Environmental Toxicology and Chemistry, Vol 6, pp 827-837, 1987 Printed In the USA Pergamon Journals Ltd 0730-7268/87 $3.00 + .OO Environmental Chemistry DEGRADATION OF SELECTED HALOGENATED ETHANES IN ANOXIC SEDIMENT-WATER SYSTEMS CHAD T. JAFVERT and N. LEE WOLFE" US. Environmental Protection Agency, Environmental Research Laboratory, Athens, Georgia 30613 (Received 13 November 1986; Accepted 14 May 1987) Abstract -The degradation of selected halogenated ethanes was studied in anoxic sediment-water suspensions at 1 to 20% sediment concentrations. Batch kinetic experiments were used to quan- tify decay. Eh measurements of all suspensions were below -100 mV (vs SHE), indicating reduced environmental conditions. Hexachloroethane (HCA), 1,1,2,2-tetrachloroethane (TTCA), 1,2- diiodoethane (DIA) and 1,2-dibromoethane (DBA) were degraded within minutes to days, but 1,Zdichloroethane (DCA) remained in the system through 35 d, at which point the study was ended. The major route of transformation of these compounds in this system was vicinal dehalogenation. Rates of disappearance followed pseudo-first-order kinetics at low reactant concentrations and high sediment concentration. The order of reactivity in the same sediment sample followed the sequence, HCA = DIA > DBA > TTCA > DCA. Keywords - Haloethanes Sediment Degradation Anoxic conditions INTRODUCTION Reductive transformations constitute an im- portant fate process for a variety of aquatic pol- lutants. For example, the classes of compounds known to undergo reduction in environmental sys- tems include nitroaromatics [I], azo compounds [2] and halogenated organics [3-121. In several of these studies, the rates of reduction of specific compounds were shown to be influenced by envi- ronmental factors, such as the redox potential and solids content, but in other studies physical and chemical properties of the compounds themselves were shown to correlate with the rates of reduction under specific conditions. The characterization of these environmental parameters and intrinsic chem- ical properties, therefore, can be an important tool in assessing the significance of reductive pathways for various classes of compounds. Reduction of halogenated hydrocarbons occurs under a variety of reduced incubation systems, including sewage sludge [3], anaerobic microbio- logical systems [7-lo], saturated aquifer material [9,10-121, reduced iron porphyrin solutions [3,5, 6,13,14] and solutions of various chemical reduc- ing agents [15,16]. The reduction of halogenated *To whom correspondence may be addressed. Mention of trade names or commercial products does not constitute endorsement or recommendation for use by the U.S. Environmental Protection Agency. organic compounds in the aquatic environment appears to result from both biotic and abiotic pro- cesses. When biological reduction is inferred and there is no induction period, it has been suggested that biologically reduced iron, in the form of iron porphyrins, is responsible for the initial reduction [3,5,61. At present, no satisfactory method is available for predicting the rates of reductive reactions in aquatic environments. Predictive methods have been developed, however, for other processes, such as hydrolysis, volatilization, sorption-desorp- tion, bioaccumulation and photolysis. The capabil- ity of such methods allows a priori determination of the significance of each of these processes for chemicals under various environmental conditions. The lack of a priori knowledge about reductive reactions can lead to the overestimation of pollu- tant concentrations in various environments and, at the same time, ignore product formation. Until predictive methods are developed for reductive pathways, experimental observation must be used to determine the fate of reducible compounds in environmental systems. This study was undertaken to investigate factors that influence the rates of reductive transforma- tions of halogenated hydrocarbons in environmental systems. The factors that were examined included both environmental variables and inherent chem- ical properties of a series of substituted compounds. 827
Transcript
Environmental Toxicology and Chemistry, Vol 6 , pp 827-837, 1987 Printed In the USA Pergamon Journals Ltd
0730-7268/87 $3.00 + .OO
Environmental Chemistry
DEGRADATION OF SELECTED HALOGENATED ETHANES IN ANOXIC SEDIMENT-WATER SYSTEMS
CHAD T. JAFVERT and N. LEE WOLFE" US. Environmental Protection Agency, Environmental Research Laboratory,
Athens, Georgia 30613
(Received 13 November 1986; Accepted 14 May 1987)
Abstract -The degradation of selected halogenated ethanes was studied in anoxic sediment-water suspensions at 1 to 20% sediment concentrations. Batch kinetic experiments were used to quan- tify decay. Eh measurements of all suspensions were below -100 mV (vs SHE), indicating reduced environmental conditions. Hexachloroethane (HCA), 1,1,2,2-tetrachloroethane (TTCA), 1,2- diiodoethane (DIA) and 1,2-dibromoethane (DBA) were degraded within minutes to days, but 1,Zdichloroethane (DCA) remained in the system through 35 d, at which point the study was ended. The major route of transformation of these compounds in this system was vicinal dehalogenation. Rates of disappearance followed pseudo-first-order kinetics at low reactant concentrations and high sediment concentration. The order of reactivity in the same sediment sample followed the sequence, HCA = DIA > DBA > TTCA > DCA.
Reductive transformations constitute an im- portant fate process for a variety of aquatic pol- lutants. For example, the classes of compounds known to undergo reduction in environmental sys- tems include nitroaromatics [I], azo compounds [2] and halogenated organics [3-121. In several of these studies, the rates of reduction of specific compounds were shown to be influenced by envi- ronmental factors, such as the redox potential and solids content, but in other studies physical and chemical properties of the compounds themselves were shown to correlate with the rates of reduction under specific conditions. The characterization of these environmental parameters and intrinsic chem- ical properties, therefore, can be an important tool in assessing the significance of reductive pathways for various classes of compounds.
Reduction of halogenated hydrocarbons occurs under a variety of reduced incubation systems, including sewage sludge [3], anaerobic microbio- logical systems [7-lo], saturated aquifer material [9,10-121, reduced iron porphyrin solutions [3,5, 6,13,14] and solutions of various chemical reduc- ing agents [15,16]. The reduction of halogenated
*To whom correspondence may be addressed. Mention of trade names or commercial products does
not constitute endorsement or recommendation for use by the U.S. Environmental Protection Agency.
organic compounds in the aquatic environment appears to result from both biotic and abiotic pro- cesses. When biological reduction is inferred and there is no induction period, it has been suggested that biologically reduced iron, in the form of iron porphyrins, is responsible for the initial reduction [3,5,61.
At present, no satisfactory method is available for predicting the rates of reductive reactions in aquatic environments. Predictive methods have been developed, however, for other processes, such as hydrolysis, volatilization, sorption-desorp- tion, bioaccumulation and photolysis. The capabil- ity of such methods allows a priori determination of the significance of each of these processes for chemicals under various environmental conditions. The lack of a priori knowledge about reductive reactions can lead to the overestimation of pollu- tant concentrations in various environments and, at the same time, ignore product formation. Until predictive methods are developed for reductive pathways, experimental observation must be used to determine the fate of reducible compounds in environmental systems.
