Page 1
Biochemical characterization of a novel iron-sulfur
flavoprotein from Methanosarcina thermophila strain TM-1
Ubolsree Leartsakulpanich
Dissertation submitted to the Faculty of the
Virginia Polytechnic Institute and State University
in partial fulfillment of the requirement for the degree of
Doctor of philosophy
in
Biochemistry
Dr. James G. Ferry, Chair
Dr. John L. Hess
Dr. Eugene M. Gregory
Dr. Timothy J. Larson
Dr. Dennis R. Dean
Dr. Robert H. White
June 1999
Blacksburg, Virginia
Keywords: Iron-sulfur flavoprotein, Methanosarcina thermophila
Copyright 1999, Ubolsree Leartsakulpanich
Page 2
ii
Ubolsree Leartsakulpanich
ABSTRACT
The iron-sulfur flavoprotein (Isf) from the acetate utilizing methanoarchaeon
Methanosarcina thermophila was heterologously produced in Escherichia coli, purified
to homogeneity, and characterized to determine the properties of the iron-sulfur cluster
and FMN. Chemical and spectroscopic analyses indicated that Isf contained one 4Fe-4S
cluster and one FMN per monomer. The midpoint potentials of the [4Fe-4S]2+/1+ center
and FMN/FMNH2 redox couple were -394 and -277 mV respectively.
The deduced amino acid sequence of Isf revealed high identity with Isf
homologues from the CO2 reducing methanoarchaea Methanococcus jannaschii and
Methanobacterium thermoautotrophicum. Extracts of H2-CO2-grown M.
thermoautotrophicum cells were able to reduce Isf from M. thermophila using either H2
or CO as the reductant. Addition of ferredoxin A to the reaction further stimulated the
rate of Isf reduction. These results suggest that Isf homologues are coupled to ferredoxin
in electron transfer chains in methanoarchaea with diverse metabolic pathways.
Reconstituted systems containing carbon monoxide dehydrogenase/acetyl-CoA
synthase complex (CODH/ACS), ferredoxin A, Isf, and the designated electron carriers
(NAD, NADP, F420, and 2-hydroxyphenazine) were used in an attempt to determine the
electron acceptor for Isf. Isf was unable to reduce any of these compounds. Furthermore,
2-hydroxyphenazine competed with Isf to accept electrons from ferredoxin A indicating
that ferredoxin A is a more favorable electron partner for 2-hydroxyphenazine. Thus, the
physiological electron acceptor for Isf is unknown.
Amino acid sequence alignment of Isf sequences revealed a conserved atypical
cysteine motif with the potential to ligate the 4Fe-4S cluster. Site-directed mutagenesis
of the cysteine residues in this motif, and the two additional cysteines in the sequence,
was used to investigate these cysteine residue as ligands for coordinating the 4Fe-4S
Page 3
iii
center of Isf. Spectroscopic and biochemical analyses were consistent with the
conserved cysteine motif functioning as ligating the 4Fe-4S center. Redox properties of
the 4Fe-4S and FMN centers revealed a role for the 4Fe-4S center in the transfer of
electrons from ferredoxin A to FMN.
Page 5
iv
FORWARD
This dissertation focuses on the characterization of the heterologously produced
iron-sulfur flavoprotein from Methanosarcina thermophila by different approaches.
Chapters 1 and 2 are intended to serve as an introduction to biological methanogenesis
and iron-sulfur proteins. Chapters 3 and 4 describe the research pertaining to the
characterization of iron-sulfur flavoprotein and a summary is presented in Chapter 5. The
studies described in Chapters 3 and 4 have or will be published as follows:
Becker D.F., Leartsakulpanich U., Surerus K.K., Ferry J.G., and Ragsdale S.W.
1998. Electrochemical and spectroscopic properties of the iron-sulfur
flavoprotein from Methanosarcina thermophila. J. Biol Chem. 273:26462-26469
Leartsakulpanich U, Antokine M.L., Golbeck J.H., and Ferry J.G. A novel [4Fe-
4S] iron-sulfur cluster binding motif in the iron-sulfur flavoprotein of
Methanosarcina thermophila (manuscript in preparation).
Page 6
v
ACKNOWLEDGEMENT
I would like to express my gratitude to my thesis advisor, Dr. James G. Ferry, for
giving me the opportunity, financial support, encouragement and guidance in my
academic career. This dissertation would be impossible with out his guidance and insight
toward my study. I would like to thank my committee members, Dr. T.J. Larson, Dr.
D.R. Dean, Dr. E.M. Gregory, and Dr. J.L. Hess, for their interest in my research and
their flexibility and understanding of my situation as an out of state student. In addition, I
would like to thank Dr. R.H. White for being substituted for Dr. Dean as another thesis
examining committee. I thank Dr. D.F. Becker, Dr. S.W. Ragsdale, and Dr. K.K. Surerus
for their contributions to the characterization of the iron-sulfur flavoprotein (Isf). I thank
M.L. Antokine and Dr. J.H. Golbeck for their assist on EPR experiments with Isf
variants. I gratefully acknowledge the financial support from MOSTE, Thailand
throughout my 5 years in the US.
I also would like to thank all the postdocs and students in Dr. Ferry's lab. Cheryl
Ingram Smith (who helped me during my research rotation and has been very helpful
since, also for her compassion and caring), Birgit Alber (who was another Hoakie, special
soul-mate, my volleyball coach, and my driver when I wanted to head south), Kerry
Smith and Rob Barber (for their muscles and advice and thousands of suggestion),
Madeline Rasche, Kavita Singh-Wissman, and Julie Maupin-Furlow (for their kindness
and patience in teaching me techniques), Sean O'Hearn (for let me know that being a
biochemist is better than being a chef, Really?) and Mike Painter, Tong Zhao, Rebecca
Miles, Christie Brosius, Birthe Borup, Prabha Iyer, Brian Tripp, Laura Lierman, and
Mark Signs, for their friendship, English teaching, and support (especially when I cannot
smile). My appreciation goes to the secretary of the department at VA Tech, Mary Jo
Smart, and several secretaries of the Ferry lab, Vonni Kladde, and Carol DeArmitt, for
taking care of things promptly, efficiently, and conveniently for me.
I want to extend my appreciation to my ex-roommates at VATech, Kitsiri
Kaewpipat and Chanpen Chanchao, together with Sunitiya Thuannadee, Charaspim
Page 7
vi
Boonyanan, and Matt Mamorino who made the years in Blacksburg an incredibly
wonderful experience for me. Also, Somboon Kiratiprayoon, Patcharin Poosanaas,
Amin Tanuminhadjo, Bill and Barb Saxton, Joanie Zhoa and HenSiong Tan, Ari and
Purwadi Purwasumato Venyi Hoa, Joy Wang, Maki Murata, Kerwin Foster, and all other
members of ICF, who supply the happiness at PSU.
Finally and the most important, I would like to express my deep gratitude to my
family, Khajon (for taking all the burden off my shoulders and letting me continue my
studies), Sirinthip (for her compassion, consideration, and love), Paramate ( for his
sharing, and prompt assistance with seemingly ceaseless energy), and particularly to my
Mom and Dad (for their unconditional and endless love, supporting guidance, generosity,
sweetness, and always always being there for me); without their inspiration,
encouragement, motivation, and optimism, I would not be able to come this far.
Thanks again to all.
Page 8
vii
TABLE OF CONTENTS
Page
Title page i
Abstract ii
Forward iv
Acknowlegement v
Table of contents vii
List of tables ix
List of figures x
Introduction xiii
Chapter 1: Methanogenesis 1
I. Microbiology 1
Methanoarchaea 1
Growth substrates 1
Ecology 2
II. Biochemistry 6
Coenzymes 6
CO2-reduction pathway 8
Acetate fermentation pathway 10
III. Bioenergetics and electron transport 14
References 18
Chapter 2: Iron-sulfur proteins 32
I. Introduction 32
II. Structural and properties of clusters 32
1[Fe] cluster type 33
[2Fe-2S] cluster type 34
[3Fe-4S] cluster type 34
[4Fe-4S] cluster type 35
III. Ligation of iron-sulfur clusters 39
IV. Function of iron-sulfur centers 41
Page 9
viii
TABLE OF CONTENTS (cont.)
Page
A. Electron transfers 41
B. Catalysis 42
C. Regulatory role 43
D. Iron storage role 44
E. Structural role 44
V. Iron-sulfur cluster assembly in proteins 45
References 46
Chapter 3: Objectives 55
Chapter 4: Electrochemical and spectroscopic properties of
the iron-sulfur flavoprotein from Methanosarcina thermophila 57
Abstract 57
Introduction 57
Materials and Methods 59
Results 63
Discussion 81
Acknowledgement 84
References 85
Chapter 5: A novel [4Fe-4S] iron-sulfur cluster binding motif in the iron-sulfur
flavoprotein of Methanosarcina thermophila 88
Abstract 88
Introduction 89
Experimental procedures 91
Results 93
Discussion 118
Acknowledgement 120
References 121
Chapter 6: Summary and future directions 124
Curriculum Vista 126
Page 10
ix
LIST OF TABLES
Page
Chapter 1
Table 1. Substrates for methanogenesis 3
Chapter 5
Table 1. EPR properties of wild-type Isf and variants 115
Table 2. Rates for reduction of FMN in wild type Isf and variants 117
Page 11
x
LIST OF FIGURES
Page
Chapter 1
Figure 1. Microbial food chain 5
Figure 2. Structure of the coenzyme of the coenzymes involved
in methanogenesis 7
Figure 3. Methanogenesis from CO2 reduction pathway 9
Figure 4. Proposed pathway for acetate conversion to CO2 and
CH4 in Methanosarcina thermophila 13
Chapter 2
Figure 1. Structures and properties of the structurally
characterized iron-sulfur centers that are
involved in biological system 36
Figure 2. Arrangement of residues involved in coordination of
[2Fe-2S] (A), [4Fe-4S] or 2[4Fe-4S] (B), [3Fe-4S] or
[4Fe-4S] (C), and 2[4Fe-4S] or [3Fe-4S] plus [4Fe-4S]
(D) clusters 38
Chapter 4
Figure 1. Corrected nucleic acid sequence and predicted amino
acid sequence of isf from M. thermophila 64
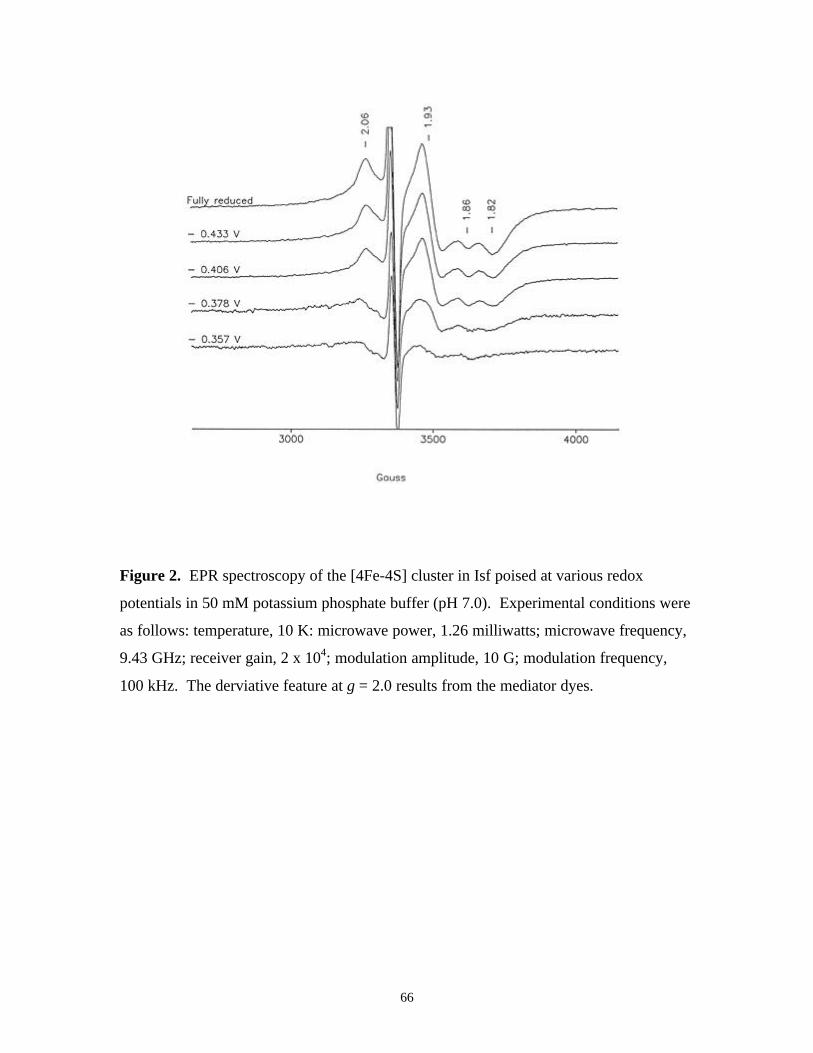
Figure 2. EPR spectroscopy of the [4Fe-4S] cluster in Isf poised at
various redox potentials in 50 mM potassium phosphate
buffer (pH 7.0) 66
Figure 3. Semilogarithmic plot of P1/2 versus 1/T, which shows a
linear relationship according to equation P1/2 = Aexp (∆-/kT) 67
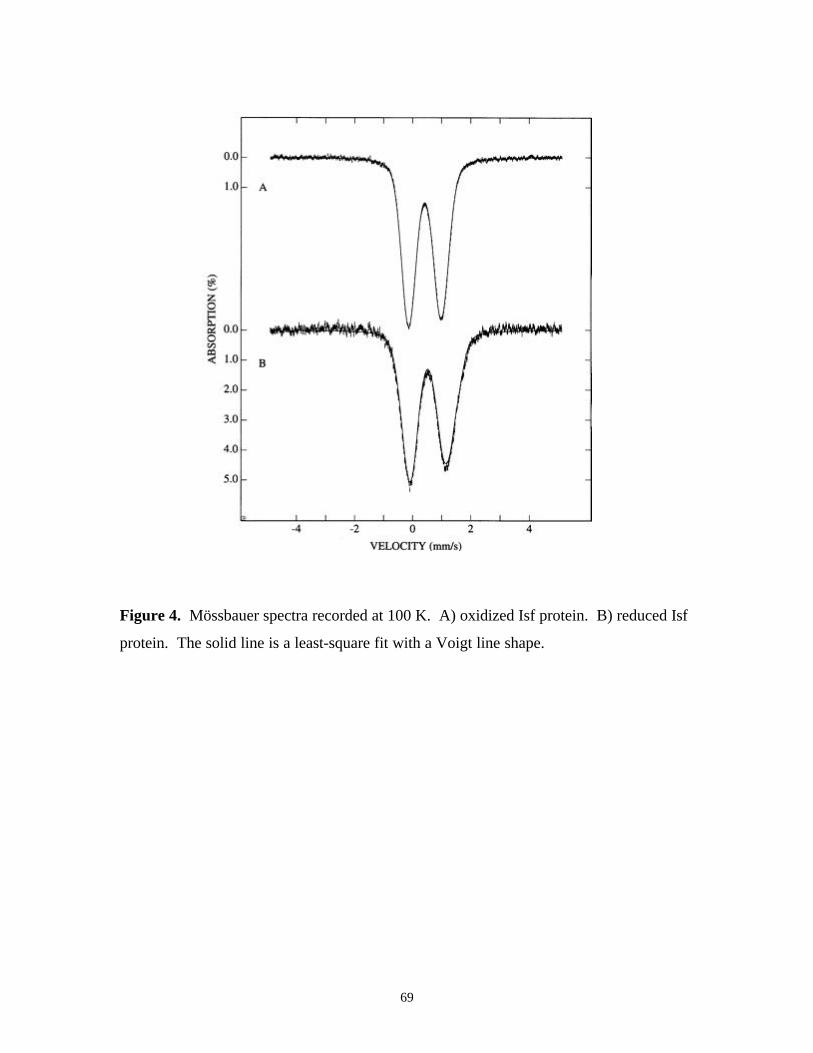
Figure 4. Mössbauer spectra recorded at 100 K. A) oxidized Isf protein.
B) reduced Isf protein 69
Figure 5. Mössbauer spectra of reduced Isf protein recorded at 4.2 K
and 450 G applied parallel (A) or 450 G applied perpendicular
(B) to the γ beam 70
Page 12
xi
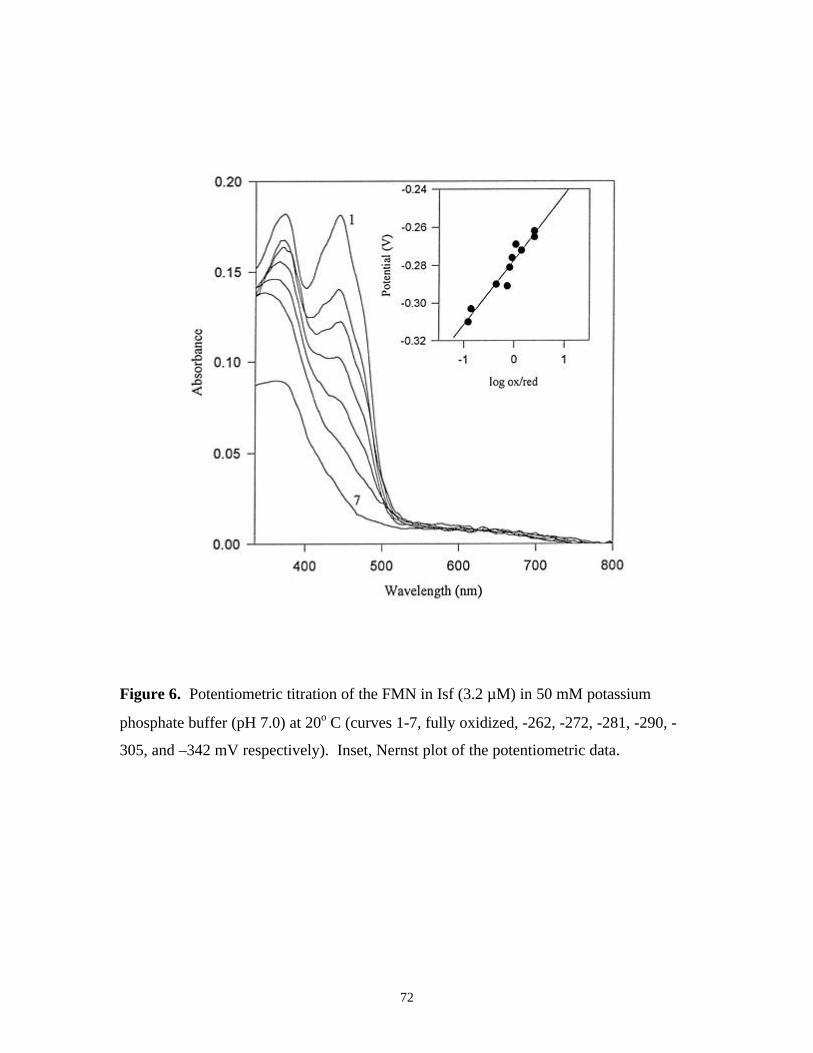
Figure 6. Potentiometric titration of the FMN in Isf (3.2 µM) in
50 mM potassium phosphate buffer (pH 7.0) at 20o C
(curves 1-7, fully oxidized, -262, -272, -281, -290, -305,
and –342 mV respectively). Inset, Nernst plot of the
potentiometric data 72
Figure 7. EPR spectrum of Isf (170 µM dimer) was recorded at
10 K following incubation for 17 min with CO and CODH
(0.5 µM) at 25o C) 73
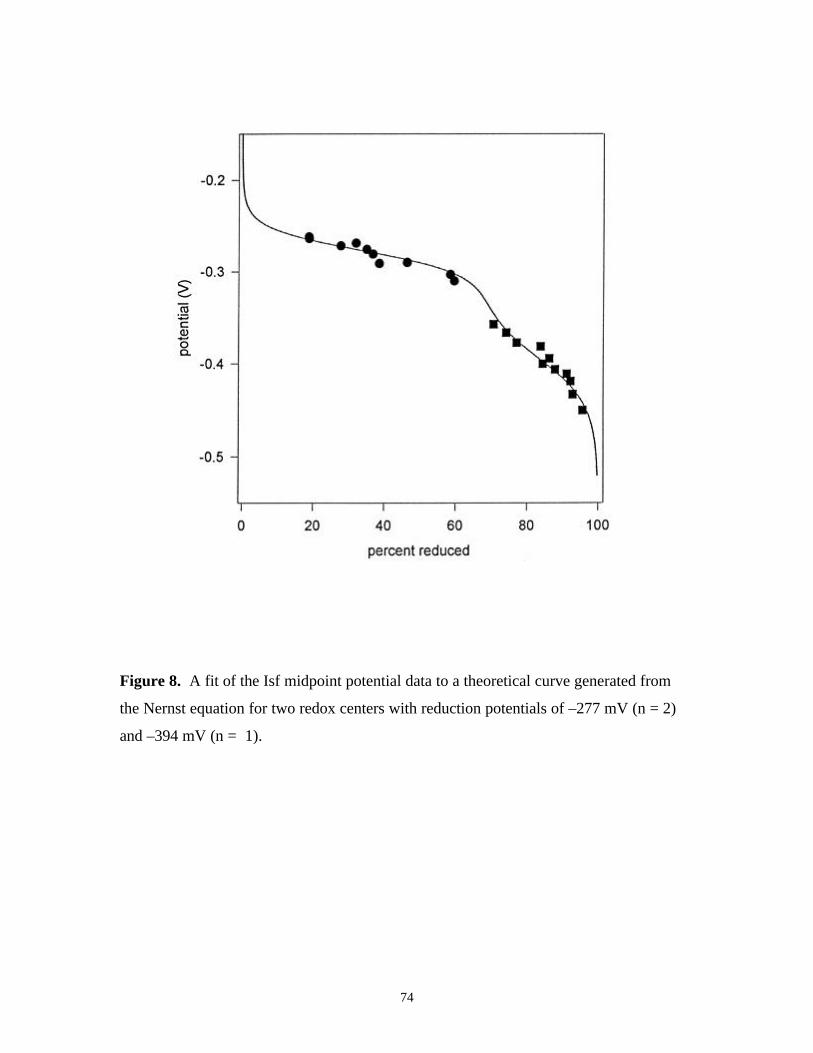
Figure 8. A fit of the Isf midpoint potential data to a theoretical
curve generated from the Nernst equation for two redox
centers with reduction potentials of –277 mV (n = 2)
and –394 mV (n = 1) 74
Figure 9. Time course for reduction of methanophenazine with
Isf from M. thermophila 76
Figure 10. Multiple amino acid sequence alignment of Isf from
M. thermophila with sequences deduced from open
reading frames identified in the genomic sequences of
M. jannashii and M. thermoautotrophicum 78
Figure 11. Time course for reduction of Isf with extract from M.
thermoautotrophicum 79
Figure 12. Time course for reduction of Isf with extract from M.
thermoautotrophicum 80
Figure 13. Proposed electron transport pathway for oxidation of CO
or the carbonyl group of acetyl-CoA 82
Chapter 5
Figure 1. Multiple amino acid sequence alignment of Isf from
M.thermophila (MST) with sequences deduced from open
reading frames identified in the genomic sequences of
M. jannaschii (MCJ), M. thermoautotrophicum (MBT),
Archaeoglobus fulgidus (AF), Chlorobium vibrioforme (CV),
Chlorobium tepidum (CT), and Clostridium difficile (CD). 94
Page 13
xii
Figure 2. Coomassie blue stained native PAGE of wild type Isf and
variants 97
Figure 3. UV-visible absorption spectra of as-purified, denatured, and
reconstituted wild type Isf 99
Figure 4. UV-visible absorption spectra of wild type Isf and alanine
variants 100
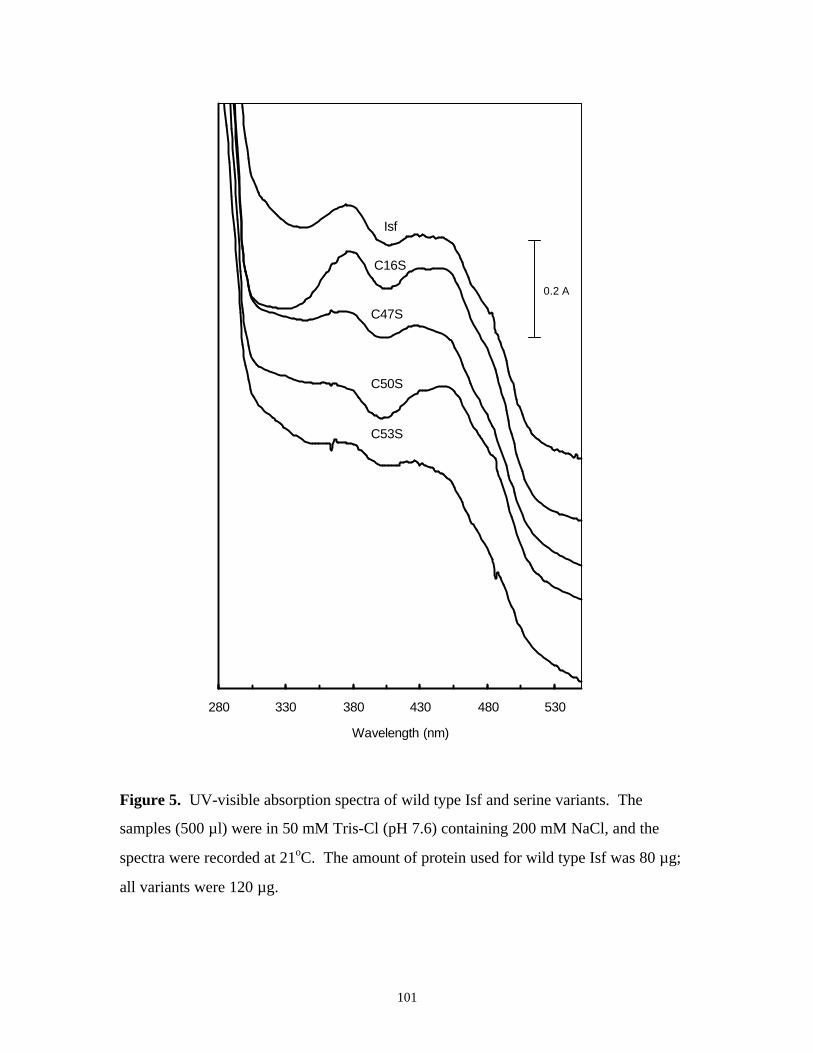
Figure 5. UV-visible absorption spectra of wild type Isf and serine
variants 101
Figure 6. EPR spectra of reduced wild type Isf with different
processes to recover iron-sulfur center 102
Figure 7. EPR spectra of C16X (X = A or S) 104
Figure 8. EPR spectra of reduced C180A with different reconstitution
processes 105
Figure 9. EPR spectrum of reduced C180S 106
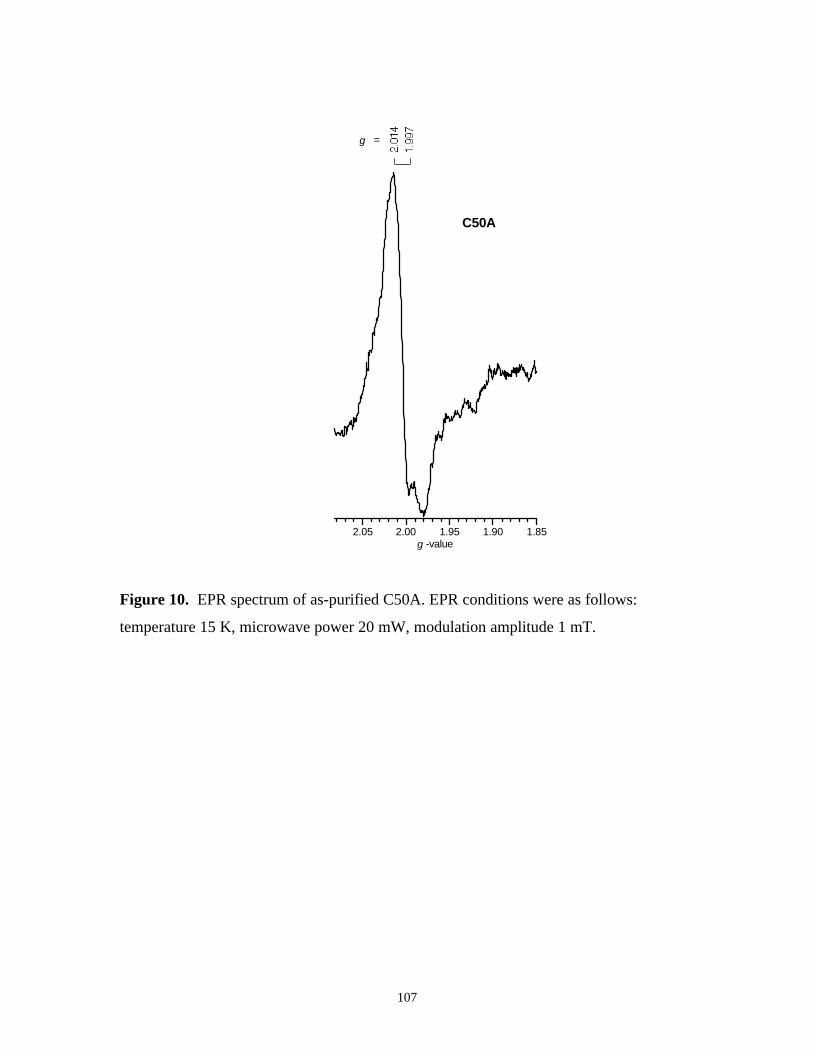
Figure 10. EPR spectrum of as-purified C50A 107
Figure 11. EPR spectra of as-purified C59A 108
Figure 12. EPR spectrum of reduced C47A 110
Figure 13. EPR spectrum of reduced C53A 111
Figure 14. EPR spectrum of reduced C47S 112
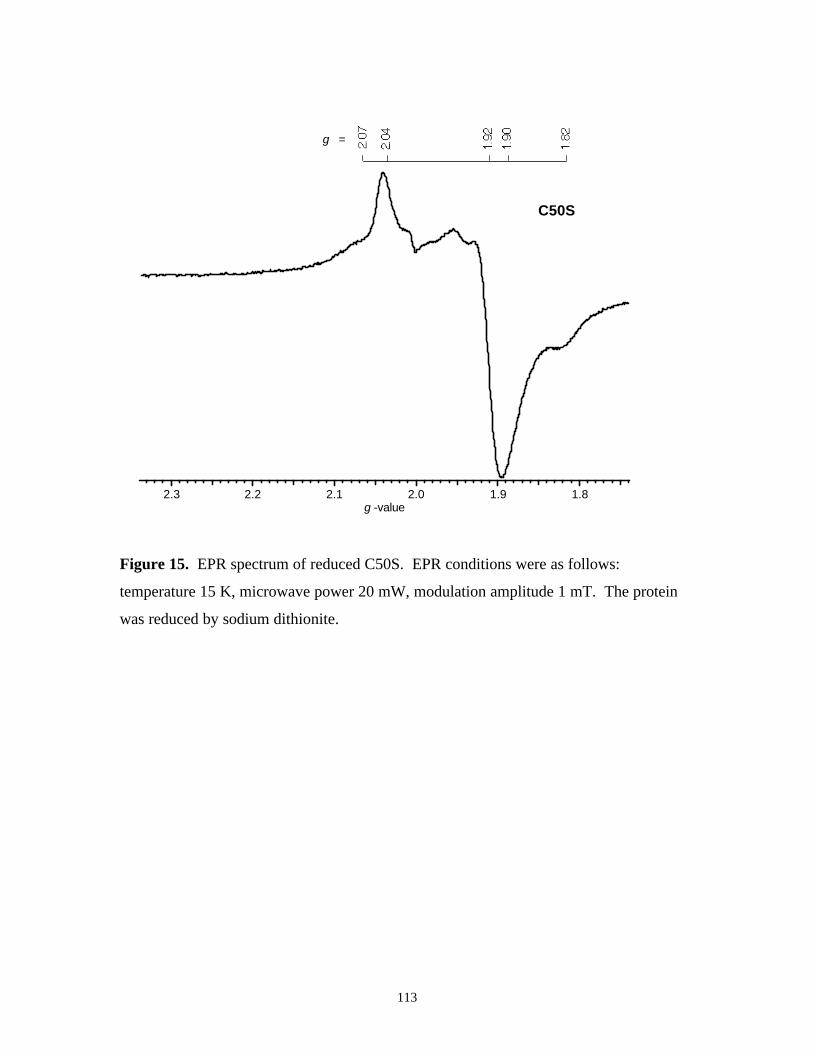
Figure 15. EPR spectrum of reduced C50S 113
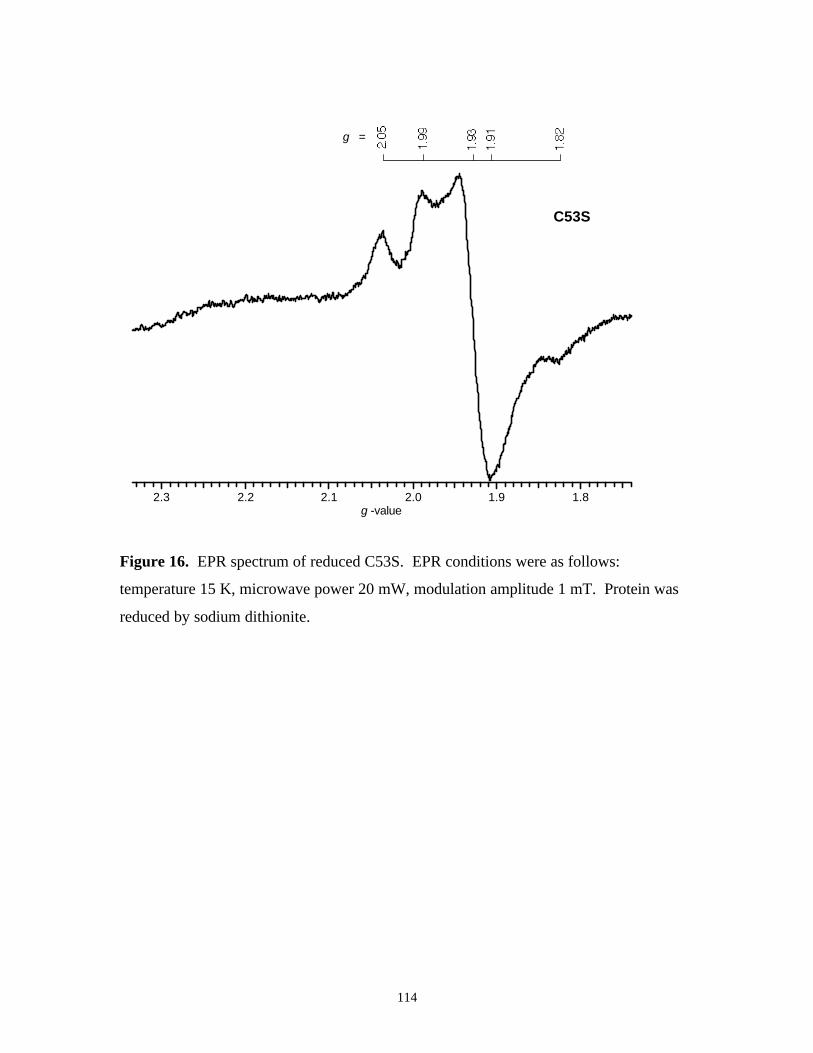
Figure 16. EPR spectrum of reduced C53S 114
Page 14
xiii
INTRODUCTION
Methanogenesis is a prominent process in the biological world, in which it
represents the final step in the carbon cycle in anaerobic environment. Methanoarchaea
have a major impact on the environment and human activities. More than 109 tons of
methane have been released into the atmosphere. Methane is produced by two major
pathways. The first is the CO2 reduction pathway in which CO2 is reduced to methane
using electrons derived from either H2 or formate. Acetate is a key product in the
decomposition of organic compounds and is the primary substrate for methane
production with two-thirds of all biological methane derived the methyl group of acetate.
