1506 Anal. Chern. 1985, 57, 1506-1511 Theoretical Interpretation of Transient Signals Obtained with Precipitate-Based Ion-Selective Electrodes in the Presence of Interfering Ions Miklds Gratzl, Ern6 Lindner, and Ern6 Pungor* Institute for General and Analytical Chemistry, Technical University of Budapest, Gelldrt tdr 4, Budapest, 1111 Hungary F Preclpltate-based Ion-selectlve electrodes respond to sudden changes In the lnterferlng Ion actlvlty with nonmonotonic, overshoot-type transient slgnals, when a certaln amount of prlmary Ion Is also present In the solution and when the re- spectlve selectlvlty factor Is much less than one. A slmpllfled and more detalled quantilatlve description of these slgnals Is presented In terms of diffusion processes In the adherent solution layer and of adsorptlon/desorptlon equillbrla on the electrode membrane surface. The valldlty of thls descrlptlon Is proved by excellent flttlngs to experlmental signals In cases of (i) lncreaslng and decreasing lnterferlng Ion actlvlty steps, (11) subsequent changes in lnterferlng Ion actlvlty, (111) inter- fering Ion actlvlty steps at different prlmary Ion actlvlty levels. The selectlvlty factor, the dlffuslon layer thlckness values, and the amount of Ions adsorbed or desorbed provldlng good flttlng were In agreement wlth the experlmental values delermlned by other methods, wlthln the respectlve experlmental and calculatlon errors. A contribution by Morf (1) has recently appeared in Ana- lytical Chemistry. In this work Morf actually responded to an earlier publication of Lindner et al. (2) in which it was demonstrated, for the first time, that precipitate-based ion- selective electrodes respond to sudden changes in interfering ion concentration (activity) by potential overshoot type transient functions (Figure 1). To support his theoretical model Morf (1) fitted the function he has derived to the experimental curves obtained by Hulanicki (3) and Lindner (2) and aimed at a general description of the phenomenon. Hulanicki (3) and Lindner (Z), however, reported on in- vestigations performed under essentially different conditions, of which only the fmt one (3) corresponded to the assumptions the theoretical model of Morf was based on. This becomes evident even when the quality of fittings published by Morf is considered (compare Figure 1 and Figure 2 in ref 1). The aim of the present paper is to clear up the incoherences concerning this phenomenon of both practical and theoretical interests, by giving the quantitative version of our earlier qualitative model. In the literature the attempts at the qualitative and quantitative interpretations of the overshoot type transients (4-14) were related first to glass electrodes (4-8) and later to ion-exchanger based liquid membrane electrodes (9, 11). When interpreting the nonmonotonic transient functions obtained with precipitate based ion-selective electrodes, we believe it would not be correct to use assumptions valid for other systems (i.e., for glass electrodes) and applied to earlier models (i.e., different mobilities of primary and interfering ions in the membrane phase (4,8,14) and layers characterized by different selectivity factors in the membrane (1, 13)) as these assumptions cannot be based on a realistic physical picture in the case of precipitate-based electrodes. Conse- quently a new model was set up in accordance with the physical properties (2). It is based on the assumption that following the sudden increase in the activity of interfering ions, primary ions are desorbed from the electrode surface in quantities depending on the concentration level of the in- terfering ions or in the opposite case, Le., when the activity of the interfering ions suddenly decreases, after the interfering ion desorption primary ions are adsorbed in certain quantities on the electrode membrane surface. Due to the desorption or adsorption of primary ions from or at the electrode surface, respectively, ionic activities in the adhering solution layer, which determine the instantaneous values of the electrode potential, temporarily differ from those in the bulk of the sample. The difference in primary ion concentration thus created by the interfering ion activity step initiates diffusion precesses toward equalization. In our opinion, the transient signals in question are the results of the following processes taking place consecutively or simultaneously at the elec- trode/solution interface (the increase of interfering ion activity is discussed here): (1) diffusion of interfering ions (electrolyte) from the bulk of the solution to the electrode membrane surface; (2) adsorption (chemisorption)of interfering ions; (3) desorption of primary ions; (4) diffusion of excess primary ions into the bulk of the sample solution; (5) change of chemical composition and morphology of the electrode membrane surface, and related diffusion processes. Of the processes described above the first three are assigned to the fast in- creasing section of transient signals (Figure 1, phase A; 0.1 s range). However, during the decreasing relaxation section (Figure 1, phase B, seconds range) and the slowest, drift type signal range (marked by C in Figure 1, minutes range), the fourth or fifth process prevail over the others, respectively. This model was sharply criticized by Morf (1) who was of the opinion that our model “would imply strongly disparate diffusion rates for the two ionic species” and “it would be in conflict with the zero current condition”. In this work (1) Morf developed his segmented membrane model (13) further in such a way that-making use of Hulanicki’s (3) equation on the apparent selectivity coefficient-he derived an equation de- scribing the time dependence of the apparent coverage factor, which is essentially also a description of the time dependence of the apparent selectivity coefficient. Before making a comparison between the two models dis- cussed above, based on experimental data, and rejecting some of Morf s statements, we consider it useful to present the description we suggest in a more detailed form, and to compare the fit of theoretical equations (derived in the course of this work and reported by Morf (1)) to experimental data. EXPERIMENTAL SECTION In this work the experimental results of our earlier publication (2) were used for the model calculations; i.e., the measuring setup (15), the electrochemical cell, and the activity steps were identical with those published earlier (2). The data were analyzed with a HP-85 type desk-top computer. 0003-2700/85/0357-1508$01.50/0 0 1985 American Chemical Society
