UNIVERSITY OF COPENHAGEN FACULTY OF SCIENCE This PhD thesis has been submitted to the PhD school of The Faculty of Science, University of Copenhagen, Denmark, 4. January 2016. PhD Thesis Thomas Thyge Thomsen, M.Sc. Biology Peptide antibiotics for ESKAPE pathogens Past, present and future perspectives of antimicrobial peptides for the treatment of serious Gram-negative and Gram-positive infections.
Transcript
U N I V E R S I T Y O F C O P E N H A G E N F A C U L T Y O F S C I E N C E
This PhD thesis has been submitted to the PhD school of The Faculty of Science, University of Copenhagen, Denmark, 4. January 2016.
PhD Thesis
Thomas Thyge Thomsen, M.Sc. Biology
Peptide antibiotics for ESKAPE pathogens
Past, present and future perspectives of antimicrobial peptides for the
treatment of serious Gram-negative and Gram-positive infections.
Academic advisors Kim Rewitz, Ph.D., Associate Professor Department of Biology Cell and Neurobiology University of Copenhagen, Denmark Anders Løbner-Olesen, Ph.D., Professor Department of Biology Functional Genomics University of Copenhagen, Denmark Assessment committee Hanne Ingmer, Professor (chair) Health Department
University of Copenhagen Volker Loeschcke, Professor Department of Biosciences – Genetics, Ecology and Evolution
Aarhus University, Denmark Gabriele Bierbaum, Professor. Dr.
Institute of Medical Microbiology, Immunology and Parasitology University of Bonn
Submitted 04.01.2016
2
ACKNOWLEDGEMENTS
I would like to give special thanks to my supervisors on this project. Kim Rewitz has provided
excellent support as my main supervisor and it would be hard to find a person with more dedication
to science and his students. He has provided expert knowledge to all work regarding Drosophila
and experimental procedures and supported me during all aspects of my PhD. Anders Løbner-
Olesen has provided excellent guidance and discussion on all my microbiological work. Anders has
maintained focused and supportive during times when the project seemed less successful, which has
helped me keep focused.
Much of the work in this thesis could not have been carried out without the work of Håvard Jenssen
and Biljana Mojsoska; Thank you guys for your work on peptide synthesis and discussion. I wish to
thank Paul Robert Hansen and Alberto Oddo for their collaborations and for excellent discussions.
Also a special thanks you to Stefano Donadio for his interest in collaborations and discussion along
the way.
Furthermore I would like to thank all the people from the two labs where I have worked. The
Rewitz Lab; Erik Thomas Danielsen, Morten Møller, Julie Lilith Hentze, Anne Færch Jørgensen,
Morten Rose and all the other students and people from the neurobiology section. ALO Lab; Jakob
Frimodt-Møller for sharing the ups and down during our PhD, Louise Bjørn, Maria Schei Haugan,
Godefroid Charbon, Henrik Jakobsen, Michaela Lederer, Rasmus Nielsen Klitgaard, Luis Clàudio
Nascimento da Silva, Susanne Kjelstrup, Linette Skov, Christopher Campion and all the other
students from the ALO lab.
Finally I wish to thank my family for support during my PhD.
3
ABSTRACT
Multi-drug resistance to antibiotics represents a global health challenge that results in increased
morbidity and mortality rates. The annual death-toll is >700.000 people world-wide, rising to ~10
million by 2050. New antibiotics are lacking, and few are under development as return on
investment is considered poor compared to medicines for lifestyle diseases. According to the WHO
we could be moving towards a post-antibiotic era in which previously treatable infections become
fatal. Of special importance are multidrug resistant bacteria from the ESKAPE group (Enterococcus
and Enterobacter). As a consequence of widespread multi-drug resistance, researchers have sought
for alternative sources of antimicrobials. Antimicrobial peptides are produced by almost all living
organisms as part of their defense or innate immune system and are therefore of interest for
development of novel antimicrobials.
This thesis aimed at developing new or improved peptide-based antimicrobials, capable of killing or
inhibiting the proliferation of important multidrug resistant bacteria. Further we sought to analyze in
vivo efficacy and toxicity by utilizing of the fruit fly Drosophila melanogaster as a whole animal
model. This was carried out by testing of antimicrobial peptides targeting Gram-positive bacteria
exemplified by the important human pathogen methicillin resistant S. aureus (MRSA).
The peptide BP214 was developed from a cecropin-mellitin hybrid peptide and proved effective in
killing colistin resistant Gram-negative A. baumannii in vitro. The molecule was improved with
regard to toxicity, as measured by hemolytic ability. Further, this peptide is capable of specifically
killing non-growing cells of colistin resistant A. baumannii, also known as persisters.
Using D. melanogaster as an in vivo efficacy model it was demonstrated that the Lantibiotic NAI-
107, currently undergoing pre-clinical studies, rescues D. melanogaster from MRSA infection with
similar efficacy to last resort antimicrobial vancomycin. Furthermore, it was shown that this
antimicrobial has similar capability to BP214 in killing non-growing cells of S. aureus. However,
for NAI-107 this is independent of genotype and underscores its potential for future development.
4
ABSTRACT – DANISH
Multiresistente bakterier er et voksende globalt sundhedsproblem. På verdensplan dør mere end
700.000 mennesker årligt som følge af infektioner med multiresistente bakterier. Udvikling af nye
antibiotika er mangelfuld fordi det for medicinal industrien bedre kan betale sig, at udvikle medicin
til behandling af livsstil sygdomme. Ifølge WHO kan vi snart befinde os i en post antibiotika æra,
hvor behandling af infektioner ikke længere er mulig. Særligt problematiske er bakterier fra
ESKAPE gruppen (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae,
Acinetobacter, Pseudomonas aeruginosa and Enterobacter). Som en konsekvens af multiresistente
bakterier har det været nødvendigt at finde alternativer til de kendte antibiotika. Næsten alle levende
organismer producere antimikrobielle peptider som forsvar eller innate immun forsvar imod
mikroorganismer hvilket gør disse stoffer interessante for udviklingen af nye antibiotika.
Dette Ph.d. projekt havde til formål at udvikle nye eller forbedrede peptid baserede antimikrobielle
stoffer, som kunne inhibere eller dræbe multiresistente bakterier. Vi ønskede at benytte banan fluen
Drosophila melanogaster som en simple model til at vurdere in vivo effektiviteten og toksiciteten af
peptider. Dette gjorde vi ved at inficere Drosophila med den Gram-positive bakterie methicillin-
resistent S. aureus (MRSA) og behandle med antimikrobielle peptider mod Gram-positive bakterier.
Med udgangspunkt i et cecropin-mellitin hybrid peptid, udviklede vi BP214, som effektivt kan slå
colistin resistente stammer af Gram-negative A. Baumannii ihjel in vitro. BP214 havde en forbedret
toksicitet profil i forhold til tidligere molekyler. Dette blev målt ved hemolytisk kapabilitet.
Endvidere har vi vist at BP214 specifikt kan slå ikke voksende (”persisters”) celler af colistin
resistente A. baumannii ihjel.
I Drosophila in vivo modellen påviser vi at peptidet NAI-107 med samme effektivitet som
vancomycin redder Drosophila fra infektioner med MRSA. Nai-107 er allerede under udvikling til
fremtidig brug som antibiotika. Vi viser yderligere at NAI-107 ligesom BP214 kan slå ikke
voksende celler af S. aureus ihjel. Ulig BP214 er denne egenskab for NAI-107 uafhængig af
bakteriernes genotype hvilket understreger dets egenskaber som fremtidens antibiotika.
5
LIST OF PAPERS
Paper I “An all-D amphipathic undecapeptide shows promising activity against colistin-resistant strains of Acinetobacter baumannii and a dual mode of action” by Alberto Oddo, Thomas TT, Susanne Kjelstrup, Ciara Gorey, Henrik Franzyk, Niels Frimodt-Møller, Anders Løbner-Olesen, and Paul Hansen [AAC01966-15].
Paper II “Modulation of backbone flexibility for effective dissociation of antibacterial and hemolytic activity in cyclic antimicrobial peptides without loss of potency” by Alberto Oddo, Thomas T. Thomsen, Hanna M. Britt, Anders Løbner-Olesen, Peter W. Thulstrup, John M. Sanderson, and Paul R. Hansen. Submitted to ACS Medicinal Chemistry Letters
Paper III “The Lantibiotic Nai-107 efficiently rescues Drosophila melanogaster from infection with methicillin-resistant Staphylococcus aureus USA300” by Thomas T. Thomsen, Biljana Mojsoska, Joao Cruz, Stefano Donadio, Håvard Jenssen, Anders Løbner-Olesen, Kim Rewitz. Submitted to Antimicrobial Agents and Chemotherapy.
Papers not included in the thesis (see appendix)
Paper I “Rapid selection of Plasmodium falciparum chloroquine resistance transporter gene and multidrug resistance gene-1 haplotypes associated with past chloroquine and present artemether-lumefantrine use in Inhambane District, southern Mozambique.” By Thomsen TT, Madsen LB, Hansson HH, Tomás EV, Charlwood D, Bygbjerg IC, Alifrangis M Am J Trop Med Hyg. 2013 Mar; 88(3):536-41. doi: 10.4269/ajtmh.12-0525. Epub 2013 Feb 4.
Paper II “Collateral Resistance and Sensitivity Modulate Evolution of High-
Level Resistance to Drug Combination Treatment in Staphylococcus aureus.” by Rodriguez de Evgrafov M, Gumpert H, Munck C, Thomsen TT, Sommer MO. Mol Biol Evol. 2015 May; 32(5):1175-85. doi: 10.1093/molbev/msv006. Epub 2015 Jan 23.
6
LIST OF FIGURES
Figure 1. Drug Development and Resistance: Adapted from (9) ...................................................... 11
Figure 2. Generalized representation of Gram-negative and Gram-positive membranes: Adapted
from (13, 14) ...................................................................................................................................... 13
LIST OF PAPERS ............................................................................................................... 6
Papers not included in the thesis (see appendix) ............................................................................................................ 6
LIST OF FIGURES .............................................................................................................. 7
Spectrum of activity ........................................................................................................................................................ 12
Genetics of resistance .................................................................................................................................................. 16
Mechanism of resistance .............................................................................................................................................. 18
Modified or synthetic: peptides and peptoids .............................................................................................................. 37
ALTERNATIVE IN VIVO MODELS ................................................................................... 39
PAPER I ............................................................................................................................ 43
8
An all-D amphipathic undecapeptide shows promising activity against colistin-resistant strains of Acinetobacter
baumannii and a dual mode of action ........................................................................................................................... 43
PAPER II ........................................................................................................................... 75
Modulation of backbone flexibility for effective dissociation of antibacterial and hemolytic activity in cyclic
antimicrobial peptides without loss of potency ............................................................................................................ 75
PAPER III .......................................................................................................................... 83
The Lantibiotic Nai-107 efficiently rescues Drosophila melanogaster from infection with methicillin-resistant
A Drosophila in vivo efficacy model of infection ......................................................................................................... 123
FUTURE PERSPECTIVES .............................................................................................. 126 The cecropin-mellitin hybrid BP214 .......................................................................................................................... 126
Lantibiotics and other Lipid II targeting antimicrobials ............................................................................................ 127
PAPER II ......................................................................................................................... 152
Collateral Resistance and Sensitivity Modulate Evolution of High-Level Resistance to Drug Combination
Treatment in Staphylococcus aureus .......................................................................................................................... 152
9
INTRODUCTION - ANTIBIOTICS
Historical overview
The first antimicrobial compounds to be mass produced and used on a large scale in
clinical settings were the Sulfonamides or “sulpha drugs”. These synthetic compounds containing a
sulfonamide group, inhibit the enzyme dihydropteroate synthetase (DHPS) involved in folate
biosynthesis, and were first synthesized by Alfred Bertheim and Paul Ehrlich in 1907 (1), however
Gerhard Domagk is credited for the discovery of the first commercially available sulfonamide used
in the clinical setting “Prontocil”, for which he later received the Nobel Prize in 1939 (2).
The Discovery of penicillin in 1928 by Alexander Fleming (3), is by many recognized
as the first true antibiotic, a term coined by Selman Waksman as a compounds produced by or
derived from microorganisms that in dilute concentration effectively inhibit the growth of or
effectively kill other microorganisms (4). Today the words antibiotics or antimicrobials are often
used interchangeably for compounds used in the treatment of bacterial, protozoan or other
infections by pathogenic microorganisms. The active compound of penicillin was isolated and set in
production thanks to ground breaking work by Howard Florey and Ernst Boris Chain (5), for which
they alongside Sir Alexander Fleming received the Nobel Prize in 1945. Devastating diseases
previously untreatable, such as streptococcal and chlamydial infections suddenly became treatable
with the introduction of penicillin. The discovery of antibiotics sparked a new era in the treatment
of infectious diseases and paved the way for modern medicine, through the golden era of antibiotics
drug discovery from the 1940-1960´s.
During these decades, there was a huge expansion in the arsenal against bacterial
infections through the continued discovery of new compounds. Gerhard Domagk and Alexander
Fleming`s work was followed by many pioneering scientists. Especially, the work of Selman
Waksman, whom paved the way for new methodologies in antibiotic discovery (6) and whom was
originally accredited for the discovery of streptomycin, the first treatment for one of human
history`s greatest pathogens, Tuberculosis. This discovery earned him the name “Father of
10
Antibiotics” and the Nobel Prize, although his PhD student Albert Schatz predominantly was the
discoverer (7).
During this relatively short period in history most of today’s known classes of
antibiotics were discovered (Figure 1. Top). With antibiotics covering some of history`s most
important human pathogens (Tuberculosis, Cholera, Malaria etc.) and it has been said that in 1969,
the then US Surgeon General William Stewart told the US Congress “that it was time to close the
books on infectious diseases" (8). Although, Stewart most likely never said such a thing, it clearly
illustrates the general assumption at the time, that infectious diseases would pose a problem no
more. Without the work of these groundbreaking microbiologists and their coworkers, modern
medicine could not have developed to the point of today. Antibiotic treatment is the foundation for
surgeries, cancer treatments and treatment of chronic diseases like diabetes and cystic fibrosis.
Without efficacious antimicrobials clinical medicine as we know it could be jeopardized.
Figure 1. Drug Development and Resistance: Adapted from (9)
Top: Introduction of major classes of antibiotics. Bottom: First time resistance to the class of antibiotic was observed in the clinical setting. Observation of resistance is not equal to loss of clinical efficacy against all clinical isolates. Not all classes or antibiotics are included.
11
Spectrum of activity
Antibiotics have historically been grouped in two groups based on their spectrum of
activity. Broad spectrum antibiotics cover bacterial species of both Gram-positive and Gram-
negative origin making them useful in treatment where the causative pathogen is unknown.
Whereas, narrow spectrum antibiotics like vancomycin used against Gram-positive bacteria are
used when the causative agent is known and therefore treatment will have less impact on the normal
flora of the patient. Narrow spectrum antibiotics can minimize unwanted side effects on the
bacterial microflora of the patient as the causative agent is known (10-12).
The spectrum of activity is highly dependent upon the structural difference of the
bacterial membrane between Gram-negative and Gram-positive bacteria (Figure 2). Although most
antimicrobials cross the bacterial membrane via passive transport through porin`s or other
transporters, the Gram-negative bacteria are generally considered more difficult to kill by
antimicrobials due to their outer membrane (13). The bacterial cell membrane is a bilayer composed
of phospholipids such as Phosphatidylglycerol, Cardiolipin (Diphosphatidylglycerol), and/or
Phosphatidylethanolamine. Gram-negative bacteria have both an inner and an outer membrane; in
the periplasmic space a thin layer of peptidoglycan (cell wall) is connected to the outer membrane
via lipoproteins (murein lipoprotein). The outer membrane of the Gram-negative bacteria is on the
inward facing side composed of phospholipids similar to the inner membrane, whereas the outward
facing side contains lipopolysaccharide (LPS). The Gram-positive bacteria on the other hand do not
contain an outer membrane. However, they do have a thick cell wall (peptidoglycan), through
which lippotechoic acids and wall techoic acids traverse. Both Gram-negative and Gram-positive
bacteria may include an S-layer (capsule) consisting of protein or glycoproteins that acts as an
additional protective layer (14). Bacterial membranes also incorporate various efflux systems for
the transport of substance such as toxic compounds and waste products, these are discussed briefly
later (15).
12
Figure 2. Generalized representation of Gram-negative and Gram-positive membranes: Adapted from (13, 14)
The Gram-negative bacterial membrane is composed of an Outer membrane covered with lipopolysaccharide (LPS) and with large porin crossing the outer membrane. Below the outer membrane is the periplasmic space on top of a thin layer of peptidoglycan, under which is found the inner cell membrane. The Gram-positive membrane is made up of a thick multiple layer peptidoglycans, with wall techoic acids and lippotechoic acids. Gram-positive bacteria contain a single underlying cell membrane. See text for detailed description.
Further, antibiotics can be either bactericidal i.e. exert their effect by killing the
bacterium or they can be bacteriostatic i.e. exert their effect by inhibiting growth of the bacteria
thereby allowing for the immune system of the host to clear the infection. These definitions are not
unambiguous and mainly apply in vitro and on organismal level, whereas the definitions become
more arbitrary in clinical setting (16). Some compounds act bactericidal against some pathogens,
while being bacteriostatic against others; exemplified by the antibiotic chloramphenicol that is
bacteriostatic against S. aureus, but bactericidal against S. pneumoniae (17).
13
Antibiotic targets
Interestingly, most of the known antibiotics inhibit relatively few pathways in the
bacterial cell; I) Folic acid synthesis, II) transcription, III) DNA replication, IV) protein synthesis
inhibitors and V) cell wall synthesis inhibitors (Figure 3). Most of these compounds were
discovered by cultivation and purification from the natural producers, or they are chemical
derivatives of such compounds (18-20). Historically new compounds have been based on the
structure of previously developed antibiotics and so, has the targets remained the same for many
new antibiotics, but with slight modification to the molecules and their affinity for the target (19,
20). An example of such development, are the generally considered broad spectrum β-lactam
antibiotics the Cephalosporin’s; divided into 1st, 2nd, 3rd, 4th and 5th generation cephalosporin’s
depending on their spectrum of activity and structural changes to the compounds (20, 21).
The sulfa drugs are inhibitors of the essential folic acid synthesis pathway in bacteria
and can be exemplified by the combination therapy of sulfamethoxazole-trimethoprim. In the folate
synthesis pathway; the dihydropteroate synthetase (DHPS) enzyme synthesizes Dihydropteroic
acid, from Para-aminobenzoic Acid (PABA) and Pteridine. Dihydropteroic acid is in turn is used to
synthesize Dihydrofolic acid by Dihydrofolate Synthetase. Finally dihydrofolate reductase (DHFR)
synthesizes Tetrahydrofolic Acid from Dihydrofolic acid. Sulfamethoxazole inhibit the DHPS
enzyme, whereas trimethoprim inhibits the DHFR enzyme (22). The Rifamycins (e.g. Rifampicin)
inhibits transcription by binding to the DNA-dependent RNA polymerase holoenzyme before the
unwinding of the DNA (closed complex) and keep it in the closed state. If the RNA polymerase has
already opened the DNA for transcription (open complex) the newly transcribed RNA blocks the
rifampicin binding site and therefore transcription continues (14). The quinolones and
fluoroquinolones (e.g. Ciprofloxacin) inhibits the DNA topoisomerase II (E. coli DNA gyrase) and
topoisomerase IV. The function of these enzymes is to introduce either negative or positive
supercoils in the DNA respectively. This is mediated through introduction of double strand breaks
which are re-ligated after introduction of the supercoil. Binding of the fluoroquinolone to the DNA
Gyrase blocks re-ligation of the DNA (23). Protein synthesis inhibitors target either the 30S
(aminoglycosides and tetracycline’s) or the 50S subunit (macrolides, chloramphenicol’s and
clindamycin) of the 70S initiator complex necessary for protein synthesis. With most of the protein
synthesis inhibiting antibiotics acting on the elongation of the polypeptide synthesis (18). The cell
14
wall inhibitors can be exemplified by the historically important β-lactam antibiotics (Penicillin’s,
cephalosporin’s, carbapenem’s and monobactam’s) targeting the conserved penicillin binding
proteins (PBPs). PBP are involved in cross linking of peptidoglycan precursor Lipid II in
peptidoglycan synthesis (24). Other inhibitors of cell wall synthesis include the glycopeptide
antibiotic vancomycin (discussed later) which inhibit peptidoglycan synthesis through a different
mechanisms than β-lactams.
Figure 3. Antibiotic targets:
An overview of the major cellular targets of most antibiotics targeting either Gram-negative or Gram-positive bacteria. Sulphonamides and Trimethoprim inhibit folic acid synthesis (22). Rifamycins (Rifampicin) inhibit DNA-dependent RNA polymerase (25). Quinolones and fluoroquinolones inhibit the DNA gyrase and topoisomerase IV (23). Protein synthesis inhibitors include Aminoglycosides, tetracycline’s, macrolides, chloramphenicol’s and clindamycin, which interact with ribosomal subunits 30S and 50S (18). The β-lactams (Penicillin’s, cephalosporin’s, carbapenem`s and monobactam`s) and glycopeptide`s inhibit cell wall synthesis (26). Not all antibiotic drug classes are represented here.
15
Antibiotic resistance
History has shown that after the introduction of new compounds, so follows the
development and dissemination of resistance (Figure 1. bottom). We are today faced with the dire
consequences of untreatable human pathogens that are capable of surviving treatment with all
known antibiotics. This can in part be attributed to decades of uncontrolled antibiotic consumption,
both from the agricultural and human setting (9, 27), but also to the mere fact that evolution and
selection is an intrinsic part of this process. Resistance evolves as a natural selective advantage,
where an organism capable of overcoming an antibiotic perturbation will flourish while other
individuals succumb to the perturbation (28). In the case of antibiotics, these compounds will
inherently drive selection of resistant microorganisms if the genetic background is present in the
population. Once a resistant pathogen has gained foothold, it can spread throughout the population,
and if this resistance profile poses no detrimental side effects, then over time such genotypes will
become continually more prevalent (28).
Genetics of resistance
Most bacteria like Escherichia coli and Staphylococcus aureus contain a single
circular chromosome (29, 30). Exceptions to this do exist, as Vibrio cholera has two chromosomes
(31) and Burkholderia cepacia has three (32). Bacteria are an intricate part of the human biology as
normal commensals of our intestinal and skin flora (12). While most bacteria are important to our
normal health, some have developed as pathogenic species or are opportunistic pathogens and
therefore the target of antibiotic treatment (33, 34). Bacteria generally have short generation times
and unprecedented abundance in nature. Therefore, the evolutionary adaptability of these organisms
is truly remarkable, which is also why, resistance development is a continuing problem.
As for all organisms, the genetic makeup is highly important in the evolutionary
process, and changes to the DNA will accommodate fitness advantages, fitness loss or be neutral
(28). Changes to the genetic makeup, may in turn change the amino acid composition of the protein
targets of antibiotics, such that the interaction of the antibiotic with its target is compromised.
Therefore, modifications to the DNA are highly important to the evolutionary processes. These are
16
driven through minor or major modifications of the genetic makeup. Point mutations, insertions or
deletions of single or multiple bases in the DNA are usually considered as minor and of less
importance to resistance development and dissemination. While, major rearrangements of the DNA
by gene duplication/deletion homologous or non-homologous recombination or inversions of
chromosomal sections have much more pronounced effect on resistance development and
dissemination (35, 36). What truly set bacteria apart from higher organisms is their unprecedented
ability to acquire new genetic material either as addition to their genomes or in the form of extra-
chromosomal genetic material such as plasmids via horizontal gene transfer [Figure 4 (35, 37)].
Acquisition of new genetic material through horizontal gene transfer is accommodated by uptake of
free DNA from the environment (Transformation; through natural competence), phage transduction
or conjugation (transfer of DNA or plasmids between bacteria), not discussed in detail here [Figure
4 (37)]. Of special importance to these processes is that the acquired material can be integrated on
the chromosome or plasmids via integrons or transposons (35). Acquired genes can then be passed
to daughter cells by vertical gene transfer or passed on to other species via horizontal gene transfer
(Fig 3) (37).
17
Figure 4. Vertical and horizontal gene transfer: Adapted from (37)
Horizontal gene transfer through phage transduction, natural competence (uptake of DNA from environment) or conjugation (here exemplified by plasmid transfer from one species to another). Genetic material can be insertion into DNA/Plasmids via integrons or transposons (tn) as exemplified here. Genes and mutations are passed to the next generation through vertical gene transfer or are passed on via horizontal gene transfer.
Mechanism of resistance
The genetic basis of resistance has to translate into bacteria survival by counteracting
the effect of the antibiotic. This can be accommodated in several ways or by combinations of these
in which changes to the genetic material or acquisition of new genetic material, results in either: i)
modification to the antibiotic target, ii) Limiting access to the antibiotic target and/or iii)
modification of the antibiotic. A generalized overview of these mechanisms can be seen in Figure.
5. Below, some examples of these mechanisms are given, they should be considered as examples
and not a comprehensive review, as resistance to many antibiotics is often accommodated through a
combination of these mechanisms.
18
Figure 5. Resistance mechanisms:
Modification of the target; changes the affinity of the antibiotic for the target, this can also be mediated through production of multiple variants of the target (not shown). Limiting access to the target; mediated through efflux of the antibiotic or lowered permeability of the cell. Modification of the antibiotic; by enzymatic activity the antibiotic can be degraded or modified to an inactive form. The genetic background for these mechanisms can be chromosomally or plasmid encoded. See text below for specific examples.
Modificationofthetarget
Resistance to several important antibiotics is accommodated through changes to the
target of the antibiotic, thereby changing the affinity of the antibiotic to the target. Rifampicin
resistance is acquired by mutations to the RNA polymerase beta-subunit (rpoB) gene in E. coli (38)
and Mycobacterium tuberculosis (25, 39). Fluoroquinolone (ex. Ciprofloxacin) resistance is
mediated through sequential acquisition of mutations in the gyrA, gyrB genes (DNA gyrase,
primarily target of fluoroquinolones in Gram-negative bacteria) and parC, parE genes
(topoisomerase IV genes, primarily target of fluoroquinolones in Gram-positive bacteria) depending
on the individual fluoroquinolone and bacterial strain (23). This mode of protection can also be
accommodated through carriage of alternative copies of the target protein, inducible when needed
and enabling the bacterium to survive. In methicillin-resistant Staphylococcus aureus (MRSA) the
mobile genetic element SCCmec (staphylococcal cassette chromosome mec) harboring the mecA
19
gene that encodes an alternative penicillin binding protein (PBP2a) induced by β-lactams and with
low affinity for β-lactam antibiotics (40, 41).
Limitingaccesstothetarget
Many antibiotics exert their effect by interaction with intracellular target (Figure 5).
Limiting access to the intracellular environment of the cell is an important determinant of antibiotic
resistance. In bacteria, efflux mechanisms are highly diverse and important resistance determinants
reviewed in detail elsewhere (15). Usually efflux is accommodated through molecule specific or
multidrug non-specific extrusion of antibiotics via passive or active transport across the membrane
(15). Resistance through efflux is highly problematic in antimicrobial and cancer treatment, but in
bacteria transporters are mainly of the passive type, in contrast to resistance to anticancer
compounds in mammalian cells (42). In bacteria transporters are highly diverse in nature and
divided into five major families; I) The major facilitator family (MF), II) The ATP-binding cassette
(ABC) family, III) the resistance-nodulating division (RND), IV) the drug/metabolite transporter
(DMT) family and V) the multidrug and toxic compound extrusion (MATE) family (15). Non-
specific multidrug-resistance determinants are usually chromosome encoded and most likely not
evolutionary intended for drug transport. Whereas efflux of antibiotics by drug specific transporters
and often found on plasmids (15). The plasmid carried tet genes of E. coli encode the membrane
associated Tet proteins (MF family), that are antiporter that extrude tetracycline in exchange of a H+
(43, 44). Likewise chloramphenicol resistance can be accommodated by carriage of the gene (cmlA)
encoding a protein transporter (MF family) that accommodates efflux of chloramphenicol (45). In
Salmonella enterica multidrug resistance to quinolones, chloramphenicol and tetracycline`s has
been shown to be mediated through overexpression of AcrAB-TolC (RND transporter) (46).
Importantly, resistance by limiting access of the antibiotic can also be mediated by
changing the permeability of the membrane. In the Gram-negative bacterium Pseudomonas
aeruginosa imipenem resistance is mediated via several mechanisms among which loss of the outer
membrane porin, OprD, is a major facilitator (47). Likewise, elevated tolerance to vancomycin in
the Gram-positive bacterium S. aureus can be mediated through thickening of the cell, as first
observed by Hiramatsu et al. (48).
20
Modificationoftheantibiotic
Resistance to antibiotic compounds by degradation or modification of the active
compound is of huge clinical importance. Chloramphenicol resistance can be mediated by carriage
of a constitutive active (E. coli) or inducible (S. aureus) chloramphenicol acetyltransferase cat gene
(49, 50). This encodes the CAT enzyme that is capable of transferring an acetyl group from Acetyl-
Coenzyme A to chloramphenicol, blocking binding of chloramphenicol to the ribosomal subunit.
Likewise aminoglycoside resistance is mediated by aminoglycoside modifying enzymes such as
acetyltransferase (AAC), adenylyl transferases (ANT) or phosphotransferases (APH) that modify
aminoglycosides (51). Finally and with almost premonition of the future Sir Alexander Flemming
himself discovered the production of β-lactamase enzymes capable of hydrolyzing β-lactam
antibiotics such as penicillin (52).
Multidrug-resistance (MDR)
Resistance development in many human pathogens has been on an unprecedented
scale, as resistance has evolved into multidrug resistance. This has led to increased global morbidity
and mortality and we are today facing the possibility of an post antibiotic era (53). Especially
bacterial strains belonging to the ESKAPE group of pathogens (Enterococcus faecium,
Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter, Pseudomonas aeruginosa and
Enterobacter) are of importance to this pandemic (54). These pathogens encompassing both Gram-
negative and Gram-positive bacteria often carry MDR determining genes residing on genetic
resistance island (RI) of complex evolutionary origin that are encoded on the chromosome or
plasmids (29, 55-57).
MDR Gram-positive bacteria:
The widespread MDR among Gram-positive bacteria can be exemplified by the
important opportunistic pathogen S. aureus. This bacterium is a normal commensal carried in 30%
of the human population (58). The bacterium causes a wide variety of infections such as soft tissue
21
and skin infections to life-threatening endocarditis (34). Prior to the introduction of penicillin, S.
aureus caused bacteremia killed 80% of patients (59). The discovery of penicillin led to its
widespread use against S. aureus, but by the mid-1940s penicillin resistance through plasmid borne
penicillinases was widespread (60). As alternative treatment, the semisynthetic penicillin
(methicillin), resistant to degradation, was marketed in 1959. By 1961 methicillin resistant S.
aureus (MRSA) had been described in England (61). The MRSA genotype is caused by carriage of
the SCCmec element of which 11 types have been described (59). The SCCmec elements all share
common features; i) they insert into the same site of the orfX gene on the chromosome of S. aureus,
ii) they contain a mecA gene (PBP2a) controlled by the regulatory genes mecI and mecR and the
cassette chromosome recombinase (ccr) which mediate excision and integration of the SCCmec
element, iii) excision and integration is mediated through recognition of the insertion site sequence
found within specific inverted and direct repeats, recognized by the ccr-encoded DNA recombinase
(59). The MRSA strains evolved from previously treatable background of methicillin sensitive S.
aureus (MSSA).
Several different lineages of MRSA have since evolved and are presently divided into
three major groups; healthcare-associated MRSA (HA-MRSA), community-associated MRSA (CA-
MRSA) and recently livestock-associated MRSA (LA-MRSA) (62, 63). While they all carry the
SCCmec cassette rendering them resistant to virtually all β-lactam antibiotics, they differ in their
ability to cause infection. HA-MRSA and LA-MRSA have both been shown to cause infection in
hospitalized patients as nosocomial infections (63, 64), although LA-MRSA is considered less
virulent in humans (62). This is in contrast to the recently emerged strains of CA-MRSA such as
USA300, capable of causing infections in otherwise healthy adults (65, 66). The differences among
MRSA lineages are mainly associated with differences in virulence between the isolates from the
different lineages. Virulence of MSSA and MRSA are dependent on multifactorial mechanism such
as; toxin production, adhesive properties of the cell and immune evasion to name a few (63). MRSA
strains are problematic because they apart from their SCCmec genotype often carry resistance
determinants to other important antibiotics for treatment of S. aureus infections. Co-carriage of
resistance to aminoglycosides by enzymatic degradation or alteration of the antibiotic has been
found in up to 70% of MRSA isolated in Europe (57). For these reasons treatment of MRSA usually
include treatment with glycopeptide antibiotics (vancomycin) and oxazolidinones such as linezolid.
22
Vancomycin resistance has been reported either as intermittent-vancomycin resistant
S. aureus (VISA) (48) or as vancomycin resistant S. aureus (VRSA) (67). Furthermore, vancomycin
resistance through horizontal gene transfer of the vanA gene cluster has been reported and this
originates from another important Gram-positive pathogen vancomycin-resistant Enterococcus
faecalis (VRE) (68-70). The vanA gene cluster encodes a ligase responsible for synthesis of an
alternative pentapeptide on the peptidoglycan precursor Lipid-II, in which the dipeptide (D-Ala-D-
Ala) is substituted for D-Ala-D-Lac (Figure 6) thereby changing affinity for vancomycin (71).
Induction of the vanA gene cluster is controlled by the VanS-VanR two-component system in
response to extracellular glycopeptide, which was first described in E. faecium (72). Ekstracellular
vancomycin is sensed by VanS (sensor kinase) when vancomycin is bound to D-Ala-D-Ala, VanS
phosphorylates the response regulator (VanR), which in turn regulate expression of the vanHAX
genes responsible for the D-Ala-D-Lac dipeptide substitution (72, 73).
Linezolid is being used increasingly as treatment for MRSA but also in the treatment
of VRE (74) and has been considered unlikely of resistance development. This is due to the
synthetic nature of the molecule whereby enzymatic degradation by preexisting enzymes in nature
would be unlikely (75). Furthermore, linezolid targets and binds to 23S rRNA of the 50S ribosomal
subunit encoded in the ribosomal DNA (rDNA) genes, which are present in multiple copies. This
has been expected to slow resistance through mutation (74). Linezolid resistance is rare (76),
however it has been reported as mutations in multiple 23S rRNA genes. No cross resistance through
mutations has been found to other protein synthesis inhibitors (77). Further linezolid resistance has
been reported through carriage of a Cfr rRNA methyltransferase that modifies the 23S rRNA at
base position A2503 by addition of a methyl group (78, 79).
23
Figure 6. Peptidoglycan synthesis inhibition in S. aureus: Adapted from (80)
Peptidoglycan precursor Lipid II is synthesized in the cytosol of S. aureus. Here depicted after initial synthesis of UDP-MurNac-pentapeptide. MraY (phospho-MurNac-pentapeptide translocase) facilitates the transfer of the UDP-MurNac-pentapeptide to the undecaprenyl phosphate (Lipid carrier), resulting in Lipid I formation. MurG (glycosyltransferase) forms the glycosylic linkage between the MurNac (N-acetylmuramic-acid) and GlcNac (N-acetyl-D-glycosamine) creating Lipid II. Lipid II in transported to the outside of the membrane, where PBP (penicillin binding protein) facilitates crosslinking of the Lipid II subunits into polymeric peptidoglycan. PBP can be blocked by Penicillin, but carriage of the mecA gene from the SCCmec element, will facilitate crosslinking in presence of penicillin, due to the lowered affinity of PBP2a to penicillin. Vancomycin, can bind to the D-ala-D-ala dipeptide, thereby inhibiting peptidoglycan synthesis by PBP2a. Carriage of the vanA gene cluster facilitates the synthesis of alternative dipeptide D-ala-D-lac with lowered affinity for vancomycin, resulting in vancomycin resistance
24
The current last resort antibiotic in use for treatment of serious VISA, VRSA, VRE
and vancomycin resistant Enterococcus faecium, is the lipopeptide antibiotic daptomycin.
Daptomycin interacts with the bacterial membrane in a Ca2+ and phosphatidylglycerol (PG)
dependent manner through electrostatic interactions (81, 82). This causes membrane instability and
mislocalization of cell division proteins (83). As for vancomycin and linezolid, resistance to
daptomycin has been reported, either in VISA strains with overlapping cross resistance to
vancomycin through cell wall thickening (84) or through changes to genes such as the mprF gene
encoding the MprF protein (lysylphosphatidylglycerol synthetase). Dysfunctional regulation of this
gene accommodates synthesis of lysylphosphatidylglycerol (L-PG) instead of the normal PG,
thereby changing the overall net charge of the bacterial membrane [(85) Figure 7].
Figure 7. Lysylphosphatidylglycerol (L-PG) synthesis: Adapted from (86)
The staphylococcal cell wall with peptidoglycan (Orange) and underlying negatively charged phosphatidylglycerol (PG) containing membrane (Black). Dysfunctional expression of the MprF protein facilitates synthesis of Lysylphosphatidylglycerol (L-PG). L-Lysine is believed to be derived from Lysyl-tRNA. L-PG renders the bacterium resistant to Daptomycin and other electrostatic interacting antimicrobials such as host innate immune defence peptides like Defensins.