However, only species of the genera Methanosarcina and Methanothrix are known to
convert acetate to methane and CO2. The study of methanogenesis has made an
enormous impact in many areas of physiology, ecology, biochemistry, molecular biology,
and evolution.
Methanosarcina thermophila strain TM-1 is a moderate thermophile in the
Archaea domain. It can utilize acetate, methanol and methylamines as growth substrates.
In the past decades, one carbon metabolism in acetate catabolism has been well
established in Ms. thermophila, but the details of the path of electron flow and energy
conservation are less well understood. The carbon monoxide dehydrogenase/acetyl CoA
synthase (CODH/ACS) enzyme complex of M. thermophila is a key enzyme in acetate
metabolism and previous studies showed that ferredoxin A accepts electrons from
CODH/ACS. The electrons are then donated to iron-sulfur flavoprotein (Isf). Isf was
partially characterized and contains iron-sulfur cluster and FMN. As a result, the
properties of the Fe-S cluster and FMN were examined. Site-directed mutagenesis was
performed in an effort to identify the iron-sulfur cluster ligands.
Page 15
1
CHAPTER 1
METHANOGENESIS
I. Microbiology.
Methanoarchaea. During the past three decades, the increasing interest in
methane-producing microorganisms has resulted in a rapid accumulation of knowledge.
The advent of 16S rRNA sequencing introduced a new classification scheme in which all
forms of life could be categorized into three "primary domains"; Eucarya, Bacteria, and
Archaea (42, 130). These three domains replaced the conventional classifications of
either the five kingdom system or the prokaryote/eukaryote dichotomy. The Eucarya
domain is comprised of plants, animals, and fungi while the Bacteria and Archaea
domains contain the prokaryotes. Methane producers, extreme halophiles, sulfate-
reducers, and extreme thermophiles are members of the Archaea (129). All
methanoarchaea belong to the Euryarcheota kingdom, and are classified into five orders,
ten families, and twenty-five genera (16).
The methanoarchaea represent the most diverse and extensively studied members
of the Archaea domain. Despite the fact that they share the common feature of methane
production, they are not closely related phylogenetically. They show diversity in: (1)
morphology (rod, coccus, spirillum, and aggregate forms) (30, 57, 61, 62, 106, 108); (2)
habitats with variable temperatures (2o to more than 100o C), pH (3 to 9.2), and salinity (1
mM to 3 M salt) (136); (3) cell wall components, such as pseudomurein (59), protein,
glycoprotein, and heteropolysaccharides (7, 68); (4) the appearance of novel cofactors
such as F430, coenzyme B, and coenzyme M in their metabolic pathways; and (5) the
ability to grow on one- and two-carbon substrates.
Growth Substrates. The sole means by which methanoarchaea obtain energy for
growth is through methanogenesis (123). They are extremely specialized in using only a
limited number of simple compounds as their growth substrates (Table 1) (136). They
require a minimum reduction potential of – 300 mV to achieve growth (52).
Page 16
2
Most methanoarchaea are able to utilize H2 and CO2 as sources of energy and
carbon (eq. 1, Table 1). However, several methanoarchaea including Methanosarcina
thermophila strain TM1 lack this ability (134). Several methanoarchaea contain formate
dehydrogenase, which allows them to use formate as a reductant (eq. 2, Table 1) (64).
Methanobacterium thermoautotrophicum and some Methanosarcina sp. are able to
oxidize CO for their growth (eq. 3, Table 1) (23). Acetate is a catabolic product of many
fermentation processes; however, only Methanosarcina and Methanosaeta species can
ferment acetate to CO2 and methane (eq. 4, Table 1) (136). The methanoarchaea in the
genus Methanosarcina are able to catabolize methyl containing compounds such as
methanol and methylamine (eqs. 5-7, Table 1). Utilization of short chain alcohols such
as ethanol has been observed in some hydrogenotrophic methanoarchaea (eq. 8, Table 1)
(128, 133). A small number of methylotrophic species utilize di-methylated sulfide (eq.
9, Table 1) (91).
Ecology. Biological methane production is a strictly anaerobic process; thus,
methanoarchaea are exclusively found in anaerobic environments, although some can
tolerate a brief exposure to O2. Methanoarchaea can be found in diverse anaerobic
habitats such as marine and freshwater sediments, hot springs, sites of geothermal
activity, and in ruminant animals. They have also been found in association with human
activities such as rice paddy fields, sewage sludge digesters, and landfills.
Methanoarchaea are restricted to only a few substrates that in nature are provided
by other microbes (136). Many such metabolic interactions among microbes in different
communities can occur (109), some examples of which are: neutralism, mutualism,
symbiosis, or competitive interaction. Environments containing sulfate (SO42-)-reducing
microbes and methanoarchaea involve competitive interactions. Sulfate reducing
microbes have been reported to grow on H2 plus CO2 and acetate; in addition, SO42-
reducers have a higher affinity for these substrates than methanoarchaea (97, 103). In
nutrient limited environments, SO42- reducers out-compete methanoarchaea for these
substrates resulting in inhibited growth of the latter (103).
Page 17
3
Table 1. Substrates for methanogenesis.
Reactants Products
1) 4H2 + HCO3- + H+ CH4 + 3H2O
2) 4HCO2- + H+ + H2O CH4 + 3HCO3
-
3) 4CO + 5H2O CH4 + 3HCO3- +3 H+
4) CH3COO- + H2O CH4 + HCO3-
5) 4CH3OH 3CH4 + HCO3- + H2O + H+
6) CH3OH + H2 CH4 + 3H2O
7) 4 (CH3) 3-NH+ + 9H2O 3CH4 + HCO3- + 4NH4
+ + 3H+
8) 2CH3CH2OH + HCO3- 2CH3COO- + CH4 + 3H2O
9) 2(CH3)2-S + 3H2O 3CH4 + HCO3- + 2H2S + H+
Page 18
4
The methanoarchaea execute the terminal step in the degradation of complex
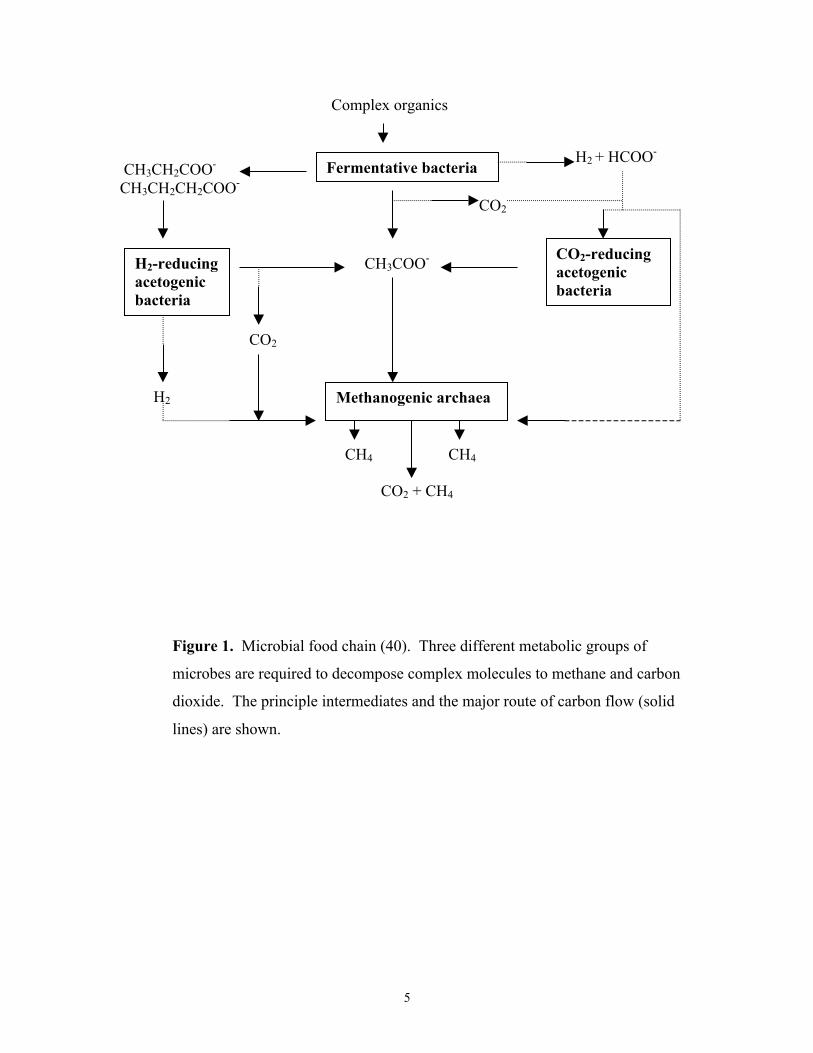
biomass to methane (Fig 1) (40), which is very important for the global carbon cycle. The
microbial degradation of biomass requires three inter-dependent metabolic groups of
microbes (37). The fermentative microorganisms degrade the large complex molecules
such as cellulose to the simple molecules H2, CO2, formate, and acetate, as well as
various fatty acids. Then, acetogens metabolize fatty acids into H2 and CO2, formate, and
acetate. Finally, methanoarchaea reduce CO2 with H2 or formate to methane, and ferment
acetate to methane and CO2. Hydrogen-producing acetogens also provide substrates for
methanoarchaea. The methanoarchaea in turn maintain a low H2 partial pressure that is
beneficial to acetogens because high concentrations of H2 inhibit the acetogens' metabolic
activity. Much of the released methane is utilized by methane oxidizing bacteria, called
methylotrophs, as their growth substrate (67). However, a large amount of methane
escapes and reaches the atmosphere where it is a major greenhouse gas (118). About 1%
annual increase of methane in the atmosphere has been observed (136), and is mainly due
to human activities.
Recent work has focused on using methanoarchaea in bioremediation. In sewage
sludge digesters, methanoarchaea and a mix of other anaerobic microorganisms degrade
organic waste into methane which can then be used as an alternative energy source (90).
In addition, studies are being performed on the ability to detoxify pollutants produced by
industry and agriculture (65, 66, 100).
Page 19
5
Complex organics
CH3CH2COO-
CH3CH2CH2COO-Fermentative bacteria
H2 + HCOO-
H2-reducingacetogenicbacteria
CH3COO-
CO2
CO2-reducingacetogenicbacteria
H2
CO2
Methanogenic archaea
CH4 CH4
CO2 + CH4
Figure 1. Microbial food chain (40). Three different metabolic groups of
microbes are required to decompose complex molecules to methane and carbon
dioxide. The principle intermediates and the major route of carbon flow (solid
lines) are shown.
Page 20
6
II. Biochemistry.
Methanoarchaea are diverse in physiology and phylogeny; however, they show
similarity in their metabolic pathways. These unique biochemical processes involve
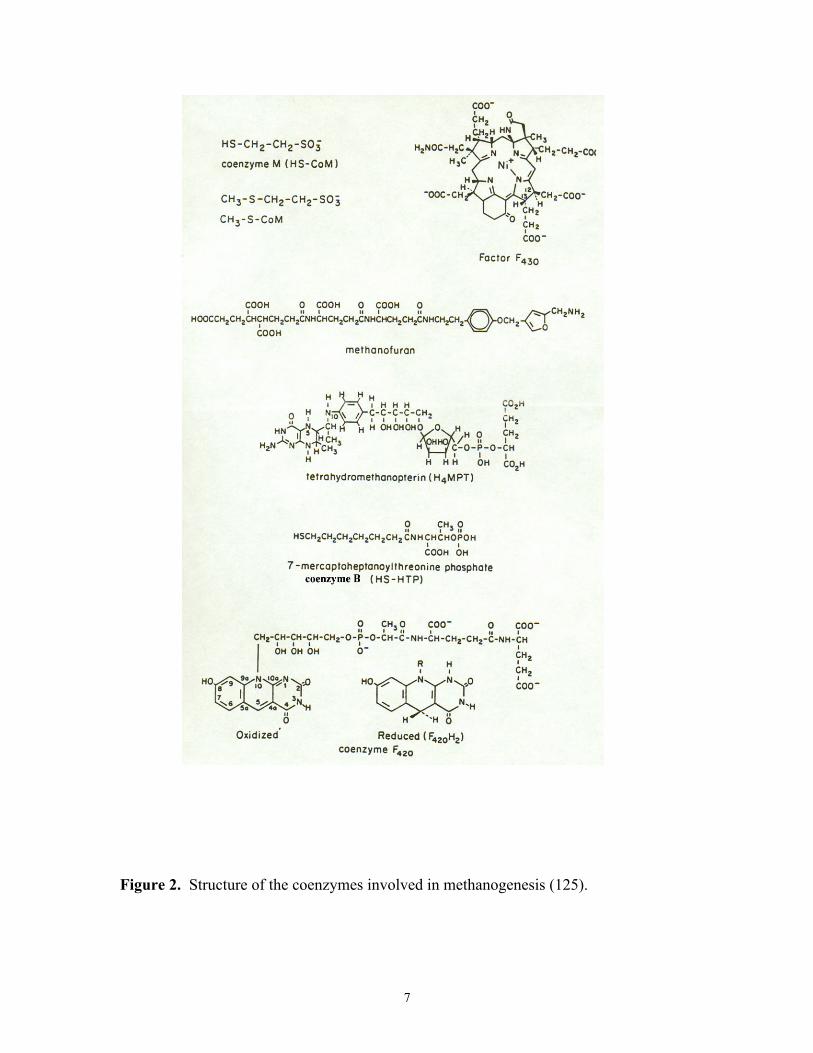
several novel coenzymes (Fig 2) (125).
Coenzymes. Methanofuran (MF), a low molecular weight C1-intermediate
carrier, has been found in methanoarchaea and a SO42- reducing archeaeon, Archaeglobus
fulgidus (60, 126). MF binds CO2 and forms formyl methanofuran in the first step of the
CO2 reduction pathway (76). Tetrahydromethanopterin (H4MPT) has a similar structure
and function to tetrahydrofolate found in the Eucarya and Bacteria domains (123).
Tetrahydrosarcinapterin (H4SPT), isolated from Methanosarcina sp., has an additional
glutamyl group which differs from H4MPT (119). H4MPT has been isolated from
methanoarchaea, Archaeoglobus fulgidus, and the methylotroph Methylobacterium
extorquens AM1 from the Bacteria domain (19). Coenzyme M is first found in
methanoarchaea and is the smallest of all coenzymes known. The structure is a thiol
attached to sulfonic acid by two methylene groups (110). Coenzyme M is methylated
and CH3CoM is further reduced to methane by CH3CoM methylreductase. This reaction
is found in all methanogenesis pathways (29). Coenzyme B is a low molecular weight,
heat stable, oxygen-sensitive compound (29). It contains a reactive thiol group which
donates electrons in the methyl reductase reaction of CH3CoM (32). Factor F430, named
for its characteristic absorption at 430 nm (46), is a Ni-porphyrin cofactor that is tightly
associated with methylreductase (24-26, 28, 127). F430 is present exclusively in the
methanoarchaea (27). F420 has structural resemblance to FMN or FAD, but it is an
obligate 2-electron carrier equivalent to NAD(P) (123). The redox potential of F420 is in a
range of -340 to –350 mV. Due to the strong fluorescence of F420, it has been used to
determine the presence of methanoarchaea in mixed cultures. Factor III is a corrinoid-
containing compound. It is a component of methyl transferases and the carbon monoxide
dehydrogenase/acetyl CoA synthase complex (78).
Page 21
7
Figure 2. Structure of the coenzymes involved in methanogenesis (125).
Page 22
8
Methanoarchaea also contain cofactors that are commonly found in Eucarya and
Bacteria such as thiamin, riboflavin, pyridoxine, biotin, niacin, panthothenate, p-amino
benzoic acid, and molybdopterin (59, 75).
CO2 reduction pathway. Our knowledge of methanogenesis from the CO2
reduction pathway is mostly derived from studies of Methanobacterium
thermoautotrophicum strains ∆H and Marburg (37). Figure 3 illustrates the CO2
reduction pathway. The process is conducted by several one-carbon (C1) intermediate
carriers (14). Electrons for reductive steps in the pathway are derived from the oxidation
of H2 by hydrogenase or formate by formate dehydrogenase (FDH) (14). Hydrogenases
are classified into 3 groups based on their metal composition: NiFe-dehydrogenase (5, 9),
NiFeSe-hydrogenase (86, 131), and Fe-hydrogenase (4). When cells are grown in
formate, FDH oxidizes formate to CO2, which then enters the pathway (107).
Molybdenum- and tungsten-containing formate dehydrogenases have been identified (11-
13, 101, 102). Tungsten-FDH has higher O2 sensitivity than Mo-FDH. FDH contains
FAD, molybdopterin, nonheme iron and acid labile sulfur (8, 56, 116). The FDH of
Methanocoocus vannielii also contains Se (58).
The CO2 reduction pathway is initiated by transferring CO2 to MF followed by the
reduction of CO2 to formyl-MF (76). The formyl group is then transferred to the C1-
carrier H4MPT to produce 5-formyl-H4MPT. Reduction of the formyl moiety proceeds
via F420H2 and involves methenyl, methylene, and methyl redox states (104). Next, the
methyl group is transferred to coenzyme M by a corrinoid-containing methyltransferase
(115). The methyl group of CH3-CoM is finally reductively demethylated to CH4 by the
enzyme complex methyl-CoM methyl reductase (MCR) (32, 33). Electrons for methyl
CoM reduction are derived from coenzyme B which, after oxidation, bonds with
coenzyme M to form the heterodisulfide CoM-S-S-CoB as a byproduct (34). Two MCR
isoenzymes have been identified. Expression of the enzymes is growth phase dependent
(88, 93) and is correlated to H2 levels in the growth medium (15, 83, 120). Recently, the
crystal structure of MCR I was determined and has helped elucidate the active site and
Page 23
9
Figure 3. The CO2 reduction pathway of methanogenesis (125). X, unknown electron
carrier; MFR, methanofuran; H4MPT, tetrahydromethanopterin; HS-CoM, coenzyme
M; HS-CoB, coenzyme B.
CO2XH2 + MFR
H2O + X
CHO-MFR
CHO-H4MPT
CH H4MPT
CH2 H4MPT
CH3-H4MPT
CH3-S-CoM
CH4
H4MPT
MFR
H2O
F420H2
F420
F420H2
F420
CoM-SH
H4MPT
CoB-SH
CoM-S-S-CoB
Page 24
10
catalytic mechanism (35). The MCR enzymes contain the F430-Ni porphyrin coenzyme in
which Ni (I) is the catalytically active form (6, 54, 99). The CoM-S-S-CoB is
regenerated to active thiol compounds by a heterodisulfide reductase (49, 51, 104).
Electrons are derived from either H2 or formate.
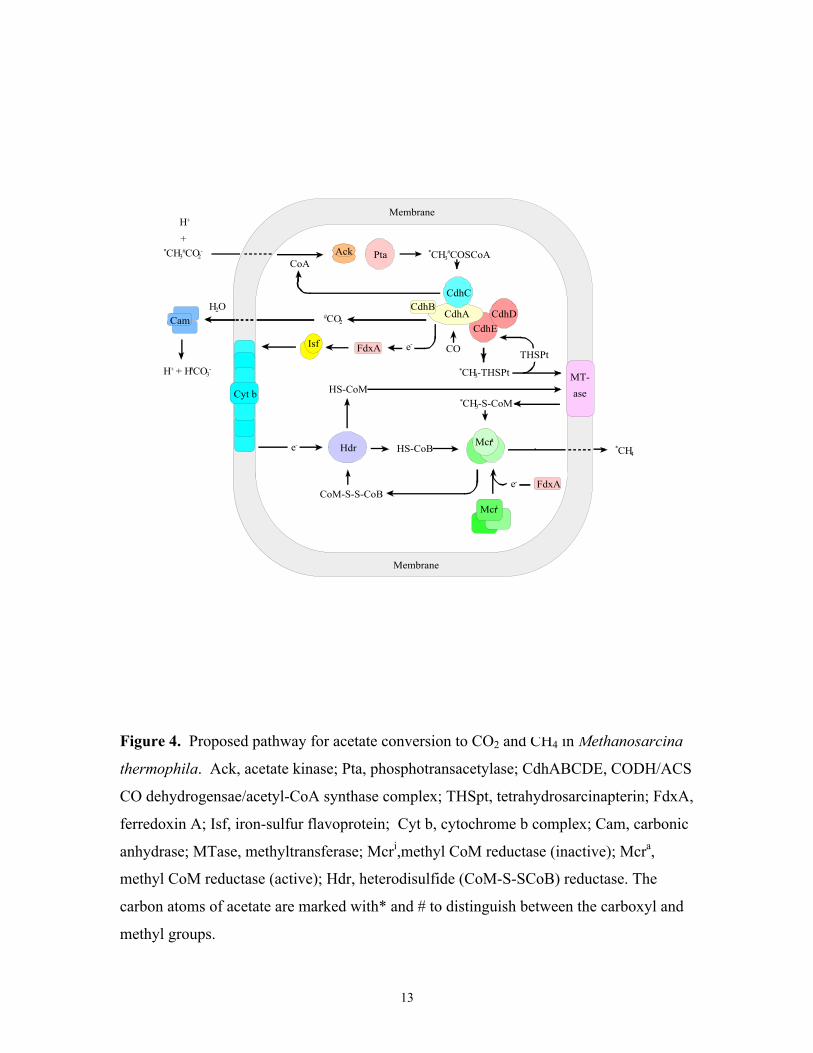
Acetate fermentation pathway. The fermentation of acetate contributes two-
thirds of all biologically produced methane. Figure 4 summarizes the pathway of acetate
conversion to methane and CO2. In summary, the methyl group of acetate is reduced to
methane by electrons derived from the oxidation of the carbonyl group to CO2. Perhaps
the best characterized acetate fermentation pathway is from Methanosarcina thermophila
TM1. Methanosarcina and Methanosaeta are capable of growth on acetate; however,
they exhibit different affinities for acetate. At high acetate concentrations
Methanosarcina predominate, whereas in acetate-limited environments Methanosaeta
out-competes Methanosarcina (82, 89, 124, 135).
The first step of the acetate fermentation pathway requires activation of acetate.
In Methanosarcina, acetate kinase (3) and phosphotransacetylase (79) activate acetate to
acetyl CoA. In Methanosaeta this reaction is catalyzed by a single enzyme, acetyl CoA
synthetase (acetate thiokinase) (55). The carbon monoxide dehydrogenase/acetyl CoA
synthase (CODH/ACS) enzyme complex is a central enzyme in the pathway.
CODH/ACS catalyzes the cleavage of the C-C and C-S bonds of acetyl CoA, the
oxidation of CO to CO2, and transfer of the methyl group to H4SPT (95, 113).
CODH/ACS enzymes are widespread in procaryotes from both the Bacteria and Archaea
domains, and play roles in oxidation of CO, synthesis of acetyl CoA, or cleavage of
acetyl CoA (38). Enzymes from Methanosarcina sp. contain 5 different subunits which
can be divided into three components (1, 43, 69). Based on the studies of CODH/ACS
from M. thermophila and Clostridium thermoaceticum, the first component, a Ni/Fe-S
enzyme comprised of two subunits, cleaves acetyl CoA and transfers the methyl moiety
to the second component (53, 77). The Ni/Fe-S component also oxidizes CO and reduces
ferredoxin (111, 112, 114). The second component, the two-subunit Co/Fe-S enzyme,
contains factor III in which the active Co (I) is methylated (1, 53). The Co/Fe-S
Page 25
11
component is a methyltransferase that transfers the methyl group from Co (III) to H4SPT
resulting in CH3-H4SPT (44, 45). The third component is very unstable and only the
truncated subunit from M. barkeri has been characterized. It appears to have acetyl
transferase activity that is responsible for binding of CoA and acetyl CoA (45). The
genes encoding the five subunits of CODH/ACS from M. thermophila cluster in an
operon (cdh) (80). In addition to the genes encoding the five subunits, a sixth open
reading frame (ORF) is co-transcribed with the cdh operon. It was suggested that this
ORF may encode a protein required for maturation of CODH/ACS (36, 39). The methyl
moiety on CH3-H4SPT is finally transferred to CoM by methyl transferase. Methyl
transferase from acetate grown cells has not been characterized. However, it is proposed
that a corrinoid-containing methyl transferase, as present in CO2-reducing
methanoarchaea, is likely to be involved (36, 37). CH3-SCoM is reductively
demethylated to methane in the same way as in the final step in the CO2 reduction
pathway.
Electrons derived from CO oxidation by the Ni/Fe-S component are used to
reduce ferredoxin (1, 114). Ferredoxin from M. thermophila contains 2 [4Fe-4S] centers
(20), which are potentially coordinated by two cysteine motifs of CXXCXXCXXXCP
(21). Electrons from ferredoxin are eventually transferred to heterodisulfide reductase
(HDR) to generate the active sulfhydryl forms of CoM and CoB, as described in the CO2-
reducing pathway. A reconstituted CO:CoM-S-S-CoB oxidoreductase system can be
established with the following purified components: ferredoxin, CODH/ACS,
membranes, and HDR (92). Electron carriers between ferredoxin and HDR have been
identified, including novel iron-sulfur flavoprotein (Isf) and membrane bound carriers.
Heterologously-produced Isf is a homodimer containing two FMN, 7-8 Fe and acid labile
S. Isf stimulates electron transfer from ferredoxin to the heterodisulfide reductase (74).
Further characterization of Isf is described in chapters 4 and 5. At present, what is known
about the electron transport chain is drawn from the CO:CoM-S-S-CoB oxidoreductase
system. The midpoint potentials (Em) determined for each component is consistent with
the electron flow as CODH →ferredoxin →Isf →cytochrome B→ heterodisulfide
reductase (36), but other components may be required. HDRs from M. thermophila and
Page 26
12
M. barkeri have been purified (72, 105), and both contain b-type hemes and 4Fe-4S
clusters. They lack FAD, which is different from the HDR of M. thermoautotrophicum.
Page 27
13
Figure 4. Proposed pathway for acetate conversion to CO2 and CH4 in Methanosarcina
thermophila. Ack, acetate kinase; Pta, phosphotransacetylase; CdhABCDE, CODH/ACS
CO dehydrogensae/acetyl-CoA synthase complex; THSpt, tetrahydrosarcinapterin; FdxA,
ferredoxin A; Isf, iron-sulfur flavoprotein; Cyt b, cytochrome b complex; Cam, carbonic
anhydrase; MTase, methyltransferase; Mcri,methyl CoM reductase (inactive); Mcra,
methyl CoM reductase (active); Hdr, heterodisulfide (CoM-S-SCoB) reductase. The
carbon atoms of acetate are marked with* and # to distinguish between the carboxyl and
methyl groups.
*CH3#COSCoA
CdhCCdhB
CdhA
CdhE
CdhD
*CH3-THSPt
THSPt
*CH3-S-CoM
*CH4
Mcra
Mcri
FdxA
FdxA
e-
e-
Cam
Isf
e- Hdr
HS-CoM
HS-CoB
CoM-S-S-CoB
MT-
ase
H+ + H#CO3-
H2O
H+
*CH3#CO2
-
+
CoA
CO
Pta
#CO2
Membrane
Membrane
Cyt b
Ack
Page 28
14
III. Bioenergetics and electron transport carriers.
As in organisms belonging to the Bacteria and Eucarya domains, ATP is a
general currency in methanoarchaea. Substrate level phosphorylation and electron
transport phosphorylation are the two major mechanisms for ATP synthesis in all
procaryotes. So far, there is no evidence for ATP synthesis by substrate level
phosphorylation in methanoarchaea (85). It has been proposed that a chemiosmotic
mechanism with electron transport-driven phosphorylation is required for ATP synthesis
(117). Several experiments have been performed to test this hypothesis. A number of
thermodynamically favorable reactions associated with ion gradient formation have been
described. Methanoarchaea use both proton and sodium gradients for ATP synthesis and
endergonic reactions. The formation of ion gradients occurs during electron transfer
through a membrane-bound pathway that results in reduction of CoB-S-S-CoM. This
reduction is dependent on H2, F420, or ferredoxin in different methanogenic pathways
(94).