This study was undertaken to investigate factors that influence the rates of reductive transforma- tions of halogenated hydrocarbons in environmental systems. The factors that were examined included both environmental variables and inherent chem- ical properties of a series of substituted compounds.
827
828 C. T. JAFVERT A N D N . LEE WOLFE
Experiments were performed in which hexachloro- ethane (HCA), 1 , I ,2,2-tetrachloroethane (TTCA), 1,2-dichloroethane (DCA), I ,2-dibromoethane (DBA), and 1,2-diiodoethane (DIA) were added to anoxic sediment-water mixtures, and the disap- pearance of compound was quantified over time. Model compounds were chosen that contained vic- inal halides, allowing comparisons to he made among the relative rates of disappearance with respect to chemical properties. Environmental fac- tors studied included sediment concentration, sedi- ment source and microbial inhibition.
MATERIALS AND METHODS Reagents
Analytical reference standards of HCA (%To), TTCA (98%), DCA (99.9%), tetrachloroethylene (TTCE, +98%) and DBA (+99%) were obtained from the U S . Environmental Protection Agency (EPA) Chemical Repository at Research Triangle Park, North Carolina. A standard of DIA (97070) was purchased from Aldrich. All samples were confirmed by GC-MS. Resazurin (98%; Aldrich), tryptone glucose extract agar (Difco) and thio- glycollate medium (Difco) were used to test for microbial growth by the sediments. Formalin (pur- chased from Baker as a 37.4% formaldehyde: 12% methanol solution) and sodium azide (Fisher) were used as metabolic inhibitors. UV-grade acetonitrile (Burdick and Jackson) was used as the solvent to make stock solutions of all halogenated com- pounds, and UV-grade hexane and isooctane (Bur- dick and Jackson) were used to extract halogenated ethanes and products from the sediment-water samples.
Sediment samples Sediment-water slurries were collected from
ponds (referred to as Bar H, Hickory Hill and Memorial Park), or from a local, slow-moving, stagnant stream (Beaver Dam) within 3.2 m of Athens, Georgia. Samples were collected under 0.30- to 0.60-m depths of water by scraping the first 5.0 to 7.5 cm of bottom sediment into glass jars. These jars were completely filled with sedi- ment (about 40% total volume) and water (about 60%) and cappcd under the water surface, allow- ing no headspace, for transport to the laboratory. The sand-siIt-clay fraction of the sediment was separated from debris by wet-sieving with a I-mm wire mesh sieve. In experiments in which only the silt-clay fraction was used, the sediment was wet- sieved through I-mm, 250-pm and 53-pm sieves in series, collecting only the less than 53-pm fraction. All sediment-water samples were stored in a nitro-
gen-atmosphere glove box unt i l use. All proce- dures in which sediment-water was exposed to an atmosphere were performed in the nitrogen-atmo- sphere glove box, except for sieving. I t was found, however, that the sediments were poised suffi- ciently that Eh measurements did not change dur- ing this procedure.
Elemental analyses were made using a Perkin- Elmer Plasma 11 inductively coupled plasma emis- sion spectrometer (ICP). Both pore water samples and acid extracts of sediment were analyzed. Sedi- ment pore water was filtered through a Millipore AW filter and acidified with 1% (v/v) concen- trated nitric acid before analysis. Sediment samples were concentrated by filtration on Whatman qual- itative filter paper. A moist portion ( I g) of each sediment was placed in a 30-ml test tube, which was then fillcd completely with deionized water and 1% (v/v) concentrated nitric acid. Both the pore water samples and the aqueous portion of the sediment samples were analyzed 1 d after acidifi- cation. Each analysis was replicated three times. The iron values for the sediments exceeded the standard values; therefore, a 1 : l O dilution was reanalyzed for iron. The ICP was calibrated prior to analysis and two quality control samples were interspersed between samples.
Kinetic experiments Kinetic experiments were performed using a
hatch method in which sediment-water aliquots were distributed into a series of test tubes and spiked with a known concentration of chemical under a nitrogen atmosphere. Time-concentration data were collected by periodically sacrificing a tube for analysis. For each experiment, 10-ml ali- quots of sediment-water were placed into 18-ml test tubes and capped using Teflon-lined septa and open-top screw caps. To ensure that there were equal portions of sediment in each tube, the stock jars of sediment-water were either vigorously stirred with a pipet prior to the removal of each aliquot or stirred using a magnetic stirring bar. Mixing with a magnetic stirring bar was found to he suffi- cient for the silt-clay sediment fraction. To per- form experiments using different sediment-to-water ratios, the stock sediment samples were ccntrifuged and the resulting supernatant pond water was used as diluent.
A stock solution of each halogenated ethane was made in acetonitrile such that 2O-pl additions of chemical into 10-ml sediment-water solutions gave the desired initial experimental concentrations. Sample tubes were spiked with chemical using a
Degradation of halogenated ethanes in sediment-water systems 829
25-pl Hamilton syringe, capped, vortex-mixed and incubated at 25°C in a waterbath. Sample tubes were periodically mixed by hand. At specific inter- vals, the tubes were extracted with 4 ml hexane by vortex-mixing at high speed for 1 to 2 min. The hexane was recovered from the tubes by centri- fuging at 1,600 g for 20 min. The hexane layer was removed from samples not analyzed on the same day as extracted, and placed in a clean tube.
Distribution coefficients The distribution of chemical between the aque-
ous phase and the solid phase was measured in conjunction with most kinetic experiments. Mea- surement was accomplished by centrifuging spiked sediment-water samples at 5,000 g for 10 min, removing the aqueous layer and extracting both phases with 4 ml hexane as described above. High- speed Corex glass centrifuge tubes (25 ml) were used and, in data analysis, corrections were made for the water remaining in the sediment phase. The values for distribution coefficients are reported as Kd in units of mol/kg sediment per mol/L of water.
Chemical analysis Hexane extracts were analyzed using a Tracor
Model 220 gas chromatograph equipped with an electron-capture detector, a Tracor 560 linearizer and a Tracor strip-chart recorder. HCA, TTCA, TTCE, DBA and DIA were separated using a 1.8-m glass column of OV-17 on Gas Chrom Q, 80/100 Mesh, run isothermally at 110°C. DCA was ana- lyzed using a 1.8-m glass column of l % SP-1000 on 60/80 mesh Carbopack B, run isothermally at 190°C. Sample concentrations were calculated by comparing peak heights of samples against stan- dards. Standard curves were made for each series of analyses; the correlation coefficients ( r 2 ) for these lines generally were greater than 0.98.
