What you should remember for today Algorithms are mechanical recipe to compute something. Structure of prokaryotic genes. Each genome has uneven codon preferences. Basic form of the Bayes Theorem (earlier this term?)
Transcript
What you should remember for today
Algorithms are mechanical recipe to compute something.
Structure of prokaryotic genes.
Each genome has uneven codon preferences.
Basic form of the Bayes Theorem (earlier this term?)
Learning outcomes for today?
1.Interpret algorithms expressed as pseudocode.
2.Describe the limitations of assuming an equivalence
between long ORFs and expressed gene products in
prokaryotes.
3.Compute and interpret the following prediction performance
measures:sensitivity, specificity and Fscore.
4.Desribes how GeneMark learns the structure of genes in
prokaryotes.
5.Explain why GLIMMER attempts to consider data from
longer kmers.
Open Reading FramesFi
ndin
g G
e nes
v.1
.0 ORF : Open Reading Frame. Since codons are words of 3 characters. Finding a START and one of three possible STOP codons constitute a reading frame.
STOP codonTAA, TGA, TAG
START codonATG (also code for protein character M)
Open Reading FramesFi
ndin
g G
e nes
v.1
.0 There are 6 possible frames : Some organisms even manage to overlap genes using two reading frames!
Sequence as in the DB
TCCAACTCGGGGTCCGCATCGCTCCGCCGGCGACCGACGAAGCCG
Three first frames TCC AAC TCG GGG TCC GCA TCG CTC CGC CGG CGA CCG ACG AAG CCG A T CCA ACT CGG GGT CCG CAT CGC TCC GCC GGC GAC CGA CGA AGC CGATC CAA CTC GGG GTC CGC ATC GCT CCG CCG GCG ACC GAC GAA GCC GA
But DNA is a double strand… 5’-TCCAACTCGGGGTCCGCATCGCTCCGCCGGCGACCGACGAAGCCG-3’ 3’-AGGTTGAGCCCCAGGCGTAGCGTGGCGGCCGCTGGCTGCTTCGGC-5’
ORF ≠ GeneFi
ndin
g G
e nes
v.1
.0
In bacteria : It is OK to assume that all genes are ORFs. Not all ORFs are genes!
● Find the longest possible sequence beginning with an ATG, and terminating by a TAA, TAG, TGA.
● There may be multiple ATG inside the gene, but only a single stop codon.
● Real genes will have a regulatory regions upstream of ORF. Use pattern detection to do this.
● Real genes are typically 100-500 codons long.
● Real genes tend to have a bias in the composition of their characters.
Control sequencesFi
ndin
g G
e nes
v.1
.0
Regulatory sequence : Promote the translation of a gene. Sequence found in 5’ of gene.
70% in Baker’s yeast (Saccharomyces cerevisiae )25% in Fruit fly (Drosophila melanogaster)13% in flowering plants.
Intron : Discontinuity in the sequence of a gene.
~150 bp exon (coding DNA) interspaced with introns (noncoding; few hundreds to thousand bp).
95% human genes have at least 1 intron.80% fruit fly5 % Yeast
A procedure to identify ORFs in prokaryotic genomes
Starting at the first base in the genome, find the position of the first START codon. Once a START codon is found, find
the position of the first STOP codon that is in the same frame. Consider all nucleotides including and inbetween the position of the first nucleotide in START and the last nuclotide in STOP
to be an ORF. Repeat from the end of this gene until the whole genome has been scanned.
Tentative Pseudocode
lastSTOP = 0start = 0stop = 0
for as long as start is smaller than the length of the genome SET start as the index of the next ATG after lastSTOP. FIND the next inframe STOP codon SET stop as the index of the third base of the STOP codon WRITE start and stop
ORFs and genes aren't the same
It is straightforward to determine the probability of and ORF to be spurious if you know its length.