25
Because of the continuous development and dissemination of resistant isolates, it is of
importance to develop new strategies for combating important Gram-positive pathogens. And while
restriction of antibiotic consumption in the UK has been shown to reduce infection rates with
bacterial infections such as MRSA (87), there is a continued need to develop new or improved
antibiotics for Gram-positive infections. The newest antibiotic for serious Gram-positive infections
is telavancin, but like so many drugs before, this is a derivative of the previously developed
antibiotic vancomycin (88).
26
MDR Gram-negative bacteria:
The Gram-negative bacterial pathogens are by far the most important and costly in our
society today, as the vast majority of nosocomial infections are caused by MDR Gram-negative
infections (89). The highly problematic strains carry Extended Spectrum β-Lactamase (ESBL) and
carbapenemase genes. These encode β-Lactamase enzymes with capacity to hydrolyze several
generation of the β-Lactam antibiotics such as penicillin’s, cephalosporin`s and the last the resort β-
lactams the carbapenem`s [(90-92) Figure 8]. Important bacterial strains encompass the
enterobacteria such as E. coli sequence type ST131 carrying the CTX-M-15 (ESBL) gene (93) and
K. pneumoniae carbapenemase (KPC) ST258 strain (91, 94). Although several inhibitors of ESBL
enzymes have been developed, such as tazobactam and clavulanic acid (95), resistance to such
inhibitors has been described in E coli. (96) and KPC (97).
Figure 8. Structure of Important β-Lactam antibiotics: Adapted from (98)
The figure shows 1. The structure of the D-ala-D-Ala dipeptide on the Lipid II peptidoglycan precursor that penicillin binding protein (PBP) recognizes for the transpeptidation reaction. 2. Overall structure of penicillin 3. Overlay of the penicillin structure with the D-Ala-D-Ala structure. With penicillin recognized as substrate, the PBP gets trapped in its acetylated form and is rendered incapable of performing the transpeptidation step (99). 4. Cephalosporin overall structure. 5. Meropenem structure (carbapenem drug). 6. Tazobactam structure; an inhibitor of several β-Lactamase enzymes (not discussed in detail). R, R1, R2 and X designate the positions where penicillin’s and cephalosporin’s have been modulated for development of new generations of antibiotics.
27
Other highly important MDR Gram-negative bacteria include P. aeruginosa and
Acinetobacter baumannii (21, 100). A. Baumannii is a relatively new problem in the hospital
settings, but is becoming a growing problem in immunocompromised patients (101). It is the
causative agents of a wide variety of infections such as skin and soft tissue infections, urinary tract
infections and life threatening pneumonias (102). Because A. baumannii is a less frequent cause of
serious infection compared to MDR E. coli, K. pneumoniae and P. aeruginosa, it is often
misdiagnosed and the success of initial antimicrobial therapy against A. baumannii is compromised
(103). The recent development of A. baumannii as an important nosocomial infection is largely
attributable to its ability to acquire resistance determining genes and the fact that it is well adapted
to survive in the environment (100). The ability of A. baumannii to acquire resistance genes can be
exemplified by the AYE strain described by Fournier et al. (104). This strain contains an 86-
kilobase resistance island that includes resistance to many β-lactams, fluoroquinolones,
tetracycline’s, aminoglycosides and more. A major part of the genes were acquired via horizontal
gene transfer (104). Such resistance islands have been associated with widespread MDR resistance
is other Gram-negative pathogens like K. pneumoniae and especially the dissemination of ESBL
and carbapenemase genes (55).
Carbapenemase resistance has led to the re-introduction of the peptide antibiotics,
colistin (polymyxin E) and polymyxin B. Discovered in 1949 (polymyxin E), but used very limited
due to their unattractive toxicity profile (105). These antimicrobial peptides are now used
exclusively as last resort antibiotics against MDR Gram-negative infections that are resistant to all
other antibiotics (105). As they were developed and approved prior to the introduction of modern
standards for clinical approval by the United States Food and Drug Administration (FDA) and
European Medicines Agency (EMA), they have not undergone the same vigorous clinical testing as
newer compounds. Therefore less is known about optimal dosing, pharmacokinetics and
pharmacodynamics (106). Polymyxins are cationic amphipathic circular peptides and kill bacteria
through disruption of the bacterial membrane (Figure 9). Specifically polymyxins disrupt membrane
integrity via electrostatic interaction with the anionic charged LPS layer of the outer membrane,
while displacing Mg+ and Ca2+, leading to cell leakage and cell lysis (107).
28
Figure 9. Polymyxin Mechanism of action: Adapted from (107)
The binding of colistin to the anionic charged LPS layer of gram-negative bacteria causes displacement of cations. Disrupting membrane stability, leads to influx of more colistin molecules and disruption of the inner bacterial membrane.
Historically it has been considered unlikely that colistin resistance would develop.
However, genomic resistance to colistin has been reported in A. baumannii and K. pneumoniae as
changes to the LPS layer (108-111). In A. Baumannii, colistin resistance is acquired through
complete loss of LPS (109) or by changes in the two-component PmrA-PmrB system (polymyxin
resistance A and B). The PmrA-PmrB two-component system is a major regulator of LPS
modifying gene products. PmrA-PmrB is normally induced by external signals such as low pH,
high Fe3+ or high Al3+. When induced the sensor kinase PmrB auto-phosphorylates and transfers the
phosphoryl group to the response regulator PmrA. Phosphorylated PmrA regulates LPS modifying
genes through DNA binding (112). Colistin resistance through mutations in PmrA-PmrB is
mediated via point mutations in pmrB (113), constitutive activation of PmrA (114) or upregulation
of pmrAB (113). These changes can lead to addition of phosphoethanolamine to the Lipid A through
expression of the pmrC gene (112, 115). In K. pneumoniae, resistance can also be mediated through
changes in the PhoP-PhoQ (nonspecific acid phosphatase) two-component system (110), involved
29
in sensing of Mg2+ and Ca2+, and which can cross talk through the PmrA-PmrB two component
system (116). Further, PhoP-PhoQ has been shown to regulate the Pagp gene responsible for
modification to Lipid A thereby changing the overall negative charge of the membrane (117). Lipid
A, being the inner most part of LPS anchoring it to the bacterial outer membrane.
However, of the outmost importance is the new discovery of plasmid mediated
colistin resistance in E. coli, encoded in the mcr-1 gene, which result in the addition of
phosphoethanolamine to the Lipid A rendering the bacterium colistin resistant (118). This
mechanism is essentially the same as in A. Baumannii PmrA-PmrB mutants (113). The discovery of
colistin resistance via horizontal gene transfer seriously underscores the foreseeable future of a post
antibiotic era. Transfer of the mcr-1 gene to already MDR carbapenemase carrying Gram-negative
pathogens could render these strains untreatable. As described, horizontal gene transfer has already
been ascribed to the rapid emergence and spread of the global MDR pandemic (35) and will
undoubtedly increase the prevalence of colistin resistance. Therefore, the continued development of
new or improved antimicrobials is of the outmost importance, especially against Gram-negative
bacteria of the ESKAPE group (106).
Peptide antibiotics: a part of the solution
The first antimicrobial peptide (AMP) to be described in detail was Gramicidin.
Discovered around the same time as penicillin (1939), by a French microbiologist René Dubos
while working with the peptide producer Bacillus brevis (119). Gramicidin was the first
commercially produced true antibiotic and proved especially efficient at killing gram-positive
bacteria. However, Gramicidin had limited applications because of its hemolytic ability and
therefore it was only applicable as topical treatment. Another such important AMP used is
bacitracin (120), like colistin it is a non-ribosomal synthesized naturally produced mixture of
antimicrobial peptides, but unlike colistin has broad spectrum activity. AMPs have been isolates
from single celled to multicellular organisms (121) and are usually composed of 10-100 amino
acids (122). They are highly diverse in structure and activity and because of their widespread
distribution in nature they have been proposed as new sources of antibiotics (122, 123).
30
Bacteriocins
Bacteriocins are a highly diverse group of AMP from bacteria. They are ribosomal
synthesized AMPs and serve as a means of inhibiting/killing closely related bacteria, while the
producer itself is immune (124). Because bacteriocins are produced intracellularly, they are usually
synthesized in an inactive pre-peptide form, which is transported through the membrane via ABC-
transporters. This inactive pre-peptide incorporates a leader sequence which is cleaved off either
intracellularly, during or after export, rendering the peptide active (125, 126). The producer strain is
immune to its own bacteriocins, via co-expression of immunity proteins (124, 127). Bacteriocins
have been divided into many different classes depending on their structure, mode of action and
spectrum of activity, but recently it was suggested to change this system to contain only 3 classes;
Class I (Lanthionine-containing bacteriocins/lantibiotics), Class II (Non-lanthionine-containing
bacteriocins and Class III, the bacteriolysins (not discussed here) (125). Bacteriocins target a variety
of cellular processes, but generally speaking the bacteriocins targeting Gram-positive bacteria act
on the bacterial membrane. These can be exemplified by the lantibiotic nisin (128) (described in the
next section) and Lactococcocin A (129, 130) (Figure 10). Whereas many Gram-negative targeting
bacteriocins target intracellular processes such as DNA replication, transcription or protein
J25 (MccJ25) inhibit the RNA polymerase (133) and microcin MccC7-51 inhibit protein synthesis
(Figure 9).
31
Figure 10. Bacteriocins mechanism of action: An overview. From (131)
The Bacteriocins Nisin and Lactococcocin both act on Gram-Positive bacteria, by pore formation. Nisin also inhibit
peptidoglycan synthesis. Nisin in known for its interaction with Lipid II (128), whereas Lactococcocin binds to units of
the mannose-phosphotransferase system (Man-PTS)(134). Microcins (MccB17, MccJ17 and MccC7-51) target DNA
gyrase, RNA polymerase and protein synthesis respectively (131). MccB17 cross the outer membrane through the porin
OmpF and are actively taken up by the inner membrane protein SbmA (135). MccJ25 binds to outer membrane receptor
FhuA (Ferrichrome receptor) and cross the inner membrane through SbmA or TonB (135). MccC7-51 like MccB17
crosses the outer membrane through OmpF porin, but utilizes a YejBEF-transporter to cross the inner membrane (136)
32
Lipid II targeting antimicrobial peptides
The Lipid II precursor of peptidoglycan synthesis is not a new target of antibiotics, as
already discussed antibiotics such as vancomycin and telavancin also utilizes this cell wall target.
However, the class of peptide antimicrobials known as lantibiotics has gained renewed interest
because of the widespread multidrug-resistance among Gram-positive bacteria (137, 138).
Lantibiotics are named so, for containing uncommon amino acids such as lanthionine
and methyllanthionine introduced via posttranslational modified ring-structures of the precursor
peptide (139, 140) (Figure 11). Lantibiotic are produced from gene clusters encoded on the
chromosome, on conjugative elements or plasmids (138). The overall structure is composed of
several genes involved in their synthesis, modification, export and immunity (designated as lan for
Lantibiotic). The lanA gene encodes the inactive pre-peptide form and modifying enzymes are
encoded in the lanBC, lanM or others. An ABC-transporters gene (LanT) encodes the transporter
and immunity is encoded in the lanI and lanH genes (141). The inactive pre-peptide incorporates a
leader sequence which is either cleaved off intracellularly by proteases encoded in the lanP gene (if
present) or other cellular proteases not encoded in the lan gene cluster (141). Several lantibiotics
have activity against important Gram-positive pathogens and especially the activity against MRSA
and VRE have sparked renewed interest in lantibiotics (137). Nisin is one of the best described
lantibiotics to date. It was discovered in 1928 from its natural producer Lactococcus lactis, but was
not isolated before the 1940s (142, 143). Nisin has never been applied to clinical settings, but it has
been used extensively as an additive by the food production industry (144). The mode of action of
nisin has been described as multimodal; by inhibition of peptidoglycan synthesis and pore
formation via binding to Lipid II (128, 145) in which aggregation of Lipid II in the membrane
seems to play an important role (146-149). Nisin initially bind through electrostatic interactions, but
this is considered of less importance to antimicrobial activity compared to Lipid II binding (145).
Several other lantibiotics have been discovered; such as mutacin 1140 (150),
planosporicin and microbisporicin also known as NAI-107 (151, 152), gallidermin, mersacidin and
many more (137, 139). Mutacin and mersacidin inhibit peptidoglycan synthesis, but do not form
pores like Nisin, however they do binds to Lipid II like most lantibiotics (149, 153, 154). NAI-107
also binds to Lipid II, with no apparent pore formation; rather it seems to disrupt membrane
33
function through interruption of protein localization thereby disorganizing the membrane (155).
These lantibiotics are just a few examples of antimicrobials from this group with growing interest
for clinical development (137, 156). The consensus between lantibiotics ability to kill by
multimodal mechanisms via Lipid II binding, their general low toxicity and that resistance
development has been slow, has been used to argue for their development as novel clinical therapies
(125, 157). Furthermore other antimicrobial peptides have been found to target Lipid II, such as the
defensin plectasin (80) and the recently discovered bacteriocin teixobactin. Teixobactin also target
the Lipid III precursor of wall techoic acid synthesis and has pronounced effect against VRSA and
VRE emphasizes the non-protein Lipid II and Lipid III precursors as a good target of novel
therapeutics (158).
Figure 11. Lantibiotics: From (155)
The Nisin-like lipid II binding motif is highlighted in green and lipid II binding motifs similar to that found in
mersacidin are marked red.
34
Host defense peptides
Innate immunity peptides of multicellular organisms or “host defense peptides” are
widespread in nature as part of almost all living organisms immune defense (121, 122, 159). Innate
immunity peptides capable of killing bacteria, such as the Cecropins from the moth Hyalophora
cecropia (160, 161), LL-37 part of the Human innate immunity (162) and defensins of invertebrate
and mammalian origin have become of interest in development of novel therapeutics (163, 164).
Currently more than 2000 antimicrobial peptides of eukaryotic origin have been reported (121,
165). These peptides, although similar in their antimicrobial activity, are highly diverse in sequence
and structure (122). Major structural classes include; α-helical peptides, β-sheet peptides, extended
peptides (enriched for certain amino acids) and looped peptides (122, 166) (Figure 12).
Figure 12. Structures of host defence peptides. Adapted from (122, 166)
A. Bovine Indolicidine (extended structure) (167). B. Bovine Lactoferricin B (β-sheet structure (168)). C. Human β-defensin-1 (mixed structure; both α-helix and β-sheet (169)). D. Drosophila melanogaster, Drosomycin (mixed structure (170)). E. Amphibian magainin (α-helix structure (171)). F. Insect Thanatin (Loop structure (172). Structures are from the Antimicrobial peptide database (http://aps.unmc.edu/AP/main.php) (121). Peptide database numbers; A. 1G89, B. 1LFC, C. 1IJV, D. 1MYN, E. 2MAG and F. 8TFV (121).
35
Innate immunity AMPs like many bacteriocins are often cationic and amphiphilic and
interacts with membranes through electrostatic interactions; creating pores that disrupt membrane
integrity (122). Several models of pore formation and membrane disruption have been proposed.
These can generally be divided into three models; i) the barrel-stave model ii) the carpet model and
iii) the toroidal model [(166, 173) Figure 13]. Furthermore, antimicrobial peptides of both
prokaryotic and eukaryotic origin and with novel mechanistic actions other than direct bacterial
killing have been reported; such as immunomodulatory peptides (174-178), anti-virulence peptides
(179, 180) and many more (121).
Figure 13. Overview of the pore formation models: Adapted from (173)
In the barrel-stave model: the peptide (hydrophilic in red and hydrophobic in blue) attach to and aggregate in the
membrane and insert into the membrane. With the hydrophobic part aligned with the lipid core region and the
hydrophilic region pointing to the centre. The carpet model: The peptides create a carpet structure by orientation in
parallel to the lipid bilayer, with the hydrophobic regions binding to the lipid surface, leading to disrupting of the
membrane. The toroidal model: the peptides aggregate and cause the membrane to bend so the membrane is disrupted
and the pore centre is lined with the peptide and head groups of the lipid bilayer.
36
Because microorganisms have co-evolved as an intricate part of the intestinal and skin
microflora of multicellular organisms (12) and some of these organisms have evolved into
pathogenic or opportunistic pathogens, they have had to co-evolve defences against host defence
peptides (181, 182). Many of these mechanisms are equivalent to the mechanisms evolved for
coping with antibiotics. Membrane modifications and AMP repulsion; membrane modifications as
protection mechanisms against peptide antimicrobials have already been discussed for A. baumannii
(PmrA-PmrB), K. pneumoniae (PhoP-PhoQ) and as production of L-PG in S. aureus. Capsule (S-
layer) production have also been shown to be important for K. pneumoniae protection against
AMPs such as lactoferrin and defensins (183). Similar to these mechanisms the dlt operon
(dltABCD genes) is responsible for D-alanine incorporation into techoic and lippotechoic acids of S.
aureus, thereby reducing the negative change of the membrane and providing protection against
AMPs such as nisin and gallidermin (184). Efflux of AMPs; In Clostridium difficile it has been
shown that the crpABC operon encoding an ABC transporter that provides resistance to gallidermin
and nisin (185) and in E. coli intrinsic resistance to the microcin J25 is provided through the global
regulatory protein Lrp (Leucine-responsive regulatory protein) which is a positive regulator of an
efflux pump YojI (ABC transporter) (186). Degradation of AMPs; The production of proteases
such as elastase from P. aeruginosa and E. faecalis or the protease aureolysin from S. aureus
provides protection from the broad spectrum human AMP LL-37 by enzymatic degradation (187,
188). Because of such mechanisms and many more, as reviewed elsewhere (181, 182, 189), AMPs
are not necessarily and readily applicable directly from the producer organisms.
Modified or synthetic: peptides and peptoids
The complexity and universally widespread distribution of peptides, of both bacterial
and mammalian origin underlines the potential of peptides for the development of novel
therapeutics. Because of the immense development of MDR resistance; research into development
and discovery of these compounds is both a necessity and an obvious course for development of
new antimicrobials. New technologies have provided ways of manipulating peptides to create
molecules of diverse structure and applicability (165, 190, 191). In this way several attempts has
been made to modify polymyxins to improve their efficacy while lowering toxicity (192).
37
Lantibiotics has also been proposed for manipulation (125). Further, hybrid molecules such as the
Cecropin-mellitin hybrids have been created (193) and peptides targeting novel pathways such as
DNA replication machinery of S. aureus (194).
Antimicrobial peptides can be chemically synthesized as linear molecules (195) or be
made cyclic either by chemical modification or by use of in vivo synthesis (196). Peptides can be
used as the basis for peptide mimetics such as peptoids [(197, 198) Figure 14]. Peptoids like
circularization of linear peptides has the advantage, that it renders the molecule less prone to
degradation by enzymatic digestion (197, 199). Similarly incorporation of D-amino acids,
glycosylation, or phosphorylation of peptides can be utilized to lower a peptides susceptibility to
protease degradation (200, 201). The possibilities of peptide synthesis seem unlimited as
technological development has provided methodologies for modifying these molecules.
Figure 14. Peptides and Peptoids; Adapted from Mojsoska et al. (197)
Chemical structure of peptides to the left and peptoid structure to the right.
Therefore, one major question remains; with the immense discovery of new molecules
and with the methodologies at hand to manipulate these into new and novel antimicrobial peptides,
why have so few antimicrobial peptides been approved for clinical therapy? The answer to this
question might just be (as described) low interest from the pharmaceutical industry. But of more
importance could be that many of the peptides that are brought into clinical development fail due to
toxicity problems. Or molecules are rushed into clinical development, where they fail before being
optimized properly and are eventually abandoned (202). The solution to this could be to have more
38
cost effective methodologies for discovering and testing of toxicity and efficacy in vivo, prior to
engaging in expensive and highly regulated mammalian in vivo experiments.
Alternative in vivo models
Clinical trials inevitably have to pass through mammalian efficacy and toxicity
models, before any antibiotic compound is approved for human trials. Mammalian infections
models are highly regulated by legislative laws. They are also expensive for development and this
poses a problem in large scale drug screening (19, 203). During the last decades, several alternative
methodologies has been developed for in vivo drug development and screening using non-
mammalian models. High throughput screening of large drug libraries was classically carried out in
vitro to discover new compounds that effectively kill/inhibit the proliferation of bacteria (204).
Classical toxicity screens are usually performed in vitro and encompass hemolysis assays and cell
proliferation assays in which immortalized cell lines are used (197, 205). However, using this
approach there is a possibility of underestimating toxicity that would have been discovered in a
whole animal context. Moy et al (206) devised a Caenorhabditis elegans in vivo model that can be
utilized for screening of large drug libraries. This method found a variety of drugs/pro-drugs
capable of killing/inhibiting growth, virulence inhibitors and immune modulators while also
screening for toxicity (206). This model is powerful for large scale screening as it is inexpensive
and C. elegans is easy to grow and rear in the laboratory. However, in this model screening is
performed by ingestion of molecules and only molecules that are not degraded or toxic through the
digestive system will be discovered.
Insect models are becoming widely used for the analysis of host-pathogen
interactions. Among these models two stand out; the greater wax moth Galleria mellonella and the
fruit fly Drosophila melanogaster. These models have applicable potential for drug development
and screening of antimicrobials. The larvae of G. mellonella have been explored as a model for
pathogenicity, through oral or direct injection of bacterial pathogens (207, 208). This model has
several advantages; it is cheap to rear and easily handled in the laboratory, no ethical clearance is
39
needed, it has a short life cycle and it can be kept at temperatures from 15-37°C (207, 209). Evans
et al. demonstrated that G. mellonella can be utilized to study virulence of Streptococcus
pneumoniae, as the model discriminates between strains with known differences in virulence (210).
In line with this Peleg et al. reported on the use of G. mellonella as a model for analysing
pathogenicity of A. baumannii and testing of antibiotics (211, 212). Several other bacterial
pathogens such as P. aeruginosa (213) and E. coli (214) have undergone similar studies in G.
mellonella.
Drosophila has previously been proposed as a good model for the discovery of
antimicrobials and screening of toxicity (215, 216). It has been extensively applied by Ross Cagan
for the discovery and screening of combinatorial chemo therapy (217, 218), and by Willoughby et
al. (219) to screen for new chemotherapeutics.
Drosophila, has been used extensively for genetic screening to unravel important
aspects of cellular biology in a whole animal context (220). Its genetic tractability through the
development of advanced methodologies for genetic manipulation has propelled it as a highly
valuable model organism for elucidating important aspects of the genetic regulation of cellular
biology (221, 222). Like G. mellonella, Drosophila has a fast generation time of only 10 days, is
cheap to rear and has no ethical constraints, making it attractive as an initial model for efficacy and
toxicity testing in whole animals.
Since the Nobel Prize winning discovery of the Toll like receptor by Jules A. Hoffman
and coworkers (223), Drosophila has provided valuable information to the control of innate
immunity and its regulation (224, 225). Initially, Drosophila was described as being bi-partite in the
IMD (immune Deficient) and the TOLL pathway (226, 227) (Figure 15), however newer evidence
points towards a more complex interaction between the two pathways. The Toll pathway is the
major regulator of the immune defense towards Gram-positive bacteria and fungi in Drosophila
(226, 228-231), while the IMD pathway mainly functions as a regulator of the immune response
against Gram-negative bacteria (224, 227, 231, 232). The Drosophila innate immune system has
highly conserved homology with its mammalian counterparts; from the recognition of PAMP
(pathogen-associated molecular patterns) by the PGRP`s (peptidoglycan recognizing proteins) to the
nuclear localization and transcription of immunity associate genes, such as innate immunity
40
peptides cecropins and defensin, by Jun N-terminal kinase (JNK), Dorsal-related immunity factor
(DIF), DORSAL etc. [Figure 15 (230, 231, 233-238)]. However, major differences are present. In
mammals the activation of TOLL relies on direct binding of the receptor to the invading
microorganism, whereas Drosophila relies on the secreted PGRP`s to translate recognition through
TOLL into cellular signal transduction and activation of immunity genes (233, 234, 236, 237).
Further, the secreted mammalian PGRPs act in direct bactericidal fashion (235). The major nuclear
activators of transcription are however conserved, although the mammalian regulatory system is
much more complex (239).
Research within the field of insect innate immunity in Drosophila has sprouted the
development of several techniques for infecting Drosophila by ingestion or injection with bacteria
(240-246). It has been demonstrated that infection with the important pathogenic bacteria Vibrio
cholera in Drosophila in many aspects compare to infection in humans (247). Others have applied
Drosophila to gain insight into virulence of P. aeruginosa (246), virulence and treatment of S.
aureus (248-250) and many more bacterial species (243). However, virulence studies for
comparison of Drosophila infection with human infection remain controversial, since some studies
have shown that non-pathogenic Gram-positive bacteria kill Drosophila (243, 251).
41
Figure 15. Drosophila Immunity:
Left side: Toll pathway activation leading to transcription of antimicrobial peptide genes such as Drosomycin
and defensin. Gram-positive bacteria are detected via binding of PGRP-SA and PGRP-SD (Peptidoglycan
Recognizing Proteins) to Lysine-type peptidoglycan and east by B-Glucan binding to GNBP (Gram negative
binding protein). Virulence factors from yeast and Gram-positive bacteria, such as proteases, are detected via
Persephone. Detection of bacteria and/or their virulence factors confers signalling through the TOLL receptor
via binding of spätzle to the receptor. Full length spätzle is cleaved by Spe (spätzle-processing enzyme) which is
induces either directly by Persephone or through a serine protease cascade by PGRP. Right side: IMD pathway
induction through PRRP-Le (-LF/LB/LCx etc.) by binding to DAP-type peptidoglycan of Gram-negative
bacteria. This binding confers signalling through IMD (Immune deficiency), which through a complex signalling
cascade lead to induction of antimicrobial peptides and stress and wound responsive genes. For more detailed
description al the steps in the signalling cascade (231).
42
Paper I
An all-D amphipathic undecapeptide shows promising activity against colistin-resistant
strains of Acinetobacter baumannii and a dual mode of action
Alberto Oddo,a‡ Thomas T. Thomsen,b‡ Susanne Kjelstrup,b Ciara Gorey,a Henrik Franzyk,a Niels
Frimodt-Møller,c Anders Løbner-Olesen,b Paul R. Hansena
Antimicrob. Agents Chemother.AAC.01966-15; Accepted manuscript posted online 16 November
2015,
‡These authors contributed equally to this work.
Department of Drug Design and Pharmacology , University of Copenhagen,
Copenhagen, Denmarka; Dept. of Biology, Section for Functional Genomics and
Center for Bacterial Stress Response (BASP), University of Copenhagen,
Copenhagen, Denmarkb; Department of Clinical Microbiology, Rigshospitalet,
Copenhagen, Denmarkc
43
An all-D amphipathic undecapeptide shows promising activity against colistin-1
resistant strains of Acinetobacter baumannii and a dual mode of action 2
3
4
5
Alberto Oddo,a#‡ Thomas T. Thomsen, b‡ Susanne Kjelstrup,b Ciara Gorey,a Henrik 6
Franzyk,a Niels Frimodt-Møller,c Anders Løbner-Olesen,b# Paul R. Hansena# 7
8
Department of Drug Design and Pharmacology , University of Copenhagen, 9
Copenhagen, Denmarka; Dept. of Biology, Section for Functional Genomics and 10
Center for Bacterial Stress Response (BASP), University of Copenhagen, 11
Copenhagen, Denmarkb; Department of Clinical Microbiology, Rigshospitalet, 12
Copenhagen, Denmarkc 13
14
15 16
#Address correspondence to Alberto Oddo, [email protected]; Anders 17
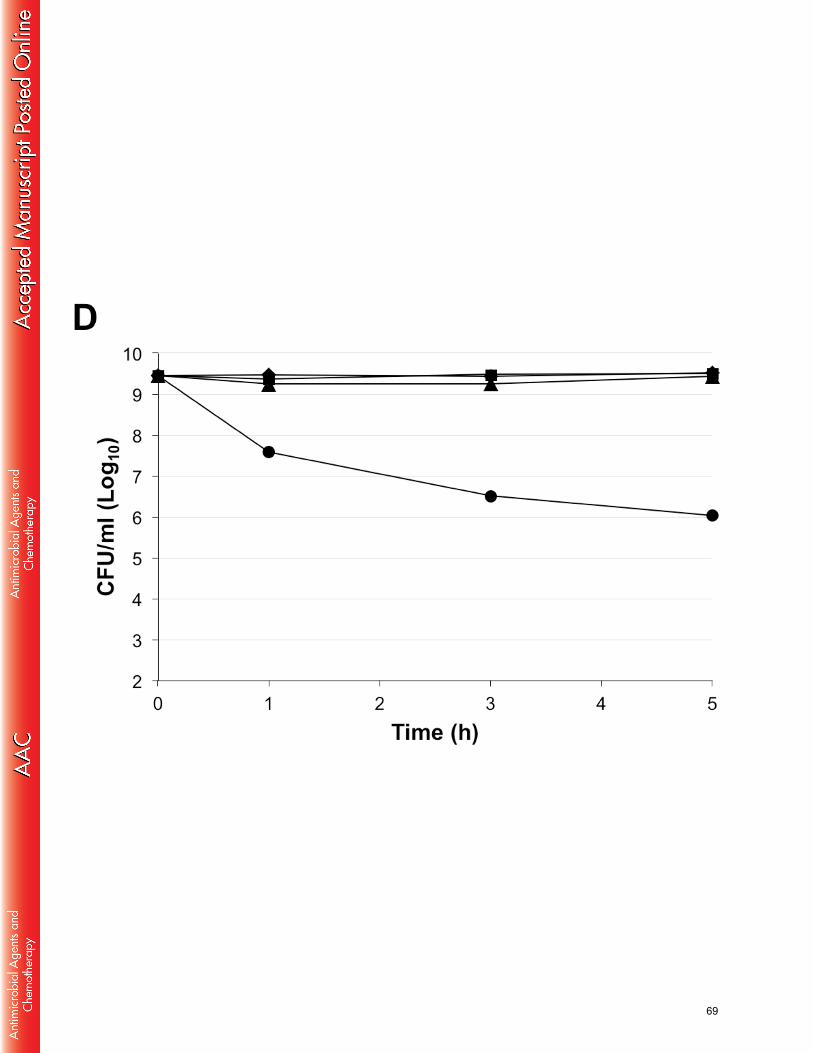
In terms of in vitro MIC, the enantiomeric pair BP203 and BP214 behaved very 317
similarly against most species (Table 1). This is expected for membrane-active 318
peptides that do not bind specifically to any target – e.g. cecropin, melittin and their 319
hybrids (33). However, moderate but consistent differences in MIC were observed 320
against several A. baumannii strains (Table 2), indeed suggesting the presence of 321
binding targets with strict chiral requirements – as it is the case for e.g. colistin and 322
56
drosocin (10, 34). Ultimately, all BP-peptides are able to kill bacteria via non-323
specific, amphipathicity-driven membrane damage; additionally, as far as A. 324
baumannii is concerned, BP214 appeared able to interact with certain structural 325
elements also in a more specific fashion. For wild-type strains and pmr mutants, the 326
high number of stereocenters in the saccharide portion of the LPS may very well 327
account for the observed enantiomeric discrimination. Moreover, being a prominent 328
feature of the cell wall, the LPS can be expected to play a major role in determining 329
the susceptibility of Gram-negative bacteria to membrane active agents in general. 330
Accordingly, LPS-deficient lpx mutants proved consistently less susceptible than 331
LPS-modified pmr mutants towards the investigated BP-peptides – again, with the 332
exception of the RW-analog. 333
However, the lpxC mutant Ab-167R proved unexpectedly very susceptible to BP214. 334
Interestingly, the same strain had been previously reported to be 100-fold less 335
susceptible to LL-37 than its parent strain, whereas other mutants proved as 336
susceptible (22). BP203 also resulted 16-fold less active than its enantiomer. While 337
the advantages of RW-BP100 can be rationalized as described above, for LL-37, 338
BP203 and BP214 the same task is more arduous without assuming the presence of 339
specific binding targets other than the LPS. The existence of such structures has been 340
hypothesized before in order to explain the higher anionic zeta potential of stationary-341
phase lpxA mutants than their parent strains (32). The lower susceptibility of Ab-176R 342
suggests that these structures might be constitutive but lost, modified or masked as a 343
consequence of lpxD mutation. 344
Further insight was provided by the observed antagonism between colistin and BP-345
peptides when challenging pmr mutants (Table 2). Due to the cationic nature of all 346
these compounds, the sequestration of BP-peptides by colistin does not appear 347
57
probable. However, previous studies have shown that, although unable to exert a 348
bactericidal effect, colistin can still effectively bind to the outer membrane of resistant 349
A. baumannii cells (28). A plausible explanation is therefore that colistin and BP-350
peptides compete for binding to the modified LPS, but the former is not able to 351
translate binding into bacterial killing. However, most BP-peptides were heavily 352
affected by the presence of colistin, confirming that the latter is a high-affinity ligand 353
also for the modified LPS. 354
Several observations support this competitive model: i) thanks to its stronger 355
cationicity and/or non-specific membrane-activity, RW-BP100 appeared unperturbed 356
by the presence of colistin; ii) the presence of colistin at high concentration raises the 357
MIC of BP-peptides for pmr mutants virtually to the same level as for the LPS-358
deficient lpx mutants – as in both cases LPS-binding is not possible; iii) this 359
antagonism was generally not observed for lpx mutants. However, the activity of 360
BP214 against Ab-167R made again an exception. This was the only case in which 361
competition between colistin and BP-peptides was observed when challenging an lpx 362
mutant. By definition, a competitive binding implies the presence of a defined target 363
available in limited quantity. This is confirmed by the activity difference between 364
BP214 and its enantiomer. Clearly, for an lpx mutant this target cannot be the LPS. 365
The competition between BP214 and colistin in the case of Ab-167R leads to 366
additional interesting considerations. To our knowledge, it has never been shown 367
before that colistin can bind other intrinsic membrane targets with high affinity. The 368
prominence of the LPS is presumably the reason why this phenomenon has not been 369
observed before. 370
From a structural perspective, the advantage of BP214 over colistin might stem from 371
the higher flexibility of linear peptides compared to macrocycles (35). Being more 372
58
rigid, colistin can bind the LPS paying a lower entropic penalty and thus with higher-373
affinity; however, this rigidity prevents it from binding to a modified partner without 374
substantial differences. The more flexible BP214 cannot bind with as high an affinity, 375
but is able of modifying its conformation easily, resulting in more robust 376
antimicrobial activity. This hypothesis however remains to be demonstrated. 377
In conclusion, under optimal conditions colistin’s activity against susceptible A. 378
baumannii strains remains unrivaled, but its performance drops dramatically in a 379
variety of other relevant scenarios. Colistin-resistant strains are becoming 380
increasingly common and virtually immune to the drug at viable concentrations. Due 381
to its toxicity to kidneys, increasing dosages constitutes a serious collateral risk for 382
patients. The advantages of slightly less active but more reliable agents should thus be 383
carefully taken into consideration. 384
BP214 is one such agent and arguably the most promising member of its family 385
identified to date. Its dual mode of action, both specific and non-specific, resulted in a 386
potent and very robust antimicrobial activity. Being composed of D-amino acids only, 387
BP214 can be expected to be proteolytically stable and potentially suitable for oral 388
administration (36). 389
Overall, BP214 displayed attractive antimicrobial properties and, most importantly, 390
its small size and chemical simplicity hold promise of ample improvement potential. 391
These characteristics make BP214 an attractive lead for the development of novel 392
antimicrobials targeting threatening Gram-negative pathogens, and especially A. 393
baumannii. 394
395
ACKNOWLEDGEMENTS 396
59
This study has been funded by the Marie Curie Actions under the Seventh Framework 397
Programme for Research and Technological Development of the EU (Grant 398
Agreement N° 289285). Financial support from the Augustinus Foundation is also 399
kindly acknowledged. The authors wish to thank Jytte M. Andersen for excellent 400
technical support. Prof. McConnell of the University of Seville, Spain, and Prof. Luis 401
Rivas of the University of Madrid, Spain, are gratefully acknowledged for providing 402
the clinical isolates mentioned in Table 2. 403
404
REFERENCES 405
1. Perez F, Hujer AM, Hujer KM, Decker BK, Rather PN, Bonomo RA. 406 2007. Global Challenge of Multidrug-Resistant Acinetobacter baumannii. 407 Antimicrob. Agents Chemother. 51:3471-3484. 408
2. Hawley JS, Murray CK, Jorgensen JH. 2008. Colistin Heteroresistance in 409 Acinetobacter and Its Association with Previous Colistin Therapy. 410 Antimicrob. Agents Chemother. 52:351-352. 411
3. Li J, Rayner CR, Nation RL, Owen RJ, Spelman D, Tan KE, Liolios L. 412 2006. Heteroresistance to Colistin in Multidrug-Resistant Acinetobacter 413 baumannii. Antimicrob. Agents Chemother. 50:2946-2950. 414
4. Abbott I, Cerqueira GM, Bhuiyan S, Peleg AY. 2013. Carbapenem 415 resistance in Acinetobacter baumannii: laboratory challenges, mechanistic 416 insights and therapeutic strategies. Expert. Rev. Anti Infect. Ther. 11:395-409. 417
5. Vila J, Pachon J. 2012. Therapeutic options for Acinetobacter baumannii 418 infections: an update. Expert Opin. Pharmacother. 13:2319-2336. 419
6. Taccone FS, Rodriguez-Villalobos H, De Backer D, De Moor V, Deviere J, 420 Vincent JL, Jacobs F. 2006. Successful treatment of septic shock due to pan-421 resistant Acinetobacter baumannii using combined antimicrobial therapy 422 including tigecycline. Eur. J. Clin. Microbiol. Infect. Dis. 25:257-260. 423
7. Valencia R, Arroyo LA, Conde M, Aldana JM, Torres MJ, Fernandez-424 Cuenca F, Garnacho-Montero J, Cisneros JM, Ortiz C, Pachon J, Aznar 425 J. 2009. Nosocomial outbreak of infection with pan-drug-resistant 426 Acinetobacter baumannii in a tertiary care university hospital. Infect. Control 427 Hosp. Epidemiol. 30:257-263. 428
8. López-Rojas R, Jiménez-Mejías ME, Lepe JA, Pachón J. 2011. 429 Acinetobacter baumannii Resistant to Colistin Alters Its Antibiotic Resistance 430 Profile: A Case Report From Spain. J. Infect. Dis. 204:1147-1148. 431
9. Vaara M. 2013. Novel derivatives of polymyxins. J. Antimicrob. Chemother. 432 68:1213-1219. 433
10. Tsubery H, Ofek I, Cohen S, Fridkin M. 2000. The Functional Association 434 of Polymyxin B with Bacterial Lipopolysaccharide Is Stereospecific: Studies 435 on Polymyxin B Nonapeptide. Biochemistry 39:11837-11844. 436
60
11. Beceiro A, Moreno A, Fernández N, Vallejo JA, Aranda J, Adler B, 437 Harper M, Boyce JD, Bou G. 2014. Biological Cost of Different 438 Mechanisms of Colistin Resistance and Their Impact on Virulence in 439 Acinetobacter baumannii. Antimicrob. Agents Chemother. 58:518-526. 440
12. Beceiro A, Llobet E, Aranda J, Bengoechea JA, Doumith M, Hornsey M, 441 Dhanji H, Chart H, Bou G, Livermore DM, Woodford N. 2011. 442 Phosphoethanolamine Modification of Lipid A in Colistin-Resistant Variants 443 of Acinetobacter baumannii Mediated by the pmrAB Two-Component 444 Regulatory System. Antimicrob. Agents Chemother. 55:3370-3379. 445
13. Moffatt JH, Harper M, Harrison P, Hale JDF, Vinogradov E, Seemann T, 446 Henry R, Crane B, St. Michael F, Cox AD, Adler B, Nation RL, Li J, 447 Boyce JD. 2010. Colistin Resistance in Acinetobacter baumannii Is Mediated 448 by Complete Loss of Lipopolysaccharide Production. Antimicrob. Agents 449 Chemother. 54:4971-4977. 450
14. Velkov T, Thompson PE, Nation RL, Li J. 2010. Structure—Activity 451 Relationships of Polymyxin Antibiotics. J. Med. Chem. 53:1898-1916. 452
15. Saugar JM, Alarcón T, López-Hernández S, López-Brea M, Andreu D, 453 Rivas L. 2002. Activities of Polymyxin B and Cecropin A-Melittin Peptide 454 CA(1-8)M(1-18) against a Multiresistant Strain of Acinetobacter baumannii. 455 Antimicrob. Agents Chemother. 46:875-878. 456
16. Saugar JM, Rodríguez-Hernández MJ, de la Torre BG, Pachón-Ibañez 457 ME, Fernández-Reyes M, Andreu D, Pachón J, Rivas L. 2006. Activity of 458 Cecropin A-Melittin Hybrid Peptides against Colistin-Resistant Clinical 459 Strains of Acinetobacter baumannii: Molecular Basis for the Differential 460 Mechanisms of Action. Antimicrob. Agents Chemother. 50:1251-1256. 461
17. Badosa E, Ferre R, Planas M, Feliu L, Besalu E, Cabrefiga J, Bardaji E, 462 Montesinos E. 2007. A library of linear undecapeptides with bactericidal 463 activity against phytopathogenic bacteria. Peptides 28:2276-2285. 464
18. Güell I, Cabrefiga J, Badosa E, Ferre R, Talleda M, Bardají E, Planas M, 465 Feliu L, Montesinos E. 2011. Improvement of the Efficacy of Linear 466 Undecapeptides against Plant-Pathogenic Bacteria by Incorporation of d-467 Amino Acids. Appl. Environ. Microbiol. 77:2667-2675. 468
19. Torcato IM, Huang Y-H, Franquelim HG, Gaspar D, Craik DJ, Castanho 469 MARB, Troeira Henriques S. 2013. Design and characterization of novel 470 antimicrobial peptides, R-BP100 and RW-BP100, with activity against Gram-471 negative and Gram-positive bacteria. Biochim. Biophys. Acta, Biomembr. 472 1828:944-955. 473
20. Zuckermann RN, Kerr JM, Kent SBH, Moos WH. 1992. Efficient method 474 for the preparation of peptoids [oligo(N-substituted glycines)] by submonomer 475 solid-phase synthesis. J. Am. Chem. Soc. 114:10646-10647. 476
21. Fernandez-Cuenca F, Pascual A, Ribera A, Vila J, Bou G, Cisneros JM, 477 Rodriguez-Bano J, Pachon J, Martinez-Martinez L. 2004. Clonal diversity 478 and antimicrobial susceptibility of Acinetobacter baumannii isolated in Spain. 479 A nationwide multicenter study: GEIH-Ab project (2000). Enferm. Infecc. 480 Microbiol. Clin. 22:267-271. 481
22. García-Quintanilla M, Pulido MR, Moreno-Martínez P, Martín-Peña R, 482 López-Rojas R, Pachón J, McConnell MJ. 2014. Activity of Host 483 Antimicrobials against Multidrug-Resistant Acinetobacter baumannii 484 Acquiring Colistin Resistance through Loss of Lipopolysaccharide. 485 Antimicrob. Agents Chemother. 58:2972-2975. 486
61
23. Fernandez-Reyes M, Rodriguez-Falcon M, Chiva C, Pachon J, Andreu D, 487 Rivas L. 2009. The cost of resistance to colistin in Acinetobacter baumannii: 488 a proteomic perspective. Proteomics 9:1632-1645. 489
24. Lewis K. 2007. Persister cells, dormancy and infectious disease. Nat. Rev. 490 Micro. 5:48-56. 491
25. Hiramatsu K, Aritaka N, Hanaki H, Kawasaki S, Hosoda Y, Hori S, 492 Fukuchi Y, Kobayashi I. 1997. Dissemination in Japanese hospitals of 493 strains of Staphylococcus aureus heterogeneously resistant to vancomycin. 494 Lancet 350:1670-1673. 495
26. Clements JM, Coignard F, Johnson I, Chandler S, Palan S, Waller A, 496 Wijkmans J, Hunter MG. 2002. Antibacterial Activities and 497 Characterization of Novel Inhibitors of LpxC. Antimicrob. Agents Chemother. 498 46:1793-1799. 499
27. Evans BA, Hamouda A, Amyes SG. 2013. The rise of carbapenem-resistant 500 Acinetobacter baumannii. Curr. Pharm. Des. 19:223-238. 501
28. Soon RL, Nation RL, Hartley PG, Larson I, Li J. 2009. Atomic Force 502 Microscopy Investigation of the Morphology and Topography of Colistin-503 Heteroresistant Acinetobacter baumannii Strains as a Function of Growth 504 Phase and in Response to Colistin Treatment. Antimicrob. Agents Chemother. 505 53:4979-4986. 506
29. Jacobs AC, Sayood K, Olmsted SB, Blanchard CE, Hinrichs S, Russell D, 507 Dunman PM. 2012. Characterization of the Acinetobacter baumannii growth 508 phase-dependent and serum responsive transcriptomes. FEMS Immunol. Med. 509 Microbiol. 64:403-412. 510
30. Fiester SE, Actis LA. 2013. Stress responses in the opportunistic pathogen 511 Acinetobacter baumannii. Future Microbiol. 8:353-365. 512
31. Pescaretti MdlM, López FE, Morero RD, Delgado MA. 2011. The 513 PmrA/PmrB regulatory system controls the expression of the wzzfepE gene 514 involved in the O-antigen synthesis of Salmonella enterica serovar 515 Typhimurium. Microbiology 157:2515-2521. 516
32. Soon RL, Nation RL, Cockram S, Moffatt JH, Harper M, Adler B, Boyce 517 JD, Larson I, Li J. 2011. Different surface charge of colistin-susceptible and 518 -resistant Acinetobacter baumannii cells measured with zeta potential as a 519 function of growth phase and colistin treatment. J. Antimicrob. Chemother. 520 66:126-133. 521
33. Wade D, Boman A, Wahlin B, Drain CM, Andreu D, Boman HG, 522 Merrifield RB. 1990. All-D amino acid-containing channel-forming antibiotic 523 peptides. Proc. Natl. Acad. Sci. U. S. A. 87:4761-4765. 524
34. Bulet P, Urge L, Ohresser S, Hetru C, Otvos L, Jr. 1996. Enlarged scale 525 chemical synthesis and range of activity of drosocin, an O-glycosylated 526 antibacterial peptide of Drosophila. Eur. J. Biochem. 238:64-69. 527
35. Liu L, Fang Y, Wu J. 2013. Flexibility is a mechanical determinant of 528 antimicrobial activity for amphipathic cationic α-helical antimicrobial 529 peptides. Biochim. Biophys. Acta, Biomembr. 1828:2479-2486. 530
Modulation of backbone flexibility for effective dissociation of antibacterial and hemolytic
activity in cyclic antimicrobial peptides without loss of potency
Alberto Oddo1,2, Thomas T. Thomsen3, Hannah M Britt2, Anders Løbner-Olesen3, Peter W
Thulstrup4, John M Sanderson2 and Paul Robert Hansen1,*
1University of Copenhagen, Department of Drug Design and Pharmacology, Universitetsparken 2,
2100, Copenhagen, Denmark 2Durham University, Department of Chemistry, South Road, DH1 3LE, Durham, United Kingdom. 3University of Copenhagen, Department of Biology, Ole Maaløes Vej 5, 2200, Copenhagen,
Denmark 4University of Copenhagen, Department of Chemistry, Universitetsparken 5, 2100, Copenhagen,
Denmark
75
76
77
78
79
80
81
82
Paper III
Submitted to Antimicrobial Agents and Chemotherapy
The Lantibiotic Nai-107 efficiently rescues Drosophila melanogaster from infection with
Key words: Antibacterial peptides; Drug screening; Antibiotics
84
Abstract
We used the fruit fly Drosophila melanogaster as a cost-effective in vivo model to evaluate the efficacy
of novel antibacterial peptides and peptoids for treatment of methicillin-resistant staphylococcus
aureus (MRSA) infections. A panel of peptides with known antibacterial activity in vivo and/or in vitro
was tested in Drosophila. Although most antibacterials that were effective in vitro failed to rescue
lethal effects of S. aureus infections in vivo, we found that two lantibiotics, Nisin and NAI-107 rescued
adult flies from fatal infections. Furthermore, NAI-107 rescued mortality of infection with the MRSA
strain USA300 with equivalent efficacy to vancomycin, a widely applied antibiotic for the treatment of
serious MRSA infections. These results establish Drosophila as a useful model for in vivo drug
evaluation of antibacterial peptides. Further, the data shows that NAI-107 has the ability to kill non-
growing stationary phase bacteria in vitro, which vancomycin is incapable of.
