Page 1
ORIGINAL PAPER
Tumor suppressor Ing1b facilitates DNA repair and preventsoxidative stress induced cell death
Anand Rotte • Gang Li • Madhuri Bhandaru
Published online: 16 November 2013
� Springer Science+Business Media New York 2013
Abstract Inhibitor of growth (ING) family of proteins
are known to coordinate with histone acetyltransferases
and regulate the key events of cell cycle and DNA repair.
Previous work from our lab showed that Ing1b regulated
the nucleotide excision repair by facilitating histone acet-
ylation and subsequent chromatin relaxation. Further, it
was also shown that Ing1b protected the cells from geno-
mic instability induced cell death by promoting ubiquiti-
nation of proliferating cell nuclear antigen (PCNA). In the
present study we explored the role of Ing1b in the repair of
oxidized DNA and prevention of oxidative stress induced
genotoxic cell death. Using HCT116 cells we show that
Ing1b protein expression is induced by treatment with
H2O2. Ing1b lacking cells showed decreased ability to
repair the oxidized DNA. PCNA monoubiquitination, a
critical event of DNA repair was blunted in Ing1b knock
down cells and augmented in Ing1b over expressing cells.
Moreover, oxidative stress induced cell death was higher in
cells lacking Ing1b whereas it was lower in Ing1b over
expressing cells. Finally we show that inhibition of histone
deacetylases, rescued the Ing1b knock down cells from
cytotoxic effects of H2O2 treatment.
Keywords Ing1b � PCNA ubiquitination � DNA
repair � Histone acetylation � Cell survival
Introduction
Oxidative stress has been implicated in wide range of dis-
orders including cancer, diabetes and neurodegeneration [1–
5]. It is defined as a condition in which there is an uncon-
trolled increase in the cellular levels of reactive oxygen
species (ROS). The source of oxidative stress can be both
exogenous like chemotherapeutic agents or UV radiation
and endogenous, as a byproduct of mitochondrial energy
metabolism. The outcome of increased cellular ROS, is
oxidation of the cell contents which include lipids, proteins
and DNA. Among the various types of oxidative modifica-
tions to DNA, 7,8-dihydro-8-oxo-guanine (8-oxoG) repre-
sents the most abundant lesion [6]. DNA lesions due to
oxidation are promptly repaired by a specialized pathway
called as ‘Base Excision Repair’ where in the oxidized base
is cleaved from the DNA strand and replaced due to the
coordinated activity of DNA glycosylase, apurinic/apyrim-
idinic endonuclease (APE1), poly(ADP-ribose) polymerase-
1 (PARP), X-ray repair cross complementing protein1
(XRCC1), proliferating cell nuclear antigen (PCNA), DNA
polymerases, and other repair factors [3, 7, 8]. The regulation
of DNA damage check point activity by oxidative stress is
not clearly understood though recent evidences suggest the
involvement of proteins regulating the check point during
replication blockade [9, 10]. The inhibitor of growth (ING)
proteins are known to regulate various biological processes
including DNA repair, cell cycle, apoptosis, and senescence
[11–20]. ING proteins are known to be components of var-
ious histone acetyltransferases and are supposed to carry out
at least part of their functions through chromatin remodeling
[21–23]. Previous work from our group showed that Ing1b
facilitated nucleotide excision repair by promoting chro-
matin accessibility to xeroderma pigmentosum, comple-
mentation group A (XPA) [14], and our previous studies also
A. Rotte (&) � G. Li � M. Bhandaru
Department of Dermatology and Skin Science, University of
British Columbia, Research Pavilion, 828 West, 10th Avenue,
Vancouver, BC V5Z 1L8, Canada
e-mail: [email protected]
123
Apoptosis (2014) 19:518–526
DOI 10.1007/s10495-013-0940-5
Page 2
showed the indispensable role of Ing1b in maintenance of
genomic stability after UV induced replication stress [23].
Cells lacking Ing1b were found to be sensitive to UV
induced stress with comparatively higher incidence of
chromatid breaks, and higher percentage of cell death [23].
Recent work by Ceruti et al., showed the significance of
Ing1b in the repair of DNA damaged due to oxidation, using
cultured cell lines and mouse embryonic fibroblasts [12].
H2O2 treatment was shown to induce Ing1b mRNA levels
and cells lacking Ing1b were reported to be more sensitive to
H2O2 induced DNA damage as seen by higher levels of c-
H2AX [12]. However, the authors did not illustrate the
possible role of Ing1b in the oxidative stress induced PCNA-
monoubiquitination. Further, the authors also did not illus-
trate the effect of Ing1b on the genotoxic stress caused by
H2O2 treatment. The present study was therefore undertaken
to demonstrate the role of Ing1b in oxidative stress induced
PCNA-monoubiquitination and repair of oxidized DNA, and
in preventing the oxidative stress induced cell death.
Materials and methods
Cell culture, antibodies, chemicals and H2O2 treatment
HCT116 cells were cultured in Dulbecco’s modified Eagle
media (DMEM) (Invitrogen, Burlington, ON, Canada)
supplemented with 10 % fetal bovine serum (Invitrogen),
100 U/ml penicillin and 100 mg/ml streptomycin (Invit-
rogen) in 5 % CO2 humidified atmosphere at 37 �C. Anti-
actin and anti-Flag antibodies were purchased from Sigma-
Aldrich (St Louis, MO, USA); and anti-PCNA from Mil-
lipore (Billerica, MA, USA); ING1b, and ORC2 from
Santa Cruz Biotechnology (Santa Cruz, CA, USA) and
mouse anti-Flag from Applied Biological Materials
(Richmond, BC, Canada). To induce oxidative stress, H2O2
was added to the culture medium of the cells at indicated
concentrations. 30 min after addition of H2O2, medium
was removed and cells were washed once with 19 PBS,
and fresh ‘complete’ medium was added to the cells.
Expression plasmid, siRNA transfections
Expression plasmids were transfected into HCT116 cells by
Effectene Transfection Reagent (Qiagen, Mississauga, ON,
Canada) according to the manufacturer’s instruction. siRNAs
were synthesized by Qiagen. ING1b siRNA sequences as
follows: 50-acccacgtactgtctgtgcaa-30. siRNA was transfected
to cells by siLenFect Lipid reagent (Bio-Rad, Mississauga,
ON, Canada) according to manufacturer’s instruction. Assays
were performed 48 h after transfection.
