Retinoblastoma Helen Dimaras 1 , Timothy W. Corson 2 , David Cobrinik 3 , Abby White 4 , Junyang Zhao 5 , Francis L.Munier 6 , David Abramson 7 , Carol Shields 8 , Guillermo Chantada 9 , Festus Njuguna 10 and Brenda L. Gallie 11 [Au: Please ensure all names are as you would like them fo 1| Department of Ophthalmology & Vision Sciences, The Hospital for Sick Children& University of Toronto, Toronto, Canada 2| Eugene and Marilyn Glick Eye Institute, Departments of Ophthalmology, Biochemistry and Molecular Biology, and Pharmacology and Toxicology, and Simon Cancer Center,Indiana University, Indianapolis, USA 3| The Vision Center and The Saban Research Institute Children's Hospital Los Angeles, Los Angeles, CA USA; and Department of Ophthalmology, Department of Biochemistry and Molecular Biology, The USC Eye Institute, and Norris Comprehensive Cancer Center, Keck School of Medicine of the University of Southern California, Los Angeles, CA USA 4| Daisy's Eye Cancer Fund, Oxford, UK 5| Beijing Tongren Eye Centre, Beijing, China Retinoblastoma Helen Dimaras 1 , Timothy W. Corson 2 , David C obrinik 3 , A bby W hite 4 , J unyang Zhao 5 , F rancis L.Munier 6 , David A bramson 7 , C arol S hields 8 , Guillermo C hantada 9 , F estus Njuguna 10 and Brenda L. Gallie 11 [A u:Please ensure allnam es are as you w ould like them forindexing (e.g.in PubM ed).] 1| Departm entofO phthalm ology & V ision Sciences, The H ospitalforSick Children& U niversity ofToronto, Toronto, Canada 2| Eugene and M arilyn G lick Eye Institute, D epartm entsofO phthalm ology, Biochem istry and M olecular Biology, and Pharm acology and Toxicology, and Sim on CancerCenter,IndianaU niversity, Indianapolis, U SA 3| The V ision Centerand The Saban Research Institute Children'sH ospitalLosA ngeles, LosA ngeles, CA U SA ;and D epartm entofO phthalm ology, Departm entofB iochem istry and M olecularBiology, The U SC Eye Institute, and N orrisCom prehensive CancerCenter, K eck SchoolofM edicineofthe U niversity ofSouthern California, LosA ngeles,CA U SA 4| D aisy'sEye CancerFund, O xford, U K 5| Beijing Tongren Eye Centre,Beijing, China 6| Jules-G onin EyeH ospital, Lausanne, Switzerland 7| M em orialSloan K ettering CancerCenter, N ew Y ork,U SA 8| W illsEye H ospital, Philadelphia, U SA 9| H ospitalJP G arrahan, BuenosA ires,A rgentina 10| M oiTeaching & ReferralH ospital, Eldoret, K enya 11| Departm entofO phthalm ology & V ision Sciences,The H ospitalforSick Children& U niversity ofToronto, 555 U niversity A ve, Toronto, O ntario M 5G 1X 8, Canada [A u:Please check and update affiliations. Every affiliation should contain the D epartm ent(w hich needs to be updated for allauthors), Institution, C ity, State and C ountry.O nly the corresponding author hasto supply the fullpostaladdress.]
Transcript
RetinoblastomaHelen Dimaras1, Timothy W. Corson2, David Cobrinik3, Abby White4, Junyang Zhao5, Francis L.Munier6, David
Abramson7, Carol Shields8, Guillermo Chantada9, Festus Njuguna10 and Brenda L. Gallie11[Au: Please ensure all names are as you would like them fo
1| Department of Ophthalmology & Vision Sciences, The Hospital for Sick Children& University of Toronto, Toronto, Canada2| Eugene and Marilyn Glick Eye Institute, Departments of Ophthalmology, Biochemistry and Molecular Biology, and Pharmacology and Toxicology, and Simon Cancer Center,Indiana University, Indianapolis, USA3| The Vision Center and The Saban Research Institute Children's Hospital Los Angeles, Los Angeles, CA USA; and Department of Ophthalmology, Department of Biochemistry and Molecular Biology, The USC Eye Institute, and Norris Comprehensive Cancer Center, Keck School of Medicine of the University of Southern California, Los Angeles, CA USA4| Daisy's Eye Cancer Fund, Oxford, UK5| Beijing Tongren Eye Centre, Beijing, China6| Jules-Gonin Eye Hospital, Lausanne, Switzerland7| Memorial Sloan Kettering Cancer Center, New York, USA8| Wills Eye Hospital, Philadelphia, USA9| Hospital JP Garrahan, Buenos Aires, Argentina10| Moi Teaching & Referral Hospital, Eldoret, Kenya
Retinoblastoma
Helen Dimaras1, Timothy W. Corson2, David Cobrinik3, Abby White4, Junyang Zhao5, Francis L.Munier6, David
Abramson7, Carol Shields8, Guillermo Chantada9, Festus Njuguna10 and Brenda L. Gallie11[Au: Please ensure all names are as you would like them for indexing (e.g. in PubMed).]
1| Department of Ophthalmology & Vision Sciences, The Hospital for Sick Children& University of Toronto, Toronto, Canada 2| Eugene and Marilyn Glick Eye Institute, Departments of Ophthalmology, Biochemistry and Molecular Biology, and Pharmacology and Toxicology, and Simon Cancer Center,Indiana University, Indianapolis, USA 3| The Vision Center and The Saban Research Institute Children's Hospital Los Angeles, Los Angeles, CA USA; and Department of Ophthalmology, Department of Biochemistry and Molecular Biology, The USC Eye Institute, and Norris Comprehensive Cancer Center, Keck School of Medicine of the University of Southern California, Los Angeles, CA USA 4| Daisy's Eye Cancer Fund, Oxford, UK 5| Beijing Tongren Eye Centre, Beijing, China 6| Jules-Gonin Eye Hospital, Lausanne, Switzerland 7| Memorial Sloan Kettering Cancer Center, New York, USA 8| Wills Eye Hospital, Philadelphia, USA 9| Hospital JP Garrahan, Buenos Aires, Argentina 10| Moi Teaching & Referral Hospital, Eldoret, Kenya 11| Department of Ophthalmology & Vision Sciences, The Hospital for Sick Children& University of Toronto, 555 University Ave, Toronto, Ontario M5G1X8, Canada [Au: Please check and update affiliations. Every affiliation should contain the Department (which needs to be updated for all authors), Institution, City, State and Country. Only the corresponding author has to supply the full postal address.]
11| Department of Ophthalmology & Vision Sciences, The Hospital for Sick Children& University of Toronto, 555 University Ave, Toronto, Ontario M5G1X8, Canada[Au: Please check and update affiliations. Every affiliation should contain the Department (which needs to be updated for all authors), Institution, City, State and Country. Only the corresponding author has to supply the full postal address.]
Abstract 173/200
[Au: Abstracts have no line/paragraph breaks]
The genetic basis of human malignancy was determined through the study of retinoblastoma, a rare cancer of
infant retina. Tumours form when both RB1 alleles mutate in a susceptible retinal cell, likely a cone
photoreceptor precursor. The tumour suppressor functions of the retinoblastoma protein, pRB, relate to cell
division and genomic stability, but the key biochemical and molecular basis of tissue specificity remain
unknown. Retinoblastoma is diagnosed in 8,000 children each year worldwide, yet patient survival is >95% in
high-income countries, but <30% globally, depending on the socio-economical context. Stakeholder
collaboration is improving outcomes by increasing awareness for earlier diagnosis, sharing expertise, and
developing guidelines. Intra-arterial and intra-vitreal chemotherapy have emerged as a promising way salvage
eyes. Ongoing international collaboration will replace the multiple different classifications of eye involvement
with standardized definitions, that will facilitate assessment of eligibility, efficacy and safety of treatments.
Heritable retinoblastoma survivors are at risk for second cancers; defining the molecular basis of RB1 retinal
specificity may explain tissue specificity of second cancers, opening the path to cancer prevention.
arterial chemotherapy, Intra-vitreal chemotherapy, Cone photoreceptor, Cancer therapy, Global health
Competing interests
[Au: Please insert the competing interest statement for each author here, or in the online submission form
with your revised manuscript.]
[Au: Pleasemakesureofficialnomenclatureisusedforprotein(Uniprot)andgene(Hugoanditalics)namesandthattheyarestyledaccordingtospecies(capitalsforhumans,onlyfirstlettercapitalformice). Also make sure that abbreviations are consistently used throughout the manuscript (N-Myc, MYCN, etc.)]
[Au: I’ve inserted HX notations at all the headings to indicate the level of heading, three of which are
allowed.]
[H1] Introduction(BLG)319/300
Retinoblastoma is a rare cancer initiated by biallelic mutation of the retinoblastoma gene (RB1) in a single
susceptible developing retinal cell. Inheritance of one mutant RB1 allele strongly predisposes to
retinoblastomatumours that form when the second RB1 allele is mutated.1 Although RB1 loss initiates cancer in
Author, 01/03/-1,
Au: we’d refer to the protein in general as RB, not its phosphorylated form ok?I NO THE COMMON USE FOR PROTIN IS pRB, as with p53 etc.
Author, 01/03/-1,
[Au: by ‘specific’ do you mean a single cell (cell of origin)?] Yes, that is what is meant.
Author, 01/03/-1,
Au: I’d suggest moving these statements out of the abstract, which should focus on what is covered in the Primer.But the primer does reach forward to the future: that is a very important function…ALSO WE CAN ADD MORE OF WHAT IS IN THE WHOLE PAPER
Author, 01/03/-1,
HD: Overall survival
Author, 01/03/-1,
HD: pRB = RB protein, not phosphorylated RB.
specific susceptible tissues, it is lost with progression in almost all human cancers.2 The principals discovered by
study of retinoblastoma led to the recognition that altered genes broadly underlie cancer initiation and
progression.
Retinoblastoma starts when the second RB1 allele is damaged in susceptible retinal cell (RB1-/-) that undergoes
limited proliferation to form a non-malignant retinoma (Figure 1).3 An intraretinal white tumour develops after
genomic changes lead to uncontrolled proliferation — the tumour is visible through the pupil (the most common
first sign) (Figures 3, 9) or blocks vision and impedes central visual fixation (the second most common sign).4
When noticed early, prompt treatment usually cures. Later diagnosis can lead to incurable invasion of the optic
nerve and brain or metastasis, sometimes cured by extensive intervention.
Rigorous clinical trials in retinoblastoma are difficult for multiple reasons: too few patients in high-income
countries; often complex presentation (two eyes withdiffering severity); too few patients to interest the
pharmaceutical industry; multidisciplinary collaboration required; and high societal value on eyes and vision
somtimes imposes considerations beyond curing the cancer. New technologies showing dramatic primary
response in the intraocular tumour have been quickly embraced, despite absence of rigorous clinical trials.
The Internet has opened many avenues for retinoblastoma: parents make the diagnosis themselves; colleagues
discuss and share patients around the globe; centres of retinoblastoma excellence can be mapped (Figure 2); and
a common database for all children no matter where they live is within reach. These developments could
empower a learning health system to achieve an evidence base for retinoblastoma care. In this Primer, we
review retinoblastoma at a time when new science, new ideas, new therapies and global collaboration are
unprecedented.
Author, 01/03/-1,
To avoid the unintended pun
Author, 01/03/-1,
[Au: please cite a reference(s). Also, if biallelic mutation is needed, that inheritance of ‘a’ (i.e., single) mutant allele also predisposes is unclear] THE BILLELIC LOSS IS ONLY IN THE TUMOUR; THE PREDISPOSITION IS ONLY ONE ALLELE.
[H1] Epidemiology (HD,GC, JZ, FN) 762/500
[H2] Globalpatients, resources and outcomes[Au: edited to fit within a 42 character limit (which includes white spaces)]
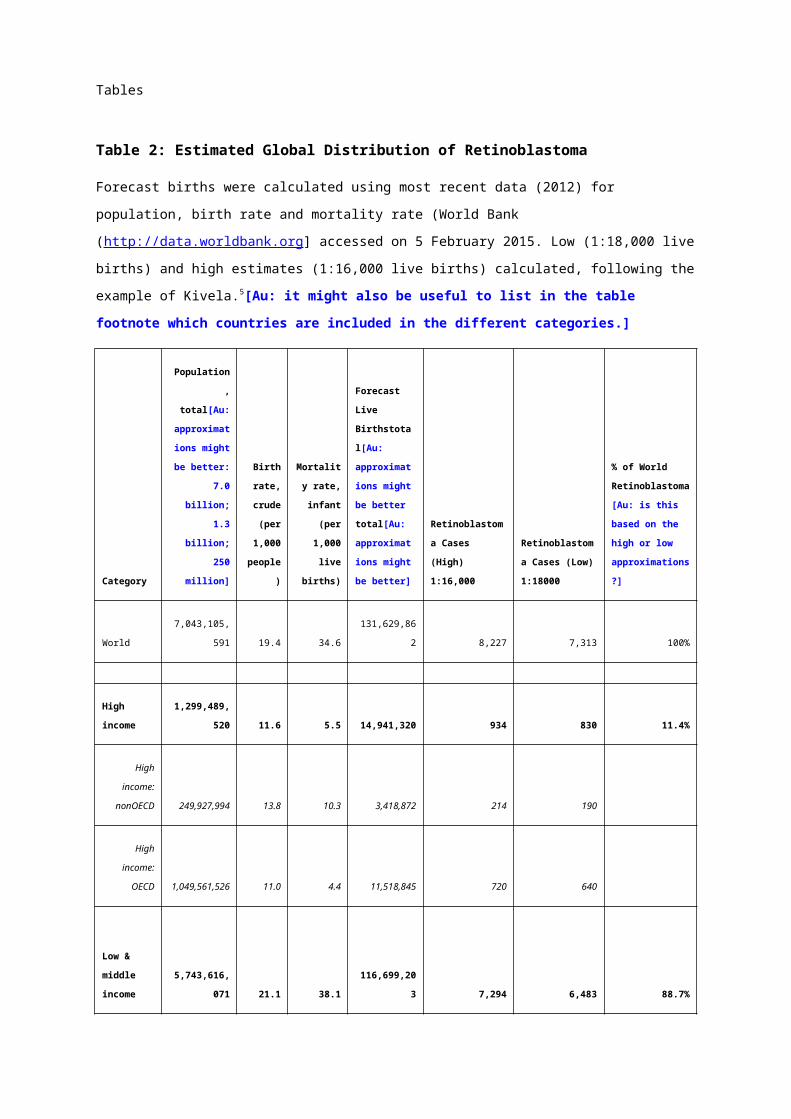
The expected number of patients with retinoblastoma annually per country can be calculated by multiplying the
global retinoblastoma incidence (1 in 16,000-18,000 live births) by forecast births (Table 2).5-7 This predicts
approximately 8,000 new cases each year.
Of all affected children, 11% reside in high-income, 69% in middle-income and 20% in low-income countries.
Although the prevalence is higher in middle- to low-income countries, most retinoblastoma treatment centres are
in middle- and high-income countries, creating a gap in healthcare access (Figure 2). Consistent with income
being a surrogate for non-economic measures of standard of living, retinoblastoma in low-income countries is
associated with low patient survival (~30%8, 9) compared with high-income countries (>95%10), but
comprehensive nation-wide data is lacking.
Poor outcome correlates with late presentation, difficulty accessing retinoblastoma-specific health care, and
socio-economic issues leading to poor compliance, including family decline to remove the affected eye and
abandonment of therapy.11-13 Without timely diagnosis and appropriate treatment, difficult-to-cure metastatic
disease may occur. Fortunately, with early diagnosis, many eyes can be safely treated to support a lifetime of
good vision, pointing to key elements for global focus: awareness, collaboration, and affordable expert care.
[H2] Solutions for global retinoblastoma
A number of initiatives address the inequality in retinoblastoma treatment between developing and developed
worlds. In 2009, the Canadian National Rb Strategy (NRbS) published the first-ever retinoblastoma clinical
practice guidelines,10 adapted by the Kenyan National Retinoblastoma Strategy (KNRbS) and published in 2014
in partnership with the Kenyan Department of Health.14 The KNRbS achieved standardization of pathology
processing and reporting to support treatment decisions and discussion of prognosis with families. Adoption of
upfront (first-line) enucleation with implants and immediate prosthetic eyes (sourced from India), parent to
parent interactions to allay uninformed fears, and standardization of information provided to parents has reduced
the rate of non-compliance with treatment.14, 15 In 2013, the Paediatric Oncology in Developing Countries
Committee of the SIOP (International Society of Paediatric Oncology) published a consensus guideline for the
management of retinoblastoma in countries with limited resources16 with clear ideas that can shape resource
development. The Mexican National Strategy17 and the Brazilian SOBOPE (Brazilian Society of Pediatric
Oncology)18 guidelines are also applicable at a national level with governmental support for treatment.
Peer-to-peer collaborations and twinning programmes have built a framework for knowledge and expertise
exchange, filling gaps in specialized training, and source donations of equipment and resources with the ultimate
aim of sustainable local capacity.19 Retinoblastoma-specific twinning programmes include partnerships between
St. Jude’s Children’s Research Hospital (USA) and the Middle-East,20 Central America21 and Mexico,17and
between the Institut Curie and a centre in Bamako, Mali.22 The Central American Association of Pediatric
Author, 03/01/-1,
: Sentences (green) moved up to minimize overlap okay
Author, 03/01/-1,
Au: Is this a worldwide guideline, or focused on the developing countries?HD: Each guideline is specific to the country of origin (Canada, Kenya) but can be adopted by other groups/nations.
Author, 03/01/-1,
Au: introductory sentence added; OK?]okay
Author, 03/01/-1,
Au: lack of family support?]Family dissolution (e.g. divorce resulting from/influenced by stress of therapy, eye removal, stigma of cancer, blame).
Author, 03/01/-1,
Au: Since this statement is referenced, it is not clear for which statement solid data are lacking.Clarified in the text
Author, 03/01/-1,
Au: survival of the eye or the patient?