Introduction
Since the golden era of antibiotic drug development during the 1940-1960`s, development and spread
of multidrug resistance have become a huge burden to societies. Today resistance to almost all known
antibiotics has emerged with the sequential introduction of new or improved antibiotics in the clinical
and agricultural setting (1, 2). Therefore, continued development of new or improved antibiotics is of
great importance to human health. However, new antibiotics are lacking and few are under
development for treatment of multidrug resistant (MDR) infectious bacteria, as drug development is
costly and success from in vitro discovery to clinical settings is limited.
85
Bacterial infections with MRSA (Methicillin Resistant Staphylococcus aureus) are no longer sporadic
in distribution and prevalence (3, 4). MRSA strains are associated with both community (CA-MRSA)
and hospital (HA-MRSA) acquired infections, with the highly β-lactam resistant USA300 CA-MRSA
clone accounting for close to 80% of all MRSA infections in the USA (5). High level β-lactam
resistance is due to acquisition of SCCmec elements (Staphylococcal Cassette Chromosome) including
the mecA gene, which encodes an alternative version of the penicillin binding protein (PBP2A), that is
inducible (6, 7) and has a lowered affinity for β-lactam antibiotics (8). Often SCCmec elements are
associated with carriage of resistance genes to other antibiotics including aminoglycoside modifying
enzymes such as acetyltransferase, adenylyltransferase or phosphotransferase (9). Due to this
resistance, MRSA treatment usually includes glycopeptide antibiotics such as vancomycin or
oxazolidinones such as linezolid. However, failure in vancomycin treatment has been reported in
vancomycin-intermediate S. aureus (VISA) (10) and vancomycin resistant S. aureus (VRSA) (11). On
the other hand, linezolid resistance is rare (12), but has been observed as mutations in the ribosomal
DNA, or through carriage of the Cfr rRNA methyltransferase gene (13, 14). Furthermore, resistance to
the last resort antibiotic daptomycin has been reported (15, 16). Given the increasing frequency of
resistance to these antibiotics, it is important to develop improved or novel therapeutics, and to
consider new strategies to contain the spread of the growing resistance problem.
Peptide based antibiotics has been proposed as the next generation of antimicrobial compounds because
of their wide distribution in nature as part of innate immunity. These molecules are often amphipathic
and interact with the bacterial membrane to disrupt its function. The cationic peptide colistin, a
bacteriocin currently used for treatment of highly resistant gram-negative infections, is part of the
polymyxins that are derived from natural producers such as Paenibacillus polymyxa (17). Another
86
bacteriocin, nisin, has been used in the food industry for decades against harmful bacteria such as S.
aureus, Listeria monocytogenes and Clostridium botulinum (18). Nisin belongs to a subgroup known as
lantibiotics, named so, for containing uncommon amino acids such as lanthionine, methyllanthionine,
didehydroalanine or didehydroaminobutyric acid (19). Nisin was described to disrupt membrane
integrity through a dual mode of action: By inhibiting cell wall synthesis through binding to the cell
wall precursor lipid-II and by subsequent pore formation (20-22), although new evidence points
towards a more complex mechanism that includes aggregation of Lipid-II (23). Peptides may be used
directly as antimicrobials or could pose as templates for development of small molecule mimetics such
as peptoids, which can accommodate improvements to toxicity and are intrinsically less prone to
degradation by proteases (24). The gap from in vitro drug screening to the large scale efficacy testing
necessary for clinical development is hampered by the expensive, labor-intensive and highly regulated
infection models in mammals. It is therefore of interest to develop improved cost-effective methods
with high predictive value for screening of antimicrobial compounds before these are put into large
scale production. Although the fruit fly Drosophila has been extensively used in drug discovery (25,
26), its application for screening of antibacterial compounds has been limited (27-29). Drosophila is a
powerful genetic model for studying disease mechanism and during the past decades it has been used
extensively in elucidating the mechanisms of innate immunity, leading to the discovery of the
conserved role of the Toll receptors (30) and the immune deficiency (IMD) pathway (31). Studies of
innate immunity in Drosophila have sprouted development of various methods for infecting flies with
important human pathogens (28, 32-34). Here, we evaluate the therapeutic potential of antimicrobial
peptides and peptoids in vivo by screening efficacy and toxicity in a Drosophila model infected with S.
aureus 8325-4 (35) and MRSA USA300 (36). Tests were performed with a range of different peptides
including the lantibiotics nisin A (37) and NAI-107 (38, 39) currently undergoing preclinical studies.
87
These are usually produced by gram-positive bacteria and characterized as ribosomally synthesized
peptides containing ring structures, introduced through the thioether containing lanthionine and
methyllanthionine residue (40). Further, a panel of amphipathic cationic peptides previously shown to
have good in vitro or in vivo efficacy were tested: GN2, GN4 (41, 42), HHC-9 (43), HHC-36 (44) and
peptoids: GN-2 Npm9, GN-2 Ntrp5-8 Nlys1-4, GN-4 (45). We found that NAI-107 rescued an otherwise
lethal infection with MRSA USA300 and with an efficacy equivalent to vancomycin. Further, nisin
provide transient protection from infection, while the majority of the peptides and peptoids show no
protection from infection or were highly toxic to the host.
Materials and methods
Bacteria and growth media
The S. aureus strains 8325-4 (35) and USA300 (36) was used as indicated in the individual
experiments. Bacterial cultures were grown in cation adjusted Müller Hinton Broth (MHB-II ) at the
indicated temperature.
Growth rate determination:
The growth rate of the S. aureus was examined at 37°C in vitro, to determine the growth period
required for obtaining balanced cultures, here defined as cultures grown exponential for no less than 6
generations. Prior to injection of bacteria into the fly in vivo model, the inoculum was prepared as
balanced cultures grown at 37°C. Since flies used in our in vivo infection model are kept at 29°C, we
88
also tested the in vitro generation at this temperature. In vitro growth rate was defined in MHB-II, by
optical density measurements at 600 nanometers (OD600) at 10 minute intervals. Further we determined
the in vivo generation time by CFU/animal measurements after infection, by counting of colony
forming units (CFU) after crushing flies infected with bacteria at various time points, and plating on
Manitol Salt Agar (MSA). This was performed in triplicate experiments; 3 individual flies were
crushed in phosphate buffered saline (PBS) and 10x dilution series were prepared, from which 10µl
was spot plated on MSA. The mean value of each experiment was determined as CFU/Fly and plotted.
Minimum inhibitory concentration
Minimum inhibitory concentrations (MIC) of all tested compounds were performed according to
protocols using the micro-broth dilution methodology (46) with minor modification. S. aureus was
grown in 10 ml MHB-II overnight at 37°C with shaking, then diluted 1:100 in fresh MHB-II and grown
to OD = 0.2-0.4. Cultures were then diluted 1:10 and grown to OD = 0.2-0.4. These steps were
performed to ensure balanced growth of cultures as explained. Finally, dilutions was made to 1 x 106
Colony Forming Units (CFU)/ml, and further diluted 1:1 in microtiter plates in MHB-II + drug,
leading to a final inoculum of 5 x 105 CFU/ml. MIC`s were determined in triplicates, if more than one
value was found, the highest was set as the MIC to be conservative.
89
Time kill assay
Time kill assays were performed as previously described (47). Minor changes were made in the
protocol, since we wanted to analyse time kill responses in both exponential and stationary phase
cultures. Exponential cultures of USA300 grown at 37°C and drugs were tested at OD600 = 0.4 and
stationary phase cultures were defined as overnight cultures grown at 37°C with shaking and with a
growth period of 16 hours prior to addition of drug. Counting of CFU were performed by spot plating
of 10 µl culture. The supernatant was removed by centrifugation and pellet resuspended in PBS before
series of 10 fold dilutions. Time point 24 hours was performed by pelleting 250 µl of culture, and
resuspending in 100 µl PBS before plating of the whole sample, for some treatment groups.
Injection assay
Injection assays was performed as previously described (33) using a nanoject-II microinjecter, but with
minor modification in preparation of bacterial inoculum to obtain balanced cultures as explained. Flies
were reared on standard bloomington formulation at 25°C under a 12:12 light:dark cycle and constant
humidity. Only adult male flies (Oregon genotype) 4-7 days old were used for injection experiments.
Initial experiments with strain 8325-4 were performed in duplicates with groups of 25-30 animals in
each experiment. For USA300 experiments were performed in triplicates (nisin only in doublicates). S.
aureus inoculum was prepared as balanced exponentially growing cultures. Inoculum was prepared by
resuspending cells in 10 mM MgSO4 at an OD600 = 0.06 and kept on ice, giving an inoculum dose of
100-400 CFU (8325-4) and 250-700 CFU (USA300) in the flies after injecting 18.4 nl. Bacterial
injections were administered in soft tissue surrounding the front legs, drug treatment was administered
90
in the lower thorax. After injection of bacteria, flies were kept at 29°C and followed for 48-96 hours to
determine mortality. Drug delivery was performed 3 hours post infection in all animals, at the
concentrations indicated for individual experiments. Flies which died within 3 hours of injection, were
considered to have died from handling and disregarded. It is important to note that when drug
concentrations were calculated, we performed a rough approximation of the fluid content of a fly. Fly
fluid content was measured by drying out 10 groups of 50 flies and comparing dry weight to wet
weight. This resulted in an average fluid content of 0.58 µl per adult fly. For simplicity and because we
assumed that the compounds would not distribute to all fluids we used 0.5 µl fluid as our measure for
calculating drug concentrations in the flies. Further we assumed rapid distribution of the compound in
the open circulatory system of Drosophila and a slow clearance of the compounds by malphigian
tubules. Therefore antimicrobial drug concentrations are given as the highest concentration obtained in
multiples of the MIC.
Statistics and graphical Plots:
Plotting of data was performed using GraphPad Prism 5. All in vivo survival plots were performed
using Caplan Meier analysis on pooled data for repetitive experiments. Statistical analysis was carried
out with GraphPad Prism 5 build in Log-Rank (Mantel cox) Test for comparison of survival curves.
Experiments with p values < 0.05 was considered significant.
91
RNA preparation and quantitative PCR:
Isolation of total RNA for quantitative PCR was prepared by the use of RNeasy Mini Kit (Qiagen)
according to manufacturer’s instructions. Biological samples were collected as 10 adult male flies
pooled for each replicate and time point. To reduce contamination with genomic DNA, all samples
were treated on-column with DNase. Total RNA concentrations were measures on a Qubit™ 3.0
fluorometer and equivalent amounts of RNA was used for cDNA synthesis for each sample. cDNA
synthesis was performed using the SuperScript III First-Strand Synthesis kit (Invitrogen) kit according
to manufacturer’s instructions. qPCR was performed on a Mx3000P qPCR System (Agilent
Technologies) platform using the following program; 95°C for10 min, followed by 45 cycles of 95°C
for 15 sec, 60°C for 15 sec and 72°C for 15 sec. Dissociation curve analysis was applied to all
reactions. Primers are described in the supplemental material (Table S1). We used Rpl23 as
housekeeping gene while performing the assay to normalize expression as previously described (48).
Compounds
Ampicillin sodium salt 99% (ROTH Art-Nr: K029.2 EG-Nr: 2007081) was used as control for efficacy
and toxicity in in vitro and in vivo experiments. Vancomycin was acquired from Hospira as
vancomycin hydrocloride for intravenous treatment (lot# 467918E01). The peptides GN-2, GN-4
HHC-9 and HHC-36 all amidated in C-terminus, nisin A and peptoids were above 95% purity and
synthesized in the lab of Håvard Jenssen, Roskilde University Denmark. NAI-107 is a complex of
congeners produced by Microbispora sp. 107891 and was prepared as previously described (49). The
92
distribution of congeners for the batch used in the curent study was as follows: A1+A2 = 80.8%, F1+F2
= 9.4%, B1+B2 = 4.
Results
Determination of the growth of S. aureus in vitro and in vivo in a Drosophila infection model
We determined the growth rate of the S. aureus strains 8325-4 and MRSA USA300 in cation adjusted
Müller Hinton Broth (MHB-II) media at 29°C, because all successive in vivo experiments were
performed at this temperature. Strain 8325-4 had a generation time of 57 minutes while USA300 had a
generation time of 44 min. The in vivo growth rate of the same strains was determined by injection of
bacteria into the flies at time 0 and samples were collected between time 0-3, 4-6 and 12 hours post
infection (Fig. 1A). Three flies were crushed and serial dilutions were made in PBS, before plating on
S. aureus selective mannitol salt agar (MSA) to determine the number of colony forming units (CFU).
USA300 had a generation time of 54 minutes, whereas 8325-4 had a generation time of 104 minutes in
vivo. Drosophila infected with USA300 died rapidly with no surviving flies after 24 hrs. Whereas flies
infected with approximately the same number of 8325-4 lived significantly longer (Fig. 1B). We
suggest that these differences in viability reflect the different in vivo growth rates of USA300 and
8325-4 bacteria.
93
Figure 1. In vivo growth rate and killing of flies by the two isolates: A. in vivo growth rate USA300 = 54 min, 8325-4 = 104 min clearly demonstrate difference in proliferation. B. USA300 kills close to 100% within 24 hours, while isolate 8325-4 kills approximately 50% within 24 hours (p < 0.0001). Slight differences are observed in starting inoculum (see materials and methods). Survival data are compiled results from all in vivo experiments presented in figures 2 and 5.
Minimum inhibitory concentrations for antimicrobial peptides and peptoids
We determined the minimal inhibitory concentrations (MIC; Table 1) for the two strains. The MIC
values for S. aureus 8325-4 of amphipathic cationic peptides GN-2, GN-4, HHC-9 and HHC-36 and
the Lantibiotic nisin were in the range of 4-10 µg/ml, while those of GN-2 and GN-4 peptoids were
higher (16-64 µg/ml). On the other hand, the MIC of NAI-107 against strain 8325-4 was only 0.06
µg/ml, showing that NAI-107 is highly efficient in inhibiting in vitro growth of S. aureus. The MIC of
NAI-107 for S. aureus USA300 was lower compared to vancomycin when determined as molar
concentrations (0.11µM versus 1.38 µM; Table 1).
94
Table 1. Minimum inhibitory concentrations (MICs) of compounds tested: The molecular weight used for calculating μM concentrations are given in the table, as well as MICs for the compounds in both μg/ml and μM. MIC for some compounds was not performed on both isolates (Na). Sequences of nisin and NAI-107 are not included as they contain ring structures making a linear sequence misleading.
Identification of nisin and NAI-107 as efficacious treatment for systemic S. aureus infections in a
Drosophila in vivo model
To evaluate the therapeutic potential of the antimicrobial peptides and peptoids, we determined their
ability to rescue flies with an otherwise lethal systemic S. aureus 8325-4 infection. In order to establish
the appropriate dosages, we made the following reasoning: Because insects are known to have an open
circulatory system, we assumed that the administered compound would be rapidly and uniformly
distributed in the hemolymph of the fly. This was determined to be 0.5 µl (see Materials and Methods)
95
and we also assumed that compound elimination through the Malpighian tubules proceeds slowly.
Under these assumptions, the highest concentration achieved for each compound can be expressed as
multiples of the MIC. For example 1xMIC nisin [10µg/ml] is equivalent to injection of 2.5 mg nisin/kg
fly; this numbers for all drugs can be seen in (Table 2). Ampicillin was chosen as control, as β-lactams
in general are considered nontoxic to the host and can be administered in high concentrations, in our
case >1000xMIC. Ampicillin efficiently promoted survival of 8325-4 infected flies (p<0.001; Fig. 2A)
when monitoring over a 70 hours period and with no detrimental lethal effects to control animals (p =
0.15); here defined as no difference in survival when comparing flies injected with 10 mM. MgSO4 to
those injected with both MgSO4 and drug.
Table 2. Antimicrobial peptide dosages: We calculated the concentration of compound injected in mg/kg Fly, based on the data in table 2. All data presented are based on 1xMIC of the compounds.
96
The two lantibiotics NAI-107 and nisin showed good efficacy in effectively rescuing or delaying
mortality of infected flies over a 96 hours period (Fig. 2 B and C). NAI-107 at 1xMIC had no positive
effect survival of the flies (Fig. 2B) and the same was found for 3xMIC (not shown). Treatment with
10xMIC of NAI-107 rescued around 20-30% of flies (p<0.001) and with no difference in the survival
of control animals (p = 0.62; not shown). We therefore tested NAI-107 at 100xMIC, and this
concentration rescued more than 70% (p < 0.0001) of the infected flies, without lethal effects to
controls (p = 0.62; Fig. 2B). Compared to NAI-107, nisin showed a difference in both efficacy and
lethality to control animals. While 1xMIC nisin delayed bacterial killing of flies (p<0.001), it produced
signs of lethal side effects (p = 0.018; Fig. 2C). Higher concentrations of 3xMIC nisin also rescued a
considerable fraction of infected animals (p = 0.0002; not shown), but showed pronounced detrimental
effects to control animals (p = 0.0056). These adverse effects were exacerbated when using 10xMIC
nisin, which resulted in the killing of 50% of control animals injected with nisin alone (p<0.0001; Fig.
2C) and also resulted in increased mortality of infected flies. Therefore, nisin was not tested at
100xMIC.
In contrast to NAI-107 and nisin, the GN-4 peptide, which possesses good in vitro efficacy against S.
aureus [Table 1; (41)], did not rescue infected flies at 1x and 3xMIC (p>0.005; Fig. 2D). When applied
at 10xMIC, GN-4 showed no adverse effects to the survival of control flies. However, the results
indicate that administration of this peptide to animals infected with bacteria may reduce the survival
because a higher number of the animals treated with the peptide after infection died, although this was
not statistically significant. The GN-4 peptoid showed pronounced detrimental side effects even at
1xMIC (Fig. 2E) therefore the peptoids were abandoned after a single experiment. The GN-2 peptide
had similar effect to that of the GN-4 peptide (supplemental data Fig. S1) and the two GN-2 peptoids
97
(supplemental Fig. S2 and S3) clearly showed lethalk effects in both control and infected animals.
Injection of peptides HHC-9 and HHC-36 in the absence of infection caused no obvious side effects,
while treatment with these peptides did not rescue infected flies (supplemental Fig. S4 and S5
respectively), but they caused a moderate decrease in survival of infected flies that may indicate
detrimental effects of peptides, although the results were somewhat ambiguous.
We also noted adverse behavioral response that could be indicating neurotoxicity in flies injected with
high concentrations of nisin, GN-2 and GN-4 along with the peptoids, but not for NAI-107. Animals
reacted to injection with these compounds by being partially paralyzed for up to 10 hours post injection
(not shown). This paralysis was not manifested as complete immobilization but as uncoordinated
movements and an inability to walk or fly.
98
Figure 2. In vivo efficacy of compounds against S. aureus 8325-4 in a Drosophila whole-animal model: Graphs show survival of flies treated with subset of peptides. Y-axis show fraction survival compared to time in Hours (x-axis). Flies were counted at time points 0, 3, 6, 12, 24, 48 – 120 hours. The individual figure legends indicate the treatment groups: Flies are either injected with MgSO4 or isolate 8325-4 at time 0, the + indicate treatment at time point 3 hours (dotted line). Flies were counted prior to injection with compound. Compound concentrations [C] are given as approximated concentration in animals.
99
Treatment with nisin and NAI-107 reduces the immune response of S. aureus infected D.
melanogaster
To further test drug efficacy of the two lantibiotics nisin and NAI-107 in vivo, we examined the
immune response of both treated and non-treated infected animals. We rationalized that infected
animals treated with these compounds would mount less of an immune response provided that bacterial
proliferation in the host was inhibited. To test this we used flies infected with S. aureus strain 8325-4.
We applied NAI-107 (100xMIC) while nisin due to its adverse effects was only applied at 3xMIC.
Samples in triplicate were taken at 6 and 12 hours post infection and processed as described in
Materials and methods. Oregon flies not exposed to infection with S. aureus served as control. As a
measure of immune response we analyzed expression of Drosomycin (Drs), Cecropin A1 (CecA1) and
Attacin-B (AttB) genes, which have all been implicated in the immune response of Drosophila to
infection by Gram-positive bacteria (50, 51). In general we observed that animals that received any
form of treatment had elevated transcription of immune response genes (Fig. 3), this is most likely
because any injection into the body of the flies, damages the tissue thereby elevating the immune
response. Further, it is highly plausible that injection of any protein like structure will elicit some
degree of immune response. Another general observation was a higher expression level of immune
responsive genes in infected untreated animals compared to animals treated with nisin and NAI-107
(Fig. 3).
The response of the three immune response genes differed. Drosomycin expression increased 30-180-
fold within 6 hours post infection and remained at that level at 12 hours (Fig 3A). Treatment with NAI-
107 and nisin decreased Drs expression approximately 10-fold relative to non-treated infected flies
after 12 hours (Fig. 3A). Expression of Cecropin A1 followed the same pattern as observed for Drs
100
except that maximal induction was only around 20-fold (Fig. 3B). The attacin B expression level was
different. Gene expression was increased considerately in all flies injected with peptides and
irrespective of a concurrent S. aureus infection (Fig. 3C). Because injection with MgSO4 did not result
in the same fold increase of attB induction (Fig. 3C) we conclude that the attB gene is initially induced
by either the pathogen or the administered peptides. The S. aureus infection further increased attB
expression to more than 1000-fold relative to the control at 12 hours. Concurrent administration of
nisin or NAI-107 reduced expression to the level observed for the peptides alone or even below (Fig.
3C).
Some compounds, including nisin, have previously been associated with immunomodulatory actions in
mice (52). Consistent with this, we observed a moderate elevation in the expression of Drosomycin,
Cecropin A1 and Attacin B in flies injected with nisin compared to MgSO4 injected control flies.
However, whether this is due to true immunomodulatory action or because of the observed toxicity is
unclear.
101
Figure 3. Induction of immune pathway genes in animals infected with S. aureus 8325-4: (A) Drosomycin. (B) Cecropin A1 (C) Attacin B were used as read out AMP genes for verification of efficacy of compounds able to rescue/prolong infection. Ribosomal protein L23 was used as reference gene. A non-infected control was used as reference of normal expression; these values were set as 1. Flies infected with S. aureus 8325-4 were sampled for qPCR, 6 and 12 hours post infection. Drug treatment was performed at 3 hours and injection of MgSO4 was used as control injection fluid.
NAI-107 kills non growing MRSA strain USA300 in vitro
After initial experiments with S. aureus strain 8325-4, we chose to test the kill-rate efficiency of the
two lantibiotics along with vancomycin against MRSA strain USA300 (36, 53). We performed time kill
experiments on exponentially growing and non-growing stationary phase USA300 cells (Fig. 4). We
tested both nisin and NAI-107 at 1x, 3x, and 10xMIC, and included NAI-107 at 100xMIC. Nisin was
not tested at higher concentrations than 10xMIC, because we had already shown Nisin to be
102
detrimental to animals at much lower levels. As control we included vancomycin in these experiments.
Treatment of exponentially growing cells (Fig. 4A) with vancomycin, showed that concentrations from
3xMIC to 10xMIC reduced the viable cell count in CFU/ml from 1*108 to 1*106 (2 log`s) within the
first 5 hours of treatment. When applying vancomycin at 10xMIC the response was more rapid and
resulted in a further decrease in CFU/ml to 1*104-1*103 (Fig. 4A). When treating exponentially
growing cells with NAI-107 10xMIC reduced CFU/ml by 3 log`s within the first 5 hours (Fig. 4C),
with a further slight decrease in CFU/ml over the next 19 hours. When applying a concentration of
100xMIC the response was more rapid, with a drop in CFU/ml of more than 6 log`s within 5 hours.
Because 100xMIC of NAI-107 is equimolar to 10xMIC vancomycin (Table 1), this demonstrates that
NAI-107 is equally or more efficient than vancomycin in killing USA300 in vitro. Treatment of
exponentially growing cells with nisin 3xMIC reduced viable counts by 4 log`s (Fig. 4E) and 10xMIC
nisin reduced viable cell count below detection. However, nisin treated cells re-grew and by 24 hours
the 10xMIC treated culture was at 1*104 CFU/ml. At 1xMIC none of the tested compounds were able
to reduce viable cell counts.
Next, we tested the compounds ability to kill stationary phase bacteria. NAI-107 at 100xMIC
efficiently killed the majority of the culture within 5 hours of treatment, i.e. the CFU count was reduced
from 1*1010 to 1*102 CFU/ml, and remained at that level until 24 hours (Fig. 4D). Nisin at 10xMIC
was somewhat less efficient and the CFU count was reduced 6 logs from 1*1010 to 1*104 CFU/ml
within the first 5 hours of treatment (Fig 4 F). However, as observed for exponentially growing cells,
the nisin treated stationary phase cells re-grew by 24 hours (Fig. 4F). Nisin was not tested at higher
concentrations due to the aforementioned lethal effects. Overall these data show NAI-107`s capability
103
to kill the cell regardless of growth state. This is in contrast to vancomycin (Fig. 4B), which at
equimolar concentrations to NAI-107 is unable to effectively kill non growing cells.
Figure 4. In vitro kill rate experiments against USA300: (A-B) vancomycin, (CD) NAI-107 and (E-F) Nisin treated exponential phase (A, C, E) and stationary phase cultures (B, D, F). Cell counts are given as log transformed colony forming units per ml (CFU), plotted against time in hours (x-axis). Right hand figure legends show control group USA300 or USA300 treated with compound at the given concentration [C]. Dotted line indicates the lower detection limit.
104
NAI-107 effeciently rescues flies from infection with USA300
We proceeded to evaluate the in vivo efficacy of lantibiotics relative to vancomycin in Drosophila
infected with USA300 (Fig. 5). Flies were treated with nisin at 1xMIC, and 10xMIC assuming liquid
content of a fly beeing approximately 0.5µl (for details see materials and methods). Although nisin did
not rescue flies over the duration of the experiment, it did delay mortality by doubling the mean
survival time (p < 0.0001) at both concentrations tested (Fig. 5A). However, mortality was increased in
the control group injected with 10xMIC relative to the MgSO4 injected control (p = 0.0008; Fig 5A). A
single dose of 100xMIC NAI-107 rescued 50-60% of USA300-infected animals over a 96 hours period
(p<0.0001), equivalent to the survival found for vancomycin treatment in equimolar concentrations, i.e.
10xMIC (p = 0.94; Fig. 5B). Positive effects on the survival of USA300 infected animals, were also
found at dosages of NAI-107 as low as 3xMIC (p<0.0001; Fig. S6). Similar to NAI-107, vancomycin
showed no adverse effect at the concentrations tested here (Fig. 5B). Taken toghether these results
demonstrate that NAI-107 delay killing of D. melanogaster by systemic USA300 infections with an
efficiency similar to vancomycin and with no apparent adverse effects. This highlights the potential of
NAI-107 as a candidate for systemically administered application.