Transcript
1506 Anal. Chern. 1985, 57, 1506-1511
Theoretical Interpretation of Transient Signals Obtained with Precipitate-Based Ion-Selective Electrodes in the Presence of Interfering Ions
Miklds Gratzl, Ern6 Lindner, and Ern6 Pungor* Institute for General and Analytical Chemistry, Technical University of Budapest, Gelldrt tdr 4, Budapest, 1111 Hungary
F
Preclpltate-based Ion-selectlve electrodes respond to sudden changes In the lnterferlng Ion actlvlty with nonmonotonic, overshoot-type transient slgnals, when a certaln amount of prlmary Ion Is also present In the solution and when the re- spectlve selectlvlty factor Is much less than one. A slmpllfled and more detalled quantilatlve description of these slgnals Is presented In terms of diffusion processes In the adherent solution layer and of adsorptlon/desorptlon equillbrla on the electrode membrane surface. The valldlty of thls descrlptlon Is proved by excellent flttlngs to experlmental signals In cases of (i) lncreaslng and decreasing lnterferlng Ion actlvlty steps, (11) subsequent changes in lnterferlng Ion actlvlty, (111) inter- fering Ion actlvlty steps at different prlmary Ion actlvlty levels. The selectlvlty factor, the dlffuslon layer thlckness values, and the amount of Ions adsorbed or desorbed provldlng good flttlng were In agreement wlth the experlmental values delermlned by other methods, wlthln the respectlve experlmental and calculatlon errors.
A contribution by Morf (1) has recently appeared in Ana- lytical Chemistry. In this work Morf actually responded to an earlier publication of Lindner et al. (2) in which it was demonstrated, for the first time, that precipitate-based ion- selective electrodes respond to sudden changes in interfering ion concentration (activity) by potential overshoot type transient functions (Figure 1). To support his theoretical model Morf (1) fitted the function he has derived to the experimental curves obtained by Hulanicki (3) and Lindner (2) and aimed at a general description of the phenomenon.
Hulanicki (3) and Lindner (Z), however, reported on in- vestigations performed under essentially different conditions, of which only the fmt one (3) corresponded to the assumptions the theoretical model of Morf was based on. This becomes evident even when the quality of fittings published by Morf is considered (compare Figure 1 and Figure 2 in ref 1).
The aim of the present paper is to clear up the incoherences concerning this phenomenon of both practical and theoretical interests, by giving the quantitative version of our earlier qualitative model. In the literature the attempts at the qualitative and quantitative interpretations of the overshoot type transients (4-14) were related first to glass electrodes (4-8) and later to ion-exchanger based liquid membrane electrodes (9, 11).
When interpreting the nonmonotonic transient functions obtained with precipitate based ion-selective electrodes, we believe it would not be correct to use assumptions valid for other systems (i.e., for glass electrodes) and applied to earlier models (i.e., different mobilities of primary and interfering ions in the membrane phase (4,8,14) and layers characterized by different selectivity factors in the membrane (1, 13)) as these assumptions cannot be based on a realistic physical picture in the case of precipitate-based electrodes. Conse-
quently a new model was set up in accordance with the physical properties (2). It is based on the assumption that following the sudden increase in the activity of interfering ions, primary ions are desorbed from the electrode surface in quantities depending on the concentration level of the in- terfering ions or in the opposite case, Le., when the activity of the interfering ions suddenly decreases, after the interfering ion desorption primary ions are adsorbed in certain quantities on the electrode membrane surface. Due to the desorption or adsorption of primary ions from or at the electrode surface, respectively, ionic activities in the adhering solution layer, which determine the instantaneous values of the electrode potential, temporarily differ from those in the bulk of the sample. The difference in primary ion concentration thus created by the interfering ion activity step initiates diffusion precesses toward equalization. In our opinion, the transient signals in question are the results of the following processes taking place consecutively or simultaneously at the elec- trode/solution interface (the increase of interfering ion activity is discussed here): (1) diffusion of interfering ions (electrolyte) from the bulk of the solution to the electrode membrane surface; (2) adsorption (chemisorption) of interfering ions; (3) desorption of primary ions; (4) diffusion of excess primary ions into the bulk of the sample solution; (5) change of chemical composition and morphology of the electrode membrane surface, and related diffusion processes. Of the processes described above the first three are assigned to the fast in- creasing section of transient signals (Figure 1, phase A; 0.1 s range). However, during the decreasing relaxation section (Figure 1, phase B, seconds range) and the slowest, drift type signal range (marked by C in Figure 1, minutes range), the fourth or fifth process prevail over the others, respectively.
This model was sharply criticized by Morf (1) who was of the opinion that our model “would imply strongly disparate diffusion rates for the two ionic species” and “it would be in conflict with the zero current condition”. In this work (1) Morf developed his segmented membrane model (13) further in such a way that-making use of Hulanicki’s (3) equation on the apparent selectivity coefficient-he derived an equation de- scribing the time dependence of the apparent coverage factor, which is essentially also a description of the time dependence of the apparent selectivity coefficient.