Knowledge of electron transfer pathways in methanogenesis is limited. Not all
electron carriers involved in this process are known; however several redox cofactors
from methanoarchaea have been identified, purified, and characterized. Although some
of these cofactors can serve as electron carriers, their physiological roles are uncertain.
Examples of known electron carriers involved in the electron transfer process are
ferredoxin, cytochromes b or c, and F420.
Ferredoxins are small, acidic redox proteins which contain clusters of non-heme
irons and acid labile sulfides. These iron-sulfur clusters are ligated to proteins by
cysteines. Clusters which serve as redox centers have been identified as [2Fe-2S], [3Fe-
4S], and [4Fe-4S] cluster types. The reduction potentials of ferredoxins range from –145
to +400 mV (81), which are suitable for a variety of redox reactions (18, 132). Unlike
several other iron-sulfur proteins, ferredoxin shows no enzyme activity. Ferredoxins
from the methanoarchaea M. thermophila strain TM1 (20, 112, 114), M. barkeri strains
MS (22, 84) and Fusaro (47), and Methanococcus thermolithotrophicus (48) have been
Page 29
15
purified and characterized. These ferredoxins have been shown to be central electron
carriers in anabolic and catabolic pathways in methanoarchaea (3, 4, 41, 47, 48, 111)
Polyferredoxins are a class of proteins containing multiple iron-sulfur clusters.
Polyferredoxin from M.thermoautotrophicum contains 12[4Fe-4S] clusters (50, 96). The
genes encoding polyferredoxin (mvh B) from M. thermoautotrophicum, Methanothermus
fervidus, and Methanococcus voltae are located in the methyl viologen (MV)
hydrogenase (mvh) operon and are conserved (50). Therefore, polyferredoxin has been
proposed to function in association with the MV-hydrogenase system; however, the
reduction of polyferredoxin by MV-hydrogenase in the presence of H2 is very low. The
second possible role for polyferredoxin is iron-storage (50, 96).
Cytochromes are ubiquitous membrane-bound electron transfer components in
nature. Only the methanoarchaea that are able to grow on methyl-containing compounds,
such as acetate, methanol and methylamine, contain cytochromes (71). This indicates
that cytochromes are not involved in methane formation from CO2 and H2, or formate. It
has been proposed that cytochromes function in the methyl oxidation of these methyl-
containing compounds (71). Two b-type cytochromes and one c-type cytochrome were
detected in methanol- and methylamine-grown cells whereas the acetate-grown cells
contain an additional b-type cytochrome (70). The midpoint potentials for b-type
cytochromes found in methanol-, methylamine- and acetate-grown cells are –325, -183,
and –253 mV, respectively (70). Recently, the membrane fraction from methanol-grown
Methanosarcina mazei Gö1 revealed two b-type cytochromes and two c-type
cytochromes (63). The midpoint potentials for the b-type cytochromes are –135 and -240
mV and -140 and -230 mV for the c-type cytochromes (63). There has been evidence
suggesting that cytochromes are involved in electron transfer between F420H2 and CoM-
S-S-CoB (63).
F420 is another required electron carrier for methanogenesis. It is an obligate two-
electron carrier in Archaea which functions in analogy to NAD and NADP in the
Eucarya and Bacteria domains. The variable amounts of F420 among methanoarchaea
may be due to different requirements for this electron carrier in diverse metabolism (31).
Page 30
16
Proteins known to interact with F420 include F420-reducing hydrogenase, NADP-F420
oxidoreductase, formate dehydrogenase, methylenetetrahydromethanopterin
dehydrogenase, methylenetetrahydromethanopterin reductase, secondary alcohol
dehydrogenase, puruvate synthase, and α-ketoglutarate synthase (29).
The FMN-containing flavoprotein, flavoprotein A, was recently purified, cloned,
and sequenced from M. thermoautotrophicum strain ∆H and Marburg (87, 121). It
copurified with the H2:heterodisulfide oxidoreductase complex. The flavoprotein A from
strain Marburg was purified as a homotetramer with a 43 kDa molecular mass per subunit
whereas the one from strain ∆H was a homodimer with a monomeric molecular mass of
45 kDa. The expression of either flavoprotein increased when cells were grown in iron
depleted media. The physiological role for flavoprotein A remains speculative, although
the FMN containing property suggests a function as an electron carrier. It may function
to substitute an essential iron-containing protein during iron starvation. Polyferredoxin
seems to be the most feasible protein for which flavoprotein A can substitute. With many
completed genome sequencing projects, a number of flavoprotein A related protein
sequences have been identified. The sequence comparisons reveal a conserved region for
FMN binding (122).
Methanophenazine is a recently discovered redox-active cofactor. It was first
isolated from the membranes of M. mazei Gö1 and shown to be very hydrophobic. The
structure is a 2-hydroxy phenazine derivative connected to a polyisoprenoid by an ether
bond. It has a molecular mass of 538 Da (2). The 2-hydroxy phenazine (a soluble
analogue of methanophenazine) is able to accept electrons from both F420-hydrogenase
and MV hydrogenase (2). Furthermore, reduced methanophenazine donates electrons to
heterodisulfide reductase from M. thermophila (10). Therefore, methanophenazine has
been proposed to play an important role in vivo in membrane-bound electron transport
systems of F420-hydrogenase: heterodisulfide oxidoreducatase and H2: heterodisulfide
oxidoreducatase. Diphenyleneiodonium chloride (DPI), a competitive inhibitor of
methanophenazine, inhibited both membrane-bound electron transport systems of M.
mazei Gö1 (17).
Page 31
17
Preliminary investigation of M. thermoautotrophicum membranes is consistent
with the presence of low potential electron carriers (73). EPR studies suggest that iron-
sulfur clusters are membrane components. Upon the addition of either F420 or CH3-CoM
to membranes, the EPR spectra changes, results which are consistent with the
involvement of membrane components in the electron transfer process (98).
Page 32
18
REFERENCES
1. Abbanat, D. R., and J.G. Ferry. 1991. Resolution of component proteins in an
enzyme complex from Methanosarcina thermophila catalyzing the synthesis or
cleavage of acetyl-CoA. Proc. Natl. Acad. Sci. USA. 88:3272-3276.
2. Abken, H.-J., M. Tietze, J. Brodersen, S. Bäumer, U. Beifuss, and U.
Deppenmeier. 1998. Isolation and characterization of methanophenazine and the
function of phenazines in membrane-bound electron transport of Methanosarcina
mazei Gö1. 180:2027-2032.
3. Aceti, D. J., and J.G. Ferry. 1988. Purification and characterization of acetate
kinase from acetate-grown Methanosarcina thermophila. J. Biol. Chem.
263:15444-15448.
4. Adams, W.W., L.E. Mortenson, J-S. Chen. 1981. Hydrogenase. Biochim.
Biophys. Acta. 594:105-176.
5. Albracht, S. P. J., E.G. Graf, and R. K.Thauer. 1982. The EPR properties of
nickel in hydrogenase from Methanobacterium thermoautotrophicum. FEBS Lett.
140:311-313.
6. Albracht, S. P. J., D. Ankel-Fuchs, R. Bocher, J. Ellermann, J. Moll, J. W.
Van der Zwaan, and R. K. Thauer. 1988. Five new EPR signals assigned to
nickel in methyl-coenzyme M reductase from Methanobacterium
thermoautotrophicum, strain Marburg. Biochim. Biophys. Acta. 941:86-102.
7. Aldrich, H. C., R.W. Robinson, and D.S. Williams. 1986. Ultrastructure of
Methanosarcina mazei. Syst. Appl. Microbiol. 7:314-319.
8. Barber, M. J., L. M. Siegel, N. L Schauer, H. D. May, and J. G. Ferry. 1983.
Formate dehydrogenase from Methanobacterium formicicum. Electron
paramagnetic resonance spectroscopy of the molybdenum and iron-sulfur centers.
J. Biol. Chem. 258:10839-10845.
9. Baron, S. F., and J. G. Ferry. 1989. Purification and properties of the
membrane-associated coenzyme F420-reducing hydrogenase from
Methanobacterium formicicum. J. Bacteriol. 171:3846-3853.
10. Baumer, S., E. Murakami, J. Brodersen, G. Gottschalk, S. W. Ragsdale, and
U. Deppenmeier. 1998. The F420H2:heterodisulfide oxidoreductase system from
Page 33
19
Methanosarcina species. 2-Hydroxyphenazine mediates electron transfer from
F420H2 dehydrogenase to heterodisulfide reductase. FEBS Lett. 428(3):295-8.
11. Bertram, P. A., M. Karrasch, R. A. Schmitz, R. Bocher, S. P. J. Albracht,
and R. K. Thauer, 1994. Formylmethanofuran dehydrogenases from
methanogenic archaea - substrate specificity, EPR properties and reversible
inactivation by cyanide of the molybdenum or tungsten iron- sulfur proteins. Eur.
J. Biochem. 220:477-484.
12. Bertram, P. A., and R. K. Thauer. 1994. Thermodynamics of the
formylmethanofuran dehydrogenase reaction in Methanobacterium
thermoautotrophicum. Eur. J. Biochem. 226:811-818.
13. Bertram, P. A., R. A. Schmitz, D. Linder, and R.K. Thauer. 1994. Tungstate
can substitute for molybdate in sustaining growth of Methanobacterium
thermoautotrophicum - identification and characterization of a tungsten
isoenzyme of formylmethanofuran dehydrogenase. Arch. Microbiol. 161:220-
228.
14. Blaut, M., V. Muller, and G. Gottschalk. 1992. Energetics of methanogenesis
studied in vesicular systems. J. Bioener. Biomemb. 24:529-546.
15. Bonacker, L. G., S. Baudner, and R. K. Thauer. 1992. Differential expression
of the two methyl-coenzyme M reductases in Methanobacterium
thermoautotrophicum as determined immunochemically via isoenzyme-specific
antisera [published erratum appears in Eur. J. Biochem. 1992 Aug 1;207(3):1129].
Eur. J. Biochem. 206(1):87-92.
16. Boone, D. R., W.B. Whitman, and P. Rouviere. 1993. Diversity and Taxonomy
of Methanogens. Methanogenesis, J.G. Ferry, ed. Chapman and Hall, New York.
35-80.
17. Brodersen, J., S. Baumer, H. J. Abken, G. Gottschalk, and U. Deppenmeier.
1999. Inhibition of membrane-bound electron transport of the methanogenic
archaeon Methanosarcina mazei Gö1 by diphenyleneiodonium. Eur. J. Biochem.
259(1-2):218-24.
18. Bruschi, M. G., F. 1988. Structure, function and evolution of bacterial
ferredoxins. FEMS Microbiol. Rev. 54:155.
Page 34
20
19. Chistoserdova, L., J. A. Vorholt, R. K. Thauer, and M. E. Lidstrom. 1998. C1
transfer enzymes and coenzymes linking methylotrophic bacteria and
methanogenic Archaea. Science. 281(5373):99-102.
20. Clements, A. P., L. Kilpatrick, W.-P. Lu, S.W. Ragsdale, and J.G. Ferry.
1994. Characterization of the iron-sulfur clusters in ferredoxin from acetate-
grown Methanosarcina thermophila. J. Bacteriol. 176:2689-2693.
21. Clements, A. P., and J.G. Ferry. 1992. Cloning, nucleotide sequence, and
transcriptional analyses of the gene encoding a ferredoxin from Methanosarcina
thermophila. J. Bacteriol. 174:5244-5250.
22. Daas, P. J. H., W. R. Hagen, J. T. Keltjens, and G. D. Vogels. 1994.
Characterization and determination of the redox properties of the 2[4Fe-4S]
ferredoxin from Methanosarcina barkeri strain MS. FEBS Lett. 356:342-344.
23. Daniels, L., G. Fuchs, R. K. Thauer, and J. G. Zeikus. 1977. Carbon monoxide
oxidation by methanogenic bacteria. J. Bacteriol. 132:118-126.
24. Diekert, G., R. Jaenchen, and R. K. Thauer. 1980. Biosynthetic evidence for a
nickel tetrapyrrole structure of factor F430 from Methanobacterium
thermoautotrophicum. FEBS Lett. 119:118-120.
25. Diekert, G., H-H. Gilles, R. Jaenchen, and R. K Thauer. 1980. Incorporation
of 8 succinate per mol nickel into factors F430 by Methanobacterium
thermoautotrophicum. Arch. Microbiol. 128:256-262.
26. Diekert, G., B. Weber, and R.K. Thauer. 1980. Nickel dependence of factor
F430 content in Methanobacterium thermoautotrophicum. Arch. Microbiol.
127:273-278.
27. Diekert, G., U. Konheiser, K. Piechulla, and R.K. Thauer. 1981. Nickel
requirement and factor F430 content of methanogenic bacteria. J. Bacteriol.
148:459-464.
28. Diekert, G., B. Klee, and R.K. Thauer. 1980. Nickel, a component of factor F430
from Methanobacterium thermoautotrophicum. Arch. Microbiol. 124:103-106.
29. DiMarco, A. A., , A. T. Bobik, and R. S. Wolfe. 1990. Unusual coenzymes of
methanogenesis. Annu. Rev. Biochem. 59:355-394.
Page 35
21
30. Doddema, H. J., J. W. M. Derksen, and G.D. Vogels. 1979. Fimbriae and
flagella of methanogenic bacteria. FEMS Microbiol. Lett. 5:135-138.
31. Eirich, L. D., G. D.Vogels, and R. S. Wolfe. 1979. Distribution of coenzyme
F420 and properties of its hydrolytic fragments. J. Bacteriol. 140:1:20-27.
32. Ellermann, J., R. Hedderich, R. Bocher, and R. K. Thauer. 1988. The final
step in methane formation. Investigations with highly purified methyl-CoM
reductase (component C) from Methanobacterium thermoautotrophicum (strain
Marburg). Eur. J. Biochem. 172:669-677.
33. Ellermann, J., S. Rospert, R. K.Thauer, M. Bokranz, A. Klein, M. Voges, and
A. Berkessel. 1989. Methyl-coenzyme-M reductase from Methanobacterium
thermoautotrophicum (strain Marburg): purity, activity and novel inhibitors. Eur.
J. Biochem. 184:63-68.
34. Ellermann, J., A. Kobelt, A. Pfaltz, and R. K. Thauer. 1987. On the role of N-
7 mercaptoheptanoyl-O-phospho-L-threonine (componentB) in the enzymatic
reduction of methyl-coenzyme M to methane (FEB 05028). FEBS Lett. 220:358-
362.
35. Ermler, U., W. Grabarse, S. Shima, M. Goubeaud, and R. K. Thauer. 1997.
Crystal structure of methyl-coenzyme M reductase: the key enzyme of biological
methane formation. Science. 278:1457-62.
36. Ferry, J., G. 1997. Enzymology of the fermentation of acetate to methane by
Methanosarcina thermophila. BioFactors. 6:25-35.
37. Ferry, J. G. 1992. Biochemistry of methanogenesis. Crit. Rev. Biochem. Mol.
Biol. 27:473-503.
38. Ferry, J. G. 1995. CO dehydrogenase. Ann. Rev. Microbiol. 49:305-333.
39. Ferry, J. G. 1999. Enzymology of one-carbon metabolism in methanogenic
pathways. FEMS Microbiol. Rev. 23:13-38.
40. Ferry, J. G. 1997. Methane: small molecule, big impact [comment]. Science.
278:1413-4.
41. Fischer, R., and R.K. Thauer. 1990. Ferredoxin-dependent methane formation
from acetate in cell extracts of Methanosarcina barkeri (strain MS). FEBS Lett.
269:368-372.
Page 36
22
42. Fox, G. E., L. J. Marrum, W. E. Balch, R. S. Wolfe, and C. R. Woese. 1977.
Classification of methanogenic bacteria by 16S ribosomal RNA characterization.
Proc. Natl. Acad. Sci. USA. 74:4537-4541.
43. Grahame, D. A., and T. C. Stadtman. 1987. Carbon monoxide dehydrogenase
from Methanosarcina barkeri. Disaggregation, purification, and physicochemical
properties of the enzyme. J. Biol. Chem. 262:3706-3712.
44. Grahame, D. A. 1991. Catalysis of Acetyl-CoA Cleavage and
tetrahydrosarcinapterin methylation by a carbon monoxide dehydrogenase-
corrinoid enzyme complex. J Biol Chem. 266:22227-22233.
45. Grahame, D. A., and E. Demoll. 1996. Partial reactions catalyzed by protein
components of the acetyl-CoA decarbonylase synthase enzyme complex from
Methanosarcina barkeri. J. Biol. Chem. 271:8352-8358.
46. Gunsalus, R. P., and R. S. Wolfe. 1978. Chromophoric factors F342 and F430 of
Methanobacterium thermoautotrophicum. FEMS Microbiol. Lett. 3:191-193.
47. Hatchikian, E. C., M. Bruschi, N. Forget, and M. Scandellari. 1982. Electron
transport components from methanogenic bacteria: the ferredoxin from
Methanosarcina barkeri (strain Fusaro). Biochem. Biophys. Res. Comm.
109:1316-1323.
48. Hatchikian, E. C., M. L. Fardeau, M. Bruschi, J.P. Belaich, A. Chapman, R.
Cammack. 1989. Isolation, characterization, and biological activity of the
Methanococcus thermolithotrophicus ferredoxin. J. Bacteriol. 171:2384-2390.
49. Hedderich, R., A. Berkessel, and R. K. Thauer. 1989. Catalytic properties of
the heterodisulfide reductase involved in the final step of methanogenesis. FEBS
Lett. 255:67-71.
50. Hedderich, R., S. P. J. Albracht, D. Linder, J. Koch, and R. K. Thauer. 1992.
Isolation and characterization of polyferredoxin from Methanobacterium-
thermoautotrophicum - The mvhB gene product of the methylviologen-reducing
hydrogenase operon. FEBS Letters. 298:65-68.
51. Hedderich, R., A. Berkessel, and R. K. Thauer. 1990. Purification and
properties of heterodisulfide reductase from Methanobacterium
thermoautotrophicum (strain Marburg). Eur. J. Biochem. 193:255-261.
Page 37
23
52. Hungate, R. E. 1967. A roll tube method for cultivation of strict anaerobes.
Methods in Microbiology, Norris J.R., and Ribbons D.W. ed, Academic Press,
New York. 2B:117-132.
53. Jablonski, P. E., W. P. Lu, S. W. Ragsdale, and J. G. Ferry. 1993.
Characterization of the metal centers of the corrinoid;iron-sulfur component of the
CO dehydrogenase enzyme complex from Methanosarcina thermophila by EPR
spectroscopy and spectroelectrochemistry. J Biol Chem. 268:325-329.
54. Jaun, B., and A. Pfaltz. 1988. Coenzyme F430 from methanogenic bacteria:
methane formation by reductive carbon-sulfur bond cleavage of methyl
sulphonium ions catalysed by F430 pentamethyl ester. J. Chem. Soc. Chem.
Commun.:293-294.
55. Jetten, M. S. M., A. J. M. Stams, and A. J. B Zehnder. 1989. Isolation and
characterization of acetyl-coenzyme A synthetase from Methanothrix soehngenii.
J. Bacteriol. 171:5430-5435.
56. Johnson, J. L., N. R. Bastian, N. L. Schauer, J. G. Ferry, and K.V.
Rajagopalan. 1991. Identification of molybdopterin guanine dinucleotide in
formate dehydrogenase from Methanobacterium formicicum. FEMS Microbiol.
Lett. 77:2-3.
57. Jones, J. B., B. Bowers, and T. C. Stadtman. 1977. Methanococcus vannielii:
ultrastructure and sensitivity to detergents and antibiotics. J. Bacteriol. 130:1357-
1363.
58. Jones, J. B, and T. C. Stadtman. 1981. Selenium-dependent and selenium-
independent formate dehydrogenases of Methanococcus vannielii. J. Biol. Chem.
256;2:656-663.
59. Jones, W. J., D. P. Nagle, and W. B. Whitman. 1987. Methanogens and the
diversity of archaebacteria. Microbiol. Rev. 51:135-177.
60. Jones, W. J., M. I. Donnelly, and R. S. Wolfe. 1985. Evidence of a common
pathway of carbon dioxide reduction to methane in methanogens. J. Bacteriol.
163(1):126-31.
Page 38
24
61. Kalmokoff, M. L., and K.F. Jarrell. 1991. Cloning and sequencing of a
multigene family encoding the flagellins of Methanococcus voltae. J. Bacteriol.
173:7113-7125.
62. Kalmokoff, M. L., K. F. Jarrell, and S. F. Koval. 1988. Isolation of flagella
from the archaebacterium Methanococcus voltae by phase separation with Triton
X-114. J. Bacteriol. 170:1752-8.
63. Kamlage, B., and M. Blaut. 1992. Characterization of cytochromes from
Methanosarcina strain Göl and their involvement in electron transport during
growth on methanol. J. Bacteriol. 174:3921-7.
64. Keltjens, J. T. 1984. Coenzymes of methanogenesis from hydrogen and carbon
dioxide. Antonie Van Leeuwenhoek. 50:383-96.
65. Kennes, C., M. C. Veiga, and L. Bhatnagar. 1998. Methanogenic and
perchloroethylene-dechlorinating activity of anaerobic granular sludge. Appl.
Microbiol. Biotechnol. 50:484-8.
66. Kennes, C., W. M. Wu, L. Bhatnagar, and J. G. Zeikus. 1996. Anaerobic
dechlorination and mineralization of pentachlorophenol and 2,4,6-trichlorophenol
by methanogenic pentachlorophenol-degrading granules. Appl. Microbiol.
Biotechnol. 44:801-6.
67. Kiene, R. P. 1991. Production and consumption of methane in aquatic systems.
Microbial production and consumption of greenhouse gases: methane, nitrogen
oxides, and halomethanes, J.E. Roger and W.B. Whitman, ed. American society
for microbiology, Washington, D.C. 111-146.
68. Kreisl, P., and O. Kandler. 1986. Chemical structure of the cell wall polymer
Methanosarcina. Syst. Appl. Microbiol. 7:293-299.
69. Krzycki, J. A., and J. G Zeikus. 1984. Characterization and purification of
carbon monoxide dehydrogenase from Methanosarcina barkeri. J. Bacteriol.
158:231-237.
70. Kuhn, W., and G. Gottschalk. 1983. Characterization of the cytochromes
occurring in Methanosarcina species. Eur. J. Biochem. 135:89-94.
Page 39
25
71. Kuhn, W., K. Fiebig, H. Hippe, R. A. Mah, B. A. Huser, and G. Gottschalk.
1983. Distribution of cytochromes in methanogenic bacteria. FEMS Microbiol.
Lett. 20:407-410.
72. Kunkel, A., M. Vaupel, S. Heim, R. K. Thauer, and R. Hedderich. 1997.
Heterodisulfide reductase from methanol-grown cells of Methanosarcina barkeri
is not a flavoenzyme. Eur. J. Biochem. 244:226-34.
73. Lancaster, J. J. R . 1986. A unified scheme for carbon and electron flow coupled
to ATP synthesis by substrate-level phosphorylation in the methanogenic bacteria.
FEBS. Lett. 199:12-18.
74. Latimer, M. T., M. H. Painter, and J. G. Ferry. 1996. Characterization of an
iron-sulfur flavoprotein from Methanosarcina thermophila. J. Biol. Chem.
271:24023-24028.
75. Leigh, J., A . 1983. Levels of water-soluble vitamins in methanogenic and non-
methanogenic bacteria. Appl. Environ. Microbiol. 45:800-803.
76. Leigh, J. A., K. L. Jr. Rinehart, and R. S. Wolfe. 1985. Methanofuran (carbon
dioxide reduction factor), a formyl carrier in methane production from carbon
dioxide in Methanobacterium. Biochemistry. 24:995-999.
77. Lu, W. P., P. E. Jablonski, M. Rasche, J. G. Ferry, and S. W. Ragsdale. 1994.
Characterization of the metal centers of the Ni-Fe-S component of the carbon-
monoxide dehydrogenase enzyme complex from Methanosarcina thermophila. J.
Biol. Chem. 269:9736-9742.
78. Ludwig, M. L., and R. G. Matthews. 1997. Structure-based perspectives on
B12-dependent enzymes. Annu. Rev. Biochem. 66:269-313.
79. Lundie, L. L., and J. G. Ferry. 1989. Activation of acetate by Methanosarcina
thermophila. purification and characterization of phosphotransacetylase. J. Biol.
Chem. 264:18392-18396.
80. Maupin-Furlow, J. A., and J. G. Ferry. 1996. Analysis of the CO
dehydrogenase/acetyl-coenzyme A synthase operon of Methanosarcina
thermophila. J. Bacteriol. 178:6849-6856.
81. Meyer, J. 1988. The evolution of ferredoxins. Trends Ecol. Evol.. 3:222-226.
Page 40
26
82. Min, H., and S.H. Zinder. 1989. Kinetics of acetate utilization by 2 thermophilic
acetotrophic methanogens - Methanosarcina Sp Strain CALS-1 and Methanothrix
Sp Strain CALS-1. Appl. Env. Microbiol. 55:488-491.
83. Morgan, R. M., T. D. Pihl, J. Nolling, and J. N. Reeve. 1997. Hydrogen
regulation of growth, growth yields, and methane gene transcription in
Methanobacterium thermoautotrophicum deltaH. J. Bacteriol. 179(3):889-98.
84. Moura, I., J. G. Moura, B. Huynh, H. Santos, J. LeGall, and A. V. Xavier.
1982. Ferredoxin from Methanosarcina barkeri: evidence for the presence of a
three-iron center. Eur. J. Biochem. 126:95-98.
85. Muller, V., M. Blaut, and G. Gottschalk. 1993. Bioenergetics of
methanogenesis. Methanogenesis, J.G. Ferry, ed. Chapman and Hall, New York.
360-406.
86. Muth, E., E. Morschel, and A. Klein. 1987. Purification and characterization of
an 8-hyroxy-5-deazaflavin-reducing hydrogenase from the archaebacterium
Methanococcus voltae. Eur. J. Biochem. 169:571-577.
87. Nolling, J., M. Ishii, J. Koch, T. D. Pihl, J. N. Reeve, R.K. Thauer, and
Hedderich, R. 1995. Characterization of a 45-kda flavoprotein and evidence for a
rubredoxin, two proteins that could participate in electron transport from H2 to
CO2 in methanogenesis in Methanobacterium thermoautotrophicum. Eur. J.
Biochem. 231:628-638.
88. Nolling, J., T. D. Pihl, A. Vriesema, and J. N. Reeve. 1995. Organization and
growth phase-dependent transcription of methane genes in two regions of the
Methanobacterium thermoautotrophicum genome. J. Bacteriol. 177:2460-2468.
89. Ohtsubo, S., K. Demizu, S. Kohno, I. Miura, T. Ogawa, and H. Fukuda. 1992.
Comparison of acetate utilization among strains of an aceticlastic methanogen,
Methanothrix-Soehngenii. Appl. Env. Microbiol. 58:703-705.
90. Oremland, R. S. 1988. Biogeochemistry of methanogenic bacteria. Biology of
anaerobic microorganisms. Zehnder A.J.B. ed. John Wiley & Son, Inc. 641-705.
91. Oremland, R. S., R. P. Kiene, I. Mathrani, M. J. Whiticar, and D.R. Boone.
1989. Description of an estuarine methylotrophic methanogen which grows on
dimethyl sulfide. Appl. Environ. Microbiol. 55:994-1002.
Page 41
27
92. Peer, C. W., M. H. Painter, M. E. Rasche, and J. G. Ferry. 1994.
Characterization of a CO:heterodisulfide oxidoreductase system from acetate-
grown Methanosarcina thermophila. J. Bacteriol. 176:6974-6979.
93. Pihl, T. D., S. Sharma, , and J.N. Reeve. 1994. Growth phase-dependent
transcription of the genes that encode the two methyl coenzyme m reductase
isoenzymes and n-5- methyltetrahydromethanopterin:coenzyme M
methyltransferase in Methanobacterium thermoautotrophicum delta H. J.
Bacteriol. 176:6384-6391.
94. Rasche, M., E, and J. G. Ferry, 1996. Methanogens and Archaea, Molecular
biology of. Encyclopedia of Molecular Biology:66-78.
95. Raybuck, S. A., S. E. Ramer, D. R. Abbanat, J. W. Peters, W. H. Orme-
Johnson, J. G. Ferry, and C. T. Walsh. 1991. Demonstration of carbon-carbon
bond cleavage of acetyl coenzyme A by using isotopic exchange catalyzed by the
CO dehydrogenase complex from acetate-grown Methanosarcina thermophila. J.
Bacteriol. 173:929-932.
96. Reeve, J. N., G. S. Beckler, D. S. Cram, P. T. Hamilton, J. W. Brown, J. A.
Krzycki, A. F. Kolodziej, L. Alex, W. H. Ormejohnson, and C.T. Walsh.
1989. A hydrogenase-linked gene in Methanobacterium thermoautotrophicum
strain H encodes a polyferredoxin. Proc. Natl. Acad. Sci. USA. 86:3031-3035.
97. Robinson, J. A., and J. M. Tiedje. 1984. Competition between sulfate-reducing
and methanogenic bacteria for H2 under resting and growing conditions. Arch.
Microbiol. 137:26-32.
98. Rogers, K. R., K. Gillies, and J. R. Lancaster Jr. 1988. Iron-sulfur centers
involved in methanogenic electron transfer in Methanobacterium
thermoautotrophicum. Biochem. Biophys. Res. Commun. 153:87-95.
99. Rospert, S., M. Voges, A. Berkessel, S. P. J. Albracht, and R. K. Thauer.
1992. Substrate-analogue-induced changes in the nickel-EPR spectrum of active
methyl-coenzyme-M reductase from Methanobacterium- thermoautotrophicum.
Eur. J. Biochem. 210:101-107.
Page 42
28
100. Schink, B. 1988. Principles and limits of anaerobic degradation: environmental
and technical aspects. Biology of anaerobic bacteria. Zehnder A.J.B. ed. Wiley
Interscience , New York. 771-846.
101. Schmitz, R. A., S. P. J. Albracht, and R. K. Thauer. 1992. A molybdenum and
a tungsten isoenzyme of formylmethanofuran dehydrogenase in the thermophilic
archaeon Methanobacterium wolfei. Eur J Biochem. 209:1013-1018.
102. Schmitz, R. A., S. P. J. Albracht, and R. K. Thauer, 1992. Properties of the
tungsten-substituted molybdenum formylmethanofuran dehydrogenase from
Methanobacterium wolfei. FEBS Lett. 11479:78-81.
103. Schonheit, P. J., K. Kristjansson, and R. K. Thauer. 1982. Kinetic mechanism
for the ability of sulfate reducers to out-compete methanogens for acetate. Arch.
Microbiol. 132:285-288.
104. Schworer, B., and R. K. Thauer. 1991. Activities of formylmethanofuran
dehydrogenase, methylenetetrahydromethanopterin dehydrogenase,
methylenetetrahydromethanopterin reductase, and heterodisulfide reductase in
methanogenic bacteria. Arch. Microbiol. 155:459-465.
105. Simianu, M., E. Murakami, J. M. Brewer, and S.W. Ragsdale. 1998.
Purification and properties of the heme and iron-sulfur containing heterodisulfide
reductase from Methanosarcina thermophila. Biochemistry. 37:10027-39.
106. Sowers, K., R,Gunsalus,R,P. 1988. Adaptation for growth at various saline
concentrations by the archaebacterium Methanosarcina thermophila. J. Bacteriol.