Subcellular fractionation
Fractionation of chromatin bound and unbound fractions
were described previously [14, 24]. Briefly, cytoplasmic
and nucleoplasmic proteins were isolated by cytoskeletal
buffer (CSK) (100 mM NaCl, 300 mM sucrose, 3 mM
MgCl2, 10 mM PIPES pH 6.8, 1 mM EGTA, 0.2 % Triton
X-100) with protease inhibitors for 15 min on ice. After
centrifugation at 900g for 5 min at 4 �C, chromatin bound
proteins in the pellet were resuspended in modified RIPA
buffer (150 mM NaCl, 50 mM Tris–HCl, pH 7.4, 1 mM
EDTA, 0.1 % NP-40, 0.25 % sodium dodecyl sulphate)
and sonicated.
Western blotting
Cells were harvested and washed with PBS thrice. Whole-
cell proteins were extracted and protein concentration was
determined by protein assay (Bio-Rad), western blot ana-
lysis was performed as described previously [25]. The
following antibodies were used for western blot: Ing1b,
ORC2, LAMP2 (1:250; Santa Cruz, CA, USA), PCNA
(1:2000 Billerica, MA, USA), Flag (1:1000, Applied Bio-
logical Materials, Richmond, BC, Canada), and actin
(1:5000; Immunechem Pharmaceuticals, Burnaby, BC,
Canada). Monoubiquitinated PCNA was detected by a
higher molecular weight band (*8–10 kDa) using PCNA
antibody. Infrared IR dye-labelled secondary antibody was
applied to the blot for 1 h at room temperature and then
signals were detected with Odyssey Infrared Imaging
System (LI-COR Biosciences, Lincoln, NE).
Cell cycle analysis
Cell cycle analysis was performed in HCT116 cells using a
protocol previously described [25]. Briefly, cells were
collected by trypsinization and pelleted by centrifugation at
5009g for 5. After overnight fixation, in 70 % ethanol at
4 �C, cell pellets were washed with 19 PBS and then
resuspended in 0.5 ml of FACS buffer (19 PBS with
0.5 mM EDTA and 0.5 % BSA) containing 25 lg/ml of
RNase A and 50 lg/ml of propidium iodide (PI) (Sigma).
After incubating the samples in the dark at room temper-
ature for 15 min, samples were analyzed by EPICS XL-
MCL flow cytometer (Beckman Coulter, Miami, FL) to
determine the percentage of subdiploid DNA. Cells in sub-
G1 phase were regarded as apoptotic cells.
Host-cell-reactivation (HCR) assay
The use of HCR assay to measure the DNA repair capacity
(DRC) has been previously described [26, 27]. The pGL3
control luciferase plasmid (Firefly) was oxidatively
Apoptosis (2014) 19:518–526 519
123
Page 3
damaged in vitro by dilution to 50 mg/ml and exposure to
the indicated concentration of H2O2 (v/v) at room tem-
perature for 1 h. Undamaged control plasmids were treated
with the vehicle solutions without exposure to the dam-
aging agents. After all treatments, the damaged or
undamaged DNA was purified by ethanol precipitation, and
resuspended in autoclaved water. pRL control luciferase
plasmid (Renilla) was used as an internal control for
transfection. To measure the DRC, HCT116 cells were first
transfected with control or Ing1b siRNA and then with
luciferase plasmids as per the standard transfection proce-
dures. 48 h after transfection, luciferase activity was
measured using dual-luciferase reporter assay kit (Pro-
mega, Madison, WI, USA). DRC (%) was calculated as the
ratio of the damaged plasmid luciferase activity to the
undamaged plasmid luciferase activity, multiplied by 100.
Statistics
All data were tested for significance using Student’s
unpaired two-tailed t test and only results with p \ 0.05
were considered statistically significant.
Results
Ing1b protein expression is induced by H2O2 treatment
Previously it was shown that DNA damage due to ultra-
violet radiation (UV-B) induced the expression of Ing1b at
the protein level [28, 29]. UV-B radiation is known to
generate reactive oxygen species (ROS) in the cell and
thereby cause oxidative damage to DNA [30]. H2O2
treatment was recently shown to induce Ing1b mRNA
levels in a variety of cell lines but the authors reported a
disconnection between mRNA and protein levels as there
was no change in Ing1b protein upon H2O2 treatment [12].
We hypothesized that induction of oxidative stress in the
cells with exogenous H2O2 treatment, could induce Ing1b
protein expression during the recovery phase. To test our
hypothesis, we treated the HCT116 cells with a previously
reported concentration of H2O2 (100 lM) for 30 min and
followed the Ing1b protein expression in recovering cells
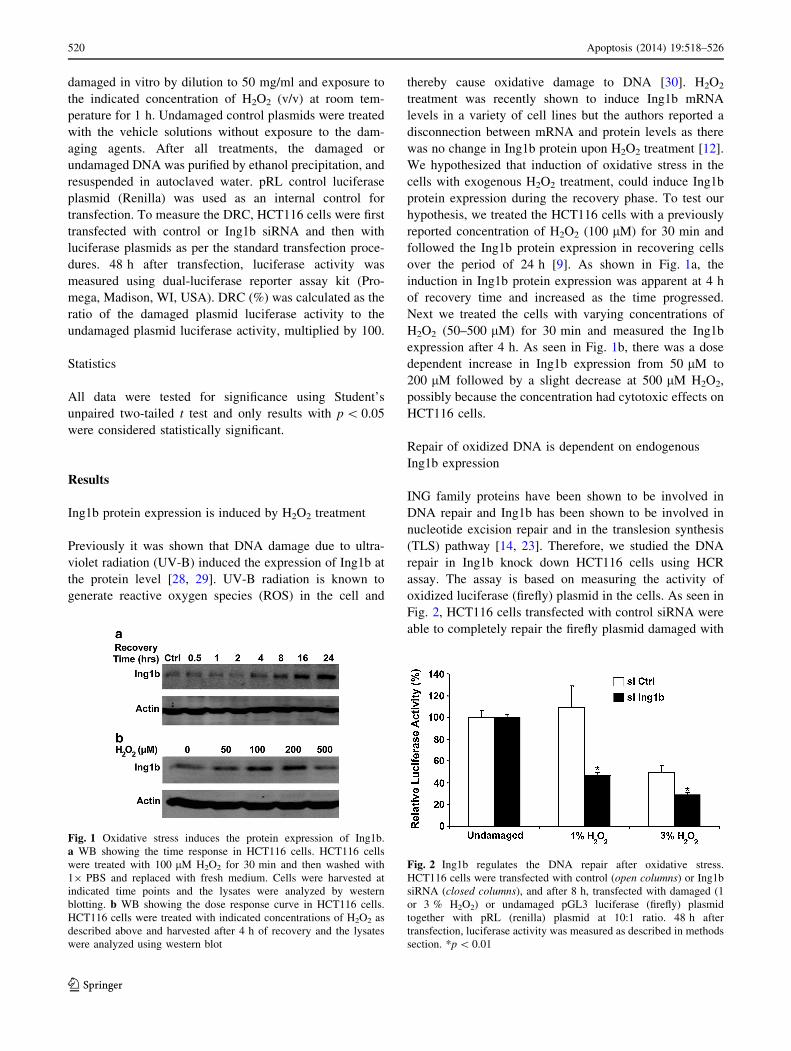
over the period of 24 h [9]. As shown in Fig. 1a, the
induction in Ing1b protein expression was apparent at 4 h
of recovery time and increased as the time progressed.
