Van Der Vliet W. A., Van Den Heuvel L. P., Willems P. H., Knoers N. V., Bindels R. J.(2002) J. Am. Soc. Nephrol. 13, 1442–1448 [PubMed: 12039972]
10. Sabath E., Meade P., Berkman J., de los Heros P., Moreno E., Bobadilla N. A., Vázquez N., Ellison D.H., Gamba G. (2004) Am. J. Physiol. Renal Physiol. 287, F195–F203 [PubMed: 15068971]
11. Ward C. L., Omura S., Kopito R. R. (1995) Cell 83, 121–127 [PubMed: 7553863]
12. Jensen T. J., Loo M. A., Pind S., Williams D. B., Goldberg A. L., Riordan J. R. (1995) Cell 83, 129–135 [PubMed: 7553864]
13. Kashlan O. B., Mueller G. M., Qamar M. Z., Poland P. A., Ahner A., Rubenstein R. C., Hughey R. P.,Brodsky J. L., Kleyman T. R. (2007) J. Biol. Chem. 282, 28149–28156 [PMCID: PMC2361386][PubMed: 17664274]
14. Buck T. M., Kolb A. R., Boyd C. R., Kleyman T. R., Brodsky J. L. (2010) Mol. Biol. Cell 21, 1047–1058 [PMCID: PMC2836957] [PubMed: 20110346]
15. Staub O., Gautschi I., Ishikawa T., Breitschopf K., Ciechanover A., Schild L., Rotin D. (1997) EMBOJ. 16, 6325–6336 [PMCID: PMC1170239] [PubMed: 9351815]
16. Valentijn J. A., Fyfe G. K., Canessa C. M. (1998) J. Biol. Chem. 273, 30344–30351[PubMed: 9804797]
17. Vembar S. S., Brodsky J. L. (2008) Nat. Rev. Mol. Cell Biol. 9, 944–957 [PMCID: PMC2654601][PubMed: 19002207]
18. Cyr D. M., Höhfeld J., Patterson C. (2002) Trends Biochem. Sci. 27, 368–375 [PubMed: 12114026]
19. Hutt D. M., Powers E. T., Balch W. E. (2009) FEBS Lett. 583, 2639–2646 [PMCID: PMC2805282][PubMed: 19708088]
20. Balch W. E., Morimoto R. I., Dillin A., Kelly J. W. (2008) Science 319, 916–919 [PubMed: 18276881]
21. Ong D. S., Kelly J. W. (2011) Curr. Opin. Cell Biol. 23, 231–238 [PMCID: PMC3078197][PubMed: 21146391]
22. Subramanya A. R., Liu J., Ellison D. H., Wade J. B., Welling P. A. (2009) J. Biol. Chem. 284, 18471–18480 [PMCID: PMC2709348] [PubMed: 19401467]
23. Mumberg D., Müller R., Funk M. (1995) Gene 156, 119–122 [PubMed: 7737504]
24. Adams A., Kaiser C., and Cold Spring Harbor Laboratory (1998) Methods in Yeast Genetics: A ColdSpring Harbor Laboratory Course Manual, 1997 Ed., Cold Spring Harbor Laboratory Press, Plainview, NY
25. Lim K. L., Chew K. C., Tan J. M., Wang C., Chung K. K., Zhang Y., Tanaka Y., Smith W., EngelenderS., Ross C. A., Dawson V. L., Dawson T. M. (2005) J. Neurosci. 25, 2002–2009 [PubMed: 15728840]
26. Bostanjoglo M., Reeves W. B., Reilly R. F., Velázquez H., Robertson N., Litwack G., Morsing P.,Dørup J., Bachmann S., Ellison D. H., Bostonjoglo M. (1998) J. Am. Soc. Nephrol. 9, 1347–1358[PubMed: 9697656]
27. Stirling C. J., Rothblatt J., Hosobuchi M., Deshaies R., Schekman R. (1992) Mol. Biol. Cell 3, 129–142 [PMCID: PMC275513] [PubMed: 1550957]
28. Brodsky J. L., Hamamoto S., Feldheim D., Schekman R. (1993) J. Cell Biol. 120, 95–102[PMCID: PMC2119491] [PubMed: 8416998]
29. Zhang Y., Nijbroek G., Sullivan M. L., McCracken A. A., Watkins S. C., Michaelis S., Brodsky J. L.(2001) Mol. Biol. Cell 12, 1303–1314 [PMCID: PMC34585] [PubMed: 11359923]
30. Fujiki Y., Hubbard A. L., Fowler S., Lazarow P. B. (1982) J. Cell Biol. 93, 97–102[PMCID: PMC2112113] [PubMed: 7068762]
31. Sullivan M. L., Youker R. T., Watkins S. C., Brodsky J. L. (2003) J. Histochem. Cytochem. 51, 545–548 [PubMed: 12642634]
32. Ahner A., Nakatsukasa K., Zhang H., Frizzell R. A., Brodsky J. L. (2007) Mol. Biol. Cell 18, 806–814[PMCID: PMC1805084] [PubMed: 17182856]