170:998-1002.
107. Sparling, R., and L. Daniels. 1986. Source of carbon and hydrogen in methane
produced from formate by Methanococcus thermolithotrophicus. J. Bacteriol.
168:1402-1407.
108. Sprott, G. D., and T. J. Beveridge. 1993. Microscopy. Methanogenesis, J.G.
Ferry, ed. Chapman and Hall, New York. 81-127.
109. Stams, A. J. 1994. Metabolic interactions between anaerobic bacteria in
methanogenic environments. Antonie Van Leeuwenhoek. 66:271-94.
110. Taylor, C. D, and R. S. Wolfe. 1974. Structure and methylation of coenzyme M
(HSCH2CH2SO3). J. Biol. Chem. 249:4879-4885.
Page 43
29
111. Terlesky, K. C., M. J. Barber, D. J. Aceti, and J. G. Ferry. 1987. EPR
properties of the Ni-Fe-C center in an enzyme complex with carbon monoxide
dehydrogenase activity from acetate-grown Methanosarcina thermophila.
Evidence that acetyl-CoA is a physiological substrate. J. Biol. Chem. 262:15392-
15395.
112. Terlesky, K. C., and J. G. Ferry. 1988. Ferredoxin requirement for electron
transport from the carbon monoxide dehydrogenase complex to a membrane-
bound hydrogenase in acetate-grown Methanosarcina thermophila. J. Biol. Chem.
263:4075-4079.
113. Terlesky, K. C., M. J. Nelson, and J. G. Ferry. 1986. Isolation of an enzyme
complex with carbon monoxide dehydrogenase activity containing a corrinoid and
nickel from acetate-grown Methanosarcina thermophila. J. Bacteriol. 168:1053-
1058.
114. Terlesky, K. C., and J. G. Ferry. 1988. Purification and characterization of a
ferredoxin from acetate-grown Methanosarcina thermophila. J. Biol. Chem.
263:4080-4082.
115. Thauer, R. K. 1990. Energy metabolism of methanogenic bacteria. Biochim.
Biophys. Acta. 1018:256-259.
116. Thauer, R. K., R. Hedderich, and R. Fischer. 1993. Reactions and enzymes
involved in methanogenesis from CO2 and H2. Methanogenesis, J.G. Ferry, ed.
Chapman and Hall, New York. 209-252.
117. Thauer, R. K., K. Jungermann, and K. Decker. 1977. Energy conservation in
chemotrophic anaerobic bacteria. Bacteriol. Rev. 41:100-80.
118. Tylor, S. C. 1991. The global methane budget. Microbial production and
consumption of greenhouse gases:, methane, nitrogen oxides, and halomethanes,
J.E. Roger and W.B. Whitman, ed. American society for microbiology,
Washington, D.C. 7-38.
119. Van Beelen, P., J. F. Labro, J. T. Keltjens, W. J. Geerts, G. D. Vogels, W. H.
Laarhoven, W. Guijt, and C. A. Haasnoot. 1984. Derivatives of methanopterin,
a coenzyme involved in methanogenesis. Eur. J. Biochem. 139:359-65.
Page 44
30
120. Vermeij, P., J. L. Pennings, S. M. Maassen, J. T. Keltjens, and G. D. Vogels.
1997. Cellular levels of factor 390 and methanogenic enzymes during growth of
Methanobacterium thermoautotrophicum delta H. J. Bacteriol. 179(21):6640-8.
121. Wasserfallen, A., K. Huber, and T. Leisinger. 1995. Purification and structural
characterization of a flavoprotein induced by iron limitation in Methanobacterium
thermoautotrophicum marburg. J. Bacteriol. 177:2436-2441.
122. Wasserfallen, A., S. Ragettli, Y. Jouanneau, and T. Leisinger. 1998. A family
of flavoproteins in the domains Archaea and Bacteria. Eur J Biochem. 254:325-
32.
123. Weiss, D. S., and R. K. Thauer. 1993. Methanogenesis and the unity of
biochemistry. Cell. 72:819-22.
124. Westermann, P., B. K. Ahring, and R.A. Mah. 1989. Threshold acetate
concentrations for acetate catabolism by aceticlastic methanogenic bacteria. Appl.
Environ. Microbiol. 55:514-515.
125. White, R. H., and D. Zhou. 1993. Biosynthesis of the coenzymes in
methanogens. Methanogenesis, J.G. Ferry, ed. Chapman and Hall, New York.
409-444.
126. White, R. H. 1988. Structural diversity among methanofurans from different
methanogenic bacteria. J. Bacteriol. 170:4594-7.
127. Whitman, W. B., and R. S. Wolfe. 1980. Presence of nickel in factor F430 from
Methanobacterium bryantii. Biochem. Biophys. Res. Commun. 92:1196-1201.
128. Widdel, F. 1986. Growth of methanogenic bacteria in pure culture with 2-
propanol and other alcohols as hydrogen donors. Appl. Environ. Microbiol.
51:1056-1062.
129. Woese, C. R, and G. E. Fox. 1978. Methanogenic bacteria. Nature. 273:101.
130. Woese, C. R., Kandler, O., and M. L. Wheelis. 1990. Towards a natural system
of organisms. Proposal for the domains archaea, bacteria, and eucarya. Proc.
Natl. Acad. Sci. U.S.A. 87:4576-4579.
131. Yamazaki, S. 1982. A selenium-containing hydrogenase from Methanococcus
vannielii. J. Biol. Chem. 257:7926-7929.
Page 45
31
132. Yoch, D. C., and R. C. Valentine. 1972. Ferredoxins and flavodoxins of
bacteria. Ann. Rev. Microbiol. 26:139-162.
133. Zellner, G., and J. Winter. 1987. Secondary alcohols as hydrogen donors for
C02-reduction by methanogens. FEMS Microbiol. Lett. 44:323-328.
134. Zinder, S. H., and R. A. Mah. 1979. Isolation and characterization of a
thermophilic strain of Methanosarcina unable to use H2-CO2 for methanogenesis.
Appl. Environ. Microbiol. 38:996-1008.
135. Zinder, S. H, S. C. Cardwell, T. Anguish, M. Lee, and M. Koch. 1984.
Methanogenesis in a thermophilic (58o C) anaerobic digestor: Methanothrix sp. as
an important aceticlastic methanogen. Appl. Environ. Microbiol. 47:796-807.
136. Zinder, S. H. 1993. Physiological ecology of methanogenesis. Methanogenesis,
J.G. Ferry, ed. Chapman and Hall, New York. 128-206.
Page 46
32
CHAPTER 2
IRON-SULFUR PROTEINS
I. Introduction.
Since their discovery in the 1960s, a vast amount of data has been accumulated
regarding iron-sulfur proteins. These proteins are abundantly found in nature and are
regarded to be an ancient component of a chemoautotrophic process in which a Ni/Fe-S
center catalyzes carbon fixation (39). Although most iron-sulfur proteins are electron
carriers, they have also been shown to possess other functions such as providing substrate
binding sites, general structural roles, iron storage, and gene regulation. Developments in
the fields of biophysical techniques, molecular biology, chemical synthesis, and computer
modeling, in conjunction with site-directed mutagenesis methods, have resulted in a rapid
accumulation of data concerning iron-sulfur proteins. Insight regarding cluster assembly,
protein stability, electronic structure, functions, and ligand coordination has been
revealed.
II. Structure and properties of clusters.
Iron-sulfur proteins are broadly classified as either simple or complex. The first
group contains only iron-sulfur clusters, whereas the latter contains additional prosthetic
groups (42). Clusters in iron-sulfur proteins, rather than single-metal sites, provide the
diversity and versatility for these proteins to function properly (76). The clusters in iron-
sulfur proteins contain Fe with at least partial S coordination (42). Their structures are
Fe2+ or Fe3+ with approximately tetrahedral coordination to S atoms of cysteine residues
and inorganic sulfides. Inorganic sulfides are sometime referred to as "acid-labile
sulfides", because the sulfur atoms have a –2 valence state that are released as H2S with
acid treatment (85). Cysteine appears to be a major residue involved in cluster bridging to
the polypeptide; nonetheless, evidence of non-cysteinyl ligands has recently emerged.
The structure/function basis for non-cysteinyl versus cysteinyl ligands has not been
determined; however, it appears that when Fe is coordinated with non-cysteinyl ligands
the cluster is involved in substrate binding and there is a shift in reduction potential (42).
Page 47
33
The properties of iron-sulfur clusters are dependent upon their electronic
configuration and capability of electron delocalization. Iron-sulfur proteins show a very
broad range of midpoint potentials. This can result from several factors including nature
of cluster ligands, hydrophobicity and charge of residues in the surrounding polypeptide
environment (61, 82). Electron paramagnetic resonance (EPR) and Mössbauer
spectroscopies are the techniques widely used to examine iron-sulfur clusters in proteins
and their properties. EPR spectroscopy is applicable to paramagnetic systems, and it is a
sensitive method which is ideal for studying many metalloproteins. Information obtained
from EPR analysis included electronic structure, metal coordination-sphere composition
and geometry (52). On the other hand, Mössbauer spectroscopy gives detailed pictures of
the chemical state of the iron atoms as well as the electron distribution in various redox
states of different iron-sulfur cluster types. Information derived from Mössbauer spectra
include the nature and arrangement of the ligand, spin of the iron atoms, spin coupling,
and the surrounding protein environment (41). To obtain an adequate Mössbauer
spectrum, the sample must contain a certain minimum quantity of the Mössbauer isotope,
which is 57Fe for iron-sulfur proteins. Due to the low natural abundance of 57Fe,
enrichment of the sample with 57Fe is always necessary (41).
[1Fe] cluster type. Basic structures of Fe-S clusters and their properties are
shown in Figure 1 (42). This cluster type, also referred to as rubredoxin-type cluster,
contains a single Fe atom bridged with four cysteines. The proteins show intense red
color when oxidized due to the charge transfer of S →Fe (17, 53). The structures of
rubredoxins from several microbial species have been solved. The EPR spectrum of the
ferric protein indicates the presence of high spin iron (S = 5/2) with g values of 4.3 and 9
(66). The oxidized rubredoxin with high spin Fe3+exhibits a small chemical shift at 0.25
mm/s at 77 oK and three quadrupole doublets in the Mössbauer spectrum (68, 71). A
large quadrupole split (3.16 mm/s at 77 oK) and small chemical shift (0.65 mm/s at 77 oK
has been observed for the Fe2+ form (68, 71). The coordinating cysteine motifs, CXXC,
at both the N- and C- terminal are conserved among these proteins (65). The midpoint
potential of the cluster is in the range of +20 to –60 mV.
Page 48
34
[2Fe-2S] cluster type. This cluster type is composed of a Fe2S2 (S-cys)4 unit.
The oxidized form ([2Fe-2S]2+) contains two Fe3+ ions. When reduced by one electron, a
mixed valence of Fe3+- Fe2+ ([2Fe-2S]1+ cluster results. This type of cluster is always
described as a “plant-type ferredoxin”, since it is present in photosynthetic organisms.
However, this cluster type has also been characterized from ferredoxins of halophiles,
and species in the genera Rhodobacter, Clostridium, and Azotobacter from the Bacteria
domain. (31). A [2Fe-2S] subclass called the Rieskie cluster describes iron-sulfur
proteins where the [2Fe-2S] clusters are coordinated by non-cysteinyl ligation from two
histidines (55). This subclass has a higher reduction potential than other [2Fe-2S]
proteins with exclusively cysteine ligands. The sequence motif for this subclass is
CXHX15-17CXXH (7, 15). All of the sequences known for coordination are shown in
Figure 2. The two Fe3+ with S = 5/2 are antiferromagnetically coupled in the oxidized
state and, thus, are EPR silent. Conversely, with the reduced state of [2Fe-2S]+, the
antiferromagnetic couple results in S = _, and the typical gav value derived from EPR is
1.96 for plant type ferredoxin and 1.90 for Rieskie proteins (81). The Mössbauer
spectrum for the oxidized proteins shows a quadrupole split with a small chemical shift
value, while the reduced proteins give two quadrpole doublets. One doublet corresponds
to the Fe3+ ion in the cluster, and the other with larger quadrupole is derived from Fe2+
ion in the cluster (41).
[3Fe-4S] cluster type. This cluster type was first interpreted as an artifact of a
degradative product of a [4Fe-4S] center (6), because [4Fe-4S] centers can be
interconverted to [3Fe-4S] centers by removal of an iron unit. However, a considerable
amount of data has shown that the [3Fe-4S] cluster is indeed a true cluster in nature for
some proteins. The core structure is Fe3S4 (S-cys)3 which forms a cube-like structure due
to a missing Fe. The [3Fe-4S] center involves one electron transfers with 1+ and 0
reduction states. In the 1+ oxidation state, all Fe3+ are antiferromagnetically coupled
which results in S = _. The EPR signal for this [3Fe-4S]1+ cluster is either axial or
rhombic with the resonance at gav values of 2.02. The Mössbauer spectrum of oxidized
ferredoxin II from Desulfovibrio gigas is a single quadrupole doublet with chemical shift
at 0.27 mm/s characteristic of the Fe3+ in tetrahedral S coordination. The reduced cluster
Page 49
35
produces a spectrum with two different intensities (2:1) of quadrupole doublet (40). The
less intense signal derived from Fe3+ is unchanged upon reduction, as indicated by the
Mössbauer parameter (40). Several sequence motifs coordinating [3Fe-4S] are shown in
Figure 2. The motifs contain two cysteines located closely to each other and another
distal cysteine. The cysteine motif coordinating the [4Fe-4S] cluster that is easily
converted to [3Fe-4S] cluster mostly contains aspartate in the place of the second
cysteine (9, 25, 42).
[4Fe-4S] cluster type. This cluster has a Fe4S4 (S-cys)4 cubane structure. It is
involved in one electron transfer reactions with three associated oxidation states, +3, +2,
and +1 (10, 11, 34). A cluster that undergoes three oxidation states in a biological system
has not been reported. These cluster types have a wide reduction potential in the range of
+450 to –700 mV. The [4Fe-4S] cluster with low reduction potential stays in the +2/+1
state, while the high reduction potential proteins (HIPIPs) stay in the +3/+2 state (31).
The low reduction potential of reduced [4Fe-4S]1+ and the oxidized HIPIPs with [4Fe-
4S]3+ are EPR active species. EPR analysis for [4Fe-4S]3+ of HIPIPs yields an axial
signal with g = 2.2 and 2.04 whereas the [4Fe-4S]1+ exhibits a rhombic type EPR with g =
20.6, 1.92, 1.88 with gav < 2. Mössbauer spectra of [4Fe-4S]2+ centers are generally
broad and also indicate almost equivalent iron atoms in the cluster. When the [4Fe-4S]2+
is oxidized or reduced, the spectra indicate an inequivalence of iron atoms in the clusters.
This feature is clearly evident from the spectra obtained in an applied magnetic field (41).
Page 50
36
Cluster type oxidation EPR Mössbauer and spin number (g) ( δ)
Figure 1. Structures, and properties of the structurally characterized iron-sulfur centers
that are involved in biological systems (42, 52).
Fe2+ S = 2 - 0.65
Fe3+ S = 5/2 4.3, 9 0.25
[2Fe-2S]+ S = _ 1.89, 1.96, 2.05 0.25, 0.55
[2Fe-2S]2+ S = 0 - 0.26
[3Fe-4S]0 S =2 - 0.30, 0.46
[3Fe-4S]+ S =1/2 1.97, 2.00, 2.02 0.27
[4Fe-4S]+ S = 1/2 or 3/2 1.88, 1.92, 2.06 0.57
[4Fe-4S]2+ S = 0 - 0.42
[4Fe-4S]3+ S = _ 2.04, 2.04, 2.12 0.31
Page 51
37
A.
N----CX4CX2CX29C----C Plant-type ferredoxins
N----CX5CX2CX36/37C----C Hydroxylase-type ferredoxins
N----CX3CX2CX36C30/31CX59C----C Biotin synthases
N----C-X2CX9CX31CX3C----C Clostridium pasteurianum ferredoxin
N----CXHX15-17CX2H----C Rieskie proteins
B.
N----CX6C----C (subunit 1) N----CX6C----C (subunit 2) [4Fe-4S] Fx in
photosystem I
N----CX34C----C (subunit 1) N----CX34C----C (subunit 2) [4Fe-4S] nitrogenase
N----C---------CX2C---------C [4Fe-4S] aconitase
N----CX2CX16CX13C----C [4Fe-4S] HIPIP
N----CX6CX2CX5C----C [4Fe-4S] Endonuclease
N----CXCX20CX20C----C [4Fe-4S] Leucine rich repeats
N----CX2CX11CX5C----C [4Fe-4S] Leucine rich variants
N----CX2CX2C---------C-P----C [4Fe-4S] ferredoxins
N----CX2CX2CX3C-P-------CX2CX2-8CX3CP----C 2[4Fe-4S] ferredoxins
C.
N----CX5C---------CP----C [3Fe-4S] ferredoxins
N----CX2DX2C---------CP----C [3Fe-4S] or [4Fe-4S]
ferredoxins
D.
N----CX2DX2CX3CP-------CX2CX2CX3CP----C 2[4Fe-4S] or [3Fe-4S] plus
[4Fe-4S] ferredoxin
N----CX7CX3CP--------- CX2CX2CX3CP----C [4Fe-4S][3Fe-4S] ferredoxins
Page 52
38
Figure 2. Arrangement of residues involved in coordination of [2Fe-4S] (A), [4Fe-4S] or
2[4Fe-4S] (B), [3Fe-4S] or [4Fe-4S] (C), and 2[4Fe-4S] or [3Fe-4S] plus [4Fe-4S] (D)
clusters. "N" and "C" at the beginning and the end the motifs represent the N and C
terminal sequence of the proteins. Dashed lines indicate variable spacing between
residues. C, D, and H within the motif are cysteine, aspartate, and histidine respectively.
Underlined and bolded residues are those considered to be ligands in each cluster based
on X-ray structure, sequence homology, site directed mutagenesis, or spectroscopic
evidence (21, 42).
Page 53
39
III. Ligation of iron-sulfur clusters
Cysteine is the major ligand in nature that coordinates iron atoms in iron-sulfur
clusters (62). Nonetheless, several different ligands, such as aspartate, histidine, or water,
have been identified. Direct information from crystal structures as well as through other
various spectroscopic studies has helped to identify the ligands involved in iron-sulfur
cluster coordination.
Aconitase is an enzyme containing a 4Fe-4S center that is not involved in redox
chemistry, but rather it is a substrate binding site (4, 49). It is an enzyme that catalyzes
the interconversion of citrate and isocitrate in the TCA cycle. The enzyme has been
characterized by different approaches. The crystallographic structures of the enzyme
alone, with substrates, and inhibitors have been determined. In the absence of substrate,
the active 4Fe-4S cluster is coordinated by 3 cysteines of the protein and an oxygen atom
from solvent is the fourth ligand (4, 74). In the presence of substrate, the crystal structure
shows a 4Fe-4S center in agreement with previous electron nuclear double resonance
(ENDOR) results. Both methods show that one Fe in the cluster has octahedral geometry
rather than tetrahedral. This octahedral geometry is due to additional ligands from the
carboxy and hydroxy groups of substrate and one water molecule (4, 49, 50).
The hydrogenase from Desulfovibrio gigas is another example of a non-cysteinyl
coordination that is shown by crystal structure. This protein contains a 4Fe-4S cluster
and Ni as prosthetic groups. The structure revealed the presence of three cysteines and
one histidine ligand to the 4Fe-4S center (83).
Albeit that the crystal structure is always a useful tool to determine ligand
identity, it is not easy to obtain the crystal structure for all proteins. Hence, other
methods often must be used. Iron-sulfur proteins are suspected of having non-cysteinyl
ligation if they have similar spectroscopic and catalytic characteristics to known non-
cysteinyl coordinated iron-sulfur proteins. Mostly, sequence comparisons with
homologues in combination with spectroscopic and site-directed mutagenesis studies
have been used to help identify the ligands for iron-sulfur clusters in proteins. For
Page 54
40
example, electron spin echo envelope modulation (ESEEM) and ENDOR spectroscopy
indicate that Rieskie proteins contain 2Fe-2S centers that are coordinated by two
cysteines and two histidines (7, 18, 26, 27). Histidines and cysteines are conserved
among Rieskie type proteins. In addition, they exhibit higher reduction potentials than a
2Fe-2S cluster coordinating solely cysteines (55, 81).
Another mixed ligand coordination has been observed in [4Fe-4S] ferredoxins
from sulfate reducers and hyperthermophilic archaea. Ligands to the iron-sulfur clusters
in these proteins are cysteines and aspartate. The [4Fe-4S] binding motif in this group is
similar to the conventional cysteine motif (CX2CX2C and a distal C) except the second
cysteine is substituted by aspartate (8, 32, 64). These ferredoxins exhibit one similar
property which is the 3Fe-4S/4Fe-4S interconversion. Even though Pyrococcus furiosus
ferredoxin contains a [4Fe-4S] center, it exhibits anomalous spectroscopic properties
from clusters ligating with cysteine exclusively (9, 13).
A number of site-directed mutagenesis studies have contributed to an
understanding of iron-sulfur cluster ligation (62). Site directed mutagenesis provides not
only identification of the ligand residue, it also provides an understanding of the protein
environment which has a direct effect upon the iron-sulfur cluster structure and
properties. Replacement of one of the ligands can result in the variant protein being
either improperly folded or a variant with no iron-sulfur clusters. The conclusion from
this kind of result is almost always that the replacement amino acid is involved in cluster
coordination and that it also influences the protein folding (62, 79). Sometimes the
variant has similar properties as wild type and, as such, this result excludes the replaced
residue as a ligand to the iron-sulfur cluster. In several incidences, the residue replacing
cysteine serves as a ligand in which case the variants can contain iron-sulfur clusters with
altered electronic, redox potential and functional properties. The term "ligand exchange"
is applied where a new residue substitutes for the missing ligand. One form of ligand
exchange is the use of serine and aspartate in place of a cysteinyl ligand. Examples of
this are [4Fe-4S] center variants of PsaC and [2Fe-2S] center variants of ferredoxin from
C. pasteurianum (20, 43, 57). Another form of ligand exchange is the use of a free
Page 55
41
cysteine from a different position to serve as a ligand rather than using the replacement
residue (23, 24, 79). This type of ligand exchange is referred to as "ligand swapping"
(23, 24). Ligand swapping often is associated with re-arrangement of the protein
conformation (23, 24, 79).
The phenomenon called "cluster conversion" occurs when the original iron-sulfur
cluster is converted into another structural type. Cluster conversions can result from
exposure to air, chemical oxidation-reduction, change of pH, and site-directed
replacement of ligands (42). A common form of cluster conversion occurs when a
cysteine ligand in a [4Fe-4S] center is replaced converting it to a [3Fe-4S] center.
Additionally, a [3Fe-4S] cluster may be converted into a [4Fe-4S] cluster by replacing a
noncysteinyl residue with cysteine that preserves the common cysteine motif (CX2CX2C
and a distal C) for coordination of a [4Fe-4S] center (1, 8, 44, 54). Another example of
cluster conversion is Clostridium pateurianum rubredoxin, in which the 1Fe cluster is
changed to a [2Fe-4S]-plant type cluster by replacing a cysteinyl ligand with alanine (58).
This result may be due to the structure around the metal site in rubredoxin which is
similar to the [2Fe-2S] Rieske protein. Another possibility is that the variant has a large
structural rearrangement allowing the additional incorporation of Fe and sulfides.
IV. Function of iron-sulfur centers.
A. Electron transfers.
Iron-sulfur clusters found in all domains of life are capable of serving several
functions. Most are electron carriers that accept, donate, and shift electrons (3).
Examples are ferredoxins and HIPIPs. Ferredoxins are well-studied proteins and their
structures from different organisms have been solved. The midpoint potentials of these
proteins are broadly varied from positive to negative values. This property allows them
to be either an electron donor or acceptor in various metabolic reactions. Examples of
enzymes and proteins that ferredoxins catalyze electron transfers are hydrogenase,
nitrogenase, cytochromes, nitrite reductase, nitrate reductase, sulfite reductase, pyruvate
oxidoreductase, and formate dehydrogenase. HIPIPs have a broad range of positive
Page 56
42
midpoint potential values, which have been suggested to be correlated to the overall
charges of the polypeptide environment (30). HIPIPs are electron carriers between the
photoreaction center and the cytochrome bc1 complex in phototrophic microbes from
Bacteria domain (36, 37, 78). They also have been suggested to transfer electrons in
thiosulfate oxidation and iron oxidation (31).
B. Catalysis.
Many enzymes use iron-sulfur clusters as their substrate binding and activation
sites. For example, enzymes that perform dehydration and hydration activities in which
iron-sulfur clusters act as a Lewis acid in the reaction (4, 42). Aconitase is the best
characterized enzyme among this group. The catalytic Fe of the [4Fe-4S] center in
aconitase shows no redox changes during the reaction (2).
The catalytic role of iron-sulfur clusters which undergo redox changes is
exemplified in the radical based mechanism reactions. Examples are anaerobic
ribonucleotide reductase (RR) and biotin synthase (63). RR is involved in the first step in
DNA synthesis, ribonucleotide reduction to 2'-deoxyribonucleotide. Escherichia coli
uses specific RRs during aerobic and anaerobic growth conditions, both of which
generate radicals required for the reaction mechanism (73). The aerobic RR is a
heterodimer in which the smaller subunits contain µ-oxo diiron centers. The metal center
is used to oxidize a tyrosyl residue to generate a tyrosyl radical. The anaerobic RR
however, contains a [4Fe-4S] center at the interface of the two small subunits. This is the
third example in which an iron-sulfur cluster is located at the interface of two
polypeptides other than nitrogenase and Fx in photosystem I. The 4Fe-4S center of the
anaerobic RR reduces S-adenosyl methionine (SAM) to give a 5'-deoxyadenosyl radical
that generates a glycyl radical. Biotin synthase catalyses the last step of the biotin
synthesis pathway (77), in which a sulfur atom in the form of a thiol derivative is inserted
into dethiobiotin (16). The aerobically purified enzyme contains a [2Fe-2S] which is
proposed to be involved in the generation of the 5'-deoxy adenosyl radical. Recent
absorption, variable temperature magnetic circular dichroism (VTMCD), and EPR
Page 57
43
spectroscopy indicated conversion of the [2Fe-2S] centers to a [4Fe-4S] center under
anaerobic conditions (16). The [4Fe-4S] center form of biotin synthase is postulated to
be involved in the radical generating reaction rather than the [2Fe-2S] cluster form. The
physiological significant of the cluster conversion may be a tool to regulate enzyme
activity due to oxidative stress (16). However, more experiments are needed to verify
this hypothesis.
C. Regulatory role
Iron-sulfur clusters also serve a regulatory role, in which they sense molecular
iron, oxygen, superoxide ion, and possibly nitric oxide concentrations (5, 22, 33, 35, 75).
For example, iron responsive binding protein (IRP) regulates the iron level in higher
organisms by controlling the gene expression of both iron storage ferritin and the
transferrin receptor (75). IRP recognizes a region called iron responsive element (IRE)
located at the 5' and 3' regions on the mRNA encoding ferritin and transferrin
respectively. When the cellular iron level is low, functional IRP binds IRE on both
ferritin and transferrin mRNA. This results in blocking ferritin translation and increasing
transferrin mRNA stability resulting in an increased production of transferrin (46, 56).
IRP is an isoform of aconitase, but contains no iron-sulfur clusters (28, 45). When iron is
limiting, IRP (apo-aconitase) increases. Conversely, when iron is abundant, holo-
aconitase with aconitase activity is increased while IRP activity is decreased. This
switching in activity of a bifunctional protein between enzyme catalysis (aconitase) and
RNA binding activity (IRP) occurs by assembly and disassembly of iron-sulfur clusters
which depends on the cellular iron level (3).
Another example of an iron-sulfur protein serving a regulatory role is fumarate
nitrate reductase regulatory protein (FNR), an oxygen sensor protein. FNR is a
transcription factor, which activates a set of genes under anaerobic conditions (80). The
functional enzyme form contains a [4Fe-4S] center. In the presence of oxygen, the
cluster is destroyed rapidly and FNR subsequently loses its DNA binding ability. Hence,
the regulatory function of FNR is controlled by the oxygen level in cells (76).
Page 58
44
There is also a group of proteins with tandem repeats of leucine and aliphatic
residues in their sequence called leucine-rich repeats (LRRs) and leucine-rich variants
(LRVs). The X-ray crystal structure for an LRV was recently obtained. The structure
shows a 4Fe-4S cluster with a distinctive motif of CXXCX11CX5C located in a small
domain (67). This cysteine motif is different from the one described for the motif in
LRRs (CXCX20CX20C). The [4Fe-4S] centers of these LRRs and LRVs are highly
susceptible to oxygen (67), hence it is postulated that those proteins may function as an
oxygen sensor; however, more evidence is needed to support this hypothesis.
Sox R is another protein where the iron-sulfur cluster is proposed to serve a
regulatory role. The Sox R system is a mechanism developed to protect cells from
superoxide and nitric oxide. The mechanism is unknown at this time; however, the iron-
sulfur center seems to have a role in sensing these compounds (35, 84).
D. Iron storage role.
Polyferredoxin from Methanobacterium thermoautotrophicum contains 12[4Fe-
4S], and is thought to function as an iron storage protein or as an electron carrier (29, 72).
The postulated physiological role for polyferredoxin as an electron acceptor of the MV-
reducing hydrogenase is obscured, since the specific rate of reduction is very slow.
E. Structural role.
Another role proposed for iron-sulfur clusters involves maintaining protein
structure (42). The [4Fe-4S] centers in endonuclease III and Mut Y, members of the base
excision repair enzyme superfamily (47, 48, 59, 60), have no catalytic role and are
resistance to oxidation and reduction (14, 19). However, the enzyme activity is
dependent upon the presence of these iron-sulfur clusters. These observations suggest the
iron-sulfur cluster may be required for structural integrity. Although the [4Fe-4S] centers
of these enzymes are not directly involved in substrate binding or catalysis, they appear
Page 59
45
to be required for efficient specific DNA binding. The iron-sulfur cluster may juxtapose
the catalytic or substrate binding sites to interact with DNA (69, 70). Another striking
feature for this superfamily of enzymes is that they contain a similar cysteine motif of
CX6CX2CX5C, which coordinates [4Fe-4S] centers (47, 48). This motif is one of the
most compact cysteine motifs known to date.
V. Iron-sulfur cluster assembly in proteins.
Understanding the in vivo formation of iron-sulfur clusters still falls behind, in
spite of the deep understanding of iron-sulfur cluster structure, reactivity, physiological
roles, and properties. Two scenarios for iron-sulfur cluster synthesis have been proposed.
The first is called spontaneous self-assembly. In the mechanism, iron-sulfur clusters are
thought to form spontaneously in the presence of ferric or ferrous salt in aqueous
mercaptoethanol with sodium sulfide (38). The logic for this process is that nature may
have synthesized iron-sulfur clusters by using the available chemistry of the geosphere
during evolution of the biosphere (52). This spontaneous process has been successful in
replicating known metalloprotein core clusters.
The other proposed scenario, assisted assembly, involves other protein
components for cluster formation and insertion into the proteins. The best characterized
are the Nif S, U, and M system from Azotobacter vinelandii. Nif S is a cysteine
desulfurase that converts L-cysteine to L-alanine and an activated sulfur (cysteinyl
persulfide). The enzyme contains pyridoxal phosphate, which is essential for catalysis
(87). Nif S is proposed to be a sulfur donor in the formation of the iron-sulfur cluster
core structure (87). The reconstitution of iron-sulfur clusters of apoproteins is facilitated
in the presence of Nif S (12, 86). The nif S gene is highly homologous and present in
different organisms, suggesting that it is a universal sulfur mobilizing agent (51, 87). Nif
U and Nif M are less well characterized, but they are hypothesized to be involved in
cluster formation.
