Frontline: Antigen co-encapsulated with adjuvants efficiently drive protective T cell immunity Antje Heit 1 , Frank Schmitz 1 , Tobias Haas 1 , Dirk H. Busch 1,2 and Hermann Wagner 1 1 Institute for Medical Microbiology, Immunology and Hygiene, Technical University of Munich, Munich, Germany 2 GSF Clinical Research group "Antigen Specific Immunotherapy", Munich, Germany Compared to "live" vaccines, the immunogenicity of "subunit" vaccines based on recombinant antigen (Ag) is poor, presumably because exogenous Ag fails to effectively access the endosomal Ag-processing pathways of Ag-presenting cells (APC). To overcome this limitation, we exploited biodegradable poly(lactic-co-glycolic) micro- spheres (MP) co-entrapping Ag and Toll-like receptor (TLR) 9 or 7 ligands as an endosomal delivery device. In vitro, microspheres were rapidly phagocytosed by APC and translocated into phago-endosomal compartments, followed by degradation of the Ag and concurrent activation of endosomal TLR. As a consequence, full maturation of and cytokine secretion by APC as well as Ag-cross-presentation ensued. In vivo, "loaded" microspheres triggered clonal expansion of primary and secondary Ag-specific CD4 and CD8 T cells. The efficacy of CD8 T cell cross-priming was comparable to that of live vectors. The potency of T cell vaccination was demonstrated by protective and therapeutic interventions using infection- and tumor-model systems. These preclinical "subunit" vaccination data thus recommend MP as a generally applicable and powerful endosomal delivery device of exogenous Ag plus TLR-based adjuvants to vaccinate for protective and therapeutic CD4 and CD8 T cell immunity. Supporting information for this article is available at http://www.wiley-vch.de/contents/jc_2040/2007/37169_s.pdf Introduction Vaccination has been regarded as one of the safest and most cost-effective strategies to prevent and treat infectious diseases [1]. Improvements, however, are required against infectious diseases, for which no or only inadequate vaccines are available, including tubercu- losis (Tb), malaria and HIV. Moreover, some chronic diseases might be prevented with novel vaccines, an example being Helicobacter pylori infection, which is Correspondence: Hermann Wagner, Institute for Medical Microbiology, Immunology and Hygiene, Trogerstrasse 30, 81675 Munich, Germany Fax: +49-89-4140-4139 e-mail: [email protected]Received 12/2/07 Revised 26/4/07 Accepted 14/6/07 [DOI 10.1002/eji.200737169] Key words: Dendritic cells T cells Vaccination Abbreviations: CpG-ODN: CpG-oligodeoxynucleotide DQ-OVA: pH-insensitive self-quenched OVA conjugate Lamp1: lysosomal-associated membrane protein-1 LLO: listeriolysin O mDC: myeloid DC MP: microspheres PLG: poly(lactic-co-glycolic) Eur. J. Immunol. 2007. 37: 2063–2074 Highlights 2063 f 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu
Transcript
Frontline:
Antigen co-encapsulated with adjuvants efficiently driveprotective T cell immunity
Antje Heit1, Frank Schmitz1, Tobias Haas1, Dirk H. Busch1,2 andHermann Wagner1
1 Institute for Medical Microbiology, Immunology and Hygiene, Technical University ofMunich, Munich, Germany
2 GSF Clinical Research group "Antigen Specific Immunotherapy", Munich, Germany
Compared to "live" vaccines, the immunogenicity of "subunit" vaccines based onrecombinant antigen (Ag) is poor, presumably because exogenous Ag fails to effectivelyaccess the endosomal Ag-processing pathways of Ag-presenting cells (APC). Toovercome this limitation, we exploited biodegradable poly(lactic-co-glycolic) micro-spheres (MP) co-entrapping Ag and Toll-like receptor (TLR) 9 or 7 ligands as anendosomal delivery device. In vitro, microspheres were rapidly phagocytosed by APCand translocated into phago-endosomal compartments, followed by degradation of theAg and concurrent activation of endosomal TLR. As a consequence, full maturation ofand cytokine secretion by APC as well as Ag-cross-presentation ensued. In vivo, "loaded"microspheres triggered clonal expansion of primary and secondary Ag-specific CD4 andCD8 T cells. The efficacy of CD8 T cell cross-priming was comparable to that of livevectors. The potency of T cell vaccination was demonstrated by protective andtherapeutic interventions using infection- and tumor-model systems. These preclinical"subunit" vaccination data thus recommend MP as a generally applicable and powerfulendosomal delivery device of exogenous Ag plus TLR-based adjuvants to vaccinate forprotective and therapeutic CD4 and CD8 T cell immunity.
Supporting information for this article is available athttp://www.wiley-vch.de/contents/jc_2040/2007/37169_s.pdf
Introduction
Vaccination has been regarded as one of the safest andmost cost-effective strategies to prevent and treatinfectious diseases [1]. Improvements, however, arerequired against infectious diseases, for which no or onlyinadequate vaccines are available, including tubercu-losis (Tb), malaria and HIV. Moreover, some chronicdiseases might be prevented with novel vaccines, anexample being Helicobacter pylori infection, which is
Correspondence: Hermann Wagner, Institute for MedicalMicrobiology, Immunology and Hygiene, Trogerstrasse 30,81675 Munich, GermanyFax: +49-89-4140-4139e-mail: [email protected]
Received 12/2/07Revised 26/4/07
Accepted 14/6/07
[DOI 10.1002/eji.200737169]
Key words:Dendritic cells � T cells
� Vaccination
Abbreviations: CpG-ODN: CpG-oligodeoxynucleotide �DQ-OVA: pH-insensitive self-quenched OVA conjugate �Lamp1: lysosomal-associated membrane protein-1 �LLO: listeriolysin O � mDC: myeloid DC � MP: microspheres �PLG: poly(lactic-co-glycolic)
Eur. J. Immunol. 2007. 37: 2063–2074 Highlights 2063
f 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu
known to lead to peptic ulcer and eventually, to gastriccancer.
Since its first description by Ramon [2], the vaccineadjuvant alum has been used to improve the immuno-genicity of non-living vaccines in humans. However,alum promotes Th2-polarizing antibody (Ab) responses[3], while control of intracellular pathogens requiresefficient Th1-polarized T effector cells [4, 5]. Therefore,the use of Toll-like receptor (TLR) ligands such as CpG-oligodeoxynucleotide (CpG-ODN) as vaccine adjuvantsmight be promising since they effectively promote Th1responses [6, 7].
Dendritic cells (DC) are essential for the induction ofrobust primary and secondary T cell responses, both invitro and in vivo [8]. Their immature state is character-ized by high antigen (Ag) uptake. Upon activation, DCshut down the Ag uptake processes, but enhanceexpression of Ag-presenting MHC class I and class IImolecules, expression of costimulatory molecules andproduction of Th1-polarizing cytokines, such as type Iinterferons (IFN) and interleukin (IL)-12 [9].
As the nucleic acid recognizing TLR3, 7, 8 and 9 areendosomally expressed in DC [10, 11], the respectiveTLR ligands need to translocate to endosomal compart-ments to activate the cells. While CD14 has recently beenshown to mediate the translocation of Poly I:C [12] toendosomal TLR3, receptors responsible for translocatingthe TLR9 ligand CpG-DNA (mimicked by immuno-stimulatory CpG-ODN) are yet ill-defined [13].
Endosomal compartments of DC not only expressTLR3, 7, 8, and 9, but in addition are organellesendowed with a molecular machinery that processesacquired exogenous Ag and presents T cell epitopes viaMHC class II or MHC class I molecules (cross-presenta-tion) [14, 15]. While soluble proteinaceous Ag is onlyweakly internalized, this restraint can be overcome bychemically linking Ag to CpG-ODN (CpG-Ag conjugate)to allow DNA receptor-mediated translocation to TLR9-expressing endosomes [16–18]. As a consequence,simultaneous TLR9 activation and Ag loading drivesDC activation together with Ag processing [19]. Hence,we proposed that concurrent endosomal translocationof TLR ligands (adjuvants) and exogenous Ag is key tobringing about robust, protective and long-lasting Ag-specific T cell vaccination [20].
The procedure to chemically link CpG-ODN toproteinaceous Ag can be inefficient, and varies betweenAg. In addition, amino acid-specific linkage maydisqualify proteins with unfavorable amino acid com-position and the applicability of CpG-Ag complexes inhumans may be restricted by cell type-specific TLR9expression [7]. It follows that a flexible endosomaldelivery device for Ag plus TLR-based adjuvants wouldhelp to translate animal vaccination protocols intoclinical applications. In the attempt to meet such
conditions, we analyzed biodegradable poly(lactic-co-glycolic) (PLG) microspheres (MP) [21–25] knownto function as an endosomal delivery vehicle [26, 27].Here we show that PLG-MP co-encapsulating TLRligands and Ag rapidly deliver their cargo intolysosomal-associated membrane protein-1 (Lamp1)-positive phago-endosomal compartments of DC. Uponbio-degradation of MP, encapsulated Ag is proteolyti-cally digested and thus conditioned for proteasome-dependent generation of T cell epitopes to be loaded onMHC class I and II molecules. In parallel, the releasedTLR ligands trigger DC activation. As a consequence,activated DC express costimulatory molecules, secretecytokines and up-regulate MHC class I- and II-restrictedAg presentation, which in turn activates MHC class I-and II-restricted Ag-specific T cells. In vivo, theimmunogenicity of "loaded" MP compared well withthat of Ag-OVA conjugates, the only subunit vaccine sofar shown to be as efficient as live vectors (i.e., Listeriamonocytogenes engineered to express OVA) [19]. MP-based vaccination not only protected towards otherwiselethal infections, but also exerted protective andtherapeutic effects towards mouse melanoma tumorcells. These data recommendMP as a promising, flexibleand effective endosomal delivery device for T cellvaccination with TLR ligand-based adjuvants combinedwith exogenous Ag.