GC-MS analyses were performed using a Hew- lett-Packard 5840 GC with a capillary column interfaced directly to a Varian CH5-DF mass spec- trometer. For HCA and TTCE analyses, a 40-m SE 54 silica capillary column was used. The run was programmed for 1 min at 75"C, followed by temperature increases of 2"C/min to 85°C. For analysis of DIA, DBA and ethylene, a 30-m DB5 capillary column was used. The column was held at -10°C for 7 min by carbon dioxide cooling, and programmed to 150°C at 4"C/min. For GC- MS product studies, relatively high concentrations (for example, 1 mM) of compound were spiked into sediment-water samples, then extracted 24 to
48 h later and analyzed. For ethylene analysis, sediment-water headspace samples were analyzed.
An Orion model 701A pH meter was used for both pH and Eh measurements. Eh was measured using a Markson 1202 combination platinum/ Ag:AgCl reference electrode. Relatively short equi- libration times (5-10 min) were required before a stable mV reading was reached. All Eh values are reported versus SHE. The platinum electrode was periodically checked against a standard ferrous- ferric solution [17]. All pH and Eh measurements were made under a nitrogen atmosphere.
Sample sterility In selected experiments, formalin (2% v/v) and
sodium azide (0.1 M) were used as inhibitors. Sediment-water samples were incubated with each of these chemicals overnight before kinetic exper- iments were performed. Autoclaved samples (20 min, three times) also were used in selected kinetic experiments. Microbial growth in these samples was tested using either (aerobic) pour plates con- taining tryptone glucose extract agar or anoxic agar tubes (Hungate roll tubes) containing thiogly- collate medium, resazurin and the respective con- centration of chemical inhibitor. The anoxic agar was prepared by adding 14.5 g of Bacto-thiogly- collate medium, 5 g of Bacto agar and 5 ml of a 1 % resazurin solution to 500 ml of distilled water. After 5 d , no growth was detected in any treated plates or tubes, whereas untreated sediment showed both aerobic and anaerobic growth.
RESULTS AND DISCUSSION
Disappearance rate constants To quantify the disappearance of pollutants
in sediment-water suspensions (Eqn. l), a simple kinetic expression (Eqn. 2) can be used:
(1) k
P + R, 4 products
where P is the specific pollutant, R, is the ith environmentally supplied reactant and k , is the rate constant for reaction between P and R,. When R, is either in great excess of the pollutant con- centration, P, or in steady-state concentration, this second-order expression follows first-order kinetics:
830 C. T. JAFVERT A N D N . LEE WOLFE
where kobs is a pseudo-first-order disappearance rate constant.
I f reduction of halogenated hydrocarbons oc- curs at the surface of sediment particles, a binding step must be included to describe the heterogene- ity of the reaction:
kl P + Ri k+, - P-R, 3 products (4)
where P-R, is the sorbed species, k , and k - , are the sorption and desorption rate constants, respec- tively, and kz is the transformation rate constant.
To solve for [PI with respect to time, two dis- tinct assumptions can be made based on [PI >> [ P-R,] . The first assumption is that the first step (formation of P-R,) is rate limiting. In this case, the kinetics of Equation 4 are described by Equa- tion 2 (or 3). The second assumption is that the second step (formation of product) is rate limiting. In this case, a prior equilibrium between P and P- R, is formed (Keq = k , / k - , ) and the kinetics are still described by Equation 3, where kobs = kzKeq R, / ( 1 + Keq R,). Pseudo-first-order kinetics, such as this, describe the majority of environmen- tal processes, including direct photolysis, hydroly- sis and volatilization from open systems.
Another distinct assumption that is often made is that the intermediate complex, P-R,, remains at pseudo-steady state throughout the reaction. This is to say that the rate of its formation, k , [PI [ R , ] , is equal to the rate of its decay, (k, + kz) [P-R,] . This assumption leads to a “hyperbolic” rate law [18,19] that is identical in form to Michaelis-Menten kinetics. A further criterion for this assumption, however, is that at high pollutant concentrations, the reactant is continuously regenerated.