Page 60
46
References
1. Aono, S., D. Bentrop, I. Bertini, C. Luchinat, and R. Macinai. 1997. The
D13C variant of Bacillus schlegelii 7Fe ferredoxin is an 8Fe ferredoxin as
revealed by 1H-NMR spectroscopy. FEBS Lett. 412:501-5.
2. Beinert, H. 1990. Recent developments in the field of iron-sulfur proteins.
FASEB J. 4:2483-91.
3. Beinert, H., R. H. Holm, and E. Munck. 1997. Iron-sulfur clusters: nature's
modular, multipurpose structures. Science. 277:653-9.
4. Beinert, H., and M. C. Kennedy. 1989. 19th Sir Hans Krebs lecture.
Engineering of protein bound iron-sulfur clusters. A tool for the study of protein
and cluster chemistry and mechanism of iron-sulfur enzymes. Eur. J. Biochem.
186(1-2):5-15.
5. Beinert, H., and P. Kiley. 1996. Redox control of gene expression involving
iron-sulfur proteins. Change of oxidation-state or assembly/disassembly of Fe-S
clusters? [comment]. FEBS Lett. 382:218-9; discussion 220-1.
6. Beinert, H., and A. J. Thomson. 1983. Three-iron clusters in iron-sulfur
proteins. Arch. Biochem. Biophys. 222:333-61.
7. Britt, R. D., K. Sauer, M. P. Klein, D. B. Knaff, A. Kriauciunas, C. A. Yu, L.
Yu, and R. Malkin. 1991. Electron spin echo envelope modulation spectroscopy
supports the suggested coordination of two histidine ligands to the Rieske Fe-S
centers of the cytochrome b6f complex of spinach and the cytochrome bc1
complexes of Rhodospirillum rubrum, Rhodobacter sphaeroides R-26, and bovine
heart mitochondria. Biochemistry. 30:1892-901.
8. Busch, J. L., J. L. Breton, B. M. Bartlett, F. A. Armstrong, R. James, and A.
J. Thomson. 1997. [3Fe-4S] <--> [4Fe-4S] cluster interconversion in
Desulfovibrio africanus ferredoxin III: properties of an Asp14 --> Cys mutant.
Biochem. J. 323:95-102.
9. Calzolai, L., C. M. Gorst, Z. H. Zhao, Q. Teng, M. W. Adams, and G. N. La
Mar. 1995. 1H NMR investigation of the electronic and molecular structure of
the four-iron cluster ferredoxin from the hyperthermophile Pyrococcus furiosus.
Page 61
47
Identification of Asp 14 as a cluster ligand in each of the four redox states.
Biochemistry. 34:11373-84.
10. Carter, C. W., Jr., S. T. Freer, N. H. Xuong, R. A. Alden, and J. Kraut. 1972.
Structure of the iron-sulfur cluster in the Chromatius iron protein at 2.25
Angstrom resolution. Cold. Spring. Harb. Symp. Quant. Biol. 36:381-5.
11. Carter, C. W., Jr., J. Kraut, S. T. Freer, R. A. Alden, L. C. Sieker, E. Adman,
and L. H. Jensen. 1972. A comparison of Fe4S4 clusters in high-potential iron
protein and in ferredoxin. Proc. Natl. Acad. Sci. U S A. 69:3526-9.
12. Chen, S., L. Zheng, D. R. Dean, and H. Zalkin. 1997. Role of NifS in
maturation of glutamine phosphoribosylpyrophosphate amidotransferase. J.
Bacteriol. 179:7587-90.
13. Conover, R. C., A. T. Kowal, W. G. Fu, J. B. Park, S. Aono, M. W. Adams,
and M. K. Johnson. 1990. Spectroscopic characterization of the novel iron-sulfur
cluster in Pyrococcus furiosus ferredoxin. J. Biol .Chem. 265:8533-41.
14. Cunningham, R. P., H. Asahara, J. F. Bank, C. P. Scholes, J. C. Salerno, K.
Surerus, E. Munck, J. McCracken, J. Peisach, and M. H. Emptage. 1989.
Endonuclease III is an iron-sulfur protein. Biochemistry. 28:4450-5.
15. Davidson, E., T. Ohnishi, E. Atta-Asafo-Adjei, and F. Daldal. 1992. Potential
ligands to the [2Fe-2S] Rieske cluster of the cytochrome bc1 complex of
Rhodobacter capsulatus probed by site-directed mutagenesis. Biochemistry.
31:3342-51.
16. Duin, E. C., M. E. Lafferty, B. R. Crouse, R. M. Allen, I. Sanyal, D. H. Flint,
and M. K. Johnson. 1997. [2Fe-2S] to [4Fe-4S] cluster conversion in
Escherichia coli biotin synthase. Biochemistry. 36:11811-20.
17. Eaton, W. A., and W. Lovenberg. 1973. The iron-sulfur complex in rubredoxin.
Iron-sulfur proteins , Academic Press, Inc. (London) Ltd., ed. Lovenberg W. 2.
18. Fee, J. A., D. Kuila, M. W. Mather, and T. Yoshida. 1986. Respiratory proteins
from extremely thermophilic, aerobic bacteria. Biochim. Biophys. Acta. 853:153-
85.
Page 62
48
19. Fu, W., S. O' Handley, R. P. Cunningham, and M. K. Johnson. 1992. The role
of the iron-sulfur cluster in Escherichia coli endonuclease III. A resonance raman
study. J. Biol. Chem. 267:16135-7.
20. Fujinaga, J., J. Gaillard, and J. Meyer. 1993. Mutated forms of a [2Fe-2S]
ferredoxin with serine ligands to the iron- sulfur cluster. Biochem. Biophys. Res.
Commun. 194:104-11.
21. Gao-Sheridan, H. S., H. R. Pershad, F. A. Armstrong, and B. K. Burgess.
1998. Discovery of a novel ferredoxin from Azotobacter vinelandii containing
two [4Fe-4S] clusters with widely differing and very negative reduction
potentials. J. Biol. Chem. 273:5514-9.
22. Gaudu, P., and B. Weiss. 1996. SoxR, a [2Fe-2S] transcription factor, is active
only in its oxidized form. Proc. Natl. Acad. Sci. U S A. 93:10094-8.
23. Golinelli, M. P., L. A. Akin, B. R. Crouse, M. K. Johnson, and J. Meyer.
1996. Cysteine ligand swapping on a deletable loop of the [2Fe-2S] ferredoxin
from Clostridium pasteurianum. Biochemistry. 35(27):8995-9002.
24. Golinelli, M. P., C. Chatelet, E. C. Duin, M. K. Johnson, and J. Meyer. 1998.
Extensive ligand rearrangements around the [2Fe-2S] cluster of Clostridium
pasteurianum ferredoxin. Biochemistry. 37:10429-37.
25. Gorst, C. M., Y. H. Yeh, Q. Teng, L. Calzolai, Z. H. Zhou, M. W. Adams, and
G. N. La Mar. 1995. 1H NMR investigation of the paramagnetic cluster
environment in Pyrococcus furiosus three-iron ferredoxin: sequence-specific
assignment of ligated cysteines independent of tertiary structure. Biochemistry.
34:600-10.
26. Gurbiel, R. J., C. J. Batie, M. Sivaraja, A. E. True, J. A. Fee, B. M. Hoffman,
and D. P. Ballou. 1989. Electron-nuclear double resonance spectroscopy of 15N-
enriched phthalate dioxygenase from Pseudomonas cepacia proves that two
histidines are coordinated to the [2Fe-2S] Rieske-type clusters. Biochemistry.
28:4861-71.
27. Gurbiel, R. J., T. Ohnishi, D. E. Robertson, F. Daldal, and B. M. Hoffman.
1991. Q-band ENDOR spectra of the Rieske protein from Rhodobactor
Page 63
49
capsulatus ubiquinol-cytochrome c oxidoreductase show two histidines
coordinated to the [2Fe-2S] cluster. Biochemistry. 30:11579-84.
28. Haile, D. J., T. A. Rouault, J. B. Harford, M. C. Kennedy, G. A. Blondin, H.
Beinert, and R. D. Klausner. 1992. Cellular regulation of the iron-responsive
element binding protein: disassembly of the cubane iron-sulfur cluster results in
high-affinity RNA binding. Proc. Natl. Acad. Sci. U S A. 89:11735-9.
29. Hedderich, R., Albracht, S. P. J., Linder, D., Koch, J., and Thauer, R.K.
1992. Isolation and characterization of polyferredoxin from Methanobacterium-
thermoautotrophicum - The mvhB gene product of the methylviologen-reducing
hydrogenase operon. FEBS Letters. 298:65-68.
30. Heering, H. A., B. M. Bulsink, W. R. Hagen, and T. E. Meyer. 1995. Influence
of charge and polarity on the redox potentials of high- potential iron-sulfur
proteins: evidence for the existence of two groups. Biochemistry. 34:14675-86.
31. Heinrich, S., and P. Rosch. 1998. The structure of iron-sulfur proteins. Prog.
Biophys. & Mol. Biol. 70:95-136.
32. Heltzel, A., E. T. Smith, Z. H. Zhou, J. M. Blamey, and M. W. Adams. 1994.
Cloning, expression, and molecular characterization of the gene encoding an
extremely thermostable [4Fe-4S] ferredoxin from the hyperthermophilic archaeon
Pyrococcus furiosus. J. Bacteriol. 176:4790-3.
33. Hentze, M. W., and L. C. Kuhn. 1996. Molecular control of vertebrate iron
metabolism: mRNA-based regulatory circuits operated by iron, nitric oxide, and
oxidative stress. Proc. Natl. Acad. Sci. U S A. 93:8175-82.
34. Herskovitz, T., B. A. Averill, R. H. Holm, J. A. Ibers, W. D. Phillips, and J. F.
Weiher. 1972. Structure and properties of a synthetic analogue of bacterial iron-
sulfur proteins. Proc. Natl. Acad. Sci. U S A. 69:2437-41.
35. Hidalgo, E., J. M. Bollinger, Jr., T. M. Bradley, C. T. Walsh, and B. Demple.
1995. Binuclear [2Fe-2S] clusters in the Escherichia coli SoxR protein and role of
the metal centers in transcription. J. Biol. Chem. 270:20908-14.
36. Hochkoeppler, A., P. Kofod, and D. Zannoni. 1995. HiPiP oxido-reductase
activity in membranes from aerobically grown cells of the facultative phototroph
Rhodoferax fermentans. FEBS Lett. 375:197-200.
Page 64
50
37. Hochkoeppler, A., D. Zannoni, S. Ciurli, T. E. Meyer, M. A. Cusanovich, and
G. Tollin. 1996. Kinetics of photo-induced electron transfer from high-potential
iron- sulfur protein to the photosynthetic reaction center of the purple phototroph
Rhodoferax fermentans. Proc. Natl. Acad. Sci. U S A. 93:6998-7002.
38. Hong, J., and J. C. Rabinowitz. 1967. Preparation and properties of Clostridial
apoferredoxins. Biochem. Biophys. Res. Commun. 29:246-52.
39. Huber, C., and G. Wachtershauser. 1997. Activated acetic acid by carbon
fixation on (Fe,Ni)S under primordial conditions. Science. 276:245-247.
40. Huynh, B. H., J. J. Moura, I. Moura, T. A. Kent, J. LeGall, A. V. Xavier, and
E. Munck. 1980. Evidence for a three-iron center in a ferredoxin from
Desulfovibrio gigas. Mössbauer and EPR studies. J. Biol. Chem. 255(8):3242-4.
41. Huynh, V. 1998. Mössbauer spectroscopy. Inorganic biochemistry summer
workshop, University of Georgia.
42. Johnson, M. K. 1994. Iron-sulfur proteins. Encyclopedia of Inorganic Chemistry,
Johnson Wiley and Sons (King, R.B., ed). 4:1896-1915.
43. Jung, Y. S., I. R. Vassiliev, J. Yu, L. McIntosh, and J. H. Golbeck. 1997.
Strains of Synechocystis sp. PCC 6803 with altered PsaC. II. EPR and optical
spectroscopic properties of FA and FB in aspartate, serine, and alanine
replacements of cysteines 14 and 51. J. Biol. Chem. 272:8040-9.
44. Kemper, M. A., H. S. Gao-Sheridan, B. Shen, J. L. Duff, G. J. Tilley, F. A.
Armstrong, and B. K. Burgess. 1998. Delta T 14/Delta D 15 Azotobacter
vinelandii ferredoxin I: creation of a new CysXXCysXXCys motif that ligates a
[4Fe-4S] cluster. Biochemistry. 37:12829-37.
45. Kennedy, M. C., L. Mende-Mueller, G. A. Blondin, and H. Beinert. 1992.
Purification and characterization of cytosolic aconitase from beef liver and its
relationship to the iron-responsive element binding protein [published erratum
appears in Proc. Natl. Acad. Sci. U S A 1993; 90:2556]. Proc. Natl. Acad. Sci. U
S A. 89:11730-4.
46. Klausner, R. D., T. A. Rouault, and J. B. Harford. 1993. Regulating the fate of
mRNA: the control of cellular iron metabolism. Cell. 72:19-28.
Page 65
51
47. Kuo, C. F., D. E. McRee, R. P. Cunningham, and J. A. Tainer. 1992.
Crystallization and crystallographic characterization of the iron- sulfur-containing
DNA-repair enzyme endonuclease III from Escherichia coli. J. Mol. Biol.
227(1):347-51.
48. Kuo, C. F., D. E. McRee, C. L. Fisher, S. F. O'Handley, R. P. Cunningham,
and J. A. Tainer. 1992. Atomic structure of the DNA repair [4Fe-4S] enzyme
endonuclease III. Science. 258:434-40.
49. Lauble, H., M. C. Kennedy, H. Beinert, and C. D. Stout. 1992. Crystal
structures of aconitase with isocitrate and nitroisocitrate bound. Biochemistry.
31:2735-48.
50. Lauble, H., M. C. Kennedy, H. Beinert, and C. D. Stout. 1994. Crystal
structures of aconitase with trans-aconitate and nitrocitrate bound. J. Mol. Biol.
237(4):437-51.
51. Leibrecht, I., and D. Kessler. 1997. A novel L-cysteine/cystine C-S-lyase
directing [2Fe-2S] cluster formation of Synechocystis ferredoxin. J. Biol. Chem.
272(16):10442-7.
52. Lippard, S. J., and J. M. Berg. 1994. Physical methods in bioinorganic
chemistry. Priciples of bioinorganic chemistry:75-100.
53. Lovenberg, W., and B. E. Sobel. 1965. Rubredoxin: a new electron transfer
protein from Clostridium pasteurianum. Proc. Natl. Acad. Sci. U S A. 54:193-9.
54. Manodori, A., G. Cecchini, I. Schroder, R. P. Gunsalus, M. T. Werth, and M.
K. Johnson. 1992. [3Fe-4S] to [4Fe-4S] cluster conversion in Escherichia coli
fumarate reductase by site-directed mutagenesis. Biochemistry. 31:2703-12.
55. Mason, J. R., and R. Cammack. 1992. The electron-transport proteins of
hydroxylating bacterial dioxygenases. Ann. Rev. Microbiol. 46:277-305.
56. Melefors, O., and M. W. Hentze. 1993. Iron regulatory factor--the conductor of
cellular iron regulation. Blood Rev. 7:251-8.
57. Meyer, J., J. Fujinaga, J. Gaillard, and M. Lutz. 1994. Mutated forms of the
[2Fe-2S] ferredoxin from Clostridium pasteurianum with noncysteinyl ligands to
the iron-sulfur cluster. Biochemistry. 33:13642-50.
Page 66
52
58. Meyer, J., J. Gagnon, J. Gaillard, M. Lutz, C. Achim, E. Munck, Y. Petillot,
C. M. Colangelo, and R. A. Scott. 1997. Assembly of a [2Fe-2S]2+ cluster in a
molecular variant of Clostridium pasteurianum rubredoxin. Biochemistry.
36:13374-80.
59. Michaels, M. L., C. Cruz, A. P. Grollman, and J. H. Miller. 1992. Evidence
that MutY and MutM combine to prevent mutations by an oxidatively damaged
form of guanine in DNA. Proc. Natl. Acad. Sci. U S A. 89:7022-5.
60. Michaels, M. L., J. Tchou, A. P. Grollman, and J. H. Miller. 1992. A repair
system for 8-oxo-7,8-dihydrodeoxyguanine. Biochemistry. 31:10964-8.
61. Moore, G. D., G. W. Pettigrew, and N. K. Roger. 1986. Factors influencing
redox potentials of electron transfer proteins. Proc. Natl. Acad. Sci. USA.
83:4998-5000.
62. Moulis, J.-M., V. Davasse, M.-P. Golinelli, J. Meyer, and I. Quinkal. 1996.
The coodination sphere of iron-sulfur clusters: lessons from site-directed
mutagenesis experiments. J. Biol. Inorg.Chem. 1:2-14.
63. Mulliez, E., M. Fontecave, J. Gaillard, and P. Reichard. 1993. An iron-sulfur
center and a free radical in the active anaerobic ribonucleotide reductase of
Escherichia coli. J. Biol. Chem. 268:2296-9.
64. Okawara, N., M. Ogata, T. Yagi, S. Wakabayashi, and H. Matsubara. 1988.
Amino acid sequence of ferredoxin I from Desulfovibrio vulgaris Miyazaki. J.
Biochem (Tokyo). 104:196-9.
65. Orme-Johnson, W. H. 1973. Iron-sulfur proteins: structure and function. Annu.
Rev. Biochem. 42:159-204.
66. Orme-Johnson, W. H., and R. H. Sands. 1973. Probing Iron-sulfur proteins
with EPR and ENDOR spectroscopy. Iron-sulfur proteins , Academic Press, Inc.
(London) Ltd., ed. Lovenberg W. 2.
67. Peters, J. W., M. H. Stowell, and D. C. Rees. 1996. A leucine-rich repeat
variant with a novel repetitive protein structural motif [letter]. Nat. Struct. Biol.
3:991-4.
Page 67
53
68. Phillips, W. D., M. Poe, J. F. Weiher, C. C. McDonald, and W. Lovenberg.
1970. Proton magnetic resonance, magnetic susceptibility and Mössbauer studies
of Clostridium pasteurianum rubredoxin. Nature. 227:574-7.
69. Porello, S. L., M. J. Cannon, and S. S. David. 1998. A substrate recognition
role for the [4Fe-4S]2+ cluster of the DNA repair glycosylase MutY.
Biochemistry. 37:6465-75.
70. Prince, R. C., and M. J. Grossman. 1993. Novel iron-sulfur clusters. Trends.
Biochem. Sci. 18:153-4.
71. Rao, K. K., R. Cammack, D. O. Hall, and C. E. Johnson. 1971. Mössbauer
effect in Scenedesmus and spinach ferredoxins. The mechanism of electron
transfer in plant-type iron-sulphur proteins. Biochem. J. 122:257-65.
72. Reeve, J. N., G. S. Beckler, D. S. Cram, P. T. Hamilton, J. W. Brown, J. A.
Krzycki, A. F. Kolodziej, L. Alex, W. H. Orme-Johnson, and C.T. Walsh.
1989. A hydrogenase-linked gene in Methanobacterium thermoautotrophicum
strain H encodes a polyferredoxin. Proc. Natl. Acad. Sci. USA. 86:3031-3035.
73. Reichard, P. 1993. From RNA to DNA, why so many ribonucleotide reductases?
Science. 260(5115):1773-7.
74. Robbins, A. H., and C. D. Stout. 1989. Structure of activated aconitase:
formation of the [4Fe-4S] cluster in the crystal. Proc. Natl. Acad. Sci. U S A.
86:3639-43.
75. Rouault, T. A., D. J. Haile, W. E. Downey, C. C. Philpott, C. Tang, F.
Samaniego, J. Chin, I. Paul, D. Orloff, and J. B. Harford. 1992. An iron-sulfur
cluster plays a novel regulatory role in the iron- responsive element binding
protein. Biometals. 5:131-40.
76. Rouault, T. A., and R. D. Klauser. 1996. Iron-sulfur clusters as biosensors of
oxidants and iron. Trends. Biochem. Sci. 21:174-177.
77. Sanyal, I., G. Cohen, and D. H. Flint. 1994. Biotin synthase: purification,
characterization as a [2Fe-2S]cluster protein, and in vitro activity of the
Escherichia coli bioB gene product. Biochemistry. 33:3625-31.
78. Schoepp, B., P. Parot, L. Menin, J. Gaillard, P. Richaud, and A. Vermeglio.
1995. In vivo participation of a high potential iron-sulfur protein as electron donor
Page 68
54
to the photochemical reaction center of Rubrivivax gelatinosus. Biochemistry.
34:11736-42.
79. Shen, B., D. R. Jollie, T. C. Diller, C. D. Stout, P. J. Stephens, and B. K.
Burgess. 1995. Site-directed mutagenesis of Azotobacter vinelandii ferredoxin I:
cysteine ligation of the [4Fe-4S] cluster with protein rearrangement is preferred
over serine ligation. Proc. Natl. Acad. Sci. U S A. 92:10064-8.
80. Spiro, S., and J. R. Guest. 1990. FNR and its role in oxygen-regulated gene
expression in Escherichia coli. FEMS Microbiol. Rev. 6:399-428.
81. Trumpower, B. L. 1990. Cytochrome bc1 complexes of microorganisms.
Microbiol. Rev. 54:101-29.
82. Tsang, H. T., C. J. Batie, D. P. Ballou, and J. E. Penner-Hahn. 1989. X-ray
absorption spectroscopy of the [2Fe-2S] Rieske cluster in Pseudomonas cepacia
phthalate dioxygenase. Determination of core dimensions and iron ligation.
Biochemistry. 28:7233-40.
83. Volbeda, A., M. H. Charon, C. Piras, E. C. Hatchikian, M. Frey, and J. C.
Fontecilla-Camps. 1995. Crystal structure of the nickel-iron hydrogenase from
Desulfovibrio gigas. Nature. 373:580-7.
84. Wu, J., W. R. Dunham, and B. Weiss. 1995. Overproduction and physical
characterization of SoxR, a [2Fe-2S] protein that governs an oxidative response
regulon in Escherichia coli. J. Biol. Chem. 270:10323-7.
85. Yoch, D. C., and R. P. Carithers. 1979. Bacterial iron-sulfur proteins.
Microbiol. Rev. 43:384-421.
86. Zheng, L., and D. R. Dean. 1994. Catalytic formation of a nitrogenase iron-
sulfur cluster. J. Biol. Chem. 269:18723-6.
87. Zheng, L., R. H. White, V. L. Cash, R. F. Jack, and D. R. Dean. 1993.
Cysteine desulfurase activity indicates a role for NIFS in metallocluster
biosynthesis. Proc. Natl. Acad. Sci. U S A. 90:2754-8.
Page 69
55
CHAPTER 3
OBJECTIVES
Methanoarchaea obtain energy for growth by coupling electron transport
phosphorylation to methane formation. An undeveloped area of research has been the
identification of electron carriers. The iron-sulfur flavoprotein (Isf) from
Methanosarcina thermophila has been proposed to be an electron carrier in the acetate
fermentation pathway. The main objectives of my research were to characterize the
properties of the iron-sulfur center and FMN in Isf, identify the ligands required for
coordination of the iron-sulfur cluster, and to identify the electron acceptor of Isf.
Isf was previously shown to contain one FMN and iron-sulfur center per subunit;
however, the properties of these redox centers were not characterized. Various proteins
posses different iron-sulfur cluster types, each showing distinct properties. Therefore, the
identity of the iron-sulfur center in Isf is important for understanding how Isf functions as
a redox protein.
Most iron-sulfur clusters are ligated to the polypeptide backbone primarily via
cysteine residues. Several amino acid motifs for iron-sulfur cluster ligation have been
reported (refer to Chapter 2). These motifs can be used as a basis for the preliminary
prediction of ligands that bind to iron-sulfur cluster types in proteins. Most iron-sulfur
clusters are coordinated by four cysteine residues. Sequence comparisons among Isf
homologues reveal a novel motif with perfectly conserved cysteines which could possibly
coordinate the iron-sulfur center in Isf (refer to Chapter 4). This putative motif is the
most compact cysteine motif known for coordinating a redox active protein.
The discovery of Isf as an electron carrier in the acetate fermentation for M.
thermophila allows the possibility for a comprehensive study of the electron transport
components in this methanoarchaeon. Determination of the midpoint potential of the
iron-sulfur center and FMN will be useful in investigating the possibility of intra- and
inter-molecular electron transfer processes in the acetate fermentation pathway. The
Page 70
56
presence of both a one-electron carrier and a two-electron carrier in the same protein
suggests a one/two electron switch function for Isf. The fact that Isf has a defined redox
potential and a possible one-two electron switch function provides a basis to identify the
electron acceptor for Isf.
Page 71
57
CHAPTER 4
ELECTROCHEMICAL AND SPECTROSCOPIC PROPERTIES OF THE
IRON-SULFUR FLAVOPROTEIN FROM Methanosarcina thermophila
ABSTRACT
Based on spectroscopic analyses, a heterologously produced iron-sulfur
flavoprotein (Isf) from Methanosarcina thermophila contains two [4Fe-4S] centers and
two flavin mononucleotide (FMN) cofactors in the homodimeric protein. The midpoint
potentials (Em) for the [4Fe-4S]2+/1+ and FMN/FMNH2 are – 394 and –277 mV
respectively. Interestingly, the semiquinone form of FMN was not detected during the
potentiometric titration; however, a small amount of red semiquinone was found in
frozen reaction mixtures of CODH/ACS and Isf. The EPR spectrum of the reduced
protein showed g values characteristic of [4Fe-4S] center with additional g values (2.06,
1.93, 1.86 and 1.82) due to microheterogeneity among Isf molecules. A variety of
physiological 2-electron acceptors were examined for the ability to oxidize Isf, but none
are able to carry out this function. Extract from H2/CO2-grown Methanobacterium
thermoautotrophicum cells catalyzed either H2 or CO-dependent reduction of M.
thermophila Isf. Furthermore, the genomic sequences of M. thermoautotrophicum and
Methanocoocus jannaschii also contain Isf homologues. These results may suggest a
general role for Isf as an electron carrier in both acetate fermenting and CO2-reducing
methanoarchaea.
INTRODUCTION
Methane production resulting from the energy-yielding metabolism of the
methanoarchaea represents the final step in the anaerobic degradation of complex
materials (7). This process is unique and involves many enzymes and coenzymes found
only in methanoarchaea (26). Methanoarchaea produce methane by two major pathways
(7). The first is the CO2-reducing pathway in which CO2 is reduced to methane using
electrons derived from either H2 or formate (equations 1 and 2). Another pathway is
Page 72
58
acetate fermentation, which contributes two-thirds of all biological methane produced in
nature (equation 3). In this pathway, acetate is cleaved in a CoA-dependent reaction and
the methyl group is reduced to methane using electrons derived from oxidation of the
carbonyl group to carbon dioxide (8).
CO2 + 4H2 à CH4 + 2H2O (1)
4 HCOO- + 4H+ à CH4 + 3 CO2 + 2 H2O (2)
CH3COO- + H+ à CH4 + CO2 (3)
Although the biochemistry and enzymology of carbon flow in these pathways are areas of
intense research, the electron transport carriers in methanogenesis are poorly understood.
Recently, a homodimeric iron-sulfur flavoprotein (Isf) was identified from an
acetate-utilizing methanoarchaeon, Methanosarcina thermophila. In the genome of M.
thermophila, the isf gene is located upstream and transcribed in the opposite direction
from the pta-ack operon encoding phosphotransacetylase and acetate kinase (14).
Comparison of the deduced Isf sequence with sequences in the available databases
suggested Isf was a novel iron-sulfur flavoprotein. Isf was hyperproduced in Escherichia
coli. The heterologously produced protein was partially characterized and in vitro
reconstitution experiments suggested Isf is a component in the electron transport chain
for methanogenesis from acetate (14). The absorption spectra and chemical analyses
suggested one iron-sulfur cluster and one FMN per subunit.
The present study reports the characterization of this iron-sulfur cluster and
unique FMN properties to offer insight into the role of Isf in electron transport.
Additionally, the context of Isf function in M. thermophila electron transport was
examined by testing several known electron carriers for interactions with Isf. Lastly,
information about the Isf homologs in other CO2-reducing methanoarchaea will be
discussed to help understand the physiological role of this electron carrier.
Portions of data contained in this section were obtained by Dr. S.W. Ragsdale and
Dr. D.F. Becker at the University of Nebraska, and Dr. K.K. Surerus at the University of
Wisconsin Milwaukee. Their contributions are noted in the appropriate sections.
Page 73
59
MATERIALS AND METHODS
Isf sequence analysis. In the process of doing site-directed mutagenesis for
ligand identification, it was discovered that the sequence at the C-terminal was incorrect
from that previously reported (14) and therefore, the gene was re-sequenced. The coding
region of isf was amplified using the Polymerase Chain Reaction (PCR) from
Methansarcina thermophila genomic DNA. The sequence of the upstream primer was 5'-
GGTGCACATATGAAAATAACAGGAAT-3' and contained the recognition sequence
for NdeI. The sequence of the downstream primer was 5'-
CAACTGGATCCATGCGATCATAAAC-3' and contained the recognition sequence for
BamHI. The PCR product was restricted with NdeI and BamHI. The resulting DNA was
cloned into the BamHI and NdeI sites of the pT7-7 overexpression vector. The construct
was checked by sequencing using the automated dideoxy method at the Penn State
University nucleic acid facility. The λ-EMBL3 (M. thermophila genomic library
containing isf , (14)) constructs were also subjected to the same procedure as described
above. Lastly, plasmid pML701 (14) was sequenced to confirm this error.
Protein production and purification. All proteins were purified anaerobically.
M. thermophila Isf was overproduced in Escherichia coli BL21(DE3) and purified as
described (14). M. thermophila ferredoxin A was purified as described (25). The
following procedure was used to obtain 57Fe-enriched Isf for Mössbauer study. The57Fe-labeled Isf was produced by growing E. coli cells in defined medium supplemented
with 57Fe at the final concentration of 20 µM. A solution of 57Fe was obtained by
dissolved 42.5 mg 57Fe in 850 µl of 2 N H2SO4 in a tube capped with a rubber stopper
that has a small plastic tubing insert at the top to let the gas out during the reaction. The
reaction solution was heat at 60-65o C for a week. The iron concentration was
determined by ferrozine using standard Fe(II) solution as a standard curve. The defined
medium composition per liter was: 745 ml deionized water; 200 ml M-solution (in a 1 l
solution contains: 42 g MOPS, 4 g tricine, 14.6 g NaCl, 8 g KOH, 2.55 g NH4Cl); 2 ml
O-solution (in a 50 ml solution contains: 2.68 g MgCl2.6H2O, 1 ml T solution (in a 100
Page 74
60
ml solution contains: 8 ml concentrate HCl, 18.4 mg CaCl2.2H20, 64 mg H3BO4, 40 mg
MnCl2.4H20, 340 mg ZnCl2, 605 mg Na2MoO4
.2H2O, 10mM 57Fe solution); 2 ml P-
solution (1 M KH2PO4); 1 ml S-solution (276 mM K2SO4); 0.5 ml of 0.2 % vitamin B
(thiamin); 20 ml of 20 % glucose; 40 ml of 3.75 % casamino acid; and 1 ml of 100
mg/ml ampicillin. The 57Fe solution was prepared by dissolving the iron metal in 2 N
H2SO4 for one week.
Cell extract of M. thermoautotrophicum used in the reduction of Isf was prepared
as follows. M. thermoautotrophicum strain Marburg was grown as described (21). Five
g (wet weight) cells resuspended in 50 mM Tris-Cl pH7.6 was lysed in a French pressure
cell at 20,000 psi. The lysate was centrifuged for 30 min at 33,000 x g, and the resulting
supernate (cell extract) was saved. All proteins were stored at –80o C until used.
