Page 1
UK Journal of Pharmaceutical and Biosciences Vol. 3(6), 30-41, 2015 RESEARCH ARTICLE
Design and In-Vitro Evaluation of Colon Targeted Prednisolone Solid Dispersion
Tablets
Sura Zuhair Mahmood Alkazzaz*, Wedad Kamal Ali
Department of Pharmaceutics, College of Pharmacy, University of Al-Mustansiriya, Baghdad-Iraq
Article Information
Received 19 September 2015
Received in revised form 26 Sep 2015
Accepted 28 Sep 2015
Abstract
In the present investigation an attempt was made to prepare colon targeted enteric coated tablet
containing different prednisolone solid dispersion formulations, to prevent ulcerative colitis,
improve the patient compliance and reduce the side effects of drug in the gastro intestinal tract
(GIT). Solid dispersion is one of the most widely used approaches to enhance the solubility as
well as dissolution rate of poorly water soluble drugs. Solid dispersions (SDs) of Prednisolone
with D-mannitol, PEG 4000 and Kollicoat IR were prepared and evaluated to deliver Prednisolone
to the colon in a pre-solubilized form. The selected formula using drug compatible excipients was
compressed into fast disintegrating tablets and then coated with Eudragit S100 (pH-responsive
polymer), several variables related to both solid dispersion preparation (carrier type and drug to
the carrier ratio) and tablet coating (coat level) were studied to show their effects on drug
solubility and dissolution. Different analytical techniques like differential scanning caloremitry
(DSC), powder x-ray diffraction (PXRD) and scanning electron microscopy (SEM) were studied to
prove the change of drug particle from crystal to amorphous form in SDs. The 1:3
Prednisolone/Kollicoat IR SDs showed the greatest improvement in the dissolution rate. The
coating level was critical for determining the duration of the lag phase. Best result was given by
the 16% coat (Eudragit S100/ Dibutyl phthalate/ talc). This coating level showed an acceptable
lag time for the proposed colonic targeting (5 h) as the selected tablet resisted pre-colonic pH
values, followed by an immediate release stage in pH 7.4. The suggested covered (coated)
tablets may provide a colonic delivery system for prednisolone with enhanced solubility and
bioavailability.
Keywords:
Prednisolone,
Colon Targeted,
Solid dispersions
*Corresponding Author:
E-mail: [email protected]
Mob.: 07901325199
1 Introduction
In the last few years, the development of the site specific drug
delivery system has been introduced to be studied with great deal of
research work which compromises numerous benefits over the
traditional drug treatments. The principle goal of the site specific
delivery is to deliver the drug in the specific organs of the body1.
Targeted drug delivery to the colon is highly required for local
treatment of a variety of bowel diseases such as ulcerative colitis,
Crohn’s disease, amebiosis, and colonic cancer2. The most critical
challenge in such drug delivery approaches is to protect the
formulation during its passage through the stomach and about first
six meters of the small intestine arriving to colon with no loss of
active ingredient by preventing the dissolution and the release till it
reached the colon3. Among the various pharmaceutical approaches
used to target drugs to the colon are pH-dependent, time-dependent
and bacterially degradable polymers4.
Prednisolone (a potent synthetic corticosteroid that available for
clinical use in 1955) making it useful for the treatment of a wide
range of inflammatory and auto-immune conditions such as Crohn’s
disease, Ulcerative colitis and Rheumatoid arthritis and others5.
Peak plasma concentrations of prednisolone are obtained after 1-2
hours of an oral dose administration, and it has a usual plasma half-
life of 2 to 4 hours6.
UK Journal of Pharmaceutical and Biosciences
Available at www.ukjpb.com ISSN: 2347-9442
Page 2
Alkazzaz et al. Design and In-Vitro Evaluation of Colon Targeted Prednisolone
UK J Pharm & Biosci, 2015: 3(6); 31
Improvement of the dissolution rates of class II water-insoluble drugs
is one of the most challenging issues of drug development, because
the enhanced dissolution rates can enhance drug oral
bioavailability7. For water-insoluble drugs the solid dispersion
technique established by Chiou and Reigelman supplies an effective
way to increase the dissolution rates of drugs8.
In the present study, efforts were made to improve the dissolution
behavior and consequently, absorption, of Prednisolone by applying
the solid dispersion technique using three different hydrophilic
carriers (PEG 4000, D- mannitol and Kollicoat IR) the solid
dispersion formulations were compressed into tablets and further
coated to deliver the drug to colon.
2 Materials and Methods
2.1 Materials
Prednisolone, D-mannitol, Crospovidone, Croscarmellose sodium,
magnesium stearate were obtained from Samara drug industry, Iraq;
Kollicoat IR was obtained from Sigma-Aldrich Co., USA; Poly
ethylene glycol (PEG4000) from Sinopharm chemical reagent Co.,
China, Eudragit S100 was purchased from Evonik Company,
Germany and Dibutyl phthalate was manufactured by Fluka
Company, UK. All other chemicals, reagents and solutions used
were of analytical grade. Marketed tablet Deltacortril® (Pfizer,
Turkey) was purchased from local pharmacy.
2.2 Methods
2.2.1 Solubility determination of Prednisolone
For the determination of solubility of Prednisolone, an excess
amount of the drug about 50 mg was added to 25 ml phosphate
buffers pH7.4. The flask was stirred for 24 hours using magnetic
stirrer and maintained at 25oC. The sample was then filtered through
0.45 μm membrane filters, suitably diluted, and analyzed by UV-
spectrophotometer at 247 for prednisolone. The study was
performed in triplicate9-11
.
2.2.2 Preparation of Prednisolone solid dispersions
Solid dispersions of prednisolone in three hydrophilic carriers at
three different weight ratios were prepared by solvent evaporation
method by using ethanol as common solvent. The calculated amount
of polymer and drug was dissolved separately in the required amount
of solvent ethanol and mixed under mechanical agitation. The
solvent was eliminated using a rotary evaporator under reduced
pressure. The solid dispersions when dried were grinded using a
mortar and pestle then passed through 0.36 mm sieve and stored in
desiccators till use12
, and the optimum one for each carrier was
compared with the physical mixture and pure Prednisolone. Solid
dispersion and physical mixture of different weight ratios are listed in
table 1.