Next we treated the cells with varying concentrations of
H2O2 (50–500 lM) for 30 min and measured the Ing1b
expression after 4 h. As seen in Fig. 1b, there was a dose
dependent increase in Ing1b expression from 50 lM to
200 lM followed by a slight decrease at 500 lM H2O2,
possibly because the concentration had cytotoxic effects on
HCT116 cells.
Repair of oxidized DNA is dependent on endogenous
Ing1b expression
ING family proteins have been shown to be involved in
DNA repair and Ing1b has been shown to be involved in
nucleotide excision repair and in the translesion synthesis
(TLS) pathway [14, 23]. Therefore, we studied the DNA
repair in Ing1b knock down HCT116 cells using HCR
assay. The assay is based on measuring the activity of
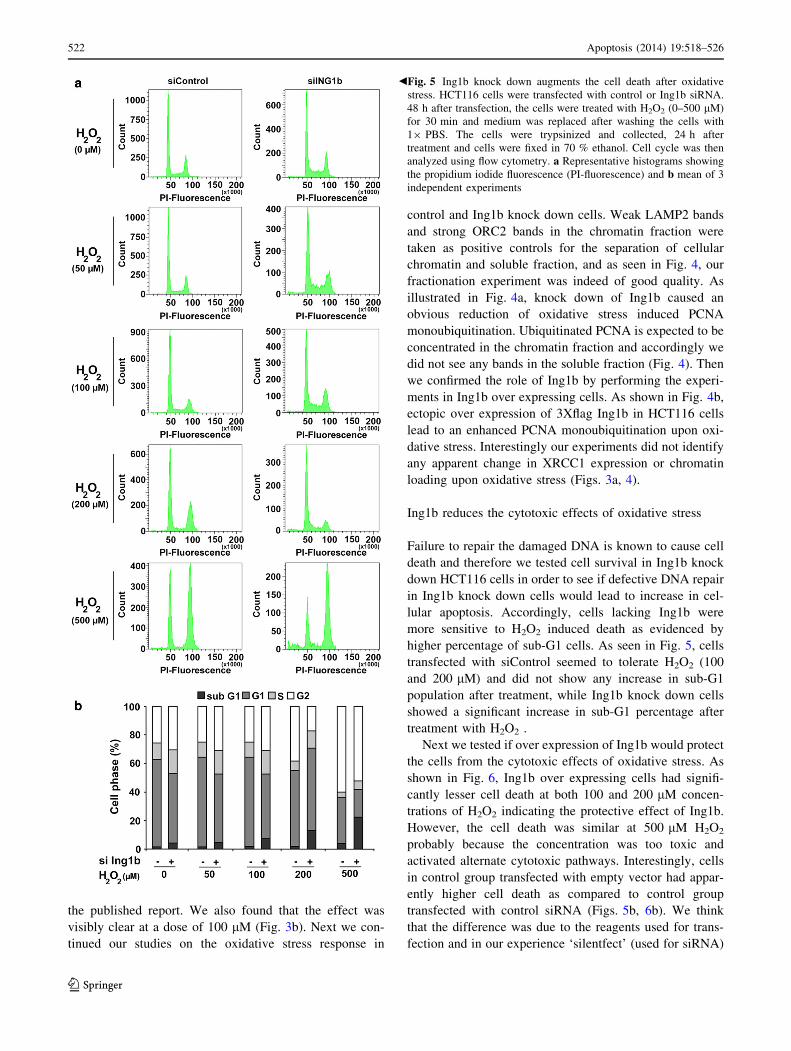
oxidized luciferase (firefly) plasmid in the cells. As seen in
Fig. 2, HCT116 cells transfected with control siRNA were
able to completely repair the firefly plasmid damaged with
Fig. 1 Oxidative stress induces the protein expression of Ing1b.
a WB showing the time response in HCT116 cells. HCT116 cells
were treated with 100 lM H2O2 for 30 min and then washed with
19 PBS and replaced with fresh medium. Cells were harvested at
indicated time points and the lysates were analyzed by western
blotting. b WB showing the dose response curve in HCT116 cells.
HCT116 cells were treated with indicated concentrations of H2O2 as
described above and harvested after 4 h of recovery and the lysates
were analyzed using western blot
Fig. 2 Ing1b regulates the DNA repair after oxidative stress.
HCT116 cells were transfected with control (open columns) or Ing1b
siRNA (closed columns), and after 8 h, transfected with damaged (1
or 3 % H2O2) or undamaged pGL3 luciferase (firefly) plasmid
together with pRL (renilla) plasmid at 10:1 ratio. 48 h after
transfection, luciferase activity was measured as described in methods
section. *p \ 0.01
520 Apoptosis (2014) 19:518–526
123
Page 4
1 % H2O2 and partly repair the plasmid damaged with 3 %
H2O2, where as the repair in cells transfected with Ing1b
siRNA was significantly blunted.