UV-visible spectroscopy and potentiometric titrations. Potentiometric
measurements were performed as previously described (23, 24). All electrochemical
potentials are reported relative to the standard hydrogen electrode. Isf (3 - 4 µM) was
titrated at 20o C in 50 mM potassium phosphate buffer (pH 7.0 - 7.05) in a solution
containing methyl viologen (0.1 mM) as the mediator dye with phenosafarin (Em = -
0.252 V, pH 7.0) (5 µM) and benzyl viologen (Em= -0.362 V, pH 7.0) (5 µM) as the
indicator dyes. The pH measured after the experiment was recorded as the pH for the
titration. The visible spectra in each experiment were obtained and stored on an Olis-14
interfaced Cary spectrophotometer. The absorbance at 480 nm was used to monitor the
amount of oxidized and reduced FMN after correcting the spectra for turbidity. The
reduction potentials reported were determined by potentiometric measurements in the
reductive direction. After each potentiometric titration of the FMN chromophore, the
iron-sulfur flavoprotein was reoxidized completely using ferrocyanide (0.1 mM) as the
mediator dye. Equilibrium of the system in the UV-visible potentiometric measurements
was considered to be obtained when the measured potential drift was less than 1 mV in 5
min; this was typically around 1-2 h. The midpoint potentials (Em) and n values were
calculated using the Nernst Equation indicated below, where E is the measured
equilibrium potential at each point in the titration and n is the number of electrons. The
Page 75
61
typical error in the reported reduction potential values was ± 2-3 mV.
E=Em+ (0.058/n) log ([ox]/[red])
All midpoint potential value determinations exhibited Nernstian behavior as indicated by
their n values.
EPR spectroscopy and potentiometric titrations. The EPR spectra were
recorded on a Bruker ESP 300E spectrometer equipped with an Oxford ITC4 temperature
controller and automatic frequency counter of a Hewlett Packard Model 5340 and Bruker
Gaussmeter. The spectroscopic parameters are given in the figure legends. Double
integration of the EPR signals was performed with copper perchlorate (1 mM) as the
standard. Isf was frozen in liquid nitrogen prior to EPR analyses.
For the power saturation studies, Isf (74 µM, pH 7.0) was reduced in the presence
of 50 mM methyl viologen with a 40-fold excess of sodium dithionite prepared freshly at
pH 9.0. The solution was immediately frozen in an EPR tube and stored in liquid
nitrogen. Spectra of the reduced [4Fe-4S] cluster were recorded at powers varying from
0.1- 200 mW at five different temperatures between 5 and 25 K. The power for half
saturation (P1/2) at each temperature was determined by a fit to a plot of log (S/P*e0.5)
versus log P using Equation 7, where S is the signal amplitude, P is the microwave power
incident on the cavity, and b is the inhomogeneity parameter. Best fits to the data were
obtained by using a b value of 1.2.
The zero field splitting constant (∆) was determined by a linear fit to a plot of ln
P1/2 versus 1/T according to Equation 8, where T is the temperature, P1/2 is the power
for half saturation, k is the Boltzmann constant, and A is a coefficient representative of
the phonon-spin coupling properties of the [4Fe-4S] cluster.
P1/2=Aexp(-∆/kT)
Potentiometric measurements of the [4Fe-4S] cluster were performed in an EPR-
0.5b*1/2 e)P/P/(1P S +=
Page 76
62
spectroelectrochemical cell with a quartz EPR tube describes previously (9). Isf samples
(80 - 160 µM) in 50 mM potassium phosphate buffer (pH 7.0) were titrated at 20o C in
the presence of the mediator dyes, 150 µM benzyl viologen (Em = -0.362 V), 150 µM
methyl viologen (Em -0.440 V), 100 µM 1,1'-trimethylene-2,2’-bipyridyl (Em = -0.540
V), and 100 µM 4,4'-dimethyl-2,2'-dipyridyl (Em = -0.586 V). The intensity of the EPR
signal with a g-value of 1.93 was monitored to determine the redox state of the [4Fe-4S]
cluster during the titration. Potentiometric measurements were performed in the
reductive and oxidative directions. The system was considered to have reached
equilibrium when the measured potential drift was less than 1 mV in 2 min.
Mössbauer spectroscopy. Mössbauer spectra were recorded on a constant
acceleration spectrophotometer, MS-1200D, using a Janis Super Varitemp cryostat model
8DT with a Lakeshore temperature controller model 340 and a 57Co gamma source. The
experiments were done at 4.2 and 100 K. The reduced Isf was generated by adding 10-
fold excess of sodium dithionite.
Reduction of methanophenazine by Isf. The assay mixtures were
ananerobically equilibrated with 1.0 atm of CO in a stoppered 1.0 ml-cuvette maintained
at 35o C. The assay mixture (700 µl) contained: 50 mM Tris-Cl (pH 7.6), 2 mM
dithiothreitol, 1 mg/l resazurine, 25 µg carbon monoxide dehydrogenase /acetyl CoA
synthetase complex (CODH/ACS), and 9 µg M. thermophila ferredoxin A. After 10 min
incubation, 180 µg Isf were added to the assay mixtures and incubated for 10 min. The
reaction was initiated by the addition of 120 µM of 2-hydroxyphenazine (1). The
absorbance at 478 nm was measured in the Hewlett Packard 8452A diode array
spectrophotometer.
Reduction of Isf by M. thermoautotrophicum cell extract. The assay mixtures
were anaerobically equilibrated with 1.0 atm of CO, H2, or N2 in a stoppered 1.0 ml-
cuvette maintained at 35o C. The assay mixture (700 µl) contained: 50 mM Tris-Cl (pH
7.6), 2 mM dithiothreitol, 1 mg/l resazurine, 180 µg M. thermoautotrophicum cell extract,
Page 77
63
and 13.5 µg M. thermophila ferredoxin A. After 10 min incubation, the reaction was
initiated by addition of 180 µg Isf. Ferredoxin A was omitted in some assays whereas
ferredoxin from Clostridium pasteurianum was substituted where indicated. The
absorbance at 476 nm was measured in the Hewlett Packard 8452A diode array
spectrophotometer.
RESULTS
Sequence analysis. Error in the previously reported isf sequence (14) was
detected during site-directed mutagenesis studies (refer to chapter 4). Figure 1 shows the
corrected sequence deduced from genomic DNA. The corrected Isf contains 191
residues, 81 fewer than previously reported. Additionally, the C-terminal residues177
KLCDVLELIQKNRDK191
in the corrected Isf sequence replace177
NSVMSWNLFRKIEIN191
in the previously reported Isf. Since the DNA library
containing isf gene was available, isf gene from this library was sequenced to see if there
was an error that may result in error in previous study. Identical sequences were found
both from genomic DNA and DNA library as reported in Figure 1. Re-sequencing
revealed the correct isf sequence in pML701 used for the heterologous production of Isf
reported here and previously (14).
Page 78
64
ATG AAA ATA ACA GGA ATT TCA GGC AGT CCA CGA AAG GGC CAG AAC 45M K I T G I S G S P R K G Q N 15
TGT GAG AAA ATA ATT GGA GCT GCT CTT GAG GTT GCA AAA GAA AGA 90C E K I I G A A L E V A K E R 30
GGG TTT GAA ACT GAT ACC GTT TTT ATC TCA AAC GAG GAG GTT GCC 135G F E T D T V F I S N E E V A 45
CCC TGC AAA GCG TGC GGG GCT TGC AGA GAT CAA GAT TTC TGT GTG 180P C K A C G A C R D Q D F C V 60
ATT GAT GAT GAT ATG GAC GAG ATA TAT GAA AAA ATG AGG GCT GCA 225I D D D M D E I Y E K M R A A 75
GAC GGT ATA ATT GTT GCA GCT CCC GTA TAT ATG GGG AAT TAT CCT 270D G I I V A A P V Y M G N Y P 90
GCC CAG CTT AAA GCC CTT TTT GAC AGG AGT GTC CTG CTT CGC CGT 315A Q L K A L F D R S V L L R R 105
AAA AAC TTT GCA CTA AAA AAT AAA GTT GGG GCA GCT CTT TCA GTT 360K N F A L K N K V G A A L S V 120
GGG GGC TCA AGA AAC GGA GGA CAG GAA AAA ACA ATT CAG TCC ATA 405G G S R N G G Q E K T I Q S I 135
CAT GAC TGG ATG CAC ATT CAC GGA ATG ATT GTA GTC GGC GAT AAT 450H D W M H I H G M I V V G D N 150
TCC CAC TTC GGT GGA ATT ACG TGG AAC CCG GCA GAA GAG GAC ACT 495S H F G G I T W N P A E E D T 165
GTT GGA ATG CAG ACA GTT TCC GAA ACT GCA AAA AAA CTC TGT GAT 540V G M Q T V S E T A K K L C D 180
GTC CTG GAA CTT ATT CAG AAA AAT AGA GAT AAA TAA CAA AAT TCA 585V L E L I Q K N R D K * 191
TAA ATT ATA TAA GTC AGG GTA GAA TAA AAC AAA AAA TAT GAA TTT 630CCG AGA AGT AAA TTA GTT ATA TTA ATC TTA TTA TAT TGC ACA TTT 675CAA ACC CTG GCA ATC CTG TGC CAC TAT GCT ATA CCA AAA AGT CAT 720TTT GTT GAT ATA AAA TCA ATA AAA TGT CCA ATA ATC AAA TAT TAT 765TGT CCT ATA TTG GTA GGA GTT CAA AAA CCC CAG GAT AAT GGA GTA 810CAT CTC CCG GTT TGA 825
Figure 1. Corrected nucleic acid sequence and predicted amino acid sequence of isf from
M. thermophila. The DNA is presented in the 5' to 3' direction. The predicted amino
acid sequence of Isf is shown in single-letter code directly below the first base of each
codon. *, initial base of translation stop codon.
Page 79
65
EPR spectroscopy of the iron-sulfur centers. Isf was purified and sent to Dr.
S.W. Ragsdale and Dr. D.F. Becker at the University of Nebraska for EPR analysis.
Their results, interpretations, and conclusions follow. The reduced heterologously
produced Isf displayed the rhombic EPR spectrum indicative of a [4Fe-4S]+1 type with g
values at 2.06, 1.93 and an unusual split signal at 1.86 and 1.82 (Fig. 2). Additional
evidence for the presence of the [4Fe-4S]1+ center is the disappearance of the g = 1.93 at
the temperatures above 25 K (20). Several lines of evidence suggest the
splitting signal observed at gmin region of EPR spectrum is derived from the
microheterogeneity within Isf molecules. For example, the presence of viologen dyes did
not contribute to this complex spectrum since the sodium dithionite-reduced sample
without dyes also give a similar spectrum. Since the flavin is diamagnetic in both the
oxidized and reduced state, this diamagnetic cofactor cannot give rise to this unusual
feature. The possibility that a strong hyperfine interaction between an unpaired electron
on the cluster and a strongly coupled proton produces a complex feature was examined.
Reduced protein after an extensive exchange with D2O also exhibited the same spectrum;
thus, this possibility was ruled out. In the power and temperature studies, the result
showed two separate and distinct species, one with g values at 2.06, 1.92, and 1.82, and
another one with g values at 2.03, 1.92, and 1.86 (refer to chapter 5). These results are
consistent with the conclusion of microheterogeneity that contributes to the unusual
spectrum.
Power saturation studies of the [4Fe-4S] cluster were examined at five difference
temperatures (5, 10, 15, 20, and 25 K). The half saturation powers (P1/2) are in the range
of 79 to 14.4 mV. The plot of P1/2 and 1/T showed a linear relationship at the
temperatures higher than 5 K (Fig. 3). The zero field splitting parameter (∆) was
observed at the values between 10 and 11.5 cm-1 (Fig. 3,inset).
Page 80
66
Figure 2. EPR spectroscopy of the [4Fe-4S] cluster in Isf poised at various redox
potentials in 50 mM potassium phosphate buffer (pH 7.0). Experimental conditions were
as follows: temperature, 10 K: microwave power, 1.26 milliwatts; microwave frequency,
9.43 GHz; receiver gain, 2 x 104; modulation amplitude, 10 G; modulation frequency,
100 kHz. The derviative feature at g = 2.0 results from the mediator dyes.
Page 81
67
Figure 3. Semilogarithmic plot of P1/2 versus 1/T, which shows a linear relationship
according to equation P1/2 = Aexp(-∆/kT). The slope (-∆/k = 1.5 +/- 0.73) yields an
estimate of 11.5 cm-1 for the zero field splitting value (∆). Inset, a nonlinear plot of P1/2
versus 1/T, which includes the data at 5 K. The slope (-∆/k = 14.1 +/- 1.8) yields an
estimate for ∆ of 9.8 cm-1.
Page 82
68
Mössbauer spectroscopy. Prior to Mössbauer spectroscopy, Isf was
heterologously produced in E. coli cultured in media enriched with 57Fe. The 57Fe-
enriched protein was purified and sent to Dr. K.K. Surerus at the University of Wisconsin
Milwaukee for Mössbauer spectroscopy. Her results, interpretations, and conclusions
follow. The as-purified Isf exhibited a single broad quadrupole doublet Mössbauer
spectrum with an average isomer shift, δ = 0.45 mm/s, and an average quadrupole
splitting, ∆EQ = 1.22 mm/s, at 4.2 K (Fig. 4a). The quadrupole splitting decreased
slightly at 100 K (∆EQ = 1.12 mm/s). Dithionite-reduced Isf exhibited a single broad
nonsymmetrical quadrupole doublet with an average isomer shift, δ = 0.55 mm/s and an
average quadrupole splitting, ∆EQ = 1.30 mm/s at 100 K (Fig. 4b). These parameters are
indicative of values for the [4Fe-4S] center with 2+ and 1+ redox states. The
nonsymmetrical line shapes observed for both as-purified and reduced proteins,
especially for reduced protein, suggest the irons within the cluster are not identical.
Another reason may due to a microheterogeneous population of iron-sulfur clusters in the
Isf molecule. The Voigt (Gaussian distribution of a Lorenzian lineshape) lineshape of the
doublet, rather than Lorenzian, indicates a microheterogenous environment for the iron
sites. These finding are consistent with the conclusion drawn from EPR spectrum.
The property of electronic spin of the iron-sulfur cluster was studied by applying
the magnetic field either parallel or perpendicular to the γ beam. Mössbauer spectra of
the reduced Isf exhibited paramagnetic hyperfine structure indicative of reduced [4Fe-
4S]1+ cluster with S = ½ (Fig. 5). The shape of the spectra and the derived hyperfine
structure are typical of that observed in other [4Fe-4S] proteins (4, 15, 17).
Page 83
69
Figure 4. Mössbauer spectra recorded at 100 K. A) oxidized Isf protein. B) reduced Isf
protein. The solid line is a least-square fit with a Voigt line shape.
Page 84
70
Figure 5. Mössbauer spectra of reduced Isf protein recorded at 4.2 K and 450 G applied
parallel (A) or 450 G applied perpendicular (B) to the γ beam. The solid line is a
theoretical fit of an S = ½.
Page 85
71
Potentiometric titrations of FMN and the [4Fe-4S] center. Potentiometric
measurements were performed by Dr. S.W. Ragsdale and Dr. D.F. Becker at the
University of Nebraska. The absorption spectrum of Isf was followed between 300 to
700 nm during potentiometric titration (Fig. 6). Reduction of FMN was monitored at 480
nm to avoid absorption interference from the [4Fe-4S] cluster. Absorption due to the
[4Fe-4S] cluster was measured at potentials below -305 mV to avoid interference from
the absorption of FMN. The FMN/FMNH2 couple showed a midpoint potential of –277
mV (Fig. 6, inset). There was no appearance of absorption around 500-600 nm, which is
generally present due to the semiquinone. This destabilization of the semiquinone is a
rare case for flavoprotein.
FMN has been shown to go through one electron reductions sequentially
generating a semiquinone and the hydroquinone. Although the semiquinone was not
detected during potentiometric titration, semiquinone may be stabilized in the
physiological system. Attempts to determine the semiquinone formation in the biological
system was performed in the mixture of CODH/ACS, Isf, and CO. The EPR spectra of
frozen reaction mixtures at different time points were observed. The maximum of only
2.5 % of semiquinone form was observed at 28 min (Fig. 7). Over the course of 50 min,
the cluster underwent reduction as the FMN was fully reduced to the hydroquinone form.
The semiquinone form occurred during this reaction is classified as the anionic or red
semiquinone, since the EPR signal yielded a line width with 16 gauss . Therefore, the
formation of semiquinone is possible under the physiological conditions.
The Em value for the +2/+1 couple state of the [4Fe-4S] center was determined
using spectroelectrochemical titration. When the data were analyzed using the Nernst
equation, the Em calculated for the [4Fe-4S]2+/1+ center was –394 mV and the slope was
53 mV (Fig. 2 and 8), results consistent with a one electron transfer carrier (the
theoretical value for a one-electron transfer is 58 mV). The redox reaction was fully
reversible since the reduction was titrated in both the oxidative and reductive directions.
Thus, the midpoint potential of the [4Fe-4S] cluster is more than 100 mV lower than that
of the FMN/FMNH2 couple (Fig. 6 and 8) and is in the range as the value reported for
Page 86
72
Figure 6. Potentiometric titration of the FMN in Isf (3.2 µM) in 50 mM potassium
phosphate buffer (pH 7.0) at 20o C (curves 1-7, fully oxidized, -262, -272, -281, -290, -
305, and –342 mV respectively). Inset, Nernst plot of the potentiometric data.
Page 87
73
Figure 7. EPR spectrum of Isf (170 µM dimer) was recorded at 10 K following
incubation for 17 min with CO and CODH (0.5 µM) at 25o C. The amount of FMN
hydroquinone and reduced iron-sulfur cluster at this time point were 46 and 3% (0/03
spin/mol), respectively. After the sample was frozen in liquid nitrogen, the spectrum was
recorded using the conditions described in figure 2)
Page 88
74
Figure 8. A fit of the Isf midpoint potential data to a theoretical curve generated from
the Nernst equation for two redox centers with reduction potentials of –277 mV (n = 2)
and –394 mV (n = 1).
Page 89
75
other low-potential [4Fe-4S] clusters (2). These results suggest electrons flow from the
4Fe-4S center to FMN.
Potential electron acceptors for Isf. A previous study indicated ferredoxin A is
a direct physiological electron donor for Isf (14); however, the physiological electron
acceptor is unknown. From the properties of redox centers in Isf described above, 2-
electron carriers are candidates to accept electrons from Isf. Isf could accept one electron
from ferredoxin A thorough the [4Fe-4S] center and two electrons are transferred to
FMN. FMN in the hydroquinone form donates the 2 electrons to the 2-electron carrier.
Several 2-electron carriers such as F420, NAD+, NADP, were included in reconstitution
electron transport assays composed of purified components of CO, CODH, ferredoxin A,
and Isf. The results showed none of these 2-electron carriers were reduced by Isf. Unless
an unknown 2-electron carrier accepts electrons from Isf, these results suggest Isf is not a
1- electron 2-electron switch.
The participation of methanophenazine as electron acceptor for Isf was examined.
2-hydroxyphenazine was a gift from Dr. U. Deppenmeier. Methanophenazine was
isolated from H2/CO2-grown Methanosarcina mazei Gö1 and shown to be involved in
reduction of CoB-S-S-CoM (1). The as-isolated methanophenazine was water insoluble,
thus 2-hydroxyphenazine (a water-soluble analogue) has been used in aqueous buffer
assays (1). The midpoint potential of 2-hydroxyphenazine is –255 mV (1). With the
assumption that the redox potential of methanophenazine is similar to this, this cofactor
should be able to act as an electron acceptor of Isf. However, the results revealed a
different data from the theoretical assumption. Reduction of 2-hydroxyphenazine in the
reconstitution electron transport system with CO, CODH/ACS, and ferredoxin A is
greater than the system containing those components and Isf (Fig. 9). In the
reconstitution system with CO, and CODH/ACS, no reduction of 2-hydroxyphenazine
was observed. These results indicate that ferredoxin A is an electron donor for both Isf
and 2-hydroxyphenazine. At this stage, we have been unable to identify the electron
acceptor for Isf.
Page 90
76
Figure 9. Time course for reduction of methanophenazine with Isf from M. thermophila.
The standard assay mixtures were anaerobically equilibrated with 1.0 atm. of CO in a
stoppered 1.0 ml-cuvette maintained at 35 o C. The standard assay mixture (700 µl)
contained: 50 mM Tris-Cl (pH 7.6), 2 mM dithiothreitol, 1 mg/l resazurine, 25 µg
CODH/ACS and 120 µM methanophenazine (♦). The assay contained all components of
the standard assay plus 9 µg M. thermophila ferredoxin A ( ). The assay contained all
components of the standard assay plus 9 µg M. thermophila ferredoxin A and 180 µg Isf
( ).
0
0.1
0.2
0.3
0.4
0.5
0 2 4 6 8 10
Minutes
A47
8
Page 91
77
Evidence for Isf homologs in phylogenetically and physiologically diverse
microbes. Two genomic sequences of the CO2-reducing methanoarchaea, M. jannaschii
and M. thermoautotrophicum, were recently completed (3, 22). M. jannaschii and M.
thermoautotrophicum are phylogenetically and physiologically distinct from M.
thermophila. Neither M. jannaschii or M. thermoautotrophicum can utilize acetate as
growth substrate, instead they evolve methane by reducing CO2 using H2 as electron
donor. Two open reading frames (ORF) from M. jannaschii, MJ0731 and MJ1083,
which encode 192 and 194 amino acid proteins have 40 and 49 % identity to Isf
from M. thermophila. The genome of M. thermoautotrophicum contains three ORFs,
MTH135, MTH1473, and MTH1595 with 41, 34, and 30 % identity to the M.
thermophila Isf. Comparisons of these Isf sequences also show a completely conserved
N-terminal cysteine motif (Fig. 10). These results suggest a general function for this
electron carrier in the methanoarchaea; thus, Isf-like proteins may be widespread electron
carriers for methanogenesis in diverse methanoarchaea. An electron carrier function of
Isf in methanoarchaea other than M. thermophila was examined to test this notion.
Extracts of H2/CO2 grown M. thermoautotrophicum were able to catalyze the reduction
of M. thermophila Isf with either H2 or CO as the electron donor (Fig. 11). These
results suggest Isf homologues are components of the electron transport chain in CO2-
reducing methanoarchaea. The reduction of Isf in the presence of CO is higher than that
of H2 suggesting Isf may be specific for electron transport coupled to CO oxidation. The
Isf reduction rate was stimulated by addition of ferredoxin A from M. thermophila. This
implies that ferredoxin A or a homologue is able to couple the oxidation of either H2 or
CO to the reduction of Isf. However, ferredoxin from C. pasteurianum cannot replace
ferredoxin A for Isf reduction in this system (Fig. 12); thus, the reaction appears to be
ferredoxin A specific.
Page 92
78
Figure 10. Multiple amino acid sequence alignment of Isf from M. thermophila with
sequences deduced from open reading frames identified in the genomic sequences of M.
jannashii and M. thermoautotrophicum. Accession numbers for the M. jannashii and M.
thermoautotrophicum sequences and percentage identity (in parenthesis) with M.
thermophila Isf are as follows: MTH135 (41%), MTH1473 (34%0, MTH1595 (30%), MJ
1083 (49%), and MJ0731 (40%). Asterisks indicate conserved cysteine residues.
Page 93
79
Figure 11. Time course for reduction of Isf with extract from M. thermoautotrophicum.
The assay mixture (700 µl) contained cell extract (180 µg protein), M. thermophila
ferredoxin (13.5 µg), 50 mM Tris (pH 7.6) and 2 mM dithiothreitol. Ferredoxin was
omitted in two of the assays (�, ♦). The assay mixtures were anaerobically equilibrated
with 1 atm. of CO (�, •), H2 ( ,,♦), or N2 (∆) in a stoppered 1.0-ml cuvette maintained at
35o C. After a 10-min incubation, the reaction was initiated be the addition of 180 µg of
Isf.
Page 94
80
Figure 12. Time course for reduction of Isf with extract from M. thermoautotrophicum.
The assay mixture (700 µl) contained: cell extract (180 µg protein), either M.
thermophila ferredoxin A (•, ) or C. pasteurianum ferredoxin (o, ∆) (13.5 µg), 50 mM
Tris (pH 7.6) and 2 mM dithiothreitol. The assay mixtures were anaerobically
equilibrated with 1 atm. of CO (•, o), or H2 ( , ∆) in a stoppered 1.0-ml cuvette
maintained at 35o C. After a 10-min incubation, the reaction was initiated by the addition
of 180 µg of Isf.
0.00
0.05
0.10
0.15
0.20
0 5 10 15Minutes
∆∆A
476
Page 95
81
DISCUSSION
The properties and physiological role of Isf was examined in this investigation.
Previous studies indicate an involvement of Isf in the electron transport chain of CO-
dependent CoM-S-S-CoB during methanogenesis of M. thermophila (14). However,
further characterization of the redox centers in Isf is necessary to provide insight into a
specific role for Isf in the electron transport chain. The iron-sulfur cluster type present in
the heterologously produced Isf was unequivocally demonstrated by EPR and Mössbauer
spectroscopy to be [4Fe-4S]. The Isf sequence reveals a completely conserved cysteine
motif which has the potential to serve as ligands for the [4Fe-4S] center; however, the
cysteine spacing in the motif is distinct from any known motifs accommodating known
[4Fe-4S] centers. The unusual cysteine motif in Isf may represent a novel class of
cysteine motif ligating the [4Fe-4S] center among the Isf-like sequences. The presence of
two negative absorption features in the EPR spectra of the reduced cluster is unusual.
The results presented here indicate that microheterogeneity within the population of Isf
molecules accounts for this atypical feature. This situation is similar to that of a class of
corinoid/iron-sulfur proteins from methanoarchaea and homoacetogenic anaerobes from
the Bacteria domain in which the reduced [4Fe-4S] cluster exhibits a broad absorption
feature in the same g value region (10, 12, 19). The Em results for the 4Fe-4S center and
FMN indicate that the intra-electron transfer from [4Fe-4S] to FMN is plausible. The Em
values from this investigation and other known Em values from other electron transfer
components are consistent with the electron flow as CODH (Em for center C and [4Fe-
4S] center = -540 and –444 mV, (16)) Õ ferredoxin (Em = -407 mV, (5)) Õ Isf [4Fe-4S] (Em
= -394 mV) Õ Isf [FMN] (Em = -277 mV) Õ an unknown electron carrier (Fig. 13). Most
flavin/Fe-S proteins stabilize four redox states: flavinox:FeSox, flavin semiquinone:FeSox,
flavin semiquinone:FeSred, and flavinred:FeSred (13, 18). Stabilization of the semiquinone
allows versatility of mediating both one-electron and two-electron transfer reactions. The
results reported here suggest only a transient stabilization of the FMN semiquinone;
however, stability of semiquinone may be increased when Isf interacts with its
physiological electron donors/acceptors.
Page 96
82
Figure 13. Proposed electron transport pathway for oxidation of CO or the carbonyl
group of acetyl-CoA. Cdh A, subunit of the CODH/ACS; FdxA, ferredoxin A; ?,
postulated unknown electron carrier. Midpoint potentials are shown in mV.
CdhA
“C” -540 mV“B” -444 mV
CO
CO2
-518 mV FdxA
4Fe4S -407mV
Isf
4Fe4S -395 mVFMN -277 mV
?
Page 97
83
Due to the high instability of the semiquinone of FMN, it is possible that the
physiological electron acceptor for Isf (which is unknown) could be a two-electron carrier
and Isf functions as a one-electron/two-electron switch. However, involvement of the
obligate two-electron carrier coenzyme F420, NAD, NADP in the electron transport chain
has been excluded. A role for the two-electron carrier methanophenazine (1) as electron
acceptor of Isf was ruled out. Indeed, methanophenazine competed with Isf for
ferredoxin A. Methanophenazine has never been isolated from acetate-grown M.
thermophila cells. Due to the reduction of methanophenazine by ferredoxin A of M.
thermophila, it is possible that this compound is present in cells and may function in
place of Isf under different growth conditions. Additional experiments are required to
confirm if the reduction of methanophenazine is physiologically significant.
The environment in the FMN binding site of Isf must be different from other
flavoproteins. To stabilize a negative charge of hydroquinone, an amino acid with
positive charge may be required. From the sequence comparisons, two positively
charged amino acids, K94 and R124, are highly conserved. These two residues may
flank the FMN binding site and would result in the thermodynamic stabilization of the
hydroquinone.
The presence of Isf-like sequences in the genomes of M. jannaschii and M.
thermoautotrophicum, and the use of either H2 or CO as electron donor for Isf reduction
by M. thermoautotrophicum cell extract, imply a physiological significance of Isf as a
primary electron carrier in different methanogenesis pathways. The greater reduction of
Isf using CO as electron donor and the ability of M. thermoautotrophicum to grow and
produce CH4 with CO as the sole energy source (6) indicate a physiological role for
CODH in the energy metabolism. Furthermore, this methanoarchaeon involves a CODH
in the synthesis of CO for incorporation into acetyl-CoA for cell carbon (11). The
CODHs from either M. thermoautotrophicum or M. jannaschii have not been purified
and, therefore, the electron acceptor is unknown. The results presented here are
consistent with ferredoxin as the electron acceptor; however, purification of the CODH is
necessary to prove this hypothesis. The implicated functions of Isf homologues in
Page 98
84
diverse methanoarchaea are also supported by the gene organization in M. jannaschii.
The MJ0731 is located adjacent to MJ0722 and MJ0728, which encode an 8Fe
ferredoxin- and CODH/ACS-like proteins (3). However, there is no such gene
arrangement found in M. thermoautotrophicum (22).
ACKNOWLEDEGEMENT
I would like to thank Dr. D.F. Becker and Dr. S.W. Ragsdale at University of
Nebraska-Lincoln for performing the EPR spectroscopy, and Dr. K.K. Surerus at
University of Wisconsin Milwaukee for Mössbauer spectroscopy. I also thank Dr. U.
Deppenmeier for the 2-hydroxymethanophanize and Dr. J. M. Bollinger for the
suggestion on 57Fe solution preparation. Work described here was partially supported
National Institutes of Health Grant No. 1-R15-GM52666-01 (KKS), and by Department
of Energy Basic Energy Sciences Grants No. DE-FG02-ER20053 (SWR) and DE-FG02-
95ER20198 (JGF). UL was supported by a MOSTE grant from Thailand.
Page 99
85
REFERENCES
1. Abken, H.-J., M. Tietze, J. Brodersen, S. Bäumer, U. Beifuss, and U.
Deppenmeier. 1998. Isolation and characterization of methanophenazine and the
function of phenazines in memebrane-bound electron transport of
Methanosarcina mazei Gö1. J.Bacteriol. 180:2027-2032
2. Bruschi, M., and F. Guerlesquin. 1988. Structure, function and evolution of
bacterial ferredoxins. FEMS Microbiol. Rev. 4:155-75.
3. Bult, C. J., O. White, G. J. Olsen, L. X. Zhou, R. D. Fleischmann, G.
G.Sutton, J. A. Blake, L. M. Fitzgerald, R. A.Clayton, J. D.Gocayne, A. R.
Kerlavage, B. A.Dougherty, J. F. Tomb, M. D. Adams, C. I. Reich, R.
Overbeek, E. F. Kirkness, K. G. Weinstock, J. M. Merric, A. Glodek, J. L.
Scott, N. S. M. Geoghagen, J. F. Weidman, J. L. Fuhrmann, D. Nguyen, T. R.
Utterback, J. M. Kelly, J. D. Peterson, P. W. Sadow, M. C. Hanna, M. D.
Cottoon, K. M. Roberts, M. A. Hurst, B. P. Kaine, M. Borodvsky, H.-P.