Before making a comparison between the two models dis- cussed above, based on experimental data, and rejecting some of Morf s statements, we consider it useful to present the description we suggest in a more detailed form, and to compare the fit of theoretical equations (derived in the course of this work and reported by Morf (1)) to experimental data.
EXPERIMENTAL SECTION In this work the experimental results of our earlier publication
(2) were used for the model calculations; i.e., the measuring setup (15), the electrochemical cell, and the activity steps were identical with those published earlier (2) . The data were analyzed with a HP-85 type desk-top computer.
0003-2700/85/0357-1508$01.50/0 0 1985 American Chemical Society
ANALYTICAL CHEMISTRY, VOL. 57, NO. 8, JULY 1985 1507
c, i x -0 . t i
... m I .I
a I V L I
Time Flgure 1. Typical nonmonotonic transient signals following interfering Ion actlvity Increase 01 decrease: (A) overshoot phase (0.1 s range), (B) relaxation phase (second range), (C) slow surface transformation phase (minute range).
THEORETICAL SECTION As already mentioned both in the introduction and in our
earlier publication (2), the transient function, following the stepwise change in the activity of interfering ions, consists of three phases of different rates (Figure 1). The quantitative description discussed in the present paper is limited to the first two sections of transient signals (Figure 1, phases A and B). Here we do not intend to deal with the slowest section of the curves (Figure 1, phase C ) where we assume chemical changes to take place a t the electrode membrane surface (2, 16,17). Neither do we want to deal here with cases where the interfering ions form, with the cations or anions of the mem- brane, precipitates of lower solubility than that of the mem- brane material itself, because in such cases: (a) the nonmo- notinic potential overshoot type transient functions discussed above cannot be observed, but rather the opposite happens; i.e., the electrode potential varies according to a monotonic, asymptotic function (e.g., in the case of AgCl based electrodes in the presence of Br- or I- ions (1,3,18)); (b) an irreversible change of the membrane material occurs, and in this respect the phenomenon is to a certain extent analogous to the pro- cesses determining the third phase of transient signals (16-18).
In our treatment, it is assumed that the sorption and ion- exchange processes are much faster than the transport pro- cesses, Le., diffusion processes arising after the change in interfering ion activity. Hence, a dynamic description is needed for the diffusion only.
The response of precipitate-based ion-selective electrodes in the presence of interfering ions can be described by the Nicolsky equation (19)
(1) E = EiO - - RT In (a{ + ~ ~ ~ a : ) F
where E is the cell potential, E? is a reference potential characteristic of the cell, and R, T , and F have their usual meanings. The activities measured by the electrode (a[ and a{) refer to the boundary solution layer being in contact with the electrode membrane surface. According to our treatment it is supposed that this boundary layer is in a thermodynamic equilibrium with the membrane surface. The theoretical selectivity factor Kij is the equilibrium constant of the basic ion exchange equilibrium
Me1 3. J- = MeJ + I- which can be calculated from the solubility products of the respective precipitates. In this chemical equation Me+ rep- resents the metal cation and I- and J- stand for the primary and interfering anions, respectively.
In connection with the theoretical selectivity factor it has to be emphasized that under the experimental conditions discussed here (Kij << l), there is practically no difference between the theoretical and apparent selectivity coefficients
0 X
iayer 1 roIut1on
t ime
c i IX .0 . t l K-- time
Flgure 2. Schematic diagrams displaying the assumptions used for the mathematical description: x , length coordinate perpendicular to the membrane surface ( x = 0 at the surface, x = 6 at the other boundary of the diffusion layer); t , time ( f = 0 at the stepwise change in the interfering ion activity). For other symbols see the text.
(no related data are found in the literature (3, 20, 21)), in contrast to the Kij >> 1 case discussed by Morf, where (as it has already been pointed out by several authors (3,20-22)) the apparent selectivity factor may vary between D//D{ and Kjj values (D{ and Dj’ are the diffusion coefficients of primary and interfering ions, respectively, in the boundary solution layer). The extension of an equation derived for the limiting case of Kij >> 1 to the opposite limiting case of Ki, << 1, however, cannot be accepted, because by doing so the over- shoot type transient signals can at most be described math- ematically with assumptions contradicting the experimental conditions (eq 15 in ref 1).
According to our model, the overall process can be described rather as follows (when a stepwise increase in interfering ion activity is considered):
(1) At time t = 0 and x = 6 the activity of interfering ions suddenly changes from a/’ to ujm (Figure 2 ) and their diffusion starts toward the electrode surface ( x = 0), where the inter- fering ions are partly drained, due to adsorption processes, although at a rate which varies with time.
(2) Parallel to the adsorption (chemisorption) of interfering ions-present in a large excess-on the electrode surface, primary ions, equivalent to the chemisorbed interfering ions, are being desorbed. The amount of adsorbed and desorbed ions is defined at any time instant by the actual activities of primary and interfering ions, according to the appropriate adsorption isotherm.
(3) Due to the adsorption of interfering ions, the primary ions have a time-dependent source. For the primary ions desorbed from the electrode surface, the bulk of the solution acts as drain at x = 6. The ion transport between the mem- brane surface ( x = 0) and the bulk of the solution ( x = 6) is controlled by diffusion.
