This work is supported by: a grant from the USDA-NSF Microbial Genome Sequencing Program, a Presidential Early Career Award for Scientists and Engineers from the NSF, and by an R01 from the NHGRI (NIH). Berkeley PHOG: PhyloFacts Orthology Group Prediction Web Server Ruchira S. Datta UC Berkeley Centre for Integrative Bioinformatics Vrije Universiteit Amsterdam June 24th, 2009
Transcript
This work is supported by: a grant from the USDA-NSF Microbial Genome Sequencing Program,
a Presidential Early Career Award for Scientists and Engineers from the NSF, and by an R01 from the NHGRI (NIH).
Berkeley PHOG:PhyloFacts Orthology Group Prediction
Web Server
Ruchira S. DattaUC Berkeley
Centre for Integrative BioinformaticsVrije Universiteit Amsterdam
June 24th, 2009
Acknowledgments• Principal Investigator
– Kimmen Sjölander• Programmer
– Chris Meacham• Research Associate
– Bushra Samad• Undergraduate Student
– Christoph Neyer
•This work is supported by: •a grant from the USDA-NSF Microbial Genome Sequencing Program,
•a Presidential Early Career Award for Scientists and Engineers from the NSF, •and by an R01 from the NHGRI (NIH).
"Nothing in Biology Makes Sense Except in the Light of Evolution"
Theodosius Dobzhansky (1973)
Genome trees: Phylogenomic reconstruction of species trees (based on multiple genes).Accomplished using gene-matrix (aka supermatrix approaches) or supertree methods.
4
Homology-based functional annotations are fraught with systematic error
Gilks et al, “Modeling the percolation of annotation errors in a database of protein sequences” Bioinformatics 2002Galperin and Koonin 1998 "Sources of Systematic Error in Functional Annotation of Genomes" In Silico Biology.Brenner, 1999 "Errors in Genome Annotation" Trends Genet.Brown & Sjölander, "Functional Classification using Phylogenomic Inference." PLoS Computational Biology, 2006
Neofunctionalization stemming from gene duplication
Domain shuffling
Percolation of annotation errors
Two key statistics: 1. Up to 25% of sequences may be mis-annotated. 2. Fewer than 3% of sequences have experimental support for their annotated function
5
Gene duplication produces protein superfamilies including paralogs with divergent functions
6
The evolution of PhyloFacts
EXFOLIATIVE TOXIN AStaphylococcus aureus
TRYPSINFusarium
Oxysporum
16%ID
UCSC Protein structure prediction in CASP2
Celera Genomics human genome annotation
Over 57K protein families and domains with predicted phylogenies, structures, functions for millions of proteins
Over 1.4M hidden Markov models for classification of user-submitted sequences to families and subfamilies
•PhyloBuilder build-your-own protein family phylogeny
•INTREPID functional site prediction server
•PhyloFacts orthology prediction
Berkeley Phylogenomics Group PhyloFacts Encyclopedias
7
Construction of genome-scale phylogenomic libraries
Cluster genome into global homology groups
Predict protein structurePredict key residues
Predict cellular localization.
Include homologs from other species
Construct HMMs for the family and for individual subfamilies.
Construct multiple sequence alignment
Construct phylogenetic trees. Overlay with annotation data. Identify subfamilies.Retrieve key literature
Deposit book in library
FlowerPower
INTREPID & Discern
SCI-PHY
SATCHMO
PHOG
(Krishnamurthy et al, Genome Biology, 2006)
8
PhyloFacts Orthology Group (PHOG) prediction
Datta et al., Nucleic Acids Research Web Server Issue, 2009http://phylofacts.berkeley.edu/orthologs/
Inputs accepted: UniProt accession or ID, GenBank ID for protein sequences only.Sequences can also be submitted in FASTA format for BLAST analysis.
Homologs: orthologs and Homologs: orthologs and paralogsparalogs
Homologs: genes that have descended from a common ancestral gene.
Gene 1
Gene2
Ancestral gene
Orthologs: the last evolutionary event separating the genes was speciation.
S
Paralogs: the last evolutionary event separating the genes was duplication.
D
Courtesy of Nir Yosef
10
Why is orthology important?
Phylogenomic inference of protein function
Prediction of biological pathways and network alignment
PPI prediction (using interlog analysis)
Reconstructing the Tree of Life
Phylogenetic profile construction
11
Many Orthology Prediction Methods(InParanoid, OrthoMCL, COGs, Reciprocal BLAST, TreeFam, etc.)
• Inputs– BLAST scores– Phylogenetic analysis – Species tree– Alignment analysis– Etc.
• Evaluation criteria– Agreement at GO– Agreement in EC number– Agreement at other annotation– Consistency with observed
interactions– Consistency with synteny
Most rigorous approach: Gene-tree species-tree reconciliation.Process is computationally expensive: Gather many homologs from many species; construct alignments; estimate protein family (gene) phylogeny; reconcile gene tree and species tree; label nodes as duplication or speciation; predict orthologs as those whose MRCA represents a speciation event.
Issues: restrict data to whole genomes results in sparse taxonomic sampling (reduced phylogenetic signal). Including sequences from partially sequenced genomes will include duplicate entries for same gene.
TreeFam-A does full phylogenetic tree reconciliation for whole genomes and follows this up with manual curation. Restricted to animal gene families.
p12
Orthology: the MRCA must correspond to a speciation event. (By this definition, the Yeast sequence is orthologous to all sequences in this example.)
Super-orthology is more restrictive than orthology: all nodes on a path between two leaves must correspond to a speciation event. (Zmasek & Eddy, 2002)
S
D
Orthology prediction using treesOrthology prediction using trees
Human, Chimp, Mouse, Rat, Fly, Worm
H1 C1 M1 R1 F1 W1 H2 C2 M2 R2 F2 W2Yeast
Super-orthologs
13
PHOG approach: Each protein can belong to multiple trees (for global homology or individual domains)
• Trees for individual domains as well as whole proteins.• Homologs from both whole and partially sequenced genomes (as well
as metagenome sequences). • These different trees provide different views of the evolutionary history
of the molecule• Different PHOG variants are available
– PHOG-O (standard definition: the MRCA can be labelled as speciation)– PHOG-S (super-orthology: all sequences in a subtree are each other’s
closest ortholog based on tree distance)– PHOG-T (thresholded variant: allows a subtree to expand past the PHOG-
S cutoff; can be tuned to a specific taxonomic distance)
14
PHOG uses tree distances to avoid ambiguous orthology calls
15
PHOG Report
16
PhyloScope view of a PhyloFacts tree, coloring super-orthology groups
Note: Standard orthology is not transitive, but super-orthology is. This is convenient for functional annotation and species phylogeny estimation.
17
Example PHOG report
18
Reciprocal Nearest Neighbor (RNN) “Orthologs”
• For a protein P in taxon (species) S, its RNN ortholog in taxon (species) T is the
protein Q in taxon T whose tree distance to P is smallest, such
that P is also the protein in S whose tree distance to Q is smallest
• Some proteins may have no RNN ortholog in a given taxon, due to gene loss or incomplete sequencing in some species
19
20
RNN vs Tree Reconciliation
• Tree reconciliation (the accepted standard for phylogenetic ortholog identification):• Comparing the gene tree to the species tree
• Issue: Requires accurate species tree
• Labelling internal nodes as duplication or speciation events• One can argue that this is the “right” thing to do• However, many parts of a species phylogeny are unresolved
• This will lead tree reconciliation to give incorrect results
• Tree reconciliation is time-consuming
• RNN is a fast alternative• RNN can give inconsistent results when genes are
missing
21
Mutually Reciprocal “Orthologs”can be incorrect (when genes from taxa are missing)
22
Add new nodes corresponding to species
Draw edges of length 0 from each observed protein sequence (leaf node) to the corresponding species
S
D
Augment the Tree
Human, Chimp, Mouse, Rat, Fly, Worm
H1 C1 M1 R1 F1 W1 H2 C2 M2 R2 F2 W2Yeast
Yeast
23
Computing RNN Orthologs
• A shortest path between nodes in a tree is V-shaped– From one node up to their most recent common ancestor down to
the other node
• Use dynamic programming– Use breadth-first search to order all nodes in the tree
• This order goes from root to leaves• Each parent is visited before any of its children
– Find the closest protein in each taxon to each node, and record its distance
• Go up from the leaves to the root• Then go down from the root to the leaves
• O(nm) where n = # nodes, m = # taxa– We have m <= n/2 as can omit single-protein taxa
24
Inparalogs & Coorthologs
• Fast computation of inparalogs:– Proteins belonging to pure subtrees
• Containing only proteins of a single taxon
• From previous method:– P0, Q0 are RNN orthologs
– P0, P1, …, Pk are inparalogs
– Q0, Q1, …, Ql are inparalogs
– Then (P0, P1, …, Pk) are co-orthologs of (Q0, Q1, …, Ql)
25
PHOG-S: SuperorthologyA PHOG-S is a maximal subtree such that all its leaves are RNN orthologs or co-
orthologs of each other
26
PHOG-T: Thresholded PHOG
• Let N be a node which is maximal such that P is the nearest protein to N in taxon T
• PHOG-S: Among all such nodes N which contain more than one species, those which are minimal, ordered by inclusion
• Let Q be the next nearest protein to N in taxon T
• Duplication distance: (dist(N,P)+dist(N,Q))/2
• Thresholded PHOG: If the duplication distance is less than the threshold, keep proceeding up the tree to the next maximal node
27
Dataset: 100 (non-homologous) human sequences from TreeFam-A, filtered to remove homologs.
PHOG-O: Standard orthology definitionPHOG-S: Super-orthologs (Zmasek & Eddy)PHOG-T: thresholded PHOGsPHOG-T(M); optimized for mousePHOG-T(Z): optimized for zebrafishPHOG-T(F): optimized for fruit fly
Ortholog prediction accuracy
Assessed vs TreeFam-A manually curated orthologs
28
Interolog analysis(infer interactions based on experimentally observed interactions in orthologs)
29
Phylogenomic inference of pathways and protein-protein interaction
30
Pathways and PPI Through PHOG
31
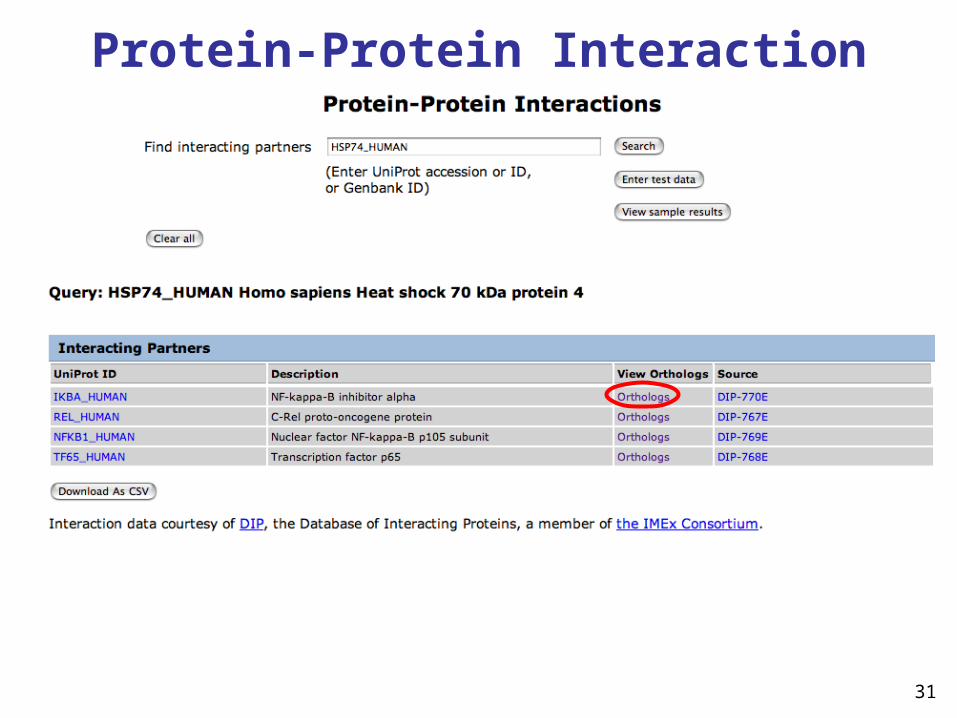
Protein-Protein Interaction
32
Interologs Through PHOG
33
Coming soon: Interactome Viewer
Dean Starrett, Ph.D
34
Postdoc and PhD student
positions!
This work is supported by: a grant from the USDA-NSF Microbial Genome Sequencing Program,
a Presidential Early Career Award for Scientists and Engineers from the NSF, and by an R01 from the NHGRI (NIH).
Thanks to the many people who have made this possibleKimmen Sjölander, Principal InvestigatorChris Meacham, ProgrammerBushra Samad, Research AssociateChristoph Neyer, Undergraduate Student
And previous group members for PhyloFacts development:Nandini Krishnamurthy, Postdoctoral ResearcherDuncan Brown, Ph.D Student