Ing1b regulates PCNA-ubiquitination upon oxidative
stress
Monoubiquitination of PCNA at K164, a known hallmark
for lesion bypass after UV induced replicative blockade,
has been recently shown to be also involved in DNA repair
after oxidative stress [9, 24]. Previous studies on PCNA-
ubiquitination upon replicative stress, have demonstrated a
regulatory role of Ing1b in the process [23]. We asked if
Ing1b had a similar regulatory role in PCNA ubiquitination
upon oxidative DNA damage. Zlatanou et al., in their
report on ‘oxidative stress induced PCNA monoubiquiti-
nation and its significance’, showed that treatment of cells
with H2O2 for 20 min followed by washing once with PBS
and replacement of medium induced a transient ubiquiti-
nation of PCNA. We reproduced the results from the study
by Zlatanou et al., in our cell line and in our laboratory
conditions [9]. As shown in Fig. 3a, H2O2 treatment led to
a transient ubiquitination of PCNA, a result consistent with
Fig. 3 a Time course of PCNA monoubiquitination. HCT116 cells were
treated with 100 lM H2O2 for 30 min and medium was replaced after
washing thecellswith19 PBS.Cellswere harvestedat indicated timepoints
and the soluble and chromatin fractions were separated as described in
‘‘methods’’ section. The proteins were analyzed using western blotting.
b Concentration response of PCNA ubiquitination. HCT116 cells were
treated as described above with indicated concentrations of H2O2 for 30 min
and cells were immediately harvested, and the proteins in chromatin fraction
were analyzed using western blotting. We do not know the origin of the
bands slightly below the monoubiquitinated PCNA band, which were of
relatively low intensity (indicated by asterisk), but we consider them related
to PCNA
Fig. 4 Oxidative stress induced ubiquitination of PCNA is dependent on
Ing1b. a Decreased PCNA ubiquitination in Ing1b knock down cells.
HCT116 cells were transfected with control or Ing1b siRNA. 48 h after
transfection, the cells were treated with 100 lM H2O2 for 30 min and the
cells were harvested at indicated time points. The whole cell extract (WCE),
soluble and chromatin fractions were isolated and analyzed by western
blotting. Asterisk-occasionally (but not always) we observed a band below
the actual Ing1b band, which we think was an unknown protein which cross
reacts with the Ing1b antibody we used. b PCNA fractionation is augmented
in Ing1b over expressing cells. HCT116 cells were transfected with empty
vector or 3XFlag Ing1b plasmid. 24 h after transfection, the cells were
treated with H2O2 as described above and analyzed by western blotting
Apoptosis (2014) 19:518–526 521
123
Page 5
the published report. We also found that the effect was
visibly clear at a dose of 100 lM (Fig. 3b). Next we con-
tinued our studies on the oxidative stress response in
control and Ing1b knock down cells. Weak LAMP2 bands
and strong ORC2 bands in the chromatin fraction were
taken as positive controls for the separation of cellular
chromatin and soluble fraction, and as seen in Fig. 4, our
fractionation experiment was indeed of good quality. As
illustrated in Fig. 4a, knock down of Ing1b caused an
obvious reduction of oxidative stress induced PCNA
monoubiquitination. Ubiquitinated PCNA is expected to be
concentrated in the chromatin fraction and accordingly we
did not see any bands in the soluble fraction (Fig. 4). Then
we confirmed the role of Ing1b by performing the experi-
ments in Ing1b over expressing cells. As shown in Fig. 4b,
ectopic over expression of 3Xflag Ing1b in HCT116 cells
lead to an enhanced PCNA monoubiquitination upon oxi-
dative stress. Interestingly our experiments did not identify
any apparent change in XRCC1 expression or chromatin
loading upon oxidative stress (Figs. 3a, 4).
Ing1b reduces the cytotoxic effects of oxidative stress
Failure to repair the damaged DNA is known to cause cell
death and therefore we tested cell survival in Ing1b knock
down HCT116 cells in order to see if defective DNA repair
in Ing1b knock down cells would lead to increase in cel-
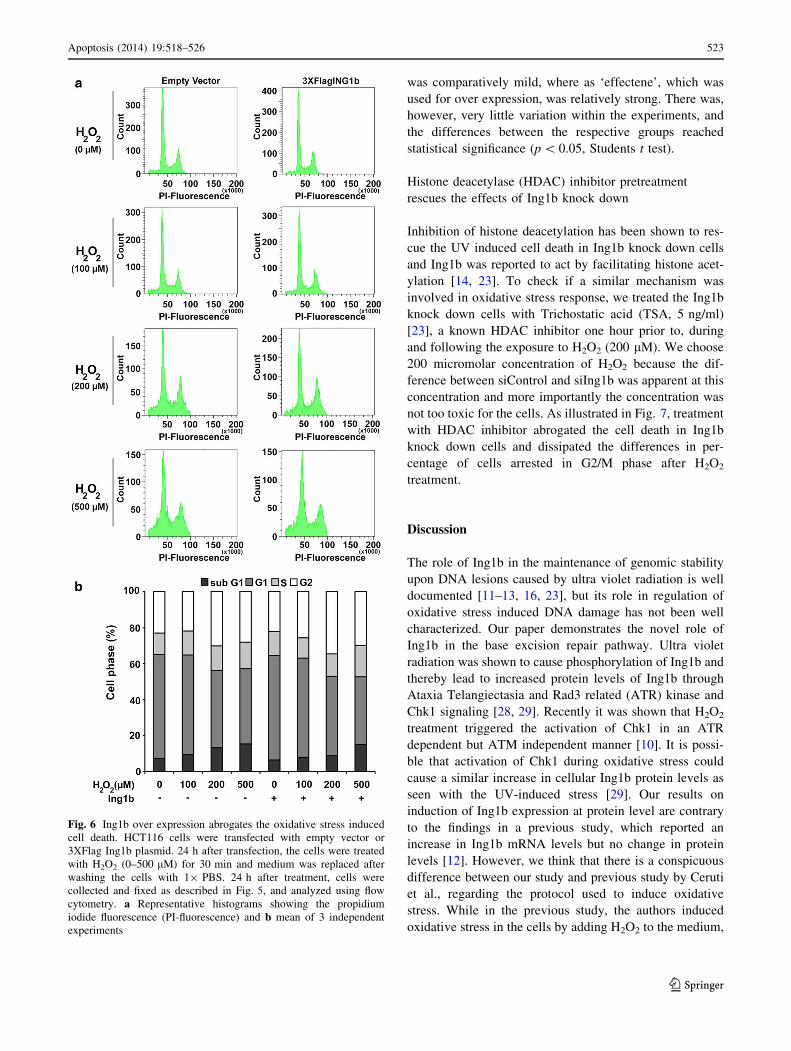
lular apoptosis. Accordingly, cells lacking Ing1b were
more sensitive to H2O2 induced death as evidenced by
higher percentage of sub-G1 cells. As seen in Fig. 5, cells
transfected with siControl seemed to tolerate H2O2 (100
and 200 lM) and did not show any increase in sub-G1
population after treatment, while Ing1b knock down cells
showed a significant increase in sub-G1 percentage after
treatment with H2O2 .