The decrease in the activity of the interfering ions can be regarded as a negative step in the activity. Thus, in such cases, the direction of diffusion as well as of the adsorption and desorption processes change their sign, but in other aspects the transient functions can be described similarly as above.
To describe the nonmonotonic transient signals, the surface activities (ai ( x = 0,t); aj ( x = 0,t)) should be inserted into the Nikolsky equation. To define the surface activities as func- tions of time, the differential equations describing the above-mentioned processes should be solved simultaneously. This task is made even more difficult by the fact that the adsorption (chemisorption) isotherm, valid in the presence of both primary and interfering ions, is not known. Moreover, the boundary condition of one of the differential equations (describing the transport of primary and interfering ions) depends on the solution of the other. Hence, to be able to
1508 ANALYTICAL CHEMISTRY, VOL. 57, NO. 8, JULY 1985
describe explicitly the whole process, it is better to start with simplified assumptions.
As first and simplest approximation, let us suppose, that the desorption of the primary ions following the stepwise increase in interfering ion activity can be regarded as in- stantaneous (although the diffusion of the interfering ions toward the electrode surface is not an instantaneous process). This assumption may be supported by the fact that, in the course of the experiments resulting in nonmonotonic transient signals, the concentration gradient of the interfering ions is several orders of magnitude larger than that of the primary ions desorbed and diffusing in the opposite direction. Moreover, the boundary conditions of the differential equa- tions used for describing the simultaneous counterflowing transport processes also differ fundamentally.
As a second assumption, in calculation of the surface ac- tivities, the amount of interfering ions drained (chemisorbed) by the electrode surface is neglected because of their large excess. This approximation is justified, moreover, by the fact that in the Nikolsky equation (eq 1) the interfering ion activity is multiplied by the weighing factor Kij << 1, and because the relative (and not the absolute) activity changes are gov- erning the electrode potentials.
In this case the task can be defined mathematically as follows (in the equations concentrations are used because in the experiments the ionic strength was kept constant):
( 2 ) c i ( ~ , t = 0 ) = Mia(%)
C i ( S , t ) = 0 (3)
(4)
a2ci(x,t)
E ( t ) = EiO - s log [ C i O + q ( x = 0,t) + (5)
dci(x,t) at = D ' 2 ax
Kijcj(x = 0,t = m)] (6)
where x is the distance from the electrode surface, x = 0 at the electrode surface and x = 6 at the other boundary of the diffusion layer, ci0 is the bulk concentration level of the primary ions and ci(x = 0,t) is the increase in the primary ion concentration with respect to cio in the vicinity of the electrode surface, due to desorption, cj(x = 0,t = m) is the final con- centration of the interfering ions following the activity change, Kij is the selectivity coefficient of the ion-selective electrode with respect to the interfering ions j measured potentio- metrically, E(t ) is the cell potential measured as function of time, S is the experimentally determined slope of the loga- rithmic calibration curve of the electrode, Mi is the quantity of primary ions forced to desorb from the electrode surface (mmol/cm2), A(x) is the Dirac-delta function, and D'is the mean diffusion coefficient of ions in the adhering solution layer.
The solution of the above described problem (eq 2-5) with fast convergence in the small time range ((Dt)1/2 << 6) is as follows:
Ci(X,t) = (aD't)'/2 M i (""-&) +
Function 7 was deduced in an elementary way with the help of the principle of reflection and superposition (23,24) . In testing this function it turned out to be convergent in every time range when a sufficient number of terms were considered.
However, in the time range (Dt)1/2 << 6 it is enough to use only one (the first) term. Next after, inserting function 7 into eq 6 with the number of terms discussed above, the poten- tial-time function thus obtained was fitted to experimental transient curves recorded in the "two-ionic" range.
Some of the necessary parameters (EO, S) were determined by prior calibration; ci0 and cj(x = 0,t = m) were given by the experimental conditions, while D'was taken to be the medium value 1.86 X 10" cm2/s (25). During fitting, the Kij, Mi, and 6 values were varied and the best solution was searched for. First the value of Kij was determined from the steady-state potential values preceding and following the interfering ion activity step, and finally the optimal values of Mi and 6 were found, based on the peak height of the overshoot signals and the relaxation rate of the curves, respectively. The results (e.g., Figure 3) seemed to justify the simplifications in the mathematical derivation mentioned above. Concerning the reliability of the fitted parameters, the experimentally found sensitivities of the transient signals with respect to Kij and 6 are reflected properly by the mathematical model, while the effect of changes in Mi on the overshoots depends on the background primary ion level.