33. Youker R. T., Walsh P., Beilharz T., Lithgow T., Brodsky J. L. (2004) Mol. Biol. Cell 15, 4787–4797[PMCID: PMC524727] [PubMed: 15342786]
34. Zhang Y., Michaelis S., Brodsky J. L. (2002) Methods Mol. Med. 70, 257–265 [PubMed: 11917528]
35. Kolb A. R., Buck T. M., Brodsky J. L. (2011) Am. J. Physiol. Renal Physiol. 301, F1–F11[PMCID: PMC3129885] [PubMed: 21490136]
36. de Jong J. C., Willems P. H., van den Heuvel L. P., Knoers N. V., Bindels R. J. (2003) J. Am. Soc.Nephrol. 14, 2428–2435 [PubMed: 14514720]
37. Hoover R. S., Poch E., Monroy A., Vázquez N., Nishio T., Gamba G., Hebert S. C. (2003) J. Am. Soc.Nephrol. 14, 271–282 [PubMed: 12538726]
38. Davidson H. W., Balch W. E. (1993) J. Biol. Chem. 268, 4216–4226 [PubMed: 8382697]
39. Lee D. H., Goldberg A. L. (1996) J. Biol. Chem. 271, 27280–27284 [PubMed: 8910302]
40. Gaczynska M., Osmulski P. A. (2005) Methods Enzymol. 398, 425–438 [PubMed: 16275348]
41. Jones E. W., Zubenko G. S., Parker R. R. (1982) Genetics 102, 665–677 [PMCID: PMC1201965][PubMed: 6764901]
42. Huyer G., Piluek W. F., Fansler Z., Kreft S. G., Hochstrasser M., Brodsky J. L., Michaelis S. (2004) J.Biol. Chem. 279, 38369–38378 [PubMed: 15252059]
43. Bordallo J., Plemper R. K., Finger A., Wolf D. H. (1998) Mol. Biol. Cell 9, 209–222[PMCID: PMC25244] [PubMed: 9437001]
44. Nishikawa S. I., Fewell S. W., Kato Y., Brodsky J. L., Endo T. (2001) J. Cell Biol. 153, 1061–1070[PMCID: PMC2174341] [PubMed: 11381090]
45. Carvalho P., Stanley A. M., Rapoport T. A. (2010) Cell 143, 579–591 [PMCID: PMC3026631][PubMed: 21074049]
46. Nakatsukasa K., Huyer G., Michaelis S., Brodsky J. L. (2008) Cell 132, 101–112[PMCID: PMC2219389] [PubMed: 18191224]
47. Sato B. K., Schulz D., Do P. H., Hampton R. Y. (2009) Mol. Cell 34, 212–222[PMCID: PMC2710143] [PubMed: 19394298]
48. Becker J., Walter W., Yan W., Craig E. A. (1996) Mol. Cell. Biol. 16, 4378–4386[PMCID: PMC231436] [PubMed: 8754838]
49. Han S., Liu Y., Chang A. (2007) J. Biol. Chem. 282, 26140–26149 [PubMed: 17631501]
50. Arroyo J. P., Lagnaz D., Ronzaud C., Vázquez N., Ko B. S., Moddes L., Ruffieux-Daidié D., Hausel P.,Koesters R., Yang B., Stokes J. B., Hoover R. S., Gamba G., Staub O. (2011) J. Am. Soc. Nephrol. 22,1707–1719 [PMCID: PMC3171941] [PubMed: 21852580]
51. Zaarour N., Demaretz S., Defontaine N., Mordasini D., Laghmani K. (2009) J. Biol. Chem. 284,21752–21764 [PMCID: PMC2755897] [PubMed: 19535327]
52. Huang P., Gautschi M., Walter W., Rospert S., Craig E. A. (2005) Nat. Struct. Mol. Biol. 12, 497–504[PubMed: 15908962]
53. Ahner A., Whyte F. M., Brodsky J. L. (2005) Arch Biochem. Biophys. 435, 32–41[PubMed: 15680904]
54. Wang X., Venable J., LaPointe P., Hutt D. M., Koulov A. V., Coppinger J., Gurkan C., Kellner W.,Matteson J., Plutner H., Riordan J. R., Kelly J. W., Yates J. R., 3rd, Balch W. E. (2006) Cell 127, 803–815[PubMed: 17110338]
55. Rubenstein R. C., Zeitlin P. L. (2000) Am. J. Physiol. Cell Physiol 278, C259–C267[PubMed: 10666020]
56. Choo-Kang L. R., Zeitlin P. L. (2001) Am. J. Physiol. Lung Cell Mol. Physiol. 281, L58–L68[PubMed: 11404246]
57. Singh O. V., Vij N., Mogayzel P. J., Jr., Jozwik C., Pollard H. B., Zeitlin P. L. (2006) J. Proteome Res.5, 562–571 [PubMed: 16512671]
58. Goldfarb S. B., Kashlan O. B., Watkins J. N., Suaud L., Yan W., Kleyman T. R., Rubenstein R. C.(2006) Proc. Natl. Acad. Sci. U.S.A. 103, 5817–5822 [PMCID: PMC1458656] [PubMed: 16585520]
59. Carvalho P., Goder V., Rapoport T. A. (2006) Cell 126, 361–373 [PubMed: 16873066]