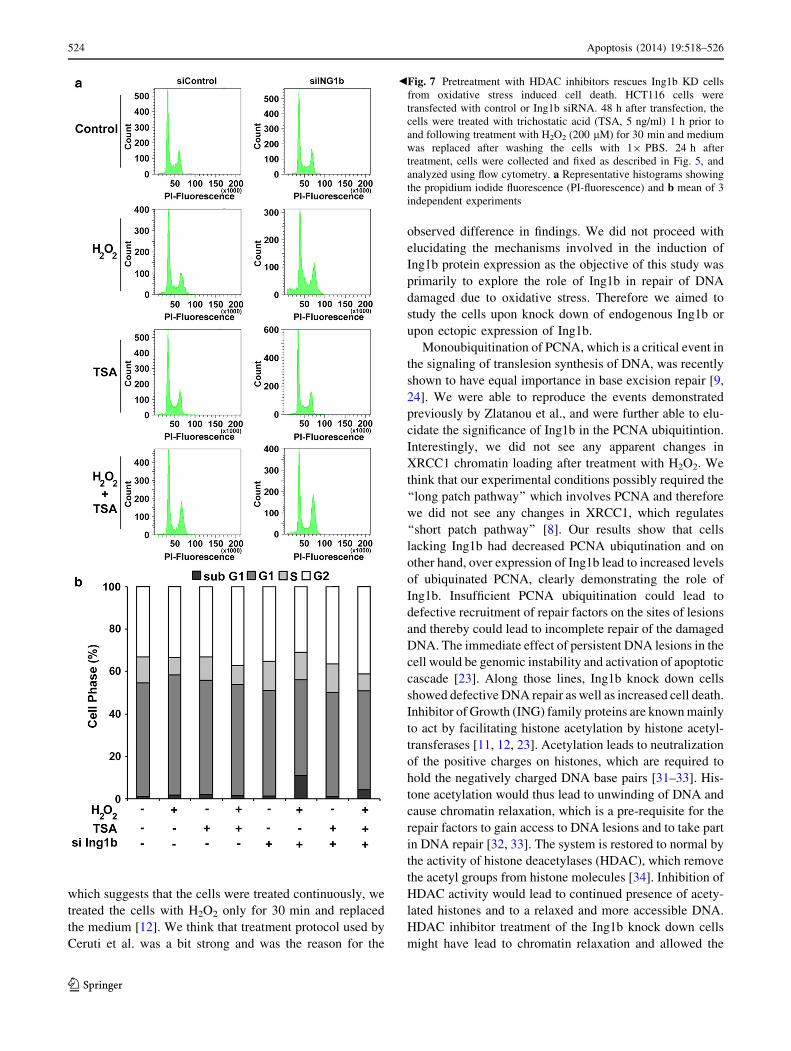
Next we tested if over expression of Ing1b would protect
the cells from the cytotoxic effects of oxidative stress. As
shown in Fig. 6, Ing1b over expressing cells had signifi-
cantly lesser cell death at both 100 and 200 lM concen-
trations of H2O2 indicating the protective effect of Ing1b.
However, the cell death was similar at 500 lM H2O2
probably because the concentration was too toxic and
activated alternate cytotoxic pathways. Interestingly, cells
in control group transfected with empty vector had appar-
ently higher cell death as compared to control group
transfected with control siRNA (Figs. 5b, 6b). We think
that the difference was due to the reagents used for trans-
fection and in our experience ‘silentfect’ (used for siRNA)
Fig. 5 Ing1b knock down augments the cell death after oxidative
stress. HCT116 cells were transfected with control or Ing1b siRNA.
48 h after transfection, the cells were treated with H2O2 (0–500 lM)
for 30 min and medium was replaced after washing the cells with
19 PBS. The cells were trypsinized and collected, 24 h after
treatment and cells were fixed in 70 % ethanol. Cell cycle was then
analyzed using flow cytometry. a Representative histograms showing
the propidium iodide fluorescence (PI-fluorescence) and b mean of 3
independent experiments
b
522 Apoptosis (2014) 19:518–526
123
Page 6
was comparatively mild, where as ‘effectene’, which was
used for over expression, was relatively strong. There was,
however, very little variation within the experiments, and
the differences between the respective groups reached
statistical significance (p \ 0.05, Students t test).
Histone deacetylase (HDAC) inhibitor pretreatment
rescues the effects of Ing1b knock down
Inhibition of histone deacetylation has been shown to res-
cue the UV induced cell death in Ing1b knock down cells
and Ing1b was reported to act by facilitating histone acet-
ylation [14, 23]. To check if a similar mechanism was
involved in oxidative stress response, we treated the Ing1b
knock down cells with Trichostatic acid (TSA, 5 ng/ml)
[23], a known HDAC inhibitor one hour prior to, during
and following the exposure to H2O2 (200 lM). We choose
200 micromolar concentration of H2O2 because the dif-
ference between siControl and siIng1b was apparent at this
concentration and more importantly the concentration was
not too toxic for the cells. As illustrated in Fig. 7, treatment
with HDAC inhibitor abrogated the cell death in Ing1b
knock down cells and dissipated the differences in per-
centage of cells arrested in G2/M phase after H2O2
treatment.