Klenk, C. M. Fraser, H. O. Smith, C. R. Woese, and J. C. Venter. 1996.
Complete genome sequence of the methanogenic archaeon, Methanococcus
jannaschii. Science. 273:1058-1073.
4. Christner, J. A., P. A. Janick, L. M. Siegel, and E. Munck. 1983. Mössbauer
studies of Escherichia coli sulfite reductase complexes with carbon monoxide and
cyanide. Exchange coupling and intrinsic properties of the [4Fe-4S] cluster. J.
Biol. Chem. 258:11157-64.
5. Clements, A. P., L. Kilpatrick, W.-P. Lu, S. W. Ragsdale, and J. G. Ferry.
1994. Characterization of the iron-sulfur clusters in ferredoxin from acetate-
grown Methanosarcina thermophila. J. Bacteriol. 176:2689-2693.
6. Daniels, L., G. Fuchs, R. K. Thauer, and J. G. Zeikus. 1977. Carbon
monoxide oxidation by methanogenic bacteria. J. Bacteriol. 132:118-126.
7. Ferry, J. G. 1992. Biochemistry of methanogenesis. Crit. Rev. Biochem. Mol.
Biol. 27:473-503.
8. Ferry, J. G. 1992. Methane from acetate. J. Bacteriol. 174:5489-5495.
9. Harder, S. R., B. A. Feinberg, and S. W. Ragsdale. 1989. A
Page 100
86
spectroelectrochemical cell designed for low temperature electron paramagnetic
resonance titration of oxygen-sensitive proteins. Anal. Biochem. 181:283-7.
10. Harder, S. R. L., W. P. Lu, B. A. Feinberg, and S.W. Ragsdale. 1989.
Spectroelectrochemical studies of the corrinoid iron-sulfur protein involved in
acetyl coenzyme-A synthesis by Clostridium thermoaceticum. Biochemistry.
28:9080-9087.
11. Hemming, A., and K. H. Blotevogel. 1985. A new pathway for CO2 fixation in
methanogenic bacteria. Trends Biochem. Sci. 10 :198-200.
12. Jablonski, P. E., W. P. Lu, S. W. Ragsdale, and J. G. Ferry. 1993.
Characterization of the metal centers of the corrinoid;iron- sulfur component of
the CO dehydrogenase enzyme complex from Methanosarcina thermophila by
EPR spectroscopy and spectroelectrochemistry. J. Biol. Chem. 268:325-329.
13. Johnson, M. K. 1994. Iron-sulfur proteins. Encyclopedia of Inorganic Chemistry,
Johnson Wiley and Sons (King, R.B., ed). 4:1896-1915.
14. Latimer, M. T., M. H. Painter, and J. G. Ferry. 1996. Characterization of an
iron-sulfur flavoprotein from Methanosarcina thermophila. J. Biol. Chem.
271:24023-24028.
15. Lindahl, P. A., E. P. Day, T. A. Kent, W. H. Orme-Johnson, and E. Munck.
1985. Mössbauer, EPR, and magnetization studies of the Azotobacter vinelandii
Fe protein. Evidence for a [4Fe-4S]1+ cluster with spin S = 3/2. J. Biol. Chem.
260:11160-73.
16. Lu, W. P., P. E. Jablonski, M. Rasche, J. G. Ferry, and S. W. Ragsdale. 1994.
Characterization of the metal centers of the Ni-Fe-S component of the carbon-
monoxide dehydrogenase enzyme complex from Methanosarcina thermophila. J.
Biol. Chem. 269:9736-9742.
17. Middleton, P., D. P. Dickson, C. E. Johnson, and J. D. Rush. 1978.
Interpretation of the Mössbauer spectra of the four-iron ferredoxin from Bacillus
stearothermophilus. Eur. J. Biochem. 88:135-41.
18. Muh, U., I. Cinkaya, S. P. Albracht, and W. Buckel. 1996. 4-Hydroxybutyryl-
CoA dehydratase from Clostridium aminobutyricum: characterization of FAD and
iron-sulfur clusters involved in an overall non-redox reaction. Biochemistry.
Page 101
87
35:11710-8.
19. Ragsdale, S. W., P. A. Lindahl, and E. Munck. 1987. Mössbauer, EPR, and
optical studies of the corrinoid;iron-sulfur protein involved in the synthesis of
acetyl-CoA by Clostridium thermoaceticum. J. Biol. Chem. 262:14289-14297.
20. Rupp, H., K. K. Rao, D. O. Hall, and R. Cammack. 1978. Electron spin
relaxation of iron-sulphur proteins studied by microwave power saturation.
Biochim. Biophys. Acta. 537(2):255-60.
21. Schonheit, P., J. Moll, and R. K. Thauer. 1980. Growth parameters (Ks, mu-
max, Ys) of Methanobacterium thermoautotrophicum. Arch. Microbiol. 127:59-
65.
22. Smith D. R. , L. A. Doucette-Stamm, C. Deloughery, H. Lee, J. Dubois, T.
Aldredge, R. Bashirzadeh, D. Blakely, R. Cook, K. Gilbert, D. Harrison, L.
Hoang, P. Keagle, W. Lumm, B. Pothier, D. Qiu, R. Spadafora, R. Vicaire,
Y. Wang, J. Wierzbowski, R. Gibson, N. Jiwani, A. Caruso, D. Bush, H.
Safer, D. Patwell, S. Prabhakar, S. McDougall, G. Shimer, A. Goyal, C. J.
Daniels, J. I. Mao, P. Rice, J. Nölling, and J. N. Reeve. 1997. The complete
genome sequence of Methanobacterium thermoautotrophicum strain ∆H:
functional analysis and comparative genomics. J. Bacteriol. 179:7135-7155.
23. Stankovich, M., and B. Fox. 1983. Redox potentials of the flavoprotein lactate
oxidase. Biochemistry. 22:4466-72.
24. Stankovich, M. T. 1980. An anaerobic spectroelectrochemical cell for studying
the spectral and redox properties of flavoproteins. Anal. Biochem. 109:295-308.
25. Terlesky, K. C., and J. G. Ferry. 1988. Purification and characterization of a
ferredoxin from acetate-grown Methanosarcina thermophila. J. Biol. Chem.
263:4080-4082.
26. White, R. H., and D. Zhou. 1993. Biosynthesis of the coenzymes in
methanogens. Methanogenesis, J.G. Ferry, ed. Chapman and Hall, New York.
409-444.
Page 102
88
CHAPTER 5
A NOVEL 4Fe-4S CLUSTER BINDING MOTIF IN THE IRON-
SULFUR FLAVOPROTEIN OF Methanosarcina thermophila
ABSTRACT
Isf (Iron-sulfur flavoprotein) from Methanosarcina thermophila has been
produced in Escherichia coli as a dimer containing two [4Fe-4S] clusters and two FMN
(flavin mononucleotide). The deduced sequence of Isf contains six cysteines (C16, C47,
C50, C53, C59, and C180), four of which (C47, C50, C53, and C59) compromise a motif
perfectly conserved among several putative Isf sequences available in the databases. The
spacing of the four conserved cysteines is highly compact and atypical of motifs
coordinating known 4Fe-4S clusters; therefore, all 6 cysteines in Isf were altered to either
alanine or serine to obtain biochemical confirmation that the motif coordinates the 4Fe-
4S cluster and to determine the influence of the protein environment on the properties of
the cluster. All except the C16S variant were produced in inclusion bodies that required
solubilization and reconstitution of the iron-sulfur cluster and FMN. The UV-visible
spectra of all variants indicated the presence of iron-sulfur clusters and FMN. The
reduced C16X (X = A or S) variants showed the same EPR spectra as wild type Isf
whereas the reduced C180X variants showed EPR spectra similar to one of the 4Fe-4S
species present in the wild type Isf spectrum. EPR spectra of the oxidized C50A and
C59A variants showed g-values characteristic of a 3Fe-4S cluster. The spectra of the
C47A and C53A variants indicated a 4Fe-4S cluster but with g-values different from wild
type. The retention of 4Fe-4S cluster in the C47A and C53A variants suggests functional
replacement of the cysteines by 2-mercaptoethanol that was present in the reconstitution
buffer. The reduced C47S, C50S, and C53S exhibited EPR spectra of 4Fe-4S centers.
EPR spectrum of both C47S and C53S revealed two different 4Fe-4S ligand species,
which could be due to the replacement of the missing ligand by serine or 2-
mercaptoethanol. Taken together with strict sequence conservation, these results indicate
that C47, C50, C53 and C59 are ligands to the 4Fe-4S cluster, a result which identifies
the most compact cysteine motif know which ligates a redox-active 4Fe-4S cluster. The
Page 103
89
results suggest C16 is important for maintaining a local conformation required for
transfer of electrons from ferredoxin A to FMN, and that C180 is essential for the overall
structural integrity of Isf. The reduction of FMN in the Isf variants by ferredoxin A was
either several-fold impaired or enhanced suggesting that the 4Fe-4S cluster serves to
transfer electrons from ferredoxin A to FMN.
INTRODUCTION
Two-thirds of the biologically produced methane in nature originates from the
methyl group of acetate in a pathway where acetate is cleaved and the methyl group is
reduced to methane with electrons derived from oxidation of the carbonyl group to
carbon dioxide (7). Much is known concerning the cleavage of acetate and one-carbon
transfer reaction (6); however, less is known regarding electron transport. Recently, a
novel iron-sulfur flavoprotein (Isf) from Methanosarcina thermophila was characterized
which participates in electron transport during the methanogenic fermentation of acetate
(2, 13). The homodimeric Isf contains two FMN molecules and two 4Fe-4S clusters
unequivocally identified by EPR and Mössbauer spectroscopy. The midpoint-potential
values of the 4Fe-4S cluster and FMN are -394 and -277 mV, respectively. These results
are the basis for a postulated role for the cluster in electron flow from ferredoxin A, the
physiological electron donor for Isf, to the 4Fe-4S cluster and then to the FMN of Isf,
however, this proposal has not been tested. The physiological electron acceptor for Isf is
unknown. The deduced sequence of Isf contains six cysteines, four of which are in a
highly compact novel motif (CX2CX2CX4-7C) that is perfectly conserved among putative
Isf-like sequences identified in the databases suggesting the motif ligates the 4Fe-4S
cluster.
The cubane [4Fe-4S] cluster is ubiquitous in proteins from all domains of life (22)
where they mainly function in electron transfer. The sulfur atom of cysteine is the
prominent protein ligand coordinated to iron atoms in these clusters. Few examples of
variations from cysteine ligation include aconitase with oxygen ligation originating from
hydroxide, water, or substrate. The 4Fe-4S cluster in the ferredoxin from Pyrococcus
Page 104
90
furiosus is ligated with oxygen from aspartate (27). An iron atom in the 4Fe-4S cluster of
hydrogenase from Desulfovibrio gigas is coordinated by a histidyl nitrogen (26). A single
motif (CX2CX2C plus a distal C in the polypeptide chain) coordinates all low potential,
redox active, 4Fe-4S clusters for which cysteine is the exclusive ligand. Possible
exceptions to this ubiquitous 4Fe-4S motif are found in the corrinoid/iron sulfur proteins
of M. thermophila and Clostridium thermoaceticum, and a putative iron-sulfur protein
from Rhodobacter capsulatus, where the sequence CX2CX4CX16C is perfectly conserved
(14); however, conclusive evidence for involvement of this motif in ligation of 4Fe-4S
clusters has not been reported. Thus, the highly conserved CX2CX2CX4-7C motif in Isf is
the most compact motif known with the potential to coordinate a 4Fe-4S motif. Although
the great majority of iron-sulfur proteins function in electron transfer reactions, the
clusters in a few function in non-redox catalysis or serve a structural role. Still other iron-
sulfur clusters bind nucleic acids or play a regulatory role (3, 10). Two of these,
endonuclease III and MutY, contain a redox inert 4Fe-4S cluster coordinated by a
compact cysteine motif (CX6CX2CX5C) (19, 20). Although the 4Fe-4S cluster of Isf has
reversible redox activity, conclusive evidence for a role in electron transfer has not been
reported.
Much has been learned regarding the coordination of iron-sulfur clusters utilizing
site-specific replacement of residues ligating the clusters. Changes in spectroscopic
properties and other characteristics have provided information regarding the polypeptide
environment of the cluster and the effects that the coordinating ligands have on the
biochemical properties of the cluster. Thus, a series of site-specific replacements in Isf
were performed to obtain biochemical evidence for the novel putative cysteine motif and
further characterize the biochemical and physiological properties of the 4Fe-4S cluster.
The results support involvement of the remarkably compact motif in coordination of the
4Fe-4S cluster that is surprisingly resistant to changes in ligation. The results also support
a role for the 4Fe-4S cluster in the transfer of electrons from the physiological electron
donor (ferredoxin A) to FMN.
Page 105
91
Portion of the results contained in this section were obtained by Dr. J.H. Golbeck
and M. L. Antokine at Pennsylvania State University. Their contributions are noted in
the appropriate sections.
EXPERIMENTAL PROCEDURES
Sequence comparisons. Microbial genomic sequence databases were searched at
http://www.tigr.org. Sequences were aligned using the program Clustal X version 1.64b.
Plasmid construction and site directed mutagenesis. Plasmid pML701, which
contains the entire gene for Isf, was used as a template to construct mutants. Site directed
mutagenesis was performed using MORPH as described by the manufacturer (5 Prime →
3 Prime, Inc. 5603 Arapahoe, Boulder, CO 80303). Each construct was confirmed for
the intended mutation by sequencing using the automated dideoxy method at the Penn
State University nucleic acid facility.
Protein production and purification. Escherichia coli BL21 (DE3) cells
transformed with derivative expression plasmids carrying the designated isf mutations
were grown on LB broth supplemented with 100 µg/ml ampicillin. Once cells reached an
A600 of about 0.8, they were induced to produce high levels of the Isf variants by addition
of 1% (final concentration, w/v) Bacto-lactose for 2 h. The cells were harvested by
centrifugation at 11,800 x g for 10 min at 4oC. The cell pellets were frozen at -70oC.
The C16S variant and wild type was purified as described (13). All other variants
were purified as follows. Approximately 5 g (wet weight) of cells were suspended in 6
volumes (w/v) of buffer A (50 mM Tris-HCl pH 7.6, 200 µg/ml lysozyme, and 2 mM
DTT) and incubated for 20 min at 21oC. Cells were lysed by two passages through a
French pressure cell at 20,000 psi. The lysate was centrifuged at 10,000 x g for 30 min at
4oC. The pellet, containing inclusion bodies, was washed twice in 30 ml buffer B (50 mM
Tris-HCl, pH 7.6, 2 M urea, 1% Triton X-100, 2 mM DTT). The protein aggregates were
solubilized in 2 ml buffer C (50 mM Tris-HCl, pH 7.6, 6 M guanidine-HCl), and
incubated for 2 h at 21oC. Insoluble protein was removed by centrifugation at 10,000 x g
Page 106
92
for 10 min at 4oC. The protein solution at this stage is termed "denatured". The soluble
fraction was then diluted 100-fold in buffer D (50 mM Tris-HCl, pH 7.6, 500 mM L-
arginine, 2 mM DTT), and incubated at 4oC for 12 h. In the following step, the sample
was concentrated using PEG 8000. The protein was dialyzed in buffer E (50 mM Tris-
HCl, pH 7.6, 250 mM L-arginine, 200 mM NaCl, 2 mM DTT) and then F (50 mM Tris-
HCl, pH 7.6, 200 mM NaCl, 2 mM DTT). The protein at this stage is defined as
"renatured". There was no apparent change in subunit size among wild type and the
variants as judged by migration in SDS-PAGE. The overall procedure resulted in
homogenous proteins as judged by SDS-PAGE.
Ethylenediaminetetraacetic acid (sodium salt) (EDTA)-treated wild type protein
was prepared by incubated 6 mg of as-purified wild type in buffer G (50 mM Tris-Cl
pH7.6, 400 mM NaCl, 2 mM EDTA and 2 mM DTT) for at 21o C. Then the EDTA-
treated protein was treated in buffer D, E, and F as stated above.
Reconstitution of iron-sulfur clusters and FMN into renatured apoprotein.
Reconstitution of iron-sulfur clusters and FMN was performed by adding 1 ml of 10 mM
FMN, 800 µl of 2-mercaptoethanol, 300 µl of 60 mM FeCl3, and 300 µl of 60 mM Na2S
to 100 ml renatured apoprotein solution (12 mg). All reagents were added drop-wise with
10 min intervals between steps, and the reconstitution reaction was incubated at 4oC for
12 h. The protein was concentrated with an ultrafiltration unit fitted with a YM 30
membrane (Amicon, Beverly, Mass.) and the unbound molecules were removed by a
PD10 gel filtration. The protein at this step is called "reconstituted". The recovery yield
after the overall processes (denatured, renatured, and reconstituted processes) was varied,
and in the range of 20-50 %.
Spectroscopy. UV-visible spectra were obtained with a Hewlett-Packard 8452A
diode array spectrophotometer. EPR signals of iron sulfur clusters were recorded with a
Bruker ECS 106 Electron Paramagnetic Resonance (EPR) X-band spectrometer operating
with an ER/4012 ST resonator and an Oxford liquid helium cryostat. The temperature
Page 107
93
was controlled using an ITY4 Oxford temperature controller. The microwave frequency
was determined with a Hewlett-Packard 5340A frequency counter.
Reduction of Isf by ferredoxin A. Experiments were carried out in a stoppered
1.0 ml-cuvette equilibrated with an atmosphere of CO. Continuous reduction of
ferredoxin A was accomplished by including catalytic amounts of CODH/acetylCoA
synthetase. All protein components except the Isf variants were anaerobically purified as
previously described (13, 23, and 24). The assay mixture contained 27 µg
CODH/acetylCoA synthetase and 9 µg ferredoxin A in anaerobic 50 mM Tris-HCl (pH
7.6) containing 500 mM sucrose, 0.1 mg/l resazurine, and 2 mM DTT. After 10 min
incubation, 180 µg of the indicated Isf variant was added to the assay mixtures to initiate
the reaction. The absorbance at 476 nm was measured to follow the reduction of FMN
without interference from reduction of the iron-sulfur cluster. Ferredoxin A and
CODH/acetyl CoA synthetase were present in catalytic amounts such that any absorption
change in these proteins did not interfere with the assay.
RESULTS
Sequence comparisons of Isf from M. thermophila with putative Isf proteins.
Figure 1 shows that metabolically diverse species contain sequences with identity to M.
thermophila Isf suggesting the possibility that this electron carrier functions in carbon
dioxide-reducing (Methanococcus jannaschii and Methanobacterium
thermoautotrophicum) and sulfate-reducing (Archaeoglobus fulgidus) Archaea, and also
in metabolically diverse procaryotes from the Bacteria domain (Chlorobium vibrioforme,
Clorobium tepidum, and Clostridium difficile). Only Isf from M. thermophila has been
characterized; thus, the sequences shown in Figure 1 are putative Isf homologs.
Nonetheless, comparison of these sequences with Isf from M. thermophila shows an
unusually compact N-terminal cysteine motif with a strictly conserved spacing of
CX2CX2CX4-7C atypical of cysteine motifs required for 4Fe-4S coordination. The strict
conservation provides a strong indication for involvement in ligation of the 4Fe-4S
cluster; however, it is possible that any one of these conserved cysteines is essential for
Page 108
94
16 47
MST ---------M KITGISGSPR KGQNCEKIIG AALEVAKERG FETDTVFISN EEVAP--CKA 49MCJ-2 ---------M KVIGISGSPR PEGNTTLLVR EALNAIAEEG IETEFISLAD KELNP--CIG 49MBT-1 MKQKEVDFMV KVIGICGSPR KNGNTEILLR EALDAAEEAG AETELVRLAG LDINP--CRA 58MBT-2 ---------- MILGICGSPR K-QATEHVLE RALSMLEDDG LETEFFTVRG KNISP--CRH 47AF-2 ---------- MIVGISGSPR R-KATEFVLG EALKMLEERG FETKFFTVRG KKISP--CQH 47MCJ-1 ---------M KVFGISGSPR L-QGTHFAVN YALNYLKEKG AEVRYFSVSR KKINF--CLH 48CV ---------M KVIGINGSPR PAGNTSIMLK TVFETLEQEG IETELIQVGG TDIKG--CRA 49CT ---------M KVIGINGSPR RAGNTSIMLK TIFEVLEDEG IETELIQVGG TNIKG--CRA 49AF-3 ---------M KLLAINGSPN K-RNTLFLLE VIAEEVKKLG HEAEIIHLKD YEIKE--CKG 48MBT-3 ---------- ---------- ------MVLE HCRDAIESHG VETDIISLRG MKIES--CRA 32AF-1 ---------M KAVGILGSPR KYGNASKMLD AALKELENSG FEVEKVHISS KKINY--CTG 49CD ---------M IITVMNGSPR KNGATSKVLT YLYKDIERLI PDVKINYFDL SEVNPSYCIG 51
50 53 59MST CGACRDQDF- -CVID-DDMD EIYEKMRAAD GIIVAAPVYM GNYPAQLKAL FDRSVLLRR- 105MCJ-2 CNMCKEEGK- -CPII-DDVD EILKKMKEAD GIILGSPVYF GGVSAQLKML MDRSRPLR-- 104MBT-1 CDSCKKTGE- -CAIE-DDLN RVVELAASAH GIIIGSPVYF GSVTAQTKMF MDRTRPLR-- 113MBT-2 CDYCLRNKE- -CVLK-DDMF PLYELLRRAA GIIIATPVYN GGVSAQIKAI MDRCRALGAE 104AF-2 CDYCLKHKE- -CRIK-DDMF ELYEMLKDAK GIVMATPVYN GGVSAQIKAV MDRCRALVAA 104MCJ-1 CDYCIKKKEG -CIHK-DDME EVYENLIWAD GVIIGTPVYQ GNVTGQLKTL MDRCRAILAK 106CV CYACIRNKNS KCSTK-DGFN EIFEKMVEAN GMILGSPVYF ADITPELKAL IDRSGFVSRT 108CT CYACIKNKNS ECSTKGDGFN EIFAKMVEAD GMILGSPTYF ADITPELKAL IDRAGFVSRT 109AF-3 CDACLKGD-- -CSQK-DDIY KVLEKMQEAD AIVIGTPTYF GNVTGIVKNL IDRSRMAR-M 103MBT-3 CLSCAKKHR- -CRID-DGLN DIIDRIRDSE GFIVATPVYF GTARGDLMAA LQRIGMVSRA 89AF-1 CGTCLAKGE- -CVQR-DDMD ELKRLVEESD AVILASPVYY LNVTAQMKTF IDRMLPYG-- 104CD CLNCYKMGK- -CINQNDKVE YIHDIITKSD GVIFGSPTYG SSVTGLFKVF TDRAHMML-- 107
MST KNFALKNKVG AALSVGGSRN GGQEKTIQSI HDWMHIHGMI VVGDNS---- HFGGI---TW 158MCJ-2 IGFQLRNKVG GAVAVGASRN GGQETTIQQI HNFFLIHSMI VVGDND-PTA HYGGT---GV 160MBT-1 SEFRLANRVG GAVTVGGSRN GGQETACRDI HSFFLIHEAA VVGNAS-PTA HYGGT---GV 169MBT-2 DYDSLRGKVG MGIAVGGDRC GGQEPALMQI HTFYILNGVI PVSGGS-FGA NLGAC---FW 160AF-2 DYDFFRGKVG MAIAVGGDRI GGQELAIQQI LTFYILNGVI PVSGGS-FGA NIGAT---FW 160MCJ-2 NPKVLRGRVG MAIAVGGDRN GGQEIALRTI HDFFIINEMI PVGGGS-FGA NLGAT---FW 162CV NGQLFRHKVG ASIVS--LRR GGGVHAYDSI NHLFQICQMF MVGSTY---W NLG-----FG 158CT NGQLFRHKVG ASVVS--LRR GGGIHAYDSI NHLFQICQMF MVGSTY---W NLG-----FG 159AF-3 GNYRLRNRVF APVVTSGLRN GGAEYAAMSL IVYALGQAML PVSIVE-NPI TTGTFPVGVI 162MBT-3 SDGFLSWKVG GPIAV--ARR GGHTATIQEL LMFYFINDMI VPGSTY-WNM VFG------- 139AF-1 HRPTLKGKYG GSIVVY-AGV GKPEEVAGYM NRVLKAWGIV PVGYAVGFGV IPGEVGDEDL 163CD ERLLYRKPCI AVTTY--ENA RGS-KAISFI KSMVLDSGGY VCGSLS---I KTG------F 155
180MST NPAE------ EDTVGMQTVS E-TAK--KLC D-----VLEL IQKNR----- -------DK- - 191MCJ-2 GKAP------ GDCKNDDIGL E-TAR--NLG K-----KVAE VVKLI----- -------KK- - 193MBT-1 GGAK------ GESADDMTGI E-TAR--NLG R-----RVAL LAARI----- -------HG- -- 202MBT-2 SRDT-L---- EVLKRTHMDS KPSKRPWACL KGSWTLKDPE ILFYS----- -------EFI -- 203AF-2 SRDT-L---- EGVKEDEEGF R-SLR--KTV K-----RFAE MLEKM----- -------EGV - 195MCJ-1 SKDRGK---- KGVEEDEEGL R-VLR--KTL N-----RFYE VLKEK----- -------RGL -- 198CV GRDG------ GEVVNDTEGM D-NMR--DLG K-----SMAF LLKKL----- -------NAS -- 192CT GRDG------ GEVVNDTEGM E-NMR--DLG H-----SMAF LLK------- ---------- -- 188AF-3 QGDAGW---- RSVKKDEIAI N-SAK--ALA KR--IVEVAE ATKNL----- -------RES -- 201MBT-3 -WAP------ GEVEDDSEGI E-TIR--RFG E-----NVAE LIKRI----- -------NGG S- 173AF-1 KKASQLGSKI AEAFESKYRM EPSDEDLELQ K-----QLLT LIKNYGHLMK ADYEFWKEKG FI 220CD NQNP------ ---------- ---------- ---------- ---------- ---------- -- 159
Figure 1. Multiple amino acid sequence alignment of Isf from M.thermophila (MST)
with sequences deduced from open reading frames identified in the genomic sequences of
M. jannaschii (MCJ), M. thermoautotrophicum (MBT), Archaeoglobus fulgidus (AF),
Page 109
95
Chlorobium vibrioforme (CV), Chlorobium tepidum (CT), and Clostridium difficile (CD).
The numbers after the abbreviated name of organisms indicate the different protein
isoforms. Database codes for each protein are as follows, MST: Genbank U50189;
MCJ-1, -2 : Genbank C64391 (MJ0731), B64435 (MJ1083); MBT-1,-2,-3: Genbank
AE000802 (MTH135), AE000908 (MTH1473), AE000919 (MTH1595); AF1-,-2,-3:
AE0010041 (AF1438), AE0009971 (AF1519), AE0009721 (AF1896); CV: EMBL
Z83933.1; CT: C tepidum gct10; CD: CD shotgun.dbs cd2h6.q1t. Cysteines (C16, 47,
50, 53, 59, and 180) in Isf of M.thermophila are numbered at the top line. Residues
conserved in at least 7 out of 10 sequences are shaded in gray. Putative FMN binding
regions in Isf are underlined.
Page 110
96
another function and other non-cysteinyl residues may ligate the 4Fe-4S cluster. Thus, we
undertook a biochemical approach to obtain experimental evidence for the proposed role
of the motif and investigate properties of the cluster dependent on the protein
environment.
Heterologous production, purification and reconstitution of wild type Isf and
variants. In the course of the investigation of cysteine ligation in the 4Fe-4S cluster, six
cysteines present in Isf (Fig. 1) were individually altered to either alanine or serine.
Except for C16S, the variants were contained in inclusion bodies. The soluble C16S was
purified the same as for wild type. The inclusion bodies were isolated by centrifugation
as a first step in purification of the remaining variants. The isolated inclusion bodies were
extracted in guanidine-hydrochloride to solubilize the proteins. At this juncture, no
discrete bands were detected by native PAGE (data not shown) suggesting the proteins
were denatured. The solubilized variants were diluted in buffer containing arginine to
prevent protein aggregation during renaturation (1, 5, and 25). After removal of the
arginine by dialysis, native PAGE indicated no discrete bands (data not shown)
suggesting the proteins had not achieved the native state. UV-visible spectra indicated the
proteins contained very low amounts of iron-sulfur clusters and FMN (data not shown);
thus, the apoproteins were incubated in the presence of ferric iron, sulfide, and FMN to
reconstitute the redox clusters. UV-visible spectroscopy (Fig. 4, 5) indicated
incorporation of flavin and iron-sulfur clusters. Native PAGE (Fig. 2) indicated a discrete
band for each variant migrating to approximately the same position as the purified wild
type, which suggested all are dimeric in accord with the initial characterization of the
wild type (13). These results suggested that either an iron-sulfur cluster or flavin, or both,
must be present to adopt a native conformation. There was no apparent change in subunit
size among wild type and the Isf variants as judged by migration in SDS-PAGE (data not
shown). A similar denaturation/renaturation/reconstitution process was performed for
wild type. As for the variants, only the reconstituted wild type exhibited a discrete band
after native PAGE (Fig. 2). There was intermittent success and low yields in the
reconstitution of both C59X and C180X (X = A or S) indicating they were unstable.
Page 111
97
Figure 2. Coomassie blue stained native PAGE of wild type Isf and variants. Top
panel contains as-purified wild type Isf (25 µg), reconstituted Isf (25 µg), and cysteine
to alanine variants (25 µg; except 18 µg for 180A). Bottom panel contains as-purified
wild type Isf (18 µg), reconstituted cysteine to serine variants (25 µg for C16S and
C50S, 18 µg for C47S, C53S, and C180S).
Page 112
98
Since the proteins showed less precipitation during refolding in the presence of 0.5 M
arginine, cofactors reconstitution in C59X and C180X in the presence of arginine were
performed. In some instances with no consistency, this method yields higher recovery of
reconstituted proteins, which is indicated in higher intensity of EPR signal (Fig. 8).
Spectroscopic characterization of reconstituted wild type Isf and variants.
The UV-visible spectra of denatured and renatured wild type Isf showed no absorbance
characteristic of either iron-sulfur clusters or FMN (Fig 3) suggesting the complete loss
of both redox components. The UV-visible spectrum of the reconstituted Isf was nearly
identical to the as-purified wild type suggesting the reconstituted protein might have
properties similar to as-purified Isf. The UV-visible absorption spectra for the
reconstituted variants were similar to the wild type (Fig. 4, 5); however, the ratio of FMN
absorbance at 378 nm relative to iron-sulfur cluster absorbance centered at 430 nm was
generally lower for all except C16A and C16S. These results suggest significantly lower
incorporation of FMN relative to iron-sulfur clusters for all variants except C16A and
C16S. The UV-visible spectrum of C59S contained no features characteristic of iron-
sulfur cluster incorporation, a result that was confirmed by EPR (data not shown).
EPR spectrum of wild type Isf, alanine and serine variants were recorded and
compared to determine cysteines that participate in ligating iron-sulfur cluster in Isf.
