Genome wide classification and characterisation of CpG sites in cancer and normal cells. Mohammadmersad Ghorbani 1 , Michael Themis 2 and Annette Payne 1* 1 Department of Computer Science, 2 Department of Biosciences, Brunel University, Uxbridge, Middlesex, UB8 3PH, UK * To whom correspondence should be addressed. [email protected]Key words: motif, pattern identification, methylation in cancer, computational analysis, pattern searching algorithm, CpG, DNA sequence Abstract This study identifies common methylation patterns across different cancer types in an effort to identify common molecular events in diverse types of cancer cells and provides evidence for the sequence surrounding a CpG to influence its susceptibility to aberrant methylation. CpGs sites throughout the genome were divided into four classes: sites that either become hypo or hyper-methylated in a variety cancers using all the freely available microarray data (HypoCancer and HyperCancer classes) and those found in a constant hypo (Never methylated class) or hyper-methylated (Always methylated class) state in both normal and cancer cells. Our data shows that most CpG sites included in the HumanMethylation450K microarray remain unmethylated in normal and cancerous cells; however, certain sites in all the cancers investigated become specifically modified. More detailed analysis of the sites revealed that majority of those in the never methylated class were in CpG islands whereas those in the HyperCancer class
Transcript
Genome wide classification and characterisation of CpG sites in cancer and normal cells.
Mohammadmersad Ghorbani1, Michael Themis2 and Annette Payne1*
1 Department of Computer Science, 2 Department of Biosciences, Brunel University, Uxbridge, Middlesex, UB8 3PH, UK
to evaluate the potential of using these motifs for classification of four classes of
CpG sites. Using three different machine learning methods and 10 fold cross
validation CpG sites were classified according to their motifs. The input matrix was
the CpG sites with their corresponding class, and the features are motifs which
appear in the flanking DNA. Similar methods have been used in previous studies [82,
83]. J48, logistic and support vector machines were used as a classification tools for
this purpose.
Acknowledgements
We acknowledge the support in kind of Brunel University and staff in the
Departments of Computer Science and Biosciences.
Figure Legends
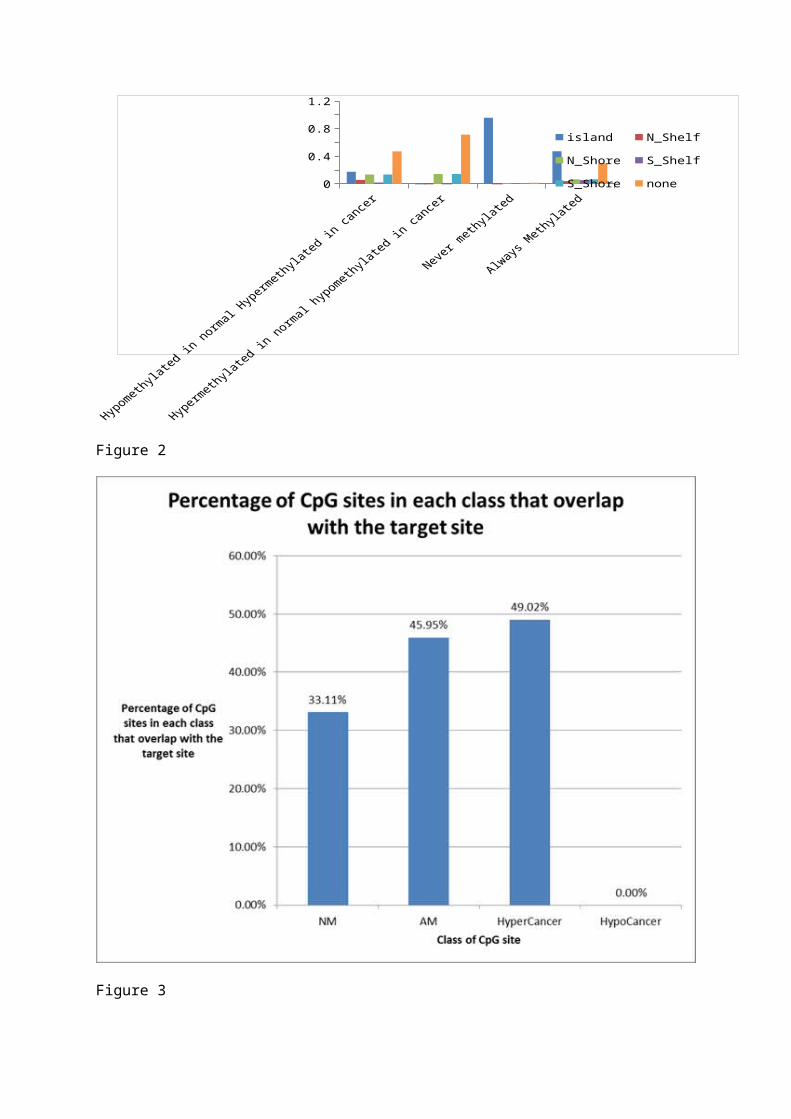
Figures 1 and 2 Graphs to show the number of CpG sites in each class and the positional relationships to CpG Islands. Figure 1 showing the number as a proportion of the total in each position relative to the CpG subdivided into classes. Figure 2 showing the number as a proportion of the total in each class, subdivided into positions relative to the CpG.
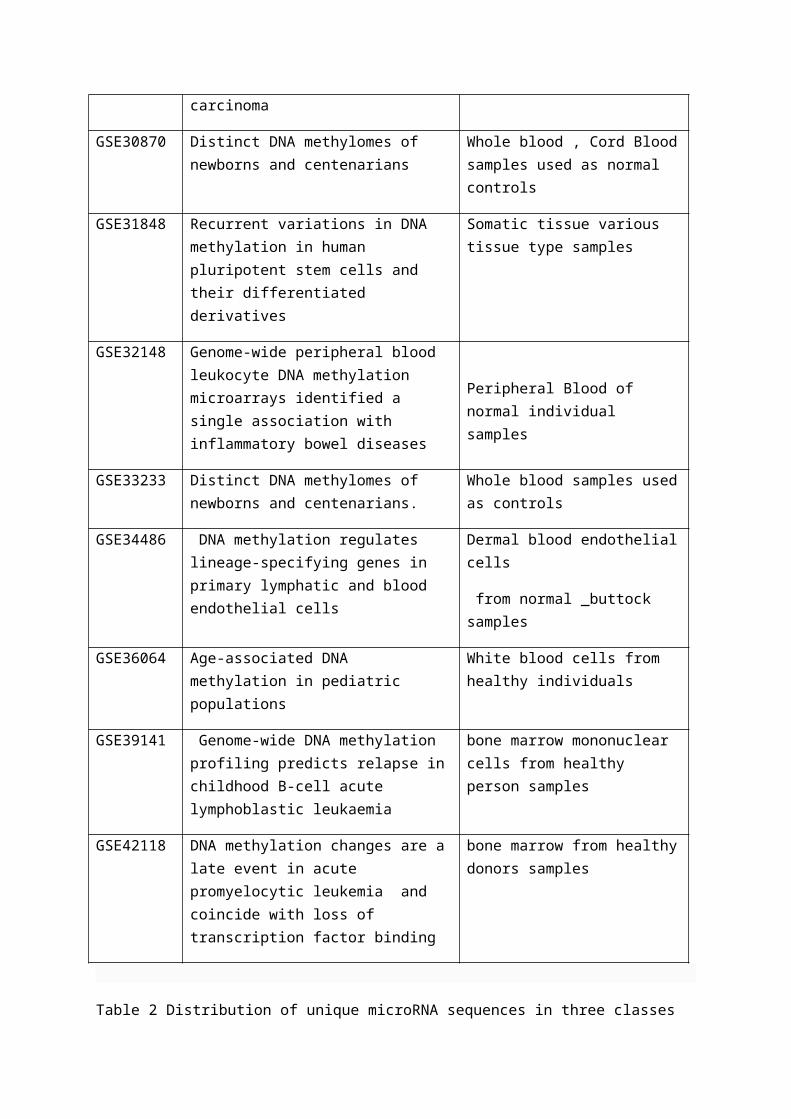
Figure 3 Graph to show the percentage of each class of CpG that are associated with a microRNA site.
Figure 4 Graph to show the number of times a particular miRNA species coding sequence occurs in the DNA sequence in the different classes of CpGs identified in this study.
Figure 5 Weka result for the most discriminative motif using the J48 algorithm to classify the CpGs into the 2 classes.
island N_Shelf N_Shore S_Shelf S_Shore0
0.0005
0.001
0.0015
0.002
0.0025
0.003
0.0035
0.004
0.0045
Hypomethylated in normal Hypermethylated in cancer
Hypermethylated in normal hypomethylated in cancer
Never methylated
Always Methylated
Figure 1
00.20.40.60.8
11.2
island N_Shelf N_Shore
S_Shelf S_Shore none
Figure 2
Figure 3
Figure 4
Figure 5
Tables
Table 1 Data series and the samples contained in them used in this study. All were obtained from GEO http://www.ncbi.nlm.nih.gov/gds/
Series Title of study Tissue and samples used
GSE20945 Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells
Primary leukaemia untreated samples
GSE29290 Evaluation of the Infinium Methylation 450K technology
Normal breast and breast cancer tumour samples
GSE30338 IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype
Glioma tumour samples
making sure only the tumour samples were included
GSE36278 Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma
Primary glioblastoma and non-neoplastic brain samples
Table 4 The genes neighbouring the CpGs found in Class HypoCancer along with their function as described by Cormine software http://www.coremine.com/medical/.
UCSC RefGene Name Function Cancer InvolvementRPTOR Androgen receptor activity,
HyperCancer normal_hypomethylated_cancer_hypermethylatedHypoCancer normal_hypermethyl_cancer_hypomethylAM always methylatedNM never methylated
References
1. Bird, A.P. and Wolffe,A.P. (1999) Methylation-induced repression— belts, braces, and chromatin. Cell, 99: 451-454.
2. Baylin S.B. (2005) DNA methylation and gene silencing in cancer Nature Clinical Practice Oncology http://lists.bilkent.edu.tr/~science/MBG523/Lectures/Epigenetics%20articles/DNA%20Methy.%20and%20Gene%20silenc.%20in%20cancer.pdf accessed 21/02/2014.
3. Heyn, H., Carmona, F.J., Gomez, A., Ferreira, H.J., Bell, J.T., Sayols, S., Ward, K., Stefansson, O.A., Moran, S., Sandoval, J., Eyfjord, J.E., Spector, T.D. And Esteller, M. (2013) DNA methylation profiling in breast cancer discordant identical twins identifies DOK7 as novel epigenetic biomarker. Carcinogenesis, 34(1): 102-108.
4. Fukushige S, Horii A. (2013) DNA methylation in cancer: a gene silencing mechanism and the clinical potential of its biomarkers. Tohoku J Exp Med., 229(3):173-85.
5. Klose R.J. and Bird A.P. (2006) Genomic DNA methylation: The mark and its mediators. Trends Biochem. Sci. 31: 89-97.
6. Das PM and Singal R. (2004) DNA methylation and cancer. Journal of Clinical Oncology, 22: 4632-4642
7. Taberlay PC, PA Jones. (2011) DNA methylation and cancer Epigenetics and Disease, - Springer. http://www.springer.com/cda/content/document/cda_downloaddocument/9783764389888-c1.pdf?SGWID=0-0-45-1004851-p174022756 (accessed 17/02/14)
8. Shames, D. S., Girard, L., Gao, B., Sato, M., Lewis, C. M., et al. (2006) A genome-wide screen for promoter methylation in lung cancer identifies novel methylation markers for multiple malignancies. PLoS Medicine, 3: e486
9. Michaelson-Cohen R, Keshet I, Straussman R, Hecht M, Cedar H, Beller U. (2011) Genome-wide de novo methylation in epithelial ovarian cancer. Int J Gynecol Cancer. 21(2): 269-79.
10.Gama-Sosa,M.A., Slagel,V.A., Trewyn,R.W., Oxenhandler,R.,Kuo,K.C., Gehrke,C.W. and Ehrlich,M. (1983) The 5-methylcytosinecontent of DNA from human tumors. Nucleic Acids Res., 11: 6883–6894.
11.Feinberg,A.P. and Vogelstein,B. (1983) Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature, 301: 89–92.
12.Feinberg, A.P., Gehrke,C.W., Kuo,K.C. and Ehrlich,M. (1988) Reducedgenomic 5-methylcytosine content in human colonic neoplasia. Cancer Res., 48: 1159–1161.
13.Cho,D.H., Thienes,C.P., Mahoney,S.E., Analau,E., Filippova,G.N. and Tapscott,S.J. (2005) Antisense transcription and heterochromatin at the DM1 CTG repeats are constrained by CTCF. Mol. Cell, 20: 483-489.
14.McKinnell,I.W., Ishibashi,J., Le Grand,F., Punch,V.G., Addicks,G.C., Greenblatt,J.F., Dilworth,F.J. and Rudnicki,M.A. (2008) Pax7 activates myogenic genes by recruitment of a histone methyltransferase complex. Nat. Cell Biol., 10: 77-84.
15.De Biase,I., Chutake,Y.K., Rindler,P.M. and Bidichandani,S.I. (2009) Epigenetic silencing in friedreich ataxia is associated with depletion of CTCF (CCCTC-binding factor) and antisense transcription. PLoS One, 4: e7914.
16.Gebhard C, Benner C, Ehrich M, Schwarzfische L (2010) General transcription factor binding at CpG islands in normal cells correlates with resistance to de novo DNA methylation in cancer cells. Cancer Res; 70(4): 1398–407.
17.An Integrated Encyclopedia of DNA Elements in the Human Genome The ENCODE Project Consortium. (2012) Nature doi: 10.1038/nature11247
18.Bock,C. and Lengauer,T. (2008) Computational epigenetics. Bioinformatics, 24: 1-10.
19.Yamada,Y. and Satou,K. (2008) Prediction of genomic methylation status on CpG islands using DNA sequence features. WSEAS Transactions on Biology and Biomedicine, 5: 153-162.
20.Ali,I. and Seker,H. (2010) A comparative study for characterisation and prediction of tissue-specific DNA methylation of CpG islands in chromosomes 6, 20 and 22. Conf. Proc. IEEE Eng. Med. Biol. Soc., 1832-1835.
21.Previti,C., Harari,O., Zwir,I. and del Val,C. (2009) Profile analysis and prediction of tissue-specific CpG island methylation classes. BMC Bioinformatics, 10: 116.
22.Glass JL1, Fazzari MJ, Ferguson-Smith AC, Greally JM. (2009) CG dinucleotide periodicities recognized by the Dnmt3a-Dnmt3L complex are distinctive at retroelements and imprinted domains. Mamm Genome. 20(9-10): 633-43.
24.Filippova,G.N., Thienes,C.P., Penn,B.H., Cho,D.H., Hu,Y.J., Moore,J.M., Klesert,T.R., Lobanenkov,V.V. and Tapscott,S.J. (2001) CTCF-binding sites flank CTG/CAG repeats and form a methylation-sensitive insulator at the DM1 locus. Nat. Genet., 28: 335-343.
25.Lienert F, Wirbelauer C, Som I, Dean A, Mohn F, Schübeler D. (2011) Identification of genetic elements that autonomously determine DNA methylation states. Nat Genet. 43(11):1091-7
27.Ooi SK, Qiu C, Bernstein E, Li K, Jia D, Yang Z, Erdjument-Bromage H, Tempst P, Lin SP, Allis CD, Cheng X, Bestor TH.: (2007) DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 448(7154):714-7.
28.Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Schöler A, van Nimwegen E, Wirbelauer C, Oakeley EJ, Gaidatzis D, Tiwari VK, Schübeler D. (2011) DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 480(7378):490-5.
29.Weber M, Hellmann I, Stadler MB, Ramos L, Pääbo S, Rebhan M, Schübeler D. (2007) Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. 39(4):457-66.
30.Yuan,G. (2011) Prediction of epigenetic target sites by using genomic DNA sequence. In Anonymous Handbook of Research on Computational and Systems Biology: Interdisciplinary Applications. IGI Global, pp. 187-201.
31.Feltus, F.A., Lee, E.K., Costello, J.F., Plass, C. And Vertino, P.M. (2006) DNA motifs associated with aberrant CpG island methylation. Genomics, 87(5): 572-579.
32.Lu,L., Lin,K., Qian,Z., Li,H., Cai,Y. and Li,Y. (2010) Predicting DNA methylation status using word composition. Journal of Biomedical Science and Engineering, 3: 672-676.
33.Ghorbani M, Taylor SJ, Pook MA, Payne A. (2013) Comparative (computational) analysis of the DNA methylation status of trinucleotide repeat expansion diseases. J Nucleic Acids.; 689798.
34.McCabe,M.T., Lee,E.K. and Vertino,P.M. (2009) A multifactorial signature of DNA sequence and polycomb binding predicts aberrant CpG island methylation. Cancer Res., 69: 282-291.
35.Hervouet,E., Vallette,F.M. and Cartron,P.F. (2009) Dnmt3/transcription factor interactions as crucial players in targeted DNA methylation. Epigenetics, 4: 487-499.
36.Leung DC, Dong KB, Maksakova IA, Goyal P, Appanah R, Lee S, Tachibana M, Shinkai Y, Lehnertz B, Mager DL, Rossi F, Lorincz MC. (2011) Lysine methyltransferase G9a is required for de novo DNA methylation and the establishment, but not the maintenance, of proviral silencing. Proc Natl Acad Sci U S A. 108(14):5718-23.
37.Ooi SK1, Wolf D, Hartung O, Agarwal S, Daley GQ, Goff SP, Bestor TH. (2010) Dynamic instability of genomic methylation patterns in pluripotent stem cells. Epigenetics Chromatin. 3(1):17.
38.Rowe HM, Friedli M, Offner S, Verp S, Mesnard D, Marquis J, Aktas T, Trono D. (2013) De novo DNA methylation of endogenous retroviruses is shaped by KRAB-ZFPs/KAP1 and ESET. Development. 140(3):519-29.
39. Iorio,M.V., Piovan,C. and Croce,C.M. (2010) Interplay between microRNAs and the epigenetic machinery: An intricate network. Biochimica Et Biophysica Acta (BBA) - Gene Regulatory Mechanisms, 1799, 694-701.
40.Vrba L, Muñoz-Rodríguez JL, Stampfer MR, Futscher BW. (2013) miRNA Gene Promoters Are Frequent Targets of Aberrant DNA Methylation in Human Breast Cancer. PLoS ONE 8(1): e54398.
41.Huang DW, Sherman BT, Lempicki RA. (2009a) Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nature Protoc.; 4(1):44-57.
42.Huang DW, Sherman BT, Lempicki RA. (2009b) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res.; 37(1):1-13.
43.Ho-Sui SJ, Mortimer J, Arenillas DJ, Brumm J, Walsh CJ, Kennedy BP and Wasserman WW. (2005) oPOSSUM: Identification of over-represented transcription factor binding sites in co-expressed genes. Nucleic Acids Res. 33(10):3154-64.
44. Inoue M1, Takahashi K, Niide O, Shibata M, Fukuzawa M, Ra C. (2005) LDOC1, a novel MZF-1-interacting protein, induces apoptosis. FEBS Lett. 579(3):604-8.
45.Hsieh YH, Wu TT, Huang CY, Hsieh YS, Liu JY. (2007) Suppression of tumorigenicity of human hepatocellular carcinoma cells by antisense
oligonucleotide MZF-1. Chin J Physiol. 50(1):9-1546.Rowland B D., Bernards R and Peeper D S. (2005) The KLF4 tumour
suppressor is a transcriptional repressor of p53 that acts as a context-dependent oncogene Nature Cell Biology 7: 1074 - 1082
47.Weisenberger D J., Campan M, Long T I., Kim M, Woods C. (2005) Analysis of repetitive element DNA methylation by MethyLight. Nucleic Acids Res. 33(21): 6823–6836
48.Walters RJ, Williamson EJ, English DR, Young JP, Rosty C, Clendenning M, Walsh MD, Parry S, Ahnen DJ, Baron JA, Win AK, Giles GG, Hopper JL, Jenkins MA, Buchanan DD. (2013) Association between hypermethylation of DNA repetitive elements in white blood cell DNA and early-onset colorectal cancer. Epigenetics. 8(7):748-55.
49.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. (2002) UCSC Genome Browser: The human genome browser at UCSC. Genome Res. 6:996-1006.
50.Bailey T. L. (2006) MEME:discovering and analysing DNA and protein sequence motifs. Nucleic Acids Res. 34
51.Lin R, Li X, Li J, Zhang L, Xu F, Chu Y, Li J. (2013) Long-term cisplatin exposure promotes methylation of the OCT1 gene in human esophageal cancer cells. Dig Dis Sci. 58(3):694-8.
52.Wang Z, Zhu S, Shen M, Liu J, Wang M, Li C, Wang Y, Deng A, Mei Q. (2013) STAT3 is involved in esophageal carcinogenesis through regulation of Oct-1. Carcinogenesis. 34(3):678-88.
53.Croce C.M. (2009) Causes and consequences of microRNA dysregulation in cancer Nat. Rev. Genet., 10: 704–714
54.Esquela-Kerscher, F.J. Slack. (2006) Oncomirs - microRNAs with a role in cancer. Nat. Rev. Cancer, 6: 259–269
55.Esteller M. (2011) Non-coding RNAs in human disease Nat. Rev. Genet.,.12: 861–874
56.Suzuki H, Maruyama R, Yamamoto E, Kai M. (2012) DNA methylation and microRNA dysregulation in cancer Molecular Oncology 6: 567–578
57.Chalitchagorn K, Shuangshoti S, Hourpai N, Kongruttanachok N, Tangkijvanich P, Thong-ngam D, et al. (2004) Distinctive pattern of LINE-1 methylation level in normal tissues and the association with carcinogenesis. Oncogene.; 23:8841-6.
58.Schulz WA. (2006) L1 retrotransposons in human cancers. J Biomed Biotechnol.:83672.
59.Estecio MR, Gharibyan V, Shen L, Ibrahim AE, Doshi K, He R, et al. (2007) LINE-1 hypomethylation in cancer is highly variable and inversely correlated with microsatellite instability. PLoS One.;2:e399.
60.Hsiung DT, Marsit CJ, Houseman EA, Eddy K, Furniss CS, McClean MD, et al. (2007) Global DNA methylation level in whole blood as a biomarker in head and neck squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev.; 16:108-14.
61.Cho NY, Kim BH, Choi M, Yoo EJ, Moon KC, Cho YM, et al. (2007) Hypermethylation of CpG island loci and hypomethylation of LINE-1 and Alu repeats in prostate adenocarcinoma and their relationship to clinicopathological features. J Pathol. 211:269-77.
62.Matsuzaki K, Deng G, Tanaka H, Kakar S, Miura S, Kim YS. (2005) The relationship between global methylation level, loss of heterozygosity, and microsatellite instability in sporadic colorectal cancer. Clin Cancer Res.; 11:8564-9.
63.Perrin D1, Ballestar E, Fraga MF, Frappart L, Esteller M, Guerin JF, Dante R. (2007) Specific hypermethylation of LINE-1 elements during abnormal overgrowth and differentiation of human placenta. Oncogene. 26(17):2518-24.
64.Pattamadilok J, Huapai N, Rattanatanyong P, Vasurattana A, Perrin D, Ballestar E, Fraga MF, Frappart L, Esteller M, Guerin JF, et al.: (2007) Specific hypermethylation of LINE-1 elements during abnormal overgrowth and differentiation of human placenta. Oncogene. 26:2518-24.
65.Pattamadilok J, Huapai N, Rattanatanyong P, Vasurattana A, Triratanachat S, Tresukosol D, et al. (2008) LINE-1 hypomethylation level as a potential prognostic factor for epithelial ovarian cancer. Int J Gynecol Cancer 18:711–7.
66.Moore LE, Pfeiffer RM, Poscablo C, Real FX, Kogevinas M, Silverman D, et al. (2008) Genomic DNA hypomethylation as a biomarker for bladder cancer susceptibility in the Spanish Bladder Cancer Study: a case-control study. Lancet Oncol. 9:359-66.
67.Smith IM, Mydlarz WK, Mithani SK, Califano JA. (2007) DNA global hypomethylation in squamous cell head and neck cancer associated with smoking, alcohol consumption and stage. Int J Cancer. 121:1724-8.
68.Subbalekha K, Pimkhaokham A, Pavasant P, Chindavijak S, Phokaew C, Shuangshoti S, et al. (2009) Detection of LINE-1s hypomethylation in oral rinses of oral squamous cell carcinoma patients. Oral Oncol. 45:184-91.
69.Karouzakis E, Gay RE, Michel BA, Gay S, Neidhart M. (2009) DNA hypomethylation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 60:3613-22.
70.Choi IS, Estecio MR, Nagano Y, Kim do H, White JA, Yao JC, et al. (2007) Hypomethylation of LINE-1 and Alu in well-differentiated neuroendocrine tumors (pancreatic endocrine tumors and carcinoid tumors). Mod Pathol. 20:802-10.
71.Roman-Gomez J, Jimenez-Velasco A, Agirre X, Castillejo JA, Navarro G, San Jose-Eneriz E, et al. (2008) Repetitive DNA hypomethylation in the advanced phase of chronic myeloid leukemia. Leuk Res. 32:487-90.
72.Lee HS, Kim BH, Cho NY, Yoo EJ, Choi M, Shin SH, et al. (2009) Prognostic implications of and relationship between CpG island hypermethylation
and repetitive DNA hypomethylation in hepatocellular carcinoma. Clin Cancer Res. 15:812-20
73.Fiala E, Ehrlich M and. Laird P W: (2005) Association between hypermethylation of DNA repetitive elements in white blood cell DNA and early-onset colorectal cancer. Nucleic Acids Research, 33, 21: 6823–6836
74.Weisenberger, D.J., Velicescu, M., Cheng, J.C., Gonzales, F.A., Liang, G., Jones, P.A. (2004) Role of the DNA methyltransferase variant DNMT3b3 in DNA methylation Mol. Cancer Res. 262–72
75.Magewu, A.N. and Jones, P.A. (1994) Ubiquitous and tenacious methylation of the CpG site in codon 248 of the p53 gene may explain its frequent appearance as a mutational hot spot in human cancer Mol. Cell. Biol. 14: 4225–4232
76. Zhipeng Wang,Shaojun Zhu,Min Shen,Juanjuan Liu, Meng Wang, Chen Li, Yukun Wang, Anmei Deng and Qibing Mei (2013) STAT3 is involved in esophageal carcinogenesis through regulation of Oct-1 Carcinogenesis 34 (3): 678-688.
77.Liao D (2009) Emerging roles of the EBF family of transcription factors in tumor suppression. Mol Cancer Res. 7(12):1893-901
78.Chen F, Song J, Di J, Zhang Q, Tian H, Zheng J. (2012) IRF1 suppresses Ki-67 promoter activity through interfering with Sp1 activation. Tumour Biol. 33(6):2217-25
79.Thompson VC. Day TK, Bianco-Miotto T, Selth LA, Han G, Thomas M, Buchanan G, Scher HI, Nelson CC; Australian Prostate Cancer BioResource, Greenberg NM, Butler LM, Tilley WD. (2012) A gene signature identified using a mouse model of androgen receptor-dependent prostate cancer predicts biochemical relapse in human disease. Int J Cancer. 131(3):662-72
80.Deutsch AJ., Angerer H, Fuchs TE, Neumeister P. (2012) The Nuclear Orphan Receptors NR4A as Therapeutic Target in Cancer Therapy. Anticancer Agents Med Chem. 12(9):1001-14
81.Du P, Zhang X, Huang C-C, Jafari N, Kibbe W A, Hou L, and Lin S M (2010) Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 11: 587.
82.Bock, C., Walter, J., Paulsen, M. and Lengauer, T. (2007) CpG Island Mapping by Epigenome Prediction, PLoS Comput Biol vol. 3, no. 6, pp. E110
83.Wrzodek, C., Büchel, F., Hinselmann, G., Eichner, J., Mittag, F. and Zell, A. (2012) Linking the Epigenome to the Genome: Correlation of Different Features to DNA Methylation of CpG Islands, PLoS ONE , vol. 7, no. 4, pp. e35327