105
Figure 5. Efficacy of Nisin and Nai-107 in vivo against USA300: (A) Nisin prolonged the lifespan at all concentrations (B) NAI-107 rescues 50-60% of flies at 100xMIC (p<0.001), comparison of vancomycin with NAI-107 produced no difference in the response between the drugs (p=0.94). Antibiotics are injected at time 3 hours post infection (dotted line).
106
Discussion
We have used Drosophila melanogaster as a model organism for testing the efficacy and adverse
effects of antimicrobial peptides. We examined several cationic antimicrobial peptides previously
reported to have either in vitro or in vivo efficacy against S. aureus. Furthermore, the two lantibiotics
nisin and NAI-107 were included. We found that both lantibiotics, can delay or even rescue lethal
injections with wild type S. aureus 8325-4 isolate, but more importantly also the MRSA USA300
isolate.
Amphipathic peptides
None of the cationic amphipathic peptides previously tested in vitro and/or in vivo against both gram-
negative and gram-positive bacteria (41) had any positive effect on the survival of S. aureus infected
flies. These peptides are believed to work through pore formation, thereby disrupting the integrity of
the bacterial membrane(s). Although this does not exclude the possibility that peptides may be effective
in mammalian models, our data do not support their use in whole-animal infections. There were
indications that the GN peptides even had negative effects on survival of infected flies. Suprisingly, the
peptoids based on GN peptides were highly detrimental to animals. The explanation for this, could be
ascribed to the peptoids having high MIC, and therefore had to be injected in higher concentrations
compared to native compounds to reach the same integer of MIC. Consistent with our findings, adverse
effects for most of the compounds in the >100 µg/ml range have previously been reported from cell
based assays (45). Two of the cationic amphipatic peptides tested here, HHC-9 and HHC-36, had
marked negative effect on the survival of infected flies. This contradicts previous data in which HHC-
107
36 was found to have in vivo efficacy against S. aureus in a well-established mouse intra-peritoneal
model (44). Although we expected the HHC compounds to be able to clear or delay infection in
Drosophila, our results indicate that they are detrimental to flies when injected at high concentrations.
The number of experiments performed previously for the HHC peptides, especially in vivo, are limited,
which makes it difficult to explain the differences observed in the two infection models. One main
difference is, however, that both bacteria and peptides are delivered systemically into circulation in the
Drosophila infection model, while both bacteria and peptides are injected into the body cavity in the
intra-peritoneal mouse model. It is not clear wether the peptides enter circulation in the mouse to the
same extent as bacteria does, and this could skew data obtained through counting of colony forming
units in peritoneal fluid only. However, we argue that injection of peptides into the hemocoel of a fly
provides access to more diverse tissues due to the fly physiology beeing less compartmentalized, which
could be a reason for the differences seen in adverse effects. Adverse effects should not be considered
as a definite rejection of compounds, since they can be used for further structure relationship studies
and development of better compounds. Despite our data arguing that the potential of several of these
compounds for systemic use are limited, these compounds may be further developed into topical usage,
as is the case for the systemically toxic peptide antibiotic bacitracin, which has been highly successful
in topical ointments (54, 55).
Lantibiotics
Lantibiotics remain of interest for development of new therapeutics. Nisin is one of the best studied
lantibiotics (19) and has recently gained new interest as a therapeutic since it was proven effective
108
against MRSA (56, 57). A newly discovered lantibiotic interesting for clinical development is NAI-107
(38). It is currently undergoing preclinical studies, and it has already proven effective in vivo against
multi-drug resistant S. aureus (39, 58).
Our results reinforces the notion that nisin may have therapeutic potential in clinical settings, although
systemic application seems limited due the observed lethality. In our Drosophila model, nisin is
detrimental even at relatively low concentrations, which contrasts previous in vivo findings from rats
(59). However, our study utilizes injection into the circulatory system of whole animals, while the
Reddy et al. study utilized administration through oral dosing, inevitably changing the bioavailability
of a compound (60). Despite the apparent side effects to flies, a single injection of nisin delays death
due to infection in doses equivalent to the MIC of the compound, demonstrating in vivo efficacy at low
doses, which may be improved by multiple dosing. Therefore, further studies are needed to address the
intricate interactions of nisin with eukaryotic cell systems. Although the bacterial target of nisin has
been characterized (22, 61, 62), the interplay of nisin with other molecules of eukaryotic cells remain
poorly understood. Perhaps nisin, because of its poor bioavailability and fast degradation (63), could be
modified chemically to address these issues (19, 64), and in this context it would be of importance to
know more about adverse effects.
Due to the low MIC of NAI-107 to S. aureus, we expected good in vivo efficacy at low doses of the
compound. However, NAI-107 only seemed to delay infection at doses around 10xMIC. Higher doses
of NAI-107, however, resulted in remarkable in vivo efficacy with no signs of detrimental side effects.
This is in accordance with previous findings that the effects of NAI-107 is concentration dependent
(58). Further, it should be acknowledged that low MIC in in vitro experiments not necessarily translates
into the same efficiencies in vivo, since pharmacodynamics and kinetics come into play.
109
Nisin was clearly less potent than NAI-107, although they both bind to lipid-II (65) and rapidly kill
bacteria. Nisin has been described to work through disruption of cell wall synthesis and pore formation,
by binding to Lipid-II (20, 21, 61), although new evidense points to nisin working through aggregation
of Lipid-II in the membrane (23). Evidence also points towards NAI-107 having a dual mode of action
through binding to the Lipid-II cell wall precursor and destabilizing membrane integrity but also
interfering with protein localization and promoting disorganization in the cell (65). Our in vitro data
support the findings that the two compounds mode of action differ; Nisin rapidly kills exponentially
growing cultures, whereas NAI-107 has a prolonged, but lower initial effect in vitro. Furthermore, in
vitro treated nisin cultures re-grow by 24 hours, which fits with the in vivo data that nisin only doubles
the life expectansy of infected flies. Nisin`s aparent side effects may be due to it`s ability to create
pores by non-specific interaction with membranes (66), which could mean that it will do so in
eukaryotic membranes as well. Our in vitro and in vivo data support that NAI-107 can be applied in
concentrations where it not only effectively kills growing bacteria, but also might prove efficient
against persistent non growing bacteria.
In conclusion, we provide evidence for the use of Drosophila as a model for in vivo efficacy testing of
antimicrobial peptides. We have clearly shown, that infected animals can be rescued by treatment with
certain antimicrobial peptides. Further, we have demonstrated Drosophila as a putative model for
assessing adverse effects of antimicrobial peptides. The Drosophila model presented here was adapted
from previously developed methodologies (33, 67) to provide researchers with a relativly cheap method
for efficacy evaluation of lead compound antimicrobials discovered through more appropriate drug
screens. To the best of our knowledge Drosophila has not previously been used for the testing of
antimicrobial peptide efficacy and toxicity. Drosophila does not allow for high throughput screening of
110
large drug libraries by injection, as this procedure is relatively labor intensive, compared to drug
screening methodologies developed in the worm Caenorhabditis elegans (68), but our method is
applicable to lead compounds discovered following such screens. Therefore, as an initial model for
efficacy testing of lead compounds Drosophila could prove interesting for further analysis, especially
regarding it as whole-animal model for toxicity screening, as classical toxicity screens usually involve
hemolysis and metabolic cell based assays performed on imortalized cell lines.
Between the compounds tested by us, the lantibiotic NAI-107 was superior to Nisin, but equivalent to
vancomycin. Nai-107`s ability to kill non growing bacteria is to our knowledge the first time this has
been reported for this particular lantibiotic.
Acknowledgements
This work was supported by the Danish Council for Independent Research | Technology and
Production Sciences (FTP) grant 11-106387 to Professor Anders Løbner-Olesen. The research was also
partially supported by the European Community's Seventh Framework Programme (FP7/2007-2013)
under grant agreement N°289285 held by Stefano Donadio and partially funded by The Federation of
European Microbiological Societies under grant agreement IT-SIMGBM2014-1. J.C.S.C. supported by
grant agreement N°289285 and IT-SIMGBM2014-1 held by João S. C. Cruz.
111
1. Clatworthy AE, Pierson E, Hung DT. 2007. Targeting virulence: a new paradigm for antimicrobial therapy. Nat Chem Biol 3:541-548.
2. Barton MD. 2014. Impact of antibiotic use in the swine industry. Curr Opin Microbiol 19c:9-15. 3. Kang CI, Song JH. 2013. Antimicrobial resistance in Asia: current epidemiology and clinical
implications. Infect Chemother 45:22-31. 4. WHO. 2014. Antimicrobial resistance: global report on surveillance 2014.257. 5. Liu C, Graber CJ, Karr M, Diep BA, Basuino L, Schwartz BS, Enright MC, O'Hanlon SJ,
Thomas JC, Perdreau-Remington F, Gordon S, Gunthorpe H, Jacobs R, Jensen P, Leoung G, Rumack JS, Chambers HF. 2008. A population-based study of the incidence and molecular epidemiology of methicillin-resistant Staphylococcus aureus disease in San Francisco, 2004-2005. Clin Infect Dis 46:1637-1646.
6. Hiramatsu K, Asada K, Suzuki E, Okonogi K, Yokota T. 1992. Molecular cloning and nucleotide sequence determination of the regulator region of mecA gene in methicillin-resistant Staphylococcus aureus (MRSA). FEBS Lett 298:133-136.
7. Katayama Y, Ito T, Hiramatsu K. 2000. A new class of genetic element, staphylococcus cassette chromosome mec, encodes methicillin resistance in Staphylococcus aureus. Antimicrob Agents Chemother 44:1549-1555.
8. Hartman BJ, Tomasz A. 1984. Low-affinity penicillin-binding protein associated with beta-lactam resistance in Staphylococcus aureus. J Bacteriol 158:513-516.
9. Schmitz FJ, Fluit AC, Gondolf M, Beyrau R, Lindenlauf E, Verhoef J, Heinz HP, Jones ME. 1999. The prevalence of aminoglycoside resistance and corresponding resistance genes in clinical isolates of staphylococci from 19 European hospitals. J Antimicrob Chemother 43:253-259.
11. Hanaki H, Labischinski H, Inaba Y, Hiramatsu K. 1998. [Increase of non-amidated muropeptides in the cell wall of vancomycin-resistant Staphylococcus aureus (VRSA) strain Mu50]. Jpn J Antibiot 51:272-280.
12. Gu B, Kelesidis T, Tsiodras S, Hindler J, Humphries RM. 2013. The emerging problem of linezolid-resistant Staphylococcus. J Antimicrob Chemother 68:4-11.
13. Fines M, Leclercq R. 2000. Activity of linezolid against Gram-positive cocci possessing genes conferring resistance to protein synthesis inhibitors. J Antimicrob Chemother 45:797-802.
14. Long KS, Poehlsgaard J, Kehrenberg C, Schwarz S, Vester B. 2006. The Cfr rRNA methyltransferase confers resistance to Phenicols, Lincosamides, Oxazolidinones, Pleuromutilins, and Streptogramin A antibiotics. Antimicrob Agents Chemother 50:2500-2505.
15. Cui L, Tominaga E, Neoh HM, Hiramatsu K. 2006. Correlation between Reduced Daptomycin Susceptibility and Vancomycin Resistance in Vancomycin-Intermediate Staphylococcus aureus. Antimicrob Agents Chemother 50:1079-1082.
16. Mishra NN, Bayer AS, Weidenmaier C, Grau T, Wanner S, Stefani S, Cafiso V, Bertuccio T, Yeaman MR, Nast CC, Yang SJ. 2014. Phenotypic and genotypic characterization of daptomycin-resistant methicillin-resistant Staphylococcus aureus strains: relative roles of mprF and dlt operons. PLoS One 9:e107426.
17. Biswas S, Brunel JM, Dubus JC, Reynaud-Gaubert M, Rolain JM. 2012. Colistin: an update on the antibiotic of the 21st century. Expert Rev Anti Infect Ther 10:917-934.
18. Cotter PD, Hill C, Ross RP. 2005. Bacteriocins: developing innate immunity for food. Nat Rev Microbiol 3:777-788.
112
19. Willey JM, van der Donk WA. 2007. Lantibiotics: peptides of diverse structure and function. Annu Rev Microbiol 61:477-501.
20. van Kraaij C, Breukink E, Noordermeer MA, Demel RA, Siezen RJ, Kuipers OP, de Kruijff B. 1998. Pore formation by Nisin involves translocation of its C-terminal part across the membrane. Biochemistry 37:16033-16040.
21. Wiedemann I, Breukink E, van Kraaij C, Kuipers OP, Bierbaum G, de Kruijff B, Sahl HG. 2001. Specific binding of nisin to the peptidoglycan precursor lipid II combines pore formation and inhibition of cell wall biosynthesis for potent antibiotic activity. J Biol Chem 276:1772-1779.
22. Breukink E, de Kruijff B. 2006. Lipid II as a target for antibiotics. Nat Rev Drug Discov 5:321-332. 23. Scherer KM, Spille JH, Sahl HG, Grein F, Kubitscheck U. 2015. The lantibiotic nisin induces lipid II
aggregation, causing membrane instability and vesicle budding. Biophys J 108:1114-1124. 24. Tan NC, Yu P, Kwon YU, Kodadek T. 2008. High-throughput evaluation of relative cell permeability
between peptoids and peptides. Bioorg Med Chem 16:5853-5861. 25. Dar AC, Das TK, Shokat KM, Cagan RL. 2012. Chemical genetic discovery of targets and anti-
targets for cancer polypharmacology. Nature 486:80-84. 26. Willoughby LF, Schlosser T, Manning SA, Parisot JP, Street IP, Richardson HE, Humbert PO,
Brumby AM. 2013. An in vivo large-scale chemical screening platform using Drosophila for anti-cancer drug discovery. Dis Model Mech 6:521-529.
27. Chamilos G, Samonis G, Kontoyiannis DP. 2011. Drosophila melanogaster as a model host for the study of microbial pathogenicity and the discovery of novel antimicrobial compounds. CurrPharmDes 17:1246-1253.
28. Ben-Ami R, Watson CC, Lewis RE, Albert ND, Arias CA, Raad, II, Kontoyiannis DP. 2013. Drosophila melanogaster as a model to explore the effects of methicillin-resistant Staphylococcus aureus strain type on virulence and response to linezolid treatment. Microb Pathog 55:16-20.
29. Tzelepis I, Kapsetaki S-E, Panayidou S, Apidianakis Y. 2013. Drosophila melanogaster: a first step and a stepping-stone to anti-infectives. Current Opinion in Pharmacology 13:763-768.
30. Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. 1996. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 86:973-983.
31. Lemaitre B, Kromer-Metzger E, Michaut L, Nicolas E, Meister M, Georgel P, Reichhart JM, Hoffmann JA. 1995. A recessive mutation, immune deficiency (imd), defines two distinct control pathways in the Drosophila host defense. Proceedings of the National Academy of Sciences of the United States of America 92:9465-9469.
32. Dionne MS, Ghori N, Schneider DS. 2003. Drosophila melanogaster is a genetically tractable model host for Mycobacterium marinum. InfectImmun 71:3540-3550.
33. Apidianakis Y, Rahme LG. 2009. Drosophila melanogaster as a model host for studying Pseudomonas aeruginosa infection. NatProtoc 4:1285-1294.
34. Atilano ML, Yates J, Glittenberg M, Filipe SR, Ligoxygakis P. 2011. Wall teichoic acids of Staphylococcus aureus limit recognition by the drosophila peptidoglycan recognition protein-SA to promote pathogenicity. PLoS Pathog 7:e1002421.
35. Novick R. 1967. Properties of a cryptic high-frequency transducing phage in Staphylococcus aureus. Virology 33:155-166.
36. McDougal LK, Steward CD, Killgore GE, Chaitram JM, McAllister SK, Tenover FC. 2003. Pulsed-field gel electrophoresis typing of oxacillin-resistant Staphylococcus aureus isolates from the United States: establishing a national database. J Clin Microbiol 41:5113-5120.
37. Mattick AT, Hirsch A. 1947. Further observations on an inhibitory substance (nisin) from lactic streptococci. Lancet 2:5-8.
113
38. Castiglione F, Cavaletti L, Losi D, Lazzarini A, Carrano L, Feroggio M, Ciciliato I, Corti E, Candiani G, Marinelli F, Selva E. 2007. A novel lantibiotic acting on bacterial cell wall synthesis produced by the uncommon actinomycete Planomonospora sp. Biochemistry 46:5884-5895.
39. Jabes D, Brunati C, Candiani G, Riva S, Romano G, Donadio S. 2011. Efficacy of the new lantibiotic NAI-107 in experimental infections induced by multidrug-resistant Gram-positive pathogens. Antimicrob Agents Chemother 55:1671-1676.
40. Ross AC, Vederas JC. 2011. Fundamental functionality: recent developments in understanding the structure-activity relationships of lantibiotic peptides. J Antibiot (Tokyo) 64:27-34.
41. Fjell CD, Jenssen H, Cheung WA, Hancock REW, Cherkasov A. 2011. Optimization of Antibacterial Peptides by Genetic Algorithms and Cheminformatics. Chemical Biology & Drug Design 77:48-56.
42. Troels Godballe BM, Hanne M. Nielsen, Håvard Jenssen. 2015. Antimicrobial activity of GN peptides and their mode of action Biopolymers doi:BIP-PEP-2015-00052.R1.
43. Fjell CD, Jenssen H, Hilpert K, Cheung WA, Pante N, Hancock RE, Cherkasov A. 2009. Identification of novel antibacterial peptides by chemoinformatics and machine learning. J Med Chem 52:2006-2015.
44. Cherkasov A, Hilpert K, Jenssen H, Fjell CD, Waldbrook M, Mullaly SC, Volkmer R, Hancock RE. 2009. Use of artificial intelligence in the design of small peptide antibiotics effective against a broad spectrum of highly antibiotic-resistant superbugs. ACS Chem Biol 4:65-74.
45. Mojsoska B, Zuckermann RN, Jenssen H. 2015. Structure-Activity Relationship Study of Novel Peptoids That Mimic the Structure of Antimicrobial Peptides. Antimicrob Agents Chemother 59:4112-4120.
46. Wiegand I, Hilpert K, Hancock RE. 2008. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat Protoc 3:163-175.
47. Maisonneuve E, Castro-Camargo M, Gerdes K. 2013. (p)ppGpp controls bacterial persistence by stochastic induction of toxin-antitoxin activity. Cell 154:1140-1150.
48. Danielsen ET, Moeller ME, Dorry E, Komura-Kawa T, Fujimoto Y, Troelsen JT, Herder R, O'Connor MB, Niwa R, Rewitz KF. 2014. Transcriptional control of steroid biosynthesis genes in the Drosophila prothoracic gland by ventral veins lacking and knirps. PLoS Genet 10:e1004343.
49. Maffioli SI, Iorio M, Sosio M, Monciardini P, Gaspari E, Donadio S. 2014. Characterization of the congeners in the lantibiotic NAI-107 complex. J Nat Prod 77:79-84.
50. Wu K, Conly J, Surette M, Sibley C, Elsayed S, Zhang K. 2012. Assessment of virulence diversity of methicillin-resistant Staphylococcus aureus strains with a Drosophila melanogaster infection model. BMC Microbiol 12:274.
51. Gordon MD, Ayres JS, Schneider DS, Nusse R. 2008. Pathogenesis of listeria-infected Drosophila wntD mutants is associated with elevated levels of the novel immunity gene edin. PLoS Pathog 4:e1000111.
52. Kindrachuk J, Jenssen H, Elliott M, Nijnik A, Magrangeas-Janot L, Pasupuleti M, Thorson L, Ma S, Easton DM, Bains M, Finlay B, Breukink EJ, Georg-Sahl H, Hancock RE. 2013. Manipulation of innate immunity by a bacterial secreted peptide: lantibiotic nisin Z is selectively immunomodulatory. Innate Immun 19:315-327.
54. Johnson BA, Anker H, Meleney FL. 1945. BACITRACIN: A NEW ANTIBIOTIC PRODUCED BY A MEMBER OF THE B. SUBTILIS GROUP. Science 102:376-377.
55. Spann CT, Taylor SC, Weinberg JM. 2004. Topical antimicrobial agents in dermatology. Dis Mon 50:407-421.
114
56. Piper C, Draper LA, Cotter PD, Ross RP, Hill C. 2009. A comparison of the activities of lacticin 3147 and nisin against drug-resistant Staphylococcus aureus and Enterococcus species. J Antimicrob Chemother 64:546-551.
57. Dosler S, Gerceker AA. 2011. In vitro activities of nisin alone or in combination with vancomycin and ciprofloxacin against methicillin-resistant and methicillin-susceptible Staphylococcus aureus strains. Chemotherapy 57(6):511-516.
58. Lepak AJ, Marchillo K, Craig WA, Andes DR. 2015. In vivo pharmacokinetics and pharmacodynamics of the lantibiotic NAI-107 in a neutropenic murine thigh infection model. Antimicrob Agents Chemother 59:1258-1264.
59. Reddy KV, Gupta SM, Aranha CC. 2011. Effect of antimicrobial Peptide, nisin, on the reproductive functions of rats. ISRN Vet Sci 2011:828736.
60. Padovan J, Ralic J, Letfus V, Milic A, Bencetic Mihaljevic V. 2012. Investigating the barriers to bioavailability of macrolide antibiotics in the rat. Eur J Drug Metab Pharmacokinet 37:163-171.
61. Breukink E, Wiedemann I, van Kraaij C, Kuipers OP, Sahl HG, de Kruijff B. 1999. Use of the cell wall precursor lipid II by a pore-forming peptide antibiotic. Science 286:2361-2364.
62. Scherer K, Wiedemann I, Ciobanasu C, Sahl HG, Kubitscheck U. 2013. Aggregates of nisin with various bactoprenol-containing cell wall precursors differ in size and membrane permeation capacity. Biochim Biophys Acta 1828:2628-2636.
63. Brand AM, de Kwaadsteniet M, Dicks LM. 2010. The ability of nisin F to control Staphylococcus aureus infection in the peritoneal cavity, as studied in mice. Lett Appl Microbiol 51:645-649.
64. Cotter PD, Hill C, Ross RP. 2005. Bacterial lantibiotics: strategies to improve therapeutic potential. Curr Protein Pept Sci 6:61-75.
65. Munch D, Muller A, Schneider T, Kohl B, Wenzel M, Bandow JE, Maffioli S, Sosio M, Donadio S, Wimmer R, Sahl HG. 2014. The lantibiotic NAI-107 binds to bactoprenol-bound cell wall precursors and impairs membrane functions. J Biol Chem 289:12063-12076.
66. Ruhr E, Sahl HG. 1985. Mode of action of the peptide antibiotic nisin and influence on the membrane potential of whole cells and on cytoplasmic and artificial membrane vesicles. Antimicrob Agents Chemother 27:841-845.
67. Ben-Ami R, Watson CC, Lewis RE, Albert ND, Arias CA, Raad II, Kontoyiannis DP. 2012. Drosophila melanogaster as a model to explore the effects of methicillin-resistant Staphylococcus aureus strain type on virulence and response to linezolid treatment. MicrobPathog.
68. Moy TI, Ball AR, Anklesaria Z, Casadei G, Lewis K, Ausubel FM. 2006. Identification of novel antimicrobials using a live-animal infection model. Proc Natl Acad Sci U S A 103:10414-10419.
115
Supplemental Material
Supplemental Figure 1. In vivo efficacy of peptides and peptoids: Graphs show survival of flies treated with GN-2 (S1), GN-2 peptoids (S2 and S3), HHC-9 (S4), HHC-36 (S5) and NAI-107 (S6). Y-axis show fraction survival compared to time in Hours (x-axis). Flies were counted at time points 0, 3, 6, 12, 24, 48 – 120 hours. Right side figure legends represent treatment groups: Flies are injected with MgSO4, bacterial isolate 8325-4 or USA300 at time 0, + indicate peptide/peptoid treatment at time point 3 hours (dotted line). Compound concentrations [C] are given as approximated concentration in animals.
116
Table S1. Primer sequences: Forward (FW) and Reverse (RV) primers are shown in right column. Gene annotations as CG numbers are indicated, according to flybase.org.
117
Discussion
Important pathogens from the ESKAPE group, pose an eminent and growing problem
globally. Resistance to colistin the last resort drug for serious Gram-negative infections has been
found in A. baumannii and K. pneumoniae caused by LPS modifications through mutation (107,
109, 110, 114, 252). However, the discovery of horizontally transferrable genetic element carrying
the mcr-1 gene in E. coli seriously jeopardizes future treatment with colistin (118). Furthermore,
evidence of failure to the important antibiotics vancomycin, linezolid and daptomycin is increasing
in the important Gram-positive bacteria E. Faecalis and S. aureus (67, 69, 74, 77, 84, 85).
Bacteriocins and host defense peptides are widespread and universally spread throughout nature as
part of organisms defenses against invading microorganisms (122). For these reasons antimicrobial
peptides are of interest for the development of the next generation of novel antibiotics.
Amphiphilic cationic peptides and peptoids
We have applied solid phase synthesis to develop the BP214 peptide into a lead
candidate with some promising characteristics in vitro (253). BP214 follow in the footsteps of other
Cecropin-mellitin hybrid molecules that effectively kill colistin resistant strains of A. baumannii
(160, 254). Several such molecules have even been tested in the mouse peritoneal sepsis model with
some efficacy (161). The BP214 peptide was developed from BP100 (195) and RW-BP100 peptides
(193). By analytical and combinatorial approach we developed a panel of lead compounds with
characteristic from both peptides (253). The BP214 peptide proved highly effective in killing
A. baumannii, while unable to kill non-growing wild type cells. BP214 is able to kill all growing
cells, irrespective of genotype. Theoretically this could have a selective capability of the compound,
not previously seen i.e. BP214 will kill all colistin resistant A. baumannii while leaving persisters of
wild type origin. If this applies to other colistin resistant Gram-negative bacteria such as E. coli or
K. pneumoniae it is contemplated if this might lower detrimental side effects to the commensal
flora.
The spectrum of activity for BP214 is more broad than narrow, as it also has a fairly
low MIC towards E. faecium [Table 1 (253)]. However, the MIC towards S. aureus is significantly
118
higher. The overall broad spectrum of the BP214 cecropin-mellitin hybrid is comparable with
previous findings that cecropins target both Gram-positive and Gram-negative species (255) and
mellitin is non-selective in nature (256, 257) and other cecropin-mellitin hybrids share these
characteristic (193). Our data seems to be in line with this even after modifications of the molecule.
Regarding the toxicity of the compound, BP214 is relatively non-toxic as measured by hemolytic
ability. However, based on our own data indicating toxicity of cationic amphiphilic molecules using
the Drosophila in vivo efficacy model, it seems likely that BP214 could be toxic to eukaryotic cells.
Other cecropin-mellitin hybrids have been reported to have relatively high hemolytic ability on
mouse blood cells (161).
In vivo efficacy of antimicrobial peptides is generally hampered by the in vivo
sequestration by blood serum and proteolytic degradation by proteases (161, 187, 201). In this
regard it is of high interest that BP214 consists of only D-amino acids that retain antimicrobial
activity while displaying low hemolytic ability (253). The D-amino acids should render the molecule
less prone to degradation (200, 258) thereby expanding a possible application to include oral
therapy. These same features are apparent for molecules such as the circular antimicrobial peptides
described in paper II, which were modulated to retain activity, while lowering toxicity in hemolytic
ability. With respect to sequestration by serum this remains to be discovered, but it seems plausible
that this issue would have to be addressed for further development.
It has previously been argued that synthetic compounds could be expected to slow
development of resistance (74). This might be further accommodated by the incorporation of D-
amino acids. As BP214 is active against A. baumannii resistant to colistin through PmrA-PmrB
mutations and loss of LPS, it seems that these highly relevant mechanisms for bacterial resistance
towards cationic molecules have been circumvented. However, it would be of interest to examine
whether the changes to the LPS in K. pneumoniae due to PhoP-PhoQ mutations associated with
increased resistance to antimicrobial peptides such as colistin, have the same impact on activity of
BP214 (110). On the negative side; BP214`s relative broad spectrum activity, but low activity to S.
aureus, might cross select for resistance genotypes such as the VISA strains or strains with
dysfunctional expression of MprF and the dlt operon (84-86, 259, 260) as these changes relate to
changes in membrane charge. The broad spectrum AMP bacitracin has been associated with
selection of highly resistant MRSA strains (261). Circular peptides as described in Paper II, with
119
efficacy towards Gram-positive bacteria such as VRE, could possibly have similar effect on
selection of Gram-positive bacteria, but because of the narrow spectrum, would most likely not
select for resistant Gram-negative bacteria.
With respect to the amphiphilic and cationic peptides and peptoids developed against
S. aureus. Previous studies reported low toxicity in cell based assays and in mouse peritoneal model
(197, 262). We find clear and not fully understood differences with regard to toxicity. As explained
we believe that some of our findings indicate neurotoxic effects, these would not necessarily have
been seen in the HeLa cell based assays (197). Since we do not know anything about
pharmacokinetics and pharmacodynamics of HHC peptides in the mouse peritoneal model (262),
we speculate that the peptides do not leave the peritoneum and enter circulation in this model
leaving toxicities unobserved. There is also the possibility that the Drosophila model described is
hyper sensitive to toxic effects, as the Drosophila circulatory system is less compartmentalized.
However, a whole animal model, intuitively encompass a more realistic physiological environment
and more complexity than cell based assays. This reasoning is based on the fact that the whole
animal model encompasses infection, host pathogen interaction and treatment in one. This is in line
with our findings that several already proven antimicrobials are non-toxic in this model (NAi-107,
vancomycin and ampicillin). Further, the peptides and peptoids are toxic, but only when injected
after infection which would by left undescribed in most cell based assays where cells are subjected
to peptides but without any pathogen interaction. Peptides might not interfere with cell viability, but
stress the cells or otherwise interfere with important cellular functions. The phenotypes described
regarding possible neurotoxicity, certainly imply that several compounds interfere with neuronal
physiology. We speculate if this could be because of the molecules general electrostatic interaction
with membranes.
Lipid-II targeting peptides
In paper III we have shown that the two Lantibiotics nisin and NAI-107 targeting the
peptidoglycan cell wall precursor Lipid II can rescue or postpone lethality of USA300 infection in a
Drosophila in vivo efficacy model. Nisin has previously been reported as non-toxic (144, 157) but
has been tested limited in vivo. One experiments showed that nisin A was unable to control
120
infection by L. monocytogenes while nisin V showed efficacy (263). Campion et al (263) described
reoccurrence of bacteria after treatment with nisin A, which is in line with our data that nisin A is
unable to rescue infection and only prolong survival. Further this is underlined by in vitro
experiments showing re-growth of nisin treated bacterial cultures. NAI-107 was shown to have
prolonged effect in vitro and in vivo, in line with previous studies, showing prolonged post
antibiotic effects in vivo (264). NAI-107 also rescued animals from infection without any observed
detrimental effects, consistent with observations by Lepak et al. demonstrating no toxicity in a
murine thigh infection model (264). In the study by Lepak et al. NAI-107 was injected at doses
ranging from 5 to 80 mg/kg, our maximum dose of NAI-107 was 16 mg/kg well within this range
and the low toxicity is comparable. NAI-107 has been found superior to several reference
compounds including vancomycin (265), contradicting our data. However, our experiments only
included injection of a single dose of each compound. The previously reported superiority might be
explained by the ability of NAI-107 to kill non-growing cells, but this will have to be further
explored. Further the Jabes et al study showed that a single dose of NAI-107 (40mg/kg) prevented
regrowth over 96 hours (265). Our data for NAI-107 in the Drosophila model seems to support
these findings, but because we used lower doses this might explain why we did not rescue all
animals. Finally, we showed that both nisin and NAI-107 has the capability of effectively killing
stationary phase cultures of S. aureus, emphasizing yet another potential for these lantibiotics. Since
this property is not found for many widely used antibiotics such as vancomycin and β-lactams it
could pose as an important characteristic against persistent infections. Several Lipid II targeting
compounds, like NAI-107 are of growing interest as the next generation of antimicrobials for
clinical therapy (137). Like the newly discovered teixobactin (158), NAI-107 efficiently kills highly
drug resistant strains such as MRSA and VRE (265). The killing of stationary phase cultures has not
been reported for teixobactin, although it might have been tested.
Nisin has been shown to be rapidly degraded by pancreatic proteases through oral
administration (144, 157). This could indicate that it would be sensitive to other proteases as well.
Indeed nisin has been shown to be degradable by aureolysin (187). So far no protease capable of
degrading NAI-107 has been found, but elevated MIC has been found for VISA and VRSA
(personal communication with Stefano Donadio). Nisin resistance in S. aureus has been described
as D-alanylation of techoic acids by changing expression or copy number of the dlt operon,
production of L-PG by MprF, upregulation of the BraRS two-component system and similar
121
systems (266-268). Generally the overall picture is that lantibiotic resistance is accommodated
through changes in the cell membrane architecture (266). These mechanisms are in many aspects
parallel to resistance mechanism to peptides acting on the cell wall of Gram-negative bacteria such
as colistin (108-110, 112, 113, 182, 252). We expect that similar mechanisms might be found for
NAI-107, but to our knowledge this has not been described. Furthermore, the resistance profiles
found for lantibiotics such as nisin emphasizes the possibility of lantibiotics selecting for cross
resistance to antibiotics such as daptomycin in Gram-positive bacteria. Although NAI-107 has been
shown to have activity against some Gram-negative bacteria (265), most lantibiotics are not active
against Gram-negative bacteria because of their outer membrane (138, 139). Therefore, cross
selection of resistance seems unlikely. However, given the historical evidence of drug resistance
development, it would be wise not to underestimate such development.
Interestingly we have also found that NAI-107 has activity against LPS deficient (108,
109) strains of A. baumannii (unpublished data), and that NAI-107 in combination with colistin has
synergistic effects against A. baumannii (unpublished data). This is consistent with observations of
Cui et al. demonstrating that vancomycin in combination with colistin have synergistic effects
against carbapenem resistant A. baumannii (269). This is most likely due to colistin permeabilizing
and accommodating penetrance of vancomycin/NAI-107 through the outer membrane, thereby
gaining access to the underlying peptidoglycan layer of Gram-negative bacteria. Although Gram-
negative bacteria have slightly different composition of the pentapeptide on the peptidoglycan
precursor Lipid II, the D-ala-D-ala binding motif of vancomycin is the same [Figure 16 (24)]. This
opens for the possibility of combining these compounds to produce broad spectrum combination
treatments using colistin or perhaps similar compounds (e.g. BP214) in combination with Lipid II
targeting antimicrobials.
122
Figure 16. Lipid II of Gram-positive and Gram-negative bacteria: Adapted from (24, 235).
Overall composition of the peptidoglycan precursor Lipid II of Gram-positive and Gram-negative bacteria. The Gram-positive bacilli have similar Lipid II to Gram-negative bacteria. The major difference between Lipid II is the substitution of L-lys with meso-DAP in the pentapeptide (235).
A Drosophila in vivo efficacy model of infection
The Drosophila in vivo model presented in paper III has several benefits compared to
the tightly regulated and expensive mammalian models normally used (270). We have provided
evidence for the use of this model for analysis of efficacy and toxicity in vivo. Because in vivo
testing of many of the compounds has been limited, it is difficult to evaluate the results by
comparison to previous studies. Several of the discrepancies found will need further evaluation of
the model in comparison to established mammalian models. However, we do show that the two
lantibiotics in this model compare to previous findings from mice, both regarding toxicity and
efficacy of NAI-107. On the other hand, we do find nisin to be toxic, which was not found by
Campion et al. (263) when tested in mice. As already discussed all the amphipathic cationic
peptides and peptoids tested were not efficacious in this model and several proved lethal. At least
for the HHC36 peptide, this is different to previous findings from mice (262). However, to evaluate
systemic toxicity in mice, we argue that HHC36 should be injected into the circulatory system
instead of the peritoneum. As a whole animal model system Drosophila seems to encompass
several important aspects of antimicrobial drug testing, but the model suffers from one major
drawback. Because Drosophila has an ideal maximum temperature of 29°C, it does not
accommodate optimal bacterial growing temperature at 37°C which is encountered by human
pathogens in the body. Finally, it should be noted as previously discussed, that this model might be
123
more sensitive to toxic/lethal effects and therefore it should be used as an addition to other models
and not as an alternative.
Conclusions
Our data support previous findings of Lipid II acting molecules as good candidates for
clinical development. Importantly, we have shown that NAI-107 unlike vancomycin and other
important antibiotics has the ability to kill non-growing bacteria in vitro and at concentrations
comparable to doses tested in mice in vivo (264). Although NAI-107 already has been tested in vivo
with good efficacy, experiments with NAI-107 provided evidence of our Drosophila models
applicability and this adds to the growing evidence of NAI-107 as a lead molecule. Because of the
low cost of the Drosophila model, we have been able to experiment on large numbers of animals
and we have been able to reproduce the data several times. Our Drosophila experiments have also
shed light on possible problems with classical toxicity screenings using cell lines. We are not
arguing that the usage of cell lines should be disregarded in the developmental process; merrily we
are arguing that an intermediate in vivo model might be useful to antimicrobial development before
undertaking expensive and labor intensive in vivo experiments on mammalian models. Especially
since it seems that many previous peptides fail in clinical trials because of toxicity issues (202). It
has certainly been argued by others that compounds are often rushed into clinical development and
therefore fail in this process (20). Especially insect models such as the one presented here or the G.
mellonella model (211) could be combined with models such as the C. elegans model for large scale
drug screening (206) and toxicity evaluation. The larvae of Drosophila can also be grown in 96 well
based systems, which could be applicable to large scale screening as performed for C. elegans.