Discussion
The role of Ing1b in the maintenance of genomic stability
upon DNA lesions caused by ultra violet radiation is well
documented [11–13, 16, 23], but its role in regulation of
oxidative stress induced DNA damage has not been well
characterized. Our paper demonstrates the novel role of
Ing1b in the base excision repair pathway. Ultra violet
radiation was shown to cause phosphorylation of Ing1b and
thereby lead to increased protein levels of Ing1b through
Ataxia Telangiectasia and Rad3 related (ATR) kinase and
Chk1 signaling [28, 29]. Recently it was shown that H2O2
treatment triggered the activation of Chk1 in an ATR
dependent but ATM independent manner [10]. It is possi-
ble that activation of Chk1 during oxidative stress could
cause a similar increase in cellular Ing1b protein levels as
seen with the UV-induced stress [29]. Our results on
induction of Ing1b expression at protein level are contrary
to the findings in a previous study, which reported an
increase in Ing1b mRNA levels but no change in protein
levels [12]. However, we think that there is a conspicuous
difference between our study and previous study by Ceruti
et al., regarding the protocol used to induce oxidative
stress. While in the previous study, the authors induced
oxidative stress in the cells by adding H2O2 to the medium,
Fig. 6 Ing1b over expression abrogates the oxidative stress induced
cell death. HCT116 cells were transfected with empty vector or
3XFlag Ing1b plasmid. 24 h after transfection, the cells were treated
with H2O2 (0–500 lM) for 30 min and medium was replaced after
washing the cells with 19 PBS. 24 h after treatment, cells were
collected and fixed as described in Fig. 5, and analyzed using flow
cytometry. a Representative histograms showing the propidium
iodide fluorescence (PI-fluorescence) and b mean of 3 independent
experiments
Apoptosis (2014) 19:518–526 523
123
Page 7
which suggests that the cells were treated continuously, we
treated the cells with H2O2 only for 30 min and replaced
the medium [12]. We think that treatment protocol used by
Ceruti et al. was a bit strong and was the reason for the
observed difference in findings. We did not proceed with
elucidating the mechanisms involved in the induction of
Ing1b protein expression as the objective of this study was
primarily to explore the role of Ing1b in repair of DNA
damaged due to oxidative stress. Therefore we aimed to
study the cells upon knock down of endogenous Ing1b or
upon ectopic expression of Ing1b.
Monoubiquitination of PCNA, which is a critical event in
the signaling of translesion synthesis of DNA, was recently
shown to have equal importance in base excision repair [9,
24]. We were able to reproduce the events demonstrated
previously by Zlatanou et al., and were further able to elu-
cidate the significance of Ing1b in the PCNA ubiquitintion.
Interestingly, we did not see any apparent changes in
XRCC1 chromatin loading after treatment with H2O2. We
think that our experimental conditions possibly required the
‘‘long patch pathway’’ which involves PCNA and therefore
we did not see any changes in XRCC1, which regulates
‘‘short patch pathway’’ [8]. Our results show that cells
lacking Ing1b had decreased PCNA ubiqutination and on
other hand, over expression of Ing1b lead to increased levels
of ubiquinated PCNA, clearly demonstrating the role of
Ing1b. Insufficient PCNA ubiquitination could lead to
defective recruitment of repair factors on the sites of lesions
and thereby could lead to incomplete repair of the damaged
DNA. The immediate effect of persistent DNA lesions in the
cell would be genomic instability and activation of apoptotic
cascade [23]. Along those lines, Ing1b knock down cells
showed defective DNA repair as well as increased cell death.
Inhibitor of Growth (ING) family proteins are known mainly
to act by facilitating histone acetylation by histone acetyl-
transferases [11, 12, 23]. Acetylation leads to neutralization
of the positive charges on histones, which are required to
hold the negatively charged DNA base pairs [31–33]. His-
tone acetylation would thus lead to unwinding of DNA and
cause chromatin relaxation, which is a pre-requisite for the
repair factors to gain access to DNA lesions and to take part
in DNA repair [32, 33]. The system is restored to normal by
the activity of histone deacetylases (HDAC), which remove
the acetyl groups from histone molecules [34]. Inhibition of
HDAC activity would lead to continued presence of acety-
lated histones and to a relaxed and more accessible DNA.
HDAC inhibitor treatment of the Ing1b knock down cells
might have lead to chromatin relaxation and allowed the
Fig. 7 Pretreatment with HDAC inhibitors rescues Ing1b KD cells
from oxidative stress induced cell death. HCT116 cells were
transfected with control or Ing1b siRNA. 48 h after transfection, the
cells were treated with trichostatic acid (TSA, 5 ng/ml) 1 h prior to
and following treatment with H2O2 (200 lM) for 30 min and medium
was replaced after washing the cells with 19 PBS. 24 h after
treatment, cells were collected and fixed as described in Fig. 5, and
analyzed using flow cytometry. a Representative histograms showing
the propidium iodide fluorescence (PI-fluorescence) and b mean of 3
independent experiments
b
524 Apoptosis (2014) 19:518–526
123
Page 8
activation of DNA repair pathways similar to the Ing1b
positive cells. Understandably, HDAC inhibitor treatment
lead to comparable cell death in control and Ing1b knock
down cells. Oxidative stress is known to cause cell cycle
arrest in G2/M phase [35] and our results showed that there
was a clear increase in G2/M population of cells at a dose of
200 lM and above. Interestingly, treatment with HDAC
inhibitors had more pronounced effects on the G2/M arrest
as seen by slight increase in G2/M percentage in control cells
and more importantly, by the virtual dissipation of difference
in percentage of cells in G2/M phase after H2O2 treatment.
Possibly, HDAC inhibition lead to a more complete activa-
tion of DNA repair pathways and enhanced the G2/M arrest
after oxidative stress. Our results are in agreement with the
previous study which reported that TSA treatment rescued
the Ing1b lacking cells from UV-C induced cytotoxicity
[23], but seem to contradict the results from the study by
Ceruti et al. which showed that TSA treatment exacerbated
the H2O2 induced DNA damage [12]. We think that our
experimental design differed from the study by Ceruti et al.,
and the dose (100 nM TSA to *16.5 nM)) and pretreatment
time (5 vs 1 h) used by them were relatively high [12]. We
speculate that treatment with higher concentration of TSA,
and for longer time led to greater degree of chromatin
relaxation in the cells prior to and during treatment with
H2O2, allowing for a greater degree of DNA damage. As
pointed earlier, we treated the cells only for 30 min, whereas
the report by Ceruti et al. indicates a continuous treatment
with H2O2, which could be another reason for difference in
the findings.