Author, 03/01/-1,
Au: standard of livingHD: okay
Author, 03/01/-1,
Au: isn’t this economical?]HD: economic. (economic = good value for money; we mean a measure relating to economics
Author, 03/01/-1,
Au: Callout changed to figure 2; OK?HD: okay
Author, 03/01/-1,
Au: I think the data in the extended table is beyond the scope of the Primer. Technically, the information in Table 1 is a new analysis and not something we would normally consider in a review article, but it is based on published data so I will leave it in. Some comments on the table are below.]HD: okay
Hematology Oncology (AHOPCA) created a cooperative group and implemented multicentre protocols for
retinoblastoma treatment,21, 23, 24 a major achievement, not yet paralleled in developed countries. The AHOPCA
funding is now 90% local. Twinning programme sbenefit from strong participation of both non-governmental
and governmental organizations. However, where government may prove volatile and unpredictable, sustainable
local capacity is a challenge. Less formal cooperation between developed and less developed countries can also
result in highly efficient programmes.25, 26
A successful national strategy has been developed in China. About 1,100 newly diagnosed cases are forecast
annually, scattered over 32 provinces,I mposing high travel costs. Before 2005, enucleation was the only
available treatment for most children. For better treatment options and follow-up, centres classified by expertise
and resources were established in 28 hospitals covering 25 provinces (over 90% of the population)27. The
improved efficiency and collaboration from 2006 to 2014 has led to standardized classification and treatment of
2,097 newly diagnosed patients with retinoblastoma on common protocols with gains in survival (unpublished
data). In Argentina, a single centre with high expertise coordinates affiliated retinoblastoma clinics. This
collaboration between governmental hospitals, local and international NGO’s with prospective protocols has
significantly improved survival in two decades.28 In addition, translational research and population-based
incidence and survival studies are reported.29, 30
In a rapidly changing world, Internet based strategies are opening up global collaboration. The One
Retinoblastoma World map (Figure 2) leads families to the nearest centre with known expertise.REFurl The
retinoblastoma-specific point-of-care database (eCancerCareRB, eCCRB) summarizes the medical record for
retinoblastoma for each child (Figures 3, 4 and 9). eCCRB on the Internet is freely accessible to every
retinoblastoma centre with local ethics and privacy approvals. The Disease-specific, electronic Patient
Illustrated Clinical Timeline (DePICT) is language-independent, well understood by parents, and supports fully
informed care choices.31 With guardian consent, fully identified data is accessible to those in the circle of care of
each child. Ultimately, the de-identified data will be fuel for a learning health system.
The search for the genetic basis of retinoblastoma provided the two-hit model of tumor suppressor gene
inactivation. In 1971, Knudson discovered that the age of diagnosis of retinoblastoma is consistent with one
rate-limiting event in bilaterally affected patients (who have heritable disease), and two events in unilaterally
affected patients with no family history (usually non-heritable).{Knudson, 1971 #591} Others proposed that
heritable retinoblastomas result from a germline mutation (‘first hit’) and an acquired somatic mutation (‘second
hit’), and non-heritable retinoblastomas arise when two somatic mutations in the same transformation
suppressor gene in a susceptible cell (Figure 4).{Comings, 1973 #21098} Chromosomal deletions in some
patients pointed to a chromosome 13q14 locus. Loss of heterozygosity of 13q14 polymorphic markers in 70% of
retinoblastoma tumours{Cavenee, 1983 #62} implied that the second hit involved the same locus. The
breakthrough came when one retinoblastoma was missing the sequence of a 13q DNA clone,{Dryja, 1986
#10404} that turned out to be a conserved, exonic sequence of RB1.{Dryja, 1986 #10404;Friend, 1986
#4934;Lee, 1987 #333;Fung, 1987 #21743}
Author, 03/01/-1,
CAN’T LOCATE Perhaps also cite:Kundson, AG, Mutation and Human Cancer, Advances in Cancer ResearchVolume 17, 1973, Pages 317–352I don’t have this yet but Knudson cited this after stating that mutation in same gene could be explanation (p 6 of ‘Chasing the Cancer Demon’)
Author, 03/01/-1,
Au: Detailed information about 1RBW added in Figure legend 2 / information abouteCCRB added to Figure legend 3HD: okay
Author, 03/01/-1,
Au:OK?]HD: okay
Author, 03/01/-1,
Au: What was the source of the data? As a Review, we should not be referencing unpublished data, but can include the odd personal communication as long as we have written consent from the owner of the dataHD: I believe this data is Junyang Zhao, author on this manuscript.
Author, 03/01/-1,
Au: do you mean ‘standard’ or ‘standardized’?
Author, 03/01/-1,
Au: Please include the URL as a numbered referencedone
RB1 is a large (190 kb) gene with 27 exons, encoding a 4.7 kb mRNA that translates into a 928 amino acid
protein, pRB. Many modifications impair pRB function, including point mutations, promoter methylation, and
small and large deletions.{Lohmann, 1999 #4359} The A/B “pocket” region40 harbours most missense
mutations.
The pRB functions that normally suppress retinoblastoma within the retina remain unknown. pRB is best known
as a cell cycle regulator that binds to E2F transcription factors to repress cell proliferation-related genes.
Hyperphosphorylation of pRB by cyclin-dependent kinases in response to mitogenic signals normally relieves
repression and promotes the G1 to S phase transition. pRB loss relieves this suppression in the absence of
mitogenic signals to enable tumourigenesis. It is tempting to speculate that pRB is primarily needed to suppress
E2F.{Dick, 2013 #10937;Lee, 2006 #11229} Several “low penetrance” RB1 mutations encode proteins with
minimal ability to bind E2F, and predispose carriers to fewer retinoblastomas than RB1 null alleles.{Lohmann,
1994 #10195} Such defective E2F-binding alleles may function to block retinoblastoma development through
an E2F-independent mechanism. pRB also up-regulates p27, implicated in cell differentiation, apoptosis, and
genomic integrity.40 Increasing pRB expression is associated with decreasing p27 during cone precursor
maturation.42 However, pRB N-terminus functions may also be important.45, 46
Biallelic RB1 inactivation is necessary to initiate most retinoblastomas, but is not sufficient; RB1-/- retinal cells
likely undergo apoptosis or limited proliferation to form the benign retinal lesion, retinoma (Figure 4).3 Further
genetic or epigenetic changes are likely needed for malignant transformation.47 Comparative genomic
hybridization studies identified common regions of DNA gain that led to several candidate retinoblastoma
oncogenes: the mitotic kinesin KIF14 and p53 regulator MDM4 (1q32), transcription factors E2F3 and DEK
(6p22), and the onco-miR clusters miR-106b~25 (7q22.1) and miR-17~92 (13q31). Consistent loss of 16q22 led
to cadherin-11 (CDH11) as a tumour suppressor gene; whole-genome sequencing identified inactivating
mutations in the transcriptional corepressor BCOR.48 Epigenetic alterations might also drive retinoblastoma
formation by inducing H3K4me3 and H3K9/14ac marks and expression of the SYK oncogene.48 Besides SYK,
many other genes and microRNAs show altered expression in retinoblastoma compared to normal retina.47 Gene
expression profiles may segregate RB1-/-retinoblastomas either into two subtypes or into a spectrum of
phenotypes correlating with histologic and cytogenetic aberrations.49, 50
Although nearly all retinoblastomas have mutation of both RB1 alleles, 1.4% of unilateral tumours have no
detectable RB1 mutation, but instead have high-level amplification of the oncogene MYCN (MYCNA).51
RB1+/+MYCNA are diagnosed at a much younger age than unilateral RB1-/- tumours and have a distinct histology,
reflecting a unique subtype. The trend of increasing MYCN copy number with age at diagnosis and the very
young age of presentation of very large tumours, places the cell of origin of RB1+/+MYCNA tumours earlier in
development than that of RB1-/-tumours.51 Another 1.5% of unilateral non-familial retinoblastomas have
apparently normal both RB1and MYCN genes; an unidentified factor or mechanism may inactivate pRB.
Author, 03/01/-1,
Au: Green sentences moved up I am not sure about the logic in this sentence, since they could just grow faster thatn RB-/- while becoming more MYCNA. Since it’s not certain this might be a place to cut 38 words.TC: I would be fine deleting this
Author, 03/01/-1,
Au: uni- and bilateral?]Yes
Author, 03/01/-1,
[Au: Paragraph removed to reduce overlap with next section Note deleted para partially restored above
Author, 03/01/-1,
Au: which ones?
Author, 03/01/-1,
Check ref formatting - FIXED
Author, 03/01/-1,
In keeping with sidelining p107/130, deleting this makes sense
Author, 03/01/-1,
They were reorganized to keep E2F issues together and non-E2F functions in a separate paragraph Au: Two paragraphs fused to improve flow and to reduce overlap; OK?]
Author, 03/01/-1,
Au: I suggest to either add this information to Figure legend 4, or to have a separate figure with the protein structure. The first option probably makes the most sense.Response: Description of RB structure seems central to understanding pathogenesis and was retained. The p107 and p130 aspects were deleted and mentioned below where more appropriateThe first half of the paragraph seems important for the mechanisms/ pathophysiology main text but if editor feels the second half is too detailed it could be dropped (as it’s not specifically about RB pathophysiology)TC: agreed. This wouldn't fit well in the Fig 4 legend. We could add a structure figure (I have one we could use) but that would add more length!
[H2] Cone circuitry sensitizes to RB1 loss
or - The retinoblastoma cell of origin
Since pRB is expressed in most if not all cells, the retina’s unique sensitivity to pRB loss is perplexing.
Intracellular signaling specific to the retinoblastoma cell-of-origin lost RB1 inactivation could explain the retinal
tropism. Detection of diverse retinal cell type markers and RNAs in retinoblastomas pointed to a pleuripotent
cell, but could also represent normal RB1+/+ cells, or aberrant expression of oncogenic transformation.{Xu, 2009
#14691;McEvoy, 2011 #16182} Rb1 plus p107, p130, or p27,53 express different retinal markers, and may not
extrapolate to humans.54
Apart from the detection of cone markers that might have beeninduced during transformation],55, 56the first
suggestion that retinoblastomas originate in cone precursors came from evidence that emerging tumourshave a
topographic distribution that mimics the horizontal visual streak characteristic of red and green cones .44Further
support came from evidence that RB1-/- retinoblastomas show consistent expression of cone photoreceptor but
not other retinal cell type-specific proteins and that maturing cone precursors have unusually high expression
ofoncoproteins (MDM2 and N-myc)that could collaborate with RB1 loss.52, 55, 56 Moreover, experimental
depletion of RB1induced cone precursor cell proliferation in vitro and development of experimental tumours
with histological, protein expression, and lack of cytogenetic changes typical of differentiated retinoblastomas 57 Proliferation depended upon high levels of N-Myc and MDM2, cone-specific transcription factors RXR and
TR2, and a down-regulation of p27 that normally occursduring cone precursor maturation.5742 These features
suggest that RB is needed to counter an oncogenic programme associated with cone precursor maturation.
Notably,smalltumours detected via optical coherence tomography (OCT) appeared to be centred in the inner
nuclear layer of the retina, not the outer nuclear layer where mature cones reside.58 However, they also extended
into the outer nuclear layer, and the smallest tumour detected to date had a significant outer nuclear layer
presence (Figure 5c).The apparent growth of early tumours within the inner nuclear layer58 could reflect a
retinoblastoma cell requirement to interact with blood vessels and retinal astrocytes, which are present only in
the inner retina and ubiquitous in tumours, and promote retinoblastoma cell growth in vitro.59Despite progress, it
remains uncertain whether cone precursors originate all RB1-/- retinoblastomas, or only highly differentiated
tumours that have few cytogenetic changes.50
[H2] Translating knowledge of pathogenesis
Understanding retinoblastoma signalling pathways could lead to treatment and prevention opportunities. For
example, pathways specific to embryoniccone precursors could be targeted postnatally .Oncoproteins such as N-
myccould be targeted60 both in RB1+/+MYCNAtumours51 and in the RB1-/- tumours that are also N-myc-dependent. 52 Furthermore, studies based on molecular discoveriesshowed thatexposing retinoblastoma-prone murine
fetuses to small molecule inhibitors of E2f or Cdk inhibited subsequent tumorigenesis without disrupting normal
retinal development53.
These new translational opportunities require cell lines and animal models that accurately reflect retinoblastoma
cell responses. Most in vitro studies have used Y79 and WERI-Rb1 cell lines, which were developed in the
1970s.61, 62Many other lines exist63 and but require further characterization. Primary retinoblastoma cellscan be
Author, 03/01/-1,
Brenda – do you agree to this version vs the original?
Author, 03/01/-1,
If needed, could delete this and cite ref 61 for all cell lines
Author, 03/01/-1,
Au: Official names: MYCN for gene, N-myc for protein.]
Author, 03/01/-1,
Delete ref?
Author, 03/01/-1,
Au: at which age would this be? any postnatal age
Author, 03/01/-1,
Au: Shortened for style.less ambiguous?]
Author, 03/01/-1,
We may be able to delete if new data is published…
Author, 03/01/-1,
Au: edit OK?revised because RXRg and TRb2 do not change with maturation; MDM2 and MYCN were previously noted to be especially high in maturing CPs; so here need only note that same holds for p27
Author, 03/01/-1,
[Au:OK?YES
Author, 03/01/-1,
is this better?)[Au: OK?]
Author, 03/01/-1,
Au: References moved to improve flow. Please check if they are relevant here. Not really, see explanations above-dc
Author, 03/01/-1,
Note: this is the preferred protein name (NCBI)Au: Is the MDM2 and NMYC expression specific to cones. Or also other cells within or outside the retina?N-Myc is unusually high in cones vs other retinal cell types (ref 49); we have not compared expression tocells outside retina but it’s probably higher in cones than cortical neural progenitors that are usually similar to RPCs
Author, 03/01/-1,
Au: Edit OK? We cannot use a dash. Would the official name be L and M cones?Either L and M or red and green is OK, not sure if there is an official name-dc
Author, 03/01/-1,
Need we cite both papers?
Author, 03/01/-1,
These early cone marker analyses were in fact the first suggestion of the cone origin, but were not conclusive for reasons given in the prior paragraph, namely that expression in the tumor could be aberrant, or reflect transformation. The cone-like topographic distribution was harder to attribute to an aberration, and more strongly suggested a cone lineage.
Author, 03/01/-1,
Au: Please briefly define/describe the highlighted phrase.]
implanted asxenografts in immunodeficient mice6448However, caution is needed, becausexenografts implanted
subretinallyor intravitreallyhave a different anatomy relative to the eye’s natural barriers, potentially affecting
drug distribution and limiting translation to patients. Genetically engineered mouse models can also be used to
assess novel treatments.65 While Rb1+/- mice do not develop retinoblastoma, retinal deletion of Rb1 (using
Pax6a, Nestin, or Chx10 promoters) in p107, p130 or p27mutant backgrounds achieves retinal tumour
formation.54 These models have been used to examine genetic interactions in vivo, such as the role of
microRNA miR-17~92 overexpression in Pax6a-Cre;Rb1lox/lox; p107-/- mice.66 However, since the mouse
tumours require different collaborating mutations and may not originate from the same retinal cell type, their
ability to predict human treatment responses is not certain. Viral oncoproteins can promote murine
retinoblastoma development, such as adenovirus E1A expressed in retinas of p53-/- mice, and Simian Virus 40
T-antigen expressed in developing Müller cellsdirected by the transgene insertion site.67, 68
Subconjunctivaltopotecan was tested in this latter model and is now in the clinic, and Cdh11 was confirmed as a
tumour suppressor by crossing a knockout into this model.69 Thus, much may be learned and translated to the
bedside with available systems while awaiting models that more precisely simulate retinoblast1oma
pathogenesis.
[H1] Diagnosis, screening and prevention(HD, GC, BLG) 1720/1500
Diagnosis is usually clear from presenting signs70 and clinical examination. Biopsies are not performed on
presumed intraocular retinoblastoma due to risk of seeding tumour outside of the eye.71-73]Differential diagnosis
for Coats disease, persistent fetal vasculature, and vitreous haemorrhage is needed.74
[H2] Clinical Diagnosis
Diagnosis is usually clear from presenting signs70and clinical examinationThe most common first sign of
retinoblastoma is leukocoria (white pupil), which is caused by light reflecting directly off the tumour(Figure 6)
Often parents notice leukocoria, but cannot convince their health advisors that there is a problem. The second
most common sign is strabismus, or misaligned eyes, due to tumour blocking central vision. Signs of advanced
disease include change in iris colour, enlarged eye due to glaucoma, and orbital cellulitis, a dangerous eye
infection.75[?]Proptosis (protrusion of the eye from the socket)[?]presents very late, but commonlyin countries
where awareness and resources are poor. Importantly, as awareness efforts are being implemented globally,
advanced retinoblastoma is declining.
Unlike most cancers for whichpathology provides definitive diagnosis, diagnosis of retinoblastoma is wholly
clinical; biopsy incurs risk of metastasis.71-73Observation of leukocoria necessitates rapid referral to an expert. A
dilated eye exam (dilation of the pupil) with indirect ophthalmoscope (retinal camera) is performed to provide a
clear view of the retina, macula and optic nerve. ?The OPTOS® (Optos, UK) ],a non-mydriatic, wide field
camera of 200°, might be useful for a rapid view of the retina, with minimal intrusion on the awake
child. Calcification, a typical characteristic of retinoblastoma , is easily detected by ultrasonography(b-scan) .
MRI is used to detect infiltration of the optic nerve and trilateral tumours (intracranial tumours associated with
RB1 mutations such as pinealoblastoma and primitive neuroectodermaltumou][.. CT scansshould beavoided if
possible owingto oncogenic toxicity of radiation in these children.
Author, 03/01/-1,
Au: I don’t think the callout to Figure 6 is OK here (leukocoria). Please checkHD: correct
Author, 03/01/-1,
Au:OK?])[ okay
Author, 03/01/-1,
Au: also of the choroid and sclera?] HD: Not as reliably as pathology.
Author, 01/03/-1,
[Au: or others too?]
Author, 01/03/-1,
Au: OK?