Results
MP as vaccine delivery system in vitro
Originally, biodegradable PLG-MP were introduced as acontrolled drug delivery system and for controlleddelivery of proteins (reviewed in [21, 23]). Morerecently this approach has been adapted to exploreco-encapsulation of Ag plus immunostimulatory mole-cules [26–28]. We analyzed whether MP co-encapsulat-ing OVA and the TLR9 ligand CpG-ODN 1826 wouldtranslocate their cargo into the phago-endosomalcompartment of APC. Mouse myeloid DC (mDC) wereco-cultured with "loaded" MP. Within 2–3 h, MP werephagocytosed (Fig. 1A) into intracellular compartmentssurrounded by Lamp1-positive membrane structures. Toascertain that MP became internalized to phago-endosomal compartments, we disrupted phagocytosingmDC after 3 h and isolated the internalized MP. About60% of purified MP were coated with Lamp1-positivemembranes (Fig. 1B, arrowhead). Since late endo-somes/lysosomes are rich in proteolytic enzymes, weassessed whether MP entrapped proteinaceous Agbecomes degraded. We used a pH-insensitive self-quenched OVA conjugate (DQ-OVA) that exhibitsbright-green fluorescence upon proteolytic degradation.
Antje Heit et al. Eur. J. Immunol. 2007. 37: 2063–20742064
f 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu
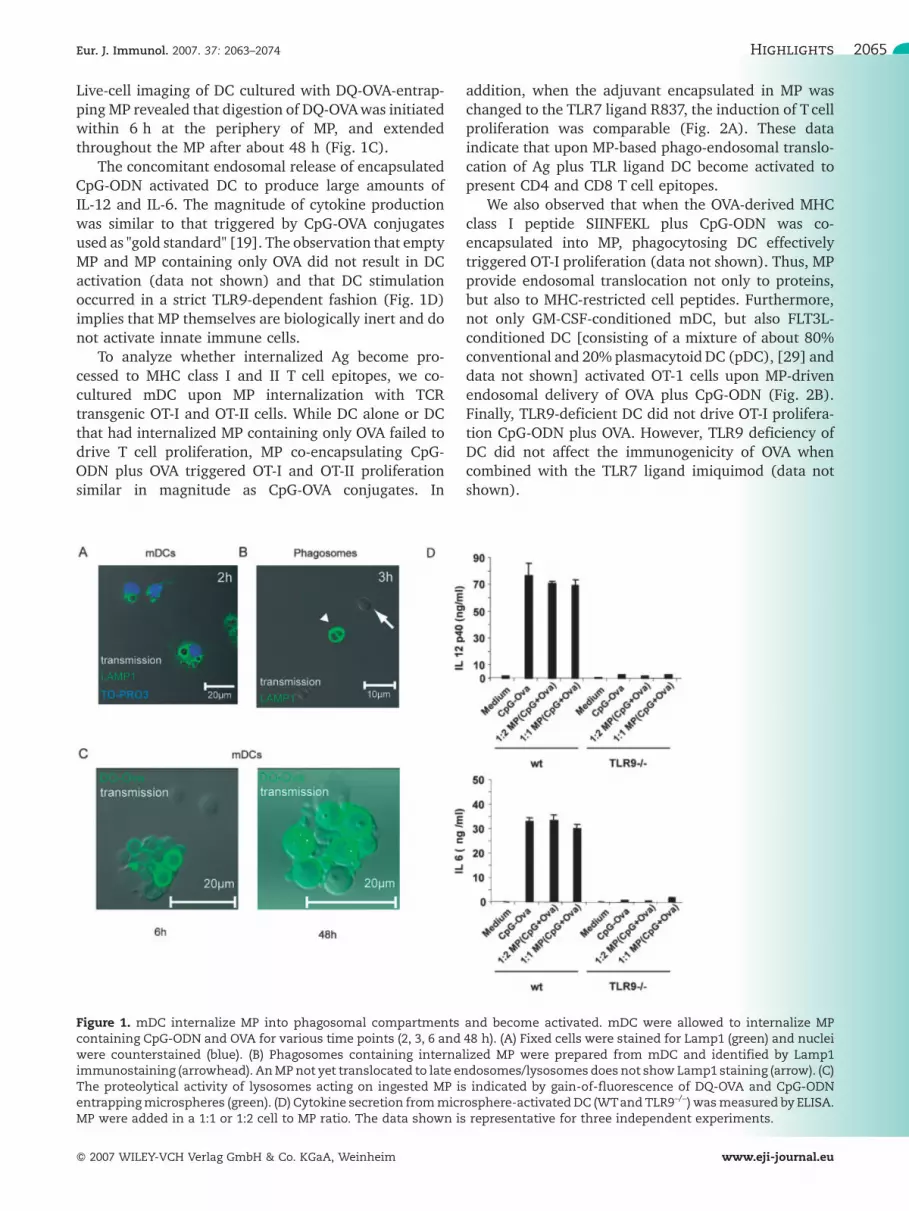
Live-cell imaging of DC cultured with DQ-OVA-entrap-ping MP revealed that digestion of DQ-OVAwas initiatedwithin 6 h at the periphery of MP, and extendedthroughout the MP after about 48 h (Fig. 1C).
The concomitant endosomal release of encapsulatedCpG-ODN activated DC to produce large amounts ofIL-12 and IL-6. The magnitude of cytokine productionwas similar to that triggered by CpG-OVA conjugatesused as "gold standard" [19]. The observation that emptyMP and MP containing only OVA did not result in DCactivation (data not shown) and that DC stimulationoccurred in a strict TLR9-dependent fashion (Fig. 1D)implies that MP themselves are biologically inert and donot activate innate immune cells.
To analyze whether internalized Ag become pro-cessed to MHC class I and II T cell epitopes, we co-cultured mDC upon MP internalization with TCRtransgenic OT-I and OT-II cells. While DC alone or DCthat had internalized MP containing only OVA failed todrive T cell proliferation, MP co-encapsulating CpG-ODN plus OVA triggered OT-I and OT-II proliferationsimilar in magnitude as CpG-OVA conjugates. In
addition, when the adjuvant encapsulated in MP waschanged to the TLR7 ligand R837, the induction of T cellproliferation was comparable (Fig. 2A). These dataindicate that upon MP-based phago-endosomal translo-cation of Ag plus TLR ligand DC become activated topresent CD4 and CD8 T cell epitopes.
We also observed that when the OVA-derived MHCclass I peptide SIINFEKL plus CpG-ODN was co-encapsulated into MP, phagocytosing DC effectivelytriggered OT-I proliferation (data not shown). Thus, MPprovide endosomal translocation not only to proteins,but also to MHC-restricted cell peptides. Furthermore,not only GM-CSF-conditioned mDC, but also FLT3L-conditioned DC [consisting of a mixture of about 80%conventional and 20% plasmacytoid DC (pDC), [29] anddata not shown] activated OT-1 cells upon MP-drivenendosomal delivery of OVA plus CpG-ODN (Fig. 2B).Finally, TLR9-deficient DC did not drive OT-I prolifera-tion CpG-ODN plus OVA. However, TLR9 deficiency ofDC did not affect the immunogenicity of OVA whencombined with the TLR7 ligand imiquimod (data notshown).
Figure 1. mDC internalize MP into phagosomal compartments and become activated. mDC were allowed to internalize MPcontaining CpG-ODN and OVA for various time points (2, 3, 6 and 48 h). (A) Fixed cells were stained for Lamp1 (green) and nucleiwere counterstained (blue). (B) Phagosomes containing internalized MP were prepared from mDC and identified by Lamp1immunostaining (arrowhead). AnMPnot yet translocated to late endosomes/lysosomes does not showLamp1 staining (arrow). (C)The proteolytical activity of lysosomes acting on ingested MP is indicated by gain-of-fluorescence of DQ-OVA and CpG-ODNentrappingmicrospheres (green). (D) Cytokine secretion frommicrosphere-activatedDC (WTand TLR9–/–) wasmeasured by ELISA.MP were added in a 1:1 or 1:2 cell to MP ratio. The data shown is representative for three independent experiments.
Eur. J. Immunol. 2007. 37: 2063–2074 Highlights 2065
f 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu
MP as delivery system in vivo
Based on these in vitro observations, we hypothesizedthat MP could be used in vivo as an endosomal deliverydevice for Ag and TLR ligand-based adjuvants. To assessthe optimal route of in vivo application (i.p., i.m., i.v. ors.c.), we measured clonal expansion (at days 8 and 13)of SIINFEKL-specific CD8 T cells in blood of C57BL/6
mice immunized with MP co-encapsulating OVA plusCpG-ODN. We observed the highest frequencies ofSIINFEKL-specific CD8 T cells in blood upon s.c.challenge (Fig. 3). In subsequent experiments wetherefore used the s.c. application route for vaccination.