2.2.3 Evaluation of the prepared solid dispersion
2.2.3.1 Determination of saturated solubility of Prednisolone in solid
dispersions
Similar procedure mentioned in (2.2.1) was used to determine the
saturation solubility of different solid dispersions in phosphate buffer
pH7.4 and compared to that of pure drug13,14
.
Table 1: Formulation code of Prednisolone solid dispersions
and physical mixtures prepared with different carriers
Formulation
Codes
Carrier
Drug:
Carrier
ratio
Method of
preparation
SD1 PEG4000 1:1 Solvent Evaporation
SD2 1:2 Solvent Evaporation
SD3 1:3 Solvent Evaporation
PM1 1:3 Physical Mixture
SD4 D- Mannitol 1:1 Solvent Evaporation
SD5 1:2 Solvent Evaporation
SD6 1:3 Solvent Evaporation
PM2 1:3 Physical Mixture
SD7 Kollicoat IR 1:1 Solvent Evaporation
SD8 1:2 Solvent Evaporation
SD9 1:3 Solvent Evaporation
PM3 1:3 Physical Mixture
2.2.3.2 In-vitro dissolution study:
The in vitro dissolution study was carried out by using USP type II
(paddle type) dissolution test apparatus (Cosmo Lab). Using 900 ml
dissolution medium (pH 7.4) at 37◦C and rotation speed of 50 rpm.
Accurately weighted amount (5mg) of pure drug and equivalent
amount from solid dispersions and physical mixtures to 5mg
prednisolone were placed in the dissolution vessel for 90min and at
appropriate time intervals (2, 5, 10, 15, 20, 30, 45, 60 and 90 min), 5
ml samples were withdrawn and replenished with the same volume
of fresh medium to keep the sink condition constant, samples then
filtered and analyzed spectrophotometrically at (247 nm for
Prednisolone). The procedure was performed in triplicate for each
run test and the mean and standard deviation were calculated15-17
.
Page 3
Alkazzaz et al. Design and In-Vitro Evaluation of Colon Targeted Prednisolone
UK J Pharm & Biosci, 2015: 3(6); 32
2.2.3.3 Determination of drug content
The drug content in each solid dispersion formulation was
determined by placing the weighted amount of solid dispersion
samples equivalent to 2mg of Prednisolone in 100ml volumetric flask
containing phosphate buffer (pH 7.4), the samples were continuously
shaking until completely dissolve them then filtered. The absorbance
of the samples was determined at λmax 247 nm, using UV-visible
spectrophotometer. Three readings were taken, and then mean and
standard deviation was calculated18
.
2.2.3.4 Selection of the best formula
The phase solubility and in vitro dissolution test were used for
selecting the best solid dispersion formula which will be subjected to
further analysis.
2.2.4 Characterization of the selected solid dispersion formula
2.2.4.1 Fourier transforms infrared spectroscopy (FTIR):
Samples of pure drug, Kollicoat IR and SD9 (equivalent to about 5
mg of Prednisolone) were ground, mixed with dry potassium bromide
and pressed in the form of discs using hydraulic press. The discs
were analyzed by FTIR spectroscopy (4000-400 cm-1)19, 20
.
2.2.4.2 Differential scanning calorimetry (DSC)
DSC was used to determine thermal behavior of Prednisolone,
Kollicoat IR, and SD9 formulations. The pure drug, polymer and solid
dispersions were examined by DSC 60 (Shimadzu, Japan), where 5-
6 mg sample was placed in aluminum pan and scan at a heating rate
of 10oC/min (in range of 0-350
oC ) with purging of dry nitrogen at a
constant rate; an empty aluminium pan was used as a reference.
Indium/Zinc standards were used to calibrate the DSC temperature
and enthalpy scale21, 22
.
2.2.4.3 Powder x-ray diffraction (PXRD)
The extent of crystallinity was determined for pure drug and prepared
solid dispersion using X-ray (Shimadzu, Japan) powder diffraction
system equipped with Cu radiation (λ=1.54060 A◦) at a voltage of (40
Kv) and a current of (30 mA).The instrument was operated in the
continuous scan mode and sample were analyzed in the range (5-
80oC) with a step size of (0.05
oC) at scanning speed of (5
oC /min)
and (2θ) axis23
.
2.2.4.4 Scanning electron microscopy (SEM):
The SEM analysis was carried out using a scanning electron
microscope (SEM Tescan Vega lll Czech). Before to examination,
mounted the sample on an aluminum stub using a double sided
adhesive tape, then coating with a thin layer of gold (approximately
20 nm) in the vacuum to make it electrically conductive. SEM
provides a high resolution images that show details of a sample
surface since a high energy beam of electrons typically from 0.5 kV
to 40 kV is used to scan the surface of sample to give image in a
raster scan pattern24
.
2.2.5 Manufacturing of colon targeted tablet of Prednisolone by direct
compression method
Tablets of pure drug and solid dispersion formulations were prepared
to evaluate the impact of solid dispersion on the release of the drug.
Tablets ingredients used in tablet formulation (Table 2) were
accurately weighed then passed through 0.36 mm sieve to get
uniform particle size. The drug and all the ingredients except
lubricants were mixed and blended for 5 min. Finally, magnesium
stearate was added, mixed for 2 min to coat the particle surface by
lubricant evenly. The blend was compressed using 6mm punch and
die on a single punch tablet machine. The formulated tablets were
stored in a tightly closed container until evaluated. Based on the
results of dissolution study the best formulation was selected among
the six formulations for further study25
.