It was, however, checked whether these assumptions "imply strongly disparate diffusion rates for the two ionic species" as stated by Morf (1). To answer this question, it is enough, to determine the quantity of primary ions desorbed from the electrode surface as a function of the time-dependent activity of the interfering ions. Thus, simultaneously two diffusion processes (of finite rate and of reverse direction) are consid- ered. In this way the similarity of diffusion rates are taken explicitly into consideration, for both the primary and in- terfering ions. The corresponding more detailed mathematical description is as follows:
c&,t = 0 ) = 0 (8) C i ( X = 6,t) = 0 (9)
In addition, the cj(x = 0,t = m) term in eq 6 has to be replaced by the cj(x = 0,t) function, describing the surface activity variation of the interfering ions which, for relatively long times ( (Dt)1 /2 >> 6) can be given as follows (22, 26-28): c;(x = 0,t) = c;(x,t =
+ 1 462 / J '- if cj(x,t = 0) = 0. Parallel to the activity increase of interfering ions at the electrode surface (according to eq 12), the primary ions adsorbed on the membrane surface are forced to desorb according to the function
a~~ = o,t) (13)
The first term on the right hand side of eq 13 is the slope of the related adsorption isotherm (being not necessarily con- stant) which correlates the actual rate of desorption with the rate of change in interfering ion activity. In the simplest case the adsorption isotherm in question is approximately linear (29) (in a wide concentration range) in the range before sat- uration, that is
aMi at acj(x = o,t) at
- =
ANALYTICAL CHEMISTRY, VOL. 57, NO. 8, JULY 1985 1509
Accordingly, making by use of eq 10 and 13 acj(x = 0,t) = o,t)
at ax = D' (15)
where K is the slope (in this case regarded as constant) of the adsorption isotherm.
Thus, the more detailed problem involving finite diffusion rates for both components can be described by eq 8,9,11,12, and 15 and by function 6 properly modified. By use of the principle of superposition, this set of equations can be solved as follows:
c;(x = 0,t) = J t c [ ( t - T ) f ( r ) dr (16)
where c i describes infinitely fast desorption and relaxation, following the stepwise increase in the interfering ion con- centration, given by eq 7 for unit amount of desorbed ions (Mi = l ) , while f ( ~ ) = dMj(t = T)/at can be given by eq 15.
Thus, according to eq 16, the more elaborate approach considering finite diffusion rates in both directions can be given as the mathematical convolution of the solutions of the separate diffusion problems; one of them corresponds to the extremely fast desorption of primary ions and their diffusion into the bulk of the sample solution, while the other corre- sponds to the diffusion of interfering ions toward the electrode surface. As in the convolution only the initial period of the relaxation of the desorbed elementary quantities play a sig- nificant role, therefore in the case of c/ it is advantageous to use the mathematical solution for relatively short times (7) which converges rapidly in the time range (Dt)lI2 << 6. In the case off, however, as the complete relaxation of the total process has to be described, it is better to apply here the solution converging quickly at large timea (12). Thus eq 7 and the derivative of eq 12 have to be inserted into eq 16. Fur- thermore, eq 16 and 12 should be combined with eq 6 to obtain the time dependence of the electrode potential in a more elaborate form E(t) =
aMi at
-K -- =
0
I
By fitting eq 17 to experimental data, the same procedure was used as in the simpler case described above. The result (e.g., Figure 4) is convincing proof to show that the earlier quali- tative model (2) does not imply strongly disparate diffusion rates of the various compounds, rather the opposite is true. Regarding these rates, a realistic picture of the signals in question can be obtained.
RESULTS AND DISCUSSION The results presented in Figures 3 and 4 prove that both
approaches based on our earlier model assumptions (2) (the simplified solution considering extremely fast desorption, and a more comprehensive model with finite fast diffusion of both the primary and interfering ions) are suitable for describing quantitatively the transient signals obtained after a fast change
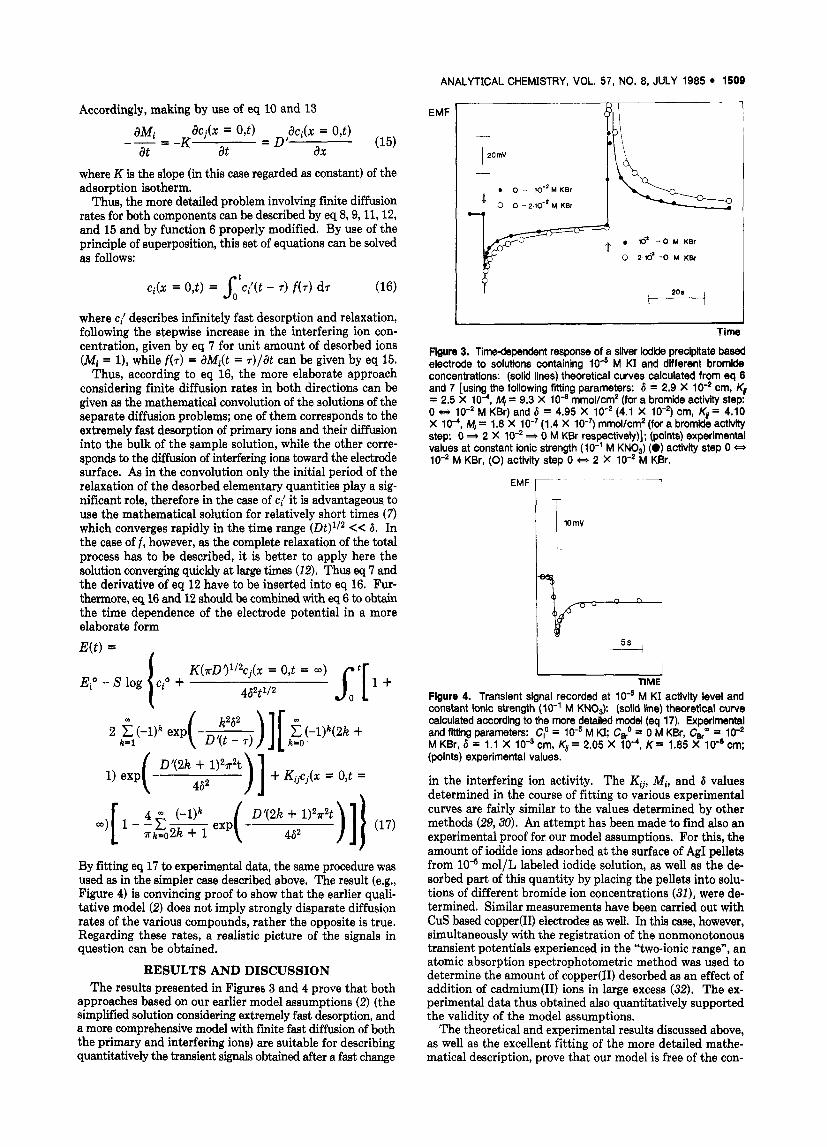