A mixture of CpG-ODN (TLR9 ligand) and OVA (Ag)promotes CD8 T cell cross-priming in a CD4-indepen-dent manner [30]. On the other hand, the activity of
Figure 2. In vitro proliferation of enriched T cells induced byMP-loaded APC. (A) Purified, naive, CFSE-labeled CD4+ or CD8+ OT-1 orOT-2 T cells were co-incubated with GM-CSF-conditioned MP-loaded DC for 3 days and analyzed by FACS. The reduction of CFSEintensity correlates with T cell division. One representative example out of three independent experiments is shown. n.d.: notdone. (B) FLT3-derived DCwere loadedwith the indicatedMP preparations and co-culturedwith CFSE-labeled CD8+ OT-1 T cells for3 days followed by analysis of the CFSE+ cells via FACS. Data from one representative out of two independent experiments isshown.
Figure 3.Analysis of the route of application. (A) Naive C57BL/6micewere injected once either s.c., i.m., i.p. or i.v. with 5 mgMP co-encapsulating CpG-ODN 1826 and OVA. After 7 and 13 days, peripheral bloodwas analyzed for the presence of SIINFEKL tetramer-specific, CD8+ T cells by FACS. Blots shown here are gated on living CD8+ cells. (B) Data from (A) are shown as mean � SD, n=3.Representative data out of two independent experiments are shown.
Antje Heit et al. Eur. J. Immunol. 2007. 37: 2063–20742066
f 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu
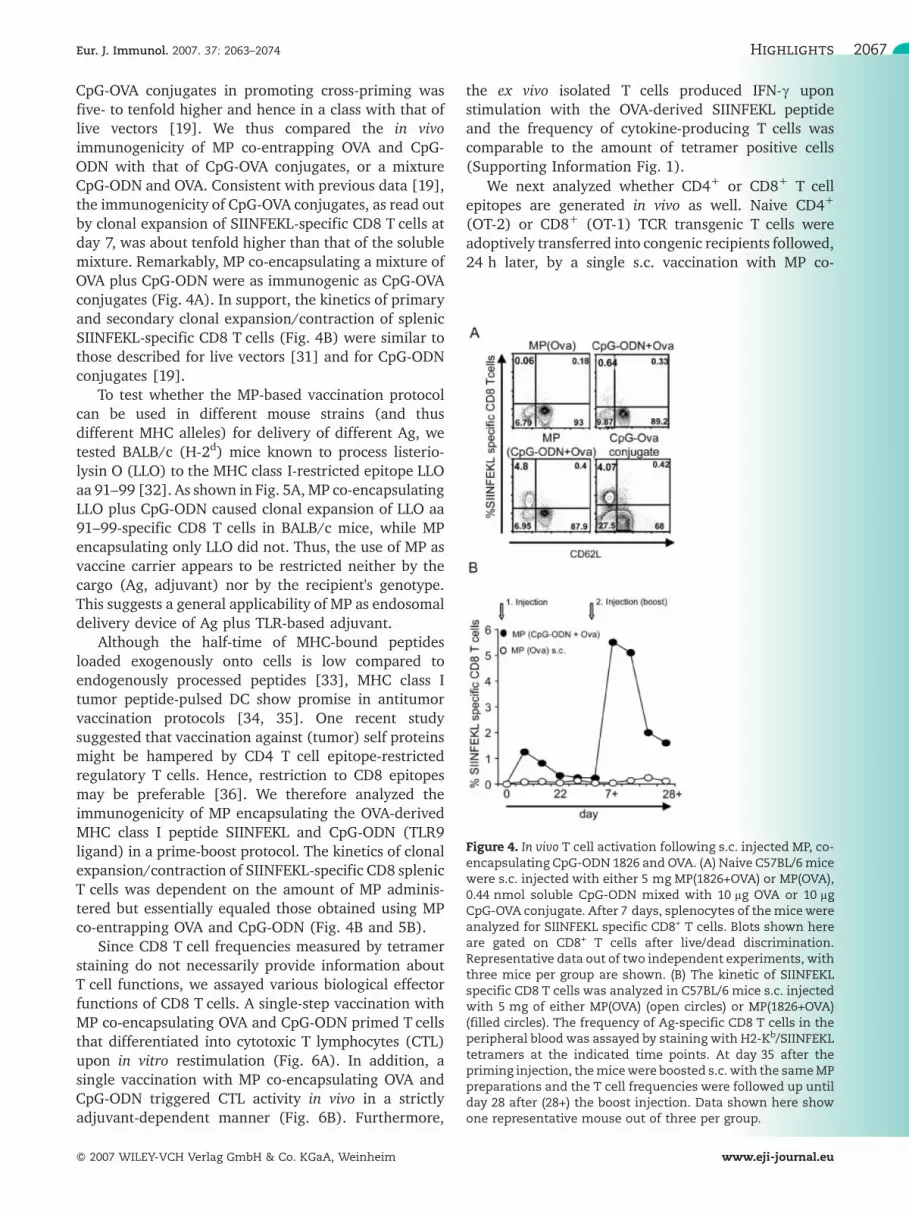
CpG-OVA conjugates in promoting cross-priming wasfive- to tenfold higher and hence in a class with that oflive vectors [19]. We thus compared the in vivoimmunogenicity of MP co-entrapping OVA and CpG-ODN with that of CpG-OVA conjugates, or a mixtureCpG-ODN and OVA. Consistent with previous data [19],the immunogenicity of CpG-OVA conjugates, as read outby clonal expansion of SIINFEKL-specific CD8 T cells atday 7, was about tenfold higher than that of the solublemixture. Remarkably, MP co-encapsulating a mixture ofOVA plus CpG-ODN were as immunogenic as CpG-OVAconjugates (Fig. 4A). In support, the kinetics of primaryand secondary clonal expansion/contraction of splenicSIINFEKL-specific CD8 T cells (Fig. 4B) were similar tothose described for live vectors [31] and for CpG-ODNconjugates [19].
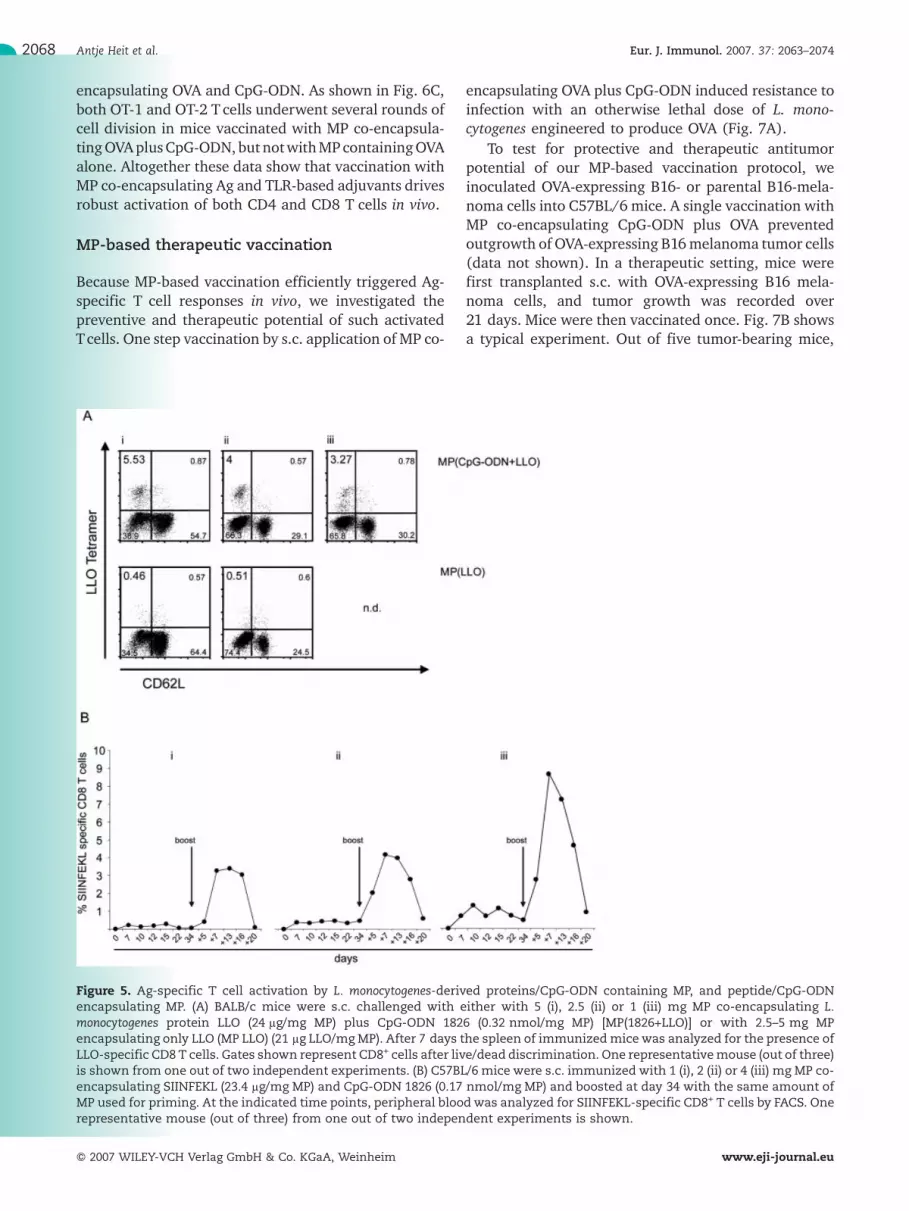
To test whether the MP-based vaccination protocolcan be used in different mouse strains (and thusdifferent MHC alleles) for delivery of different Ag, wetested BALB/c (H-2d) mice known to process listerio-lysin O (LLO) to the MHC class I-restricted epitope LLOaa 91–99 [32]. As shown in Fig. 5A,MP co-encapsulatingLLO plus CpG-ODN caused clonal expansion of LLO aa91–99-specific CD8 T cells in BALB/c mice, while MPencapsulating only LLO did not. Thus, the use of MP asvaccine carrier appears to be restricted neither by thecargo (Ag, adjuvant) nor by the recipient's genotype.This suggests a general applicability of MP as endosomaldelivery device of Ag plus TLR-based adjuvant.