Table 2: Formulation ingredient of pure and solid dispersions
tablet of Prednisolone
Ingredient F1
F2
SD3
F3
SD3
F4
SD3
F5
SD6
F6
SD18
Prednisolone 5 - - - - -
Solid Dispersion - 20 20 20 20 20
Cros-carmellose Na 2 2 - - 2 2
Cros-povidone - - 2 - - -
Magnesium stearate 1 1 1 1 1 1
Avicel pH302 92 77 77 79 77 77
Total weight 100 100 100 100 100 100
2.2.5.1 Pre-compression parameters evaluation
Various micromeritic parameters like angle of repose, bulk density,
tap density, Carr’s (Compressibility) Index (CI), and Hausner’s ratio
were measured for solid dispersions powders26
.
2.2.5.2 Post-compression parameters evaluation
Thickness of tablets prepared was calculated using Vernier caliper27
,
the hardness of the tablets was determined using electrical hardness
tester. It is expressed in Kg/cm2. The hardness test was performed in
which five tablets from each formula were tested randomly and the
average reading ± sd was recorded28
. The friability test was done by
Page 4
Alkazzaz et al. Design and In-Vitro Evaluation of Colon Targeted Prednisolone
UK J Pharm & Biosci, 2015: 3(6); 33
placing 20 pre-weighed tablets in the friabilator which was then
operated for 25 rpm for 4 minutes; the tablets were then dusted and
reweighed. Tablets that lose a maximum of not more than 1% of their
weight are generally considered acceptable29
. In addition, weight
variation was performed according to the USP specifications.
2.2.5.3 Content Uniformity test
Content uniformity was done by weighing and powdering 20 tablets.
Weigh accurately a quantity of the powder equivalent to (5 mg of
Prednisolone) and transferred to 100 mL volumetric flasks containing
50 mL of phosphate buffer pH 7.4. The flasks were shaken to
solubilize the drug. The volume was completed to 100 mL with the
buffer, allowed to stand for 24 h to make sure complete solubility of
the drug. The solution was filtered, and 1 mL of the filtrate liquid was
suitably diluted and analyzed for prednisolone content
spectrophotometrically at 247 nm30-32
.
2.2.5.4 In-vitro disintegration study
The in-vitro disintegration study of the uncoated tablets was
determined using the disintegration test apparatus as per USP
specifications. One tablet was placed in each of the six tubes of the
basket, the disc was add to each tube and running the apparatus
using 900 ml of phosphate buffer pH 7.4 maintained at 37ºC. The
time in seconds for complete disintegration of the tablets with no
palpable mass remaining in the apparatus was measured and
recorded33
.
2.2.5.5 In-vitro dissolution study
The in vitro dissolution study was carried out as mentioned
previously in section (2.2.3.2) except that one tablet of each
prepared formula was placed in the dissolution vessel instead of
powdered sample for 90min
2.2.5.5.1 Effect of different superdisintegrants addition on uncoated
tablet release profile
Formulas F2SD3 and F3SD3 were designed to study the effect of
different superdisintegrants addition on drug release of uncoated
tablet compared with one without the addition of superdisintegrant
(F4SD3), where 2% Croscarmellose, 2%Crospovidone and no
superdisintegrant were used respectively.
2.2.6 Eudragit S100 coating of tablets for pH dependent release
For minimizing the drug release in upper GIT (stomach and small
intestine) Eudragit S100 was selected as the pH dependent coating
polymer. A 6% w/v Eudragit S100 coating solution was prepared
using the mixture of isopropyl alcohol and acetone with the addition
of 1% plasticizer- Dibutyl phthalate and used to coat tablet of
optimized formula using dip coating method34
.
2.2.6.1 Evaluation of the prepared coated tablets
Tablet of optimum formula was coated and around the selected core
tablet formula and the resulted coated tablets evaluated for
thickness, hardness and friability in the same way for uncoated
tablets.
2.2.6.2 Disintegration test for enteric coated tablets
The disintegration test was carried out for all the formulations
according to British Pharmacopeia method for enteric-coated tablets.
0.1N HCL and 7.4 pH phosphate buffer was used as disintegrating
media. Six tablets were used in each case35
.
2.2.6.3 In-vitro drug release study of coated tablets of Prednisolone
Similar dissolution conditions mentioned for powder, and uncoated
tablets were used. For simulating the gastric fluid in stomach, the
dissolution was accomplished in 0.1 N HCl (pH 1.2) for 2hr, in the
phosphate buffer (pH 6.8) to simulate the small intestinal fluid for
three hours and for another two hours in phosphate buffer (pH 7.4),
simulating the colonic environment. Sample aliquots withdrawn at
specific time intervals, were analyzed at 247 nm using UV-Vis
Spectrophotometer34,36
.
2.2.6.4 Drug-excipient interactions
The physicochemical compatibilities of the drug and the used
excipients were tested by FTIR. Pure Prednisolone, selected core
and press coated tablets (which were previously grinded); were
mixed thoroughly with potassium bromide. The potassium bromide
discs were prepared by compressing the powder at a pressure in a
hydraulic press and analyse in the ranges (4000- 400 cm-1)37
.
2.2.6.5 Statistical analysis
The results of the experiments are given as a mean values ±
standard deviation (SD) and were analyzed according to one-way
analysis of variance (ANOVA) at which significant results (p<0.05)
and non-significant (p>0.05).