We think that the upregulation of Ing1b protein levels is
interesting feature of our findings as we see the effect at 4 h
of recovery after exposure to H2O2. The kinetics of the repair
mechanisms shown by previous reports and our studies,
showed that the presence of Ing1b was essential at very initial
stages (at time T = 0 h) [9]. The protein upregulation was
thus not part of the DNA repair mechanism and was sort of an
additional effect of the pathway. However, its significance
need not be underestimated because Ing1b is known to cause
cell cycle arrest and induce cellular senescence [13, 17].
Oxidative stress is also known to induce senescence in cells
and it would be interesting to study the role Ing1b in this
process [2, 26]. We speculate that cells lacking Ing1b would
have more initial cell death under stress conditions, but
would have more oncogenic mutations and lesser activation
of senescence which is considered as a delayed effect of
oxidative stress. Further, we believe that the mechanisms
regulating the cellular handling of intracellular reactive
oxygen species (ROS) are particularly important for cells
where intracellular ROS acts as a second messenger [36–38].
We think it would be interesting to study role of Ing1b in
survival of immune cells, where activated cells need to tol-
erate the intracellular ROS.
To summarize our paper describes an upregulation of
Ing1b in cells exposed to oxidative stress, elucidates the
critical role played by Ing1b in the repair of oxidized DNA
and demonstrates the decreased survival in cells lacking
Ing1b.
Acknowledgments We thank Dr Ronald P C Wong for the valuable
discussions during the course of the project. This project was sup-
ported by grants from Canadian Institute of Health Research (MOP-
93810).
Conflict of interest The authors declared that they have no conflict
of interest.
References
1. Drews G, Krippeit-Drews P, Dufer M (2010) Oxidative stress and
beta-cell dysfunction. Pflugers Arch 460(4):703–718. doi:10.
1007/s00424-010-0862-9
2. Bokov A, Chaudhuri A, Richardson A (2004) The role of oxi-
dative damage and stress in aging. Mech Ageing Dev
125(10–11):811–826. doi:10.1016/j.mad.2004.07.009
3. Maynard S, Schurman SH, Harboe C, de Souza-Pinto NC, Bohr
VA (2009) Base excision repair of oxidative DNA damage and
association with cancer and aging. Carcinogenesis 30(1):2–10.
doi:10.1093/carcin/bgn250
4. Gonfloni S, Maiani E, Di Bartolomeo C, Diederich M, Cesareni G
(2012) Oxidative Stress, DNA Damage, and c-Abl Signaling: at
the Crossroad in Neurodegenerative Diseases? Int J Cell Biol
2012:683097. doi:10.1155/2012/683097
5. Limberg JK, Harrell JW, Johansson R, Eldridge MW, Proctor LT,
Sebranek JJ, Schrage WG (2013) Microvascular function in
younger adults with obesity and metabolic syndrome: role of
oxidative stress. Am J Physiol Heart Circ Physiol. doi:10.1152/
ajpheart.00291.2013
6. Neeley WL, Essigmann JM (2006) Mechanisms of formation,
genotoxicity, and mutation of guanine oxidation products. Chem
Res Toxicol 19(4):491–505. doi:10.1021/tx0600043
7. Scott TL, Rangaswamy S, Wicker CA, Izumi T (2013) Repair of
oxidative DNA damage and cancer—recent progress in DNA
base excision repair. Antioxid Redox Signal. doi:10.1089/ars.
2013.5529
8. Wilson DM 3rd, Bohr VA (2007) The mechanics of base excision
repair, and its relationship to aging and disease. DNA Repair
(Amst) 6(4):544–559. doi:10.1016/j.dnarep.2006.10.017
9. Zlatanou A, Despras E, Braz-Petta T, Boubakour-Azzouz I, Po-
uvelle C, Stewart GS, Nakajima S, Yasui A, Ishchenko AA,
Kannouche PL (2011) The hMsh2–hMsh6 complex acts in con-
cert with monoubiquitinated PCNA and Pol eta in response to
oxidative DNA damage in human cells. Mol Cell 43(4):649–662.
doi:10.1016/j.molcel.2011.06.023
10. Willis J, Patel Y, Lentz BL, Yan S (2013) APE2 is required for
ATR-Chk1 checkpoint activation in response to oxidative stress.
Proc Natl Acad Sci USA 110(26):10592–10597. doi:10.1073/
pnas.1301445110
11. Aguissa-Toure AH, Wong RP, Li G (2011) The ING family
tumor suppressors: from structure to function. Cell Mol Life Sci
68(1):45–54. doi:10.1007/s00018-010-0509-1
12. Ceruti JM, Ogara MF, Menendez C, Palmero I, Canepa ET
(2013) Inhibitor of growth 1 (ING1) acts at early steps of multiple
DNA repair pathways. Mol Cell Biochem 378(1–2):117–126.
doi:10.1007/s11010-013-1601-2
Apoptosis (2014) 19:518–526 525
123
Page 9
13. Jafarnejad SM, Li G (2012) Regulation of p53 by ING family
members in suppression of tumor initiation and progression.
Cancer Metastasis Rev 31(1–2):55–73. doi:10.1007/s10555-011-
9329-5
14. Kuo WH, Wang Y, Wong RP, Campos EI, Li G (2007) The
ING1b tumor suppressor facilitates nucleotide excision repair by
promoting chromatin accessibility to XPA. Exp Cell Res
313(8):1628–1638. doi:10.1016/j.yexcr.2007.02.010
15. Li G, Piche B (2010) ING2 in cell cycle regulation. Cell Cycle
9(19):3846. doi:10.4161/cc.9.19.13382
16. Li J, Wang Y, Wong RP, Li G (2009) The role of ING tumor
suppressors in UV stress response and melanoma progression.