Although the half-time of MHC-bound peptidesloaded exogenously onto cells is low compared toendogenously processed peptides [33], MHC class Itumor peptide-pulsed DC show promise in antitumorvaccination protocols [34, 35]. One recent studysuggested that vaccination against (tumor) self proteinsmight be hampered by CD4 T cell epitope-restrictedregulatory T cells. Hence, restriction to CD8 epitopesmay be preferable [36]. We therefore analyzed theimmunogenicity of MP encapsulating the OVA-derivedMHC class I peptide SIINFEKL and CpG-ODN (TLR9ligand) in a prime-boost protocol. The kinetics of clonalexpansion/contraction of SIINFEKL-specific CD8 splenicT cells was dependent on the amount of MP adminis-tered but essentially equaled those obtained using MPco-entrapping OVA and CpG-ODN (Fig. 4B and 5B).
Since CD8 T cell frequencies measured by tetramerstaining do not necessarily provide information aboutT cell functions, we assayed various biological effectorfunctions of CD8 T cells. A single-step vaccination withMP co-encapsulating OVA and CpG-ODN primed T cellsthat differentiated into cytotoxic T lymphocytes (CTL)upon in vitro restimulation (Fig. 6A). In addition, asingle vaccination with MP co-encapsulating OVA andCpG-ODN triggered CTL activity in vivo in a strictlyadjuvant-dependent manner (Fig. 6B). Furthermore,
the ex vivo isolated T cells produced IFN-c uponstimulation with the OVA-derived SIINFEKL peptideand the frequency of cytokine-producing T cells wascomparable to the amount of tetramer positive cells(Supporting Information Fig. 1).
We next analyzed whether CD4+ or CD8+ T cellepitopes are generated in vivo as well. Naive CD4+
(OT-2) or CD8+ (OT-1) TCR transgenic T cells wereadoptively transferred into congenic recipients followed,24 h later, by a single s.c. vaccination with MP co-
Figure 4. In vivo T cell activation following s.c. injected MP, co-encapsulating CpG-ODN 1826 and OVA. (A) Naive C57BL/6micewere s.c. injected with either 5 mg MP(1826+OVA) or MP(OVA),0.44 nmol soluble CpG-ODN mixed with 10 lg OVA or 10 lgCpG-OVA conjugate. After 7 days, splenocytes of themicewereanalyzed for SIINFEKL specific CD8+ T cells. Blots shown hereare gated on CD8+ T cells after live/dead discrimination.Representative data out of two independent experiments, withthree mice per group are shown. (B) The kinetic of SIINFEKLspecific CD8 T cells was analyzed in C57BL/6 mice s.c. injectedwith 5 mg of either MP(OVA) (open circles) or MP(1826+OVA)(filled circles). The frequency of Ag-specific CD8 T cells in theperipheral blood was assayed by staining with H2-Kb/SIINFEKLtetramers at the indicated time points. At day 35 after thepriming injection, themicewere boosted s.c. with the sameMPpreparations and the T cell frequencies were followed up untilday 28 after (28+) the boost injection. Data shown here showone representative mouse out of three per group.
Eur. J. Immunol. 2007. 37: 2063–2074 Highlights 2067
f 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu
encapsulating OVA and CpG-ODN. As shown in Fig. 6C,both OT-1 and OT-2 T cells underwent several rounds ofcell division in mice vaccinated with MP co-encapsula-tingOVAplusCpG-ODN,but notwithMPcontainingOVAalone. Altogether these data show that vaccination withMP co-encapsulating Ag and TLR-based adjuvants drivesrobust activation of both CD4 and CD8 T cells in vivo.
MP-based therapeutic vaccination
Because MP-based vaccination efficiently triggered Ag-specific T cell responses in vivo, we investigated thepreventive and therapeutic potential of such activatedTcells. One step vaccination by s.c. application of MP co-
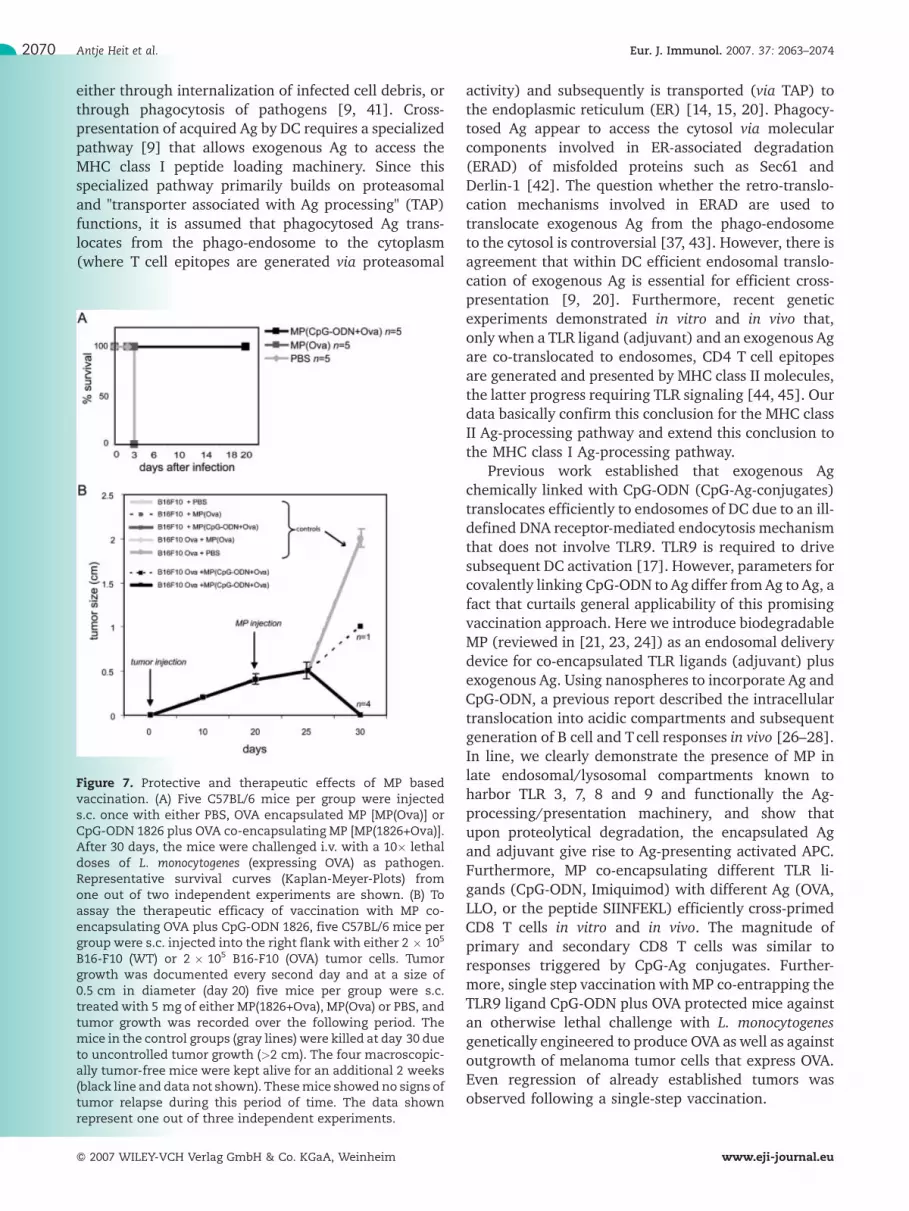
encapsulating OVA plus CpG-ODN induced resistance toinfection with an otherwise lethal dose of L. mono-cytogenes engineered to produce OVA (Fig. 7A).