3 Results and Discussions
The measured solubility of Prednisolone in phosphate buffer pH7.4
(215±0.005 μg/ml) indicates that the drug is a very slightly soluble
compound in this buffer. Solubility studies revealed a linear increase
in drug solubility in the presence of an increasing carrier
concentration this is because hydrophilic carriers are known to
interact with drug molecules, mainly by electrostatic forces and
occasionally by other types of forces like intermolecular hydrogen
bonds38,39
. The solubility enhancement of the various carriers was in
the order of Kollicoat IR > PEG4000> D-Mannitol. The markedly
higher solubility of Prednisolone in Kollicoat IR may be attributed to
the higher solubilizing capacity as it is non-ionic polymer and its
Page 5
Alkazzaz et al. Design and In-Vitro Evaluation of Colon Targeted Prednisolone
UK J Pharm & Biosci, 2015: 3(6); 34
solubility is pH-independent40
. The content of Prednisolone was
determined in all the prepared formulas and was found to range from
98-101% of the theoretical calculated content which is within the
limits of the official monographs of Prednisolone preparations of the
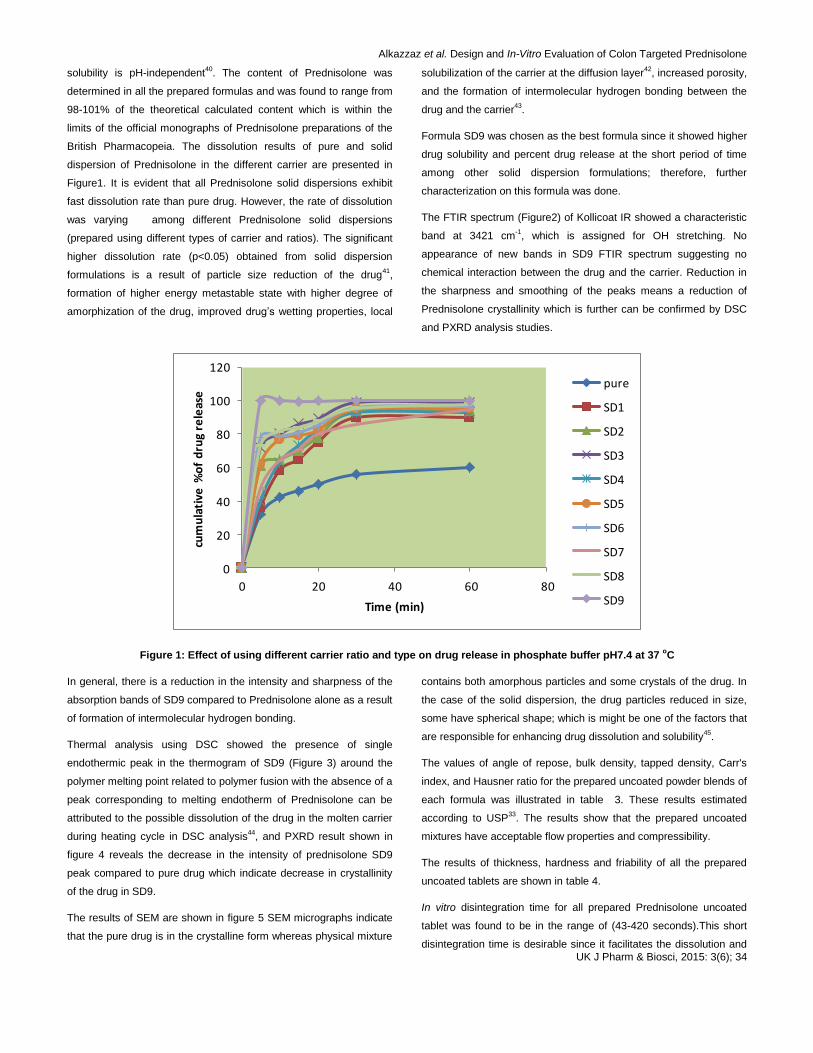
British Pharmacopeia. The dissolution results of pure and solid
dispersion of Prednisolone in the different carrier are presented in
Figure1. It is evident that all Prednisolone solid dispersions exhibit
fast dissolution rate than pure drug. However, the rate of dissolution
was varying among different Prednisolone solid dispersions
(prepared using different types of carrier and ratios). The significant
higher dissolution rate (p<0.05) obtained from solid dispersion
formulations is a result of particle size reduction of the drug41
,
formation of higher energy metastable state with higher degree of
amorphization of the drug, improved drugʼs wetting properties, local
solubilization of the carrier at the diffusion layer42
, increased porosity,
and the formation of intermolecular hydrogen bonding between the
drug and the carrier43
.
Formula SD9 was chosen as the best formula since it showed higher
drug solubility and percent drug release at the short period of time
among other solid dispersion formulations; therefore, further
characterization on this formula was done.
The FTIR spectrum (Figure2) of Kollicoat IR showed a characteristic
band at 3421 cm-1, which is assigned for OH stretching. No
appearance of new bands in SD9 FTIR spectrum suggesting no
chemical interaction between the drug and the carrier. Reduction in
the sharpness and smoothing of the peaks means a reduction of
Prednisolone crystallinity which is further can be confirmed by DSC
and PXRD analysis studies.
0
20
40
60
80
100
120
0 20 40 60 80
cum
ula
tive
%o
f d
rug
rele
ase
Time (min)
pure
SD1
SD2
SD3
SD4
SD5
SD6
SD7
SD8
SD9
Figure 1: Effect of using different carrier ratio and type on drug release in phosphate buffer pH7.4 at 37 oC
In general, there is a reduction in the intensity and sharpness of the
absorption bands of SD9 compared to Prednisolone alone as a result
of formation of intermolecular hydrogen bonding.
Thermal analysis using DSC showed the presence of single
endothermic peak in the thermogram of SD9 (Figure 3) around the
polymer melting point related to polymer fusion with the absence of a
peak corresponding to melting endotherm of Prednisolone can be
attributed to the possible dissolution of the drug in the molten carrier
during heating cycle in DSC analysis44
, and PXRD result shown in
figure 4 reveals the decrease in the intensity of prednisolone SD9
peak compared to pure drug which indicate decrease in crystallinity
of the drug in SD9.
The results of SEM are shown in figure 5 SEM micrographs indicate
that the pure drug is in the crystalline form whereas physical mixture
contains both amorphous particles and some crystals of the drug. In
the case of the solid dispersion, the drug particles reduced in size,
some have spherical shape; which is might be one of the factors that
are responsible for enhancing drug dissolution and solubility45
.
The values of angle of repose, bulk density, tapped density, Carr's
index, and Hausner ratio for the prepared uncoated powder blends of
each formula was illustrated in table 3. These results estimated
according to USP33
. The results show that the prepared uncoated
mixtures have acceptable flow properties and compressibility.