lastSTOP = 0start = 0stop = 0
while start != 1: # Find the first ATG after lastSTOP start = genome.find("ATG", lastSTOP)
# Find the first inframe STOP codon for i in range(start, len(genome), 3): codon = genome[i:i+3] if codon in ["TAA","TGA","TAG"]: stop = i + 2 break
print start, stop, stop start
Tentative Pseudocode II
lastSTOP = 0start = 0stop = 0minGenLen = 300
for as long as start is smaller than the length of the genome SET start as the index of the next ATG after lastSTOP. FIND the next inframe STOP codon SET stop as the index of the third base of the STOP codon
IF (stop start) IS LARGER THAN minGenLen WRITE start and stop
SET lastSTOP to stop OTHERWISE INCREMENT lastSTOP by 1
lastSTOP = 0start = 0stop = 0
while start != 1: # Find the first ATG after lastSTOP start = genome.find("ATG", lastSTOP)
# Find the first inframe STOP codon for i in range(start, len(genome), 3): codon = genome[i:i+3] if codon in ["TAA","TGA","TAG"]: stop = i + 2 break
# Is the ORF long enough? if stop start >= minGeneLength: print start, stop, stop start lastSTOP = stop + 1 else: lastSTOP = lastSTOP + 1
The Confusion Matrix
True Positives(TP)
False Positive(FP)
False Negative(FN)
True Negative(TN)
Real Codon Not Real Codon
Predicted as Codon
Ignored as Codon
Sensitivity
(TP)(TP) (FP)
(FN)(FN) (TN)
Real Not Real
Predicted
Ignored
The proportion of real codons that are correctly classified as such.
Sn = TP / (TP + FN)
Specificity
(TP)(TP) (FP)(FP)
(FN) (TN)
Real Not Real
Predicted
Ignored
The proportion of predicted codons that are real ORFboundaries.
Sp = TP / (TP + FP)
Fscore
(TP)(TP) (FP)(FP)
(FN)(FN) (TN)
Real Not Real
Predicted
Ignored
The harmonic mean of Sn andSp.
F = 2 * Sn * Sp / (Sn + Sp)
An example
100100 5050
1010 1000
Real Not Real
Predicted
Ignored
Sn = 100 / (100 + 10) = 0.909
Sp = 100 / (100 + 50) = 0.666
F = (2 * .9 * .66) / (.9 + .66)F = 0.769
This classifier is thus more sensitive than specific.
Using Signal
Given two ORFs of exactly the same length. One is a gene while the other is an “accident”. Tell them apart.
● There can be a difference in signal.● This difference will be genomedependent.● The classifier needs to learn what is this difference.
● This must be done without assuming that the ORF is known.
Open Reading FramesFi
ndin
g G
e nes
v.1
.0 When given a fragment of sequences, it can be in either 6 frames, or NOT in a gene. The classification is thus a 7way classification problem.
Sequence as in the DB
TCCAACTCGGGGTCCGCATCGCTCCGCCGGCGACCGACGAAGCCG
Three first frames TCC AAC TCG GGG TCC GCA TCG CTC CGC CGG CGA CCG ACG AAG CCG A T CCA ACT CGG GGT CCG CAT CGC TCC GCC GGC GAC CGA CGA AGC CGATC CAA CTC GGG GTC CGC ATC GCT CCG CCG GCG ACC GAC GAA GCC GA
But DNA is a double strand… 5’-TCCAACTCGGGGTCCGCATCGCTCCGCCGGCGACCGACGAAGCCG-3’ 3’-AGGTTGAGCCCCAGGCGTAGCGTGGCGGCCGCTGGCTGCTTCGGC-5’
Training set
Dataset for which we already know the classification and is assumed to be a uniform sample of the data to be classified in the future.
● BLAST results of known genes (prior knowledge).● Sample from long ORFs.
GeneMark
Is a Markov Model based approach.
A statistically averse personmay prefer the term: kmer
An approach that is somewhatsimilar to the de Bruijn graphs.
GeneMark training
Principle
Store the probability of allPossible hexamer (6mer) inEach possible frames, andIn noncoding regions.