I f isolation of reduction reactions from other processes is shown through product identification or relative rate analysis, the factors influencing the rate of reduction of compounds can be de- termined, and the applicability of each of these assumptions can be determined. In the kinetic experiments described, hydrolysis reactions can be shown to be insignificant pathways for the time frame of these,experiments through the use of dis- tilled water blanks. Furthermore, a major product of HCA decay was shown to be TTCE by both GC-MS analysis and GC retention time, whereas ethylene was found in the headspace of sediment- water samples spiked with either DBA or DIA after 24 h. From Table 1 [20-241, the Henry’s Law constants of the chemicals studied indicate that only DBA should volatilize to any appreciable extent. Moreover, only 8% of DBA should be found in the vapor phase, indicating that the observed disappearance rate constants, kobs, ob- tained from data based on the decay of 1 to 2 half- lives. are the result of reduction reactions.
Disappearance kinetics of halogenated ethanes in anoxic sediments
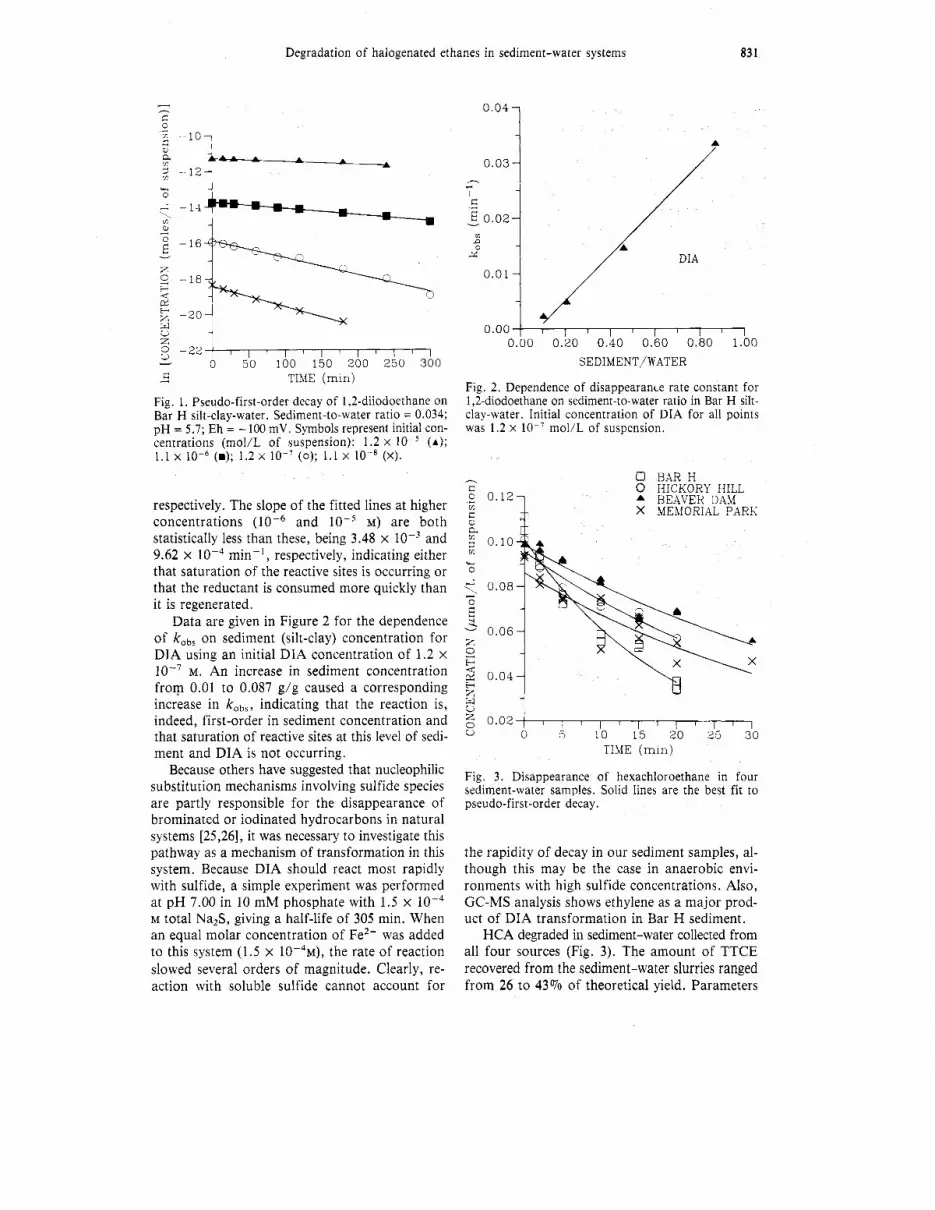
The kinetics of decay of HCA and DIA were studied to determine the suitability of each of the above assumptions. The decay of DIA was stud- ied in a silt-clay-water slurry from Bar H Pond by varying both DIA concentration and sediment concentration. Using a sediment-to-water ratio (S/W) of 0.034 (g/g), the decay of DIA followed pseudo-first-order kinetics at concentrations of lo-* to lo-’ M. The first-order fit of data at these concentrations, shown in Figure 1, gives /cobs val- ues of 8.75 x min-l and 9.48 x lop3 min-’,
Table I . Physical constants of selected halogenated ethanes
Compound
Dimensionless Bond dissociation Electron affinity
(H; C,/C,,) at 25°C Kcal/mol) halogen (Kcal/mol)” Henry’s Constant energy (C-X; of associated
Hexachloroet hane 0.049b I , I ,2,2-Terrachloroethane 0.019b 1,2-Dichloroethane 0.040b
I ,2-Diiodoethane 0.040d I ,2-Dibromoethane 0.102d
72‘ 74.3‘ 77.9‘ 69.8‘ <47’
83(C1) 83(CI) 83(CI) 77.5(Br) 70.5(1)
”From ref. 20. bCalculated from vapor pressure and water solubility data at 2 5 T , by Dilling [21]. ‘From ref. 22. dMeasured in distilled water at 25°C. ‘From ref. 23. ‘From ref. 24.
Degradation of halogenated ethanes in sediment-water systems 83 1
m
v E 0.02-
9 e
0.01 -
0.00 I I I I I I I I I
respectively. The slope of the fitted lines at higher concentrations and M) are both statistically less than these, being 3.48 x and 9.62 x min-', respectively, indicating either that saturation of the reactive sites is occurring or that the reductant is consumed more quickly than it is regenerated.
Data are given in Figure 2 for the dependence of kobs on sediment (silt-clay) concentration for DIA using an initial DIA concentration of 1.2 X
lo-' M. An increase in sediment concentration from 0.01 to 0.087 g/g caused a corresponding increase in kobs, indicating that the reaction is, indeed, first-order in sediment concentration and that saturation of reactive sites at this level of sedi- ment and DIA is not occurring.
Because others have suggested that nucleophilic substitution mechanisms involving sulfide species are partly responsible for the disappearance of brominated or iodinated hydrocarbons in natural systems [25,26], it was necessary to investigate this pathway as a mechanism of transformation in this system. Because DIA should react most rapidly with sulfide, a simple experiment was performed at p H 7.00 in 10 mM phosphate with 1.5 x M total Na,S, giving a half-life of 305 min. When an equal molar concentration of Fez+ was added to this system (1.5 x l O P 4 ~ ) , the rate of reaction slowed several orders of magnitude. Clearly, re- action with soluble sulfide cannot account for
z
0 BAR H 0 HICKORY HILL A BEAVER DAM X MEMORIAL PARK
g 0 . 0 2 2 , 0 5 10 15 20 25 30
TIME (min) u
Fig. 3 . Disappearance of hexachloroethane in four sediment-water samples. Solid lines are the best fit to pseudo-first-order decay.
the rapidity of decay in our sediment samples, al- though this may be the case in anaerobic envi- ronments with high sulfide concentrations. Also, GC-MS analysis shows ethylene as a major prod- uct of DIA transformation in Bar H sediment.
HCA degraded in sediment-water collected from all four sources (Fig. 3). The amount of TTCE recovered from the sediment-water slurries ranged from 26 to 43% of theoretical yield. Parameters
832 C. T. JAFVERT AND N . LEE WOLFE
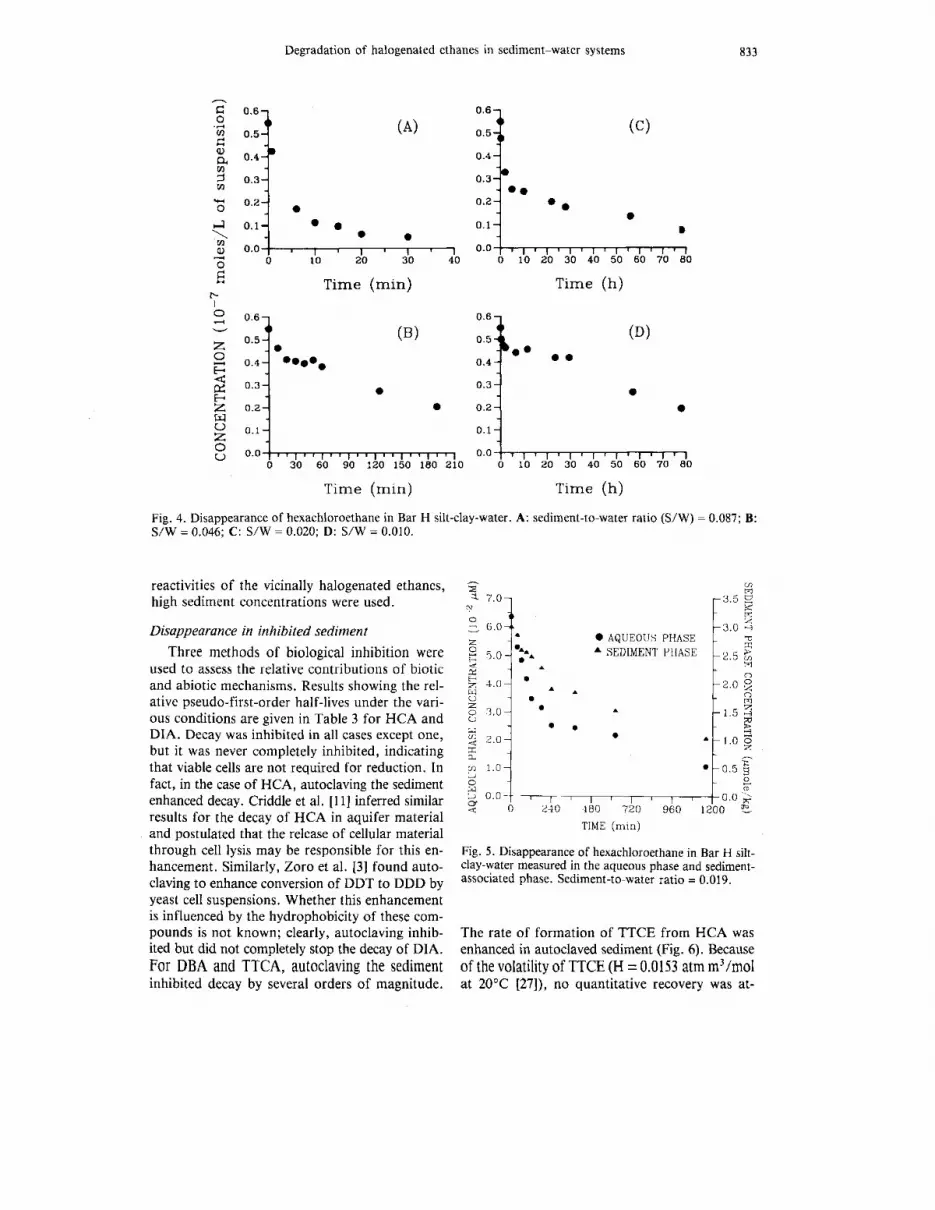
characterizing the sediment are given in Table 2. Pseudo-first-order rate constants (/cob\) are given as indicators of relative reactivity of the sediment. No attempt was made in these experiments to nor- malize sediment-to-water ratios (S/W) because the sediment varied greatly in particle size. Memorial Park contained the most sand per dry weight; Bar H contained the least. Experiments performed in water separated from sediment by centrifugation resulted in no decay over the same time period as in other experiments (less than 1 d), indicating that the reductant was associated with the sediment. The distribution of H C A between the sediment and water phases (Kd) was measured in diluted sediment-water mixtures. Although this may have affected Kd values, dilution was necessary because of the rapidity with which H C A decayed in con- centrated sediments. T h e Kd values presented in Table 2 were measured over a 4-h period, with no apparent change.