The results of thickness, hardness and friability of all the prepared
uncoated tablets are shown in table 4.
In vitro disintegration time for all prepared Prednisolone uncoated
tablet was found to be in the range of (43-420 seconds).This short
disintegration time is desirable since it facilitates the dissolution and
Page 6
Alkazzaz et al. Design and In-Vitro Evaluation of Colon Targeted Prednisolone
UK J Pharm & Biosci, 2015: 3(6); 35
releases of the drug from the tablet. In general disintegration of
tablets achieved through overcoming the cohesive strength of tablets
using different types and amount of Disintegrants in tablet
formulation.
A
B
C
Figure 2: FTIR spectra of A- Prednisolone, B- Kollicoat IR and C - SD9
Figure 6 shows dissolution profiles of Prednisolone from 4 different
SD tablets in the phosphate buffer (pH 7.4) compared to marketed
one. These results are similar to those obtained from powdered
Prednisolone, which indicates that the enhancement in the
dissolution of drug from solid dispersion samples is maintained after
manufacturing these samples into tablets.
The result of in vitro dissolution (F2, F3 and F4) uncoated tablet
formula which was designed to show the effect of Croscarmellose
sodium, Crospovidone and without addition of any disintegrants
respectively on the drug release from the uncoated tablet shown in
figure7. There was significant difference (p<0.05) in the initial release
of drug from these formulation among these formulas the F2 give the
best result of 100% release due to the rapid increase in dissolution of
Prednisolone with the use of Croscarmellose sodium may be
attributed to rapid swelling and disintegration of tablet into apparently
primary small particles while Crospovidone disintegrates the tablets
quickly but into larger masses of accumulated particles. It exhibits
high capillary activity and marked hydration with a little tendency to
gel formation46, 47
.
The developed formulation of coated tablet F6 was studied for its
physical properties like thickness, hardness, friability and weight
variation and the result was as follow thickness (3.66±0.002 mm),
hardness (7±0.005 Kg/cm2), friability (0.18%) and weight variation
(116±0.5 mg). The content uniformity test was done for the selected
coated tablet formula and the result was 99.95%. This result agrees
with the requirements of the United States pharmacopeia.
The coated tablet met pharmacopeial (BP/USP) requirements for the
enteric performance test in the 0.1N HCl for 2hr. tablet disintegrate in
phosphate buffer solution pH7.4 after 20±0.02 min. Also we found
that tablet coated at higher levels had longer disintegration times
than that coated at lower levels at the same medium as for 16% and
19% coat level was 20 and 60min respectively48
.
Page 7
Alkazzaz et al. Design and In-Vitro Evaluation of Colon Targeted Prednisolone
UK J Pharm & Biosci, 2015: 3(6); 36
Figure3: DSC thermograms of A - pure Prednisolone, B - Kollicoat IR and C - SD9 (1:3 Kollicoat IR)
Prednisolone
SD9
Figure 4: X-ray diffraction (XRD) patterns of Pure Prednisolone and SD9
The requirement for in vitro release pattern selected for the colon
targeting was no drug release up to the end of 5hrs to achieve this
different Eudragit S100 coating level was examined, and the best
result was using 16% coating level.
The concentration of polymer in solution and the % coating level was
related to the drug release directly. Percent of drug release versus
time plot illustrations that the dissolution rate was in reverse
Page 8
Alkazzaz et al. Design and In-Vitro Evaluation of Colon Targeted Prednisolone
UK J Pharm & Biosci, 2015: 3(6); 37
proportional to the coating level applied. The percentage of drug released for different coating level showed significant differences36
.
Figure 5: SEM of A-pure drug, B- physical mixture and C-SD9
Table 3: Pre-compression physical parameters for core powder blend
Formula
Angle of
repose
(Degree)
Bulk density
(g/cm3)
Tapped density
(g/cm3)
Carr's
Index
Hausner
ratio
Type of flow
F1 33.78±0.66 0.323±0.04 0.37±0.01 12.7 1.15 Good
F2 31.21±0.51 0.323±0.01 0.364±0.06 11.26 1.13 Good
F3 31.21±0.51 0.323±0.01 0.364±0.06 11.26 1.13 Good
F4 31.15±0.63 0.364±0.04 0.408±0.01 10.78 1.12 Good