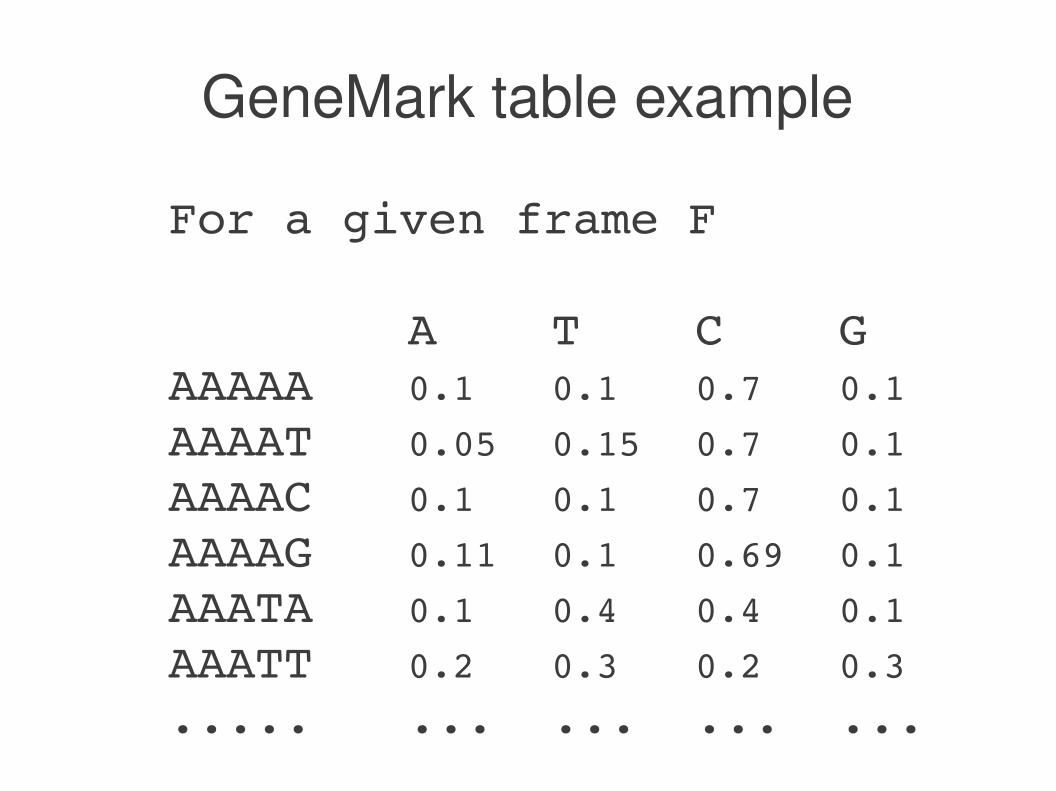
GeneMark table example
For a given frame F
A T C GAAAAA 0.1 0.1 0.7 0.1
AAAAT 0.05 0.15 0.7 0.1
AAAAC 0.1 0.1 0.7 0.1
AAAAG 0.11 0.1 0.69 0.1
AAATA 0.1 0.4 0.4 0.1
AAATT 0.2 0.3 0.2 0.3
..... ... ... ... ...
Training for GeneMarkDERIVE probabilities for frame F
bndrs <==CREATE A list of gene boundaries sampling the frame F. probs <== CREATE a table enumerating each possible kmersASSOCIATE a count of 0 for A,T,C,G for each kmers.
for each gene boundary pairs in bndrs orf <== SLICE the matching genome sequence for each hexamer in orf Nuc <== Last nucleotide of the word prefix <== First k nucleotides ADD 1 to the count of nucleotide Nuc occuring after prefix
CONVERT all counts into probabilities
GeneMark table example
For a given frame F
A T C GAAAAA 0.1 0.1 0.7 0.1
AAAAT 0.05 0.15 0.7 0.1
AAAAC 0.1 0.1 0.7 0.1
AAAAG 0.11 0.1 0.69 0.1
AAATA 0.1 0.4 0.4 0.1
AAATT 0.2 0.3 0.2 0.3
..... ... ... ... ...
Classifying with GeneMarkFind the frame F with the highest posterior probability
INPUT: 1 sequence s of lenght 12, 7 tables of hexamer frequencies T = {F1,F2,F3,F4,F5,F6,NC} a sequence sOUTPUT: a coding frame or the answer noncoding
bestFrame = NonebestProb = 0.0
for each frame F in [1,2,3,4,5,6,nc]: freq <== GET table in T which correspond to F Lkl <== EVALUATE the likelihood of s given freq prior <== SET to 0.5 if freq is NC, or 1/12 otherwise P <== EVALUATE the posterior probability of frame given s if P > bestProb: bestFrame = FR bestProb = P
RETURN bestFrame
Classifying with GeneMark
Principle
For a sequence s of length 12: Compute the likelihood of s in all 7 frames
Bayesian Posterior Probability(that the segment is in frame 3, given sequence s)
s = abcdefghi
Point of discussion about GeneMark
● 7way classification will pick up on genes, regardless of frames. ●Supervised training – is the training set appropriate?● Is 6mer appropriate?
● What if we use longer Kmers? ● What if we use shorter Kmers?
Glimmer 3 – State of the art
● Interpolated Markov Model● Training set based on long ORFs. ● Consider all kvalues from 2 to 8.● Attempt to compute the likelihood with k = 8 IF the data is not too sparse (400 instances counted).● Use interpolation to when the data for k is different than for k1.● 95% Accuracy !