BP214 was shown to encompass several interesting characteristics such low hemolytic
ability and incorporation of D-amino acids while retaining activity against A. baumannii and to a
certain extent also E. coli and K. pneumoniae. Further, the evidence points to BP214 interacting
with the membrane via mechanisms that are not solely dependent on LPS. For this reason the
molecule retains its activity against LPS modified A. baumannii resistant to the last resort antibiotic
colistin. Finally, BP214 is capable of killing non-growing cells of colistin resistant A. baumannii,
implying that AMP resistance through LPS modification renders the cell more susceptible to certain
124
cell disrupting compounds like BP214 when in stationary phase. This discovery could be interesting
for future drug development.
125
Future perspectives
The cecropin-mellitin hybrid BP214
BP214 remains as an interesting candidate for further development. As we have only
scratched the surface of this cecropin-mellitin hybrid. The data presented in paper I, does provide
interesting and positive insight for further development of this molecule. It would be of interest to
continue working with BP214 as this molecule might give insight into future aspects of
antimicrobial peptide development. Especially since we are now experiencing what can only be
expected to be the beginning of colistin treatment failure (118). This discovery certainly adds to the
growing body of evidence that we could be moving towards a post-antibiotic era and for this reason
it is of huge importance to develop new antibiotics with novel applications.
We are hoping to undertake a larger project for further investigation of BP214 as a
lead candidate. We would like to do more structure activity analysis, to determine the optimal
length of BP214 by synthesis of C and N terminal truncated versions of the molecule. Furthermore,
alanine scanning or incorporation of non proteinogenic amino acids (including peptoids) could be
further explored for optimization. Addition of sidechain and the distribution of these could be
another means of creating molecules with improved characteristics. However, such work would
have to be in collaboration with other people such as Professor Paul Robert Hansen with whom we
have collaborated on paper I and II.
For BP214 to have wide applicability it would need to have activity against other
Gram-negative bacteria (E. coli, K. pneumoniae and P. aeruginosa). Such compound would also
have to undergo vigorous in vitro and in vivo toxicity and efficacy testing. This could be performed
using hemolysis and cell proliferation assays, but with the addition of the Drosophila or G.
mellonella models as intermediary in vivo platforms. It would be of high interest to include analysis
of BP214 having synergistic effects with molecules such as NAI-107 and vancomycin. Further, the
molecule might be optimized to kill non-growing cells regardless of genotype which might increase
its impact as a therapeutic option. If BP214 could be optimized to incorporate these characteristics
in a lead molecule it might be tested for in vivo efficacy on mammalian models such as the, mouse
126
peritoneal sepsis model (161). Further, the investigation of toxicity in both insect and a mammalian
model might explain some of the major differences found in paper III.
Because BP214 most likely interact with bacterial membranes through electrostatic
interactions and selectively kill non-growing colistin resistant A. baumannii, it seems that the
colistin resistance genotype causes collateral damage to the membrane when cells are in stationary
phase. Because it is often difficult to describe the interaction of AMP with membranes, it might be
possible to gain insight into these mechanisms through resistance development studies. By an
evolutionary approach where bacterial cells are grown at continually increasing concentration of
BP214 we could select for tolerance/resistance to BP214. Such mutants can be subjected to full
genome sequencing and compared to wild type cells. Such studies would be of general interest
given that peptide antibiotic`s such as colistin and polymyxin are used increasingly, and in this
respect it is of interest to know how resistance to peptide antimicrobials might force bacterial cells
to fundamentally change the overall architecture of the bacterial membrane. Studying the
toxicology and efficacy of peptide antimicrobials is also of importance for future development and
understanding of peptide based antimicrobials.
Lantibiotics and other Lipid II targeting antimicrobials
For the future, it seems evident that peptide based antibiotics and especially the
lantibiotics and/or other Lipid II targeting antibiotics will become useful in clinical therapy for
treatment of highly drug resistant strains such as USA300 and VRE. As NAI-107 is patented by
Naicons (Naicons Srl. Milan, Italy) we can only hope to be part of future development through
continued collaboration with Stefano Donadio. Our findings that NAI-107 kills stationary phase
bacteria might be utilized for future development. It has already been proposed that lantibiotics has
potential for bioengineering of new compounds (125, 131) and in this respect it could be important
to determine the characteristics that make NAI-107 efficacious against stationary phase cultures. As
many lantibiotics have been demonstrated to function through binding of Lipid II, it would be
interesting to understand what governs activity against stationary phase cultures. It would also be of
interest to try and understand how future resistance might develop. Resistance development to nisin
has been described as slow. It would be interesting to undertake evolutionary studies in which S.
127
aureus are grown at continually increasing concentrations of NAI-107 and as for BP214 evaluate
resistance through full genome sequencing. Teixobactin was described as killing bacteria without
detectable resistance (158), but this statement seems overestimated given the historical evidence of
resistance development.
It seems evident that the continued overuse of last resort antibiotics such colistin (118)
has to be managed on a global scale. Colistin is used increasingly in clinical medicine (105) and in
agricultural settings (118), driving selection of resistance determinants. New and novel antibiotics
are desperately needed to avoid a problematic post antibiotic era (53), which is moving continually
closer. Antimicrobial peptides such as lantibiotics or other host defense peptides have been
proposed as the solution (122, 125, 131, 262). However, the latest antibiotics approved, such as
telavancin, are representatives of older drug classes. This might be because previous attempts of
peptide development has been rushed (202). Therefore we need to further understand the biology of
these molecules; their interaction with membranes, resistance development and their toxicities, so
that we may develop them into next generation of antimicrobials. Especially how these compounds
relate to previously developed antibiotics. Do they select for similar resistance profiles or do they
force collateral damage to the cell and can this be further explored as a means of antibiotic
development.
128
BIBLIOGRAPHY
1. Williams KJ. The introduction of ‘chemotherapy’ using arsphenamine – the first magic bullet. Journal of the Royal Society of Medicine. 2009;102(8):343-8. 2. Otten H. Domagk and the development of the sulphonamides. The Journal of antimicrobial chemotherapy. 1986;17(6):689-96. 3. Fleming A. On the antibacterial action of cultures of a penicillium, with special reference to their use in the isolation of B. influenzae. 1929. Bulletin of the World Health Organization. 2001;79(8):780-90. 4. Waksman SA. What is an antibiotic or an antibiotic substance? Mycologia. 1947;39(5):565-9. 5. Ligon BL. Penicillin: its discovery and early development. Seminars in pediatric infectious diseases. 2004;15(1):52-7. 6. Waksman SA, Woodruff HB. The Soil as a Source of Microorganisms Antagonistic to Disease-Producing Bacteria. Journal of bacteriology. 1940;40(4):581-600. 7. Wainwright M. Streptomycin: discovery and resultant controversy. History and philosophy of the life sciences. 1991;13(1):97-124. 8. Spellberg B. Dr. William H. Stewart: mistaken or maligned? Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2008;47(2):294. 9. Clatworthy AE, Pierson E, Hung DT. Targeting virulence: a new paradigm for antimicrobial therapy. Nature chemical biology. 2007;3(9):541-8. 10. Acar J. Broad- and narrow-spectrum antibiotics: an unhelpful categorization. Clinical microbiology and infection : the official publication of the European Society of Clinical Microbiology and Infectious Diseases. 1997;3(4):395-6. 11. Blaser M. Antibiotic overuse: Stop the killing of beneficial bacteria. Nature. 2011;476(7361):393-4. 12. Willing BP, Russell SL, Finlay BB. Shifting the balance: antibiotic effects on host-microbiota mutualism. Nature reviews Microbiology. 2011;9(4):233-43. 13. Tommasi R, Brown DG, Walkup GK, Manchester JI, Miller AA. ESKAPEing the labyrinth of antibacterial discovery. Nature reviews Drug discovery. 2015;advance online publication. 14. Slonczewski J, Foster, J. W., & Gillen, K. M. Microbiology: An evolving science. 2nd edition ed: New York London: W.W. Norton & Co.; 2009. 1100 p. 15. Poole K. Efflux-mediated antimicrobial resistance. The Journal of antimicrobial chemotherapy. 2005;56(1):20-51. 16. Pankey GA, Sabath LD. Clinical relevance of bacteriostatic versus bactericidal mechanisms of action in the treatment of Gram-positive bacterial infections. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2004;38(6):864-70. 17. Rahal JJ, Jr., Simberkoff MS. Bactericidal and bacteriostatic action of chloramphenicol against memingeal pathogens. Antimicrobial agents and chemotherapy. 1979;16(1):13-8. 18. Wilson DN. Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nature reviews Microbiology. 2013;12(1):35-48. 19. Nathan C. Antibiotics at the crossroads. Nature. 2004;431(7011):899-902. 20. Fischbach MA, Walsh CT. Antibiotics for emerging pathogens. Science (New York, NY). 2009;325(5944):1089-93.
129
21. Bassetti M, Merelli M, Temperoni C, Astilean A. New antibiotics for bad bugs: where are we? Annals of clinical microbiology and antimicrobials. 2013;12:22. 22. Masters PA, O'Bryan TA, Zurlo J, Miller DQ, Joshi N. Trimethoprim-sulfamethoxazole revisited. Archives of internal medicine. 2003;163(4):402-10. 23. Redgrave LS, Sutton SB, Webber MA, Piddock LJ. Fluoroquinolone resistance: mechanisms, impact on bacteria, and role in evolutionary success. Trends Microbiol. 2014;22(8):438-45. 24. Typas A, Banzhaf M, Gross CA, Vollmer W. From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nature reviews Microbiology. 2012;10(2):123-36. 25. Goldstein BP. Resistance to rifampicin: a review. The Journal of antibiotics. 2014;67(9):625-30. 26. Waxman DJ, Strominger JL. Penicillin-binding proteins and the mechanism of action of beta-lactam antibiotics. Annual review of biochemistry. 1983;52:825-69. 27. Barton MD. Impact of antibiotic use in the swine industry. Current opinion in microbiology. 2014;19c:9-15. 28. Andersson DI, Hughes D. Antibiotic resistance and its cost: is it possible to reverse resistance? Nature reviews Microbiology. 2010;8(4):260-71. 29. Lowy FD. Antimicrobial resistance: the example of Staphylococcus aureus. The Journal of clinical investigation. 2003;111(9):1265-73. 30. Nielsen O, Lobner-Olesen A. Once in a lifetime: strategies for preventing re-replication in prokaryotic and eukaryotic cells. EMBO reports. 2008;9(2):151-6. 31. Val ME, Soler-Bistue A, Bland MJ, Mazel D. Management of multipartite genomes: the Vibrio cholerae model. Current opinion in microbiology. 2014;22:120-6. 32. Agnoli K, Schwager S, Uehlinger S, Vergunst A, Viteri DF, Nguyen DT, et al. Exposing the third chromosome of Burkholderia cepacia complex strains as a virulence plasmid. Molecular microbiology. 2012;83(2):362-78. 33. Tenaillon O, Skurnik D, Picard B, Denamur E. The population genetics of commensal Escherichia coli. Nature reviews Microbiology. 2010;8(3):207-17. 34. Lowy FD. Staphylococcus aureus infections. The New England journal of medicine. 1998;339(8):520-32. 35. Mazel D. Integrons: agents of bacterial evolution. Nature reviews Microbiology. 2006;4(8):608-20. 36. Serres MH, Kerr AR, McCormack TJ, Riley M. Evolution by leaps: gene duplication in bacteria. Biology direct. 2009;4:46. 37. Levy SB, Marshall B. Antibacterial resistance worldwide: causes, challenges and responses. Nature medicine. 2004;10(12 Suppl):S122-9. 38. Jin DJ, Gross CA. Mapping and sequencing of mutations in the Escherichia coli rpoB gene that lead to rifampicin resistance. Journal of molecular biology. 1988;202(1):45-58. 39. Taniguchi H, Aramaki H, Nikaido Y, Mizuguchi Y, Nakamura M, Koga T, et al. Rifampicin resistance and mutation of the rpoB gene in Mycobacterium tuberculosis. FEMS microbiology letters. 1996;144(1):103-8. 40. Katayama Y, Ito T, Hiramatsu K. A new class of genetic element, staphylococcus cassette chromosome mec, encodes methicillin resistance in Staphylococcus aureus. Antimicrobial agents and chemotherapy. 2000;44(6):1549-55. 41. Hartman BJ, Tomasz A. Low-affinity penicillin-binding protein associated with beta-lactam resistance in Staphylococcus aureus. Journal of bacteriology. 1984;158(2):513-6. 42. Borges-Walmsley MI, McKeegan KS, Walmsley AR. Structure and function of efflux pumps that confer resistance to drugs. The Biochemical journal. 2003;376(Pt 2):313-38.
130
43. McMurry L, Petrucci RE, Jr., Levy SB. Active efflux of tetracycline encoded by four genetically different tetracycline resistance determinants in Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America. 1980;77(7):3974-7. 44. Butaye P, Cloeckaert A, Schwarz S. Mobile genes coding for efflux-mediated antimicrobial resistance in Gram-positive and Gram-negative bacteria. Int J Antimicrob Agents. 2003;22(3):205-10. 45. Bissonnette L, Champetier S, Buisson JP, Roy PH. Characterization of the nonenzymatic chloramphenicol resistance (cmlA) gene of the In4 integron of Tn1696: similarity of the product to transmembrane transport proteins. Journal of bacteriology. 1991;173(14):4493-502. 46. Baucheron S, Tyler S, Boyd D, Mulvey MR, Chaslus-Dancla E, Cloeckaert A. AcrAB-TolC directs efflux-mediated multidrug resistance in Salmonella enterica serovar typhimurium DT104. Antimicrobial agents and chemotherapy. 2004;48(10):3729-35. 47. Pai H, Kim J, Kim J, Lee JH, Choe KW, Gotoh N. Carbapenem resistance mechanisms in Pseudomonas aeruginosa clinical isolates. Antimicrobial agents and chemotherapy. 2001;45(2):480-4. 48. Hiramatsu K, Hanaki H, Ino T, Yabuta K, Oguri T, Tenover FC. Methicillin-resistant Staphylococcus aureus clinical strain with reduced vancomycin susceptibility. The Journal of antimicrobial chemotherapy. 1997;40(1):135-6. 49. Shaw WV, Brodsky RF. Characterization of chloramphenicol acetyltransferase from chloramphenicol-resistant Staphylococcus aureus. Journal of bacteriology. 1968;95(1):28-36. 50. Shaw WV. Enzymatic chlorampheicol acetylation and R factor induced antibiotic resistance in Enterobacteriaceae. Antimicrob Agents Chemother (Bethesda). 1966;6:221-6. 51. Ramirez MS, Tolmasky ME. Aminoglycoside modifying enzymes. Drug resistance updates : reviews and commentaries in antimicrobial and anticancer chemotherapy. 2010;13(6):151-71. 52. CHAIN EPAE. An Enzyme from Bacteria able to Destroy Penicillin. Nature. 1940:1. 53. WHO. Antimicrobial resistance: global report on surveillance 2014. 2014:257. 54. Rice LB. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. The Journal of infectious diseases. 2008;197(8):1079-81. 55. Toleman MA, Walsh TR. Combinatorial events of insertion sequences and ICE in Gram-negative bacteria. FEMS microbiology reviews. 2011;35(5):912-35. 56. Ellington MJ, Reuter S, Harris SR, Holden MT, Cartwright EJ, Greaves D, et al. Emergent and evolving antimicrobial resistance cassettes in community-associated fusidic acid and meticillin-resistant Staphylococcus aureus. Int J Antimicrob Agents. 2015. 57. Schmitz FJ, Fluit AC, Gondolf M, Beyrau R, Lindenlauf E, Verhoef J, et al. The prevalence of aminoglycoside resistance and corresponding resistance genes in clinical isolates of staphylococci from 19 European hospitals. The Journal of antimicrobial chemotherapy. 1999;43(2):253-9. 58. Noble WC, Valkenburg HA, Wolters CH. Carriage of Staphylococcus aureus in random samples of a normal population. The Journal of hygiene. 1967;65(4):567-73. 59. Peacock SJ, Paterson GK. Mechanisms of Methicillin Resistance in Staphylococcus aureus. Annual review of biochemistry. 2015;84:577-601. 60. Rountree PM, Freeman BM. Infections caused by a particular phage type of Staphylococcus aureus. The Medical journal of Australia. 1955;42(5):157-61. 61. Jevons MP, Coe AW, Parker MT. Methicillin resistance in staphylococci. Lancet. 1963;1(7287):904-7.
131
62. Price LB, Stegger M, Hasman H, Aziz M, Larsen J, Andersen PS, et al. Staphylococcus aureus CC398: host adaptation and emergence of methicillin resistance in livestock. mBio. 2012;3(1). 63. Otto M. MRSA virulence and spread. Cell Microbiol. 2012;14(10):1513-21. 64. Camoez M, Sierra JM, Pujol M, Hornero A, Martin R, Dominguez MA. Prevalence and molecular characterization of methicillin-resistant Staphylococcus aureus ST398 resistant to tetracycline at a Spanish hospital over 12 years. PloS one. 2013;8(9):e72828. 65. McDougal LK, Steward CD, Killgore GE, Chaitram JM, McAllister SK, Tenover FC. Pulsed-field gel electrophoresis typing of oxacillin-resistant Staphylococcus aureus isolates from the United States: establishing a national database. Journal of clinical microbiology. 2003;41(11):5113-20. 66. CDC. Methicillin-resistant staphylococcus aureus infections among competitive sports participants--Colorado, Indiana, Pennsylvania, and Los Angeles County, 2000-2003. MMWR Morbidity and mortality weekly report. 2003;52(33):793-5. 67. Hanaki H, Labischinski H, Inaba Y, Hiramatsu K. [Increase of non-amidated muropeptides in the cell wall of vancomycin-resistant Staphylococcus aureus (VRSA) strain Mu50]. The Japanese journal of antibiotics. 1998;51(4):272-80. 68. Rossi F, Diaz L, Wollam A, Panesso D, Zhou Y, Rincon S, et al. Transferable vancomycin resistance in a community-associated MRSA lineage. The New England journal of medicine. 2014;370(16):1524-31. 69. Zhu W, Murray PR, Huskins WC, Jernigan JA, McDonald LC, Clark NC, et al. Dissemination of an Enterococcus Inc18-Like vanA plasmid associated with vancomycin-resistant Staphylococcus aureus. Antimicrobial agents and chemotherapy. 2010;54(10):4314-20. 70. CDC. Staphylococcus aureus resistant to vancomycin--United States, 2002. MMWR Morbidity and mortality weekly report. 2002;51(26):565-7. 71. Perichon B, Courvalin P. VanA-type vancomycin-resistant Staphylococcus aureus. Antimicrobial agents and chemotherapy. 2009;53(11):4580-7. 72. Arthur M, Molinas C, Courvalin P. The VanS-VanR two-component regulatory system controls synthesis of depsipeptide peptidoglycan precursors in Enterococcus faecium BM4147. Journal of bacteriology. 1992;174(8):2582-91. 73. Kwun MJ, Novotna G, Hesketh AR, Hill L, Hong HJ. In vivo studies suggest that induction of VanS-dependent vancomycin resistance requires binding of the drug to D-Ala-D-Ala termini in the peptidoglycan cell wall. Antimicrobial agents and chemotherapy. 2013;57(9):4470-80. 74. Meka VG, Gold HS. Antimicrobial resistance to linezolid. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2004;39(7):1010-5. 75. Moellering RC. Linezolid: the first oxazolidinone antimicrobial. Annals of internal medicine. 2003;138(2):135-42. 76. Gu B, Kelesidis T, Tsiodras S, Hindler J, Humphries RM. The emerging problem of linezolid-resistant Staphylococcus. The Journal of antimicrobial chemotherapy. 2013;68(1):4-11. 77. Fines M, Leclercq R. Activity of linezolid against Gram-positive cocci possessing genes conferring resistance to protein synthesis inhibitors. The Journal of antimicrobial chemotherapy. 2000;45(6):797-802. 78. Long KS, Poehlsgaard J, Kehrenberg C, Schwarz S, Vester B. The Cfr rRNA methyltransferase confers resistance to Phenicols, Lincosamides, Oxazolidinones, Pleuromutilins, and Streptogramin A antibiotics. Antimicrobial agents and chemotherapy. 2006;50(7):2500-5.
132
79. Kehrenberg C, Schwarz S, Jacobsen L, Hansen LH, Vester B. A new mechanism for chloramphenicol, florfenicol and clindamycin resistance: methylation of 23S ribosomal RNA at A2503. Molecular microbiology. 2005;57(4):1064-73. 80. Schneider T, Kruse T, Wimmer R, Wiedemann I, Sass V, Pag U, et al. Plectasin, a fungal defensin, targets the bacterial cell wall precursor Lipid II. Science (New York, NY). 2010;328(5982):1168-72. 81. Eliopoulos GM, Willey S, Reiszner E, Spitzer PG, Caputo G, Moellering RC, Jr. In vitro and in vivo activity of LY 146032, a new cyclic lipopeptide antibiotic. Antimicrobial agents and chemotherapy. 1986;30(4):532-5. 82. Jung D, Rozek A, Okon M, Hancock RE. Structural transitions as determinants of the action of the calcium-dependent antibiotic daptomycin. Chemistry & biology. 2004;11(7):949-57. 83. Pogliano J, Pogliano N, Silverman JA. Daptomycin-mediated reorganization of membrane architecture causes mislocalization of essential cell division proteins. Journal of bacteriology. 2012;194(17):4494-504. 84. Cui L, Tominaga E, Neoh HM, Hiramatsu K. Correlation between Reduced Daptomycin Susceptibility and Vancomycin Resistance in Vancomycin-Intermediate Staphylococcus aureus. Antimicrobial agents and chemotherapy. 2006;50(3):1079-82. 85. Friedman L, Alder JD, Silverman JA. Genetic changes that correlate with reduced susceptibility to daptomycin in Staphylococcus aureus. Antimicrobial agents and chemotherapy. 2006;50(6):2137-45. 86. Staubitz P, Neumann H, Schneider T, Wiedemann I, Peschel A. MprF-mediated biosynthesis of lysylphosphatidylglycerol, an important determinant in staphylococcal defensin resistance. FEMS microbiology letters. 2004;231(1):67-71. 87. Lawes T, Lopez-Lozano JM, Nebot CA, Macartney G, Subbarao-Sharma R, Dare CR, et al. Effects of national antibiotic stewardship and infection control strategies on hospital-associated and community-associated meticillin-resistant Staphylococcus aureus infections across a region of Scotland: a non-linear time-series study. Lancet Infect Dis. 2015. 88. Karlowsky JA, Nichol K, Zhanel GG. Telavancin: Mechanisms of Action, In Vitro Activity, and Mechanisms of Resistance. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2015;61 Suppl 2:S58-68. 89. Livermore DM. Fourteen years in resistance. Int J Antimicrob Agents. 2012;39(4):283-94. 90. Poirel L, Nordmann P. Carbapenem resistance in Acinetobacter baumannii: mechanisms and epidemiology. Clinical microbiology and infection : the official publication of the European Society of Clinical Microbiology and Infectious Diseases. 2006;12(9):826-36. 91. Pitout JD, Nordmann P, Poirel L. Carbapenemase-Producing Klebsiella pneumoniae, a Key Pathogen Set for Global Nosocomial Dominance. Antimicrobial agents and chemotherapy. 2015;59(10):5873-84. 92. Nordmann P, Dortet L, Poirel L. Carbapenem resistance in Enterobacteriaceae: here is the storm! Trends in molecular medicine. 2012;18(5):263-72. 93. Nicolas-Chanoine MH, Bertrand X, Madec JY. Escherichia coli ST131, an intriguing clonal group. Clinical microbiology reviews. 2014;27(3):543-74. 94. Gootz TD. The global problem of antibiotic resistance. Critical reviews in immunology. 2010;30(1):79-93. 95. Harris PN, Tambyah PA, Paterson DL. beta-lactam and beta-lactamase inhibitor combinations in the treatment of extended-spectrum beta-lactamase producing Enterobacteriaceae: time for a reappraisal in the era of few antibiotic options? Lancet Infect Dis. 2015;15(4):475-85.
133
96. Canton R, Morosini MI, de la Maza OM, de la Pedrosa EG. IRT and CMT beta-lactamases and inhibitor resistance. Clinical microbiology and infection : the official publication of the European Society of Clinical Microbiology and Infectious Diseases. 2008;14 Suppl 1:53-62. 97. Bradford PA, Bratu S, Urban C, Visalli M, Mariano N, Landman D, et al. Emergence of carbapenem-resistant Klebsiella species possessing the class A carbapenem-hydrolyzing KPC-2 and inhibitor-resistant TEM-30 beta-lactamases in New York City. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2004;39(1):55-60. 98. Macheboeuf P, Contreras-Martel C, Job V, Dideberg O, Dessen A. Penicillin binding proteins: key players in bacterial cell cycle and drug resistance processes. FEMS microbiology reviews. 2006;30(5):673-91. 99. Macheboeuf P, Lemaire D, Teller N, Martins Ados S, Luxen A, Dideberg O, et al. Trapping of an acyl-enzyme intermediate in a penicillin-binding protein (PBP)-catalyzed reaction. Journal of molecular biology. 2008;376(2):405-13. 100. Peleg AY, Seifert H, Paterson DL. Acinetobacter baumannii: emergence of a successful pathogen. Clinical microbiology reviews. 2008;21(3):538-82. 101. Perez F, Hujer AM, Hujer KM, Decker BK, Rather PN, Bonomo RA. Global challenge of multidrug-resistant Acinetobacter baumannii. Antimicrobial agents and chemotherapy. 2007;51(10):3471-84. 102. Maragakis LL, Perl TM. Acinetobacter baumannii: epidemiology, antimicrobial resistance, and treatment options. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2008;46(8):1254-63. 103. Shorr AF, Zilberberg MD, Micek ST, Kollef MH. Predictors of hospital mortality among septic ICU patients with Acinetobacter spp. bacteremia: a cohort study. BMC infectious diseases. 2014;14:572. 104. Fournier PE, Vallenet D, Barbe V, Audic S, Ogata H, Poirel L, et al. Comparative genomics of multidrug resistance in Acinetobacter baumannii. PLoS genetics. 2006;2(1):e7. 105. Falagas ME, Kasiakou SK. Colistin: the revival of polymyxins for the management of multidrug-resistant gram-negative bacterial infections. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2005;40(9):1333-41. 106. Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, et al. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2009;48(1):1-12. 107. Bialvaei AZ, Samadi Kafil H. Colistin, mechanisms and prevalence of resistance. Curr Med Res Opin. 2015;31(4):707-21. 108. Garcia-Quintanilla M, Pulido MR, Moreno-Martinez P, Martin-Pena R, Lopez-Rojas R, Pachon J, et al. Activity of host antimicrobials against multidrug-resistant Acinetobacter baumannii acquiring colistin resistance through loss of lipopolysaccharide. Antimicrobial agents and chemotherapy. 2014;58(5):2972-5. 109. Moffatt JH, Harper M, Harrison P, Hale JD, Vinogradov E, Seemann T, et al. Colistin resistance in Acinetobacter baumannii is mediated by complete loss of lipopolysaccharide production. Antimicrobial agents and chemotherapy. 2010;54(12):4971-7. 110. Jayol A, Nordmann P, Brink A, Poirel L. Heteroresistance to Colistin in Klebsiella pneumoniae Associated with Alterations in the PhoPQ Regulatory System. Antimicrobial agents and chemotherapy. 2015;59(5):2780-4. 111. Li J, Rayner CR, Nation RL, Owen RJ, Spelman D, Tan KE, et al. Heteroresistance to colistin in multidrug-resistant Acinetobacter baumannii. Antimicrobial agents and chemotherapy. 2006;50(9):2946-50.
134
112. Chen HD, Groisman EA. The biology of the PmrA/PmrB two-component system: the major regulator of lipopolysaccharide modifications. Annual review of microbiology. 2013;67:83-112. 113. Beceiro A, Llobet E, Aranda J, Bengoechea JA, Doumith M, Hornsey M, et al. Phosphoethanolamine modification of lipid A in colistin-resistant variants of Acinetobacter baumannii mediated by the pmrAB two-component regulatory system. Antimicrobial agents and chemotherapy. 2011;55(7):3370-9. 114. Adams MD, Nickel GC, Bajaksouzian S, Lavender H, Murthy AR, Jacobs MR, et al. Resistance to colistin in Acinetobacter baumannii associated with mutations in the PmrAB two-component system. Antimicrobial agents and chemotherapy. 2009;53(9):3628-34. 115. Lee H, Hsu FF, Turk J, Groisman EA. The PmrA-regulated pmrC gene mediates phosphoethanolamine modification of lipid A and polymyxin resistance in Salmonella enterica. Journal of bacteriology. 2004;186(13):4124-33. 116. Groisman EA. The pleiotropic two-component regulatory system PhoP-PhoQ. Journal of bacteriology. 2001;183(6):1835-42. 117. Bishop RE, Gibbons HS, Guina T, Trent MS, Miller SI, Raetz CR. Transfer of palmitate from phospholipids to lipid A in outer membranes of gram-negative bacteria. The EMBO journal. 2000;19(19):5071-80. 118. Liu Y-Y, Wang Y, Walsh TR, Yi L-X, Zhang R, Spencer J, et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. The Lancet Infectious Diseases. 119. Van Epps HL. Rene Dubos: unearthing antibiotics. The Journal of experimental medicine. 2006;203(2):259. 120. Johnson BA, Anker H, Meleney FL. BACITRACIN: A NEW ANTIBIOTIC PRODUCED BY A MEMBER OF THE B. SUBTILIS GROUP. Science (New York, NY). 1945;102(2650):376-7. 121. Wang Z, Wang G. APD: the Antimicrobial Peptide Database. Nucleic acids research. 2004;32(Database issue):D590-2. 122. Jenssen H, Hamill P, Hancock RE. Peptide antimicrobial agents. Clinical microbiology reviews. 2006;19(3):491-511. 123. Fjell CD, Hiss JA, Hancock RE, Schneider G. Designing antimicrobial peptides: form follows function. Nature reviews Drug discovery. 2012;11(1):37-51. 124. Cotter PD, Hill C, Ross RP. Bacteriocins: developing innate immunity for food. Nature reviews Microbiology. 2005;3(10):777-88. 125. Cotter PD, Hill C, Ross RP. Bacterial lantibiotics: strategies to improve therapeutic potential. Current protein & peptide science. 2005;6(1):61-75. 126. Oppegard C, Rogne P, Emanuelsen L, Kristiansen PE, Fimland G, Nissen-Meyer J. The two-peptide class II bacteriocins: structure, production, and mode of action. Journal of molecular microbiology and biotechnology. 2007;13(4):210-9. 127. Yang SC, Lin CH, Sung CT, Fang JY. Antibacterial activities of bacteriocins: application in foods and pharmaceuticals. Frontiers in microbiology. 2014;5:241. 128. Wiedemann I, Breukink E, van Kraaij C, Kuipers OP, Bierbaum G, de Kruijff B, et al. Specific binding of nisin to the peptidoglycan precursor lipid II combines pore formation and inhibition of cell wall biosynthesis for potent antibiotic activity. The Journal of biological chemistry. 2001;276(3):1772-9. 129. van Belkum MJ, Kok J, Venema G, Holo H, Nes IF, Konings WN, et al. The bacteriocin lactococcin A specifically increases permeability of lactococcal cytoplasmic membranes in a voltage-independent, protein-mediated manner. Journal of bacteriology. 1991;173(24):7934-41.
135
130. Holo H, Nilssen O, Nes IF. Lactococcin A, a new bacteriocin from Lactococcus lactis subsp. cremoris: isolation and characterization of the protein and its gene. Journal of bacteriology. 1991;173(12):3879-87. 131. Cotter PD, Ross RP, Hill C. Bacteriocins - a viable alternative to antibiotics? Nature reviews Microbiology. 2013;11(2):95-105. 132. Vizan JL, Hernandez-Chico C, del Castillo I, Moreno F. The peptide antibiotic microcin B17 induces double-strand cleavage of DNA mediated by E. coli DNA gyrase. The EMBO journal. 1991;10(2):467-76. 133. Mukhopadhyay J, Sineva E, Knight J, Levy RM, Ebright RH. Antibacterial peptide microcin J25 inhibits transcription by binding within and obstructing the RNA polymerase secondary channel. Molecular cell. 2004;14(6):739-51. 134. Diep DB, Skaugen M, Salehian Z, Holo H, Nes IF. Common mechanisms of target cell recognition and immunity for class II bacteriocins. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(7):2384-9. 135. Mathavan I, Beis K. The role of bacterial membrane proteins in the internalization of microcin MccJ25 and MccB17. Biochemical Society transactions. 2012;40(6):1539-43. 136. Vondenhoff GH, Blanchaert B, Geboers S, Kazakov T, Datsenko KA, Wanner BL, et al. Characterization of peptide chain length and constituency requirements for YejABEF-mediated uptake of microcin C analogues. Journal of bacteriology. 2011;193(14):3618-23. 137. Breukink E, de Kruijff B. Lipid II as a target for antibiotics. Nature reviews Drug discovery. 2006;5(4):321-32. 138. Dischinger J, Basi Chipalu S, Bierbaum G. Lantibiotics: promising candidates for future applications in health care. International journal of medical microbiology : IJMM. 2014;304(1):51-62. 139. Willey JM, van der Donk WA. Lantibiotics: peptides of diverse structure and function. Annual review of microbiology. 2007;61:477-501. 140. Schnell N, Entian KD, Schneider U, Gotz F, Zahner H, Kellner R, et al. Prepeptide sequence of epidermin, a ribosomally synthesized antibiotic with four sulphide-rings. Nature. 1988;333(6170):276-8. 141. Chatterjee C, Paul M, Xie L, van der Donk WA. Biosynthesis and mode of action of lantibiotics. Chemical reviews. 2005;105(2):633-84. 142. Mattick AT, Hirsch A. Further observations on an inhibitory substance (nisin) from lactic streptococci. Lancet. 1947;2(6462):5-8. 143. Rogers LA, Whittier EO. LIMITING FACTORS IN THE LACTIC FERMENTATION. Journal of bacteriology. 1928;16(4):211-29. 144. Delves-Broughton J, Blackburn P, Evans RJ, Hugenholtz J. Applications of the bacteriocin, nisin. Antonie van Leeuwenhoek. 1996;69(2):193-202. 145. van Kraaij C, Breukink E, Noordermeer MA, Demel RA, Siezen RJ, Kuipers OP, et al. Pore formation by nisin involves translocation of its C-terminal part across the membrane. Biochemistry. 1998;37(46):16033-40. 146. Scherer K, Wiedemann I, Ciobanasu C, Sahl HG, Kubitscheck U. Aggregates of nisin with various bactoprenol-containing cell wall precursors differ in size and membrane permeation capacity. Biochimica et biophysica acta. 2013;1828(11):2628-36. 147. Tol MB, Morales Angeles D, Scheffers DJ. In vivo cluster formation of nisin and lipid II is correlated with membrane depolarization. Antimicrobial agents and chemotherapy. 2015;59(6):3683-6.
136
148. Scherer KM, Spille JH, Sahl HG, Grein F, Kubitscheck U. The lantibiotic nisin induces lipid II aggregation, causing membrane instability and vesicle budding. Biophysical journal. 2015;108(5):1114-24. 149. Hasper HE, Kramer NE, Smith JL, Hillman JD, Zachariah C, Kuipers OP, et al. An alternative bactericidal mechanism of action for lantibiotic peptides that target lipid II. Science (New York, NY). 2006;313(5793):1636-7. 150. Smith L, Zachariah C, Thirumoorthy R, Rocca J, Novak J, Hillman JD, et al. Structure and dynamics of the lantibiotic mutacin 1140. Biochemistry. 2003;42(35):10372-84. 151. Castiglione F, Cavaletti L, Losi D, Lazzarini A, Carrano L, Feroggio M, et al. A novel lantibiotic acting on bacterial cell wall synthesis produced by the uncommon actinomycete Planomonospora sp. Biochemistry. 2007;46(20):5884-95. 152. Castiglione F, Lazzarini A, Carrano L, Corti E, Ciciliato I, Gastaldo L, et al. Determining the structure and mode of action of microbisporicin, a potent lantibiotic active against multiresistant pathogens. Chemistry & biology. 2008;15(1):22-31. 153. Smith L, Hasper H, Breukink E, Novak J, Cerkasov J, Hillman JD, et al. Elucidation of the antimicrobial mechanism of mutacin 1140. Biochemistry. 2008;47(10):3308-14. 154. Brotz H, Bierbaum G, Leopold K, Reynolds PE, Sahl HG. The lantibiotic mersacidin inhibits peptidoglycan synthesis by targeting lipid II. Antimicrobial agents and chemotherapy. 1998;42(1):154-60. 155. Munch D, Muller A, Schneider T, Kohl B, Wenzel M, Bandow JE, et al. The lantibiotic NAI-107 binds to bactoprenol-bound cell wall precursors and impairs membrane functions. The Journal of biological chemistry. 2014;289(17):12063-76. 156. Oppedijk SF, Martin NI, Breukink E. Hit 'em where it hurts: The growing and structurally diverse family of peptides that target lipid-II. Biochimica et biophysica acta. 2015. 157. Reddy KV, Gupta SM, Aranha CC. Effect of antimicrobial Peptide, nisin, on the reproductive functions of rats. ISRN veterinary science. 2011;2011:828736. 158. Ling LL, Schneider T, Peoples AJ, Spoering AL, Engels I, Conlon BP, et al. A new antibiotic kills pathogens without detectable resistance. Nature. 2015;517(7535):455-9. 159. Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415(6870):389-95. 160. Saugar JM, Rodriguez-Hernandez MJ, de la Torre BG, Pachon-Ibanez ME, Fernandez-Reyes M, Andreu D, et al. Activity of cecropin A-melittin hybrid peptides against colistin-resistant clinical strains of Acinetobacter baumannii: molecular basis for the differential mechanisms of action. Antimicrobial agents and chemotherapy. 2006;50(4):1251-6. 161. Lopez-Rojas R, Docobo-Perez F, Pachon-Ibanez ME, de la Torre BG, Fernandez-Reyes M, March C, et al. Efficacy of cecropin A-melittin peptides on a sepsis model of infection by pan-resistant Acinetobacter baumannii. European journal of clinical microbiology & infectious diseases : official publication of the European Society of Clinical Microbiology. 2011;30(11):1391-8. 162. Wang G, Hanke ML, Mishra B, Lushnikova T, Heim CE, Chittezham Thomas V, et al. Transformation of Human Cathelicidin LL-37 into Selective, Stable, and Potent Antimicrobial Compounds. ACS chemical biology. 2014. 163. Ganz T, Selsted ME, Szklarek D, Harwig SS, Daher K, Bainton DF, et al. Defensins. Natural peptide antibiotics of human neutrophils. The Journal of clinical investigation. 1985;76(4):1427-35. 164. Verma C, Seebah S, Low SM, Zhou L, Liu SP, Li J, et al. Defensins: antimicrobial peptides for therapeutic development. Biotechnology journal. 2007;2(11):1353-9.