F5 24.52±0.62 0.385±0.02 0.417±0.03 7.67 1.08 Excellent
F6 33.17±0.64 0.345±0.03 0.408±0.04 15.44 1.18 Good
The selected tablet formula was used to study the effect of coat level
(thickness) on lag time of colon targeted coated tablet. The lag time
of coated tablet which have tablet thickness 3.66mm was 5 hours
and 20 min while for the same tablet formula which have tablet
thickness 3.75mm was 7 hours as shown in figure 8. These results
showed that as the thickness of the coat increased, the lag time
increased since the time required to complete the erosion of the
outer shell would be longer. The same results were reported with
other related studies49
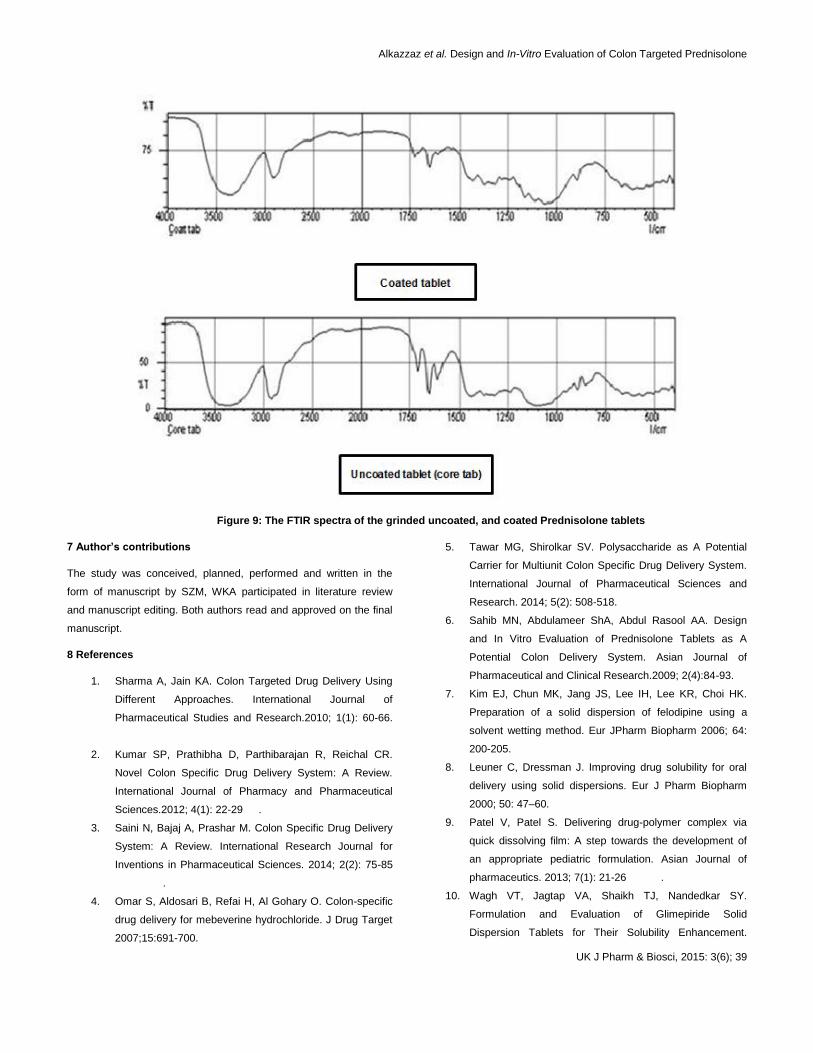
. The FTIR spectra results showed that the
drug bands didn't change significantly in the FTIR spectra of the
grinded uncoated, and selected coated tablets as shown in figure 9
and the small shifting in the absorption bands was listed in table 5.
These results indicating that there is no significance evidence of
chemical interaction between drug and polymer, which confirm the
stability of drug.
4 Conclusion
Amorphous solid dispersions of prednisolone were successfully
prepared by solvent evaporation method. The presence of
amorphous form in SDs was confirmed by DSC, PXRD and SEM,
and was reflected in the significant improvement in rate as well as
the extent of in vitro drug dissolution. Proper selection of the
Page 9
Alkazzaz et al. Design and In-Vitro Evaluation of Colon Targeted Prednisolone
UK J Pharm & Biosci, 2015: 3(6); 38
Eudragit®S100 coat level is essential to deliver prednisolone to the
colon. The optimized prednisolone colon targeted tablets could be
promising in reducing the drug dose and improving its bioavailability
based on the protection from the intestinal metabolism. Such a
delivery system could be applied for similar water insoluble drugs
liable to intestinal enzymatic degradation. Additional studies are
needed to assess its performance in vivo.
Table 4: Post compression parameter of Prednisolone tablets
Formula Thickness
(mm)
Hardness
(kg/ cm2)
Friability
(%)
Weight
variation
(mg)
F1 3.19± 0.01 4.5± 0.02 0.46±0.04 99±0.04
F2 3.19±0.03 5.02±0.04 0.1±0.05 99.5±0.01
F3 3.19±0.02 5.8±0.01 0.2±0.03 100±0.02
F4 3.21±0.01 5.5±0.02 0.25±0.05 98.5±0.01
F5 3.19±0.01 4±0.03 0.51±0.02 98±0.02
F10 3.20±0.01 4±0.03 0.6±0.01 99.7±0.01
Figure 6: Dissolution profiles of Prednisolone from 4 different
uncoated tablets in phosphate buffer (pH 7.4) compared to
marketed one
5 Acknowledgements
We are grateful for the cooperation of Iraqi Ministry of Science and
Technology for doing the required analytical measurements and Ibn-
Sena center for drug research (Baghdad/Iraq) for providing
chemicals from Samara drug industry.
6 Competing interests
Authors have no competing interests.
Figure 7: Effect of different superdisintegrant addition on
uncoated tablet for F2, F3 and F4 in phosphate buffer pH 7.4
Figure 8: In- vitro release of uncoated tablet and Effect of coat
thickness on percent of drug release in different dissolution
media
Table 5: Characteristic absorption bands of Prednisolone
Characteristic
Group
Pure
Prednisolone
cm-1
Prednisolone
core tablet
cm-1
Prednisolone
coat tablet
cm-1
-OH 3454 cm-1 3379.4 cm
-1 3354 cm
-1
Sp3 C-H 2982 cm-1 2910 cm
-1 2910 cm
-1
-C=O 1654 cm-1 1654 cm
-1 1654 cm
-1
Aromatic C=C 1610 cm-1 1612 cm
-1 1610 cm
-1
C-H bend 1446 cm-1 1437 cm
-1 1431 cm
-1
OH bend 1348 cm-1 1371 cm
-1 1371 cm
-1
-C-O 1236 cm-1 1240 cm
-1 1242 cm
-1
Aromatic C=C bend 893 cm-1 893 cm
-1 895 cm
-1
Page 10
Alkazzaz et al. Design and In-Vitro Evaluation of Colon Targeted Prednisolone
UK J Pharm & Biosci, 2015: 3(6); 39
Figure 9: The FTIR spectra of the grinded uncoated, and coated Prednisolone tablets
7 Author’s contributions
The study was conceived, planned, performed and written in the
form of manuscript by SZM, WKA participated in literature review
and manuscript editing. Both authors read and approved on the final
manuscript.
8 References
1. Sharma A, Jain KA. Colon Targeted Drug Delivery Using
Different Approaches. International Journal of
Pharmaceutical Studies and Research.2010; 1(1): 60-66.
2. Kumar SP, Prathibha D, Parthibarajan R, Reichal CR.
Novel Colon Specific Drug Delivery System: A Review.