Author, 01/03/-1,
Au: Sentence moved up to group all eye examinations together. Is OPTOS already used, or is this still experimental?]
Author, 01/03/-1,
Au: OK?],okay
Author, 03/01/-1,
Au: Edit OK?]okay
Author, 03/01/-1,
Au: Does this imply that the tumour is extra-ocular? Is this linked to orbital retinoblastoma?Yes
Author, 03/01/-1,
Au: edit OK? How is the infection linked to the tumour?]Pathophysiology is unknown? Ref inserted Retinoblastoma associated orbital cellulitis Paul B Mullaney, Zeynel A Karcioglu, Antonio M Huaman, Saleh Al-MesferBr J Ophthalmol1998;82:517–521
Author, 03/01/-1,
[Au: Is this always visible, or only in photographs?].
Author, 03/01/-1,
Moved from above.
Author, 03/01/-1,
HD: Moved below
Author, 03/01/-1,
Au: reference? Ref inserted HD: Karcioglu ZA. Fine needle aspiration biopsy (FNAB) for retinoblastoma. Retina 2002;22:707e10. Also historical papers indicating seeding can occur after biopsy: Sanders TE, Smith ME. Biopsy of intraocular tumors. A reevaluation. IntOphthalmolClin 1972;12:163–176. Karcioglu ZA, Gordon RA, Karcioglu GL. Tumor seeding in ocular fine needle aspiration biopsy. Ophthalmology 1985; 92:1763–1767. Glasgow BJ, Brown HH, Zargoza AM, Foos RY. Quantita- tion of tumor seeding from fine needle aspiration of ocular melanomas. Am J Ophthalmol 1988;105:538–546.
Author, 03/01/-1,
HD: This is sentence is redundant as it is mentioned below.
Author, 03/01/-1,
[Au: Sentences (green) moved from management section as a short introduction; OK?] HD: moved to section below
Author, 03/01/-1,
Au: How was the cell-specificity achieved?
Author, 03/01/-1,
Brenda and Tim – can you add your ref here?Betty: this is Pajovic IOVS 2011 REF INSERTED
Author, 03/01/-1,
This needs reference?
Author, 03/01/-1,
Au: reference? Cobrinik in animal models of brain tumors – added
Author, 03/01/-1,
Au: OK?the original seems more clear to me
Author, 01/03/-1,
Au: edits OK? revised as above
Author, 03/01/-1,
Au: SYK was not mentioned above in the pathogenesis. What is its role in retinoblastoma?– it is mentioned above
Differential diagnosis for Coats disease, persistent fetal vasculature, and vitreous haemorrhage is
needed.74Detailed retinal examination under general anaesthesia almost always confirms the diagnosisand
facilitates classification of severity of intraocular disease.The hand-held fundus camera (RetCam®, Clarity,
USA ) is used to view and record the whole retina (Figures 7, 8), with views spanning all the retina fields with
the wide field lens (130° or 120°). Scleral depression(physical manipulation of the sclera) to visualize
theoraserrata(the junction between the retina and the ciliary body)is critical for accurate classification of the
retinoblastoma , andoptic nerve arterial pulsations are watched for as indication of excessive pressure on the
eye.When available,high frequency (50 MHz)ultrasound biomicroscopymonitors,76and OCT can discover
invisible tumours in infants with familial disease (Figures 5).77Intravenous fluorescein angiogram with the
RetCam®can evaluate vascular abnormalities, suspicious residual or recurrent tumour, new vessel activity in
scars, and areas of non-perfusion. Very tiny intra-retinal tumours not yet vascularized are better discovered with
OCT. Imaging supports eye classification and cancer staging, documents treatment responses, supports
consultation with colleagues, and helps parents understand treatment options.
[H2] Classification and Staging
The Reese-Ellsworth classification78for intraocular retinoblastoma predicted outcomes of external beam
radiation. When intravenous chemotherapy, (IVC)systemic chemotherapy andfocal therapy became the
common primary treatment to salvage eyes, new schemes were developed (Figure 7, Extended Data 2). An
international collaboration, led byMurphree, developedthe International Classification of Intraocular
Retinoblastoma (IIRC) which better predicted responses to IVC than did the Reese-Ellsworth classification
.79Eyes are classified by the IIRC with groups ranging from A to E, with E being the most severe. However,
subsequent modifications of the IIRC group definitions without an overt change to the Classification Scheme
name (Figure 7).The subsequent Shields version of the IIRC 80, 81 results in discrepant classification of 25% of
the most severely involved eyes when compared to the original IIRC definitions (Figure 7).82, 83Subsequently, the
Children’s Oncology Group (COG) used a variation84 with minor measurement differences with the IIRC
(Figure 7).
Similarly, for extraocular retinoblastoma, a number of staging systems co-exist and none has gained uniform
acceptance. The St Jude Staging System, reported decades ago, is still used, especially in Latin America. In a
coordinated action with the originalMurphreeIIRC 79collaboration, the International Retinoblastoma Staging
System focused on the overall staging of cancer including [retinoblastoma (Extended Data 2).85 The American
Joint Committee on Cancer (AJCC)/International Union Against Cancer (UICC) staging system (TNM, defined
by primary tumour (T), lymph node extension (N), and distant metastasis (M))for retinoblastoma86 is not widely
used.There is a strong need for a single, collaborative, consensus classification of eyes and cancer staging.
[H2] Pathology
Retinoblastomas consist of small, round cells that stain blueon haematoxylin staining. Well-differentiated
regions form rosette structures: Flexner-Wintersteiner rosettes, highly indicative of retinoblastoma; and Homer
Wright rosettes, common in diverse neural cancers (Figure 4)[.87Retinomas feature more differentiated
photoreceptor-like clusters of cells termed fleurettes (Figure 4).3
Author, 03/01/-1,
HD: This should also be indicated in the figure.
Author, 03/01/-1,
Au: Are the different rosettes visible in the histological images – inserts Figure 4? If so, please specify in legend and add arrows to point them out in the images.HD: BRENDA, TIM: only FW rosettes are visible, where is the HW image?
Author, 03/01/-1,
Au:OK?] okay
Author, 03/01/-1,
Au: Edit OK?okay
Author, 03/01/-1,
Au:OK?]okay
Author, 03/01/-1,
Au: or IRC?]
Author, 03/01/-1,
Au: differences between?](Figure 7).[Au: It would be good to include a short sentence along the lines of: ‘The tumours are classified from A to E, with E being the most severe. The exact definitions depend on the specific scale used.’ This will make is clear when these gradings come back in the next section.]
Author, 03/01/-1,
Au: What do you mean with ‘discrepant classification’?Were 25% of the RB classified wrongly using these updated criteria or did the classification depend on the investigator?](
Author, 03/01/-1,
Au: IRC or IIRC?]
Author, 03/01/-1,
[Au:OK?]
Author, 03/01/-1,
okay Au: We didn’t receive the ‘Extended data 2’]
Author, 03/01/-1,
Au: shortened for style, OK?]okay
Author, 03/01/-1,
: Callout to Figure 1 required here?okay
Author, 03/01/-1,
Au: Are both the RetCam and the scleral depression done under anesthesia?]yesHD: Yes, in the context we are speaking about here.
Author, 01/03/-1,
Au:OK?] ;[Au: please provide a short explanation for the highlighted terms
Author, 03/01/-1,
Please check callouts. Especially Figure 7 looks wrong here. HD: Figures 5c, 9, and 10 contain RetCam images, but their purpose is different. Calling out to figures here may be confusing to the reader.
Author, 03/01/-1,
Au:OK OK.
Author, 03/01/-1,
HD: Au: is needed to confirm? Bremda?
Author, 03/01/-1,
Au: I would avoid using this abbreviation. Non-standard abbreviations reduce readability, especially for the non-specialist reader okay
Author, 03/01/-1,
Moved from intro
Histological study of the enucleated eye is the only way to evaluate high-risk features (tumour invasion into the
optic nerve, post lamina cribosa, cut end of nerve; invasion of uvea ≥3 mm dimension; or both optic nerve and
uveal invasion)and establish pathological staging. []88-92 High-risk features are observed in 17% of Group D and
24% of Group E eyes90using Shields classification, and 15% Group D and 50% Group E88using the Murphree
classification.79 The documentation of high-risk pathological features is important to recognize need foradjuvant
systemic chemotherapy to reduce risk of metastatic relapse.93
The preparation and examination of the enucleated eye have been optimized to completely evaluate risk
features.92, 94Pathology specimens are staged using the pathology TNM (pTNM) and the International
Retinoblastoma Staging System (Extended Data 2).86, 92Still, too frequently retrospective examination finds a
previously unnoticed risk after metastatic disease is diagnosed. Highly retinoblastoma-specific stains such as
staining for the cone-rod homeobox transcription factor (CRX; not normally seen outside the eye)95 and N-
glycosylated ganglioside (NeuGc-GM3)96 mayfacilitate observation of a few tumour cells lurking in a high-risk
location.
[H2] Genetic Diagnosis
The majority (94%) of patients withretinoblastoma are the first individuals to be diagnosed in a family, only141
out of 2,141 patients(6%) indicated thatone or more family members were previously diagnosed with
retinoblastomawhen tested for RB1 mutations,(data from Impact Genetics, January 2015). Approximately 50%
of people with retinoblastoma carry one RB1 mutation in their constitutional cells (100% ofbilateral and 15% of
loss or somatic amplification of the MYCN oncogene) with no additional cancer risks.
Knowledge of the patient’sRB1mutation enables precise screening of relatives and subsequent generations. The
mutation can be detected prenatally from DNA in amniotic fluid when amniocentesis can be safely performed
during pregnancy, and, if necessary, labour can be induced near term to detect and treat retinoblastoma tumours
early. Without genetic testing, it is recommended that children who are at risk( already one eye affected ,and/or
positive family history) continue to undergo multiple exams under anaesthesia.Whengenetic tests canconfirm
that no mutant RB1 gene was inherited , 85% of patients with unilateral retinoblastoma will test negative with
<1% residual risk for undetectable low level mosaicism, which informs the decision to reducetheintensity of
surveillance and accurate determination of cancer risks of family members.10Pre-implantation genetic diagnosis
offers the family the option to selectunaffected embryoswhen the mutation ina parent is identified.98, 99
Characterization ofunique RB1 mutations of the tumourDNA (if available) can providea highly sensitive marker
to screen CSF, bone marrow and harvested stem cells for minimal residual disease. Markers that have been used
are the RB1tumour mutation (when mutation is not present or different from the germline alleles),100the
signature of post-RB1 genomic gains and losses 101and CRX expression.95
Best outcomes in familial retinoblastoma depend on the effectiveness of the healthcare team to identify and
counsel families. Parentsneed appropriate genetic counselling and understanding of the risks and actions for
each at-risk pregnancy. Alarmingly, a study of retinoblastoma in several developing countries observed that
familial cases were diagnosed later than non-familial probands.102 The authors inferred that the probandsdid not
Author, 03/01/-1,
Au: Is CRX commonly mutated in retinoblastoma? Or do you mean that CRX is used as a marker for eye cells, which shouldn’t be present in CSF, bone marrow, stem cells?].HD: Need to read the in press paperStill can’t locate pdf
Author, 03/01/-1,
ReplacedShould be replaced with Bowles Genes Chromosomes Cancer 2006
Author, 03/01/-1,
Au:OK?
Author, 03/01/-1,
[Au: OK?okay
Author, 03/01/-1,
Au: How are samples taken? Amniotic fluid? At which age? Maybe you can elaborate on this in the prevention sections.]
Author, 03/01/-1,
u: I would delete the callout to the figures hereokay
Author, 03/01/-1,
Au:non-germ? somatic?]HD: Both germ and somatic
Author, 03/01/-1,
Au: Please include this as a numbered reference in the reference list
Author, 03/01/-1,
[Au:OK?]94[Au: NeuGc-GM3 is specific for tumours but isn’t CRX also expressed in normal tissue? Is it a specific form or an abnormal location that is detected?]HD: Isn’t this more important for outlook? Not standard of pathology yet. I haven’t seen the in press paper so I can’t comment.
Author, 03/01/-1,
Au:OK?]okay
understood the risks to their children, but they also might be frightened that there would be no treatment
available. Alternatively, socioeconomic or geographic barriers may have reduced desired healthcare access.
Clearly, study of social determinants of health, such as health seeking behaviour, perceptions of medical care,
and sociocultural issues related to cancer inheritancewould inform counselling approaches that meet the needs
of families.103
[H2] Screening
[H3] Screening througheye examination[Au: edits OK? Dashes are not allowed]
To detect retinoblastoma as early as possible,vision screening and eye examination have been developed.
General recommendations for childhood vision screening with effective training to detect signs of
retinoblastoma do occasionally identify a child with retinoblastoma . However, onenegativechildhood screening
test doesnot mean this child will remain tumour-free since a tumourmight appear later, or may have been in the
periphery and not visible (false negative)
Leukocoria is most often first noted by parents (Figure 6), rarely first by physicians.Too often health workers
fail to take the parent’s complaint seriously, due to lack of awareness[Au: reference?]. Photoleukocoria refers
to the appearance of leukocoria on flash photographs (Figure 6). Awareness campaigns bring children to
attention, but are lose effectiveness with time. An innovative study called PhotoRed in India trained healthcare
professionals to used flash photography to identify childhood eye diseases, including retinoblastoma.104Another
pioneering project is developing software to enable cameras to detect photoleukocoria.105A camera’s Red Eye
Reduction technology constricts pupils with a pre-photograph flash, limiting photoleukocoria detection. Red-eye
and pet-eye correction tools also enable unsuspecting parents to remove photoleukocoria. The global imaging
industry could play a role in early diagnosis of retinoblastoma.
[H3] Second Cancer Surveillance
Individuals with germline RB1 mutation and/or have been treated with radiotherapyhave an elevated risk of
developing specific second cancers, including leiomyosarcoma, osteosarcoma, melanoma, lung and bladder
cancer.106 Surveillance screening for second cancers is a pressing need in the opinion of retinoblastoma
survivors. The first study to evaluate annual whole body MRI surveillance for individuals with predisposing
RB1 mutation showed it was feasible to detect second cancers but with modest sensitivity(5 out of 25 patients
screened).107 This is an important area for further research so that early intervention can reduce mortality from
second cancers, as has been demonstrated in Li-Fraumeni syndrome.108
[H2] Prevention
Lifestyle counselling educates survivors on ways to mitigate their second cancer risk. In addition to being
vigilant about reporting unexplained lesions, they are encouraged to avoid an unnecessary radiation and
carcinogens (e.g. cigarettes and excessive alcohol). The extent to which these ideaswill prevent second cancers
is unknown.
Author, 03/01/-1,
Au: Maybe the information about prenatal genetic screening can be added here HD: Prenatal genetic testing does not prevent RB…but it does screen out non-affected individuals. Does it really belong in Prevention?[Au: The next paragraph overlaps with the outlook section; hence deleted.]
Author, 03/01/-1,
Au: what was the value?]
Author, 03/01/-1,
Au:reference?]. anecdotal
Author, 03/01/-1,
Au: metastases, or other primary tumours HD: Second cancers are OTHER primary tumors.
Author, 03/01/-1,
Au: how much?
Author, 03/01/-1,
anecdotal
Author, 03/01/-1,
okay
Author, 03/01/-1,
Au: edits ok?].
Author, 03/01/-1,
[Au: reference?]
Author, 03/01/-1,
[Au: Title inserted, OK?][Au: Given the previous subsection dealt with screening in genetically at-risk individuals, I wonder whether we should specify that here we’re describing screening in the general population? Alternatively, below I suggest integrating the genetic screening section with the prevention section.]
[H1] Management(FLM, CS, DA, GC, FN, JZ, BLG) 4012/3000
Management of retinoblastoma depends on extent of disease at diagnosis (classification of intraocular disease,
stage of systemic disease), status of the opposite eye, overall health of the child, socioeconomic opportunitiesfor
the family, and accesstoexpert care.70, 109
[H2] Intraocular retinoblastoma
[H3] Primary Treatment
Choice of primary treatment is based on likelihood of cure (patient survival), eye salvage and ultimate vision–
weighed against short term and long term complications of treatment.109 In order of approximate frequency of
global use, primary treatmentsfor intraocular disease include enucleation, intravenous chemotherapy (IVC) with
focal therapy (laser, cryotherapy), intra-arterial chemotherapy (IAC) with focal therapy, and focal therapy
whentumours aresmall at diagnosis. External beam radiotherapy (EBRT)is no longer recommended for primary
intraocular retinoblastoma, since radiation incurs a very high risk of second cancers on persons carrying an
RB1mutation, especially in the first year of life.110, 111The preferred primary treatment depends on the severity of
disease of each affectedeye(Figure 8).Bilateral retinoblastoma in much of the developing world is best treated
with bilateral enucleation, where there is lack of expertise, equipment and resources, and especially when there
are difficulties with close monitoring.4, 16Manybilaterally enucleated retinoblastoma survivors lead active,
productive and satisfying lives because they were treatedby timely surgery as infants.
[H3] Second-Line (Salvage) Therapy
Second line salvage means initiating a new plan of therapies, to make a second attempt to save an eye that has
failed the first plan. All retinoblastoma treatments involve multiple modalities, and a range of modalities is
appropriate for second line therapy. However, each subsequent plan has a lower success rate112 and long drawn
out attempts to salvage an eye incur high costs of many kinds for the child and family.113, 114
Second line treatments have included focal therapies including peri-ocular chemotherapy,115 repeated systemic
and tantalum ring localization122 or whole-eye123Criteria for secondary enucleation are not well defined but are
dominated by refractory subretinal and vitreous seeding,116, 124 complications that suggest risk of extraocular
extension123such as vitreous haemorrhage and secondary neovascular glaucomaand socio-economic and
psychological fatigue to save an eye with poor vision.11479 eyes, high risk pathology is uncommon. However,
secondarily enucleated eyes showing scleral invasion many developextra-ocular relapse;125and Group E79eyes
treated with neo-adjuvant systemic chemotherapy before enucleation have significant risk for death from
retinoblastoma.126
Of Group D eyes treated with systemic chemotherapy and focal therapy 53% failed (required salvage EBRT
and/or enucleation).121127128and a combination of repeat IAC and intravitreal chemotherapy (IViC) may playan
important partin saving eyes that have failed IVC.128-130 However, extensive treatments to save an eyeincrease
risk for metastases.112, 131, 132
Author, 01/03/-1,
[Au: or isn’t this sure yet?]