137
165. da Costa JP, Cova M, Ferreira R, Vitorino R. Antimicrobial peptides: an alternative for innovative medicines? Applied microbiology and biotechnology. 2015;99(5):2023-40. 166. Nguyen LT, Haney EF, Vogel HJ. The expanding scope of antimicrobial peptide structures and their modes of action. Trends in biotechnology. 2011;29(9):464-72. 167. Rozek A, Friedrich CL, Hancock RE. Structure of the bovine antimicrobial peptide indolicidin bound to dodecylphosphocholine and sodium dodecyl sulfate micelles. Biochemistry. 2000;39(51):15765-74. 168. Hwang PM, Zhou N, Shan X, Arrowsmith CH, Vogel HJ. Three-dimensional solution structure of lactoferricin B, an antimicrobial peptide derived from bovine lactoferrin. Biochemistry. 1998;37(12):4288-98. 169. Hoover DM, Chertov O, Lubkowski J. The structure of human beta-defensin-1: new insights into structural properties of beta-defensins. The Journal of biological chemistry. 2001;276(42):39021-6. 170. Landon C, Sodano P, Hetru C, Hoffmann J, Ptak M. Solution structure of drosomycin, the first inducible antifungal protein from insects. Protein science : a publication of the Protein Society. 1997;6(9):1878-84. 171. Gesell J, Zasloff M, Opella SJ. Two-dimensional 1H NMR experiments show that the 23-residue magainin antibiotic peptide is an alpha-helix in dodecylphosphocholine micelles, sodium dodecylsulfate micelles, and trifluoroethanol/water solution. Journal of biomolecular NMR. 1997;9(2):127-35. 172. Mandard N, Sodano P, Labbe H, Bonmatin JM, Bulet P, Hetru C, et al. Solution structure of thanatin, a potent bactericidal and fungicidal insect peptide, determined from proton two-dimensional nuclear magnetic resonance data. European journal of biochemistry / FEBS. 1998;256(2):404-10. 173. Brogden KA. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nature reviews Microbiology. 2005;3(3):238-50. 174. Scott MG, Dullaghan E, Mookherjee N, Glavas N, Waldbrook M, Thompson A, et al. An anti-infective peptide that selectively modulates the innate immune response. Nature biotechnology. 2007;25(4):465-72. 175. Niyonsaba F, Madera L, Afacan N, Okumura K, Ogawa H, Hancock RE. The innate defense regulator peptides IDR-HH2, IDR-1002, and IDR-1018 modulate human neutrophil functions. JLeukocBiol. 2013;94(1):159-70. 176. Kindrachuk J, Jenssen H, Elliott M, Nijnik A, Magrangeas-Janot L, Pasupuleti M, et al. Manipulation of innate immunity by a bacterial secreted peptide: lantibiotic nisin Z is selectively immunomodulatory. Innate immunity. 2013;19(3):315-27. 177. Nijnik A, Madera L, Ma S, Waldbrook M, Elliott MR, Easton DM, et al. Synthetic cationic peptide IDR-1002 provides protection against bacterial infections through chemokine induction and enhanced leukocyte recruitment. Journal of immunology (Baltimore, Md : 1950). 2010;184(5):2539-50. 178. Finlay BB, Hancock RE. Can innate immunity be enhanced to treat microbial infections? NatRevMicrobiol. 2004;2(6):497-504. 179. Gottschalk S, Gottlieb CT, Vestergaard M, Hansen PR, Gram L, Ingmer H, et al. The Amphibian Antimicrobial Peptide Fallaxin Analogue, FL9, affects Virulence Gene Expression and DNA replication in Staphylococcus aureus. Journal of medical microbiology. 2015. 180. Mansson M, Nielsen A, Kjaerulff L, Gotfredsen CH, Wietz M, Ingmer H, et al. Inhibition of virulence gene expression in Staphylococcus aureus by novel depsipeptides from a marine photobacterium. Marine drugs. 2011;9(12):2537-52.
138
181. Nawrocki KL, Crispell EK, McBride SM. Antimicrobial Peptide Resistance Mechanisms of Gram-Positive Bacteria. Antibiotics (Basel, Switzerland). 2014;3(4):461-92. 182. Band VI, Weiss DS. Mechanisms of Antimicrobial Peptide Resistance in Gram-Negative Bacteria. Antibiotics (Basel, Switzerland). 2015;4(1):18-41. 183. Campos MA, Vargas MA, Regueiro V, Llompart CM, Alberti S, Bengoechea JA. Capsule polysaccharide mediates bacterial resistance to antimicrobial peptides. Infection and immunity. 2004;72(12):7107-14. 184. Peschel A, Otto M, Jack RW, Kalbacher H, Jung G, Gotz F. Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. The Journal of biological chemistry. 1999;274(13):8405-10. 185. McBride SM, Sonenshein AL. Identification of a genetic locus responsible for antimicrobial peptide resistance in Clostridium difficile. Infection and immunity. 2011;79(1):167-76. 186. Socias SB, Vincent PA, Salomon RA. The leucine-responsive regulatory protein, Lrp, modulates microcin J25 intrinsic resistance in Escherichia coli by regulating expression of the YojI microcin exporter. Journal of bacteriology. 2009;191(4):1343-8. 187. Sieprawska-Lupa M, Mydel P, Krawczyk K, Wojcik K, Puklo M, Lupa B, et al. Degradation of human antimicrobial peptide LL-37 by Staphylococcus aureus-derived proteinases. Antimicrobial agents and chemotherapy. 2004;48(12):4673-9. 188. Schmidtchen A, Frick IM, Andersson E, Tapper H, Bjorck L. Proteinases of common pathogenic bacteria degrade and inactivate the antibacterial peptide LL-37. Molecular microbiology. 2002;46(1):157-68. 189. Nizet V. Antimicrobial peptide resistance mechanisms of human bacterial pathogens. Current issues in molecular biology. 2006;8(1):11-26. 190. Fjell CD, Jenssen H, Hilpert K, Cheung WA, Pante N, Hancock RE, et al. Identification of novel antibacterial peptides by chemoinformatics and machine learning. Journal of medicinal chemistry. 2009;52(7):2006-15. 191. Fjell CD, Jenssen H, Cheung WA, Hancock REW, Cherkasov A. Optimization of Antibacterial Peptides by Genetic Algorithms and Cheminformatics. Chemical Biology & Drug Design. 2011;77(1):48-56. 192. Vaara M. Novel derivatives of polymyxins. The Journal of antimicrobial chemotherapy. 2013;68(6):1213-9. 193. Torcato IM, Huang YH, Franquelim HG, Gaspar D, Craik DJ, Castanho MA, et al. Design and characterization of novel antimicrobial peptides, R-BP100 and RW-BP100, with activity against Gram-negative and Gram-positive bacteria. Biochimica et biophysica acta. 2013;1828(3):944-55. 194. Kjelstrup S, Hansen PM, Thomsen LE, Hansen PR, Lobner-Olesen A. Cyclic peptide inhibitors of the beta-sliding clamp in Staphylococcus aureus. PloS one. 2013;8(9):e72273. 195. Badosa E, Ferre R, Planas M, Feliu L, Besalu E, Cabrefiga J, et al. A library of linear undecapeptides with bactericidal activity against phytopathogenic bacteria. Peptides. 2007;28(12):2276-85. 196. Scott CP, Abel-Santos E, Wall M, Wahnon DC, Benkovic SJ. Production of cyclic peptides and proteins in vivo. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(24):13638-43. 197. Mojsoska B, Zuckermann RN, Jenssen H. Structure-Activity Relationship Study of Novel Peptoids That Mimic the Structure of Antimicrobial Peptides. Antimicrobial agents and chemotherapy. 2015;59(7):4112-20.
139
198. Chongsiriwatana NP, Patch JA, Czyzewski AM, Dohm MT, Ivankin A, Gidalevitz D, et al. Peptoids that mimic the structure, function, and mechanism of helical antimicrobial peptides. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(8):2794-9. 199. Tan NC, Yu P, Kwon YU, Kodadek T. High-throughput evaluation of relative cell permeability between peptoids and peptides. Bioorganic & medicinal chemistry. 2008;16(11):5853-61. 200. Wade D, Boman A, Wahlin B, Drain CM, Andreu D, Boman HG, et al. All-D amino acid-containing channel-forming antibiotic peptides. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(12):4761-5. 201. Jenssen H, Aspmo SI. Serum stability of peptides. Methods in molecular biology. 2008;494:177-86. 202. Fox JL. Antimicrobial peptides stage a comeback. Nature biotechnology. 2013;31(5):379-82. 203. Butler MS, Blaskovich MA, Cooper MA. Antibiotics in the clinical pipeline in 2013. The Journal of antibiotics. 2013;66(10):571-91. 204. Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nature reviews Drug discovery. 2007;6(1):29-40. 205. Troels Godballe BM, Hanne M. Nielsen, Håvard Jenssen. Antimicrobial activity of GN peptides and their mode of action Biopolymers. 2015. 206. Moy TI, Ball AR, Anklesaria Z, Casadei G, Lewis K, Ausubel FM. Identification of novel antimicrobials using a live-animal infection model. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(27):10414-9. 207. Ramarao N, Nielsen-Leroux C, Lereclus D. The insect Galleria mellonella as a powerful infection model to investigate bacterial pathogenesis. Journal of visualized experiments : JoVE. 2012(70):e4392. 208. Glavis-Bloom J, Muhammed M, Mylonakis E. Of model hosts and man: using Caenorhabditis elegans, Drosophila melanogaster and Galleria mellonella as model hosts for infectious disease research. Advances in experimental medicine and biology. 2012;710:11-7. 209. Junqueira JC. Galleria mellonella as a model host for human pathogens: recent studies and new perspectives. Virulence. 2012;3(6):474-6. 210. Evans BA, Rozen DE. A Streptococcus pneumoniae infection model in larvae of the wax moth Galleria mellonella. European journal of clinical microbiology & infectious diseases : official publication of the European Society of Clinical Microbiology. 2012;31(10):2653-60. 211. Peleg AY, Jara S, Monga D, Eliopoulos GM, Moellering RC, Jr., Mylonakis E. Galleria mellonella as a model system to study Acinetobacter baumannii pathogenesis and therapeutics. Antimicrobial agents and chemotherapy. 2009;53(6):2605-9. 212. Yang H, Chen G, Hu L, Liu Y, Cheng J, Li H, et al. In vivo activity of daptomycin/colistin combination therapy in a Galleria mellonella model of Acinetobacter baumannii infection. Int J Antimicrob Agents. 2015;45(2):188-91. 213. Krezdorn J, Adams S, Coote PJ. A Galleria mellonella infection model reveals double and triple antibiotic combination therapies with enhanced efficacy versus a multidrug-resistant strain of Pseudomonas aeruginosa. Journal of medical microbiology. 2014;63(Pt 7):945-55. 214. Ciesielczuk H, Betts J, Phee L, Doumith M, Hope R, Woodford N, et al. Comparative virulence of urinary and bloodstream isolates of extra-intestinal pathogenic Escherichia coli in a Galleria mellonella model. Virulence. 2015;6(2):145-51.
140
215. Tzelepis I, Kapsetaki S-E, Panayidou S, Apidianakis Y. Drosophila melanogaster: a first step and a stepping-stone to anti-infectives. Current Opinion in Pharmacology. 2013;13(5):763-8. 216. Chamilos G, Samonis G, Kontoyiannis DP. Drosophila melanogaster as a model host for the study of microbial pathogenicity and the discovery of novel antimicrobial compounds. CurrPharmDes. 2011;17(13):1246-53. 217. Dar AC, Das TK, Shokat KM, Cagan RL. Chemical genetic discovery of targets and anti-targets for cancer polypharmacology. Nature. 2012;486(7401):80-4. 218. Das TK, Cagan RL. A Drosophila approach to thyroid cancer therapeutics. Drug discovery today Technologies. 2013;10(1):e65-71. 219. Willoughby LF, Schlosser T, Manning SA, Parisot JP, Street IP, Richardson HE, et al. An in vivo large-scale chemical screening platform using Drosophila for anti-cancer drug discovery. Disease models & mechanisms. 2013;6(2):521-9. 220. St Johnston D. The art and design of genetic screens: Drosophila melanogaster. Nature reviews Genetics. 2002;3(3):176-88. 221. Danielsen ET, Moeller ME, Dorry E, Komura-Kawa T, Fujimoto Y, Troelsen JT, et al. Transcriptional control of steroid biosynthesis genes in the Drosophila prothoracic gland by ventral veins lacking and knirps. PLoS genetics. 2014;10(6):e1004343. 222. Musselman LP, Fink JL, Narzinski K, Ramachandran PV, Hathiramani SS, Cagan RL, et al. A high-sugar diet produces obesity and insulin resistance in wild-type Drosophila. Disease models & mechanisms. 2011;4(6):842-9. 223. Nelson B, Freisinger T, Ishii K, Okado K, Shinzawa N, Fukumoto S, et al. Activation of Imd pathway in hemocyte confers infection resistance through humoral response in Drosophila. Biochemical and biophysical research communications. 2013;430(3):1120-5. 224. Kaneko T, Silverman N. Bacterial recognition and signalling by the Drosophila IMD pathway. Cell Microbiol. 2005;7(4):461-9. 225. Lemaitre B. The road to Toll. NatRevImmunol. 2004;4(7):521-7. 226. Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell. 1996;86(6):973-83. 227. Lemaitre B, Kromer-Metzger E, Michaut L, Nicolas E, Meister M, Georgel P, et al. A recessive mutation, immune deficiency (imd), defines two distinct control pathways in the Drosophila host defense. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(21):9465-9. 228. Ashok Y. Drosophila toll pathway: the new model. SciSignal. 2009;2(52):jc1. 229. Valanne S, Wang JH, Ramet M. The Drosophila Toll signaling pathway. JImmunol. 2011;186(2):649-56. 230. Aggarwal K, Silverman N. Positive and negative regulation of the Drosophila immune response. BMBRep. 2008;41(4):267-77. 231. Reynolds JRaSE. Insect Infection and Immunity. Reynolds JRaSE, editor. OXFORD University Press: OXFORD University Press; 2009. 254 p. 232. Kleino A, Silverman N. The Drosophila IMD pathway in the activation of the humoral immune response. Developmental & Comparative Immunology. 2014;42(1):25-35. 233. Bischoff V, Vignal C, Boneca IG, Michel T, Hoffmann JA, Royet J. Function of the drosophila pattern-recognition receptor PGRP-SD in the detection of Gram-positive bacteria. NatImmunol. 2004;5(11):1175-80. 234. Gendrin M, Zaidman-Remy A, Broderick NA, Paredes J, Poidevin M, Roussel A, et al. Functional Analysis of PGRP-LA in Drosophila Immunity. PLoS One. 2013;8(7):e69742.
141
235. Royet J, Dziarski R. Peptidoglycan recognition proteins: pleiotropic sensors and effectors of antimicrobial defences. Nature reviews Microbiology. 2007;5(4):264-77. 236. Bosco-Drayon V, Poidevin M, Boneca IG, Narbonne-Reveau K, Royet J, Charroux B. Peptidoglycan sensing by the receptor PGRP-LE in the Drosophila gut induces immune responses to infectious bacteria and tolerance to microbiota. Cell HostMicrobe. 2012;12(2):153-65. 237. Neyen C, Poidevin M, Roussel A, Lemaitre B. Tissue- and ligand-specific sensing of gram-negative infection in drosophila by PGRP-LC isoforms and PGRP-LE. JImmunol. 2012;189(4):1886-97. 238. Atilano ML, Yates J, Glittenberg M, Filipe SR, Ligoxygakis P. Wall teichoic acids of Staphylococcus aureus limit recognition by the drosophila peptidoglycan recognition protein-SA to promote pathogenicity. PLoS Pathog. 2011;7(12):e1002421. 239. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124(4):783-801. 240. Vodovar N, Vinals M, Liehl P, Basset A, Degrouard J, Spellman P, et al. Drosophila host defense after oral infection by an entomopathogenic Pseudomonas species. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(32):11414-9. 241. Apidianakis Y, Rahme LG. Drosophila melanogaster as a model for human intestinal infection and pathology. DisModelMech. 2011;4(1):21-30. 242. Cheng LW, Portnoy DA. Drosophila S2 cells: an alternative infection model for Listeria monocytogenes. Cell Microbiol. 2003;5(12):875-85. 243. Panayidou S, Ioannidou E, Apidianakis Y. Human pathogenic bacteria, fungi, and viruses in Drosophila: Disease modeling, lessons, and shortcomings. Virulence. 2014;5(2). 244. Garcia-Lara J, Needham AJ, Foster SJ. Invertebrates as animal models for Staphylococcus aureus pathogenesis: a window into host-pathogen interaction. FEMS ImmunolMedMicrobiol. 2005;43(3):311-23. 245. Nehme NT, Liegeois S, Kele B, Giammarinaro P, Pradel E, Hoffmann JA, et al. A model of bacterial intestinal infections in Drosophila melanogaster. PLoSPathog. 2007;3(11):e173. 246. Apidianakis Y, Rahme LG. Drosophila melanogaster as a model host for studying Pseudomonas aeruginosa infection. NatProtoc. 2009;4(9):1285-94. 247. Blow NS, Salomon RN, Garrity K, Reveillaud I, Kopin A, Jackson FR, et al. Vibrio cholerae infection of Drosophila melanogaster mimics the human disease cholera. PLoS Pathog. 2005;1(1):e8. 248. Wu K, Conly J, Surette M, Sibley C, Elsayed S, Zhang K. Assessment of virulence diversity of methicillin-resistant Staphylococcus aureus strains with a Drosophila melanogaster infection model. BMC microbiology. 2012;12:274. 249. Atilano ML, Pereira PM, Vaz F, Catalao MJ, Reed P, Grilo IR, et al. Bacterial autolysins trim cell surface peptidoglycan to prevent detection by the Drosophila innate immune system. eLife. 2014;3:e02277. 250. Ben-Ami R, Watson CC, Lewis RE, Albert ND, Arias CA, Raad II, et al. Drosophila melanogaster as a model to explore the effects of methicillin-resistant Staphylococcus aureus strain type on virulence and response to linezolid treatment. MicrobPathog. 2012. 251. Jensen RL, Pedersen KS, Loeschcke V, Ingmer H, Leisner JJ. Limitations in the use of Drosophila melanogaster as a model host for gram-positive bacterial infection. Letters in applied microbiology. 2007;44(2):218-23. 252. Lopez-Rojas R, Jimenez-Mejias ME, Lepe JA, Pachon J. Acinetobacter baumannii resistant to colistin alters its antibiotic resistance profile: a case report from Spain. The Journal of infectious diseases. 2011;204(7):1147-8.
142
253. Alberto Oddo TT, Susanne Kjelstrup, Ciara Gorey, Henrik Franzyk, Niels Frimodt-Møller, Anders Løbner-Olesen, and Paul Hansen. An all-D amphipathic undecapeptide shows promising activity against colistin-resistant strains of Acinetobacter baumannii and a dual mode of action. AAC01966-15R1. 2015. 254. Rodriguez-Hernandez MJ, Saugar J, Docobo-Perez F, de la Torre BG, Pachon-Ibanez ME, Garcia-Curiel A, et al. Studies on the antimicrobial activity of cecropin A-melittin hybrid peptides in colistin-resistant clinical isolates of Acinetobacter baumannii. The Journal of antimicrobial chemotherapy. 2006;58(1):95-100. 255. Ekengren S, Hultmark D. Drosophila cecropin as an antifungal agent. Insect biochemistry and molecular biology. 1999;29(11):965-72. 256. Asthana N, Yadav SP, Ghosh JK. Dissection of antibacterial and toxic activity of melittin: a leucine zipper motif plays a crucial role in determining its hemolytic activity but not antibacterial activity. The Journal of biological chemistry. 2004;279(53):55042-50. 257. Habermann E. Bee and wasp venoms. Science (New York, NY). 1972;177(4046):314-22. 258. Rabanal F, Grau-Campistany A, Vila-Farres X, Gonzalez-Linares J, Borras M, Vila J, et al. A bioinspired peptide scaffold with high antibiotic activity and low in vivo toxicity. Sci Rep. 2015;5:10558. 259. Cazares-Dominguez V, Cruz-Cordova A, Ochoa SA, Escalona G, Arellano-Galindo J, Rodriguez-Leviz A, et al. Vancomycin Tolerant, Methicillin-Resistant Staphylococcus aureus Reveals the Effects of Vancomycin on Cell Wall Thickening. PloS one. 2015;10(3):e0118791. 260. Mishra NN, Bayer AS, Weidenmaier C, Grau T, Wanner S, Stefani S, et al. Phenotypic and genotypic characterization of daptomycin-resistant methicillin-resistant Staphylococcus aureus strains: relative roles of mprF and dlt operons. PloS one. 2014;9(9):e107426. 261. Suzuki M, Yamada K, Nagao M, Aoki E, Matsumoto M, Hirayama T, et al. Antimicrobial ointments and methicillin-resistant Staphylococcus aureus USA300. Emerging infectious diseases. 2011;17(10):1917-20. 262. Cherkasov A, Hilpert K, Jenssen H, Fjell CD, Waldbrook M, Mullaly SC, et al. Use of artificial intelligence in the design of small peptide antibiotics effective against a broad spectrum of highly antibiotic-resistant superbugs. ACS chemical biology. 2009;4(1):65-74. 263. Campion A, Casey PG, Field D, Cotter PD, Hill C, Ross RP. In vivo activity of nisin A and nisin V against Listeria monocytogenes in mice. BMC microbiology. 2013;13:23. 264. Lepak AJ, Marchillo K, Craig WA, Andes DR. In vivo pharmacokinetics and pharmacodynamics of the lantibiotic NAI-107 in a neutropenic murine thigh infection model. Antimicrobial agents and chemotherapy. 2015;59(2):1258-64. 265. Jabes D, Brunati C, Candiani G, Riva S, Romano G, Donadio S. Efficacy of the new lantibiotic NAI-107 in experimental infections induced by multidrug-resistant Gram-positive pathogens. Antimicrobial agents and chemotherapy. 2011;55(4):1671-6. 266. Draper LA, Cotter PD, Hill C, Ross RP. Lantibiotic resistance. Microbiology and molecular biology reviews : MMBR. 2015;79(2):171-91. 267. Kawada-Matsuo M, Yoshida Y, Zendo T, Nagao J, Oogai Y, Nakamura Y, et al. Three distinct two-component systems are involved in resistance to the class I bacteriocins, Nukacin ISK-1 and nisin A, in Staphylococcus aureus. PloS one. 2013;8(7):e69455. 268. Hiron A, Falord M, Valle J, Debarbouille M, Msadek T. Bacitracin and nisin resistance in Staphylococcus aureus: a novel pathway involving the BraS/BraR two-component system (SA2417/SA2418) and both the BraD/BraE and VraD/VraE ABC transporters. Molecular microbiology. 2011;81(3):602-22.
143
269. Garnacho-Montero J, Amaya-Villar R, Gutiérrez-Pizarraya A, Espejo-Gutiérrez de Tena E, Artero-González ML, Corcia-Palomo Y, et al. Clinical Efficacy and Safety of the Combination of Colistin plus Vancomycin for the Treatment of Severe Infections Caused by Carbapenem-Resistant <b><i>Acinetobacter baumannii</i></b>. Chemotherapy. 2013;59(3):225-31. 270. Needham AJ, Kibart M, Crossley H, Ingham PW, Foster SJ. Drosophila melanogaster as a model host for Staphylococcus aureus infection. Microbiology. 2004;150(Pt 7):2347-55.
144
Appendix: Papers not Included in Thesis
Paper I
Rapid Selection of Plasmodium falciparum Chloroquine Resistance Transporter
Gene and Multidrug Resistance Gene-1 Haplotypes Associated with Past
Chloroquine and Present Artemether-Lumefantrine Use in Inhambane District,
Southern Mozambique
Thomas T. Thomsen, Laura B. Madsen, Helle H. Hansson, Elsa V. E. Toma´ s, Derek Charlwood,
Ib C. Bygbjerg, and Michael Alifrangis. Am. J. Trop. Med. Hyg., 88(3), 2013, pp. 536–541
Rapid Selection of Plasmodium falciparum Chloroquine Resistance Transporter Gene
and Multidrug Resistance Gene-1 Haplotypes Associated with Past Chloroquine and Present
Artemether-Lumefantrine Use in Inhambane District, Southern Mozambique
Thomas T. Thomsen, Laura B. Madsen, Helle H. Hansson, Elsa V. E. Tomas, Derek Charlwood,Ib C. Bygbjerg, and Michael Alifrangis*
Centre for Medical Parasitology, Department of International Health, Immunology and Microbiology, and Centre for Health Researchand Development, Faculty of Life Sciences, University of Copenhagen, Copenhagen, Denmark; Department of Infectious Disease,
Abstract. Chloroquine (CQ) use in Mozambique was stopped in 2002 and artemether-lumefantrine (AL) wasimplemented in 2008. In light of no use of CQ and extensive use of AL, we determined the frequency of molecularmarkers of Plasmodium falciparum drug resistance/tolerance to CQ and AL in persons living in Linga-Linga, an isolatedpeninsula and in Furvela village, which is located 8 km inland. The P. falciparum chloroquine resistance transporter geneCVMNK wild type increased in frequency from 43.9% in 2009 to 66.4% in 2010 (P £ 0.001), and combined P. falciparummultidrug resistance gene 1 N86-184F-D1246 haplotype increased significantly between years (P = 0.039). The combina-tion of P. falciparum chloroquine resistance transporter gene CVMNK and P. falciparummultidrug resistance gene NFDincreased from 24.3% (2009) to 45.3% in (2010, P = 0.017). The rapid changes observed may largely be caused bydecreased use of CQ and large-scale use of AL. In the absence of a clear AL-resistance marker and the (almost)continent-wide use of AL in sub-Saharan Africa, and when considering CQ reintroduction, continued monitoring ofthese markers is needed.
INTRODUCTION
Malaria remains one of the major killers in the tropical andsub-tropical world today, even though recent years haveshown progress in its control. The World Health Organizationestimated a decrease in malaria cases from 244 million to216 million during 2005–and 2010, and estimated mortalitydecreased from 781,000 in 2009 to 655,000 in 2010.1,2 This sig-nificant decrease in malaria-associated morbidity and mortal-ity is largely attributed to large-scale malaria control effortssuch as distribution of insecticide-treated nets, indoor residualspraying, intermittent preventive treatment in vulnerablegroups, and implementation of highly efficacious artemisinin-based combination therapies (ACT) for the treatment ofuncomplicated Plasmodium falciparum malaria in mostmalaria-endemic countries. The large-scale improvements arehighly dependent on continued reliability of efficacious ACTs.However, P. falciparum ACT resistance has emerged along
theThailand-Cambodia andThailand-Myanmar borders,3–6 andit might eventually be found in Africa, as happened with chloro-quine (CQ) and sulfadoxine-pyrimethamine (SP).7–9 ResistancetoCQinmalaria-endemicAfrica becameas prevalent asmalariaand thedrug has not been officially used for several years inmostmalaria-endemic countries. Depending on fitness costs in theparasites associated with acquired drug resistance, the latentperiod without a certain drug pressure may result in thereemergence of drug-sensitiveP. falciparum parasites.10
Resistance to CQ is mainly associated with a single nucleo-tide polymorphism (SNP) in the P. falciparum chloroquineresistance transporter (Pfcrt) gene, resulting in an amino acidchange from threonine to lysine mutation at codon 76(K76T).11,12 There are three main haplotypes in codons 72–76
of the Pfcrt gene, resulting in wild type CVMNK and CQ-resistant haplotypes CVIET and SVMNT.13 In Africa, theCVIET haplotype is the dominant mutant haplotype.14
By monitoring the temporal prevalence of Pfcrt K76,Kublin and others showed the reemergence of fully CQ-sensitive parasite populations after several years since cessationof CQ use in Malawi.15 Since this study, studies in Tanzania,16
Kenya,17 Senegal,18 and Mozambique19 have shown similartrends of reemergence of CQ sensitivity, and it is tempting toconsider reintroduction of CQ in combination with anotherantimalarial drug in areas where CQ resistance has decreasedand possibly reserved for malaria treatment of targetedpopulations, such as pregnant women, as has been suggestedby others.20
Another marker of antimalarial drug resistance is the P.
falciparum multidrug resistance gene-1 (Pfmdr-1) implicatedin resistance/tolerance to almost all antimalarial drugs includ-ing CQ, amodiaquine (AQ) and most importantly, theartemisinins. It has recently been shown that certain combi-nations of SNPs in the Pfmdr-1 gene, mainly at codons 86,184, and 1246, are emerging in areas where the ACT drugcombination artemether-lumefantrine (AL) is being widelyused21,22 and suggested that certain Pfmdr-1 haplotypes maybe markers of emergence of ACT tolerance.23
The Ministry of Health of Mozambique introduced SP-AQin late 2002 to replace CQ monotherapy as first-line treat-ment against uncomplicated malaria.24 In 2006, this combina-tion was replaced with the ACT combination artesunate–SP.However, already in 2008, the policy was changed to ALbecause of widespread SP resistance in the country.19
Chloroquine resistance in Mozambique was reported forthe first time in 1983, followed by a number of studiesreporting it throughout most of the country.25,26 In 1999,before abandonment of CQ, a study found that the PfcrtK76T mutation was prevalent in 90% of infected children inMozambique.27 In 2001–2002, a trial conducted in southernMozambique estimated a clinical efficacy for CQ of only
*Address correspondence to Michael Alifrangis, Centre for MedicalParasitology, Institute for International Health, Immunology andMicrobiology, CSS, Øster Farimagsgade 5, Building 22+23, PO Box2099, 1014 Copenhagen K, Denmark. E-mail: [email protected]
536 146
47%.26 Another study conducted in the same district in 2002–2003 demonstrated a frequency of the mutant CVIET haplo-type to be > 90%.24 Since CQ was officially abandoned in2002, the CQ drug pressure has most likely waned in subse-quent years. However, since the 4-aminoquinoline analog AQ(combined with SP) replaced CQ, this may have ensuredsome level of sustained drug pressure.In a recent report by Raman and others over a five-year
period (2006–2010), the prevalence of the Pfcrt K76T muta-tion was determined in children living in Gaza Province insouthern Mozambique.19 Overall, there was a strikingdecrease in the prevalence of the K76T mutation from > 95%in the four zones of Gaza Province in 2006 to 17.5–37.3% in2010. The study also examined the prevalence of SNPs in thePfmdr1-gene, but only regarding the N86Ymutation, in whicha reduction was observed from > 70% to 25.8–48.8%.19 Otherstudies from Mozambique have, to the best of our knowledge,not assessed SNP prevalence changes in the Pfmdr-1 gene.Therefore, temporal changes in selection of polymorphismsin this gene remains to be elucidated. This need is especiallyimportant in light of the suggested relationship between cer-tain Pfmdr-1 haplotypes and emergence of ACT tolerance.We therefore analyzed the distribution and investigatedshort-term temporal change of SNPs in the Pfcrt codons 72–76 and Pfmdr-1 codons 86, 184, and 1246 in persons living inLinga Linga, an isolated peninsula of Mozambique and in thevillage of Furvela located 8 km inland from Linga Linga.
MATERIALS AND METHODS
Study site. The peninsula of Linga Linga (23°43¢1.29²S,35°24¢15.04²E) is located in Inhambane District and 500 kmnorth of Maputo and opposite the district capital ofMorrumbene, which is 6 km west (across the MorrumbeneBay). The residents are mainly fishermen or involved inthe artisanal manufacture of raffia baskets, hats and bags.Furvela Village is 8 km west of Linga Linga on the mainland.Furvela has approximately 4,500 inhabitants, and Linga Lingais somewhat smaller with approximately 1,000 inhabitants. Atthe onset of the study in 2007, there was no health center onthe peninsula proper, but one was established in 2009. Other-wise, the nearest health centers were situated in the village ofCoche, 5 km north of Linga Linga, or in Morrumbene. Theproject received ethical clearance from the National BioethicsCommittee of Mozambique (reference 123/CNBS/06) onAugust 2, 2006.Sample collection and preparation. After an initial census,
an all-age malaria prevalence survey was performed. Sevenlocations in Linga Linga based on local knowledge were cho-sen for establishment of the survey. At each location, resi-dents were informed the day before the survey. In addition, asurvey of school-age children was undertaken. After informedconsent was obtained, survey teams collected cross-sectionalsamples from as many volunteers as possible, including smallchildren whose parents consented. Blood samples were col-lected in March–April 2009 and April 2010 in the village ofLinga Linga, and in May 2010 in the village of Furvela. Asimilar protocol was adopted in the latter village,28–30 and fivelocations were used as sites for the survey.Finger prick blood was used for preparation of thick
and thin blood films and added to 1.5-mL Eppendorf tubescontaining EDTA (Militom-14; VWR-Bie & Berntsen,
Denmark). Blood samples were allowed to separate intoserum and blood clot until clear separation was observed.Plasma was transferred into Eppendorf tubes and the bloodclot was used for various molecular analyses of the parasites.Blood slides stained with 5% Giemsa for 20 minutes wereread by technicians at the malaria reference laboratory inMaputo. Two hundred fields were examined before a slidewas declared negative. Numbers of parasites per 500 leuko-cytes were counted and converted to densities per microliterof blood, assuming a density of 8,000 leukocytes/mL. Onlyblood slide–positive samples for P. falciparum were used formolecular analysis. The age of donors ranged from 1 to 79 years,and the degree of P. falciparum positivity varied markedlybetween years andwhen age groups were compared.DNA extraction and SNP analysis of Pfcrt and Pfmdr-1
genes. DNA was extracted by using the NucleoSpin GenomicDNA Bloodpure Kit (Macherey-Nagel, Duren, Germany).Extraction was performed according to the manufac-turer’s instructions.Pfcrt genotyping was performed by using a nested polymer-
ase chain reaction, followed by sequence-specific oligonucle-otide probe (SSOP)–enzyme-linked immunosorbent assay asdescribed.31 A set of P. falciparum laboratory isolates wereused for positive controls: 3D7 and HB3 as CVMNK controls,FCR3 and DD2 as CVIET controls, and 7G8 as an SVMNTcontrol. Genotyping of Pfmdr-1 SNPs was performed by usingpublished polymerase chain reaction–restriction fragmentlength polymorphism protocols,32,33 with minor modificationsas described23 and 3D7 (N86-Y184-D1246), FCR3 (86Y-Y184-1246D), DD2 (86F-184Y-1246D), and 7G8 (N86-184F-1246Y)used as positive controls. Blood donors from Denmark whowere never exposed to malaria were used as P. falciparum-negative controls.Statistical analysis. Statistical analysiswas performed in2 + 2
contingency tables, and chi-square test statistics or Fisher’sexact test were applied when appropriate. For analysis ofPfcrt haplotypes, samples were considered to be mixed, butas containing a majority haplotype, when the optical density(OD) value of the weakly reacting Pfcrt SSOP was less thanhalf the OD value of the strongly reacting Pfcrt SSOP. Con-versely, if the OD value of the weakly reacting Pfcrt SSOPwas higher than half the OD value of the strongly reactingPfcrt SSOP, the infection was categorized as mixed with nodominant haplotype. To analyze for a possible temporalchange in the frequency of the Pfcrt CVMNK haplotype, allinfections containing CVMNK only or as the majority inmixed CVMNK/CVIET infections were tested against singleCVIET infections. The analysis of temporal change in theprevalence of Pfcrt CVMNK haplotype were performed bycomparing all infections containing CVMNK including allmixed CVMNK/CVIET haplotype infections against singleCVIET infections.Prevalence of Pfmdr-1 SNPs was examined individually for
codons 86, 184, and 1246 where the genotypes N86, 184F, andD1246 including mixed infections were compared against sin-gle 86Y, Y184, and 1246Y genotype infections, respectively.For frequency analysis, all mixed infections were omitted.Possible changes in frequency of constructed 86–184–1246haplotypes were analyzed by excluding infections with oneor more mixed genotype. Finally, analysis of the temporalfrequency of constructed Pfcrt-Pfmdr-1 haplotypes wasperformed by omitting all mixed Pfmdr-1 infections and for
SELECTION OF PFCRT AND PFMDR-1 HAPLOTYPES IN MOZAMBIQUE 537
147
Pfcrt, mixed infections where a majority haplotype could notbe determined.