60. Ravid T., Kreft S. G., Hochstrasser M. (2006) EMBO J. 25, 533–543 [PMCID: PMC1383530][PubMed: 16437165]
61. Hassink G., Kikkert M., van Voorden S., Lee S. J., Spaapen R., van Laar T., Coleman C. S., Bartee E.,Früh K., Chau V., Wiertz E. (2005) Biochem. J. 388, 647–655 [PMCID: PMC1138973][PubMed: 15673284]
62. Mueller B., Klemm E. J., Spooner E., Claessen J. H., Ploegh H. L. (2008) Proc. Natl. Acad. Sci. U.S.A.105, 12325–12330 [PMCID: PMC2527910] [PubMed: 18711132]
63. Shmueli A., Tsai Y. C., Yang M., Braun M. A., Weissman A. M. (2009) Biochem. Biophys. Res.Commun. 390, 758–762 [PMCID: PMC3014991] [PubMed: 19835843]
64. Knoers N. V., Levtchenko E. N. (2008) Orphanet J. Rare Dis. 3, 22. [PMCID: PMC2518128][PubMed: 18667063]
Figures and Tables
FIGURE 1.
NCC is an integral membrane protein and resides in the ER when expressed in yeast. Yeast lysates containing NCCwere subjected to sucrose gradient centrifugation in the absence (A) or presence (B) of 2 mM MgCl in the buffer. Thegradients were fractionated from the top (fraction 1) and analyzed for ER and plasma membrane (PM) containingfractions by immunoblotting for Kar2/BiP (ER) and Pma1 (PM). C, yeast membrane fractions were treated with Na COor a buffer control and subjected to high speed centrifugation. Pellet (P) and supernatant (S) fractions were immunoblottedfor NCC and for the integral membrane protein Sec61 and the soluble, peripheral ER protein, Pdi1.
2
2 3
FIGURE 2.
NCC degradation is proteasome-dependent and requires ER associated E3 ubiquitin ligases. Cycloheximide chasereactions were performed as described under “Experimental Procedures” at 30 °C, and lysates were immunoblotted withanti-HA antibody to measure NCC stability over time. A, a pdr5Δ yeast strain was treated for 20 min with 50 µM MG-132(closed squares) or DMSO (open squares) before a cycloheximide chase reaction. Data represent the means ± S.E. ofthree experiments. B, cycloheximide chase reactions were performed in wild type, hrd1Δ, doa10Δ, or hrd1Δdoa10Δ yeaststrains. Data represent the mean ± S.E. for 7–9 experiments. In both A and B, representative immunoblots are also shownin the bottom panels. C, NCC expressed in either wild type or hrd1Δdoa10Δ mutant yeast strains was immunoprecipitated(IP) from lysates with anti-HA-agarose beads and analyzed by immunoblotting (WB) with either anti-HA or anti-ubiquitinantibody. The result from an immunoprecipitation using cells containing an empty expression vector is shown as anegative control.
FIGURE 3.
The cytoplasmic Hsp70, Ssa1, is required for efficient NCC degradation. Cycloheximide chase reactions wereperformed as described under “Experimental Procedures,” and lysates were immunoblotted with anti-HA antibody. Cellswere grown at 26 °C and shifted to 37 °C for 10 min before cycloheximide was added. A, wild type cytoplasmic Hsp70SSA1 or ssa1–45 mutant yeast is shown. B, wild type ER luminal Hsp70 KAR2 or kar2–1 mutant yeast is shown. C, wildtype cytoplasmic Hsp40 HLJ1YDJ1 or hlj1Δydj1–151 mutant yeast is shown. D, wild type ER luminal Hsp40 SCJ1JEM1or scj1Δjem1Δ mutant yeast is shown. Closed squares represent data from mutant strains, and open squares represent datafrom the isogenic wild type strains. Data represent the mean ± S.E. for 6–11 experiments for each strain. In each partrepresentative immunoblots are also shown in the bottom panels.