To test for protective and therapeutic antitumorpotential of our MP-based vaccination protocol, weinoculated OVA-expressing B16- or parental B16-mela-noma cells into C57BL/6 mice. A single vaccination withMP co-encapsulating CpG-ODN plus OVA preventedoutgrowth of OVA-expressing B16melanoma tumor cells(data not shown). In a therapeutic setting, mice werefirst transplanted s.c. with OVA-expressing B16 mela-noma cells, and tumor growth was recorded over21 days. Mice were then vaccinated once. Fig. 7B showsa typical experiment. Out of five tumor-bearing mice,
Figure 5. Ag-specific T cell activation by L. monocytogenes-derived proteins/CpG-ODN containing MP, and peptide/CpG-ODNencapsulating MP. (A) BALB/c mice were s.c. challenged with either with 5 (i), 2.5 (ii) or 1 (iii) mg MP co-encapsulating L.monocytogenes protein LLO (24 lg/mg MP) plus CpG-ODN 1826 (0.32 nmol/mg MP) [MP(1826+LLO)] or with 2.5–5 mg MPencapsulating only LLO (MP LLO) (21 lg LLO/mgMP). After 7 days the spleen of immunizedmicewas analyzed for the presence ofLLO-specific CD8 T cells. Gates shown represent CD8+ cells after live/dead discrimination. One representativemouse (out of three)is shown from one out of two independent experiments. (B) C57BL/6 mice were s.c. immunized with 1 (i), 2 (ii) or 4 (iii) mg MP co-encapsulating SIINFEKL (23.4 lg/mg MP) and CpG-ODN 1826 (0.17 nmol/mg MP) and boosted at day 34 with the same amount ofMP used for priming. At the indicated time points, peripheral blood was analyzed for SIINFEKL-specific CD8+ T cells by FACS. Onerepresentative mouse (out of three) from one out of two independent experiments is shown.
Antje Heit et al. Eur. J. Immunol. 2007. 37: 2063–20742068
f 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu
one-step vaccination caused complete tumor regressionin four mice, while one mouse displayed retarded tumorgrowth.
Discussion
The results presented here advocate biodegradable PLG-MP as a powerful endosomal/lysosomal delivery devicefor vaccination protocols that are based on exogenous("subunit") proteinaceous Ag plus ligands for endoso-mally expressed TLR. The power of MP co-entrapping Agplus TLR-based adjuvants relies on combined endosomaltranslocation of its cargo since endosomal translocationof Ag plus adjuvant triggers DC maturation to profes-sional APC as well as activation of its MHC class I and IIprocessing machinery. Because activation/maturation
of APC prior to Ag delivery paralyzes APC for Ag uptakeand thus for cross-priming [37], classical vaccinationprotocols based on mixed Ag/adjuvants formula mayhave limitations. In addition, practical restrictions in thepreparation of CpG-Ag conjugates, i.e., the limitedcombinatorial options of Ag and adjuvants, can becircumvented by the use of "loaded" MP as endosomaldelivery device. In vivo MP-based vaccination triggeredclonal expansion of Ag-specific MHC class I-restrictedCD8 T cells similar in kinetics and magnitude as CpG-Agconjugates [19]. Indeed MP-based single step vaccina-tion conferred protective and even therapeutic immu-nity in an infection and tumor model system.
CD8+ T cell responses can be efficiently generatedagainst pathogens that fail to infect DC, as well ascellular pathogens that never reach the cytosol of DC[38–40]. DC therefore acquire pathogen-derived Ag
Figure 6. Functional analysis of T cells induced by loadedMP. (A) OfMP co-encapsulating OVA plus CpG-ODN 1828 [MP(1826+OVA)]orMPencapsulating onlyOVA [MP(Ova)], 5 mgwere injected s.c. intoC57BL/6mice. After 7 days, the spleen cellswere restimulatedin vitro and the CTL activity was assayed in a 51Cr-release assay. Filled, black symbols represents SIINFEKL peptide-pulsed cells;gray and unfilled symbols depict unpulsed EL4 target cells. (B) The in vivo CTL activity was assayed in mice immunized s.c. with5 mg MP co-encapsulating OVA plus CpG-ODN 1826 [MP(1826+Ova)] or 5 mg MP encapsulating only OVA [MP(Ova)] as detailed inMaterials and methods. Lytic activity was compared to mice s.c. challenged with 10 lg 1668-OVA conjugate. Data of onerepresentative mouse (out of four) are shown. (C) Immunogenic Ag presentation to CD4 T cells and cross-presentation toCD8 T cellswas analyzed in vivo by assaying proliferation of adoptively transferred CFSE-labeled CD8+ (OT-1) or CD4+ (OT-2) T cells.One representative mouse (out of three) from at least two independent experiments is shown.
Eur. J. Immunol. 2007. 37: 2063–2074 Highlights 2069
f 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu
either through internalization of infected cell debris, orthrough phagocytosis of pathogens [9, 41]. Cross-presentation of acquired Ag by DC requires a specializedpathway [9] that allows exogenous Ag to access theMHC class I peptide loading machinery. Since thisspecialized pathway primarily builds on proteasomaland "transporter associated with Ag processing" (TAP)functions, it is assumed that phagocytosed Ag trans-locates from the phago-endosome to the cytoplasm(where T cell epitopes are generated via proteasomal
activity) and subsequently is transported (via TAP) tothe endoplasmic reticulum (ER) [14, 15, 20]. Phagocy-tosed Ag appear to access the cytosol via molecularcomponents involved in ER-associated degradation(ERAD) of misfolded proteins such as Sec61 andDerlin-1 [42]. The question whether the retro-translo-cation mechanisms involved in ERAD are used totranslocate exogenous Ag from the phago-endosometo the cytosol is controversial [37, 43]. However, there isagreement that within DC efficient endosomal translo-cation of exogenous Ag is essential for efficient cross-presentation [9, 20]. Furthermore, recent geneticexperiments demonstrated in vitro and in vivo that,only when a TLR ligand (adjuvant) and an exogenous Agare co-translocated to endosomes, CD4 T cell epitopesare generated and presented by MHC class II molecules,the latter progress requiring TLR signaling [44, 45]. Ourdata basically confirm this conclusion for the MHC classII Ag-processing pathway and extend this conclusion tothe MHC class I Ag-processing pathway.
Previous work established that exogenous Agchemically linked with CpG-ODN (CpG-Ag-conjugates)translocates efficiently to endosomes of DC due to an ill-defined DNA receptor-mediated endocytosis mechanismthat does not involve TLR9. TLR9 is required to drivesubsequent DC activation [17]. However, parameters forcovalently linking CpG-ODN to Ag differ fromAg to Ag, afact that curtails general applicability of this promisingvaccination approach. Here we introduce biodegradableMP (reviewed in [21, 23, 24]) as an endosomal deliverydevice for co-encapsulated TLR ligands (adjuvant) plusexogenous Ag. Using nanospheres to incorporate Ag andCpG-ODN, a previous report described the intracellulartranslocation into acidic compartments and subsequentgeneration of B cell and Tcell responses in vivo [26–28].In line, we clearly demonstrate the presence of MP inlate endosomal/lysosomal compartments known toharbor TLR 3, 7, 8 and 9 and functionally the Ag-processing/presentation machinery, and show thatupon proteolytical degradation, the encapsulated Agand adjuvant give rise to Ag-presenting activated APC.Furthermore, MP co-encapsulating different TLR li-gands (CpG-ODN, Imiquimod) with different Ag (OVA,LLO, or the peptide SIINFEKL) efficiently cross-primedCD8 T cells in vitro and in vivo. The magnitude ofprimary and secondary CD8 T cells was similar toresponses triggered by CpG-Ag conjugates. Further-more, single step vaccination with MP co-entrapping theTLR9 ligand CpG-ODN plus OVA protected mice againstan otherwise lethal challenge with L. monocytogenesgenetically engineered to produce OVA as well as againstoutgrowth of melanoma tumor cells that express OVA.Even regression of already established tumors wasobserved following a single-step vaccination.
Figure 7. Protective and therapeutic effects of MP basedvaccination. (A) Five C57BL/6 mice per group were injecteds.c. once with either PBS, OVA encapsulated MP [MP(Ova)] orCpG-ODN 1826 plus OVA co-encapsulating MP [MP(1826+Ova)].After 30 days, the mice were challenged i.v. with a 10� lethaldoses of L. monocytogenes (expressing OVA) as pathogen.Representative survival curves (Kaplan-Meyer-Plots) fromone out of two independent experiments are shown. (B) Toassay the therapeutic efficacy of vaccination with MP co-encapsulating OVA plus CpG-ODN 1826, five C57BL/6 mice pergroup were s.c. injected into the right flank with either 2 � 105
B16-F10 (WT) or 2 � 105 B16-F10 (OVA) tumor cells. Tumorgrowth was documented every second day and at a size of0.5 cm in diameter (day 20) five mice per group were s.c.treated with 5 mg of either MP(1826+Ova), MP(Ova) or PBS, andtumor growth was recorded over the following period. Themice in the control groups (gray lines) were killed at day 30 dueto uncontrolled tumor growth (>2 cm). The four macroscopic-ally tumor-free mice were kept alive for an additional 2 weeks(black line and data not shown). Thesemice showed no signs oftumor relapse during this period of time. The data shownrepresent one out of three independent experiments.