These observations on the decay of H C A are consistent with the results of Wade and Castro [13], who found that the reduction of H C A to TTCE occurred easily by reaction with iron(1l)deu- teroporphyrins and with chromous sulfate [ 161. Likewise, Criddle et al. [ I I] found that H C A re- duction occurred in the presence of aquifer ma- terial. Experiments by Castro and Kray [15,16] clearly showed the rapidity with which vicinal dehalogenation occurs with respect to reductive dehalogenation, where one halogen is replaced by hydrogen. This ease of reduction of vicinal halides has been postulated by Kray and Castro to occur through the stabilization of the intermediate by bridging between the neighboring halogen and car- bon from which the initial halogen was removed.
Experiments designed to elucidate an appropri- ate kinetic expression for the decay of H C A were inconclusive. Figure 4 shows the disappearance of H C A from Bar H silt-clay at four different
silt-clay concentrations. At the highest sediment concentration (0.08 g/g) decay was rapid, n i th a half-life of less than 5 min. At the lowest sediment concentration (0.01 g/g) decay occurred over a period of days. In all cases, however, decay was initially very rapid followed by slower decay over a period of approximately 5 h. At the lower sedi- ment concentrations this xas followed by yet a slower decay rate. I t is likely that these transitions are the result of the consumption of sediment- supplied reactants, each having a different reactiv- i t y to HCA.
Because HCA appreciably adsorbs to sediment, the influences of sorption-desorption were inves- tigated by measuring the residual concentrations of HCA in both the sediment and aqueous phases after separating the phases by centrifugation dur- ing an experiment. Figure 5 shows the results of analysis of H C A in the separated phases over time at a silt-clay concentration of 0.019 g/g. Initially, 52% of H C A was associated with the sediment. The measured Kd steadily increased during this experiment from 47.1 a t t = 5 min to 96 at t = 1,160 min. Analysis of the data in Figure 5 for the first 240 min suggests that the disappearance of HCA from the sediment phase is slower than from the aqueous phase. For the aqueous phase, kobs = 3.76 x l op3 min-’, whereas for the sediment phase, kobs = 1.75 x min-I. However, data from similar experiments suggest that the rate of disappearance is the same. As is also evident from Figure 4 using the same sediment concentration, after the decay of I to 2 half-lives, the rate of decay is inhibited. I t is apparent that for both HCA and DIA, saturation or depletion of reactive species associated with the sediment can occur at low ratios of sediment t o compound. A t high ratios of sediment to compound, however, pseudo- first-order kinetic expressions may be appropriate. Therefore, to facilitate comparisons of chemical
Table 2. Disappearance of hexachloroethane and appearance of tetrachlorotheylene (TTCE) in four sediments
Initial Sediment concn. concn. (g/ml)
Sediment S/W” pH (mV) suspension) min-’) as TTCEb Kd measurement Eh (mol/L of kob\ % recovery for Kd
Bar H 0.08 6.72 - 181 1.1 x 5.0 k 0.34 43% (f = 20 min) 80 k 14 (n = 10) 0.0039 Beaver Dam 0.20 6.65 -250 0.96 x lo-’ 2.0 +_ 0.17 42% ( I = 30 min) 20 k 2 (n = 11) 0.029 Hickory Hill 0.1 I 6.30 - 165 1.0 x 2.6 k 0.20 26% (I = 20 min) 9.0 k 2 (n = 2) 0.077 Memorial Park 0.055 6.70 -172 0.93 x 1.9 k 0.16 33% (I = 50 min) 35 k 13 (n = 12) 0.02
“S/W = ratio of sediment (sand-silt-clay fraction) to water (g/g). bDuration of experiment in parentheses. ‘Number of determinations in parentheses.
Degradation of halogenated cthanes in sediment-watcr systems 833
0 .d
0.4
2 0.3
i; 0.2
* * . cl 0.1
0.0 0 10 20 30 40 -
2 Time (min) c I
(D)
z 0.2 W
0 30 60 90 120 150 180 210
0.6
0.3 ::;:, ~;, , , , , m , , , ,,~
0.2
0.1
0.0 0 10 20 30 40 50 60 70 80
T i m e (h)
0.1 L- 0.0 O 10 20 30 40 50 60 70 80
T i m e (min) T i m e (h)
Fig. 4. Disappearance of hexachloroethane in Bar H silt-clay-water. A: sediment-to-water ratio (S/W) = 0.087; B: S/W = 0.046; C: S/W = 0.020; D: S/W = 0.010.
reactivities of the vicinally halogenated ethanes, high sediment concentrations were used.