International Journal of Pharmacy and Pharmaceutical
Sciences.2012; 4(1): 22-29 .
3. Saini N, Bajaj A, Prashar M. Colon Specific Drug Delivery
System: A Review. International Research Journal for
Inventions in Pharmaceutical Sciences. 2014; 2(2): 75-85
.
4. Omar S, Aldosari B, Refai H, Al Gohary O. Colon-specific
drug delivery for mebeverine hydrochloride. J Drug Target
2007;15:691-700.
5. Tawar MG, Shirolkar SV. Polysaccharide as A Potential
Carrier for Multiunit Colon Specific Drug Delivery System.
International Journal of Pharmaceutical Sciences and
Research. 2014; 5(2): 508-518.
6. Sahib MN, Abdulameer ShA, Abdul Rasool AA. Design
and In Vitro Evaluation of Prednisolone Tablets as A
Potential Colon Delivery System. Asian Journal of
Pharmaceutical and Clinical Research.2009; 2(4):84-93.
7. Kim EJ, Chun MK, Jang JS, Lee IH, Lee KR, Choi HK.
Preparation of a solid dispersion of felodipine using a
solvent wetting method. Eur JPharm Biopharm 2006; 64:
200-205.
8. Leuner C, Dressman J. Improving drug solubility for oral
delivery using solid dispersions. Eur J Pharm Biopharm
2000; 50: 47–60.
9. Patel V, Patel S. Delivering drug-polymer complex via
quick dissolving film: A step towards the development of
an appropriate pediatric formulation. Asian Journal of
pharmaceutics. 2013; 7(1): 21-26 .
10. Wagh VT, Jagtap VA, Shaikh TJ, Nandedkar SY.
Formulation and Evaluation of Glimepiride Solid
Dispersion Tablets for Their Solubility Enhancement.
Page 11
Alkazzaz et al. Design and In-Vitro Evaluation of Colon Targeted Prednisolone
UK J Pharm & Biosci, 2015: 3(6); 40
Journal of Advanced Scientific Research. 2012; 3(4): 36-
41.
11. Habeeba P, Madhavanb N, Gladisa K, Anithaa Y,
Mohammed Sh, Raghunathc P. Formulation, Optimization
And Evaluation Of Solid Dispersion Tablets Of
Aceclofenac Using Kollidon 30. International Journal of
Biopharmaceutics. 2013; 4(1): 10-17.
12. Nikghalb LA, Singh G, Singh G, Kahkeshan KF. Solid
Dispersion: Methods and Polymers to increase the
solubility of poorly soluble drugs. Journal of Applied
Pharmaceutical Science. 2012; 2 (10): 170-175 .
13. Sambhakar S, Singh B, Madan K, Monalisha, Kashyap N,
Mayle S. Solid dispersions: A tool for improving the
solubility and dissolution of metronidazole. International
Journal of Drug Delivery.2013; 5(1):94-9.
14. Chaitanya P, Penta J, Devadasu VR, Venisetty RK,
Vemula SK. Ezetimibe Solid Dispersions: Formulation,
Development and In vitro Evaluation. American Journal of
Advanced Drug Delivery. 2014; 2(1): 90-103.
15. Varma V, Sowmya C, Tabasum SG. Formulation and
Evaluation of Piroxicam Solid Dispersion with Suitable
Carrier. Research Journal of Pharmaceutical, Biological
and Chemical Sciences. 2012; 3(3): 929-940 .
16. Yadav PS, Kumar V, Singh UP, Bhat HR, Mazumder B.
Physicochemical characterization and in vitro dissolution
studies of solid dispersions of ketoprofen with PVP K30
and D-mannitol. Saudi Pharmaceutical Journal. 2013; 21:
77–84.
17. Chivate N, Chivate A, Shinde Sh, Saboji J. Improvement of
Bioavailability and Solubility of Telmisartan by Solid
Dispersion Technique using Various Carriers. International
Journal of Pharmaceutical Sciences Review and
Research.2013; 19(2): 36-41.
18. Wahab A, Khan A, Khan GM. Preparation and Evaluation
of Solid Dispersions of Ibuprofen Using Glucosamine HCl
as a Carrier. British Journal of Pharmaceutical Research.
2013; 3(4): 722-733.
19. KS, GR, Harikiran L, Krishna K Srinivas A. Dissolution
Enhancement Of A Poorly Water Soluble Drug Using
Water Soluble Carriers. Journal of Advanced
Pharmaceutical Sciences. 2011; 1(1): 42-46 .
20. Ibrahim MA, Shazly GA, El-Badry M. Albendazole
Microparticles Prepared by Spray Drying Technique:
Improvement of Drug Dissolution. Tropical Journal of
Pharmaceutical Research. 2014; 13 (12): 1963-1970.
21. Sarkar MdR, Monjur-Al-Hossain ASM, Sultana R, Faroque
ABM. Improvement Solubility Of Atorvastatin Calcium
Using Solid Dispersion Technique. International Journal of
Pharmaceutical Sciences and Research.2014; 5(12):
5405-5410.
22. Arun PK, Narayanan N, Rajalakshmi G. Preparation and
Evaluation of Solid Dispersion of Terbinafine
Hydrochloride. International Journal of Pharmaceutical
Sciences Review and Research. 2010; 3(1): 130-134.
23. Palanisamy M, Khanam J. Solid dispersion of
prednisolone: solid state characterization and
improvement of dissolution profile. Drug Development and
Industrial Pharmacy. 2011; 37(4): 373–386.
24. Weli AM, Saddar E, Hiremath JG, Balamurugan M.
Preparation and characterization of Benzathine Penicillin
G solid dispersions using different hydrophilic carriers.
International Journal of Drug Delivery. 2013; 5(4): 420-
429.
25. Rajbanshi K, Bajracharya R, Shrestha A, Thapa P.
Dissolution enhancement of aceclofenac tablet by solid
dispersion technique. International Journal of Pharma
Sciences and Research.2014; 5(4): 127-139.