FIGURE 4.
NCC ubiquitination is compromised in ssa1–45/Hsp70 mutant yeast. A, NCC was immunoprecipitated (IP) from wildtype SSA1 and ssa1–45 mutant yeast after a 15-min temperature shift to 37 °C, and the precipitated proteins wereimmunoblotted (WB) with anti-HA or anti-ubiquitin antibody. A representative blot is shown. B, relative ubiquitination ofNCC immunoprecipitated from wild type and ssa1–45 mutant yeast is shown. The graph represents the mean ± S.E. ofseven precipitated samples, each from a distinct clone.
FIGURE 5.
NCC is an ERAD substrate in mammalian cells. A, pulse-chase analysis of NCC proteasomal degradation in HEK293cells is shown. Top, 24 h post-transfection cells transiently expressing HA-NCC were pretreated with 10 µM MG-132 (+)or DMSO vehicle (−) for 4 h. Cells were then pulsed with S for 30 min, and the radiolabeled cotransporter was isolatedfrom cell extracts after the indicated chase period by immunoprecipitation. Treatment with MG-132 or DMSO was
35
maintained through the entire pulse and chase periods. Bottom, quantification of the progressive decay in NCC abundanceduring the 90-min chase period, expressed as a percentage of NCC abundance at time zero. B, polyubiquitination of NCCis shown. 24 h post-transfection whole cell lysates of HEK293 cells transiently expressing untagged NCC, HA-taggedubiquitin, or both constructs were subjected to immunoprecipitation (IP) with anti-NCC antibody and immunoblotted(WB) with anti-HA antibody. Immunoprecipitates are shown on the left, and whole cell lysates verifying NCC expressionare shown on the right. The molecular weight corresponding to core glycosylated NCC is indicated with an arrowhead. Alow abundance 75-kDa proteolytic fragment of NCC that is ubiquitinated is indicated with an asterisk. Oligomeric NCC isindicated with brackets. Representative of five experiments.
FIGURE 6.
An Hsp70 chaperone complex associates with NCC and accelerates its ERAD in mammalian kidney epithelial cells.A, shown is the effect of Hsp70 expression on NCC maturation. MDCK cells transiently expressing NCC with humanHsp70 (+) or vector pcDNA3.1 (−) (4 µg of DNA total per 10-cm well) for 36 h were lysed, and whole cell lysates (20µg) were subjected to SDS-PAGE and immunoblotting with the indicated antibodies. Core and mature NCC glycoformsare indicated with arrowheads. Note that the Hsp70 antibody is highly specific for the human protein and does not cross-react with the endogenous canine Hsp70 in MDCK cells. Results are representative of five experiments. B, pulse-chaseanalysis of Hsp70 effects on NCC degradation in MDCK cells is shown. Top, cells transiently expressing HA-NCC in thepresence of Hsp70 or pcDNA3.1 vector for 24 h were metabolically labeled for 30 min with [ S]methionine and[ S]cysteine and chased for various times as indicated. Radiolabeled HA-NCC immunoprecipitates were subjected toSDS-PAGE and phosphorimaging analysis as described under “Experimental Procedures.” Bottom, shown is a plot ofNCC abundance, measured by densitometry at various points during the time course and expressed as a percent of thestarting material at time zero. *, p < 0.018 by Student t test; n = 4. C, reciprocal co-immunoprecipitation of Hsp70 andNCC is shown. Top left, whole cell lysates of MDCK cells transiently expressing HA-NCC with either myc-Hsp70 (+) orpMO-myc vector (−) were immunoprecipitated (IP) with a polyclonal anti-myc antibody and immunoblotted (IB) withanti-HA antibody. Bottom left, 10% of the whole cell lysate inputs for the immunoprecipitation were immunoblotted forHA-NCC and myc-Hsp70 with the indicated antibodies. Results are representative of four experiments. Top right, wholecell lysates of MDCK cells transiently expressing myc-tagged Hsp70 with either HA-NCC (+) or pcDNA3.1 vector (−)were subjected to immunoprecipitation with polyclonal anti-NCC antibody and immunoblotted with anti Hsp70. Bottomright, 10% inputs of the whole cell lysates used in the immunoprecipitation were immunoblotted with the indicatedantibodies. Results are representative of three experiments.
2
35
35
Articles from The Journal of Biological Chemistry are provided here courtesy of American Society forBiochemistry and Molecular Biology