Antje Heit et al. Eur. J. Immunol. 2007. 37: 2063–20742070
f 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu
The CpG-OVA conjugates have been demonstrated todramatically reduce serum borne pro-inflammatorycytokines observed in mice vaccinated with Ag admixedto CpG-ODN. Although the amount of Ag and adjuvantadministered by MP exceeds that of CpG-OVA con-jugates, no adverse side effects were observed upon invivo use. In addition, titrating the amounts of MP(encapsulating either SIINFEKL peptide or LLO protein)revealed robust Tcell induction evenwith reduced dosesof MP. Since MP encapsulation fundamentally alters Agdelivery [27], the question of how much Ag per MP isrequired for the induction of optimal T cell responsesneeds to be addressed.
MP co-encapsulating the CD8 T cell epitope SIIN-FEKL together with CpG-ODN also triggered robustsecondary responses. These data may be of interest in aclinical setting aiming at antitumor responses usingpeptide-pulsed DC [34, 35], or for vaccination protocolsagainst tumor Ag, a situation in which CD4 regulatoryT cells may compromise the vaccination efficacy [36].
In mice, TLR9 is expressed by most innate immunecells, while in humans its expression is restricted to pDCand B cells [11]. As TLR7 and 8 are expressed by themajority of human APC, TLR7 and 8 ligands mayrepresent the adjuvant of choice for MP-based humanvaccination protocols. Natural single-stranded (ss) RNAhas been shown to act as ligand for TLR7 and 8 [46, 47].Due to their sensibility to RNases, these ligands are notideal for use in TLR-based vaccination protocols.However, when complexed with cationic lipids (toprotect from extracellular degradation and to enhanceendosomal translocation) both DNA or RNA activateinnate immune cells via TLR9 or TLR7, respectively [47,48]. When mixed with Ag, such complexes also triggerrobust cross-priming [49]. It is therefore of interest toevaluate the efficacy MP co-entrapping ssRNA and Ag[25]. On the other hand, synthetic TLR7 agonists such asImiquimod (R837) may be an attractive alternative asadjuvants for use in humans, since in mice MP co-encapsulating Ag plus Imiquimod activate immature DCto professional APC (Fig. 2B).
Apart from combined endosomal delivery of TLRagonists (adjuvant) and exogenous Ag in DC, a factorfavoring the potential clinical use of biodegradable MPas an endosomal delivery device relates to its safety andgeneral applicability. In humans, these polymers have along history as a safe material [50]. Their breakdownproducts are biologically inert and the relative ease inproduction meets the requirements of GMP guidelines.In line with others [27], our data therefore advocate MPco-encapsulating exogenous Ag and TLR ligands (ad-juvants) for subunit vaccines to drive activation ofprotective CD4 T and CD8 T cells in humans.
Materials and methods
Reagents and CpG-OVA conjugate
OVA (chicken egg OVA) was purchased from Sigma-Aldrichand DQ-OVA from Invitrogen (Germany). The listeriolysinpeptide LLO aa 91–99 and the OVA-derived peptide SIINFEKL(OVA aa 257–264) were custom synthesized by Biosyntan,Germany. Phosphothioate-stabilized immunostimulatory CpG-ODNwas purchased from TIBMolBiol (ODN1668) or was a giftfrom Coley Pharmaceuticals (Hilden Germany) (ODN1826).The immunostimulatory CpG sequences used as ODN are: ODN1668: 50-TCCATGACGTTCCTGATGCT-30 and ODN 1826: 50-TCCATGACGTTCCTGACGTT-30. Of note, in vitro and in vivocomparison of CpG-ODN 1668 and CpG-ODN 1826 yieldedcomparable results. R837 (Imiquimod) was purchased fromInvivogen (France). 1668 CpG-ODN was covalently linked toOVA essentially using a cross-linker (sulfo-maleimidobezoyl-N-hydroxy succinimide ester) (Pierce) as described [51]. Thebatch used in this study contained two to three CpG-ODNmolecules per OVA molecule, with 10 lg CpG-OVA complexcontaining 0.44 nmol CpG.
Mice, cells, reagents
C57BL/6 and BALB/c mice were obtained from HarlanWinkelmann, TCR transgenic OT-1 C57BL/6 mice and OT-2C57BL/6 mice or TLR9–/– mice were derived from in-housebreeding stocks. All mice were kept under specific pathogen-free conditions and used at 6–12 weeks of age. All experimentsinvolving animals have been carried out according to federalregulations.
DC were differentiated from bone marrow cells, harvestedfrom femur and tibia of mice. For generation of GM-CSF-conditioned DC, bone marrow cells were incubated withrecombinant murine GM-CSF (Peprotech) for 7 days. FLT3LDC were differentiated in the presence of human FLT3L (R&DSystems) for 8 days essentially as described previously [52].
Murine EL4 (H-2b) thymoma and B16-F10 melanoma celllines were purchased from the American Type CultureCollection (Manassas, VA). OVA-expressing B16 melanomacells [B16-F10(OVA)] were generated by electroporation ofB16 cells with the OVA-expressing plasmid pcOVA [52],selection with Neomycin (G418) and subsequent cloning. Ahigh-level OVA-expressing B16 F10 melanoma clone (asdetermined by Western blot) was used for the tumorexperiments.
Preparation of MP
All MP were prepared using a solvent/evaporation technique,as described previously [53]. Briefly, the microparticles weregenerated by emulsifying 200 mg polymer (PLG Resomer 502,50:50 lactide:glycolide ratio, Boehringer Ingelheim, Germany)in 15 mLmethylene chloride (Merck, Germany) to 200 lL Ag/adjuvant solution (5–30 mg protein or 20 mg peptidedissolved in water) at high speed using an IKA homogenizer.This primary organic phase was then mixed with 50 mL of anaqueous phase containing 3% polyvinyl alcohol (Mowiol40–88; Sigma-Aldrich Chemicals, Germany) to form a stable
Eur. J. Immunol. 2007. 37: 2063–2074 Highlights 2071
f 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu
oil-in-water emulsion, followed by stirring at 15 000 rpm atroom temperature for 4–5 h, allowing the methylene chlorideto evaporate. The resulting microparticles were washed threetimes in low-tox water (Sigma) to remove non-encapsulatedcomponents and lyophilized. The particle size ranged from1–30 lm as measured using reference beads (MolecularProbes) in a FACSCalibur (Becton Dickinson). Microspheresbatches were only used when more then 90% of MP weresmaller than 10 lm. The amount of entrapped Ag and adjuvantwas measured by an encapsulation assay: 10 mg lyophilizedmicroparticles were incubated with 0.1% SDS/0.1 MNaOH for12 h at room temperature. After centrifugation (1500 rpm,10 min), the amount of Ag was assayed with the BCA proteinassay kit (Pierce, USA) following the instructions of themanufacturer. The amount of DNA was measured by spectro-scopy. All OVA encapsulating MP used here were "loaded" in arange of 40–45 lg Ag and about 1 lg (0.2 nmol) DNA per mgMP, the amount of LLO and SIINFEKL are noted in therespective figure legends.
In vivo priming
In vivo priming for Ag-specific T cells was performed byinjection of 10 lg ODN 1668-OVA conjugates or 5 mg MPdiluted in 100 lL PBS s.c. at the tail base of mice.
Surface staining and cytokine measurement
H2-Kb/SIINFEKL or H2-Kd/LLO multimer reagents weregenerated as previously described [31]. In short, 200 lLvenous blood or spleen cells were collected and erythrocyteswere lysed using ammonium chloride. Cells (1 � 106–6 � 106)were stained for each sample. For live/dead discrimination,cells were incubated with ethidium monoazide (MolecularProbes), followed by MHC multimer and surface markerstaining for 45 min at 4�C. The following mAbwere used: anti-CD8 (clone 3B5; Caltag Laboratories), anti-CD62L (cloneMEL-14; BD Pharmingen). Data were acquired on a FACSCaliburand analyzed with FlowJo (Tree Star) software.
To analyze activation of APC by CpG-OVA complex, bonemarrow-derived DC generated in GM-CSF-conditioned med-ium were incubated with 10 lg/mL CpG-OVA conjugates orMP (MP:cell ratio was 2:1) for 24 h, and thereafter thesupernatant were collected for ELISA. Cytokine release (IL-6and IL-12p40; R&D Systems) was assayed as instructed by themanufacturer.
Intracellular cytokine staining and cytotoxicitymeasurement
Splenocytes were incubated for 2 h in the presence of theCD8 T cell epitope SIINFEKL (peptide) and brefeldin A(GolgiPlug; BD Pharmingen) was added for the last 3 h.Intracellular cytokine staining for IFN-c (FITC-labeled; BDPharmingen) was performed using the Cytofix/Cytoperm kit(BD Pharmingen) according the manufacturer's recommend-ations.
For in vivo cytotoxicity assay, splenocytes from naiveC57BL/6 mice were either loaded or not with the peptideSIINFEKL and stained with carboxyfluorescein diacetate
succinimidyl ester (CFSE; Molecular Probes) at concentrationsof 5 lM (SIINFEKL-loaded cells) or 0.5 lM (unloaded cells).Equal numbers of either cells were injected i.v. into primedanimals. After 5 and 21 h, blood was collected from the tailvein and analyzed for CFSE-positive cells.