26. Reddy V, Syed M, Rao DS. Formulation and Evaluation of
Colon Targeted Oral Drug Delivery System for Meloxicam.
Scholars Academic Journal of Pharmacy. 2015; 4(1): 1-9.
27. Disha SN, Raju S, Govind RS, Mithilesh, Dev JA. An
Investigation on Colon Drug Delivery System for
Satranidazole Tablet. International Journal of Research in
Pharmacy and Science. 2013; 3(2): 161-182.
28. Ramesh KA, Shabaraya AR, Mohd A, Kamath KK.
Formulation and evaluation of pulsatile drug delivery
system containing indomethacin using natural polymers.
International Research Journal of Pharmacy 2013, 4
(2):111-114.
29. Hrushikesh D, Hariprasanna RC, Upendra K. Design and
development of fast dissolving tablets containing
ziprasidone by solid dispersion method.Journal of
Pharmaceutical Science and Bioscientific Research. 2012;
2(1): 18-24.
30. Hashem M, Shaker DS, Nasr M, Saad IE, Ragaey R. Guar
gum and hydroxy propyl methylcellulose compressed
coated tablets for colonic drug delivery: in vitro and in vivo
evaluation in healthy human volunteers. Drug Discoveries
& Therapeutics. 2011; 5(2):90-95.
31. Mehta R, Chawla A, Sharma P, Pawar P. Formulation and
in vitro evaluation of Eudragit S‑100 coated naproxen
matrix tablets for colon targeted drug delivery system.
Journal of Advanced Pharmaceutical Technology &
Research. 2013; 4(1): 31-41.
32. Abdul Hadi M, Rao NGR, Rao AS. Formulation and
Evaluation of pH-Responsive Mini-Tablets for Ileo-Colonic
Page 12
Alkazzaz et al. Design and In-Vitro Evaluation of Colon Targeted Prednisolone
UK J Pharm & Biosci, 2015: 3(6); 41
Targeted Drug Delivery. Tropical Journal of
Pharmaceutical Research. 2014; 13 (7): 1021-1029
33. United states pharmacopea, electronic copy, 2007.
34. Girotra P, Singh SK. Formulation Optimization for Colon
Targeted Delivery of Katira Gum Matrix Tablets Containing
Azathioprine. International Journal of Pharmaceutical
Sciences and Drug Research. 2013; 5(4): 133-140.
35. Bhawna G, Lovekesh N, Shailendra KS. Formulation and
Gamma Scintigraphic Evaluation of Colon Targeted Drug
Delivery Systems Of Tinidazole In Healthy Human
Volunteers. Journal of Pharmaceutical and Biomedical
Sciences. 2011; 7(16): 1-9.
36. Chauhan CS, Naruka PS, Rathore RS, Badadwal V.
Formulation and evaluation of Prednisolone tablet for
colon targeted drug delivery system. Journal of Chemical
and Pharmaceutical Research. 2010; 2(4): 993-998.
37. Badhana S, Garud N, Akanksha G. Colon specific drug
delivery of mesalamine using eudragit S100-coated
chitosan microspheres for the treatment of ulcerative
colitis. International Current Pharmaceutical Journal. 2013;
2(3): 42-48.
38. Sambhakar S, Singh B, Madan K, Monalisha, Kashyap N,
Mayle S. Solid dispersions: A tool for improving the
solubility and dissolution of metronidazole. International
Journal of Drug Delivery.2013; 5(1):94-98.
39. Dua K, Pabreja K, Ramana MV. Preparation,
Characterization and In Vitro Evaluation of Aceclofenac
Solid Dispersions. Ars Pharmaceutica. 2010; 51 (1): 57-76.
40. Fouad EA, El-Badry M, Neau SH, Alanazi FK, Alsarra IA.
Technology evaluation: Kollicoat IR. Expert Opin. Drug
Deliv. 2011; 8(5): 693-703.
41. Sharma D, Soni M, Kumar S, Gupta GD. Solubility
enhancement-eminent role in poorly soluble drugs. Res J
Pharm and Tech. 2009; 2(2): 220-224.
42. Yasser S, Hussain SSN, Tayyab AM, Romana R, Aasma
S, Talib H, Madeeha M. Effects of drug-polymer
dispersions on solubility and in vitro diffusion of artemisinin
across a polydimethylsiloxane membrane. Chinece
science Bulletin. 2012; 57 (14): 1685-1692.
43. Sonali D, Tejal S, Vaishali T, Tejal G. Silymarin-solid
dispersions: Characterization and influence of preparation
methods on dissolution. Acta Pharm. 2010; 60: 427-443.
44. Ahuja N, Katare OP, Singh B. Studies on dissolution
enhancement and mathematical modeling of drug release
of a poorly water-soluble drug using water-soluble carriers.
Europ J Pharm and Biopharm. 2007; 65: 26-38.
45. Ibrahim MA. Tenoxicam-Kollicoat IR a Binary Systems:
Physicochemical and Biological Evaluation. Acta Poloniae
Pharmaceutica - Drug Research. 2014; 71 (4): 647-659.
46. Mukesh PR, Fattesingh US, Chaitali VM, Sham AP, Shilpa
PCh. Formulation And Development Of Enteric Coated
Ornidazole Tablet For Colon Targeted Drug Delivery
System. Journal of Drug Discovery and
Therapeutics.2013; 1 (9): 1-5.
47. Setty CM, Prasad DVK, Gupta VRM, Sa B. Development
of fast dispersible aceclofenac tablets: Effect of
functionality of superdisintegrants. Indian Journal of
pharmaceutical Sciences. 2008; 70(2): 180-185
48. Abha, Kaur LP. Superdisintegrants: An Arising Exemplar in
Orodispersible Tablets. International Journal of Drug
Research and Technology. 2015; 5 (1): 1-12.
49. Sarfaraz Md, Prasad Y, Reddy SR, Doddayya H, Udupi
RH. Development and evaluation of press coated time-
release Tablet of Nifedipine. Asian Journal of
Pharmaceutical Research. 2011; 1(3):58-63.