Disappearance in inhibited sediment Three methods of biological inhibition were
used to assess the relative contributions of biotic and abiotic mechanisms. Results showing the rel- ative pseudo-first-order half-lives under the vari- ous conditions are given in Table 3 for HCA and DIA. Decay was inhibited in all cases except one, hut it was never completely inhibited, indicating that viable cells are not required for reduction. In fact, in the case of HCA, autoclaving the sediment enhanced decay. Criddle et al. [ I 11 inferred similar results for the decay of HCA in aquifer material and postulated that the release of cellular material through cell lysis may be responsible for this en- hancement. Similarly, Zoro et al. [3] found auto- claving to enhance conversion of DDT to DDD by yeast cell suspensions. Whether this enhancement is influenced by the hydrophobicity of these com- pounds is not known; clearly, autoclaving inhih- ited hut did not completely stop the decay of DIA. For DBA and TTCA, autoclaving the sediment inhibited decay by several orders of magnitude.
TIME (nnn)
Fig. 5 . Disappearance of henachlaroethane in Bar H silt- clay-water measured in the aqueous phase and sediment- associated phase. Sediment-to-water ratio = 0.019.
The rate of formation of TTCE from HCA was enhanced in autoclaved sediment (Fig. 6) . Because of the volatility of TTCE (H = 0.0153 atm m'/mol a t 20°C [27]), no quantitative recovery was at-
834 C. T. JAFVSRT AND N. LEE WOLFK
Table 3 . Relative rates of disappearance o l hexachloroethane and diidoethane in sterilized or inhibited sediment-water systems'
'Bar-H silt-clay fraction; sediment-to-water ratio = 0.0895.
shed light on the extent of reductive processes
Because it has often been postulated that the 0.08 Autoclaved reduction of various halogenated hydrocarbons in
environmental systems results from reactions with 0.06 reduced iron [3,5,13,14], the sediments were ana-
lyzed for metals as previously described. Acid- soluble iron was found to be a major component of all four sediments, ranging from 2 to 4 mg/g moist sediment. The concentration of acid-soluble iron was found to be 10 times greater than that of any other metal. Aaueous concentrations of iron
- C
a Ci HCA' Autoclaved A HCA Natural involving pollutants in the environment. 3 I X k , Natural
0 0.10
P
.. 0
0 - Ei 0 - 0.04 z c
c z 2 0.02
4 o 0 O ranged from 0.5 to 2.0 mg/L. Because Eh values " 0 i n 20 30 for these sediments were low (less than -150 mV)
Y x u g 0.00 I I I ~~ ..
TIME ( r n i n j ~ anc, pH values were between 6 and 7, presumably a considerable amount of this iron should exist in reduced forms. Although homogeneous aqueous solutions of iron porphyrins have been shown
= 6.65; of autoclave = 6.23. Eh of react with various organic halides, heterogeneous forms of iron or other metals have not been scru- tinized thoroughly for reactivity.
Re'o1ive disappearance rates Of
Using the same sediment sample, the rates of disappearance of five vicinally halogenated ethanes were determined. Table 4 presents the relative order of disappearance of the compounds studied: HCA = DIA > DBA > TTCA > DCA. Figure 7 shows that the decay of four of these compounds fits the pseudo-first-order model reasonably well, using 7.5% solids. After 35 d, no apparent decay of DCA had occurred. This was anticipated, based on work by Bouwer and McCarty [S], who found only a 63% concentration reduction of DCA after 25 weeks when incubated under methanogenic conditions at 35°C.
Two parameters-strength of the carbon-hal-
Fig. 6. Decay of hcxachloroethane in &aver Dam sediment-water with formation of tetrachloroethylene (TTCE). Sediment-to-water ratio = 0.2: pH of natural
natural and autoclaved sediment = -250 mV.
tempted. Coextraction of TTCE from solution with HCA, however, indicated that the rate of product formation was highly dependent on the rate of decay of HCA.
The use of chemical inhibitors gave contrasting results. In each case, however, inhibition occurred to some extent. Formalin is generally known as a protein denaturing agent and sodium azide as a metabolic inhibitor interfering with electron trans- port. The specific effects that each of these have on strictly chemical reduction is not known and it is possible that the addition of inhibitors may have an adverse effect on abiotic degradation. Future investigations of specific reduction-enzyme inhibi- tors would be useful. The use in kinetic experi- ments of isolated soil enzyme suspensions also may
Degradation of halogenated ethanes in sediment-water systems 835
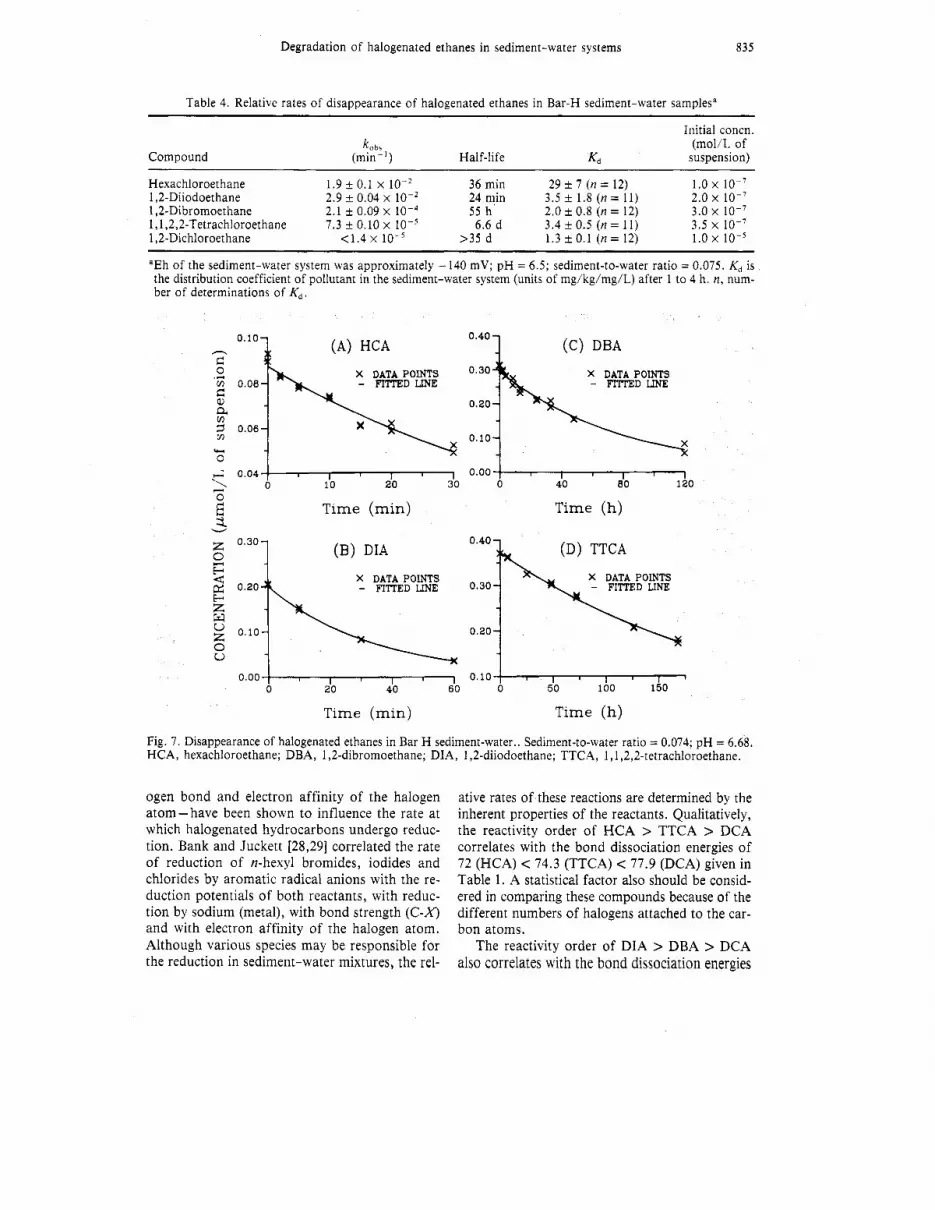
Table 4. Relative rates of disappearance of halogenated ethanes in Bar-H sediment-water samples"
Compound
~~ ~~
Initial concn. (mol/L of
Half-life Kd suspension)
Hexachloroethane 1.9 f 0.1 X lo-* 36 min 29 t 7 (n = 12) 1.0 x 1,2-Diiodoethane 2.9 t 0.04 x 24 min 3.5 * 1.8 (n = 1 1 ) 2.0 x 1 0 - ~ 1,2-Dibromoethane 2.1 f 0.09 x 10-4 55 h 2.0 t 0.8 (n = 12) 3.0 x 1 0 - ~ 1,1,2,2-TetrachIoroethane 7.3 f 0.10 x 10-5 6.6 d 3.4 t 0.5 (n = 11) 3.5 x 10-7 1,2-Dichloroethane < i .4 x 10-5 >35 d 1.3 k 0.1 (n = 12) 1.0 x 10-5