Time Flgure 3. Time-dependent response of a silver iodide precipitate based electrode to solutlons containing M KI and different bromide concentratlons: (solid lines) theoretical curves calculated from eq 6 and 7 [using the following fltting parameters: 6 = 2.9 X lo-* cm, Kt = 2.5 X lo4, M, = 9.3 X 10" mmol/cm2 (for a bromide activity step: 0 t) lo-' M KBr) and 6 = 4.95 X lo-' (4.1 X lo-') cm, Kl= 4.10 X lo4, M, = 1.8 X lO-'(1.4 X lo-') mmol/cm2 (for a bromkle activity step: 0 * 2 X lo-' * 0 M KBr respectively)]; (points) experimental values at constant ionic strength (lo-' M KN03) (0) activity step 0 Q
lo-' M KBr, (0) activity step 0 Q 2 X lo-' M KBr. - -~ EMF r l i
TIME Flgure 4. Transient signal recorded at M KI actlvlty level and constant ionic strength (lo-' M KN03): (solid Ilne) theoretical curve calculated accordlng to the more detailed model (eq 17). Experimental and fitting parameters: C," = M KI; Ceo = 0 M KBr, Cwm = lo-' M KBr, 6 = 1 . 1 X cm, Kl = 2.05 X lo4, K = 1.85 X lo-' cm; (points) experimental values.
in the interfering ion activity. The Kij, Mi, and 6 values determined in the course of fitting to various experimental curves are fairly similar to the values determined by other methods (29,30). An attempt has been made to find also an experimental proof for our model assumptions. For this, the amount of iodide ions adsorbed at the surface of AgI pellets from mol/L labeled iodide solution, as well as the de- sorbed part of this quantity by placing the pellets into solu- tions of different bromide ion concentrations (31), were de- termined. Similar measurements have been carried out with CuS based copper(I1) electrodes as well. In this case, however, simultaneously with the registration of the nonmonotonous transient potentials experienced in the "two-ionic range", an atomic absorption spectrophotometric method was used to determine the amount of copper(I1) desorbed as an effect of addition of cadmium(I1) ions in large excess (32). The ex- perimental data thus obtained also quantitatively supported the validity of the model assumptions.
The theoretical and experimental results discussed above, as well as the excellent fitting of the more detailed mathe- matical description, prove that our model is free of the con-
1510 ANALYTICAL CHEMISTRY, VOL. 57, NO. 8, JULY 1985
b I I I
T i m
Flgure 5. Transient si nais measured (0) and calculated (solid line) with eq 6 and 7 at 10- M KI activity level and for a bromide activity step: 0 H 10-1 M KBr. Fitted parameters values: 6 = 8 X cm, Ku = 7.9 X M, = 2.5 X 10“ (4.5 X lo-*) mmoi/cm2. The fltted 6 and K values are identical for activity increase and decrease but slightly different for M, (value in parentheses).
9
tradictions Morf had claimed it had. Thus, the description presented here also “invokes simultaneous (counter) flows of primary and interfering ions coupled by the zero current condition” (1) which in the present case means that the ad- sorption-desorption processes are balanced, i.e., the quantities of adsorbed and desorbed substances are identical in our model.
Our model clears up the concentration dependence of the transient signals (see also Figure 3). In the presence of low primary ion activities, the potential overshoots also increase with increasing interfering ion activity step. Moreover, our model helps to explain, simply, the experimental finding that the potential overshoot is more limited (Figure 5) if the background primary ion concentration level and the interfering ion concentration step are 10-fold higher as well.
At high primary ion background concentrations the relative concentration change is smaller. Furthermore, near the saturation range of the adsorption isotherm (30) (at high primary ion activities) the desorbed amount of primary ions cannot increase parallel to the increase of interfering ion activity (29, 30).
Moreover, our description can validate phenomena when the interfering ion activity is increased in several subsequent steps (Figure 6). In such cases the primary ions are desorbed from the surface also in several steps; consequently, more potential overshoots can be observed successively in the same direction, because as long as the saturation range of the ad- sorption isotherm is not reached, the increase of interfering ion activities continues to force new quantities of primary ions to desorb from the electrode surface, which results in new transients.
The model presented here explains clearly also the effect of the direction of activity change, namely, that the ion-se- lective electrodes respond with nonmonotonic transient po- tential overshoots not only to the appearance of interfering ions but also to their disapparance ( 2 , 4 , 9). In accordance with the results of others (2, 4,9, 11) it was found that the potential overshoot (AEs) is larger in the case of decreasing interfering ion activity steps than in the case of increasing ones (A&). Furthermore AE, - AE2 is also larger than AEl (where AE2 is the steady state potential difference between the po- tentials obtained in the presence and in the absence of in- terfering ions, respectively) (2). This can be explained as follows: At decreasing interference, simultaneously with the related diffusion processes, the desorption of the interfering ions and adsorption of primary ions take place at the electrode surface. In such cases the relative concentration decrease of
Time($) Flgure 6. Transient signals recorded with a precipitate based iodide electrode at the M KI level following subsequent changes in the interfering ion activity: (solid line) theoretical curve calculated with eq 6 and 7; (points) experimental values measured at constant ionic strength (10-1 M KNO,). Bromide activity steps were 0 * lo4 M KBr and M KBr =+ I O - * M KBr. Fitted parameters were 6 = 3.2 X lo-, cm, Ku = 5.2 X lo-‘, and M, = 2.5 X mmoi/cm* for Cem = cm, 6 = 2.5 X IO-‘, and M, = 1.8 X lo-’ mmol/cm2 for Ce = lo-’ M dBr.
primary ions in the boundary layer (of small volume) is much larger than the relative concentration increase in the opposite case, at otherwise identical absolute quantities of adsorbed or desorbed ion amounts. As the electrode potential change measured is a function of relative concentration changes, it is evident that greater potential overshoot can be measured at decreasing activity steps. According to Figures 3 and 5 this tendency can quantitatively be described by our model. At the same time it is not clear how this phenomenon can be interpreted by the apparent selectivity factor treatment (I).
Based on the facts discussed above, it can be stated that in the case of precipitate based ion selective electrodes and for Kij << 1 the interpretation and mathematical formulation of the nonmonotonic transients with the help of the time dependence of apparent selectivity or coverage factor can be formal at most. Additional arguments supporting this statement are that under the experimental conditions applied (Kij << 1) the equilibrium of the apparent coverage factor (seq) is also practically zero ( I ) , and that the transformation mechanism of the surface layer and its kinetics can be fun- damentally different (16) depending on whether Kij >> 1 or Kij << 1. On the other hand, the time dependence of apparent coverage factor can be well used in cases where the surface activities of primary and interfering ions are determined by solubility and precipitate formation reactions. In such cases the liberated or adsorbed quantity can be neglected in com- parison with the quantity of ions formed in the course of chemical reactions (Figure 1 in ref 1).
Summing up, it can be stated that the two different de- scriptions share several features: one presents a realistic picture for cases of Kij >> 1 (1) while the other for Kij << 1 (2) is describing dynamic characteristics of ion-selective electrodes in the presence of interfering ions. Moreover, both descriptions are in formal agreement with Cammann’s elec- trode kinetic model (12) according to which, following the changes in interfering ion activity the “preequilibrium current flowing through the cell (in the direction of decreasing chemical potential) necessary for the restoration of new equilibrium, can often be many times larger than that flowing under equilibrium conditions”. The “preequilibrium current” being several times larger than the exchange current density of the interfering ions can be interpreted as the current generated in a chemical reaction (1,13) or of those adsorbed or desorbed, respectively, on or from the electrode surface.
M KBr and 6 = 3.2 X
Anal. Chem. 1985, 57, 1511-1517 1511
Naturally, during the preequilibrium phase, the “zero-current membrane condition” is not a precondition.
(19) Nicolsky, B. P. Zh. Flz. Khim. 1937, 70, 495. (20) Pungor, E.; Tbth, K.; HrabBczy-PPII, A. Pure Appl. Chem. 1979, 57,
19 13- 1980.
LITERATURE CITED (1) Mod, W. E. Anal. Chem. 1983, 55, 1165-1168. (2) Lindner, E.; Tbth, K.; Pungor, E. Anal. Chem. 1982, 54, 202-207. (3) Hulanicki, A.; Lewenstam, A. Anal. Chem. 1981, 53, 1401-1405. (4) Rechnitz, G. A.; Kugler, G. C. Anal. Chem. 1967, 39, 1682. (5) Rechnitz, G. A. “Ion-Selective Electrodes”; Durst, R. A., Ed.; US. Na-
tlonal Bureau of Standards: Washington, DC, 1969; NBS Spec. Pubi. 314, Chapter 9.
(6) Tomita, T. “Glass Microelectrodes”; Lavalle, M., Schane, 0. F., Her- bert, N. C., Eds.; Wiley: New York, 1969; Chapter 8.
(7) Grundfest, H. ”Glass Mlcroelectrodes”; Lavalle, M., Schane, 0. F., Herbert, N. C., Eds.; Wlley: New York, 1969; Chapter 10.
(8) Karlberg. B. J. Electroanal. Chem. 1973, 42, 115. (9) Bagg, J.; Vinen, R. Anal. Chem. 1972, 44, 1773.
(10) Mathis, D. E.; Stover, F. S.; Buck, R. P. J. Membr. Sci. 1979, 4 , 395. (11) Reinsfelder, R. E.; Schuitz, F. A. Anal. Chim. Acta 1973, 65, 425. (12) Cammann. K. “Das Arbeiten mk ionenselektlven Elektroden”; Springer
Verlag: Berlln, Heidelberg, New York, 1977. (13) Morf, W. E. Anal. Lett. 1977, 70, 87. (14) Beiljustln, A. A.; Valova, I. V.; Ivanovskaja, I. S. “Ion-Selectlve
Electrodes”; Pungor, E., Ed.; Akademiai Kiadb: Budapest, 1978; p 235.