For in vitro cytotoxicity experiments, spleen cell suspensionswere cocultured with SIINFEKL-loaded (1 lM) irradiated(15 Gy) syngenic spleen cells plus 5 U/mL rIL-2. At day 7,cytolytic activity was measured by the 51Cr-release assayessentially as described [17]. Untreated EL4 cells served asspecificity control. Radioactivity was measured with a Perkin-Elmer gamma counter. Specific lysis was calculated accordingto the formula: percentage of specific lysis = [(cpm/sample –cpm/spontaneous release)/(cpm/maximum release – cpm/spontaneous release)]�100. The spontaneous release rangedfrom 5% to 15%.
In vivo and in vitro proliferation assay
For in vivo proliferation, naive CD8+ or CD4+ T cells fromspleens of OT-1 or OT-2 mice were positively selected bypurification on an MACS (Miltenyi Biotec) using anti-CD4(RM4–5, BD Pharmingen) FITC-labeled Ab and secondarystaining with anti-FITC beads (Miltenyi Biotec) or anti-CD8(Ly-2) beads (Miltenyi Biotec). Approximately 1 � 107 pur-ified T cells were labeled with CFSE and transferred by i.v.injection into naive mice. One day later, the mice werechallenged s.c. with 5 mg MP. Three days later, 200 lL bloodfrom the tail vein was analyzed for CFSE+ cells.
For in vitro proliferation the purified, CFSE-labeled cellswere co-cultured with MP-loaded GM-CSF-conditioned DC for3 days. Data were acquired on a FACSCalibur and analyzedwith FlowJo software.
Preparation of phagosomes and confocal microscopy
Phagosomes were prepared and stained essentially asdescribed [54]. Briefly, DC were allowed to ingest micro-spheres at a 2:1 MP:cell ratio for 3 h (pulse) and, afterwashing, the cells were removed by scraping and disruptedwith a dounce homogenizer (20 strokes at 4�C) in homo-genization buffer [10 mM HEPES, pH 7.2, 0.25 M sucrose,Complete protease inhibitor (Roche)]. Then, the phagosomescontaining microspheres were cleared from cellular debris bycentrifugation (500 � g, 5 min) and resuspended in 3.7%formaldehyde/PBS (containing protease inhibitors) for fixa-tion (15 min, room temperature). After extensive washing(three times), the phagosomes were blocked with stainingbuffer (PBS, 10% normal goat serum (Invitrogen), 0.1% TritonX-100) for 15 min and stained first with the primary Ab [ratanti-mouse Lamp1 (BD Pharmingen), 1:400, 1 h] and there-after with the secondary Ab [goat anti-rat IgG-Alexa488(Molecular Probes), 30 min] in staining buffer. After threewashing steps, the phagosomes were spun onto microscopeslides and mounted with ProLong Gold (Molecular Probes).
For preparation of cells, DC seeded on 8-well microscopyslides (BD Falcon) were pulse/chasedwithMP, fixedwith 3.7%formaldehyde/PBS, blocked/permeabilized with staining buf-fer and stained with anti-Lamp1/anti-rat Alexa488. To mark
Antje Heit et al. Eur. J. Immunol. 2007. 37: 2063–20742072
f 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu
the nucleus, TO-PRO3 (1:1000, Molecular Probes) was addedto the secondary Ab.
Living cell microscopy was performed in 8-well ibiTreat(ibidi, Germany) slides, where cells received DQ-OVA MP forthe indicated time points.
The confocal microscopes used were a Zeiss LSM500equipped with an Ar-laser (488 nm line) and a He/Ne-laser(633 nm) line, and a Leica TCS SP5 equipped with an Ar-laser(488 nm line). The objectives used were a 40�/1.2NA oil-objective (for fixed cells and phagosomes) and a 40�/1.25NAwater-objective (for living cell microscopy). Living cells wereacquired in a 37�C incubation chamber. The images acquiredwere imported in to PhotoshopCS.
Infection experiments and tumor challenge
Infection experiments were performed by i.v. injection of Lm-OVA (kindly provided by H. Shen, University of PennsylvaniaMedical School, Philadelphia, PA). A dose of 2 � 104 i.v.-injected LM-OVA represents the LD50 for C57BL/6 mice.Survival curves were drawn by the Kaplan-Meier method. B16-F10 or B16-F10 (OVA) cells (2 � 105) were injected in 50 lLPBS s.c. at the right flank of mice. At tumor sizes of more than2 cm the mice were killed.
Acknowledgements: We acknowledge the excellenttechnical assistance of Monika Hammel, Inge Broschand Martina Koffler. We thank Carsten Kirschning andJulia Heit for critically reading the manuscript. Thiswork was supported by the Deutsche Forschungsge-meinschaft (H. Wagner and D. Busch) and by ColeyPharmceutical Group, GmbH (Langenfeld, Germany).
References
1 Jodar, L., Duclos, P., Milstien, J. B., Griffiths, E., Aguado, M. T. andClements, C. J., Ensuring vaccine safety in immunization programmes–aWHO perspective. Vaccine 2001. 19: 1594–1605.
2 Ramon, G., Procedres pour acroitre la production des antitoxins. Ann.Institut Pasteur 1926. 40: 1–10.
3 Gupta, R. K., Aluminum compounds as vaccine adjuvants. Adv. Drug Deliv.Rev. 1998. 32: 155–172.
4 Pamer, E. G., Immune responses to Listeria monocytogenes. Nat. Rev.Immunol. 2004. 4: 812–823.
5 Zammit, D. J., Cauley, L. S., Pham, Q. M. and Lefrancois, L., Dendriticcells maximize the memory CD8 Tcell response to infection. Immunity 2005.22: 561–570.
6 Krieg, A. M., CpG motifs in bacterial DNA and their immune effects. Annu.Rev. Immunol. 2002. 20: 709–760.
7 Wagner, H., Bacterial CpG DNA activates immune cells to signal infectiousdanger. Adv. Immunol. 1999. 73: 329–368.
8 Banchereau, J. and Steinman, R. M., Dendritic cells and the control ofimmunity. Nature 1998. 392: 245–252.
9 Guermonprez, P., Valladeau, J., Zitvogel, L., Thery, C. and Amigorena,S., Antigen presentation and T cell stimulation by dendritic cells. Annu. Rev.Immunol. 2002. 20: 621–667.
10 Akira, S. and Takeda, K., Toll-like receptor signalling. Nat. Rev. Immunol.2004. 4: 499–511.
11 Wagner, H., The immunobiology of the TLR9 subfamily. Trends Immunol.2004. 25: 381–386.
12 Lee, H. K., Dunzendorfer, S., Soldau, K. and Tobias, P. S.,Double-strandedRNA-mediated TLR3 activation is enhanced by CD14. Immunity 2006. 24:153–163.
13 Hacker, H., Mischak, H., Hacker, G., Eser, S., Prenzel, N., Ullrich, A. andWagner, H., Cell type-specific activation of mitogen-activated proteinkinases by CpG-DNA controls interleukin-12 release from antigen-present-ing cells. EMBO J. 1999. 18: 6973–6982.
14 Ackerman, A. L., Kyritsis, C., Tampe, R. and Cresswell, P., Earlyphagosomes in dendritic cells form a cellular compartment sufficient forcross presentation of exogenous antigens. Proc. Natl. Acad. Sci. USA 2003.100: 12889–12894.
15 Guermonprez, P., Saveanu, L., Kleijmeer, M., Davoust, J., Van Endert, P.and Amigorena, S., ER-phagosome fusion defines an MHC class I cross-presentation compartment in dendritic cells. Nature 2003. 425: 397–402.
16 Cho, H. J., Takabayashi, K., Cheng, P. M., Nguyen, M. D., Corr, M., Tuck,S. and Raz, E., Immunostimulatory DNA-based vaccines induce cytotoxiclymphocyte activity by a T-helper cell-independent mechanism. Nat.Biotechnol. 2000. 18: 509–514.
17 Heit, A., Maurer, T., Hochrein, H., Bauer, S., Huster, K. M., Busch, D. H.andWagner, H., Cutting edge: Toll-like receptor 9 expression is not requiredfor CpG DNA-aided cross-presentation of DNA-conjugated antigens butessential for cross-priming of CD8 T cells. J. Immunol. 2003. 170:2802–2805.
18 Wille-Reece, U., Flynn, B. J., Lore, K., Koup, R. A., Kedl, R. M.,Mattapallil, J. J., Weiss, W. R. et al., HIV Gag protein conjugated to a Toll-like receptor 7/8 agonist improves the magnitude and quality of Th1 andCD8+ T cell responses in nonhuman primates. Proc. Natl. Acad. Sci. USA2005. 102: 15190–15194.
19 Heit, A., Schmitz, F., O'Keeffe, M., Staib, C., Busch, D. H., Wagner, H. andHuster, K. M., Protective CD8 T cell immunity triggered by CpG-proteinconjugates competes with the efficacy of live vaccines. J. Immunol. 2005.174: 4373–4380.
20 Wagner, H., Heit, A., Schmitz, F. and Bauer, S., Targeting split vaccines tothe endosome improves vaccination. Curr. Opin. Biotechnol. 2004. 15:538–542.