aEh of the sediment-water system was approximately - 140 mV; pH = 6.5; sediment-to-water ratio = 0.075. Kd is the distribution coefficient of pollutant in the sediment-water system (units of mg/kg/mg/L) after 1 to 4 h. n, num- ber of determinations of K d .
X DATA POINTS X DATA POINTS 0'30 - F I T T E D U N E - FITI'EDLWE
7 0.06- II)
0 4 0.04) 0 . 0 0 1 '\ 0 10 20 30 0 40 80 120 c 2 Time (min) 3. v
Time (h)
0 . 0 0 1 , 0 . 1 0 . 1 t 0 20 40 60 0 50 100 150
Time (min) Time (h)
Fig. 7. Disappearance of halogenated ethanes in Bar H sediment-water.. Sediment-to-water ratio = 0.074; pH = 6.68. HCA, hexachloroethane; DBA, 1,2-dibromoethane; DIA, 1,2-diiodoethane; TTCA, 1,1,2,2-tetrachIoroethane.
ogen bond and electron affinity of the halogen atom-have been shown to influence the rate at which halogenated hydrocarbons undergo reduc- tion. Bank and Juckett [28,29] correlated the rate of reduction of n-hexyl bromides, iodides and chlorides by aromatic radical anions with the re- duction potentials of both reactants, with reduc- tion by sodium (metal), with bond strength (C-X) and with electron affinity of the halogen atom. Although various species may be responsible for the reduction in sediment-water mixtures, the rel-
ative rates of these reactions are determined by the inherent properties of the reactants. Qualitatively, the reactivity order of HCA > TTCA > DCA correlates with the bond dissociation energies of 72 (HCA) < 74.3 (TTCA) < 77.9 (DCA) given in Table 1. A statistical factor also should be consid- ered in comparing these compounds because of the different numbers of halogens attached to the car- bon atoms.
The reactivity order of DIA > DBA > DCA also correlates with the bond dissociation energies
836 C. 7. J A F V E R T A N D N. LEE WOLFE
given in Table 1 , although the complicating factor of the changing electron affinity of the associated halogen also should affect the magnitude of reac- tivity. From transition state theory, a decreased bond dissociation energy should stabilize the transi- tion state and increase the rate of reaction [28,29]. Also, as the electronegativity of the leaving group increases, the rate of reaction should increase. In the sequence DIA > DBA > DCA, the differences in bond dissociation energies are greater than the differences in halogen electronegativities.
SUMMARY Transformations of vicinally halogenated eth-
anes were studied in anoxic sediment-water sus- pensions. After 35 d, no measurable decay of DCA had occurred. H C A , TTCA, DIA and DBA all degraded in anoxic sediment-water samples within minutes to days. Product studies indicated that reduction by vicinal dehalogenation was the major fate process. With the use of metabolic inhibitors it was shown that viable cells are not required for reduction t o occur, and no lag times were observed, indicating that either strictly chemical or biolog- ically mediated processes were responsible for decay.
Transformation of H C A to TTCE occurred in sediment-water collected from four different local sources. Although the chemical and biological nature of these sediments may not be as variable as that of sediment collected from various geo- graphical regions, the results suggest that under certain conditions reduction is an important fate process for this class of compounds.
Decay of DIA followed pseudo-first-order kinet- ics at lo-’ and lo-* M concentrations in experi- ments using 3.4% silt-clay suspensions. Calculating pseudo-first-order reaction rate constants such as this allows comparisons to be made among the rel- ative rates of decay in a given environmental system and also allows the quantitative characterization of parameters that influence reaction rates. Caution is warranted, however, when attempting to quantita- tively relate reactivities of species in media as diverse and complex as saturated sediments. For example, sediment concentration was shown to influence the rate of decay of both DIA and H C A , although H C A may exhibit specificity toward a re- actant(s) that DIA does not, as evidenced in the sterility experiments. Clearly, properties of the sed- iment-reactants and properties inherent to the com- pounds, such as bond dissociation energies and electron affinity of the associated halide, influence
reactivity. By using the same ssdiment-water sus- pension to determine rates of disappearancc, the reactive sequence H C A = DIA > DBA > TTCA > DCA was observed.
Considerable work has been done recently to characterize microbial reduction of halogenated organic compounds. The mos[ difficult problem in analyzing these environmental degradation reac- tions lies in separating biological reactions from strictly chemical reactions. No technique cleanly eliminates.one reaction type from the other. There- fore, inferences must be d rann from numerous inhibition experiments to determine the mode or modes of reaction. Clearly, for several cnviron- mentally significant compounds, abiotic reduction is either extremely slow or energetically infeasible. For reactive compounds such as those studied here, however, abiotic reducrion is possible and the characterization of these reactions may lead to a better understanding of reductive reactions of the more persistent compounds through predictive techniques.