Curr Drug Targets 10(5):455–464
17. Li N, Li Q, Cao X, Zhao G, Xue L, Tong T (2011) The tumor
suppressor p33ING1b upregulates p16INK4a expression and
induces cellular senescence. FEBS Lett 585(19):3106–3112.
doi:10.1016/j.febslet.2011.08.044
18. Wang J, Chin MY, Li G (2006) The novel tumor suppressor
p33ING2 enhances nucleotide excision repair via inducement of
histone H4 acetylation and chromatin relaxation. Cancer Res
66(4):1906–1911. doi:10.1158/0008-5472.CAN-05-3444
19. Wang Y, Wang J, Li G (2006) Leucine zipper-like domain is
required for tumor suppressor ING2-mediated nucleotide exci-
sion repair and apoptosis. FEBS Lett 580(16):3787–3793. doi:10.
1016/j.febslet.2006.05.065
20. Thakur S, Feng X, Qiao Shi Z, Ganapathy A, Kumar Mishra M,
Atadja P, Morris D, Riabowol K (2012) ING1 and 5-azacytidine
act synergistically to block breast cancer cell growth. PLoS One
7(8):e43671. doi:10.1371/journal.pone.0043671
21. Doyon Y, Cayrou C, Ullah M, Landry AJ, Cote V, Selleck W,
Lane WS, Tan S, Yang XJ, Cote J (2006) ING tumor suppressor
proteins are critical regulators of chromatin acetylation required
for genome expression and perpetuation. Mol Cell 21(1):51–64.
doi:10.1016/j.molcel.2005.12.007
22. Vieyra D, Loewith R, Scott M, Bonnefin P, Boisvert FM, Cheema
P, Pastyryeva S, Meijer M, Johnston RN, Bazett-Jones DP,
McMahon S, Cole MD, Young D, Riabowol K (2002) Human
ING1 proteins differentially regulate histone acetylation. J Biol
Chem 277(33):29832–29839. doi:10.1074/jbc.M200197200
23. Wong RP, Lin H, Khosravi S, Piche B, Jafarnejad SM, Chen DW,
Li G (2011) Tumour suppressor ING1b maintains genomic stability
upon replication stress. Nucleic Acids Res 39(9):3632–3642.
doi:10.1093/nar/gkq1337
24. Kannouche PL, Wing J, Lehmann AR (2004) Interaction of
human DNA polymerase eta with monoubiquitinated PCNA: a
possible mechanism for the polymerase switch in response to
DNA damage. Mol Cell 14(4):491–500
25. Wang Y, Li G (2006) ING3 promotes UV-induced apoptosis via
Fas/caspase-8 pathway in melanoma cells. J Biol Chem
281(17):11887–11893. doi:10.1074/jbc.M511309200
26. Qiao Y, Spitz MR, Guo Z, Hadeyati M, Grossman L, Kraemer
KH, Wei Q (2002) Rapid assessment of repair of ultraviolet DNA
damage with a modified host-cell reactivation assay using a
luciferase reporter gene and correlation with polymorphisms of
DNA repair genes in normal human lymphocytes. Mutat Res
509(1–2):165–174
27. Wang S, Gong Z, Chen R, Liu Y, Li A, Li G, Zhou J (2009) JWA
regulates XRCC1 and functions as a novel base excision repair
protein in oxidative-stress-induced DNA single-strand breaks.
Nucleic Acids Res 37(6):1936–1950. doi:10.1093/nar/gkp054
28. Garate M, Campos EI, Bush JA, Xiao H, Li G (2007) Phos-
phorylation of the tumor suppressor p33(ING1b) at Ser-126
influences its protein stability and proliferation of melanoma
cells. FASEB J 21(13):3705–3716. doi:10.1096/fj.07-8069com
29. Garate M, Wong RP, Campos EI, Wang Y, Li G (2008)
NAD(P)H quinone oxidoreductase 1 inhibits the proteasomal
degradation of the tumour suppressor p33(ING1b). EMBO Rep
9(6):576–581. doi:10.1038/embor.2008.48
30. Kielbassa C, Roza L, Epe B (1997) Wavelength dependence of
oxidative DNA damage induced by UV and visible light. Carci-
nogenesis 18(4):811–816
31. Roth SY, Denu JM, Allis CD (2001) Histone acetyltransferases.
Annu Rev Biochem 70:81–120. doi:10.1146/annurev.biochem.
70.1.81
32. Reed SH (2011) Nucleotide excision repair in chromatin: damage
removal at the drop of a HAT. DNA Repair (Amst)
10(7):734–742. doi:10.1016/j.dnarep.2011.04.029
33. Yu S, Teng Y, Waters R, Reed SH (2011) How chromatin is
remodelled during DNA repair of UV-induced DNA damage in
Saccharomyces cerevisiae. PLoS Genet 7(6):e1002124. doi:10.
1371/journal.pgen.1002124
34. Sengupta N, Seto E (2004) Regulation of histone deacetylase
activities. J Cell Biochem 93(1):57–67. doi:10.1002/jcb.20179
35. Gambino V, De Michele G, Venezia O, Migliaccio P, Dall’Olio
V, Bernard L, Minardi SP, Della Fazia MA, Bartoli D, Servillo G,
Alcalay M, Luzi L, Giorgio M, Scrable H, Pelicci PG, Migliaccio
E (2013) Oxidative stress activates a specific p53 transcriptional
response that regulates cellular senescence and aging. Aging Cell
12(3):435–445. doi:10.1111/acel.12060
36. Bauer M, Goldstein M, Christmann M, Becker H, Heylmann D,
Kaina B (2011) Human monocytes are severely impaired in base
and DNA double-strand break repair that renders them vulnerable
to oxidative stress. Proc Natl Acad Sci USA 108(52):21105–21110.
doi:10.1073/pnas.1111919109
37. Bauer M, Goldstein M, Heylmann D, Kaina B (2012) Human
monocytes undergo excessive apoptosis following temozolomide
activating the ATM/ATR pathway while dendritic cells and
macrophages are resistant. PLoS One 7(6):e39956. doi:10.1371/
journal.pone.0039956
38. Bhandaru M, Yang W, Rotte A, Pasham V, Lang F (2012)
Regulation of Na?/H? exchanger in dendritic cells by Akt2.
Pflugers Arch 463(2):355–363. doi:10.1007/s00424-011-1015-5
526 Apoptosis (2014) 19:518–526
123