The reduced as-purified Isf exhibited an EPR spectrum with g-values of 2.06, 2.03, 1.92,
1.86, 1.81 (Fig. 6, Table 1), results that are nearly identical to a previous report (2) in
which the authors attributed the complexity of the spectrum to heterogeneity of the
sample (refer to chapter 3). We were able to distinguish two distinct species based on
power and temperature dependencies, one with g values of 2.06, 1.92, and 1.81 and
another with g values of 2.03, 1.92, and 1.86 (Fig. 6). The ratio of these species varied in
different Isf preparations suggesting the as-purified protein exists in two distinct
conformational states. Incubation of as purified Isf with ethylenediaminetetraacetic acid
sodium salt (EDTA) resulted in nearly complete destruction of 4Fe-4S center as followed
by EPR (Fig. 6). However, the overwhelming majority of the center reconstituted to the
g = 2.03, 1.92, and 1.81 species. This implies that under experimental conditions, this Isf
Page 113
99
Figure 3. UV-visible absorption spectra of as-purified, denatured, and reconstituted wild
type Isf. The denatured protein was in 50 mM Tris-Cl (pH 7.6) containing 6 M
guanidine-HCl. The as purified, renatured, and reconstituted proteins were in 50 mM
Tris-Cl (pH 7.6) containing 200 mM NaCl. The spectra were recorded at 21o C. The
amount of protein used for as-purified Isf was 80 µg, all other were 150 µg.
280 330 380 430 480 530
Wavelength (nm)
as-purified
reconstituted
0.1 A
renatured
denatured
Page 114
100
Figure 4. UV-visible absorption spectra of wild type Isf and alanine variants. The
samples (500 µl) were in 50 mM Tris-Cl (pH 7.6) containing 200 mM NaCl, and the
spectra were recorded at 21oC. The amount of protein used for wild type Isf was 80 µg;
all variants were 120 µg.
280 330 380 430 480 530
Wavelength (nm)
Isf
C16A
C47A
C50A
C53A
0.2 A
C59A
C180A
Page 115
101
Figure 5. UV-visible absorption spectra of wild type Isf and serine variants. The
samples (500 µl) were in 50 mM Tris-Cl (pH 7.6) containing 200 mM NaCl, and the
spectra were recorded at 21oC. The amount of protein used for wild type Isf was 80 µg;
all variants were 120 µg.
280 330 380 430 480 530
Wavelength (nm)
0.2 A
Isf
C16S
C47S
C50S
C53S
Page 116
102
Figure 6. EPR spectra of reduced wild type and reconstituted Isf. EPR conditions were
as follows: temperature 15 K, microwave power 20 mW, modulation amplitude 1 mT.
Proteins were reduced by sodium dithionite.
Page 117
103
conformation is energetically more favorable and it could be achieved more easily.
Reconstitution of wild type Isf that had been denatured and renatured also exhibited a
4Fe-4S EPR spectrum identical to wild type with evidence for both species. This result
indicated that the procedure for reconstituting the Isf apoprotein yielded a 4Fe-4S cluster
with an environment identical to the wild type.
The reduced C16A and C16S variants exhibited EPR spectra with g-values of
2.06, 2.04, 1.92, 1.86, 1.82 (Fig. 7, Table 1) characteristic of wild type Isf with two
distinct 4Fe-4S species. The reduced C180A and C180S variants showed EPR spectra
(Fig. 8, 9, Table 1) with line shapes and g-values (2.07, 2.04, 1.93, 1.86, 1.81) nearly
identical to one of the 4Fe-4S species present in the wild-type Isf spectrum. A minor
contribution of the other species was also observed as a shoulder at g 2.07. These results
strongly indicate that C16 and C180 do not participate in ligation of the 4Fe-4S cluster of
Isf. This conclusion is further supported by sequence comparisons showing that C16 and
C180 are not conserved with putative Isf proteins (Fig. 1). The predominance of one
4Fe-4S species in the EPR spectra of the C180 variants suggests one of the conformations
possible for wild type Isf is preferred in these variants. This proposal is consistent with
the instability of the C180A and C180S variants which suggests that C180 is important
for the overall structural integrity of the protein.
Cysteines 47, 50, 53 and 59 are strictly conserved among putative Isf sequences
(Fig. 1) as a compact motif CX2CX2CX4-7C suggesting the motif is essential and,
therefore, a candidate for ligation of the 4Fe-4S cluster. EPR spectroscopy of the reduced
C50A or C59A variants detected no [4Fe-4S]1+ cluster; however, spectra of the oxidized
variants had linewidths and g-values typical for [3Fe-4S]2+ clusters (Fig. 10, 11, Table 1).
These results strongly indicate that C50 and C59 are involved in ligation of the 4Fe-4S
cluster in Isf. The results also show that other ligands cannot substitute for C50 and C59
to preserve the 4Fe-4S cluster in these variants. A [4Fe-4S]1+ cluster was detected in
both of the reduced C47A and C53A variants by EPR spectroscopy; however, there were
significant differences in the line widths and g-values between the spectra of the two
Page 118
104
2.3 2.2 2.1 2.0 1.9 1.8g -value
C16A/S
C16S C16A
g =
Figure 7. EPR spectra of reduced C16X (X = A or S). EPR conditions were as follows:
temperature 15 K, microwave power 20 mW, modulation amplitude 1 mT. Proteins were
reduced by sodium dithionite.
Page 119
105
2.3 2.2 2.1 2.0 1.9 1.8g -value
C180A
+Arg -Arg
g =
Figure 8. EPR spectra of reduced C180A. (+ Arg) The protein was denatured, refolded
as described in experimental procedures. However, the reconstitution process was
performed immediately after refolding with no dialysis to remove arginine. (–Arg) The
protein was denatured, refolded, dialyzed to remove arginine, and then reconstituted.
EPR conditions were as follows: temperature 15 K, microwave power 20 mW,
modulation amplitude 1 mT. Proteins were reduced by sodium dithionite.
Page 120
106
2.3 2.2 2.1 2.0 1.9 1.8g -value
C180S
g =
Figure 9. EPR spectrum of reduced C180S. EPR conditions were as follows:
temperature 15 K, microwave power 20 mW, modulation amplitude 1 mT. The protein
was reduced by sodium dithionite.
Page 121
107
2.05 2.00 1.95 1.90 1.85
g -value
C50A
g =
Figure 10. EPR spectrum of as-purified C50A. EPR conditions were as follows:
temperature 15 K, microwave power 20 mW, modulation amplitude 1 mT.
Page 122
108
2.2 2.1 2.0 1.9
g -value
C59A
g =
Figure 11. EPR spectra of as-purified C59A. EPR conditions were as follows:
temperature 15 K, microwave power 1.26 mW, modulation amplitude 1 mT.
Page 123
109
variants. The line widths and g-values for both variants were also significantly different
from the spectra of either of the two species present in the reduced form of as-isolated
wild-type Isf (Fig. 12, 13, Table 1). A low-intensity [3Fe-4S]2+ EPR signal was detected
in the oxidized C47A and C53A variants. These data strongly indicate that one or more
ligands to the 4Fe-4S cluster changed in these variants, a result suggesting that C47 and
C53 are ligands to the 4Fe-4S cluster in wild type Isf. The EPR results obtained for the
C47A, C50A, C53A, and C59A variants, combined with strict conservation of the
CX2CX2CX4-7C motif in putative Isf sequences from diverse species (Fig. 1), strongly
suggest involvement of the motif in ligation of the 4Fe-4S cluster in Isf.
Although other residues could replace cysteine as a ligand to the 4Fe-4S clusters
in C47A and C53A, we consider it most likely that 2-mercaptoethanol is an external
thiolate ligand in these variants for the following reasons. The buffer used for
reconstitution of the variants contained 2-mercaptoethanol which has been shown to
serve as an external ligand to the 4Fe-4S cluster in the C51D and C14G variants of PsaC
from Photosystem I in Synechocystis sp. PCC 6803 (11, 28). The reconstitution
conditions used in this work were nearly identical to the conditions used to reconstitute
iron-sulfur clusters in PsaC. Thiolate ligands in C53A and C47A are expected to occupy
different positions in the coordination sphere of the 4Fe-4S cluster consistent with
differences in the EPR spectra recorded for these variants. Thiolate ligation is also
consistent with differences in the EPR signals of the C53A and C47A variants compared
to wild-type Isf.
The EPR spectra of the reduced C47S, C50S, and C53S variants indicated the
presence of 4Fe-4S clusters (Fig. 14-16, Table 1). The EPR spectra of C47S, C50S and
C53S suggest the presence of two different ligand species. These features could
potentially derive from the use of substituted serine and 2-mercaptoethanol presence in
the reconstitution system. The role of introduced serine residues in ligation of iron-sulfur
clusters has been determined for several proteins by substitution with alanine in which
case the native cluster does not assemble if the replaced cysteine or serine residues are
Page 124
110
2.3 2.2 2.1 2.0 1.9 1.8g -value
C47A
g =
Figure 12. EPR spectrum of reduced C47A. EPR conditions were as follows:
temperature 15 K, microwave power 20 mW, modulation amplitude 1 mT. The protein
was reduced by sodium dithionite.
Page 125
111
2.3 2.2 2.1 2.0 1.9 1.8g -value
C53A
g =
Figure 13. EPR spectrum of reduced C53A. EPR conditions were as follows:
temperature 15 K, microwave power 20 mW, modulation amplitude 1 mT. The protein
was reduced by sodium dithionite.
Page 126
112
2.3 2.2 2.1 2.0 1.9 1.8g -value
C47S
g =
Figure 14. EPR spectrum of reduced C47S. EPR conditions were as follows:
temperature 15 K, microwave power 20 mW, modulation amplitude 1 mT. The protein
was reduced by sodium dithionite.
Page 127
113
2.3 2.2 2.1 2.0 1.9 1.8g -value
C50S
g =
Figure 15. EPR spectrum of reduced C50S. EPR conditions were as follows:
temperature 15 K, microwave power 20 mW, modulation amplitude 1 mT. The protein
was reduced by sodium dithionite.
Page 128
114
2.3 2.2 2.1 2.0 1.9 1.8g -value
C53S
g =
Figure 16. EPR spectrum of reduced C53S. EPR conditions were as follows:
temperature 15 K, microwave power 20 mW, modulation amplitude 1 mT. Protein was
reduced by sodium dithionite.
Page 129
115
Table 1. EPR properties of wild-type Isf and variants.
Protein g values Iron-sulfur center type
As-purified wild-type 2.06, 1.92, 1.81
2.03, 1.92, 1.86
[4Fe-4S]
Reconstituted wild-type 2.06, 1.92, 1.81
2.03, 1.92, 1.86
[4Fe-4S]
C16X
X = A or S
2.06, 1.92, 1.82
2.04, 1.92, 1.86
[4Fe-4S]
C47A
C47S
2.05, 1.93, 1.89
2.05, 1.94, 1.86, 1.80
[4Fe-4S]
[4Fe-4S]
C50A
C50S
2.01,1.99
2. 07, 2.04,1.92, 1.90,1.82
[3Fe-4S]
[4Fe-4S]
C53A
C53S
2.03,1.91,1.89
2.05, 1.99, 1.93, 1.91, 1.82
[4Fe-4S]
[4Fe-4S]
C59A 2.01,1.99 [3Fe-4S]
C180X
X = A or S
2.04, 1.93, 1.86
With minor contribution from
2.07, 1.93, 1.81
[4Fe-4S]
Page 130
116
required ligands (16, 17). Thus, conversion of the 3Fe-4S cluster of C50A to a 4Fe-4S
cluster by replacement with serine suggests that serine can substitute for C50 in Isf.
Functional characterization of Isf variants. It has been proposed that electron
flow is from reduced ferredoxin A to the 4Fe-4S cluster of Isf and then to FMN based
only on midpoint potential values (2); thus, the ability of ferredoxin A to reduce FMN in
the stable variants was investigated to test this hypothesis (Table 2). The reduction of
FMN was followed at A476 to avoid interference due to reduction of the iron-sulfur
clusters. The as- purified and reconstituted Isf were reduced at similar rates (Table 2)
consistent with the spectroscopic characterizations indicating that the reincorporated 4Fe-
4S and FMN were functionally similar to as-purified wild-type Isf. The C47, C50, and
C53 variants showed either several-fold lower or higher rates compared to wild type
(table 2), a result which suggests that the 4Fe-4S cluster is required to transfer electrons
from ferredoxin A to FMN. This result is consistent with results suggesting that the
reconstitution of FMN into apo-protein is diminished by substitution of residues in the
motif (C47, C50, C53, and C59) coordinating the 4Fe-4S cluster. Local conformational
changes in the environment of the 4Fe-4S cluster could potentially influence the
reconstitution of FMN if it were adjacent to the 4Fe-4S cluster for electron transfer.
The FMN reduction in C47A and C50S was decreased relative to wild type
demonstrating that the 4Fe-4S clusters in these variants are able to function, albeit less
effectively, with the ligands that replaced C47 and C50. The several-fold higher rates
relative to wild type Isf for variants C53A and C47S is unexplained; however, the results
clearly indicate that the 4Fe-4S clusters in these variants are fully functional suggesting
that the ligands replacing C53 and C47 conserve essential properties of the wild type
4Fe-4S cluster. Midpoint potentials of 3Fe-4S clusters are generally less negative than
4Fe-4S clusters, the Em range for 3Fe-4S is +80 to – 420 mV while the 4Fe-4S center is
+80 to – 700 mV. Thus, it is hypothesized that the lower rate of FMN reduction
exhibited by C50A could possibly be influenced by the redox potential of the 3Fe-4S
cluster in this variant similar to that predicted for the 4Fe-4S cluster in the corrinoid/iron-
Page 131
117
Table 2. Rates for reduction of FMN in wild type Isf and variants.
Protein cluster type Rate a
As-purified wild type 4Fe-4S 0.52 ± 0.03
Reconstituted wild type 4Fe-4S 0.59 ± 0.08
C16A 4Fe-4S 0.27 ± 0.01
C47A 4Fe-4S 0.14 ± < 0.01
C50A 3Fe-4S 0.14 ± < 0.01
C53A 4Fe-4S 1.53 ± 0.01
C16S 4Fe-4S 0.49 ± < 0.01
C47S 4Fe-4S 1.38 ± 0.26
C50S 4Fe-4S 0.21 ± < 0.01
C53S 4Fe-4S 0.50 ± 0.09
a:Change in absorbance at 476 nm/min/µmole FMN with ferredoxin A as the electron
donor (see Materials and Methods).
Page 132
118
sulfur protein from C. thermoaceticum (15). Although the results suggest C16 is not
involved in ligation of the 4Fe-4S cluster, reduction of FMN in C16A was impaired
suggesting this residue indirectly influences the transfer of electrons from ferredoxin to
FMN.
DISCUSSION
The data presented here provides biochemical confirmation of a novel and
unusually compact motif (CX2CX2CX4-7C) for ligation of the 4Fe-4S cluster. Sequence
comparisons (Fig. 1) suggest that the spacing between the first and the second cysteines
in this motif is rigid while spacing between the third and the fourth cysteines is somewhat
variable; however, additional site-directed mutagenesis experiments are necessary to test
this hypothesis. This motif differs from those in ferredoxins (typically CX2CX2C and a
distal C) and high potential iron proteins (typically CX2CX16CX13C) where the ligands to
the 4Fe-4S clusters are more dispersed in the primary structure. Other examples of highly
compact motifs ligating a 4Fe-4S cluster are endonuclease III and MutY (CX6CX2CX5C);
however, these clusters are resistant to oxidation and reduction (19, 20) and function
instead to position basic residues for interaction with the phosphate backbone of DNA.
Another compact cysteine motif (CX2CX11CX5C) ligating a redox resistant 4Fe-4S
cluster is present in the LRR protein for which the physiological function is unknown
(18). Still other novel regulatory and enzymatic roles have been described for iron-sulfur
clusters (3, 10). The results reported here are consistent with a role for the 4Fe-4S cluster
in transfer of electrons from the physiological electron donor (ferredoxin A) to FMN;
however, alternate or additional roles cannot be ruled out.
This investigation has also provided insight into iron-sulfur cluster and FMN self-
assembly in Isf in vivo. The present work showed that Isf variants could be refolded in
the presence of arginine to a conformational state such that the variant could be
reconstituted by treating the protein with ferric ion, sulfide, and FMN. It is proposed that
arginine helps to reshuffle molecules trapped in non-productive reactions, which results
in increased refolding efficiency (4). Unfortunately, we were unable to identify
Page 133
119
conditions to produce an appreciable yield of the reconstituted C59X and C180X (X = A
or S) variants. Refolding in the presence of ferric ion, sulfide, and FMN was performed
for these variants; however, the majority of the protein precipitated during concentration
and buffer exchange. Nevertheless, we were successful in characterizing these variant
spectroscopically (see Results). Replacement of cysteines in the proposed 4Fe-4S binding
motif (C47, C50, C53, and C59) suggests that integrity of the 4Fe-4S cluster is important
for reconstitution of FMN. Changes in these residues resulted in poor incorporation of
FMN relative to the iron-sulfur cluster.
The results presented here show that the biochemical and physiological properties
of the iron-sulfur cluster are remarkably stable to changes in ligation at the same time it is
very sensitive to the ligand environment. All of the serine variants contained a 4Fe-4S
cluster, except C59S and all of the alanine variants contained an iron-sulfur cluster, two
of which were of the 3Fe-4S type. Furthermore, the FMN of all the variants was reduced
by ferredoxin A at a significant rate compared to wild type, a result demonstrating that
the iron-sulfur clusters were competent in transferring electrons from ferredoxin A to
FMN. This resiliency of the 4Fe-4S cluster to changes in the ligation environment may be
a consequence of the unusually compact nature of the motif coordinating the cluster.
Examples of 4Fe-4S clusters bridging between protein subunits have previously
been shown for the nitrogenase Fe-protein and the Fx cluster in photosystem I. The
possibility that the cysteines of the motif which ligate the 4Fe-4S centers are shared
between subunits can not be completely ruled out based on available experimental data.
However, we see no evidence of spin coupling between two clusters, although that effect
is distance dependent.
The N-terminal half of the deduced sequence of Isf contains regions
(Fig. 1, underlined residues) with identity to the flavin-binding domain of flavodoxins
(13). The unusually compact nature of the cysteine motif coordinating the 4Fe-4S cluster
obviates the need for a remote cysteine suggesting the possibility that Isf could have
evolved by insertion of a small ancestral 4Fe-4S protein, containing the compact motif,
Page 134
120
into the N-terminal half of an ancestral flavodoxin. A search of the databases revealed no
additional sequences with significant identity to residues 23-85 in the M. thermophila Isf
sequence (Fig. 1) that includes the cysteine motif.
In addition to the motif ligating the 4Fe-4S cluster, two other cysteines (C16 and
C180) are present in the Isf sequence (Fig. 1). Although the evidence suggests C16 and
C180 are not involved in coordination of the 4Fe-4S cluster, the C16A variant was
impaired in the ability to catalyze ferredoxin A-dependent reduction of FMN. A direct
role for C16 in electron transfer was ruled out by the observation that C16A is partially,
and C16S fully, competent in electron transfer from ferredoxin A to FMN compared with
wild type Isf. Sequence comparisons (Fig. 1) indicate that a threonine residue in Isf
homologs may replace C16 of the M. thermophila Isf. This finding suggests that a
hydroxyl or sulfhydryl group is important for maintaining a conformation required for
interaction of ferredoxin A with the 4Fe-4S cluster of Isf or intramolecular electron
transfer from the cluster to FMN. The results obtained for C180 suggest this residue is
not involved in iron-sulfur cluster ligation, however, instability of the C180X (X = A or
S) variants suggests that this cysteine is required only for the overall structural integrity
of the protein.
ACKNOWLEDGEMENTS
We would like to thank M.L. Antokine and Dr. J.H. Golbeck at Pennsylvania
State University for performing the EPR spectroscopy. We thank Dr. R.C. Thauer for
suggestion on the arginine refolding method and a special thank you to R.D. Miles for her
critical reading of the manuscript. Work described here was partially supported by the
Department of Energy Basic Energy Sciences Grant No. DE-FG02-95ER20198 (JGF)
and MCB-9723661 (JHG). UL was supported by MOSTE grant from Thailand.
Page 135
121
REFERENCES
1. Ahn, J. H., Y. P. Lee, and J. S. Rhee. 1997. Investigation of refolding condition
for Pseudomonas fluorescens lipase by response surface methodology. J.
Biotechnol. 54:151-60.
2. Becker, D. F., U. Leartsakulpanich, K. K. Surerus, J. G. Ferry, and S. W.
Ragsdale. 1998. Electrochemical and spectroscopic properties of the iron-sulfur
flavoprotein from Methanosarcina thermophila. J. Biol. Chem. 273:26462-9.
3. Beinert, H., R. H. Holm, and E. Munck. 1997. Iron-sulfur clusters: nature's
modular, multipurpose structures. Science. 277(5326):653-9.
4. Buchner, J., I. Pastan, and U. Brinkmann. 1992. A method for increasing the
yield of properly folded recombinant fusion proteins: single-chain immunotoxins
from renaturation of bacterial inclusion bodies. Anal. Biochem. 205:263-70.
5. Buchner, J., and R. Rudolph. 1991. Renaturation, purification and
characterization of recombinant Fab- fragments produced in Escherichia coli.
Biotechnology (N Y). 9:157-62.
6. Ferry, J. G. 1997. Enzymology of the fermentation of acetate to methane by
Methanosarcina thermophila. Biofactors. 6:25-35.
7. Ferry, J. G. 1992. Methane from acetate. J Bacteriol. 174:5489-95.
8. Golinelli, M. P., L. A. Akin, B. R. Crouse, M. K. Johnson, and J. Meyer.
1996. Cysteine ligand swapping on a deletable loop of the [2Fe-2S] ferredoxin
from Clostridium pasteurianum. Biochemistry. 35:8995-9002.
9. Golinelli, M. P., C. Chatelet, E. C. Duin, M. K. Johnson, and J. Meyer. 1998.
Extensive ligand rearrangements around the [2Fe-2S] cluster of Clostridium
pasteurianum ferredoxin. Biochemistry. 37:10429-37.
10. Johnson, M. K. 1998. Iron-sulfur proteins: new roles for old clusters. Curr. Opin.
Chem. Biol. 2:173-81.
11. Jung, Y. S., I. R. Vassiliev, F. Qiao, F. Yang, D. A. Bryant, and J. H. Golbeck.
1996. Modified ligands to FA and FB in photosystem I. Proposed chemical rescue
of a [4Fe-4S] cluster with an external thiolate in alanine, glycine, and serine
mutants of PsaC. J. Biol. Chem. 271:31135-44.
Page 136
122
12. Kemper, M. A., H. S. Gao-Sheridan, B. Shen, J. L. Duff, G. J. Tilley, F. A.
Armstrong, and B. K. Burgess. 1998. Delta T 14/Delta D 15 Azotobacter
vinelandii ferredoxin I: creation of a new CysXXCysXXCys motif that ligates a
[4Fe-4S] cluster. Biochemistry. 37:12829-37.
13. Latimer, M. T., M. H. Painter, and J. G. Ferry. 1996. Characterization of an
iron-sulfur flavoprotein from Methanosarcina thermophila. J. Biol. Chem.
271:24023-8.
14. Maupin-Furlow, J. A., and J. G. Ferry. 1996. Analysis of the CO
dehydrogenase/acetyl-coenzyme A synthase operon of Methanosarcina
thermophila. J. Bacteriol. 178:6849-56.
15. Menon, S., and S. W. Ragsdale. 1998. Role of the [4Fe-4S] cluster in reductive
activation of the cobalt center of the corrinoid iron-sulfur protein from
Clostridium thermoaceticum during acetate biosynthesis. Biochemistry. 37:5689-
98.
16. Meyer, J., J. Fujinaga, J. Gaillard, and M. Lutz. 1994. Mutated forms of the
[2Fe-2S] ferredoxin from Clostridium pasteurianum with noncysteinyl ligands to
the iron-sulfur cluster. Biochemistry. 33:13642-50.
17. Moulis, J.-M., V. Davasse, M.-P. Golinelli, J. Meyer, and I. Quinkal. 1996.
The coodination sphere of iron-sulfur clusters: lessons from site-directed
mutagenesis experiments. J. Biol. Inorg.Chem. 1:2-14.
18. Peters, J. W., M. H. Stowell, and D. C. Rees. 1996. A leucine-rich repeat
variant with a novel repetitive protein structural motif [letter]. Nat. Struct. Biol.
3:991-4.
19. Porello, S. L., M. J. Cannon, and S. S. David. 1998. A substrate recognition
role for the [4Fe-4S]2+ cluster of the DNA repair glycosylase MutY.
Biochemistry. 37:6465-75.
20. Prince, R. C., and M. J. Grossman. 1993. Novel iron-sulfur clusters. Trends.
Biochem. Sci. 18:153-4.
21. Shen, B., D. R. Jollie, T. C. Diller, C. D. Stout, P. J. Stephens, and B. K.
Burgess. 1995. Site-directed mutagenesis of Azotobacter vinelandii ferredoxin I:
Page 137
123
cysteine ligation of the [4Fe-4S] cluster with protein rearrangement is preferred
over serine ligation. Proc. Natl. Acad. Sci U S A. 92:10064-8.
22. Sticht H, R. P. 1998. The structure of iron-sulfur proteins. Prog. Biophys. Mol.
Biol. 70:95-136.
23. Terlesky, K. C., and J. G. Ferry. 1988. Purification and characterization of a
ferredoxin from acetate-grown Methanosarcina thermophila. J. Biol. Chem.
263:4080-2.
24. Terlesky, K. C., M. J. Nelson, and J. G. Ferry. 1986. Isolation of an enzyme
complex with carbon monoxide dehydrogenase activity containing corrinoid and
nickel from acetate-grown Methanosarcina thermophila. J. Bacteriol. 168:1053-8.
25. Tsumoto, K., K. Shinoki, H. Kondo, M. Uchikawa, T. Juji, and I. Kumagai.
1998. Highly efficient recovery of functional single-chain Fv fragments from
inclusion bodies overexpressed in Escherichia coli by controlled introduction of
oxidizing reagent--application to a human single-chain Fv fragment. J. Immunol.
Methods. 219:119-29.
26. Volbeda, A., M. H. Charon, C. Piras, E. C. Hatchikian, M. Frey, and J. C.
Fontecilla-Camps. 1995. Crystal structure of the nickel-iron hydrogenase from
Desulfovibrio gigas. Nature. 373:580-7.
27. Zhou, Z. H., and M. W. Adams. 1997. Site-directed mutations of the 4Fe-
ferredoxin from the hyperthermophilic archaeon Pyrococcus furiosus: role of the
cluster- coordinating aspartate in physiological electron transfer reactions.
Biochemistry. 36:10892-900.
28. Antonkine, M. L., C. Falzone, A. Hansen, F. Yang, and J. H.Golbeck.
Chemical rescue of site-modified ligands to the iron-sulfur clusters of PsaC in
Photosystem I. Proceedings of XIth international congress in photosynthesis,
Budapest, 1998, Kluwer academic publishers, (in press).
Page 138
124
CHAPTER 6
SUMMARY AND FURTURE DIRECTIONS
Advances in understanding the biochemical reactions involved in carbon
transformations during methanogenesis have inspired investigations of how electrons are
transported to generate energy for the cell. The characterization of a novel redox protein
(Isf) in this study provides a better understanding of electron transport in the acetate
fermentation pathway.
Information derived from the sequence of the heterologously produced iron-sulfur
flavoprotein (Isf) from the archaeon Methanosarcina thermophila shows many striking
features distinct from any known iron-sulfur proteins or flavoproteins. The sequence
reveals a novel cysteine motif that has been shown by site-directed mutagenesis and
spectroscopic analyses to accommodate a 4Fe-4S center. An unusual higher stability of
hydroquinone relative to semiquinone prompts an investigation of the environment in the
FMN binding site that results in hydroquinone formation. The three-dimensional
structure of this archaeal Isf is being solved using X-ray crystallography in collaboration
with Dr. C. Bremnane and Prof. D. Rees at Caltech. Since Isf of M. thermophila is the
prototype for this protein family and there is no significant sequence identity with known
proteins, the structure may help to elucidate the environment of redox centers. The
structure may support the proposed functions of these two cofactors by showing if both of
them are able to participate in electron transfer as expected. This will lead to a better
understanding of the mechanism for electron transfer.
The heterologously produced Isf has been studied, but the native Isf from M.
thermophila has not yet been purified. Thus, it is not possible to propose with confidence
that they share the same properties. Attempts to identify Isf from M. thermophila using
Western blots were not successful. This protein may be expressed only at very low
levels and only under certain growth conditions. Northern blot analysis is an alternative
approach to examine the expression and regulation of the isf gene.
Page 139
125
Although the spectroscopic techniques and site-directed mutagenesis reported
here provide structural information for Isf, the function of Isf remains an issue. The
involvement of Isf in electron transport CO-dependent CoM-S-S-CoB reduction has been
shown, and there is evidence that ferredoxin A is a direct electron donor for Isf. In
contrast, the physiological oxidative partner is not known. Passing M. thermophila cell
extract through an affinity column to which Isf is bound may be used to identify proteins
that interact with Isf and possibly function as its electron acceptor.
Biochemical characterization of Isf-like sequences from other metabolically
diverse microbes may reveal their roles in these microbes. These sequences may provide
an opportunity to study evolutionary convergence of Isf.
Page 140
126
Curriculum Vista UBOLSREE LEARTSAKULPANICH
Department of Biochemistry and Anaerobic Microbiology Virginia Polytechnic Institute and State University Blacksburg, VA 24061
204 S.Frear Bld. 447 W. Clinton Ave., # 408BMB, Penn State University State CollegeUniversity Park PA 16801PA 16802-4500 [email protected]
PERSONAL
Date of birth: 2/10/71 (Bangkok, Thailand)Place of birth: Bangkok, Thailand
EDUCATION:
Aug 1993 - present:Doctoral candidate, Department of Biochemistry and Anaerobic MicrobiologyVirginia Polytechnic Institute and State University (VPI&SU), Blacksburg, VA.
Aug 1995 - present:Department of Biochemistry and Molecular BiologyPennsylvania State University, University Park, PA.In absentia from Virginia Polytechnic Institute and State University
Apr 1992 – 1993MS. candidate, Department of Biochemistry, Mahidol University,Bangkok, Thailand
Jun 1988 - 1992B.S. Biochemistry, Chulalongkorn University, Bangkok, Thailand
PROFESSIONAL EXPERIENCE
Jun 1994 - Aug 1994:Graduate Teaching Assistant, Department of Biochemistry and AnaerobicMicrobiology. VPI&SU.Laboratory instructor for BAM5104 Advanced Methods of Biochemical Analysis
Apr 1991 – May 1991:Student training at Chareonpokapun group (CP), Samut Prakarn, Thailand
HONORS AND AWARDS:
Royal Thai Scholarship for Ministry of Science, Technology and Energy(MOSTE)
Page 141
127
National Science and Development Agent scholarship (NSDA)The first rang student of the 1992 MS. candidate class, Mahidol UniversityThe first range student for 1992 MS. entrance examination, Mahidol UnversityBS. with Second class honor degree, Chulalongkorn University
PUBLICATIONS:
Becker D.F., Leartsakulpanich U., Surerus K.K., Ferry J.G., and S.W. Ragsdale.1998. Electrochemical and spectroscopic properties of the iron-sulfurflavoprotein from Methanosarcina thermophila. J. Biol Chem. 273:26462-26469
Leartsakulpanich U, Antokine M.L., Golbeck J.H., and J.G Ferry. A novel [4Fe-4S] iron-sulfur cluster binding motif in the iron-sulfur flavoprotein ofMethanosarcina thermophila (in preparation)
ABSTRACTS AND PRESENTATIONS:
Inorganic Biology Summer Workshop (IBSW 98), Athens, GAJul 25 – Aug 5, 1998Leartsakulpanich U, Antokine M.L., Golbeck J.H., and J.G. Ferry. 1998. Ligandsto the 4Fe-4S center in the iron-sulfur flavoprotein from Methanosarcinathermophila and proposed physiological function
Penn State Sixteenth Summer Symposium in Molecular Biology MicrobialStructural Biology: Novel Enzymes from Diverse MicrobesAug 7-9, 1997Leartsakulpanich U., Becker D.F., Ragsdale S.W., Borup B., Aldrich H.C., andJ.G.Ferry. Characterization of the iron-sulfur flavoprotein (Isf) fromMethanosarcina thermophila.