Author, 01/03/-1,
[Au: Please provide an explanation for the highlighted terms. They could be added as bullet point definitions in a box.]
Author, 03/01/-1,
[Au: Since the management section is over the agreed word count at present and to reduce overlap, I have added some of the information to the figure legend 8 and to the separate sections discussing the techniques (indicated in green), but this will need shortening (see the legend at the end of the document).]
Author, 03/01/-1,
[Au: EBRT is not discussed, but mentioned quite often in the manuscript; consider adding a short paragraph. Stem cell therapy is also mentioned in one of the figure legends, without explanation. Worthwhile to mention?]
Despite advances in the eye-salvage therapiesforadvanced retinoblastoma, the major cause of failure has
remainedvitreous seeding. Pharmacokinetic studies show poor vitreous levels of drugs administered by either
systemic chemotherapy or IAC.133 The highest drug bioavailability in the vitreous is achieved by IViC using a
safety-enhanced injection technique in carefully selected eligible eyes.134 Following control of the source of
seeds, IViC achieved two-year Kaplan-Meier estimates of 98.5% control of target seeds and 90.4% event-free
ocular survival.134-136134The efficacy of IViC has eliminated the need for EBRT and decreased patient exposure to
salvage systemic or IAC. The toxicity of IViC is limited to technique-dependent localized peripheral salt-and-
pepper retinopathy.137
Combinations of new administration routes can target salvage therapies to the site of relapse. For relapse
confined to the retina and/or vitreous, salvage therapy can consist of focal therapy and/or IViC, as long as
whole-eye therapy is not required. Conversely, eyes with relapse touching the optic nerve head and/or vision-
critical regions such as the maculo-papillary bundle, and eyes with diffuse retinal/subretinal recurrence,
represent good indications for IAC, which might achieve better visual outcome than focal treatments. However,
therapies that have already failed to control the intraocular tumour are unlikely to succeed as salvage therapy for
the same eye.
[H2] Ocular Therapies
[H3] Enucleation
Enucleation is a first-line therapy for the majority of eyes with retinoblastoma globally; it is the fastest and
cheapest treatment for theseeyes138, 139. Since the majority of children with intraocular retinoblastoma have
Group D or E diseaseat diagnosis, and more than 50% have unilateral disease with other eye unaffected, cure
can be achieved with enucleation (Figures 3 and 8). All IIRC79 Group E eyes require enucleationbecause,by
definition, they carry risk of extraocular extension; however, this can only be confirmed by thepathological
assessment of the enucleated eye. Best cosmetic outcome is achieved by replacement of the volume of the eye
with an implant buried in the orbit, and provision of a prosthetic eye, worn in the conjunctival sac. Many
different reconstruction techniques are used worldwide.140 Comparative studies have shown enhanced prosthetic
eye motility with the myoconjunctival approach, which is affordable world-wide.141, 142Complex integrated
implants are commonly used but have a higher rate of infection and extrusion and are more costly. Provision of
a temporary prosthetic eye at the time of enucleation has a positive psychological impact on families,143
observed in Kenya to help the closefamily accept enucleation for their child.
[H3] Intravenous chemotherapy and focal therapy
Since 1996 first-line therapy to control IIRC Groups B, C and D diseasehas been IVC with different
combinations, doses, schedules, and durations of carboplatin, etoposide and vincristine (CEV) followed by focal
therapy to consolidate the chemotherapy responses(Table 3).124, 144, 145Sometimes high dose acute cyclosporineis
added to modulate multidrug resistance.146, 147Eyes in groups B and C do well with CEV and focal therapy. With
follow-up of 54 months, 47% of IIRC79Group D eyes121 and 47% of Reese-Ellsworth Group V124 avoided
enucleation or EBRT(Figure 9).81
Fundamental principles for systemic cancer therapy apply to retinoblastoma: optimized outcomes are achieved
Author, 03/01/-1,
Au: Table 3 has not been provided
Author, 03/01/-1,
.[Au: Please define the highlighted terms; they can be added to the box.]
Author, 03/01/-1,
[Au: in the developed countries? Or also worldwide?]
Author, 03/01/-1,
Au: Callout for figure 3 removed from the epidemiology section, please add here and adapt numbering.
by high dose intensity and combination of several agents with complementary mechanisms of action.This is
illustrated by the reduced effectiveness of single agent, low dose carboplatin for retinoblastoma.148 Acute
toxicities of IVC for retinoblastoma are as for other paediatric cancers, including short-term transient
pancytopenia (reduction in red blood cells, white blood cells and platelets), hair loss, vincristine-induced
neurotoxicity, and infections. Long term toxicities include carboplatin-induced ototoxicity (toxicity to the
ear),149 second non-ocular cancer risk with alkylating agents150, 151 and secondary acute myeloid leukaemia
following intense chemotherapy including topoisomerase inhibitors, doxorubricin and alkylating agents.152, 153
IVC alone rarely eradicates the retinoblastoma entirely, and focal therapy with repeated examinations
under[Au:general?]anaesthesiais very important (Figure 9).154-156Tumours in the macular region, which threaten
vision,are particularly at risk of recurrence without focal therapy.157
Following control of retinoblastoma with IVC, 50% of patients have visual acuity (acuteness and clearness of
vision) at 5-years of 20/20-20/40; 67% have 20/200 or better.158, 159Foveal involvement with tumour or subretinal
fluid at presentation contribute to poor vision. There is no documented local toxicity of IVC to the eye.
[H3] Intra-arterial chemotherapy
Intra-arterial chemotherapy [Au: IViC deleted since it is not discussed in this section; OK?]has had a great
impact on retinoblastoma management as it enables saving of eyes that were previously deemed unsalvageable.
IAC is usually performed by ophthalmic artery chemosurgery (OAC, also called superselective ophthalmic
artery infusion)[Au: OK?]whereby, after the patient has been administered heparin,a micro-catheter isinserted
into the femoral artery and passed up to the orifice of the ophthalmic artery (but not into the artery) where drugs
or combination of drugs (typically melphalan, carboplatin, and [Au:OK?]topotecan) areinfused in a pulsatile
fashion over many minutes.160Alternatively, a catheter with an inflatable balloon near the tip can be used to
ensure selective treatment to the eye.During the procedure, theinternal carotid artery is temporarily occluded by
inflating the tip and melphalan is injected over a few seconds below the balloon with the intention of having the
drug directedinto the ophthalmic artery(so-called selective ophthalmic treatment).131Cannulation via OAC has
been successful in almost 99% of cases.OAC is usually performed via the internal carotid artery but sometimes
also through the external carotid artery and [Au:OK?]through the middle meningeal artery (15% of cases); in
10% of the procedures a modification of the balloon technique is used.161Also, tandem therapy in which both
eyes are treatedin the same, one hour session has successfully been performed (so-called tandem
therapy).162OAC is currently performed in more than 30 countries worldwide of whichnearly half in developing
nations.163
Of the more than 2,500 infusions and >800 patients in the literature using OAC, only two patients have died of
metastatic retinoblastoma[Au: over all groups, A-E?].In the USA, patient survival was 100% at 5 years[Au:
over all groups, A-E?].129 However, death from metastatic diseasecan occur more than five years after any
treatment. The longest follow-up study described forselective ophthalmic treatmentin Japan (including death
from metastatic retinoblastoma and second cancers) showedan overall survival of 95% at 15 years.131However,
the 8 deaths from metastases of 343 patients cannot be clearly assigned to IAC alone since only a minority of
Author, 03/01/-1,
AW: Significantly more likely with IAC, as shown by the kids who have died. Would NRDP like links to their online blog stories?
Author, 03/01/-1,
AW: I find this very offensive. We know of kids in the USA who have died, even treated in NYC. Clearly not included in his published data. How can this be addressed appropriately?
Author, 03/01/-1,
AW: Where are the 8 kids who died in Japan from metastatic Rb? Are they carefully selecting their references to not include this data?
Author, 03/01/-1,
AW: Wow, that really concerns me!
Author, 03/01/-1,
[Au: what is the deciding factor(s) over which artery is used?]
Author, 03/01/-1,
AW: “Almost 99%”? Be more specific. If it’s 98.7%, say 98.7%, with the reference.
Author, 03/01/-1,
[Au: you initially had this in inverted commas. Is this because ‘a few seconds’ is wholly up to interpretation by the physician or because a set time has not been recommended?]
Author, 03/01/-1,
[Au: Since this section is quite long, I would focus on the current state-of-the-art without the historical context. This is also the overall goal of the Primer (to avoid historical discussion and focus on the current).]
Author, 03/01/-1,
AW: I think the intro should set the stage of controversy – yes it has saved eyes (not necessarily sight) that might not have been saved before, but it has also killed curable children. Survival is likely going down, not up, as a result of it.
Author, 03/01/-1,
.[Au: How does this compare to the normal values?]
Author, 03/01/-1,
Au: edits OK?]
Author, 03/01/-1,
[Au: definitions added for the non-specialist reader; OK?]
the patients received IAC exclusively.On the other hand, similar figures of deaths from metastasis have been
reported with other conservative therapeutic modalities.156
Many patients treated with IAC have advanced-stage disease and would have been candidates for enucleation
but for cultural reasons, families (and often physicians) refused this option.Despite this, ocular survival exceeds
all other approaches for advanced-stage retinoblastoma(which represent the majority of eyes at diagnosis
worldwide). It should be stressed that a comprehensive analysis of the published literature on eye survival
following IAC is confounded by the concomitant use of different classifications79, 80, 84 (Figure 7, Extended Data
2), which prevents a clear-cut comparison of the results between centres, especially regarding Group D and E
cases. We need a universally endorsed single classification in order to avoid confusion regarding eligibility and
salvage rates for new treatments. Ocular survival with the selective ophthalmic treatment65% for Group C
and45% for Group D in one Japanese study [Au:OK?].131 Using OAC in the USA, ocular survival was 96% in
both Groups B and C, and 94% in Group D.129, 164 Eyes with neovascular glaucoma, pthisisbulbi (a shrunken,
non-functional eye) , and anterior chamber involvement were universally enucleatedup front. Ocular survival is
highest in treatment-naive eyes with extensive retinal detachment (Figure 10a). With eyes with >50% retinal
detachments, ocular survival (Kaplan-Meier) was demonstrated to be87.9% at two years and 76% had complete
retinal reattachment as a result of IAC alone.165 The most common reason for second-line [Au:OK?]enucleation
of eyes with retinoblastoma in developed countries has always been the presence of extensive vitreous
seeding.166 Only 20% of such eyes can be salvaged with EBRT.167With OAC, 74% of eyes with seeding have
been salvaged(Figure 10b).112, 129, 130With the selective ophthalmic treatment, 58% of children with foveal
tumours retained a visual acuity of >0.01[Au: unit necessary? Earlier in the article, the 20/20 etc scale was
used for acuity. Please be consistent with which scale you use to avoid confusion] and for those without
foveal tumours 51% retained visual acuity of >0.5 with 36% >1.0 (2). Electroretinographic[Au:
OK?]monitoring of OAC patients160, 168-171 demonstrated remarkable stability of.OAC decreases the appearance
of subsequent, new (usually peripheral) intra-ocular tumours that commonly develop after systemic
chemotherapy or radiation in genetic[Au: patients with heritable retinoblastoma? All cancers are
genetic.]cases resulting in fewer overall treatments for the children.172[Au: Paragraph in Green moved up.]
Complications following OAC are currently few and include both short-term and long-term effects.In an
analysis of 198 catheterizations of the ophthalmic[Au: OAC technique?]artery in 70 consecutive eyes with
retinoblastoma, minor transient complications included transient eyelid oedema (5%), blepharoptosis (5%), and
choroidalischaemia (2%), and optic neuropathy (< 1%).129These complications are minimized at experienced
centres.
Historically, consolidation therapy (continued treatment once remission has been achieved)[Au: definition for
nonexperts OK?]was necessary in the majority of irradiated eyes and in nearly 100% of the eyes initially
treated with systemic chemotherapy so it was initially used in almost all of the original patients treated with
OAC. Subsequently, experience has shown that consolidation was not needed in 23-33% of cases.173, 174
Author, 03/01/-1,
[Au: What are the criteria for consolidation in the ~60% of patients that need it?]
Author, 03/01/-1,
[Au: Paragraph with details of drugs and outcomes deleted to reduce overlap and shorten the section.]
Author, 03/01/-1,
[Au: please provide short definitions.].
Author, 03/01/-1,
AW: I would contest this.
Author, 03/01/-1,
AW: This is not true. Social media evidence tells of many families whose children had anterior chamber involvement and / or glaucoma etc continuing to receive last-ditch treatments.
Author, 03/01/-1,
[Au: definition included for the non-specialist reader, OK?]
Author, 03/01/-1,
AW: I find this very hard to believe. What about sight salvage?
Author, 03/01/-1,
AW: I don’t quite understand this statement – are they saying eye salvage is now more common than enucleation for advanced stage Rb? NO HE MEANS IAC SAVES MORE ADVANCED EYES THAT OTHER KINDS OF TREATMENTS FOR ADVANCED EYES.
Author, 03/01/-1,
AW: So they mention these children here, but not in their reference above to the children treated with IAC who died – either they mention them there or not at all. They can’t have it both ways. Those 8 kids all had late stage Rb and it seems obvious the IAC treatment delaying enucleation was the cause of death.
Although this procedure has been performed in children as young as three weeks of age, most centres withhold
cannulation until the patient is at least 3 months of age and 6-7 kg in weight because of concerns about repeated
puncture of the femoral artery. As a result, these very young children are given single agent (carboplatin) IVC in
modest doses (18.7 mg/kg) as an outpatient until they attain the 6-7 kg/3 month goal when they are suitable for
OAC. This approach is called “bridge therapy” and 94.7% of such eyes (Kaplan-Meier) have been salvaged
without the need of radiotherapy.175
[H3] Focal therapy [Au: Please add references to this section.]
Focal therapy is the local application of anti-cancer therapy – thermo-, cryo-, radio-, or chemotherapy – to the
eye, under direct visualization through the pharmacologically dilated pupil. This approach is useful for primary
treatment of IIRC Group A cancersand consolidation therapy for residual or recurrent small-volume, active
tumours after systemic or IAC. In general, focal therapy is repeated monthly until the tumour is completely
atrophic or calcified.
Transpupillary thermotherapyinvolves laser treatment through the dilated pupil. Photocoagulation treatment
with 532 nm, 810 nm or continuous wave 1064 nm laser is directly applied by multiple short (0.7 s) burns to
small volume active or suspicious tumour, starting at a sub-coagulation power intensity and increasing to attain
white, opaque coagulation. Both laser treatments are repeated monthly until the tumour is flat, atrophic or
calcified.Cryotherapy is performed to freezethetumour through the sclera with a nitrous oxide probe; the tumour
is directly visualized and duration of freeze judged to completely eliminatethe tumour. Since tumour cells die
when thawing, one minute is allowed for each thaw between freezing cycles. Cryotherapy effectively destroys
small primary tumour(s) or recurrences in the periphery of the retina.Plaque radiotherapy involves the position
of a radioactive probe on the eye to delivertrans-scleral radiotherapy withan apex dose of 35 Gy over 4-7 days.
[Au: edits OK?]Plaque focal radiation has not been associated with second primary tumours and is effective for
treatment of a single primary or recurrenttumour in a location that will not compromise vision.
Paraocularchemotherapy has been usefulunder conjunctiva or in tenon’s capsule for small volume recurrences
and vitreous seeds.176-178
[H3] Intravitreal Chemotherapy
Vitreous seeds are the major cause of failure (enucleationor external beam radiation) of primary treatments.
IViC is adjunctive to many other treatments, initiated after source of the seeds is controlled, with promising
results. IViC using a safety-enhanced injection technique in carefully selected eligible eyes has shown excellent
responses with the most difficult to control form of retinoblastoma.134, 135, 137, 179, 180[Au: Sentence in green
moved up]The procedure involves lowering theintraocular pressure with an anterior chamber paracentesis or by
digital massage after induction of anesthesia.Intravitrealmelphalanis injected through the conjunctival, sclera,
and pars plana with a small-gauge needle. On needle withdrawal, the injection site is sealed and sterilized with
cryotherapy and the eye is shaken gently to distribute the drug though the vitreous. Three classes of vitreous
seeds have been identified with significantly different median times to regression, mean number of injections
and cumulative and mean melphalan dose.135Ultrasound biomicroscopymay be used to evaluate the otherwise
hidden ciliary region behind the iris,76 to confirm that the injection site is tumour-free prior to IViC treatment.
Author, 01/03/-1,
Au: specifics on dose and needle gauge removed as it’s too detaile for the Primer]
Author, 03/01/-1,
[Au: Specifics such as wavelength and level deleted from this sentence, as the next sentences describe that they can vary depending on protocol)]
[H2] Extraocular retinoblastoma
[H3] Extraocular at presentation
Retinoblastoma may present with evident extraocular disease, especially in low-income countries. Children with
orbital retinoblastoma, which may be massive and disfiguring, benefit from up-front adjuvant chemotherapy.
The preferred chemotherapeutic agents are carboplatin, etoposide and vincristine, as for intra-ocular
retinoblastoma; other agents that are useful include cisplatin, cyclophosphamide and anthracyclines.16Those who
present with overt extraocular disease have a low chance of survival, especially in low-income settings.
Chemotherapy followed byenucleation, orbital radiation, adjuvant chemotherapy, intrathecal chemotherapy and
high dose chemotherapy with stem cell rescue have potential for cure.