RESULTS
Sample collection. In 2009 and 2010, of 435 and 385 sam-ples collected from donors in Linga Linga, 159 (36.6%) and108 (28.1%) were P. falciparum positive by microscopy,respectively. In addition, 336 samples were collected inFurvela in 2010, of which 111 (33.0%) were P. falciparum
positive by microscopy.Frequency and prevalence of codon72–76 haplotypes of
the Pfcrt gene in study sites of Mozambique in 2009–2010. Ofthe P. falciparum-positive sample set, 136 (85.5%) and 195(91.1%) samples were successfully haplotyped at codon 72–76 of the Pfcrt gene in samples from 2009 (Linga Linga only)and 2010 (Linga Linga, n = 97 and Furvela, n = 98), respec-tively. For the 2010 samples, no significant difference infrequency and prevalence of the Pfcrt haplotypes betweenLinga Linga and Furvela was observed (c2 = 0.01, P = 0.91and c2 = 1.07, P = 0.30 for comparison of frequency andprevalence, respectively), wherefore the samples from thetwo villages were pooled.The frequency of P. falciparum infections carrying the Pfcrt
wild type CVMNK haplotype (including mixed CVMNK/CVIET infections in which CVMNK was the majority haplo-type) versus mutant CVIET haplotype infections showed asignificant increase of CVMNK haplotype from 43.9% in2009 to 66.4% in 2010 (c2 = 13.1, P £ 0.001) (Figure 1A).Likewise, the prevalence of infections carrying the Pfcrt wildtype CVMNK haplotype including mixed CVMNK/CVIETinfections versus pure mutant CVIET haplotype infectionsincreased significantly from 60.0% in 2009 to 74.9% in 2010(c2 = 8.02, P = 0.005) (Figure 1B).Prevalence and frequency of SNPs at codons 86, 184,
and 1246 of the Pfmdr-1 gene. The temporal prevalence ofSNPs at codons 86, 184, and 1246 was analyzed by comparingthe distribution from 2009 and 2010 for codons 86 and 184(Figure 2A and B). Except for codon 184 (see below), thedata from Linga Linga and Furvela in 2010 were pooledbecause of a lack of significance between the settings. Preva-lence of P. falciparum infections carrying the N86 wild type(including mixed 86N/Y infections) increased significantlyfrom 64.7% in 2009 to 84.1% in 2010 (c2 = 16.3, P £ 0.001),and prevalence of the D1246 wild type genotype remained> 98% (c2 = 0.163, P = 0.67). For the 184F mutant type(including mixed 184F/Y infections), the prevalence was21.5% in Linga Linga in 2009, which increased to 34.3% in2010 (c2 = 4.41, P = 0.036) and to 51.0% in Furvela.
The frequency of the Pfmdr-1 genotypes (disregardingmixed genotype infections) from Linga Linga and Furvela in2010 was pooled because data was not significant betweenthe settings. The frequency of the N86 wild type increasedsignificantly from 52.9% to 73.0% (c2 = 8.93, P = 0.003),whereas for the 184F mutant type, only an insignificantincrease from 18.5% to 26.3% was seen (c2 = 2.07, P =0.150), and no change was observed in D1246, which remainedstable at 98% between the years (c2 = 0.161, P = 0.688).Frequency of constructed haplotypes at codon 86, 184,
and 1246 of the Pfmdr-1 gene. The construction of Pfmdr-186–184–1246 haplotypes (excluding mixed SNPs at one ormore codons) showed several different haplotypes and tem-
poral changes in the distribution (Figure 2C). The frequencyof the single mutant 86Y–Y184–D1246 (YYD) haplotypedecreased significantly from 47.8% in 2009 to 24.5% in 2010(c2 = 10.32, P = 0.001), whereas the frequency of the singlemutant NFD haplotype increased significantly between theyears (c2 = 4.27, P = 0.039).
Frequency of combined Pfcrt-Pfmdr-1 haplotypes. ThePfcrt haplotypes (CVMNK or CVIET) were combined withthe constructed Pfmdr-1 haplotypes omitting samples thatwere mixed with no clear majority infection (for Pfcrt), ormixed or negative in one or more of the Pfmdr-1 codons. Ofthe remaining 165 samples (2009: n = 76, 2010: n = 89), analy-sis showed a significant increase in infections carrying thePfcrt-Pfmdr-1 combination CVMNK-NFD from 24.3% in2009 to 45.3% in 2010 (c2 = 5.66, P = 0.017).
DISCUSSION
The use of CQ to treat uncomplicatedmalaria inMozambiquewas officially abandoned in 2002. Most likely, as everywhereelse in the malaria-endemic world where CQ has beenreplaced by other antimalarial drugs, some informal use ofCQ has subsequently been ongoing because of the low price
Figure 1. A, Frequency and B, Prevalence of Plasmodiumfalciparum chloroquine resistance transporter gene codon 72–76haplotypes, CVMNK (wild type) and CVIET (mutant type) inLinga Linga (2009) and Linga Linga with Furvela village (2010)of Mozambique.
538 THOMSEN AND OTHERS
148
ofCQandgood fever compliance. Furthermore, inMozambique,the analog 4-aminoquinoline amodiaquine combined with SPreplaced CQ, for a few years, which might have impacted Pfcrtand Pfmdr-1 haplotypes. However, because of improvedmalaria diagnostics such as the use of rapid diagnostic tests,and since 2008 better treatment options, e.g., ACTs, and possi-
bly lower malaria prevalence, CQ drug pressure woulddecrease. Given that there are fitness costs for malaria para-sites associated with CQ resistance,34 it is expected that theprevalence of sensitive parasites in vivo will increase.Although the validity of the Pfcrt 76T mutation as a predic-
tive marker of CQ treatment failure remains doubtful becauseof confounding factors such as host immunity, monitoringthe emergence of wild type Pfcrt 76K parasites in indigenousP. falciparum populations more adequately illustrates thetemporal advancement of parasite sensitivity to CQ. A studyin southern Mozambique in 2001 and 2003 reported frequen-cies of the mutant Pfcrt CVIET haplotype > 90% at the timeof official abandonment of CQ.24 In the present study, from aremote setting in Mozambique, the frequency of the PfcrtCVMNK wild type increased from 44% to 66% within a sin-gle year. This finding is consistent with the recent study byRaman and others, in which the prevalence of the pure K76wild type in the southern province of Gaza, Mozambique,increased from < 5% at baseline in 2006 to 65–80% in 2010.19
Thus, both studies confirms the trend of a substantial increasein P. falciparum susceptibility to CQ in Mozambique, simi-larly to other studies conducted in other parts of the eastAfrican region such as Malawi,15 Kenya,35 and Tanzania.16
However, recent studies in 2009–2010 in Mwanza, Tanzania,and Iganga, Uganda found a striking difference of 59.5% and0% in the prevalence of Pfcrt CVMNK wild types, respec-tively.36 Thus, the re-emergence of CQ susceptibility appearsto evolve at different rates probably because of co-varyingfactors such as treatments given (also dependent on differ-ences in diagnostic practices and transmission intensity) andthe continued use of CQ and/or related drugs maintaining thedrug pressure on Pfcrt, e.g., amodiaquine. In adition, the ACTdrug combination AL has been shown to select for Pfcrt
wild types.37 Therefore, large-scale implementation of AL asfirst-line treatment in most sub-Saharan countries may as wellfacilitate re-emergence of CQ sensitivity in P. falciparum.
In this study we also describe a selection of N86 and 184Fand the combined N86–184F–D1246 Pfmdr-1 haplotypeNFD. This finding is similar to our previous findings inTanzania, where the N86 and 184F prevalence increased sig-nificantly over a five-year period.23 Recently, Baliraine andRosenthal determined the prevalence of single Pfmdr-1 N86,184F, and D1246 and combined NFD haplotype before eitherAL, artesunate-AQ, or AQ + SP treatment, and comparedwith prevalence up to 120 days after treatment in primarilynew infections.38 Only in the AL group, the prevalence of theN86, 184F and D1246, but as well the NFD haplotype weremuch higher compared with pre-treatment prevalence38 indi-cating a survival advantage of these parasites. This findingdoes not indicate an immediate potential risk of clinical fail-ures after AL treatment; AL still remains highly efficacious inAfrica. However, the NFD haplotype in particular may beconsidered as a marker of increased tolerance to AL.When combining the Pfcrt and the Pfmdr-1 haplotypes, the
present study showed a strong selection of the Pfcrt-Pfmdr-1CVMNK-NFD haplotype. This finding might be caused bydecreased use of CQ. However, we propose that selection ofthis particular haplotype is as well largely a consequence oflarge-scale AL use. Baring in mind the small scale of thisstudy and several confounding factors such as impact of otherdrugs, our findings are only indicative. Therefore, there is acontinued need and urgency to monitor these two markers in
Figure 2. Prevalence of Plasmodium falciparum multidrug resis-tance gene-1 codon 86 and 184 genotypes and frequency of the com-bined codon 86–184–1246 haplotypes in Linga Linga (2009) and LingaLinga with Furvela village (2010) of Mozambique. A, Prevalenceof codon 86 genotypes. B, Prevalence of codon184 genotypes. C, Fre-quency of 86–184–1246 haplotypes (excluding mixed infections).
SELECTION OF PFCRT AND PFMDR-1 HAPLOTYPES IN MOZAMBIQUE 539
149
the light of a possible reintroduction of CQ in combinationwith another drug or alone for vulnerable groups such aspregnant women and because of the (almost) African-wideuse of AL, in the absence of a better molecular marker forAL resistance.
ReceivedAugust 27, 2012.Accepted for publicationDecember 15, 2012.
Published online February 4, 2013.
Acknowledgments: We thank the study participants, including theirparents or guardians; the village leaders of Linga Linga and Furvela;District Health Authorities of Morrumbene; and the MOZDANteam for their assistance during the surveys; and Ulla Abildtrup(Centre for Medical Parasitology, Copenhagen, Denmark) for excel-lent technical assistance.
Authors’ addresses: Thomas T. Thomsen, Section for Functional Geno-mics, Department of Biology, University of Copenhagen, Ole MaaløesVej 5, 2200 Copenhagen N, Denmark, E-mail: [email protected]. Laura B. Madsen, Helle H. Hansson, Ib C. Bygbjerg, andMichael Alifrangis, Centre for Medical Parasitology, Institute forInternational Health, Immunology and Microbiology, CSS, ØsterFarimagsgade 5, 1014 Copenhagen K, Denmark, E-mails: [email protected], [email protected], [email protected], and [email protected]. Elsa V. E. Tomas, Mozambican-Danish Rural Malaria Project,Morrumbene, InhambaneProvince,Mozambique, E-mail: [email protected]. Derek Charlwood, Centre for Health Research andDevelopment, Faculty of Life Sciences, University of Copenhagen,Frederiksberg, Denmark, and Instituto Nacional de Saude, AvenidaEduardo Mondalane, Maputo, Mozambique, E-mail: [email protected].
REFERENCES
1. World Health Organization, 2011. World Malaria Report. Avail-able at: http://www.who.int/malaria/world_malaria_report_2011/9789241564403_eng.pdf. Accessed November 18, 2012.
2. World Health Organization, 2010.World Malaria Report. Availableat: http://whqlibdoc.who.int/publications/2010/9789241564106_eng.pdf. AccessedNovember 17, 2012.
3. Noedl H, Se Y, Schaecher K, Smith BL, Socheat D, Fukuda MM,2008. Evidence of artemisinin-resistant malaria in westernCambodia. N Engl J Med 359: 2619–2620.
4. Noedl H, Se Y, Sriwichai S, Schaecher K, Teja-Isavadharm P,Smith B, Rutvisuttinunt W, Bethell D, Surasri S, Fukuda MM,Socheat D, Chan TL, 2010. Artemisinin resistance in Cambodia:a clinical trial designed to address an emerging problem insoutheast Asia. Clin Infect Dis 51: e82–e89.
5. Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, LwinKM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, SilamutK, Imwong M, Chotivanich K, Lim P, Herdman T, An SS,Yeung S, Singhasivanon P, Day NP, Lindegardh N, SocheatD, White NJ, 2009. Artemisinin resistance in Plasmodiumfalciparum malaria. N Engl J Med 361: 455–467.
6. Phyo AP, Nkhoma S, Stepniewska K, Ashley EA, Nair S,McGready R, ler Moo C, Al-Saai S, Dondorp AM, Lwin KM,Singhasivanon P, Day NP, White NJ, Anderson TJ, Nosten F,2012. Emergence of artemisinin-resistant malaria on the westernborder of Thailand: a longitudinal study. Lancet 379: 1960–1966.
7. Roper C, Pearce R, Nair S, Sharp B, Nosten F, Anderson T, 2004.Intercontinental spread of pyrimethamine-resistant malaria.Science 305: 1124.
9. Wootton JC, Feng X, Ferdig MT, Cooper RA, Mu J, Baruch DI,Magill AJ, SuXZ, 2002. Genetic diversity and chloroquine selec-tive sweeps in Plasmodium falciparum.Nature 418: 320–323.
10. Laufer MK, Thesing PC, Eddington ND, Masonga R,Dzinjalamala FK, Takala SL, Taylor TE, Plowe CV, 2006.Return of chloroquine antimalarial efficacy in Malawi. N EnglJ Med 355: 1959–1966.
11. Fidock DA, Nomura T, Talley AK, Cooper RA, Dzekunov SM,Ferdig MT, Ursos LM, Sidhu AB, Naude B, Deitsch KW,Su XZ, Wootton JC, Roepe PD, Wellems TE, 2000. Muta-tions in the P. falciparum digestive vacuole transmembraneprotein PfCRT and evidence for their role in chloroquineresistance. Mol Cell 6: 861–871.
12. Djimde A, Doumbo OK, Cortese JF, Kayentao K, Doumbo S,Diourte Y, Dicko A, Su XZ, Nomura T, Fidock DA, WellemsTE, Plowe CV, Coulibaly D, 2001. A molecular marker forchloroquine-resistant falciparum malaria. N Engl J Med 344:257–263.
13. Warhurst DC, 2003. Polymorphism in the Plasmodiumfalciparum chloroquine-resistance transporter protein linksverapamil enhancement of chloroquine sensitivity with theclinical efficacy of amodiaquine. Malar J 2: 31.
14. Ariey F, Fandeur T, Durand R, Randrianarivelojosia M, JambouR, Legrand E, Ekala MT, Bouchier C, Cojean S, DucheminJB, Robert V, Le Bras J, Mercereau-Puijalon O, 2006. Inva-sion of Africa by a single pfcrt allele of south east Asian type.Malar J 5: 34.
15. Kublin JG, Cortese JF, Njunju EM, Mukadam RA, Wirima JJ,Kazembe PN, Djimde AA, Kouriba B, Taylor TE, Plowe CV,2003. Reemergence of chloroquine-sensitive Plasmodiumfalciparum malaria after cessation of chloroquine use inMalawi. J Infect Dis 187: 1870–1875.
16. Alifrangis M, Lusingu JP, Mmbando B, Dalgaard MB,Vestergaard LS, Ishengoma D, Khalil IF, Theander TG,Lemnge MM, Bygbjerg IC, 2009. Five-year surveillance ofmolecular markers of Plasmodium falciparum antimalarialdrug resistance in Korogwe District, Tanzania: accumulationof the 581G mutation in the P. falciparum dihydropteroatesynthase gene. Am J Trop Med Hyg 80: 523–527.
17. Mang’era CM, Mbai F, Omedo IA, Mireji PO, Omar SA, 2012.Changes in genotypes of Plasmodium falciparum humanmalaria parasite following withdrawal of chloroquine in Tiwi,Kenya. Acta Trop 123: 202–207.
18. Ndiaye M, Faye B, Tine R, Ndiaye JL, Lo A, Abiola A,Dieng Y, Ndiaye D, Hallett R, Alifrangis M, Gaye O,2012. Assessment of the molecular marker of Plasmodiumfalciparum chloroquine resistance (Pfcrt) in Senegal afterseveral years of chloroquine withdrawal. Am J Trop MedHyg 87: 640–645.
19. Raman J, Mauff K, Muianga P, Mussa A, Maharaj R, Barnes KI,2011. Five years of antimalarial resistance marker surveillancein Gaza Province, Mozambique, following artemisinin-basedcombination therapy roll out. PLoS ONE 6: e25992.
20. Frosch AE, Venkatesan M, Laufer MK, 2011. Patterns of chloro-quine use and resistance in sub-Saharan Africa: a system-atic review of household survey and molecular data. MalarJ 10: 116.
21. Lekana-Douki JB, Dinzouna Boutamba SD, Zatra R, Zang EdouSE, Ekomy H, Bisvigou U, Toure-Ndouo FS, 2011. Increasedprevalence of the Plasmodium falciparum Pfmdr1 86N geno-type among field isolates from Franceville, Gabon afterreplacement of chloroquine by artemether-lumefantrine andartesunate-mefloquine. Infect Genet Evol 11: 512–517.
22. Zeile I, Gahutu JB, Shyirambere C, Steininger C, MusemakweriA, Sebahungu F, Karema C, Harms G, Eggelte TA,Mockenhaupt FP, 2012. Molecular markers of Plasmodiumfalciparum drug resistance in southern highland Rwanda. ActaTrop 121: 50–54.
23. Thomsen TT, Ishengoma DS, Mmbando BP, Lusingu JP,Vestergaard LS, Theander TG, Lemnge MM, Bygbjerg IC,Alifrangis M, 2011. Prevalence of single nucleotide polymor-phisms in the Plasmodium falciparum multidrug resistancegene (Pfmdr-1) in Korogwe District in Tanzania before andafter introduction of artemisinin-based combination therapy.Am J Trop Med Hyg 85: 979–983.
24. Enosse S, Magnussen P, Abacassamo F, Gomez-Olive X,Ronn AM, Thompson R, Alifrangis M, 2008. Rapid increaseof Plasmodium falciparum dhfr/dhps resistant haplotypes,after the adoption of sulphadoxine-pyrimethamine as first linetreatment in 2002, in southern Mozambique. Malar J 7: 115.
25. Schwalbach J, Schapira A, Suleimanov G, 1985. Chloroquine-resistant malaria in Mozambique. Lancet 2: 897–898.
540 THOMSEN AND OTHERS
150
26. Abacassamo F, Enosse S, Aponte JJ, Gomez-Olive FX, Quinto L,Mabunda S, Barreto A,Magnussen P, Ronn AM, Thompson R,Alonso PL, 2004. Efficacy of chloroquine, amodiaquine,sulphadoxine-pyrimethamine and combination therapy withartesunate in Mozambican children with non-complicatedmalaria.TropMed Int Health 9: 200–208.
27. Mayor AG, Gomez-Olive X, Aponte JJ, Casimiro S, Mabunda S,Dgedge M, Barreto A, Alonso PL, 2001. Prevalence of theK76T mutation in the putative Plasmodium falciparum chloro-quine resistance transporter (pfcrt) gene and its relationto chloroquine resistance in Mozambique. J Infect Dis 183:1413–1416.
28. Kampango A, Cuamba N, Charlwood JD, 2011. Does moonlightinfluence the biting behaviour of Anopheles funestus? Med VetEntomol 25: 240–246.
29. Charlwood JD, 2011. Studies on the bionomics of maleAnophelesgambiae Giles and male Anopheles funestus Giles from south-ern Mozambique. J Vector Ecol 36: 382–394.
31. Alifrangis M, Enosse S, Pearce R, Drakeley C, Roper C, KhalilIF, Nkya WM, Ronn AM, Theander TG, Bygbjerg IC, 2005. Asimple high-throughput method to detectPlasmodium falciparumsingle nucleotide polymorphisms in the dihydrofolate reductase,dihydropteroate synthase, andP. falciparum cloroquine resistancetransporter genes using ploymerase chain reaction and enzymelinked immunosorbent assay-based technology. Am J Trop MedHyg 72: 155–162.
32. Duraisingh MT, Roper C, Walliker D, Warhurst DC, 2000.Increased sensitivity to the antimalarials mefloquine andartemisinin is conferred by mutations in the pfmdr1 gene ofPlasmodium falciparum. Mol Microbiol 36: 955–961.
33. Humphreys GS, Merinopoulos I, Ahmed J,Whitty CJ, MutabingwaTK, Sutherland CJ, Hallett RL, 2007. Amodiaquine andartemether-lumefantrine select distinct alleles of the Plasmodiumfalciparummdr1 gene inTanzanian children treated for uncompli-catedmalaria.AntimicrobAgents Chemother 51: 991–997.
35. Mwai L, Ochong E, Abdirahman A, Kiara SM, Ward S, KokwaroG, Sasi P, Marsh K, Borrmann S, Mackinnon M, Nzila A, 2009.Chloroquine resistance before and after its withdrawal inKenya. Malar J 8: 106.
36. Kamugisha E, Bujila I, Lahdo M, Pello-Esso S, Minde M,Kongola G, Naiwumbwe H, Kiwuwa S, Kaddumukasa M,Kironde F, Swedberg G, 2012. Large differences in prevalenceof Pfcrt and Pfmdr1 mutations between Mwanza, Tanzania andIganga, Uganda-a reflection of differences in policies regardingwithdrawal of chloroquine? Acta Trop 121: 148–151.
37. Sisowath C, Petersen I, Veiga MI, Martensson A, Premji Z,Bjorkman A, Fidock DA, Gil JP, 2009. In vivo selection ofPlasmodium falciparum parasites carrying the chloroquine-susceptible pfcrt K76 allele after treatment with artemether-lumefantrine in Africa. J Infect Dis 199: 750–757.
38. Baliraine FN, Rosenthal PJ, 2011. Prolonged selection of pfmdr1polymorphisms after treatment of falciparum malaria withartemether-lumefantrine inUganda. J Infect Dis 204: 1120–1124.
SELECTION OF PFCRT AND PFMDR-1 HAPLOTYPES IN MOZAMBIQUE 541
151
Paper II
Collateral Resistance and Sensitivity Modulate Evolution of High-Level Resistance
to Drug Combination Treatment in Staphylococcus aureus
Mari Rodriguez de Evgrafov,a Heidi Gumpert,a Christian Munck,a Thomas T. Thomsen,a and
aDepartment of Systems Biology, Technical University of Denmark, DK-2800 Lyngby, Denmark bThe Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, DK-2970 Hørsholm,
Denmark
Corresponding author: Morten O.A. Sommer, Department of Systems Biology, Technical University of Denmark, DK-
Collateral Resistance and Sensitivity Modulate Evolutionof High-Level Resistance to Drug Combination Treatmentin Staphylococcus aureus
Mari Rodriguez de Evgrafov,1 Heidi Gumpert,1 Christian Munck,1 Thomas T. Thomsen,1 andMorten O.A. Sommer*,1,2
1Department of Systems Biology, Technical University of Denmark, Lyngby, Denmark2The Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, Hørsholm, Denmark
As drug-resistant pathogens continue to emerge, combination therapy will increasingly be relied upon to treat infectionsand to help combat further development of multidrug resistance. At present a dichotomy exists between clinical practice,which favors therapeutically synergistic combinations, and the scientific model emerging from in vitro experimentalwork, which maintains that this interaction provides greater selective pressure toward resistance development than otherinteraction types. We sought to extend the current paradigm, based on work below or near minimum inhibitoryconcentration levels, to reflect drug concentrations more likely to be encountered during treatment. We performed aseries of adaptive evolution experiments using Staphylococcus aureus. Interestingly, no relationship between druginteraction type and resistance evolution was found as resistance increased significantly beyond wild-type levels.All drug combinations, irrespective of interaction types, effectively limited resistance evolution compared with mono-treatment. Cross-resistance and collateral sensitivity were found to be important factors in the extent of resistanceevolution toward a combination. Comparative genomic analyses revealed that resistance to drug combinations wasmediated largely by mutations in the same genes as single-drug-evolved lineages highlighting the importance of thecomponent drugs in determining the rate of resistance evolution. Results of this work suggest that the mechanismsof resistance to constituent drugs should be the focus of future resistance evolution work.
Key words: resistance evolution, antibiotic resistance, drug combinations.
IntroductionAntibiotic resistance poses a severe threat to public health(Read et al. 2011; World Health Organization 2012). Left unre-solved antibiotic resistance will increase the cost of healthcare,threaten medical advancement, scale back progress againstcertain infectious diseases and lead to greater morbidity andmortality (World Health Organization 2012). The increasingpresence of antibiotic-resistant organisms has led to greaternumbers of treatment failures for Gram-positive pathogens,such as methicillin-resistant Staphylococcus aureus, vancomy-cin-resistant Enterococcus, and multidrug-resistant tuberculo-sis (Cornaglia 2009; Woodford and Livermore 2009). Theproblem posed by resistant organisms is exacerbated by lim-ited development of new antibiotics (Cottarel andWierzbowski 2007; Fischbach 2011; Thaker et al. 2013).However, the arrival of new antibiotics provides only short-term relief as resistance quickly follows (Clatworthy et al.2007; Read et al. 2011). Thus, the long-term key to controllingthis threat lies in managing the unavoidable resistanceadaptation (Read et al. 2011).
Combination therapy, the concurrent use of two or moredrugs, is one such resistance management strategy, which hasproven instrumental in prolonging the useful lifespan of
antibiotics (Cottarel and Wierzbowski 2007; Read et al.2011) as well as improving treatment outcomes in a varietyof diseases, such as TB and HIV (Gilliam et al. 2006; Lennoxet al. 2009; Huang et al. 2012; Vilcheze and Jacobs 2012;Freedberg et al. 2013). Combination therapy relies upon spon-taneous resistance being rare and multiplicative so the likeli-hood of an organism gaining resistance to multiple drugs in asingle instance is less than the prospect of resistance to anyone of the component drugs acting alone (Fischbach 2011).This reasoning assumes that resistance acquisition is an inde-pendent event for each component of the mixture.
A major goal of resistance evolution research has been thesearch for the most effective yet resistance limiting combina-tions or treatment strategies (Yeh et al. 2006; Chait et al. 2007;Hegreness et al. 2008; Michel et al. 2008; Bollenbach et al.2009; Torella et al. 2010; Imamovic and Sommer 2013;Pena-Miller et al. 2013). Outcomes of nearly a decadeworth of experimental in vitro work have suggested thatdrug interactions (Chait et al. 2007; Hegreness et al. 2008;Michel et al. 2008; Torella et al. 2010; Palmer and Kishony2013; Pena-Miller et al. 2013) are a key factor in limiting ordriving resistance evolution, particularly during the earlystages of resistance development. Specifically, combinations
� The Author 2015. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution. All rights reserved. For permissions, pleasee-mail: [email protected]
with antagonistic or suppressive interactions, where drugs in amixture interfere with each other and the overall therapeuticeffect is less than the component drugs working alone, havebeen shown to slow down resistance adaption better thanthose that act in a synergistic manner, where treatmentoutcomes are better than what would be expected fromsumming the effect of the component drugs acting alone(Chait et al. 2007; Hegreness et al. 2008; Michel et al. 2008;Torella et al. 2010; Pena-Miller et al. 2013). The rationale forthis hypothesis is that the mutations conferring resistance toa single drug will have a more pronounced effect on thefitness of the organism in the presence of a synergisticcombination because of the cooperative interaction of thecomponents in the mixture (Hegreness et al. 2008; Michelet al. 2008). However, results of the in vitro work conflictwith clinical practice where synergistic combinations arethe preferred treatment regime (Cottarel and Wierzbowski2007).
There are caveats to the paradigm that has emerged fromthese findings. These include the absence of the role of epis-tasis in driving resistance evolution (Trindade et al. 2009; Halland MacLean 2011; Borrell et al. 2013) as well as the founda-tion being based on experimental work performed at or nearWT minimum inhibitory concentration (MIC) levels (Yehet al. 2006; Chait et al. 2007; Michel et al. 2008; Pena-Milleret al. 2013). Recent work has suggested that a better under-standing of epistasis among relevant resistance conferringmutations could lead to the design of better treatment reg-imens (Trindade et al. 2009; Borrell et al. 2013). Moreover,clinically relevant resistance associated with treatment failuresusually occurs in association with concentrations substantiallygreater than WT MIC levels (Anon 2013). Finally, emphasis onresistance adaptation at or near WT MIC levels may not ac-curately reflect the phenomena observed during the treat-ment of chronic bacterial infections, such as TB or cysticfibrosis. Despite the progress made through the aforemen-tioned laboratory experiments, there is still a great need for abetter understanding of the evolution of multidrug resistance(Palmer and Kishony 2013) before allowing these findings toshape or change therapeutic strategies aiming to control re-sistance evolution.
We proposed testing the generality of the current para-digm by extending the concentration range and adaptationtime frame considered while using the same model organismand drug combinations originally used to construct it(Hegreness et al. 2008; Michel et al. 2008). We hypothesizedthat at elevated concentrations resistance evolution is drivenby response to individual component drugs rather than druginteractions. To test our hypotheses, we evolved populationsof S. aureus strain Newman, a medically relevant Gram-pos-itive species, in the presence of six different antibiotics andfive different combinations. The drugs and combinationsused are well characterized, are clinically relevant, and havediverse modes of action (table 1). We performed genomicsequencing to determine the mutations involved in resistanceadaptation. Finally, we considered the role of mutations inresistance toward drug combinations.
Results
Classification of Selected Drug Combinations
Drug combinations are characterized according to the epi-static interactions between their component drugs. The frac-tional inhibitory concentration index (FICI) is used to describethese interactions and is based on the Loewe additivity zerointeraction theory (Berenbaum 1978). The index, determinedfor a given effect level, is the sum of the fractional inhibition ofeach drug in a combination relative to the drug acting alone.The interactions of each of our drug combinations weretested using the WT strain prior to commencing the resis-tance adaption experiments. The interaction types at aneffect level of 90% were as follows: doxycycline–erythromycin(FICI 0.58� 0.04), doxycycline–ciprofloxacin (FICI = 0.81�0.14), and fusidic acid–erythromycin (FICI = 0.75� 0.15)were synergistic, ciprofloxacin–ampicillin was additive(FICI = 0.99� 0.11) and fusidic acid–amikacin was antagonis-tic (FICI = 1.69� 0.1). Previous work performed in Escherichiacoli, and performed again here (supplementary data S1,Supplementary Material online) characterized the interactionbetween doxycycline and ciprofloxacin as strongly antagonis-tic (Yeh et al. 2006; Toprak et al. 2011; L�az�ar et al. 2013);however, this combination was found to be synergisticwhen tested in our S. aureus strain Newman, underscoringthe dependence of drug epistatic interactions on the specifictarget organism.
Resistance Evolution of Populations
A wild-type (WT) S. aureus strain Newman population waschallenged and adapted in three replicate lineages designatedas A, B, and C to increasing concentrations of six individualantibiotics and five antibiotic combinations (table 1). An ad-ditional three replicate lineages, also designated as A, B and C,were passaged in media only. Adaptation was performed ac-cording to the following protocol (fig. 1). Briefly, the WTorganism was inoculated into 12 different conditions withincreasing concentrations of antibiotic(s) and allowed togrow for 18 h. At the end of the growth period, optical density(OD) measurements were taken and the most resistant cul-ture from each replicate was reinoculated in fresh media atthe drug concentration it was selected from. The reculturedorganisms were then used as inoculum for the next resistancechallenge. A total of five resistance evolution periods, referredto as exposures, were performed. A total of ten inoculations(fresh media tube and exposure), equivalent to an averagecumulative number of cell divisions (CCD) of 1.16� 1013 (Leeet al. 2011), were performed. A total of 36 lineages (18 singledrug, 15 combination, and 3 media only evolved) were yieldedthrough the evolution process.
Adaptation to single agents increased steadily with eachexposure (fig. 2) for most populations and after five exposuresfour of six single-drug-evolved populations were able to growin concentrations of at least 10mg/ml (supplementary dataS1, Supplementary Material online). Lineages evolved toerythromycin and amikacin developed resistance quicklyand were able to grow in antibiotic concentrations greater
1176
de Evgrafov et al. . doi:10.1093/molbev/msv006 MBE at R
than 100mg/ml. Adaptation to doxycycline and ampicillinwas much slower, with populations tolerating less than3mg/ml after five exposures. Adaptation by four of the fivecombination-evolved populations (ciprofloxacin–ampicillin,fusidic acid–amikacin, doxycycline–erythromycin, and doxy-cycline–ciprofloxacin) was similar to their slowest evolvingsingle drug counterparts, whereas lineages evolved to thefusidic acid–erythromycin combination were approximately10� less than their slowest evolving single drug counterpart(fig. 2 and supplementary data S1, Supplementary Materialonline).
Resistance Profiles of Adapted Lineages
Following resistance adaptation, four isolates from each of theadapted populations were profiled for their individual resis-tances. Results show that all isolates exhibited a substantialincrease in resistance following five exposures (fig. 3 and sup-plementary data S1, Supplementary Material online). In manycases, the IC90 values of the isolates were 100� greater than
the WT value and in the case of the fusidic acid isolates morethan a 1,000� larger. Exceptions to this trend were observedin the ampicillin, ciprofloxacin–ampicillin, and fusidicacid–erythromycin isolates where IC90 values were only10–30� the WT value. Increased resistance differed amongisolates evolved to the same drug(s) and in some cases thisdifference was considerable (fig. 3). We attributed the differ-ences observed within a given drug(s) group to be the resultof genotypic changes acquired by the isolates throughadaption.
The fusidic acid–amikacin isolates (antagonistic interac-tion, supplementary data S1, Supplementary Material online)had the greatest increase in resistance improvement followedclosely by isolates adapted to doxycycline–ciprofloxacin (syn-ergistic interaction, supplementary data S1, SupplementaryMaterial online). Isolates evolved to ciprofloxacin–ampicillin(additive interaction, supplementary data S1, SupplementaryMaterial online) had the least resistance improvement, anaverage of 11� the WT MIC value. These results contrastwith previous reports based on sub-MIC adaptations, which
X
Exposure 1 Exposure 2 Exposure 3
Wild TypeS. aureusNewman
Concentra�on
Y
X+Y
FIG. 1. Adaptation of Staphylococcus aureus to individual drugs and drug pairs. An overnight culture of WT S. aureus was used to inoculate microtiterplates containing different drugs or combinations with increasing concentrations or media only. Three replicate populations were recreated for eachcondition. The highest concentration where growth was present was recultured in fresh media and then used to inoculate the next concentrationchallenge, referred here to as exposure. A total of five exposures were performed for each condition.
Table 1. Antibiotics Used and Their Modes of Action.
Antibiotic Name Abbreviation Class Target
Amikacin AMI Aminoglycoside 30S ribosome
Ampicillin AMP Beta lactam Cell wall
Ciprofloxacin CPR Quinolone DNA synthesis
Erythromycin ERY Macrolide 50S ribosome
Doxycycline DOX Tetracycline 30S ribosome
Fusidic acid FUS Other Protein synthesis
Combination Abbreviation Interaction
Amikacin and fusidic acid FUS-AMI Antagonistic
Ampicillin and ciprofloxacin CPR-AMP Additive
Ciprofloxacin and doxycycline DOX-CPR Synergistic
Erythromycin and doxycycline DOX-ERY Synergistic
Erythromycin and fusidic acid FUS-ERY Synergistic
1177
Resistance Evolution Dependent on Collateral Resistance . doi:10.1093/molbev/msv006 MBE at R
FIG. 2. Change in drug tolerance during adaptation. Each bar is an average of three replicate lineages and reflects the average concentration that theadapted population can grow in following exposure to ever increasing concentrations. Populations are grouped according to drug pairs: (A) FUS-ERY,(B) CPR-AMP, (C) DOX-ERY, (D) FUS-AMI, and (E) DOX-CPR. Dashed lines represent clinical breakpoints, taken from the EUCAST website, for eachindividual drug. There is no established clinical breakpoint value for ampicillin used on Staphylococcus aureus.
FIG. 3. Gain in IC90 value of the most evolved lineages following resistance adaptation. Isolates are grouped according to drug pairs: (A) FUS-ERY, (B)CPR-AMP, (C) DOX-ERY, (D) FUS-AMI, and (E) DOX-CPR. Each column is an average of four biological replicates. Error bars reflect the SEM of thereplicates. Differences within a drug(s) group suggest that resistance adaptation is a complex process. Adaptation of the combination-evolved isolatesmirrors that of the least evolved single drug isolates.
1178
de Evgrafov et al. . doi:10.1093/molbev/msv006 MBE at R
suggest that antagonistic or suppressive combinations limitresistance evolution best (Hegreness et al. 2008; Michel et al.2008; Pena-Miller et al. 2013). In general, the extent of resis-tance attained by the combination-evolved isolates was sim-ilar to that of the slowest evolved corresponding single drugisolates, highlighting the importance of individual compo-nents in resistance evolution during combination therapy.We quantified these observations using the evolvabilityindex (Munck et al. 2014), which describes how resistanceevolution toward an individual drug is impacted as a resultof being used in a combination compared with being usedalone. The evolvability index is determined by taking the av-erage of the relative change in resistance development foreach component drug of a drug combination-evolved lineageand dividing it by the relative change in resistance develop-ment in the single-drug-evolved lineages (Munck et al. 2014)(eq. 2). An evolvability index value of 1 signifies that thecombination-evolved isolate developed resistance to thesame extent as the individual drug-evolved isolates did tothe component drugs. A value greater than 1 indicates thatthe combination-evolved isolate evolved to be more resistantthan its corresponding single-drug-evolved isolates, whereas avalue of less than 1 means that the combination-evolvedisolates evolved less than the single-drug-evolved isolates. Itis important to note that the evolvability index assumes thatthe exposure time to each component or combination is thesame. Comparisons where this is not the case are not accuratemeasures of resistance evolution. Nevertheless, this simplifi-cation provides a clear and quantitative means to comparehow different combinations drive resistance adaptationacross experiments and organisms.