Acknowledgetnenr- We thank the National Research Council for its partial funding of rhis research. We also thank Dr. Robert V . Moore for performing elemenLal analyses of sediment samples, Dr. John McGuire for per- forming the GC-MS analyses, and Dr. Eric Weber for assistance in collecting the sediment samples.
REFERENCES
I . Willis, G.H., R.C. Wander and L.M. Southwick. 1974. Degradation of trifluralin in soil suspensions as related to redox potential. J . Environ. Qua/. 31262-265.
2. Dubin, P. and K.L. Wright. 1975. Reduction of azo food dyes in cultures of Proreirs vulgaris. Xenobio- rica 5:563-571.
3 . Zoro, J.A., J.M. Hunter, G. Eglinton and G.C. Ware. 1974. Degradation of p.p’-DDT in reducing environments. Narure 247:235-236.
4 . Quirke, J.M.E., A.S.M. Marei and G. Eglinton. 1979. The degradation of DDT and its degradative products by reduced iron(1I)porphyrins and ammo- nia. Chemosphere 3: 15 I - l j j .
5 . Klecka, G.M. and S.J. Gonsior. 1984. Reductive dechlorination of chlorinated inethanes and ethanes by reduced iron(l1)porphyrins. Chemosphere 13:
6 . Caslro, C.E. 1964. The rapid oxidation of iron(I1)porphyrins by alkyl halides. A possible mode of intoxication of organisms b? alkyl halides. J. Am. Chem. SOC. 86:23 10-231 1 ,
7 . Bouwer, E.T., B.E. Riltman and P.L. McCarty. 198 I . Anaerobic degradation of halogenated I - and 2-carbon organic compounds. Environ. Sci. Techno/.
8 . Bouwer, E.T. and P.L. McCarty. 1983. Transforma-
391-402.
151596-599.
Degradation of halogenated ethanes in sediment-water systems 837
tions of I - and 2-carbon halogenated aliphatic or- ganic compounds under methanogenic conditions. Appl. Environ. Microbiol. 45: 1286-1294.
9. Parsons, F., G. Barrio-Lage and R. Rice. 1985. Biotransformation of chlorinated organic solvents in static microcosms. Environ. Toxicol. Chem. 4:739- 742.
10. Barrio-Lage, G. , F.Z. Parsons, R.S. Nassar and P.A. Lorenzo. 1986. Sequential dehalogenation of chlorinated ethenes. Environ. Sci. Technol. 20:96-99.
11. Criddle, C.S., P.L. McCarty, M.C. Elliott and J.F. Barker. 1986. Reduction of hexachloroethane to tetrachloroethylene in groundwater. J . Contam. Hydrol. 1 : 133- 142.
12. Parsons, F.Z., P.R. Wood and J. Demarco. 1984. Transformations of tetrachloroethene and trichlo- roethene in microcosms and groundwater. J . Am. Water Works Assoc. 1636-59.
13. Wade, R.S. and C.E. Castro. 1973. Oxidation of iron(I1)porphyrins by alkyl halides. J . Am. Chem.
14. Holmstead, R.L. 1976. Studies of the degradation of mirex with an iron(I1)porphyrin model system. J. Agric. Food Chem. 24:620-624.
15. Castro, C.E. and W.C. Kray, Jr. 1963. The cleavage of bonds by low valent transition metal ions. The homogeneous reduction of alkyl halides by chro- mous sulfate. J . Am. Chem. SOC. 85:2768-2773.
16. Kray, W.C., Jr. and C.E. Castro. 1964. The cleav- age of bonds by low-valent transition metal ions. The homogeneous dehalogenation of vicinal dihalides by chromous sulfate. J . Am. Chem. SOC. 86:4603- 4608.
17. Light, T.S. 1972. Standard solution for redox poten- tial measurements. Anal. Chem. 44:1038-1039.
18. Hamaker, J.W. 1972. Decomposition: Quantitative aspects. In C. Goring and J .W. Hamaker, eds., Organic Chemicals in the Soil Environment. Marcel Dekker, New York, NY, pp. 253-340.
19. Barrio-Lage, G., F.Z. Parsons and R.S. Nassar. 1987. Kinetics of the depletion of trichloroethene. Environ. Sci. Technol. 21:366-370.
20. Kebarle, P. 1972. Ions and ion-solvent molecule interactions in the gas phase. In M. Szwarc, ed., Ions
SOC. 95:226-230.
and Ion Pairs in Organic Reactions, Vol I . Wile) Interscience, New York, NY, pp. 27-83.
21. Dilling, W.L. 1977. Interphase transfer processes. 11. Evaporation rates of chloro methanes, ethanes, ethylenes, propanes, and propylenes from dilute aqueous solutions. Comparisons with theoretical pre- dictions. Environ. Sci. Technol. 11:405-409.
22. Goldfinger, P. and G. Martens. 1961. Elementary rate constants in atomic chlorination reactions. Part 3: Bond dissociation energies and entropies of the activated state. Faraday SOC. Trans. 51 (pt. 2):2220- 2225.
23. Wentworth, W.E., R. George. and H. Keith. 1969. Dissociative thermal electron attachment to some ali- phatic chloro, bromo, iodo compounds. J . Chem.
24. Vedeneyev, V.I. , L.V. Gurich, V.N. Kondrat’yev, V.A. Medvedev and Ye.L. Frankevich. 1966. Bond Energies, Ionization Potentials and Electron Affini- ties. Edward Arnold Ltd., London.
25. Thayer, J.S., G.J. Olson and F.E. Brinckman. 1984. Iodomethane as a potential metal mobilizing agent in nature. Environ. Sci. Technol. 18~726-729.
26. Schwarzenbach, R.P., W. Giger, C. Schaffner and 0. Wanner. 1985. Groundwater contamination of volatile halogenated alkanes: Abiotic formation of volatile sulfur compounds under anaerobic condi- tions. Environ. Sci. Technol. 19:322-327.
27. Mabey, W.R., J.H. Smith, R.T. Podoll, H.L. John- son, T. Mill, T.W. Chou, J. Gates, I.W. Partridge, H. Jaber and D. Vandenberg. 1982. Aquatic fate processes data for organic priority pollutants. E P A 440/4-81-014. U.S. Environmental Protection Agen- cy, Washington, DC.
28. Bank, S. and D.A. Juckett. 1975. Reactions of aro- matic radical anions. 11: Kinetic studies of the reac- tion of sodium naphthalene and anthracene with n-hexyl bromide and chloride. J . Am. Chem. SOC.
29. Bank, S. and D.A. Juckett. 1976. Reactions of aro- matic radical anions. 12: Kinetic studies of the reac- tion of radical anions of varying reduction potential with n-hexyl bromides, iodides, and chlorides. J. Am. Chem. SOC. 98:7742-7746.