[H3] Adjuvant therapy for high-risk pathology
Extraocular retinoblastoma can develop despite initial diagnosis of intraocular disease. Recognition of high-risk
pathological features of primarily enucleated eyes followed by adjuvant chemotherapy with tight surveillance
for metastatic diseasehas good outcomes.89, 91 Enthusiasm to salvage eyes increases metastatic risk by masking
primary extraocular disease, and potentially continuing attempted salvage too long so that extraocular disease
develops that was not there at primary diagnosis.126, 132 Bone marrow metastasis without central nervous system
involvementhas potential for cure with extensive therapy including stem cell transplant. However, extension of
retinoblastoma into the brain has a very low likelihood of cure.
[H2] Palliation [Au: should this be a subheading under ‘Management’ in general, or under ‘Extraocular
retinoblastoma’? I think the former, but wasn’t certain]
Palliation includes pain management, symptom relief, nutritional support, and psychosocial support for the child
and families.16Untreated retinoblastoma is highly sensitive to most chemotherapy agents. Children presenting
with orbital retinoblastoma are usually in severe pain and discomfort that may be alleviated with judicious use
of anticancer therapy even when no curative intent is pursued. These children often present with severe
emaciation needing prompt medical treatment. Easily available, moderate intensity chemotherapy should be
offered to these children since life prolongation will be likely achieved and their quality of life will significantly
improve. Options include the combination of cyclophosphamide (which may also be administered orally) and
vincristine, or carboplatin and etoposide, which will seldom cause severe toxicity. Radiotherapy may also be
helpful, especially for a CNS relapse or for the treatment of massive orbital extension. EBRT and chemotherapy
are useful for pain control in palliation but are often unavailable in low-income countries.[Au: references?]
[H1] Quality of life(AW, HD, BLG) 881/500
[Au: I think this section would benefit from a short introduction that outlines what the major
QOL issues in retinoblastoma survivors are. For example, radiotherapy on a young brain can
lead to developmental issues, cognitive decline, etc. This will set up the rest of the section nicely
and need only be a sentence or two.]
Author, 03/01/-1,
Au: Please provide a short definition]
“Quality of life” describes the level of physical, emotional and psychological wellbeing experienced by an
individual. Cancer, its treatment and effects on the developing body, brain and mind can significantly decrease
Quality of Life, with implications for treatment decisions, supportive and long-term follow up care.
[H2] Measured life-long impact
Analysis of life-long treatment impact shows that 181, 182in short and long-term verbal memory, verbal learning
and verbal reasoning abilitiescompared with those diagnosed at over 1 year of age.181, 182 In bilaterally affected
children, whole brain radiation exposure at any age was significantly associated with poorer verbal
memory[Au:reference?].181, 182In children followed up from diagnosis to age 5 years, trajectories of
developmental functioningdeclined over time.182 However, verbal IQ of age-matched persons with the early
infancy form of retinoblastoma was significantly above[Au: Explanation for the higher IQ?]normal sighted
and non-retinoblastoma blind persons.183No explanation has been identified for this finding, though hypotheses
of a genetic cause have been proposed. There is altered development of visual, auditory and multisensory brain
morphology in adults who lost one eye to retinoblastoma in early life, suggesting that the remaining eye
acquired increased contralateral visual cortical connections.184, 185181182There is clinical and animal evidence that
repeated anaesthesia of young children may impair neurocognitive development.138, 186, 187Repeat anaesthesia is
necessary in eye salvage and in very young children to discover and treat small tumours. Going forward, one
outcome to record in retinoblastoma clinical studies is number of examinations under anaesthesiaandage.
[H2] Direct insight from survivors[Au: This section contains 22 references. Please try to delete some,
since the reference list needs to be cut to 200 references. ]
Most important are insights from the retinoblastoma survivors themselves. Social media brings retinoblastoma
parents and survivorsinto peer-support communities. Research processes that deepen understanding of quality of
lifefollowing retinoblastoma are needed to learn from these valuable evidence sources.
[H3] Coping during treatmentand psychosocial outcomes
A mother describes her son’s response to radiotherapy aged 16 months: “In the beginning he was extremely
combative. At the end of the treatment course, he was a broken child, withdrawn and passively accepting what
was happening to him. The long term damage caused took years of therapy to start to heal"[Au: source
reference?].Children’s perception of pain and medical interventions changes over time.188-194Repeated
procedures cause anticipation anxiety and intolerance of even minimally invasive experiences and mild pain.
The child’s initial strong emotions may be suppressed as the child gives up, and re-emerge as depression, post-
traumatic stress or developmental trauma disorder.113, 195-201at any age and with any treatment help children thrive
during treatment, reduce treatment costs, ease family stress and improve long-term mental health(Figure 11).5,
106, 202-211
Retinoblastoma treatment is often the child’s only life experience, forming the centerpiece of their earliest
memories. While adult survivors may not remember being anaesthetized, manydescribe acute fear of their
Author, 03/01/-1,
[Au: For reasons highlighted in the Figure section, we prefer not to use this photograph because we would need to obtain permission from each person in it. However, I suggest we leave it in for now and later in the process we can draw a figure that shows a puppet being used to educate the children, and we can illustrate some of the other notes in the legend (e.g., the children wearing stethoscopes etc).]
Author, 03/01/-1,
AW: Child life interventions are very specific discipline of medical care, not interchangeable with other programs.
Author, 03/01/-1,
AW: As noted in the para above, from the Retinoblastoma support community on Facebook. These are closed communities for parents and survivors only. How would you like this to be referenced other than as indicated in the para above?
Author, 03/01/-1,
[Au: regardless of age at exposure?]
Author, 03/01/-1,
[Au: reference? Any reason attributed to this?]
Author, 03/01/-1,
[Au: in terms of IQ? Please specify]
mouth and nose being covered. One adult describes how the scent and taste of strawberries makes her nauseous
– her mask was always coated with strawberry scent.
Extended isolation during therapy may impact social functioning. While most adult survivors perform well
socially, many report low confidence and intense anxiety, especially in large groups and crowded environments.
Most survivors are high cognitive performers.181 However, reduced vision causes some children to become
frustrated by their inability to keep up with peers, damaging self-esteem and confidence.212-214
[H2] Radiotherapy late effects
While radiotherapy is now rarely used for retinoblastoma, thousands of adult survivors live with its long-term
effects. Many feel neglected and demoralized by lack of follow up and prospective management. Facial
deformity causes low self-confidence and social anxiety. Reconstructive surgery is a painful process that may
impact remaining vision, but its cosmetic effects can dramatically improve quality of lie. Dry eye is very
painful, and corneal vascularization reduces already limited vision. Use of ocular lubricants may prevent
complications, and should be started early before pain and vision loss occur.Chronic primary headaches,
hormone dysfunction and seizures also impact quality of lifeafter radiotherapy.[Au: reference?]
[H2] Second cancer risk
Individuals at risk of second primary cancers require life-long oncology follow up.150, 215, 216Lack of agreed
protocols causes confusion, frustration and fear as adults struggle to access informed follow up care,
compounded by primary doctors who are not awareof late effects and lifelong implications of RB1
mutation.Full information about the patient’scancer history, genetic status and life-long risks will empower
survivors to be advocates for their own and their children’s health.217-220
180211-213
[H2] Family planning
Many adult survivors have little knowledge of retinoblastoma genetics, genetic counselling or testing, their
status, or of options for their baby. Cost and availability of genetic testing and pre-implantation genetic
diagnosis is often prohibitive.Lack of an agreed screening protocol for at-risk babies causes anxiety among
survivor-parents. Profound anger and guilt about somehow being responsible for the child’s cancer is amplified
when diagnosis is delayed by inadequate screening. Agreed screening protocols for at-risk children will reduce
survivor-parent anxiety and enhance early diagnosis to achieve minimally invasive therapy.
[H1] Outlook(All) 1332/1000
Retinoblastoma is curable if diagnosed early. We propose that within 10 years retinoblastoma can be azero-
death cancer. Our anti-retinoblastoma arsenal will consist not only of curative treatments, but also measures to
prevent development of disease in predisposed individuals. To achieve this vision, critical efforts are needed to
Author, 03/01/-1,
Au: there are no references to support this section. Is there evidence from related oncology fields we can cite that will give readers a chance to follow up?]AW: Evidence from the Adult Rb Survivor community.
Author, 03/01/-1,
AW: Reported frequently in Adult Rb Survivors group, as described above
Author, 03/01/-1,
[Au: Section on ‘Treatment choices’ deleted to remove overlap with the management section and to reduce word count of this section.][
build upon the current state-of-the-art for retinoblastoma as presented in this Primer.Now, we dream big with
key action items, responding to the immediate obvious targets for improvements, and setting the stage for
innovative discoveries in retinoblastoma.
[H2] Patient-centered, global research focus[Au:OK?]
Retinoblastoma will benefit from clinical science at the level that has achieved dramatic change for pediatric
cancer. Because most centres have too few affected children, or many children and inadequate resources to
study them, we have lagged behind in systematic accumulation of evidence on which to base therapy. [Au:
Deleted description of AHOPCA to avoid repetition]Today we are faced with sweeping change in treatment
of retinoblastoma usingIAC and salvage of unresponsive vitreous seeds by IViC. However, we do not have full
data on which to judge eligibility, long-term effectiveness or outcomes.
We have opportunities now to learn from initiatives and innovations everywhere, and to listen to the patient’s
voice to become an inclusive, participatory global network. Patients and families, empowered by social media,
will be heard in the research approaches, making them relevant, ethical, and appropriate. Stakeholders from low-
and-middle income countries will be active innovators, assuring that protocols are feasible and acceptable. For
example, AHOPCA created and implemented multicentre protocols for retinoblastoma treatment,21, 23, 24 a major
achievement not yet paralleled in developed countries.
Equality for retinoblastoma will come by education (Internet access), with shared collaborative care coordinated
online. Retinoblastoma expertise will be developed locally (proportional to the local burden of retinoblastoma)
but derived globally, with best practices drawing on rigorous molecular and clinical research. Retinoblastoma
clinical trialswill be collaborative, high enrolment studies. Effective innovations will include all outcomes and
sustainability (cost, uptake by health ministries, social acceptability of therapy, etc.).
The global burden of retinoblastoma shows the inherent inequity in how patients are diagnosed and cared for
around the world. Documentation of human and material resources required for optimal care, drawn from
established clinical care guidelines, represent the first step towards achieving health equity. National strategies
in areas currently underrepresented in the retinoblastoma literature are poised to fill the knowledge gap.
Progress in therapy is critically dependent on achieving consensus on a standard classification of intraocular
disease. The ongoing AJCC/UICC international survey of features of eyes at diagnosis, treatments given and
outcomes will be used to derive one evidence-based classification and replace the current clinical staging
schemes that confuse analysis or eligibility or outcomes.79, 80, 86
We are poised to collect uniform data on all children with retinoblastoma. The eCCRB point-of-care database
summarizes the health record from diagnosis, including features of eyes and patient, treatments, complications,
and outcomes, and is available to all retinoblastoma centres. eCCRB also supports the caregivers by graphic
display of information otherwise out of reach, ensuring high motivation for good data quality. The next phase of
this Internet project (aiming at 2020) will combine the 1RBW map with eCCRB de-identified data. “Real-time”
analysis of outcomes is planned to feedback to treating doctors, a “learning health system”,221 to assist in
selection of evidence-based care for the next affected child.
Author, 03/01/-1,
HD: I think this is better suited as a conclusing paragraph.
[H2] Cause of every death from retinoblastoma
We propose to determine the most likely cause and key risk factors of every death from retinoblastoma.Similar
in concept to the groundbreaking Million Death Study,222 this information will guide us to target preventable
deaths quickly, by simply developing awareness of the disease, clinical research, and strengthening of health
systems.223The 1RBW.org map documents staff, equipment [Au:OK?], and spaces, and provides an alternative
system of global collaboration, perhaps unique to small neglected diseases that fall through the cracks even in
the most developed health care system.Implementation of this knowledge can help tostreamlinereferral and the
speed of pathology reporting.
[H2Targetedtherapies
Our knowledge of the retinoblastoma cell of origin and its unique sensitivities is steadily increasing, and may
reveal strategies to prevent tumorigenesis. Moreover, ongoing work to define molecular lesions in
retinoblastoma will continue to yield novel therapeutic targets. Advances in therapeutic development and
prevention depend on these molecular studies, but we do not yet have preclinical models that accurately reflect
retinoblastoma pathogenesis and treatment.
One of the earliest targeted therapies developed for retinoblastoma, based on preclinical information generated
by transgenic mice,wasNutlin-3.224 Nutlin-3 targets the negative regulation of p53 by MDM2/MDM4, resulting
in p53-mediated apoptotic cell death. Nutlin-3 showed promising activity in combination with topotecan for
retinoblastoma control in preclinical models. This drug combination is currently undergoing evaluation in a
prospective study in combination with local chemotherapy.As mentioned, gene expression and epigenetic
analyses of retinoblastomas identified the spleen tyrosine kinase (SYK) as required for retinoblastoma cell
survival.48Although the SYK antagonist R406was considered a promising candidate based onpreclinical studies,
its ocular pharmacokinetic profile following peri-ocular administration rendered it unfavourable for clinical
use.225 Preclinical studies have sought to identify new molecular targets and new agents to block the drivers of
tumourigenesis in retinoblastoma, and to evaluate pharmacokinetics of new agents alone or in combination226
before clinical deployment. However, few translational studies have actually resulted in significant changes in
retinoblastoma clinical practice.227 Innovative delivery systems to the vitreous to targetvitreous seeding have
been evaluated and include devices for sustained release preparations or metronomic administration
viaperiocular or intravitreal routes,episcleral implants228 or nanoparticles229. They are not currently used in
clinical practice albeit for restricted indications. Other agents such as those targeting the tumour vasculature230
or hypoxia231 have been evaluated in preclinical models but have not yet progressed to clinical use. Alsonew
biomarkers for molecular dissemination of retinoblastoma outside the eye may lead to earlier diagnosis and
targeted threatments[Au: OK?].232
Improved understanding of retinoblastoma pathogenesis (Figure 1) could yield additional agents that with
appropriate clinical evaluation may significantly change future treatment.
Author, 01/03/-1,
Clarify?
Author, 01/03/-1,
[Au: This section still needs a display item. Could you add a schematic overview of the molecular pathways that could potentially be targeted? I have attached a figure as example, which we used to show the potential targets in Huntington disease. We could do something similar. Please note that the figures will be redrawn after peer-review. At this stage, we just need to make sure that they are clear – but they do not have to be nice. You can either send me a sketch in PowerPoint (I can clean it up for you) or a hand-drawn sketch (I can make the powerpoint for you; whatever most convenient for you.]
Author, 01/03/-1,
[Au: Please include clinical trial information (NCT number) in the reference list]I couldn’t find this trial in clinicaltrials.gov!
Author, 01/03/-1,
Au: shortened for style (max. 42 characters)]
Author, 01/03/-1,
HD “staff, stuff, systems and spaces’ is the terminology used by Paul Farmer re: global health capacity: http://www.pih.org/blog/for-ebola-countries-need-tools-to-treat-patients-in-their-communities.
Author, 03/01/-1,
[Au: shortened for style (max. 42 characters)]Maybe this should be rolled into the previous H2 section since it’s so short?
Author, 03/01/-1,
Au: I have copied the outstanding questions to a box; OK?]
[Au: QoL section deleted to remove overlap.]
Display Item Legends
Box 1: [Au: Please consider including a box with explanations for the specialist terms.]
Complex integrated implants
EBRT and tantalum ring localization
brachytherapy
myoconjunctival approach
oraserrata
orbital retinoblastoma
salt and pepper retinopathy
Scleral depression
Stem cell therapy?
proton beam radiotherapy
Box 2: Outstanding research questions [Au: Titles OK?]
Patient-centered, global research focus
Address the "reluctance of healthworkers to confer the true risks"(such as in Jordan where
doctors do not want to destroy families) and choice of families who know and understand the
risks not to followthrough because of the same stigma.
Define treatment success: cure (life), salvage of vision (one eye), salvage of eye (vision and
cosmetic), fewest examinations under anaesthesia (cumulative toxicitiyof anaesthesia)
Meta-analysis of all children treated primarily with IAC.
Develop a “retinoblastoma index” relating levels of awareness, access to centres with the
necessary resources and expertise, and application of evidence-based care, as predictors of
outcome.
Pathogenesis
What is the genomic progression post RB1 loss?
What molecular changes promote (early) metastasis?
Are there other RB1+/+ subtypes beyond MYCN-amplified retinoblastoma?
What dictates the tissue specificity of RB1 loss leading to cancer?
What are the unique molecular features of second cancers in RB1 mutation carriers?
Figure 1| Progression of retinoblastoma. a) Biallelic loss of RB1 may block differentiation of a cone precursor
cell, as itfails to localize normally. b) Genomic instability might leadto an intraretinalbenign retinoma. Inset: a
small retinoblastoma visualized by optical coherence tomography[Au: OK? Or do RB1-/- cells always form
retinomas.] [Au: Which kind of scan is shown in the insert? Please add information to the legend.] . c)
This tumourcan progress to form a retinoblastoma if additional mutations are acquired [Au:OK? Or do all the
retinomas progress to retinoblastomas], and seeds under the retina and into the vitreous; d) invades past the
lamina cribrosa of the optic nerve, uvea, or sclera to constitute high risk pathologic features. e) Finally, the
tumor mightextend extraocularly into orbit, brain (direct or subarachnoid to CSF) or metastasizes to bone
marrow.[Au: Can we add percentages of to the figure, showing how many % of patients will progress to
the next stage – if data are available? How many % of RB1 carriers, for example, will progress to develop
retinoblastoma?][Au: It would be great if we could have an extra panel, preferably panel A, showing the
normal eye anatomy, with labels for all the relevant anatomical terms mentioned in this legend, or the
manuscript. ]
Figure 2 | Global Retinoblastoma Prevalence. Heat Map of estimated distribution of the global retinoblastoma
patient population as assembled by the One Retinoblastoma World (1RBW) initiative. The majority of cases
reside in low and middle income countries. The 1RBW Map of Treatment Centers (www.1rbw.org) connects
affected families to expert care close to home, providing rich global data for anyone to use. Without prior
awareness of retinoblastoma, parents now commonly self-diagnose on the Internet, and the 1RBW map connects
them to experts without delays. Images from www.1rbw.org.[Au: I think only panel A should be sufficient. It
is quite clear that most cases are in the middle/lower income countries.]
Figure 3 | Triplets with retinoblastoma tumours.31 These triplets developed retinoblastomas in all six eyes
illustrating the full expressivity and penetrance of the RB1-/- germline mutations that result in no protein, such as
the mutation carried by each triplet. The eCCRB timelines reveal that all eyes had individualized therapy, with
choices considering the overall impact on each child. Each child lost one eye, and each has a normal vision eye
with disease control in less than one year. The number of eye exams under anaesthesia was least for the child
who had primary enucleation. OD, right eye; OS, left eye. Eye involvement at diagnosis is indicated in the
International Intraocular classification;79 the eye labelled “Group 0” had no tumour at initial diagnosis but
develop a tumour 6 weeks later.
Figure 4| Genetic origins of retinoblastoma. Three genetic subtypes of retinoblastoma are known. Heritable
retinoblastoma patients have a constitutive[Au: inactivating?YES]inactivatingmutation (M1) in the RB1tumor
suppressor gene in all cells of their body. A second, somatic mutation (M2) in a susceptible retinal cell can lead
to benign retinoma. Further genetic and/or epigenetic events (M3…Mn) are required to transition to
retinoblastoma. Non-heritable, RB1 mutant retinoblastomas follow a similar progression, except both M1 and
M2 occur in a susceptible retinal cell. MYCN-amplified (MYCNA) retinoblastoma is a rare, non-heritable
retinoblastoma subtype driven by amplification of MYCN with normal RB1; other changes in these tumours
remain uncharacterized.Retinoma histology shows distinct photoreceptor-like fleurettes, while RB1-/-
retinoblastoma can show Flexner-Wintersteiner (indicated) and Homer Wright rosettes. Conversely,
MYCNAretinoblastoma show a distinct histology with rounded nuclei and prominent nucleoli.[Au: Can you
please provide a short explanation for the histological images?]
Author, 03/01/-1,
[Au: Paragraph in green moved from the epidemiology section] I HAVE MOVED THIS PARAGRAPH BACK TO EPIDEMIOLOGY WHERE ITS POINT IS NEEDED.
Author, 03/01/-1,
.[Au: Is this an original figure, or do we need a reference? Also, because these patients are identifiable, we need to obtain consent to publish their images. I would argue that including their photographs is not essential to understanding the key information in the figureand remove the photograph.] THIS IS AN ORIGINAL FIGURE AND WE HAVE THE CONSENT. WE ALL THINK THE PHOTOGRAPH CONTRIBUTES GREATLY TO THE UNDERSTANDING OF MANY ASPECTS, FOR WHICH FIGURE 3 I REFENCED FROM MANY PLACES IN THE PAPER.
[Au: Where should this text go? Please take into account that legends should not exceed 200 words.]Retinoblastomas
are thought to originate in developing retinal cells that have biallelicRB1 inactivation. The prominent expression of cone-
specific proteins in retinoblastoma cells combined with high-level pRB, MYCN, and MDM2 in cone precursors and the
proliferative response of RB1-depleted cone precursors have provided compelling evidence of a cone precursor
origin34,38,39.Although cone precursors are among the earliest post-mitotic retinal cell types produced, they lie dormant for
several weeks to months prior to expressing pRB, cone phototransduction proteins, and the oncoproteins that may sensitize
to pRB loss. 34,39Hence, retinoblastomas have been proposed to arise following cell cycle re-entry of this normally post-
mitotic retinal cell type. 38
The figure displays the locations of maturing cone precursors relative to other cell types in the developing retina. Because
the retina develops in a central-to-peripheral direction, the central retina is developmentally far more advanced than the
periphery, and different retinal cell maturation states can be viewed in a single histologic section. Panels ( a) and (b) display
the different histologic appearance and pRB expression patterns in the relatively mature and post-mitotic central retina ( left)
and in the immature far periphery (right).In the central retina, postmitotic cone precursors in the outer nuclear layer highly
express pRB (red) and cone arrestin (green).However, in the immature peripheral retina, pRB is most highly expressed in the
retinal progenitor cells in the neuroblastic layer, but neither pRB nor cone arrestin is detected in immature post-mitotic cone
precursors. 39
Despite that maturing cone precursors but not other retinal cell types proliferate in response to pRB depletion 38, and that cone
precursors are located in the outer nuclear layer, the smallest and presumably earliest retinoblastomas that have been
detected in infants using optical coherence tomography have been centered in the inner nuclear layer (c).This poses a
challenge to the cone precursor origin model, yet might be consistent with a migration of RB1-deficient cone precursors from
the outer nuclear layer to the inner nuclear layer, where they could interact with astrocytes and blood vessels that enable
tumorigenesis.41
Figure 6 | Photoleukocoria led the parents to diagnose retinoblastoma using the Internet (a). The left eye
was enucleated. The other eye was normal at diagnosis and developed later a small tumour treated with only
laser. b, Subsequent MRI revealed thathe also had trilateral retinoblastoma. He was treated with systemic
chemotherapy, intrathecal(given directly into the cerebro-spinal fluid) chemotherapy and high dose
Author, 03/01/-1,
NOTE THIS AFFECTED IS THE LEFT ONE
Author, 03/01/-1,
THESE PICTURES ARE VERY IMPORTANT TO CONVEY THE MEANING. WE HAVE FULL CONSENT FOR USING THE FULLY IDENTIFIED IMAGES, AS FOR THE OTHER PICTURES.
Author, 03/01/-1,
We suggest a box dedicated to explaining the cone origin, if space permits.TC: Much as I like this text, I think it probably goes into more detail than is needed and duplicates some of what is in the text and main figure legend. I would suggest deletion.
Author, 01/03/-1,
[Au: Isn’t it visible in the right panel?] NO the point is the layer of the retina it arose in.
chemotherapy with hematopoietic stem cell transplant, c,The child is disease-free at age 8 years.d, His treatment
summary is illustrated in the DePICT view of eCancerCareretinoblastoma.
Figure 7 | Many different classification schemes for intraocular retinoblastoma confound comparison of
outcomes.The features listed determine the overall classification, ranging from small tumours not threatening
vision (“A”) to tumours clinically noted to have features suggesting potential spread outside the eye (“E”). Most
importantly, size of tumour alone does not make an eye dangerous by Murphree, Children’s Oncology Group
(COG), or TNM classification; but any eye with tumour >50% of eye volume is E (advanced-stage disease)by
Shields classification.The consequence is widespread confusion in the literature undermining clinical research,
since studies using the different classifications cannot be compared for outcomes.
Figure 8 | Primary treatment choices for each eye based on the International Intraocular Retinoblastoma
Classification (Murphree). Treatment depends on the combined severity of each of the affected eyes (Eye 1 /
Eye 2); the preferred option for each eye is depicted in the blue boxes. Group A eyes can be treated with laser or
cryotherapy(focal therapy) alone.. Group B and C eyes require several cycles of systemic or intra-arterial
chemotherapy followed by focal therapy(Group B) and intravitrealchemotherapy(IViC) with melphalan(Group
C). Isolated single tumours inGroup B or C eyes may occasionally be appropriate for primary radioactive plaque
therapy. For safety of the child, it is important that all eyes with features suggesting imminent extraocular
extension be removed to enable accurate pathological examination to determine if risk ofmetastasisrequires
adjuvant chemotherapy.
Figure 9 | Good responses to intravenous chemotherapy.a,Before (Left) 4 cycles of carboplatin, etoposide
and vincristine (CEV) + cyclosporine (CSA) chemotherapy and laser consolidation for Group B/ D[Au: Please
specify what the rows are: upper row: B level eye and lower row: D level eye?]; and after (Right) showing
good response and recovering fovea at edge of calcification.b, Before (Left) 4 cycles of CEV + CSA
chemotherapy and laser consolidation for Group D/ D; and after (Right) showing good response with minimal
focal therapy (15 years follow-up).
Figure 10 | Resolution of retinoblastoma with intra-arterial chemotherapy (IAC).a, Before (Left) and after
(Right) IAC for a retinoblastoma eye with extensive retinal detachment; b, Before (Left) and after (Right) IAC
for a retinoblastoma eye with extensive vitreous seeding; c,Before (Left) and after (Right) IAC treatment of an
eye with macular tumour.
Figure 11 | Child Life promotes effective coping through play, preparation, education, positive-touch and
self-expression activities, based on natural child development. Child Life helps children and their families
cope with challenging healthcare issues, hospitalization and therapeutic interventions. Even in palliation, play is
the job of a child. These children play doctor and nurse, wearing gloves and stethoscopes while they examine
Puppet Kevin who had one eye removed for retinoblastoma. Play (medical play and just general play) promotes
a sense of mastery for children within their medical experience. Photo taken at the Sally Test Pediatric Centre,
Moi Teaching and Referral Hospital, Eldoret, Kenya.
Author, 01/03/-1,
BG TO CHANE ALL TO AA
Author, 03/01/-1,
[Au: This wasn’t attached in the submission, so I cannot assess if it’s necessary. However, Figure 7 already contains detailed information, at least for the scope of the primer.] FINE WE REMOVED EXTENDED DATA 2
Author, 03/01/-1,
[Au: Has this figure been published before? If so, please add a reference. You will also need to fill in some paperwork to request permission of the original publisher. I will send this after the peer review process.]NO THIS IS AN ORIGINAL FIGURE FOR NRDP
Author, 03/01/-1,
[Au: This has not been discussed in the management section as a potential therapy for RB so a short explanation might be required.]
Tables
Table 2: Estimated Global Distribution of Retinoblastoma
Forecast births were calculated using most recent data (2012) for population, birth rate and mortality rate
(World Bank (http://data.worldbank.org] accessed on 5 February 2015. Low (1:18,000 live births) and high
estimates (1:16,000 live births) calculated, following the example of Kivela.5[Au: it might also be useful to list
in the table footnote which countries are included in the different categories.]
Category
Population,
total[Au:
approximations
might be
better: 7.0
billion; 1.3
billion; 250
million]
Birth
rate,
crude
(per
1,000
people)
Mortality
rate, infant
(per 1,000
live births)
Forecast Live
Birthstotal[Au:
approximations
might be better
total[Au:
approximations
might be
better]
Retinoblastoma
Cases (High)
1:16,000
Retinoblastoma
Cases (Low)
1:18000
% of World
Retinoblastoma[Au:
is this based on the
high or low
approximations?]
World 7,043,105,591 19.4 34.6 131,629,862 8,227 7,313 100%
High income 1,299,489,520 11.6 5.5 14,941,320 934 830 11.4%
High income:
nonOECD 249,927,994 13.8 10.3 3,418,872 214 190
High income:
OECD 1,049,561,526 11.0 4.4 11,518,845 720 640
Low &
middle
income 5,743,616,071 21.1 38.1 116,699,203 7,294 6,483 88.7%
Middle
income 4,913,580,797 19.2 33.7 91,300,546 5,706 5,072 69.4%
Upper middle
income 2,390,210,774 14.8 16.3 34,737,317 2,171 1,930 26.4%
Lower middle
income 2,523,370,023 23.4 45.3 56,490,388 3,531 3,138 42.9%
Low income 830,035,274 32.3 54.5 25,373,890 1,586 1,410 19.3%
screening and prevention (B.G., H.D., J.Z.); Management (B.G., F.M., D.A., C.S., G.C.); Quality of life (B.G.,
A.W.); Outlook (B.G., H.D., T.C., D.C., F.M.); overview of Primer (B.G.).
[Au: I noticed some discrepancies between the information in the online submission system and the
manuscript – with regard to author contributions. I took the info of the submission system. Please check
very carefully and make changes (with tracked changes on) so that I can update the submission system.
Also, Festus Njuguna does not have any contributions in the submission system; please add information
here.]
Competing interests
In the interests of transparency, the Nature Clinical Reviews journals have a competing financial
interests policy. A detailed explanation of the policy can be found in the Nature website
(http://www.nature.com/nature/submit/policies/competing/index.html). Each author is required to
disclose any relationship, financial or otherwise, that could be perceived as a conflict of interest.
[Au: We need to have competing interested information before we can send the manuscript for peer-
review. Please add to the system or declare that none of the authors have conflicts.]
References242/200[Au: Try to cut down to 200 reference; especially the QOL section has quite a lot of reference. ]
1. Knudson, A.G. Two genetic hits (more or less) to cancer. Nat Rev Cancer 1, 157-62 (2001).
2. Hanahan, D. & Weinberg, R.A. The hallmarks of cancer. Cell 100, 57-70 (2000).
3. Dimaras, H. et al. Loss of RB1 induces non-proliferative retinoma: increasing genomic instability correlates with progression to retinoblastoma. Hum Mol Genet 17, 1363-72 (2008).
4. Dimaras, H. et al. Retinoblastoma. Lancet 379, 1436-46 (2012).
5. Kivela, T. The epidemiological challenge of the most frequent eye cancer: retinoblastoma, an issue of birth and death. Br J Ophthalmol 93, 1129-31 (2009).
6. Broaddus, E., Topham, A. & Singh, A.D. Incidence of retinoblastoma in the USA: 1975-2004. Br J Ophthalmol 93, 21-3 (2009).
7. Seregard, S., Lundell, G., Svedberg, H. & Kivela, T. Incidence of retinoblastoma from 1958 to 1998 in Northern Europe: advantages of birth cohort analysis. Ophthalmology 111, 1228-32 (2004).
8. Nyawira, G., Kahaki, K. & Kariuki-Wanyoike, M. Survival among retinoblastoma patients at the Kenyatta National Hospital, Kenya. Journal of Ophthalmology of Eastern Central and Southern Africa, 15-19 (2013).
9. Dean, M. et al. Increased incidence and disparity of diagnosis of retinoblastoma patients in Guatemala. Cancer Lett 351, 59-63 (2014).
10. National Retinoblastoma Strategy Canadian Guidelines for Care / Stratégie thérapeutique du rétinoblastome guide clinique canadien. Can J Ophthalmol 44, S1-88 (2009).
11. Kumar, A., Moulik, N.R., Mishra, R.K. & Kumar, D. Causes, outcome and prevention of abandonment in retinoblastoma in India. Pediatr Blood Cancer 60, 771-5 (2013).
12. Gichigo, E.N., Kariuki-Wanyoike, M.M., Kimani, K. & Nentwich, M.M. [Retinoblastoma in Kenya : Survival and prognostic factors.]. Ophthalmologe (2014).
13. Moreno, F. et al. A population-based study of retinoblastoma incidence and survival in Argentine children. Pediatr Blood Cancer 61, 1610-5 (2014).
14. (2014).
15. Dimaras, H., White, A. & Gallie, B.L. in Ophthalmology Rounds (Toronto, 2008).
16. Chantada, G. et al. SIOP-PODC recommendations for graduated-intensity treatment of retinoblastoma in developing countries. Pediatr Blood Cancer 60, 719-27 (2013).
17. Perez-Cuevas, R. et al. Scaling up cancer care for children without medical insurance in developing countries: The case of Mexico. Pediatr Blood Cancer 60, 196-203 (2013).
18. de Castro Junior, C.G. & Macedo, C.R. Brazilian Society of Pediatric Oncology - SOBOPE: 30 years of history, a lot in the present, full of the future. Rev Bras Hematol Hemoter 33, 326-7 (2011).
19. Naseripour, M. "Retinoblastoma survival disparity": The expanding horizon in developing countries. Saudi J Ophthalmol 26, 157-61 (2012).
20. Qaddoumi, I. et al. Team management, twinning, and telemedicine in retinoblastoma: a 3-tier approach implemented in the first eye salvage program in Jordan. Pediatr Blood Cancer 51, 241-4 (2008).
21. Wilimas, J.A. et al. Development of retinoblastoma programs in Central America. Pediatr Blood Cancer 53, 42-6 (2009).
22. Traore, F. et al. [Retinoblastoma: inventory in Mali and program to develop early diagnosis, treatments and rehabilitation]. Bull Cancer 100, 161-5 (2013).
23. Luna-Fineman, S. et al. Retinoblastoma in Central America: report from the Central American Association of Pediatric Hematology Oncology (AHOPCA). Pediatr Blood Cancer 58, 545-50 (2012).
24. Barr, R.D. et al. Asociacion de Hemato-Oncologia Pediatrica de Centro America (AHOPCA): a model for sustainable development in pediatric oncology. Pediatr Blood Cancer 61, 345-54 (2014).
25. Waddell, K.M. et al. Improving survival of retinoblastoma in Uganda. Br J Ophthalmol (2015).
26. Waddell, K.M. et al. Clinical features and survival among children with retinoblastoma in Uganda. Br J Ophthalmol 99, 387-90 (2015).
27. One Retinoblastoma World is a global network with the bold idea that all children with retinoblastoma can have equal opportunity to optimal care. We aim for global collaboration to deliver coordinated, evidence-based retinoblastoma care. (Toronto, 2015).
28. Schvartzman, E. et al. Results of a stage-based protocol for the treatment of retinoblastoma. J Clin Oncol 14, 1532-6 (1996).
29. Chantada, G.L. et al. Impact of chemoreduction for conservative therapy for retinoblastoma in Argentina. Pediatr Blood Cancer 61, 821-6 (2014).
30. Chantada, G.L. et al. Results of a prospective study for the treatment of unilateral retinoblastoma. Pediatr Blood Cancer 55, 60-6 (2010).
31. Chiu, H.H., Dimaras, H., Downie, R. & Gallie, B. Breaking down barriers to communicating complex retinoblastoma information: can graphics be the solution? Canadian Journal of Ophthalmology (In Press).
32. Knudson, A.G. Mutation and cancer: statistical study of retinoblastoma. Proceedings of the National Academy of Science, USA 68, 820-823 (1971).
33. Comings, D.E. A general theory of carcinogenesis. Proc Natl Acad Sci U S A 70, 3324-8 (1973).
34. Cavenee, W.K. et al. Expression of recessive alleles by chromosomal mechanisms in retinoblastoma. Nature 305, 779-784 (1983).
35. Dryja, T.P., Friend, S. & Weinberg, R.A. Genetic sequences that predispose to retinoblastoma and osteosarcoma. Symp Fundam Cancer Res 39, 115-9 (1986).