21 Jiang, W., Gupta, R. K., Deshpande, M. C. and Schwendeman, S. P.,Biodegradable poly(lactic-co-glycolic acid) microparticles for injectabledelivery of vaccine antigens. Adv. Drug Deliv. Rev. 2005. 57: 391–410.
22 Jilek, S., Ulrich, M., Merkle, H. P. and Walter, E., Composition and surfacecharge of DNA-loaded microparticles determine maturation and cytokinesecretion in human dendritic cells. Pharm. Res. 2004. 21: 1240–1247.
23 O'Hagan, D. T., Singh, M. and Ulmer, J. B., Microparticles for the deliveryof DNA vaccines. Immunol. Rev. 2004. 199: 191–200.
24 O'Hagan, D. T., Singh, M. and Ulmer, J. B., Microparticle-basedtechnologies for vaccines. Methods 2006. 40: 10–19.
25 Westwood, A., Elvin, S. J., Healey, G. D., Williamson, E. D. and Eyles, J.E., Immunological responses after immunisation of mice with microparticlescontaining antigen and single stranded RNA (polyuridylic acid). Vaccine2006. 24: 1736–1743.
26 Diwan, M., Elamanchili, P., Cao, M. and Samuel, J., Dose sparing of CpGoligodeoxynucleotide vaccine adjuvants by nanoparticle delivery. Curr. DrugDeliv. 2004. 1: 405–412.
27 Shen, H., Ackerman, A. L., Cody, V., Giodini, A., Hinson, E. R., Cresswell,P., Edelson, R. L. et al., Enhanced and prolonged cross-presentationfollowing endosomal escape of exogenous antigens encapsulated inbiodegradable nanoparticles. Immunology 2006. 117: 78–88.
28 Diwan, M., Tafaghodi, M. and Samuel, J., Enhancement of immuneresponses by co-delivery of a CpG oligodeoxynucleotide and tetanus toxoidin biodegradable nanospheres. J. Control Release 2002. 85: 247–262.
29 Schmitz, F., Heit, A., Guggemoos, S., Krug, A., Mages, J., Schiemann, M.,Adler, H. et al., Interferon-regulatory-factor 1 controls Toll-like receptor 9-mediated IFN-beta production in myeloid dendritic cells. Eur. J. Immunol.2007. 37: 315–327.
30 Sparwasser, T., Vabulas, R. M., Villmow, B., Lipford, G. B. and Wagner,H., Bacterial CpG-DNA activates dendritic cells in vivo: T helper cell-
Eur. J. Immunol. 2007. 37: 2063–2074 Highlights 2073
f 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu
independent cytotoxic T cell responses to soluble proteins. Eur. J. Immunol.2000. 30: 3591–3597.
31 Busch, D. H., Pilip, I. M., Vijh, S. and Pamer, E. G., Coordinate regulationof complex T cell populations responding to bacterial infection. Immunity1998. 8: 353–362.
32 Vijh, S. and Pamer, E. G., Immunodominant and subdominant CTLresponses to Listeria monocytogenes infection. J. Immunol. 1997. 158:3366–3371.
33 Audran, R., Peter, K., Dannull, J., Men, Y., Scandella, E., Groettrup, M.,Gander, B. and Corradin, G., Encapsulation of peptides in biodegradablemicrospheres prolongs their MHC class-I presentation by dendritic cells andmacrophages in vitro. Vaccine 2003. 21: 1250–1255.
34 Nestle, F. O., Alijagic, S., Gilliet, M., Sun, Y., Grabbe, S., Dummer, R.,Burg, G. and Schadendorf, D., Vaccination of melanoma patients withpeptide- or tumor lysate-pulsed dendritic cells. Nat. Med. 1998. 4: 328–332.
35 Thurner, B., Haendle, I., Roder, C., Dieckmann, D., Keikavoussi, P.,Jonuleit, H., Bender, A. et al., Vaccination with mage-3A1 peptide-pulsedmature, monocyte-derived dendritic cells expands specific cytotoxic T cellsand induces regression of some metastases in advanced stage IV melanoma.J. Exp. Med. 1999. 190: 1669–1678.
36 Maksimow, M., Miiluniemi, M., Marttila-Ichihara, F., Jalkanen, S. andHanninen, A., Antigen targeting to endosomal pathway in dendritic cellvaccination activates regulatory T cells and attenuates tumor immunity.Blood 2006. 108: 1298–1305.
37 Wilson, N. S., Behrens, G. M., Lundie, R. J., Smith, C. M., Waithman, J.,Young, L., Forehan, S. P. et al., Systemic activation of dendritic cells by Toll-like receptor ligands or malaria infection impairs cross-presentation andantiviral immunity. Nat. Immunol. 2006. 7: 165–172.
38 Inaba, K., Inaba, M., Naito, M. and Steinman, R. M., Dendritic cellprogenitors phagocytose particulates, including bacillus Calmette-Guerinorganisms, and sensitize mice to mycobacterial antigens in vivo. J. Exp. Med.1993. 178: 479–488.
39 Romero, P., Maryanski, J. L., Corradin, G., Nussenzweig, R. S.,Nussenzweig, V. and Zavala, F., Cloned cytotoxic T cells recognize anepitope in the circumsporozoite protein and protect against malaria. Nature1989. 341: 323–326.
40 Szalay, G., Hess, J. and Kaufmann, S. H., Presentation of Listeriamonocytogenes antigens by major histocompatibility complex class Imolecules to CD8 cytotoxic T lymphocytes independent of listeriolysinsecretion and virulence. Eur. J. Immunol. 1994. 24: 1471–1477.
41 Inaba, K., Turley, S., Yamaide, F., Iyoda, T., Mahnke, K., Inaba, M., Pack,M. et al., Efficient presentation of phagocytosed cellular fragments on themajor histocompatibility complex class II products of dendritic cells. J. Exp.Med. 1998. 188: 2163–2173.
42 Ackerman, A. L., Giodini, A. and Cresswell, P., A role for the endoplasmicreticulum protein retrotranslocation machinery during crosspresentation bydendritic cells. Immunity 2006. 25: 607–617.
43 Hatsuzawa, K., Tamura, T., Hashimoto, H., Hashimoto, H., Yokoya, S.,Miura, M., Nagaya, H. and Wada, I., Involvement of syntaxin 18, anendoplasmic reticulum (ER)-localized SNARE protein, in ER-mediatedphagocytosis. Mol. Biol. Cell 2006. 17: 3964–3977.
44 Blander, J. M. and Medzhitov, R., Toll-dependent selection of microbialantigens for presentation by dendritic cells. Nature 2006. 440: 808–812.
45 Yarovinsky, F., Kanzler, H., Hieny, S., Coffman, R. L. and Sher, A., Toll-like receptor recognition regulates immunodominance in an antimicrobialCD4+ T cell response. Immunity 2006. 25: 655–664.
46 Diebold, S. S., Kaisho, T., Hemmi, H., Akira, S. and Reis e Sousa, C.,Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 2004. 303: 1529–1531.
47 Heil, F., Hemmi, H., Hochrein, H., Ampenberger, F., Kirschning, C.,Akira, S., Lipford, G. et al., Species-specific recognition of single-strandedRNA via toll-like receptor 7 and 8. Science 2004. 303: 1526–1529.
48 Yasuda, K., Rutz, M., Schlatter, B., Metzger, J., Luppa, P. B., Schmitz, F.,Haas, T. et al., CpG motif-independent activation of TLR9 upon endosomaltranslocation of "natural" phosphodiester DNA. Eur. J. Immunol. 2006. 36:431–436.
49 Zaks, K., Jordan, M., Guth, A., Sellins, K., Kedl, R., Izzo, A., Bosio, C. andDow, S., Efficient immunization and cross-priming by vaccine adjuvantscontaining TLR3 or TLR9 agonists complexed to cationic liposomes. J.Immunol. 2006. 176: 7335–7345.
50 Langer, R., Drug delivery and targeting. Nature 1998. 392: 5–10.
51 Heit, A., Huster, K. M., Schmitz, F., Schiemann, M., Busch, D. H. andWagner, H., CpG-DNA aided cross-priming by cross-presenting B cells. J.Immunol. 2004. 172: 1501–1507.
52 Spies, B., Hochrein, H., Vabulas, M., Huster, K., Busch, D. H., Schmitz, F.,Heit, A. and Wagner, H., Vaccination with plasmid DNA activates dendriticcells via Toll-like receptor 9 (TLR9) but functions in TLR9-deficient mice. J.Immunol. 2003. 171: 5908–5912.
53 Lima, K. M., Santos, S. A., Lima, V. M., Coelho-Castelo, A. A., Rodrigues,J. M., Jr. and Silva, C. L., Single dose of a vaccine based on DNA encodingmycobacterial hsp65 protein plus TDM-loaded PLGA microspheres protectsmice against a virulent strain ofMycobacterium tuberculosis.Gene Ther. 2003.10: 678–685.
54 Ramachandra, L., Sramkoski, R. M., Canaday, D. H., Boom, W. H. andHarding, C. V., Flow analysis of MHC molecules and other membraneproteins in isolated phagosomes. J. Immunol. Methods 1998. 213: 53–71.
Antje Heit et al. Eur. J. Immunol. 2007. 37: 2063–20742074
f 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu