SeciAlto •Batmis •There are a lot ofDynMaPp O S A •
ContextMap •-as?n1 •-RUM a_read mappers out there! StampydrFAST •-Bismark •-•-
MapSplice a- REAL a--BS-Seeker a-- - B S - S e e k e r 2 - ••SupersplatliceMapRAT • - B R A T - S W -•-BFAST •-
segemeht •-GNUMAP •-GenomeMapper •-mrFAST • • - mrsFAST m r s FA S T- L i l t r a - -• - - - -PerM • - - - - - - --RNA-Mate • - - - X-Mate a- - - - SBSMAP • - - - - S p l a z e rRazerS • --•- -MicroRazerS - • - - • RazerS3SHRIMP a — —• SHR1MP2 -•BWA s - - • BWA-SWCloudBurst •ProbeMatch • • W H A M - •
TopHat a- T o p H a t 2 -•-Bowlie •- B o w t i e 2 •-MOM 4-PASS •- P A S S - b i s - -•Slider • - - -Slider-II-()PALMA •SOCS "-MAO •SegMap •ZOOM •PalMaN a-RMAP •SOAP • —SOAP2- -•BWT-SW • - - S O A P S p l i c e - -•
From ebi.ac.uk/~nf/hts_mappers/From eloi.ac.uki-ntiGnotdrni et Atft.- 2 c 1 4 . 1.5auppl 9:512hitk.,:,www.bicrileckentrakuoiryt41-2105/75.•9•512
HISATJAGuaIR - -Bw •A-PSSM - - - -M0-A1K
ApproachARYANA: Aligning Reads by Vet Another
Milad Gnoliimi • r, Arjean kba:: ' ', Ali Sharifiviv:1-• .44, Harritireza (..hitsaz Merio. . ..ignit5.AbstractPitTsburgh , PA, 1..,'S A 31 March - OS April 20.4
iert)m Ric:COM8-Seq: Fourth Annual RkC(....V/111 Satellite 'Workshop or) Massively Parallel SequencingMotivation: Although there are
'•'--Aarly cihretent aigorithms anc software rook br Nigning sequencing reacio s rgappeo s,Fo./pnce search is far from soiven Strong Interest in fast alignrrien:- is hest 1.1,1pc7e0 in the S V or .7tm foraigorithms ',V- rh beperri on fast a rid accurate alignment.
anclitiort de now? assembty of neat-Generaton iPet. enring lng readequites fast overiap-layriur-concensustie Innoczmve competition on a going a roller:ton of reads to a giver database df reference genomes. In
-f_ultra- • -Contribution: I'le introt-Lre ARvANA. a fast gappec rear! aligner developed on Me biss of iilleA incleA•ingnisastr,_cture with a co-ripletely neoo a ighrrent eng OP that rh.akes it signrfiramly faster than 7hree other aligner's:
Sowtie2, BMA anti SegAirt), w tn comparable Gen -t,c-.: ty ant: acruracy. Instead of thp orne-consurning t-haricraciong:vac:et:ores ''L,!• handhing rntsrnatrtx5,s, ARYANIA come; with thp sese-anO-exten0 aigorIMmir framework ano a5lonificantly IrnPrOved mth
efficiency by Integrong riNpi algorithmic tetirnidt.el incluongdynamAr seer: seteCtion,nin 'ectional spec eltensiort reset-4.rep hash tables ano gap-filling cAnynn•nir brogsarnming. As thp reac length _ - -
increases ARYA-V/A•.!T Itioeflorny in terms of speed ana ahgnment rate becomes more evelent. This is in perfect',lakes At par)/ to deveion mission-specie Nigners for other appiications using ARVANA engine.harmony 4vith the iFeli lit'ngth trena as :he seci4enclnig Technologies evohie Ihe algorithmc plaTform of ARYANA
introduction
Availability: ARYAN.4 compip7e source rexie can he obrairteil from kittp.//gitbubcOrnlar)'ana-aligner
i:vt-ty liv:ns cell carries a hatA4 offnre consisting or several used a laborious hierarchil process to divide the gertorne
thnuNand itl r
billitms of characteni with answers to many into srnalier. coveg tam while the Celera (;i-siolnics firmvital qumlions_ .1-11.mnin efforts to decipher that hook has replaced that b rin
gained increasing :rloitivntlint since /953 WhtiL the double ware applied to the data geneated front bhoelly shreddedhelical structure 011)NA was discovered- 'twenty years (shotgun) whole gentorte 17,.ti:. 'the automated Sanger
Liter. W.. Gilbert and A. Maxarn react the nrst 2,1-tit...It-atter r
method was the gold standard fin- about two dettleN, asword of the book [I]. svhen II Sanger and his tsolleasties the.first *-ene.,-ntieor or 021i/A xecitiencing. until iecreasing
application of labeled dideoxynucleotide triphosphatex volome of en-or free genomir information can%ed miler-were dmeloping anothm sequenting method based on the demand for la.,,t and inexpensive methods to produce high
I I
that act ;IS chain terminators in a PC.R rmclior: /2,3...
gence of new technologies. the so tailed Nett-Geno-rainn Idrearn of reading the hunzari honk f e was rtallaed hyAbout three decades after the firn ONA vegurnLing, Sequericisv OVG,S)
.-1, paradigrn shih in both the experimental technititieli 2 0 1 3 2 0 1 4 2 0 1 5completion of the t 3 I li t h e frulnan genrmre profect (4-61, rhe and computational Inettulth octurred
rit:ctir;s1P, eye ive Sanger mate-paired reads t -, -41t7 to
• coeirsgt:,-,1,vi, i,),:kly•ieri?itt,ari,
relmenre gerunnes, such as the human genotr , or more
hvananli J-Ktruto a' V are Sarrt-tunnow tr-eas, tat,
t tore-.4.0,7 f4,,ati,
than 2000 prokitryotex- toilvar), nes and Archaea. lamg,
to the NGS tec:hnologies and also ;Availability of finished2001 2 0 0 0 Wattled Central '''''..•„Nzvoetr - - - - — -ec the crtPrta 44..0 ,,,,t,:.0.,. a.,....„.0,,,elun.:06,z, kx...,0_,-;:t:eC—rnOrdo.Ercfo;CerretnseS:0;xa:13'stect'AL:i.deelat;,,13,17,a5Vt. GISrbtco,„.-"•amoeue? aro% x,,,, (-1'sYl't “:""Mort$ Fttec r,... -0 -?D14 ',1C.4,Tr'l elow:ccrseitv..43P.Ittfrtfct 'NI a 61 Lt)&-. ACUIS ark* a rnkozo imat, re :errra o' rPt .v•nit
el, A
(611;Bloinformatics
Filter for uniquely mapped reads
SAMtools Picard GATK Unix
Filter for uniquely mapped reads
SAMtoolsPicardGATKUnix
Filter for high quality alignments
SAMtools Picard GATK Unix
Filter for high quality alignments
SAMtoolsPicardGATKUnix
Data suitable for final analysis
Data suitable forfinal analysis
Some questions you should ask yourself…Some questions you should ask yourself..
Wis for 'Why?'is for 'Why?
Why are each of these steps needed?Why are each of these steps needed?
Why should I use tool 'X' at this step?Why should I use tool X' at this step?
Wis for 'What?'is for 'What?'
What is the effect on running each step?What is the effect on running each step?
What is a good result?What is a good result?
The effect of applying many 'bioinformatics axes'
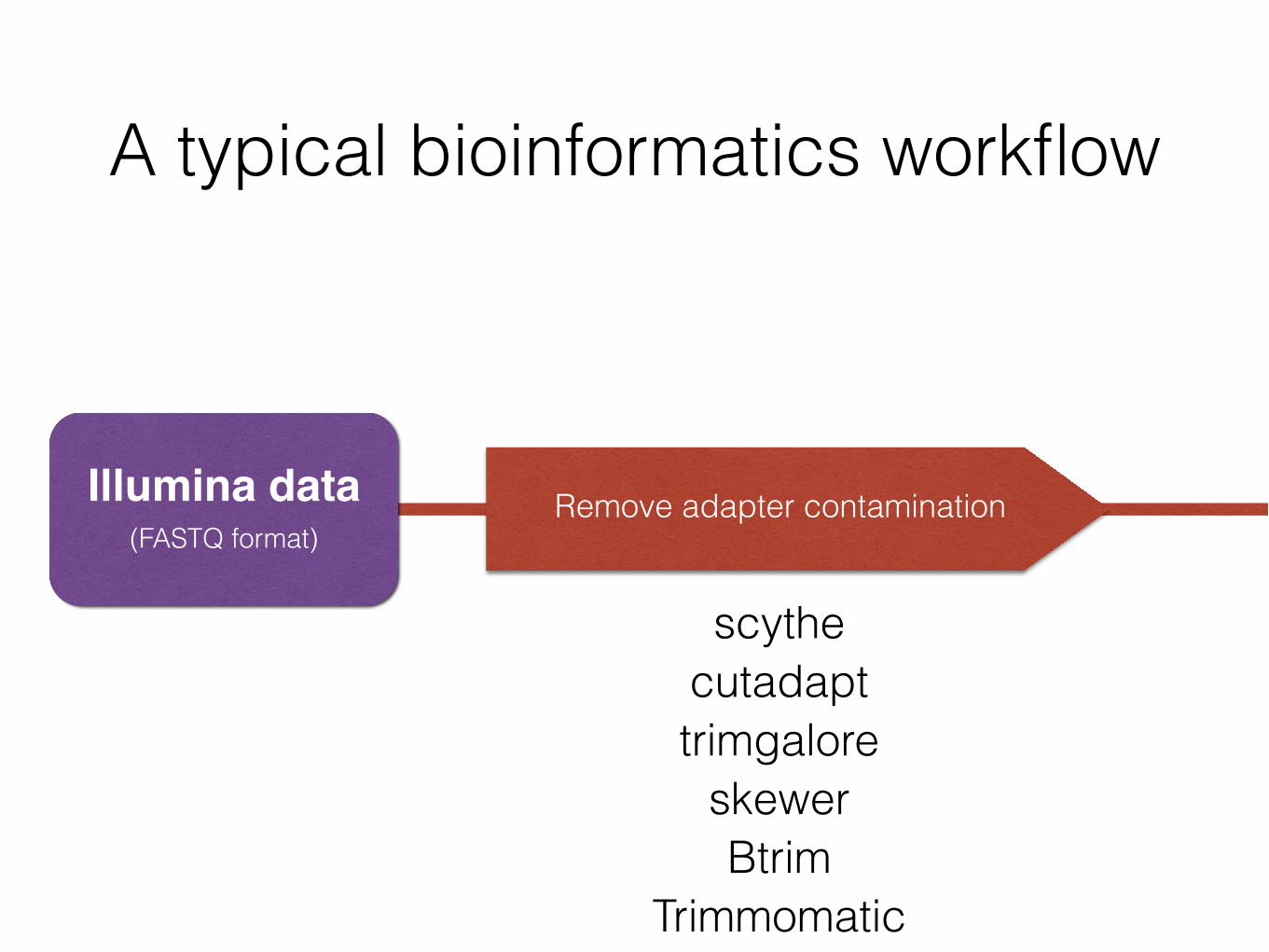
Illumina data(FASTQ format)
2 FASTQ files
Files are ~6.5 GB
52.5 million reads total
The effect of applying many1bloinformatics axes'
IIlumina data(FASTQ format)
2 FASIQ files52.5 million reads total
Files are ,-,64.5 GB
Remove adapters & trim
50.1 million reads
Remove adapters & trim
50.1 million reads
Align to transcriptome with Bowtie
35.8 million reads map
Align to transcriptome with Bowtie
35.8 million reads map
Filter for uniquely mapped reads
31.4 million reads align uniquely
Filter for uniquely mapped reads
31.4 million reads align uniquely
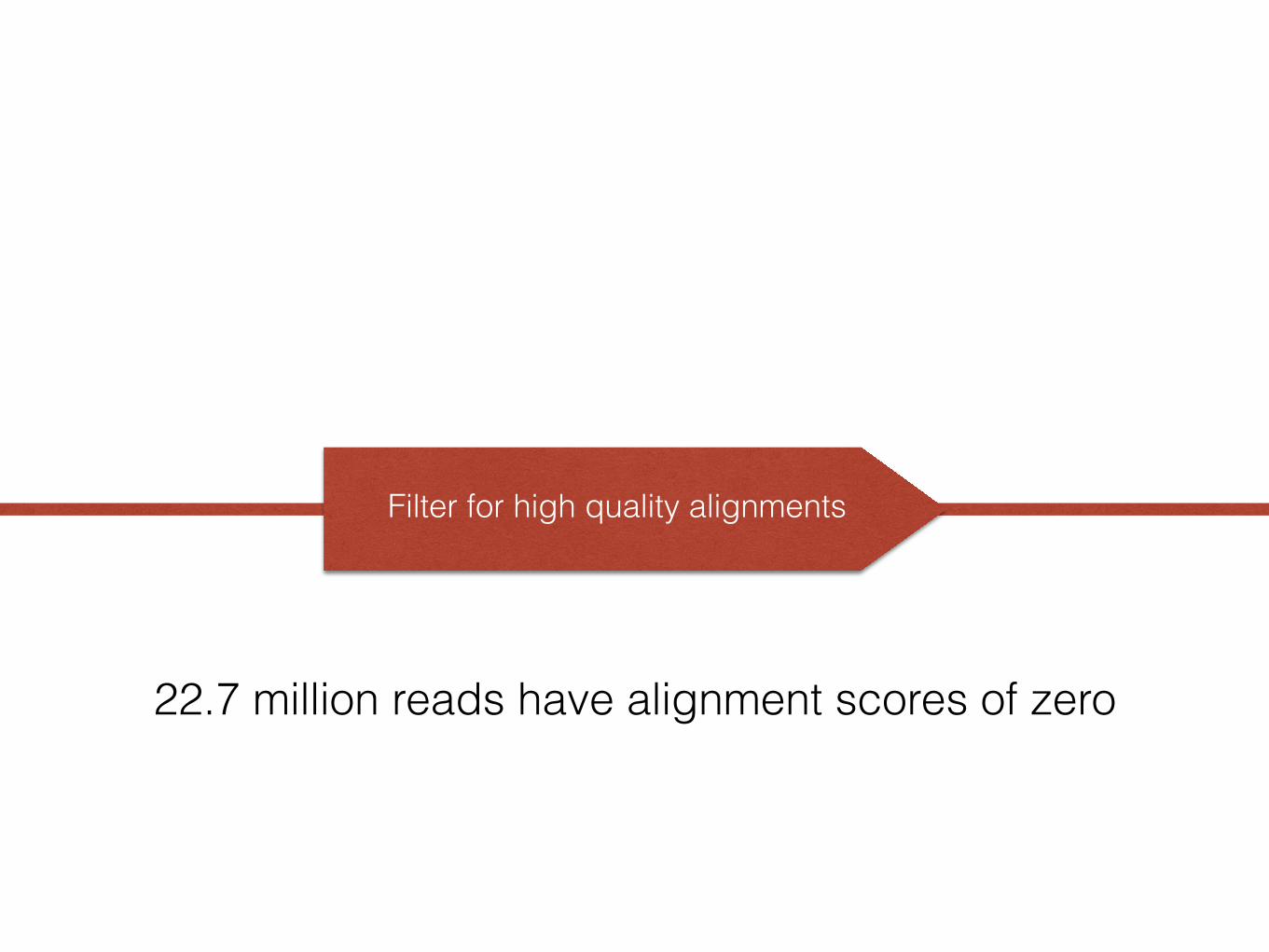
Filter for high quality alignments
22.7 million reads have alignment scores of zero
Filter for high quality alignments
22.7 million reads have alignment scores of zero
Data suitable for final analysis
Reduced data from 52.5 to 22.7 million reads
Data suitable forfinal analysis
Reduced data from 52.5 to 22.7 million reads
It can be helpful to know how the different steps in a workflow reduce your data
It can be helpful to know how the differentsteps in a workflow reduce your data
One final tip…One final tip...
ls -ltris l t r
Run this command after every step of a workflowRun this command afterevery step of a workflow
Let's you see whether output files were actually created
Let's you see whether output fileswere actually created
Let's you see whether output files contain any data
Let's you see whether output filescontain any data
Most recently modified files will be at bottom of your terminal windowMost recently modified files will beat bottom of your terminal window