36. Lee, W.H. et al. Human retinoblastoma susceptibility gene: cloning, identification, and sequence. Science 235, 1394-9 (1987).
37. Friend, S.H. et al. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 323, 643-6 (1986).
38. Fung, Y.K. et al. Structural evidence for the authenticity of the human retinoblastoma gene. Science 236, 1657-61 (1987).
39. Lohmann, D.R. RB1 gene mutations in retinoblastoma. Hum Mutat 14, 283-288 (1999).
41. Hurford, R.K., Jr., Cobrinik, D., Lee, M.H. & Dyson, N. pRB and p107/p130 are required for the regulated expression of different sets of E2F responsive genes. Genes Dev 11, 1447-63 (1997).
42. Lee, T.C., Almeida, D., Claros, N., Abramson, D.H. & Cobrinik, D. Cell cycle-specific and cell type-specific expression of Rb in the developing human retina. Invest Ophthalmol Vis Sci 47, 5590-8 (2006).
43. Lohmann, D.R., Brandt, B., Hopping, W., Passarge, E. & Horsthemke, B. Distinct RB1 gene mutations with low penetrance in hereditary retinoblastoma. Human Genetics 94, 349-354 (1994).
44. Munier, F.L., Balmer, A., van Melle, G. & Gailloud, C. Radial asymmetry in the topography of retinoblastoma. Clues to the cell of origin. Ophthalmic Genet 15, 101-6 (1994).
45. Goodrich, D.W. How the other half lives, the amino-terminal domain of the retinoblastoma tumor suppressor protein. J Cell Physiol 197, 169-80 (2003).
46. Cook, R. et al. Direct Involvement of Retinoblastoma Family Proteins in DNA Repair by Non-homologous End-Joining. Cell Rep 10, 2006-18 (2015).
47. Theriault, B.L., Dimaras, H., Gallie, B.L. & Corson, T.W. The genomic landscape of retinoblastoma: a review. Clin Experiment Ophthalmol 42, 33-52 (2014).
48. Zhang, J. et al. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature 481, 329-34 (2012).
49. McEvoy, J. et al. Coexpression of normally incompatible developmental pathways in retinoblastoma genesis. Cancer Cell 20, 260-75 (2011).
50. Kapatai, G. et al. Gene expression profiling identifies different sub-types of retinoblastoma. Br J Cancer 109, 512-25 (2013).
51. Rushlow, D.E. et al. Characterisation of retinoblastomas without RB1 mutations: genomic, gene expression, and clinical studies. Lancet Oncol 14, 327-34 (2013).
52. Xu, X.L. et al. Retinoblastoma has properties of a cone precursor tumor and depends upon cone-specific MDM2 signaling. Cell 137, 1018-31 (2009).
53. Sangwan, M. et al. Established and new mouse models reveal E2f1 and Cdk2 dependency of retinoblastoma, and expose effective strategies to block tumor initiation. Oncogene 31, 5019-28 (2012).
54. Cobrinik, D. in Animal Models of Brain Tumors (eds. Martinez-Murillo, R. & Martinez, A.) 141-152 (Springer, New York, 2013).
55. Bogenmann, E., Lochrie, M.A. & Simon, M.I. Cone cell-specific genes expressed in retinoblastoma. Science 240, 76-8 (1988).
56. Hurwitz, R.L., Bogenmann, E., Font, R.L., Holcombe, V. & Clark, D. Expression of the functional cone phototransduction cascade in retinoblastoma. J Clin Invest 85, 1872-8 (1990).
57. Xu, X.L. et al. Rb suppresses human cone-precursor-derived retinoblastoma tumours. Nature 514, 385-8 (2014).
58. Rootman, D.B. et al. Hand-held high-resolution spectral domain optical coherence tomography in retinoblastoma: clinical and morphologic considerations. Br J Ophthalmol 97, 59-65 (2013).
59. Xu, X.L. et al. Tumor-associated retinal astrocytes promote retinoblastoma cell proliferation through production of IGFBP-5. Am J Pathol 177, 424-35 (2010).
60. Hook, K.E. et al. An integrated genomic approach to identify predictive biomarkers of response to the aurora kinase inhibitor PF-03814735. Mol Cancer Ther 11, 710-9 (2012).
61. Reid, T.W. et al. Characteristics of an established cell line of retinoblastoma. J National Cancer Inst 53:1347-360 (1974).
62. McFall, R.C., Sery, T.W. & Makadon, M. Characterization of a new continuous cell line derived from a human retinoblastoma. Cancer Res 37, 1003-10 (1977).
63. Gallie, B.L., Trogadis, J. & Han, L.-P. in Human Cell Culture (ed. Masters, J.) 361-374 (1999).
64. Gallie, B.L., Albert, D.M., Wong, J.J., Buyukmihci, N. & Puliafito, C.A. Heterotransplantation of retinoblastoma into the athymic "nude" mouse. Invest Ophthalmol Vis Sci 16, 256-9 (1977).
65. Pacal, M. & Bremner, R. Insights from animal models on the origins and progression of retinoblastoma. Curr Mol Med 6, 759-81 (2006).
66. Conkrite, K. et al. miR-17~92 cooperates with RB pathway mutations to promote retinoblastoma. Genes Dev 25, 1734-45 (2011).
67. Windle, J.J. et al. Retinoblastoma in transgenic mice. Nature 343, 665-9. (1990).
68. Pajovic, S. et al. The TAg-RB murine retinoblastoma cell of origin has immunohistochemical features of differentiated Muller glia with progenitor properties. Invest Ophthalmol Vis Sci 52, 7618-24 (2011).
69. Marchong, M.N. et al. Cdh11 acts as a tumor suppressor in a murine retinoblastoma model by facilitating tumor cell death. PLos Genetics 6, e1000923 (2010).
70. Abramson, D.H. et al. Presenting signs of retinoblastoma. J Pediatr 132, 505-8 (1998).
71. Karcioglu, Z.A. Fine needle aspiration biopsy (FNAB) for retinoblastoma. Retina 22, 707-10 (2002).
72. Sanders, T.E. & Smith, M.E. Biopsy of intraocular tumors: a reevaluation. Int Ophthalmol Clin 12, 163-76 (1972).
73. Karcioglu, Z.A., Gordon, R.A. & Karcioglu, G.L. Tumor seeding in ocular fine needle aspiration biopsy. Ophthalmology 92, 1763-7 (1985).
74. Shields, C.L. et al. Lesions simulating retinoblastoma (pseudoretinoblastoma) in 604 cases: results based on age at presentation. Ophthalmology 120, 311-6 (2013).
75. Mullaney, P.B., Karcioglu, Z.A., Huaman, A.M. & al-Mesfer, S. Retinoblastoma associated orbital cellulitis. Br J Ophthalmol 82, 517-21 (1998).
76. Moulin, A.P., Gaillard, M.C., Balmer, A. & Munier, F.L. Ultrasound biomicroscopy evaluation of anterior extension in retinoblastoma: a clinicopathological study. Br J Ophthalmol 96, 337-40 (2012).
77. Rootman, D.B. et al. Hand-held high-resolution spectral domain optical coherence tomography in retinoblastoma: clinical and morphologic considerations. Br J Ophthalmol (2012).
78. Reese, A.B. & Ellsworth, R.M. The evaluation and current concept of retinoblastoma therapy. Trans Am Acad Ophthalmol-Otolaryngol, 164-172 (1963).
79. Murphree, A.L. Intraocular retinoblastoma: the case for a new group classification. Ophthalmology Clinics of North America 18, 41-53 (2005).
80. Shields, C.L. & Shields, J.A. Basic understanding of current classification and management of retinoblastoma. Curr Opin Ophthalmol 17, 228-34 (2006).
81. Shields, C.L. et al. The International Classification of Retinoblastoma predicts chemoreduction success. Ophthalmology 113, 2276-80 (2006).
82. Novetsky, D.E., Abramson, D.H., Kim, J.W. & Dunkel, I.J. Published international classification of retinoblastoma (ICRB) definitions contain inconsistencies--an analysis of impact. Ophthalmic Genet 30, 40-4 (2009).
83. Mallipatna, A., Dimaras, H., Héon, E. & Gallie, B. Published International Classification of Retinoblastoma (Icrb) Definitions Contain Inconsistencies: An Analysis of Impact. Evidence-Based Ophthalmology 10, 183-185 (2009).
84. Group, C.s.O. (2011).
85. Chantada, G. et al. A proposal for an international retinoblastoma staging system. Pediatr Blood Cancer 47, 801-5 (2006).
86. Finger, P.T. et al. in AJCC Cancer Staging Manual. (eds. Edge, S.B., Byrd, D.R., Carducci, M.A. & Compton, C.C.) 561-568 (Springer, New York, NY, 2010).
87. Eagle, R.C., Jr. The pathology of ocular cancer. Eye (Lond) 27, 128-36 (2013).
88. Wilson, M.W., Qaddoumi, I., Billups, C., Haik, B.G. & Rodriguez-Galindo, C. A clinicopathological correlation of 67 eyes primarily enucleated for advanced intraocular retinoblastoma. The British journal of ophthalmology 95, 553-8 (2011).
89. Kaliki, S. et al. Postenucleation adjuvant chemotherapy with vincristine, etoposide, and carboplatin for the treatment of high-risk retinoblastoma. Arch Ophthalmol 129, 1422-7 (2011).
90. Kaliki, S. et al. High-risk retinoblastoma based on international classification of retinoblastoma: analysis of 519 enucleated eyes. Ophthalmology 120, 997-1003 (2013).
91. Sullivan, E.M. et al. Pathologic risk-based adjuvant chemotherapy for unilateral retinoblastoma following enucleation. J Pediatr Hematol Oncol 36, e335-40 (2014).
92. Sastre, X. et al. Proceedings of the consensus meetings from the International Retinoblastoma Staging Working Group on the pathology guidelines for the examination of enucleated eyes and evaluation of prognostic risk factors in retinoblastoma. Arch Pathol Lab Med 133, 1199-202 (2009).
93. Kim, J.W. Retinoblastoma: evidence for postenucleation adjuvant chemotherapy. Int Ophthalmol Clin 55, 77-96 (2015).
94. Grossniklaus, H.E., Kivëla, T., Harbour, J.W. & Finger, P.T. (College of American Pathologists, 2011).
95. Torbidoni, A.V. et al. The cone-rod homeobox transcription factor (CRX) mRNA as a molecular marker in metastatic retinoblastoma. JAMA Ophthalmology (In press).
96. Torbidoni, A.V. et al. Immunoreactivity of the 14F7 Mab raised against N-Glycolyl GM3 Ganglioside in retinoblastoma tumours. Acta Ophthalmol (2014).
97. Gallie, B., Erraguntla, V., Heon, E. & Chan, H. in Pediatric Ophthalmology and Strabismus (eds. Taylor, D. & Hoyt, C.) 486-504 (ELSEVIER, 2004).
98. Xu, K. et al. Preimplantation genetic diagnosis for retinoblastoma: the first reported liveborn. Am J Ophthalmol 137, 18-23 (2004).
99. Dommering, C.J., Moll, A.C., Imhof, S.M., de Die-Smulders, C.E. & Coonen, E. Another liveborn after preimplantation genetic diagnosis for retinoblastoma. Am J Ophthalmol 138, 1088-9 (2004).
100. Dimaras, H. et al. Retinoblastoma CSF metastasis cured by multimodality chemotherapy without radiation. Ophthalmic Genet 30, 121-6 (2009).
101. Bowles, E. et al. Profiling genomic copy number changes in retinoblastoma beyond loss of RB1. Genes Chromosomes Cancer 46, 118-29 (2007).
102. Chantada, G.L. et al. Familial retinoblastoma in developing countries. Pediatr Blood Cancer 53, 338-42 (2009).
103. He, L.Q. et al. Developing clinical cancer genetics services in resource-limited countries: the case of retinoblastoma in Kenya. Public Health Genomics 17, 221-7 (2014).
104. Patel, T. et al. Consumer Digital Cameras: A feasible strategy for the early detection of childhood blindness. Association for Research in Vision and Ophthalmology (ARVO) (2012).
105. Abdolvahabi, A. et al. Colorimetric and longitudinal analysis of leukocoria in recreational photographs of children with retinoblastoma. PLoS One 8, e76677 (2013).
106. McCarthy, M. et al. Comfort First: an evaluation of a procedural pain management programme for children with cancer. Psychooncology 22, 775-82 (2013).
107. Friedman, D.N. et al. Whole-body magnetic resonance imaging (WB-MRI) as surveillance for subsequent malignancies in survivors of hereditary retinoblastoma: a pilot study. Pediatr Blood Cancer 61, 1440-4 (2014).
108. Villani, A. et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: a prospective observational study. Lancet Oncol 12, 559-67 (2011).
109. Shields, C.L. et al. Retinoblastoma frontiers with intravenous, intra-arterial, periocular, and intravitreal chemotherapy. Eye (Lond) 27, 253-64 (2013).
110. Eng, C. et al. Mortality from second tumors among long-term survivors of retinoblastoma. J Natl Cancer Inst 85, 1121-8 (1993).
111. MacCarthy, A. et al. Non-ocular tumours following retinoblastoma in Great Britain 1951 to 2004. Br J Ophthalmol 93, 1159-62 (2009).
112. Francis, J.H. et al. Efficacy and Toxicity of Second-Course Ophthalmic Artery Chemosurgery for Retinoblastoma. Ophthalmology (2015).
113. Hamama-Raz, Y., Rot, I. & Buchbinder, E. The coping experience of parents of a child with retinoblastoma-malignant eye cancer. J Psychosoc Oncol 30, 21-40 (2012).
114. Soliman, S.E., Dimaras, H., Souka, A.A., Ashry, M.H. & Gallie, B.L. Socioeconomic and Psychological Impact of Treatment for Unilateral Intraocular Retinoblastoma. French Journal of Ophthalmology (In Press).
115. Manjandavida, F.P., Honavar, S.G., Reddy, V.A. & Khanna, R. Management and outcome of retinoblastoma with vitreous seeds. Ophthalmology 121, 517-24 (2014).
116. Gunduz, K. et al. Causes of chemoreduction failure in retinoblastoma and analysis of associated factors leading to eventual treatment with external beam radiotherapy and enucleation. Ophthalmology 111, 1917-24 (2004).
117. Shields, C.L. et al. Iodine 125 plaque radiotherapy as salvage treatment for retinoblastoma recurrence after chemoreduction in 84 tumors. Ophthalmology 113, 2087-92 (2006).
118. Abouzeid, H. et al. (106)Ruthenium brachytherapy for retinoblastoma. Int J Radiat Oncol Biol Phys 71, 821-8 (2008).
119. Pica, A. et al. Preliminary experience in treatment of papillary and macular retinoblastoma: evaluation of local control and local complications after treatment with linear accelerator-based stereotactic radiotherapy with micromultileaf collimator as second-line or salvage treatment after chemotherapy. Int J Radiat Oncol Biol Phys 81, 1380-6 (2011).
120. Chan, M.P., Hungerford, J.L., Kingston, J.E. & Plowman, P.N. Salvage external beam radiotherapy after failed primary chemotherapy for bilateral retinoblastoma: rate of eye and vision preservation. Br J Ophthalmol 93, 891-4 (2009).
121. Berry, J.L. et al. Long-term outcomes of Group D eyes in bilateral retinoblastoma patients treated with chemoreduction and low-dose IMRT salvage. Pediatr Blood Cancer 60, 688-93 (2013).
122. Munier, F.L. et al. New developments in external beam radiotherapy for retinoblastoma: from lens to normal tissue-sparing techniques. Clin Experiment Ophthalmol 36, 78-89 (2008).
123. Mouw, K.W. et al. Proton radiation therapy for the treatment of retinoblastoma. Int J Radiat Oncol Biol Phys 90, 863-9 (2014).
124. Shields, C.L. et al. Chemoreduction plus focal therapy for retinoblastoma: factors predictive of need for treatment with external beam radiotherapy or enucleation. Am J Ophthalmol 133, 657-64 (2002).
125. Chantada, G.L. et al. Risk factors for extraocular relapse following enucleation after failure of chemoreduction in retinoblastoma. Pediatr Blood Cancer 49, 256-60 (2007).
126. Zhao, J. et al. Pre-enucleation chemotherapy for eyes severely affected by retinoblastoma masks risk of tumor extension and increases death from metastasis. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 29, 845-51 (2011).
127. Abramson, D.H. & Schefler, A.C. Update on retinoblastoma. Retina 24, 828-48 (2004).
128. Schaiquevich, P. et al. Intra-arterial chemotherapy is more effective than sequential periocular and intravenous chemotherapy as salvage treatment for relapsed retinoblastoma. Pediatr Blood Cancer 60, 766-70 (2013).
129. Shields, C.L. et al. Intra-arterial chemotherapy for retinoblastoma in 70 eyes: outcomes based on the international classification of retinoblastoma. Ophthalmology 121, 1453-60 (2014).
130. Abramson, D.H. et al. Intra-arterial chemotherapy for retinoblastoma in eyes with vitreous and/or subretinal seeding: 2-year results. Br J Ophthalmol 96, 499-502 (2012).
131. Suzuki, S., Yamane, T., Mohri, M. & Kaneko, A. Selective ophthalmic arterial injection therapy for intraocular retinoblastoma: the long-term prognosis. Ophthalmology 118, 2081-7 (2011).
133. Schaiquevich, P. et al. Pharmacokinetic analysis of topotecan after superselective ophthalmic artery infusion and periocular administration in a porcine model. Retina 32, 387-95 (2012).
134. Munier, F.L. et al. Intravitreal chemotherapy for vitreous disease in retinoblastoma revisited: from prohibition to conditional indications. Br J Ophthalmol 96, 1078-83 (2012).
135. Francis, J.H. et al. The classification of vitreous seeds in retinoblastoma and response to intravitreal melphalan. Ophthalmology (2015).
136. Shields, C.L. et al. Intravitreal melphalan for persistent or recurrent retinoblastoma vitreous seeds: preliminary results. JAMA Ophthalmol 132, 319-25 (2014).