(15) Llndner, E.; Tbth, K.; Pungor, E. Anal. Chem. 1982, 54, 72-76. (16) Jaenlcke, W.; Haase, M. 2. Electrochem. 1959, 63, 521-532. (17) Schwab, G.-M. KolloidZ. 1942, 707, 204. (18) Rhodes, R. K.; Buck, R. P. Anal. Chim. Acta 1980, 713, 67-78.
(2 1) Umezawa, K.; Umezawa, Y. “Selecthrlty Coefficients for Ion-Selective Electrodes”; University of Tokyo Press: Tokyo, 1983.
(22) Hulanlckl, A.; Lewenstam, A. Talanta 1977, 24, 171-175. (23) Crank, J. “The Mathematics of Diffusion”; Oxford University Press:
Oxford, 1956. (24) Carslaw, H. S.; Jaeger, J. C. “Conduction of Heat in Soilds”; Oxford
Universlty Press: Oxford, 1947. (25) Robinson, R. A.; Stokes, R. H. “Electrolyte Solutions”; Butterworth:
London, 1955. (26) Markovic, P. L.; Osburn, J. 0. AICHE J. 1973, 79, 564. (27) Buck, R. P. “Ion-Selective Electrodes in Analytical Chemlstry”; Freis-
er, H. Ed.; Plenum: New York, 1978; Chapter 1. (28) Morf, W. E.; Lindner, E.; Simon, W. Anal. Chem. 1975, 47, 1596. (29) Hulanlckl, A.; Lewenstam, A.; Maj-Zurawska, M. Anal. Chim’. Acta
1979, 107, 121. (30) Hargnyi, E. G.; Tbth, K.; Pblos, L.; Pungor, E. Anal. Chem. 1982, 54,
1094. (31) Lindner, E.; Farkas, A.; HarsPnyi, E. G.; TBth, K.; Pungor, E., In prepa-
ration. (32) Hardnyl, E. 0.; Tbth, K.; Pungor, E. Paper presented on the “Fourth
Scientific Sectlon on Ion-Selective Electrodes”, 8-12 October 1984, MPtrafured, Hungary.
RECEIVED for review November 16,1984. Accepted February 25, 1985.
Calibration of Ionized Calcium and Magnesium with Ligand Mixtures for Intracellular Ion-Selective Electrode Measurements
Matthias Otto Department of Chemistry, Bergakademie Freiberg, 9200 Freiberg, German Democratic Republic
Peter M. May, Kevin Murray,’ and J. D. R. Thomas* Department of Applied Chemistry, University of Wales Institute of Science and Technology, P.O. Box 13, Cardiff CFl 3XF, Wales, United Kingdom
Standard free calcium Ion concentrations from IO-’ M to M may be obtained by means of a single calibrating tltratlon with a calcium solution of a mixture of calclum-buffering iig- ands, namely, ethylene glycol bls(&amlnoethyl ester)-N,N,- N’,N’-tetraacetic acid (EGTA), N-( 2-hydroxyethy1)ethylene- diaminetrlacetic acid (HEDTA), and nltrllotriacetic acid (NTA). The accuracy of the procedure is evaluated by computer simulations. PVC matrlx membrane ion-selective electrodes (ISEs) based on organophosphate and neutral carrier mate- rials have been used to Investigate the effectiveness of the callbration procedure with submicromolar levels of caiclum ions In the presence of millimolar levels of magnesium ions. The neutral carrier calcium ISE may be used down to lo-’ M [Ca2+] In the presence of millimolar levels of magnesium. A PVC matrix membrane organophosphate based electrode can be used to determine the ionized magnesium.
Ionized calcium and magnesium are present in single cells a t submicromolar and millimolar concentration levels, re-
’ Present address: Nat ional Chemical Research Laboratory, CSIR, Pretoria, South Africa.
spectively (1). Such concentrations can be determined with ion-selective electrodes (ISEs) (1-4); the Ca2+ ion determi- nation is made possible by the sensitivity and selectivity of available calcium ISEs at micromolar levels while the Mg2+ ion determination is made with the less specific divalent ion electrodes since the calcium ion levels are sufficiently low to avoid interferences.
The accuracy and precision of the determination of calcium ions in the submicromolar range are dependent on the se- lectivity of calcium ISEs in the presence of relatively high magnesium levels and on factors controlling the level of the calcium ions themselves. To obtain standards of ionized calcium concentrations down to M, methods have been proposed that are based either on the serial dilution of solu- tions containing the metal ion with a single complexing ligand, such as, NTA, EDTA (5-8), or EGTA (2-4,8, 9) or on the variation of pH in a solution of fixed metal to ligand con- centrations (6, 10-12). Calibrations by these methods are laborious and the conditions are not applicable to those found in vivo, since both the pH and the concentration of calcium buffering ligands vary only slightly in the cell. In vivo con- ditions are more closely approached by a recent method where free calcium ion concentrations are adjusted by adding in- crements of total calcium to a 1 X lo3 M EGTA solution a t
0003-2700/85/0357-1511$01.50/0 0 1985 American Chemical Society