All but three of our combination-evolved isolates hadevolvability index values of less than 1 meaning that overallthe combinations were effective at limiting resistance evolu-tion relative to their constituent drugs alone (fig. 4 and sup-plementary data S1, Supplementary Material online). Isolateswith evolvability index values greater than 1 were theciprofloxacin–ampicillin isolate C (2.3) and the doxycy-cline–ciprofloxacin isolates B (1.38) and C (1.59). Each of
these isolates had component IC90 values that greatly ex-ceeded those of the corresponding single-drug-evolved iso-lates (supplementary fig. S1, Supplementary Material online).Elevated evolvability index values were also determined forfusidic acid isolate B (0.96) and doxycycline–erythromycinisolates B (0.86) and C (0.84) and were likely due to strongresistance to one component drug (supplementary fig. S1,Supplementary Material online). The smallest evolvabilityindex values (<0.2) belonged to the fusidic acid–erythromy-cin isolates, which suggests that this combination limited re-sistance evolution best.
WT epistatic drug interactions were not found to be sig-nificantly correlated to the extent of resistance evolutionobserved. One explanation could be that drug interactionsare not static but rather affected by resistance evolution.To assess the evolutionary stability of the epistatic druginteractions, we determined the FICI values for our combina-tions for the evolved isolates. These data show that changes inthe drug interaction profiles had taken place (supplementaryfig. S2, Supplementary Material online). For example, the in-teraction between doxycycline and ciprofloxacin postadapta-tion became antagonistic in each of the three replicateisolates. A similar shift was observed for two of the threefusidic acid–erythromycin isolates. The interaction betweenfusidic acid and erythromycin remained synergistic; however,the FICI values increased as a result of adaptation. An incon-clusive interaction existed between ciprofloxacin and ampi-cillin following adaptation with one isolate demonstratingsynergism whereas another displayed antagonism. FICIvalues for fusidic acid and amikacin decreased slightly belowthe WT value for two of the three isolates; however, the thirdisolate showed strong antagonism between the two drugs.These findings are in agreement with a recent study of E. coliexposed to erythromycin and doxycycline showing that druginteractions are strongly modulated by evolution (Pena-Milleret al. 2013). Drug interactions can predict resistance evolutionfor sub-MIC adaptation; however, our data suggest that theseinteractions change in response to resistance adaptationcausing their reliability as resistance evolution predictors tobecome less certain.
Instead, we decided to investigate the role of cross-resis-tance in driving resistance evolution as there appeared to be arelationship between the resistance evolution of combinationisolates and their corresponding constituent drug isolates.Moreover, cross-resistance has been suggested to play an im-portant role in rates of adaptation (Szybalski 1954; Hegrenesset al. 2008; Michel et al. 2008; Yeh et al. 2009; Imamovic andSommer 2013; L�az�ar et al. 2013, 2014; Oz et al. 2014). Usingthe same combination pairings all single-drug-evolved isolateswere exposed to the other respective component drug, thatis, lineages evolved to drug A were exposed to drug B to testfor cross-resistance in combination AB.
Overall, adaptation to a single antibiotic frequently re-sulted in the cross-resistance to another (fig. 5). The amika-cin-evolved isolates had strong (410�WT) cross-resistanceto fusidic acid and in the case of one replicate the IC90 valuewas nearly 100 times that of the WT. Isolates evolved toampicillin displayed limited to negligible cross-resistance or
0.5 1.0 1.50.01
0.1
1
10 FUS-ERY
CPR-AMP
FUS-AMI
DOX-CPR
DOX-ERY
WT FICI
Evol
vabi
lity
Inde
x Va
lue
FIG. 4. Evolvability index values for each drug combination isolate. Theevolvability index quantifies how being used in a combination impactedthe resistance evolution to the individual component drugs of acombination. Values are grouped according to WT drug interaction.FUS-ERY, DOX-CPR, and DOX-ERY were all synergistic, CPR-AMP wasadditive, and FUS-AMI was antagonistic. Variation among replicateswithin the same drug pair reflects the individuality of resistanceadaptation.
1179
Resistance Evolution Dependent on Collateral Resistance . doi:10.1093/molbev/msv006 MBE at R
collateral sensitivity (Imamovic and Sommer 2013; L�az�ar et al.2013) to ciprofloxacin and vice versa. The ciprofloxacin-evolved isolates, however, did display considerable(30�WT) cross-resistance to doxycycline. Adaptation todoxycycline resulted in strong (410�WT IC90) cross-resis-tance to both erythromycin and ciprofloxacin. Isolatesevolved to erythromycin displayed strong (410�WT IC90)cross-resistance to doxycycline and moderate (<5�WT IC90)cross-resistance to fusidic acid. The extent of cross-resistancedisplayed by isolates evolved to ciprofloxacin, doxycycline,and erythromycin is consistent with the elevated evolvabilityindices calculated for the corresponding combinations.Finally, adaptation to fusidic acid resulted in collateral sensi-tivity to erythromycin with IC90 values well below the WTvalue. This collateral sensitivity likely explains the compara-tively slow evolution of resistance observed for isolatesevolved to the fusidic acid–erythromycin combination. Thefusidic acid-evolved isolates also displayed moderate(<5�WT IC90) cross-resistance to amikacin. The combina-tions for which the component drugs did not confer collateralsensitivity exhibited significantly higher evolvability indexvalues (P< 0.05, Mann–Whitney), suggesting that collateralsensitivity interactions between component drugs are impor-tant for determining resistance evolution toward drugcombinations.
Whole-Genome Sequence Analysis
To explore the molecular basis of the drug resistance observedin our experiments, we sequenced the genomes of our mostevolved isolates (18 from the single-drug-evolved isolates, 15from the combination-evolved isolates, and 3 from the mediaonly-evolved isolates) and our ancestral WT. The sequencedisolates were then analyzed in groups based on the drug(s)they were evolved to. In general, the resistance phenotypesobserved in the isolates could be readily explained by thepresence of expected resistance mutations in their genomes.
An overlap of canonical resistance mutations was observedin both the combination-evolved and single-drug-evolved iso-lates (fig. 6 and supplementary data S2, SupplementaryMaterial online). For example, two of three fusidic acid–eryth-romycin-evolved isolates (A and B) and two of three eryth-romycin-evolved isolates (A and B) had mutations in the rplDgene, which codes for ribosomal protein L4. Mutations in thisgene have previously been associated with macrolide resis-tance in several bacterial species (Tait-Kamradt et al. 2000;Canu et al. 2002; Zaman et al. 2007), including S. aureus(Prunier et al. 2002). The mutations observed in the rplDgene of all four isolates are well-documented amino acid sub-stitutions (Canu et al. 2002; Diner and Hayes 2009) that resultin the alteration of the macrolide-binding site (Gregory andDahlberg 1999; Gabashvili et al. 2001; Diner and Hayes 2009).The resistance conferred by these mutations, however, variedconsiderably (fig. 3 and supplementary fig. S1, SupplementaryMaterial online) and appeared to be a function of quantity.Both erythromycin isolate B and fusidic acid–erythromycinisolate B had multiple single nucleotide polymorphism (SNPs)in the rplD gene, whereas erythromycin isolate A and fusidicacid–erythromycin isolate A each had only one SNP.Erythromycin isolate C had no ribosomal protein mutationsbut attained considerable resistance to erythromycin throughan alternate means.
Mutations in the fusA gene, known to confer fusidic acidresistance in S. aureus (Besier et al. 2003), were observed in allisolates evolved to fusidic acid as well as the amikacin-evolvedisolates. fusA gene mutations have previously been found toconfer aminoglycoside resistance in S. aureus (Norstr€om et al.2007). The fusA gene mutations observed in the amikacin-evolved lineages conferred both high levels of amikacin andfusidic acid resistance, highlighting how cross-resistance canundermine the effect of drug combinations (figs. 2 and 5). Itshould be noted that fusidic acid and amikacin do not shareoverlapping binding sites. Fusidic acid binds to elongationfactor G in complex with the ribosome (Turnidge andCollignon 1999), whereas amikacin binds to the 30S ribosome(Wright 2007).
The ciprofloxacin-, ampicillin-, and ciprofloxacin–ampicil-lin-evolved isolates shared a mix of well-documented canon-ical and lesser-known mutations. For example, all threeisolates evolved to ampicillin and ciprofloxacin–ampicillinisolate B had mutations in the pbpA gene, which codes forpenicillin-binding protein 1 (Wada and Watanabe 1998).Ciprofloxacin–ampicillin isolates A and C had mutations inan uncharacterized transport protein (NWMN600), which
0
1
2
AMI AMP CPR DOX ERY FUS
Lineage evolved to
log(
Fol
d IC
90 c
hang
e) Tested against
AMI
AMP
CPR
DOX
ERY
FUS
FIG. 5. Single-drug-evolved isolates tested for cross-resistance to theircorresponding component drug. All single-drug-evolved isolates weretested to their corresponding component drug to test for cross-resis-tance or sensitivity. Each column is an average of four biological repli-cates and represents the gain or loss in WT IC90 value by drugs adaptedto drug A tested against drug B. Error bars reflect the SD of the repli-cates. The isolates tested are listed below the x axis, whereas the drugthey are tested against is given in the legend. Isolates evolved to fusidicacid displayed considerable sensitivity to erythromycin and moderatecross-resistance to amikacin. Isolates evolved to amikacin had strongcross-resistance to fusidic acid.
1180
de Evgrafov et al. . doi:10.1093/molbev/msv006 MBE at R
may have helped provide resistance to ampicillin in the ab-sence of mutations in penicillin-binding proteins (supplemen-tary fig. S1, Supplementary Material online). Correspondingly,all isolates evolved to ciprofloxacin and ciprofloxacin–ampi-cillin had mutations in the parC gene, which codes for DNAtopoisomerase IV subunit A, and is known to confer low-levelresistance to ciprofloxacin (Janoir et al. 1996). The ciproflox-acin-evolved isolates had additional mutations in the gyrAgene, which is responsible for higher levels of quinolone re-sistance (Ferrero et al. 1995). When parC and gyrA mutationsare both present an organism has high-level quinoloneresistance (Janoir et al. 1996; Kaneko et al. 2000) (fig. 3).The deficiency of gyrA gene mutations manifested inthe tolerance of ciprofloxacin by the ciprofloxacin–ampicillin-evolved lineages (supplementary fig. S1,Supplementary Material online). Reduced fitness was notobserved for most of the isolates (supplementary data S1,Supplementary Material online).
All isolates evolved to doxycycline and its correspondingcombinations, with the exception of one, had mutations inthe rpsJ gene, which codes for the 30S ribosomal protein S10.Doxycycline targets the 30S ribosomal subunit and inhibitsthe binding of aminoacyl-transfer RNA (tRNA) to the mRNAribosome complex. Ribosomal protein S10 is involved in thebinding of tRNA to the ribsosome (Yaguchi et al. 1980) andmutations in this gene have previously been shown to conferhigh level tetracycline resistance in Neisseria gonorrhoeae (Huet al. 2005). Doxycycline–ciprofloxacin isolate A was the onlyisolate without an rpsJ gene mutation. This isolate had theleast resistance to doxycycline (supplementary fig. S1,Supplementary Material online) of all the doxycycline com-bination-evolved isolates. Moreover, the overall IC90 im-provement by this isolate was 10� less than other tworeplicate isolates.
It should be noted that a variety of auxiliary mutationswere observed in both the single-drug- and combination-drug-evolved isolates and appear to support the principaltarget mutations. These supplementary mutations were as-sessed and grouped according to function (supplementarydata S2, Supplementary Material online). Instances ofshared auxiliary mutations between the single-drug- andcombination-evolved isolates were limited; however, the nu-merical distribution of these mutations was approximatelyequal among all sequenced isolates. Many of the auxiliarymutations were part of a larger stress response network,which likely participated in or aided resistance. For example,all isolates evolved to ciprofloxacin–ampicillin had mutationsin the relA gene, which initiates the stringent response underenvironmental stress. This controls the production of thealarmone ppGpp, which in turn serves as a regulator of avariety of metabolic pathways and processes and has beenshown to play an essential role in decreased sensitivity topenicillin (Kusser and Ishiguro 1985, 1987; Rodionov andIshiguro 1995; Wu et al. 2010) and quinolones (Viducicet al. 2006). relA mutations were also observed in fusidicacid–amikacin isolate A and erythromycin isolate B.
In spite of the auxiliary mutations observed in the evolvedstrains, mutations associated with resistance to individualdrugs dominated the mutations found in the combination-evolved isolates. Speed of resistance development bycombination-evolved lineages was a function of how thesemutations interacted to cause either cross-resistance orcross-sensitivity. In the case of the doxycycline–ciprofloxacin-and doxycycline–erythromycin-evolved isolates, the muta-tions required for resistance to the constituent drugs resultedin considerable cross-resistance between the single-drug-evolved isolates and culminating in elevated evolvabilityvalues for the combination-evolved isolates. A similar
FIG. 6. Primary target genes affected by resistance adaptation. The most evolved isolates were sequenced and compared with the ancestral WT and themedia adapted lineages to identify mutations resulting from resistance adaptation. Canonical resistance mutations were observed in both the single-drug- and combination-evolved isolates. Mutations associated with resistance to individual drugs dominated the mutations observed in the combi-nation-evolved isolates.
1181
Resistance Evolution Dependent on Collateral Resistance . doi:10.1093/molbev/msv006 MBE at R
situation was observed for the fusidic acid–amikacin-evolvedisolates, where the same single resistance mutation was re-quired for both constituent drugs resulting in cross-resistancebetween the single-drug-evolved isolates. In contrast, adapta-tion to fusidic acid and erythromycin resulted in strong crosssensitivity and was reflected in the reduced evolvability valuesof the combination-evolved isolates. Our findings stress theimportance of collateral effects in limiting resistanceevolution and not drug interactions.
DiscussionWe sought to extend the current scientific paradigm by ex-panding the concentration ranges considered to envelopeconcentrations likely to be encountered during clinical treat-ment. The motivation for this pursuit stems from the factthat treatment failure typically occurs at elevated concentra-tions. We pursued our study using the same drugs, combina-tions, and organism previously employed to develop theexisting model for predicting resistance evolution based ondrug interactions. We hypothesized that at concentrationsabove WT MIC, resistance evolution to drug combinationswould be driven by the constituent drugs and collateral sen-sitivity interactions.
We were unable to reproduce the expected correlationbetween resistance evolution, as measured by evolvability,and drug interactions, as assessed by the fractional inhibitorycombination index, at drug concentrations above WT MIC.We hypothesize that this is due to the fact that drug inter-actions are modulated by resistance evolution. The dynamicnature of drug interactions challenges their use as reliablepredictors of long-term resistance evolution.
Results of our experimental evolution and genome se-quencing work suggest that the evolutionary responses toindividual constituent drugs are better predictors of resis-tance evolution. A drug pair where adaptation to one con-stituent drug confers cross-resistance to the other or whereboth constituent drugs share the same resistance mutationswill undermine the effect of the combination and will likelyhave greater resistance evolution due to cross-resistance. Incontrast, a pair where resistance evolution to one constituentresults in collateral sensitivity to the other will have slower orreduced evolution due to the incompatibility of the individualresistance profiles. Finally, in between these two poles is thecase where resistance to constituent drugs is unrelated/inde-pendent. Resistance to this drug pair is achieved in a mea-sured fashion by individually acquiring mutations for each ofcomponent drugs.
In conclusion, we find that above WT MIC levels, individualconstituent drugs and their associated resistance mutationsare reliable predictors of a combination’s potential resistanceevolution. Mutations associated with resistance to oneconstituent drug of a combination have the power toeither promote or obstruct resistance to another componentin the same combination. We suggest that rather than con-tinuing to focus on drug interactions, further research shouldconsider the mutations that will arise from resistance adap-tation and pursue those combinations with diverging
evolutionary trajectories, as these combinations will likelylimit resistance evolution best.
Materials and Methods
Bacteria and Reagents
A drug sensitive S. aureus strain Newman was adapted to fiveantibiotics: Amikacin sulfate (Sigma), ampicillin sodium salt(Sigma), ciprofloxacin hydrochloride (AppliChem), erythro-mycin (Sigma), fusidic acid sodium salt (Sigma), and doxycy-cline hyclate (TCI) and the following drug pair combinations:fusidic acid–amikacin, fusidic acid–erythromycin, ampicillin–ciprofloxacin, doxycycline–ciprofloxacin, and doxycycline–erythromycin. Drug stock solutions were prepared weekly.All evolution and MIC experiments were performed using amodified Luria broth (LB) media. The salt content was re-duced to 4 g/l instead of 5 g/l.
Evolution of Antibiotic Resistance
A WT IC90 was established for each antibiotic. Drug paircombinations were a 1:1 IC90 mixture of the componentdrugs. WT IC90s were also established for each drug pair.All evolution experiments began one dilution step belowtheir respective IC90 concentration. Evolution experimentsinvolved challenging a WT organism with increasing concen-trations, in steps of the square root of 2, of individual drugs ordrug combinations. All evolution experiments were per-formed in triplicate in a modified Luria–Bertani (LB) brothin microtiter plates. Each experiment included both negativeand positive control wells. The positive control was the inoc-ulating strain in LB media only. Following an 18-h growthperiod at 37 �C, the microtiter plates were measured forOD at wavelength 600 nm (OD600). The value of the exper-imental positive control was used to normalize the evolutiondata. A cut off of 60% inhibition was used to determine thestarting concentration of the next experiment. This concen-tration was referred to as the experimental MIC. The 60%inhibition value was chosen based on pre-experimental workthat found that this value consistently ensured a resistantpopulation was used in subsequent exposure experiments.The replicate with the best growth at the experimentalMIC concentration was used as seed material for the nextexperiment. The selected seed was added to fresh LB mediacontaining the appropriate drug(s) concentration and al-lowed to grow over night. The overnight culture was thenused to inoculate the next challenge experiment. A portion ofthis culture was saved. The challenge process was repeated atotal of five times for each individual drug and drug combi-nation. The same adaptation procedure was used for themedia only evolved populations.
IC90 Determination
Following adaptation, isolates of the adapted populationswere profiled for their individual resistances. IC90 determina-tion was performed according to standardized methods(Andrews 2001). Briefly, lineages from the fifth exposurewere plated on nonselective media and allowed to grow over-night. Four individual isolates were then randomly selected
1182
de Evgrafov et al. . doi:10.1093/molbev/msv006 MBE at R
from each plate and grown in nonselective liquid media for4–5 h before being used to inoculate additional IC90 experi-ments. All single-drug isolates were tested against the agentthey had been adapted to as well as their corresponding drugcombination and matching component drug. Combination-evolved isolates were tested against the combination towhich they had been adapted and the commensurate com-ponent drugs. In both the population and single isolate ex-periments, the inoculum size for each well was approximately104 cells. All IC90 experiments were performed in 96-wellmicrotiter plates in quadruplicate using 2-fold dilutionsteps. Positive, isolate in LB media only, and negative controlswere included in each test. Inoculated plates were placed onan orbital shaker (300 rpm) and incubated at 37 �C for at least16 h. After the allotted growth period, OD600 was read on aBioTek Epoch plate reader.
Calculation of CCD
Using the equation set forth by Lee et al (2011), n is thenumber of generations for each growth step. In our case,there are two growth steps—the resistance experiment andthe test tube pregrowth period prior to each resistance ex-periment. n values were calculated for each evolved lineageand the two growth steps.
We performed growth kinetic experiments that allowed usto calculate a generation time (G in min�1) for each strain.These values were then used to determine the number ofgenerations for each strain in an 8-h period (assumed loggrowth phase) or n.
In the Lee equation, CCD is
XM
I¼1
N0ð2N � 1Þ; ð1Þ
where N0 is the initial number of cells in each well or test tubeduring evolution. We used representative values of N0, reflect-ing each growth condition, for each strain to calculate theCCD for the test tube and resistance experiment periods. Thesubsequent CCD values were multiplied by 5 to reflect thenumber of evolution periods for each growth condition. ACCD value was calculated for each replicate lineage (supple-mentary data S1, Supplementary Material online). The aver-age CCD value in the text comes from adding the two growthconditions together.
Data Analysis
The OD600 data were analyzed using Excel and Prism
(GraphPad Software). Briefly, negative control values were
subtracted from all growth wells yielding dose–response
values. These data were then normalized by the positive con-
trol data and then used to determine the fraction of inhibi-
tion, calculated as: 1� normalized dose response of strain X.
Inhibition data were plotted in Prism and IC90 read from
graph.
Calculation of Evolvability Index
The evolvability index assesses how resistance evolutiontoward a combination compares with individual drug resis-tance evolution. The index is determined by summing a com-bination-evolved strain’s resistance to each of its componentdrugs relative to the resistance development of the corre-sponding single-drug-evolved lineages and then taking an av-erage. Each individual fraction can be used to assess howresistance evolution to an individual component isimpacted as a result of being used in a combination.The evolvability index is calculated as:
Evolvability Index ¼1
n
IC90 A½ �AB
IC90½A�Aþ
IC90½B�AB
IC90½B�B
� �; ð2Þ
where the n is the number of components in a mixture and isused to determine an average value. IC90[A]AB refers to theIC90 of the AB-evolved lineage tested against drug A.
Sequencing
Genomic DNA from our most evolved strains and WT wasisolated using either an UltraClean Microbial DNA IsolationKit (MoBio Laboratories, Inc.) or a modified chloroform/phenol extraction method. Briefly, lysostaphin in conjunctionwith proteinase K was used to disrupt the cell wall. The ex-tracted DNA was sheared into 200-bp fragments using aCovaris E210 and barcoded libraries were constructed forIllumina or IonTorrent sequencing. Illumina sequencing wasperformed by Partners HealthCare Center for PersonalizedGenetic Medicine (Cambridge, Massachusetts) and bySequencing, Informatics and Modeling Group at The NovoNordisk Foundation Center for Biosustainability, TechnicalUniversity of Denmark (Hørsholm, Denmark). IonTorrent se-quencing was performed by DTU Multi-Assay Core (KongensLyngby, Denmark). All reads were aligned to S. aureus subsp.aureus str. Newman (NC_009641.1) using Bowtie2 version2.0.0-b6 with the default options (Langmead and Salzberg2012). An average of 99.6% (minimum 97.5%) of thegenome was covered with an average read coverage of125� 40 (CI95) (supplementary data S2, SupplementaryMaterial online), as determined using BEDTools (Quinlanand Hall 2010). Variant calling for SNPs and INDELs wasdone using SAMTools version 0.1.17 with the –B,-L 1,000options(Li et al. 2009). Only SNPs with a phred score of atleast 30 and where at least 80% of the reads aligned at the sitehad the variant were used. INDELs were verified by aligningconstructed contigs around INDEL sites to the referencegenome (Zerbino and Birney 2008; Li and Durbin 2009).The BioCyc database collection (Karp et al. 2005) was usedto identify and annotate mutation sites.
Supplementary MaterialSupplementary data S1 and S2 and figures S1 and S2 areavailable at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
1183
Resistance Evolution Dependent on Collateral Resistance . doi:10.1093/molbev/msv006 MBE at R
The authors thank Elizabeth Rettedal for discussion andadvice and Gautam Dantas for input on the manuscript.This work was supported by the Danish Free ResearchCouncils for Health and Disease. M.O.A.S. further acknowl-edges support from the Novo Nordisk Foundation, theLundbeck Foundation, and the European Union FP7-HEALTH-2011-single-stage grant agreement 282004, EvoTAR.
ReferencesAndrews J. 2001. Determination of minimum inhibitory concentrations.
J Antimicrob Chemother. 48:5–16.Berenbaum MC. 1978. A method for testing for synergy with any
number of agents. J Infect Dis. 137:122–130.Besier S, Ludwig A, Brade V, Wichelhaus TA. 2003. Molecular analysis of
fusidic acid resistance in Staphylococcus aureus. Mol Microbiol. 47:463–469.
Bollenbach T, Quan S, Chait R, Kishony R. 2009. Nonoptimal microbialresponse to antibiotics underlies suppressive drug interactions. Cell139:707–718.
Borrell S, Teo Y, Giardina F, Streicher EM, Klopper M, Feldmann J, M€ullerB, Victor TC, Gagneux S. 2013. Epistasis between antibiotic resistancemutations drives the evolution of extensively drug-resistant tuber-culosis. Evol Med Public Health. 2013:65–74.
Canu A, Malbruny B, Coquemont M, Davies TA, Appelbaum PC,Leclercq R. 2002. Diversity of ribosomal mutations conferring resis-tance to macrolides, clindamycin, streptogramin, and telithromycinin Streptococcus pneumoniae. Antimicrob Agents Chemother. 46:125–131.
Chait R, Craney A, Kishony R. 2007. Antibiotic interactions that selectagainst resistance. Nature 446:668–671.
Clatworthy AE, Pierson E, Hung DT. 2007. Targeting virulence: a newparadigm for antimicrobial therapy. Nat Chem Biol. 3:541–548.
Cornaglia G. 2009. Fighting infections due to multidrug-resistant Gram-positive pathogens. Clin Microbiol Infect. 15:209–211.
Cottarel G, Wierzbowski J. 2007. Combination drugs, anemerging option for antibacterial therapy. Trends Biotechnol. 25:547–555.
Diner EJ, Hayes CS. 2009. Recombineering reveals a diverse collection ofribosomal proteins L4 and L22 that confer resistance to macrolideantibiotics. J Mol Biol. 386:300–315.
European Committee on Antimicrobial Susceptibility (EUCAST). 2013.Breakpoint tables for interpretation of MICs and zone diameters.3rd ed. The European Committee on Antimicrobial Susceptibility.V€axj€o, Sweden. [cited 2013 Aug 26]. Available from: http://www.eucast.org.
Ferrero L, Cameron B, Crouzet J. 1995. Analysis of gyrA and grlAmutations in stepwise-selected ciprofloxacin-resistant mutantsof Staphylococcus aureus. Antimicrob Agents Chemother. 39:1554–1558.
Freedberg KA, Losina E, Weinstein MC, Paltiel AD, Cohen CJ, Seage GR,Craven DE, Zhang H, Kimmel AD, Goldie SJ. 2013. The cost effec-tiveness of combination antiretroviral therapy for HIV disease. NEngl J Med. 344:824–831.
Gabashvili IS, Gregory ST, Valle M, Grassucci R, Worbs M, Wahl MC,Dahlberg AE, Frank J. 2001. The polypeptide tunnel system in theribosome and its gating in erythromycin resistance mutants of L4and L22. Mol Cell. 8:181–188.
Gilliam BL, Chan-Tack KM, Qaqish RB, Rode RA, Fantry LE, Redfield RR.2006. Successful treatment with atazanavir and lopinavir/ritonavircombination therapy in protease inhibitor-susceptible and proteaseinhibitor-resistant HIV-infected patients. AIDS Patient Care STDs 20:745–759.
Gregory ST, Dahlberg AE. 1999. Erythromycin resistance mutations inribosomal proteins L22 and L4 perturb the higher order structure of23 S ribosomal RNA. J Mol Biol. 289:827–834.
Hall AR, MacLean RC. 2011. Epistasis buffers the fitness effects of rifam-picin- resistance mutations in Pseudomonas aeruginosa. Evolution 65:2370–2379.
Hegreness M, Shoresh N, Damian D, Hartl D, Kishony R. 2008.Accelerated evolution of resistance in multidrug environments.Proc Natl Acad Sci U S A. 105:13977–13981.
Hu M, Nandi S, Davies C, Nicholas RA. 2005. High-level chromosomallymediated tetracycline resistance in Neisseria gonorrhoeae resultsfrom a point mutation in the rpsJ gene encoding ribosomal proteinS10 in combination with the mtrR and penB resistance determi-nants. Antimicrob Agents Chemother. 49:4327–4334.
Huang T-S, Kunin CM, Yan B-S, Chen Y-S, Lee SS-J, Syu W. 2012.Susceptibility of Mycobacterium tuberculosis to sulfamethoxazole,trimethoprim and their combination over a 12 year period inTaiwan. J Antimicrob Chemother. 67:633–637.
Imamovic L, Sommer MOA. 2013. Use of collateral sensitivity networksto design drug cycling protocols that avoid resistance development.Sci Transl Med. 5:204ra132.
Janoir C, Zeller V, Kitzis M-D, Moreau NJ, Gutmann L. 1996. High-levelfluoroquinolone resistance in Streptococcus pneumoniae requiresmutations in parC and gyrA. Antimicrob Agents Chemother. 40:2760–2764.
Kaneko A, Sasaki J, Shimadzu M, Kanayama A, Saika T, Kobayashi I. 2000.Comparison of gyrA and parC mutations and resistancelevels among fluoroquinolone-resistant isolates and laboratory-derived mutants of oral streptococci. J Antimicrob Chemother. 45:771–775.
Karp PD, Ouzounis CA, Moore-Kochlacs C, Goldovsky L, Kaipa P, Ahr�enD, Tsoka S, Darzentas N, Kunin V, L�opez-Bigas N. 2005. Expansion ofthe BioCyc collection of pathway/genome databases to 160 ge-nomes. Nucleic Acids Res. 33:6083–6089.
Kusser W, Ishiguro EE. 1985. Involvement of the relA gene in theautolysis of Escherichia coli induced by inhibitors of peptidoglycanbiosynthesis. J Bacteriol. 164:861–865.
Kusser W, Ishiguro EE. 1987. Suppression of mutations conferring pen-icillin tolerance by interference with the stringent control mecha-nism of Escherichia coli. J Bacteriol. 169:4396–4398.
Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie2. Nat Methods. 9:357–359.
L�az�ar V, Nagy I, Spohn R, Cs€orgo00 B, Gy€orkei �A, Nyerges �A, Horv�ath B,V€or€os A, Busa-Fekete R, Hrtyan M, et al. 2014. Genome-wide analysiscaptures the determinants of the antibiotic cross-resistance interac-tion network. Nat Commun. 5:1–12.
L�az�ar V, Pal Singh G, Spohn R, Nagy I, Horv�ath B, Hrtyan M, Busa-FeketeR, Bogos B, M�ehi O, Cs€orgo00 B, et al. 2013. Bacterial evolution ofantibiotic hypersensitivity. Mol Syst Biol. 9.
Lee D-H, Feist AM, Barrett CL, Palsson BØ. 2011. Cumulative number ofcell divisions as a meaningful timescale for adaptive laboratory evo-lution of Escherichia coli. PLoS One 6:e26172.
Lennox JL, DeJesus E, Lazzarin A, Pollard RB, Ramalho Madruga JV, BergerDS, Zhao J, Xu X, Williams-Diaz A, Rodgers AJ, et al. 2009. Safety andefficacy of raltegravir-based versus efavirenz-based combinationtherapy in treatment-naive patients with HIV-1 infection: amulticentre, double-blind randomised controlled trial. Lancet 374:796–806.
Li H, Durbin R. 2009. Fast and accurate short read alignment withBurrows–Wheeler transform. Bioinformatics 25:1754–1760.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G,Abecasis G, Durbin R and 1000 Genome Project Data ProcessingSubgroup. 2009. The Sequence Alignment/Map format andSAMtools. Bioinformatics 25:2078–2079.
Michel JB, Yeh PJ, Chait R, Moellering RC, Kishony R. 2008. Drug inter-actions modulate the potential for evolution of resistance. Proc NatlAcad Sci U S A. 105:14918.
Munck C, Gumpert HK, Wallin AIN, Wang HH, Sommer MOA. 2014.Prediction of resistance development against drug combinations
1184
de Evgrafov et al. . doi:10.1093/molbev/msv006 MBE at R
by collateral responses to component drugs. Sci Transl Med. 6:262ra156.
Norstr€om T, Lannergard J, Hughes D. 2007. Genetic and phenotypicidentification of fusidic acid-resistant mutants with the small-colony-variant phenotype in Staphylococcus aureus. AntimicrobAgents Chemother. 51:4438–4446.
Oz T, Guvenek A, Yildiz S, Karaboga E, Tamer YT, Mumcuyan N, OzanVB, Senturk GH, Cokol M, Yeh P, et al. 2014. Strength of selectionpressure is an important parameter contributing to the complexityof antibiotic resistance evolution. Mol Biol Evol. 31:2387–2401.
Palmer AC, Kishony R. 2013. Understanding, predicting and manipulat-ing the genotypic evolution of antibiotic resistance. Nat Rev Genet.14:243–248.
Pena-Miller R, Laehnemann D, Jansen G. 2013. When the most potentcombination of antibiotics selects for the greatest bacterial load: thesmile-frown transition. PLoS Biol. 11:e1001540.
Prunier A-LA, Malbruny BB, Tand�e DD, Picard BB, Leclercq RR. 2002.Clinical isolates of Staphylococcus aureus with ribosomal mutationsconferring resistance to macrolides. Antimicrob Agents Chemother.46:3054–3056.
Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities forcomparing genomic features. Bioinformatics 26:841–842.
Read AF, Day T, Huijben S. 2011. The evolution of drug resistance andthe curious orthodoxy of aggressive chemotherapy. Proc Natl AcadSci U S A. 108:10871–10877.
Rodionov DG, Ishiguro EE. 1995. Direct correlation between overproduc-tion of guanosine 30,50-bispyrophosphate (ppGpp) and penicillintolerance in Escherichia coli. J Bacteriol. 177:4224–4229.
Szybalski W. 1954. Genetic studies on microbial cross resistance to toxicagents: IV. Cross resistance of Bacillus megaterium to forty-four mi-crobial drugs1. Appl Microbiol. 2:57.
Tait-Kamradt A, Davies T, Appelbaum PC, Depardieu F, Courvalin P,Petitpas J, Wondrack L, Walker A, Jacobs MR, Sutcliffe J. 2000. Twonew mechanisms of macrolide resistance in clinical strains ofStreptococcus pneumoniae from Eastern Europe and NorthAmerica. Antimicrob Agents Chemother. 44:3395–3401.
Thaker MN, Wang W, Spanogiannopoulos P, Waglechner N, King AM,Medina R, Wright GD. 2013. Identifying producers of antibacterialcompounds by screening for antibiotic resistance. Nat Biotechnol. 31:922–927.
Toprak E, Veres A, Michel J-B, Chait R, Hartl DL, Kishony R. 2011.Evolutionary paths to antibiotic resistance under dynamically sus-tained drug selection. Nat Genet. 44:101–105.
Torella JP, Chait R, Kishony R. 2010. Optimal drug synergy in antimicro-bial treatments. PLoS Comput Biol. 6:e1000796.
Trindade S, Sousa A, Xavier KB, Dionisio F, Ferreira MG, Gordo I. 2009.Positive epistasis drives the acquisition of multidrug resistance. PLoSGenet. 5:e1000578.
Turnidge J, Collignon P. 1999. Resistance to fusidic acid. Int J AntimicrobAgents. 12:S35–S44.
Viducic D, Ono T, Murakami K, Susilowati H, Kayama S, Hirota K,Miyake Y. 2006. Functional analysis of spoT, relA and dksA geneson quinolone tolerance in Pseudomonas aeruginosa under nongrow-ing condition. Microbiol Immunol. 50:349–357.
Vilcheze C, Jacobs WR. 2012. The combination of sulfamethoxazole,trimethoprim, and isoniazid or rifampin is bactericidal and preventsthe emergence of drug resistance in Mycobacterium tuberculosis.Antimicrob Agents Chemother. 56:5142–5148.
Wada A, Watanabe H. 1998. Penicillin-binding protein 1 ofStaphylococcus aureus is essential for growth. J Bacteriol. 180:2759–2765.
Woodford N, Livermore DM. 2009. Infections caused by Gram-positivebacteria: a review of the global challenge. J Infect. 59:S4–S16.
World Health Organization. 2012. The evolving threat of antimicrobialresistance: options for action. Geneva: World Health Organization.[cited 2013 Aug 26]. Available from: http://whqlibdoc.who.int/publications/2012/9789241503181_eng.pdf.
Wright G. 2007. Mechanisms of aminoglycoside antibiotic resistance. In:Richard G Wax, Harry Taber, Abigail A Salyers, Kim Lewis, editors.Bacterial resistance to antimicrobials, 2nd ed. Boca Raton: CRC Press.p. 71–101.
Wu J, Long Q, Xie J. 2010. (p) ppGpp and drug resistance. J Cell Physiol.224:300–304.
Yaguchi M, Roy C, Wittmann HG. 1980. The primary structure of pro-tein S10 from the small ribosomal subunit of Escherichia coli. FEBSLett. 121:113–116.
Yeh P, Tschumi AI, Kishony R. 2006. Functional classification of drugs byproperties of their pairwise interactions. Nat Genet. 38:489–494.
Yeh PJ, Hegreness MJ, Aiden AP, Kishony R. 2009. Drug interactions andthe evolution of antibiotic resistance. Nat Rev Microbiol. 7:460–466.
Zaman S, Fitzpatrick M, Lindahl L, Zengel J. 2007. Novel mutations inribosomal proteins L4 and L22 that confer erythromycin resistancein Escherichia coli. Mol Microbiol. 66:1039–1050.
Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short readassembly using de Bruijn graphs. Genome Res. 18:821–829.
1185
Resistance Evolution Dependent on Collateral Resistance . doi:10.1093/molbev/msv006 MBE at R