137. Munier, F.L. Classification and management of seeds in retinoblastoma. Ellsworth Lecture Ghent August 24th 2013. Ophthalmic Genet 35, 193-207 (2014).
138. Rappaport, B.A., Suresh, S., Hertz, S., Evers, A.S. & Orser, B.A. Anesthetic neurotoxicity--clinical implications of animal models. N Engl J Med 372, 796-7 (2015).
139. Mallipatna, A.C., Sutherland, J.E., Gallie, B.L., Chan, H. & Heon, E. Management and outcome of unilateral retinoblastoma. J AAPOS 13, 546-50 (2009).
140. Mourits, D.L., Hartong, D.T., Bosscha, M.I., Kloos, R.J. & Moll, A.C. Worldwide enucleation techniques and materials for treatment of retinoblastoma: an international survey. PLoS One 10, e0121292 (2015).
141. Shome, D., Honavar, S.G., Raizada, K. & Raizada, D. Implant and prosthesis movement after enucleation: a randomized controlled trial. Ophthalmology 117, 1638-44 (2010).
142. Yadava, U., Sachdeva, P. & Arora, V. Myoconjunctival enucleation for enhanced implant motility. result of a randomised prospective study. Indian J Ophthalmol 52, 221-6 (2004).
143. Vincent, A.L., Webb, M.C., Gallie, B.L. & Heon, E. Prosthetic conformers: a step towards improved rehabilitation of enucleated children. Clin Experiment Ophthalmol 30, 58-9 (2002).
144. Gallie, B.L. et al. Chemotherapy with focal therapy can cure intraocular retinoblastoma without radiotherapy. Arch Ophthalmol 114, 1321-8 (1996).
145. Kingston, J.E., Hungerford, J.L., Madreperla, S.A. & Plowman, P.N. Results of combined chemotherapy and radiotherapy for advanced intraocular retinoblastoma. Archives of Ophthalmology 114, 1339-1343 (1996).
146. Chan, H.S. et al. Combining cyclosporin with chemotherapy controls intraocular retinoblastoma without requiring radiation. Clin Cancer Res 2, 1499-508 (1996).
147. Chan, H.S.L. (ClinicalTrials.gov, 2005).
148. Dunkel, I.J. et al. A phase II trial of carboplatin for intraocular retinoblastoma. Pediatr Blood Cancer (2007).
149. Jehanne, M. et al. Analysis of ototoxicity in young children receiving carboplatin in the context of conservative management of unilateral or bilateral retinoblastoma. Pediatr Blood Cancer 52, 637-43 (2009).
150. Wong, J.R. et al. Risk of subsequent malignant neoplasms in long-term hereditary retinoblastoma survivors after chemotherapy and radiotherapy. J Clin Oncol 32, 3284-90 (2014).
151. Draper, G.J., Sanders, B.M. & Kingston, J.E. Second primary neoplasms in patients with retinoblastoma. Br J Cancer 53, 661-71 (1986).
152. Gombos, D.S. et al. Secondary acute myelogenous leukemia in patients with retinoblastoma: is chemotherapy a factor? Ophthalmology 114, 1378-83 (2007).
153. Smith, M.A. et al. Secondary leukemia or myelodysplastic syndrome after treatment with epipodophyllotoxins. J Clin Oncol 17, 569-77 (1999).
154. Shields, C.L. et al. Factors predictive of recurrence of retinal tumors, vitreous seeds, and subretinal seeds following chemoreduction for retinoblastoma. Arch Ophthalmol 120, 460-4 (2002).
155. Gombos, D.S., Kelly, A., Coen, P.G., Kingston, J.E. & Hungerford, J.L. Retinoblastoma treated with primary chemotherapy alone: the significance of tumour size, location, and age. Br J Ophthalmol 86, 80-3 (2002).
156. Lee, V. et al. Globe conserving treatment of the only eye in bilateral retinoblastoma. Br J Ophthalmol 87, 1374-80 (2003).
157. Shields, C.L. et al. Macular retinoblastoma managed with chemoreduction: analysis of tumor control with or without adjuvant thermotherapy in 68 tumors. Arch Ophthalmol 123, 765-73 (2005).
158. Demirci, H., Shields, C.L., Meadows, A.T. & Shields, J.A. Long-term visual outcome following chemoreduction for retinoblastoma. Arch Ophthalmol 123, 1525-30 (2005).
159. Narang, S., Mashayekhi, A., Rudich, D. & Shields, C.L. Predictors of long-term visual outcome after chemoreduction for management of intraocular retinoblastoma. Clin Experiment Ophthalmol 40, 736-42 (2012).
160. Abramson, D.H., Dunkel, I.J., Brodie, S.E., Kim, J.W. & Gobin, Y.P. A phase I/II study of direct intraarterial (ophthalmic artery) chemotherapy with melphalan for
161. Klufas, M.A. et al. Intra-arterial chemotherapy as a treatment for intraocular retinoblastoma: alternatives to direct ophthalmic artery catheterization. AJNR Am J Neuroradiol 33, 1608-14 (2012).
162. Abramson, D.H., Dunkel, I.J., Brodie, S.E., Marr, B. & Gobin, Y.P. Bilateral superselective ophthalmic artery chemotherapy for bilateral retinoblastoma: tandem therapy. Arch Ophthalmol 128, 370-2 (2010).
163. Grigorovski, N. et al. Use of intra-arterial chemotherapy for retinoblastoma: results of a survey. Int J Ophthalmol 7, 726-30 (2014).
164. Abramson, D.H. et al. Ophthalmic artery chemosurgery for less advanced intraocular retinoblastoma: five year review. PLoS ONE 7, e34120 (2012).
165. Palioura, S. et al. Ophthalmic artery chemosurgery for the management of retinoblastoma in eyes with extensive (>50%) retinal detachment. Pediatr Blood Cancer 59, 859-64 (2012).
166. Abramson, D.H. Retinoblastoma in the 20th century: past success and future challenges the Weisenfeld lecture. Invest Ophthalmol Vis Sci 46, 2683-91 (2005).
167. Abramson, D.H., Beaverson, K.L., Chang, S.T., Dunkel, I.J. & McCormick, B. Outcome following initial external beam radiotherapy in patients with Reese-Ellsworth group Vb retinoblastoma. Arch Ophthalmol 122, 1316-23 (2004).
168. Brodie, S.E. et al. ERG monitoring of retinal function during systemic chemotherapy for retinoblastoma. Br J Ophthalmol 96, 877-80 (2012).
169. Brodie, S.E., Pierre Gobin, Y., Dunkel, I.J., Kim, J.W. & Abramson, D.H. Persistence of retinal function after selective ophthalmic artery chemotherapy infusion for retinoblastoma. Doc Ophthalmol 119, 13-22 (2009).
171. Francis, J.H. et al. Electroretinogram monitoring of dose-dependent toxicity after ophthalmic artery chemosurgery in retinoblastoma eyes: six year review. PLoS One 9, e84247 (2014).
172. Abramson, D.H. et al. Ophthalmic artery chemosurgery for retinoblastoma prevents new intraocular tumors. Ophthalmology 120, 560-5 (2013).
173. Gobin, Y.P., Dunkel, I.J., Marr, B.P., Brodie, S.E. & Abramson, D.H. Intra-arterial Chemotherapy for the Management of Retinoblastoma: Four-Year Experience. Arch Ophthalmol (2011).
174. Abramson, D.H. Chemosurgery for retinoblastoma: what we know after 5 years. Arch Ophthalmol 129, 1492-4 (2011).
175. Gobin, Y.P. et al. Combined, sequential intravenous and intra-arterial chemotherapy (bridge chemotherapy) for young infants with retinoblastoma. PLoS ONE 7, e44322 (2012).
176. Abramson, D.H., Frank, C.M. & Dunkel, I.J. A phase I/II study of subconjunctival carboplatin for intraocular retinoblastoma. Ophthalmology 106, 1947-50 (1999).
177. Mulvihill, A. et al. Ocular motility changes after subtenon carboplatin chemotherapy for retinoblastoma. Arch Ophthalmol 121, 1120-4 (2003).
179. Ghassemi, F. & Amoli, F.A. Pathological findings in enucleated eyes after intravitreal melphalan injection. Int Ophthalmol 34, 533-40 (2014).
180. Ghassemi, F., Shields, C.L., Ghadimi, H., Khodabandeh, A. & Roohipoor, R. Combined intravitreal melphalan and topotecan for refractory or recurrent vitreous seeding from retinoblastoma. JAMA Ophthalmol 132, 936-41 (2014).
181. Brinkman, T.M. et al. Cognitive function and social attainment in adult survivors of retinoblastoma: a report from the St. Jude Lifetime Cohort Study. Cancer 121, 123-31 (2015).
182. Willard, V.W. et al. Developmental and adaptive functioning in children with retinoblastoma: a longitudinal investigation. J Clin Oncol 32, 2788-93 (2014).
183. Tobin, M., Hill, E. & Hill, J. Retinoblastoma and superior verbal IQ scores? British Journal of Visual Impairment 28, 7-18 (2010).
184. Kelly, K.R., McKetton, L., Schneider, K.A., Gallie, B.L. & Steeves, J.K. Altered anterior visual system development following early monocular enucleation. Neuroimage Clin 4, 72-81 (2014).
185. Kelly, K.R., DeSimone, K.D., Gallie, B.L. & Steeves, J.K. Increased cortical surface area and gyrification following long-term survival from early monocular enucleation. Neuroimage Clin 7, 297-305 (2015).
186. Ing, C. et al. Long-term differences in language and cognitive function after childhood exposure to anesthesia. Pediatrics 130, e476-85 (2012).
187. Rappaport, B., Mellon, R.D., Simone, A. & Woodcock, J. Defining safe use of anesthesia in children. N Engl J Med 364, 1387-90 (2011).
188. Khin Hla, T. et al. Perception of pediatric pain: a comparison of postoperative pain assessments between child, parent, nurse, and independent observer. Paediatr Anaesth 24, 1127-31 (2014).
189. Romsing, J., Dremstrup Skovgaard, C., Friis, S.M. & Henneberg, S.W. Procedure-related pain in children in a Danish University Hospital. A qualitative study. Paediatr Anaesth 24, 602-7 (2014).
190. Fox, J.K., Halpern, L.F., Dangman, B.C., Giramonti, K.M. & Kogan, B.A. Children's anxious reactions to an invasive medical procedure: The role of medical and non-medical fears. J Health Psychol (2014).
191. Chen, E., Zeltzer, L.K., Craske, M.G. & Katz, E.R. Children's memories for painful cancer treatment procedures: implications for distress. Child Dev 71, 933-47 (2000).
192. Silver, G.H., Kearney, J.A., Kutko, M.C. & Bartell, A.S. Infant delirium in pediatric critical care settings. Am J Psychiatry 167, 1172-7 (2010).
193. Caes, L. et al. The relationship between parental catastrophizing about child pain and distress in response to medical procedures in the context of childhood cancer treatment: a longitudinal analysis. J Pediatr Psychol 39, 677-86 (2014).
194. Dahlquist, L.M. & Pendley, J.S. When distraction fails: parental anxiety and children's responses to distraction during cancer procedures. J Pediatr Psychol 30, 623-8 (2005).
195. van Dijk, J. et al. Behavioural functioning of retinoblastoma survivors. Psychooncology 18, 87-95 (2009).
196. van Dijk, J. et al. Coping strategies of retinoblastoma survivors in relation to behavioural problems. Psychooncology 18, 1281-9 (2009).
197. Cloitre, M. et al. A developmental approach to complex PTSD: childhood and adult cumulative trauma as predictors of symptom complexity. J Trauma Stress 22, 399-408 (2009).
198. van der Kolk, B. Developmental Trauma Disorder. Psychiatric Annuals, 401-408 (2005).
199. Kaplan, L.M., Kaal, K.J., Bradley, L. & Alderfer, M.A. Cancer-related traumatic stress reactions in siblings of children with cancer. Fam Syst Health 31, 205-17 (2013).
200. Buchbinder, D. et al. Psychological outcomes of siblings of cancer survivors: a report from the Childhood Cancer Survivor Study. Psychooncology 20, 1259-68 (2011).
201. Gold, J.I. in RESEARCHLA Blog (Children's Hospital Los Angeles The Saban Research Institute, Los Angeles).
202. Committee on Hospital, C. & Child Life, C. Child life services. Pediatrics 133, e1471-8 (2014).
203. Care, C.o.H. & Council, C.L. Child life services. Pediatrics 133, e1471-8 (2014).
204. Landier, W. & Tse, A.M. Use of complementary and alternative medical interventions for the management of procedure-related pain, anxiety, and distress in pediatric oncology: an integrative review. J Pediatr Nurs 25, 566-79 (2010).
205. Shockey, D.P. et al. Preprocedural distress in children with cancer: an intervention using biofeedback and relaxation. J Pediatr Oncol Nurs 30, 129-38 (2013).
206. Wohlheiter, K.A. & Dahlquist, L.M. Interactive versus passive distraction for acute pain management in young children: the role of selective attention and development. J Pediatr Psychol 38, 202-12 (2013).
207. Willis, D. & Barry, P. Audiovisual interventions to reduce the use of general anaesthesia with paediatric patients during radiation therapy. J Med Imaging Radiat Oncol 54, 249-55 (2010).
208. O'Callaghan, C., Sexton, M. & Wheeler, G. Music therapy as a non-pharmacological anxiolytic for paediatric radiotherapy patients. Australas Radiol 51, 159-62 (2007).
209. Post-White, J. et al. Massage therapy for children with cancer. J Pediatr Oncol Nurs 26, 16-28 (2009).
210. Sposito, A.M. et al. Coping Strategies Used by Hospitalized Children With Cancer Undergoing Chemotherapy. J Nurs Scholarsh (2015).
211. Hildenbrand, A.K., Clawson, K.J., Alderfer, M.A. & Marsac, M.L. Coping with pediatric cancer: strategies employed by children and their parents to manage cancer-related stressors during treatment. J Pediatr Oncol Nurs 28, 344-54 (2011).
212. van Dijk, J. et al. Restrictions in daily life after retinoblastoma from the perspective of the survivors. Pediatr Blood Cancer 54, 110-5 (2010).
213. Friedman, D.N. et al. Long-term medical outcomes in survivors of extra-ocular retinoblastoma: the Memorial Sloan-Kettering Cancer Center (MSKCC) experience. Pediatr Blood Cancer 60, 694-9 (2013).
214. van Dijk, J. et al. Quality of life of adult retinoblastoma survivors in the Netherlands. Health Qual Life Outcomes 5, 30 (2007).
215. MacCarthy, A. et al. Second and subsequent tumours among 1927 retinoblastoma patients diagnosed in Britain 1951-2004. Br J Cancer 108, 2455-63 (2013).
216. Shinohara, E.T., DeWees, T. & Perkins, S.M. Subsequent malignancies and their effect on survival in patients with retinoblastoma. Pediatr Blood Cancer 61, 116-9 (2014).
217. Ford, J.S., Chou, J.F. & Sklar, C.A. Attendance at a survivorship clinic: impact on knowledge and psychosocial adjustment. J Cancer Surviv 7, 535-43 (2013).
218. Schulz, C.J., Riddle, M.P., Valdimirsdottir, H.B., Abramson, D.H. & Sklar, C.A. Impact on survivors of retinoblastoma when informed of study results on risk of second cancers. Med Pediatr Oncol 41, 36-43 (2003).
219. Rowland, E. & Metcalfe, A. Communicating inherited genetic risk between parent and child: a meta-thematic synthesis. Int J Nurs Stud 50, 870-80 (2013).
220. Clarke, S.A., Sheppard, L. & Eiser, C. Mothers' Explanations of Communicating Past Health and Future Risks to Survivors of Childhood Cancer. Clinical Child Psychology and Psychiatry 13, 157-170 (2008).
221. Grossmann, C., Sanders, J. & English, R.A. KNOWLEDGE GENERATION IN A LEARNING HEALTH SYSTEM: Workshop Summary (National Academies Press, Washington, DC 20001, 2013).
222. Jha, P. et al. Prospective study of one million deaths in India: rationale, design, and validation results. PLoS Med 3, e18 (2006).
223. Farmer, P. Who Lives and Who Dies? Medical Examiner (2015).
224. Laurie, N.A. et al. Inactivation of the p53 pathway in retinoblastoma. Nature 444, 61-6 (2006).
225. Pritchard, E.M. et al. Pharmacokinetics and efficacy of the spleen tyrosine kinase inhibitor r406 after ocular delivery for retinoblastoma. Pharm Res 31, 3060-72 (2014).
226. Mahida, J.P. et al. A synergetic screening approach with companion effector for combination therapy: application to retinoblastoma. PLoS ONE 8, e59156 (2013).
227. Taich, P. et al. Clinical pharmacokinetics of intra-arterial melphalan and topotecan combination in patients with retinoblastoma. Ophthalmology 121, 889-97 (2014).
228. Carcaboso, A.M. et al. Episcleral implants for topotecan delivery to the posterior segment of the eye. Invest Ophthalmol Vis Sci 51, 2126-34 (2010).
229. Kang, S.J., Durairaj, C., Kompella, U.B., O'Brien, J.M. & Grossniklaus, H.E. Subconjunctival nanoparticle carboplatin in the treatment of murine retinoblastoma. Arch Ophthalmol 127, 1043-7 (2009).
230. Jockovich, M.E., Murray, T.G., Escalona-Benz, E., Hernandez, E. & Feuer, W. Anecortave acetate as single and adjuvant therapy in the treatment of retinal tumors of LH(BETA)T(AG) mice. Invest Ophthalmol Vis Sci 47, 1264-8 (2006).
231. Pina, Y. et al. Focal, periocular delivery of 2-deoxy-D-glucose as adjuvant to chemotherapy for treatment of advanced retinoblastoma. Invest Ophthalmol Vis Sci 51, 6149-56 (2010).
232. Laurent, V.E. et al. Detection of minimally disseminated disease in the cerebrospinal fluid of children with high-risk retinoblastoma by reverse transcriptase-polymerase chain reaction for GD2 synthase mRNA. Eur J Cancer 49, 2892-9 (2013).