calcium signaling?JAMES W. PUTNEY, JR., HARUO TAKEMURA, ARLENE R. HUGHES, DEBRA A. HORSTMAN,AND OLE THASTRUP*
Calcium Regulation Section, Laboratory of Cellular and Molecular Pharmacology, National Institute of Environmental
Health Sciences, Research Triangle Park, NC 27709, USA and *Depathl2enl of Clinical Chemistry, University Hospital,
Rigshospitalet, DK-2100 Copenhagen, Denmark
ABSTRACT
Activation of a variety of cell surface receptors results inthe phospholipase C -catalyzed hydrolysis of the minor
plasma membrane phospholipid phosphatidylinositol4,5-bisphosphate, with concomitant formation of mositol1,4,5-trisphosphate and diacyiglycerol. There is strong
evidence that inositol 1,4,5-trisphosphate stimulates Ca2
release from intracellular stores. The Ca2-releasing ac-
tions of inositol 1,4,5-trisphosphate are terminated by itsmetabolism through two distinct pathways. Inositol 1,4,5-
trisphosphate is dephosphorylated by a 5-phosphataseto inositol 1,4-bisphosphate; alternatively, inositol 1,4,5-
trisphosphate can also be phosphorylated to inositol1,3,4,5-tetrakisphosphate by a 3-kinase. Although themechanism of Ca2 mobilization is understood, the pre-cise mechanisms involved in Ca2 entry are not known;
the proposal that inositol 1,4,5-trisphosphate secondarilyelicits Ca2 entry by emptying an intracellular Ca2 poolis considered.-PUTNEY, J. W., JR.; TAKEMURA, H.;HUGHES, A. R.; HORSTMAN, D. A.; ThASTRUP, 0. How
do inositol phosphates regulate calcium signaling?
MORE THAN 30 YEARS HAVE passed since the discovery ofreceptor-stimulated turnover of inositol-containing phos-pholipids (1, 2). Today the effect and importance of thisphenomenon on a multitude of biological systems iswidely appreciated. Since the first paper by Hokin andHokin (1), the inositol lipids, and their metabolites havehad a tumultuous history of interest, neglect, rekindledinterest, controversy, and finally acceptance as importantintermediaries in intracellular signaling. Michell’s (3)hypothesis that the turnover of phosphoinositides coupledreceptors to cellular calcium mobilization stimulated con-siderable research and also provoked criticism; however,this highly original (albeit somewhat abstract) idea prob-ably served as the single catalyst for the discoveries that
followed, ultimately resulting in our current understandingof this system. After Michell’s hypothesis was introduced(3), there was some delay in understanding the preciserole of inositol lipid turnover, which was caused in part bya lack of knowledge of the actual biochemical pathwaysinvolved. An important finding was that the initial reac-tion in receptor-stimulated phosphoinositide turnover didnot involve the major known inositide, phosphatidylino-sitol, but rather a minor phosphorylated derivative, phos-phatidylinositol 4,5-bisphosphate (4, 5). Berridge (6) real-ized that the water-soluble product of this reaction,inositol 1,4,5-trisphosphate [(1,4,5)IP3],’ was a likely can-didate for a second messenger to activate the release ofCa2 from intracellular stores. He and collaborators inEngland and Germany soon demonstrated that this mo-lecule had the predicted biological activity: (1,4,5)1P3 inmicromolar concentrations rapidly released Ca2 from anonmitochondrial store in permeabiized pancreatic aci-nar cells (7). This was quickly confirmed in a number ofdifferent laboratories with preparations from a varietyof cells (8).
Today (1,4,5)1P3 is recognized as an important secondmessenger that signals the release of intracellular Ca2.In recent years two related questions have emerged, so-
lutions to which represent the next important steps inunderstanding this area of biological regulation. Thisreview focuses on those questions and on recent studies
that are beginning to provide answers. The first questionis a direct consequence of the recent discovery of the com-plex metabolism of (l,4,5)1P3: specifically, what (if any)are the roles of the other inositol polyphosphates formedin activated cells? The second question actually predatesthe discovery of intracellular Ca2 release by (l,4,5)1P3,and only recently has its relationship to inositol phos-phate metabolism been clearly established: What is themechanism by which agonists that activate phosphoinosi-tide turnover stimulate the entry of extracellular Ca2into cells?
1Abbreviations: HPLC, high performance liquid chromatography;
EGTA, [ethylenebis(oxyethylenenitrilo)]tetraacetic acid. Inositol phos-phates are abbreviated as (l,4,5)1P3 for D-myo-inositol 1,4,5-trisphos-phate, etc.
Just 6 years ago, it was shown that inositol tris- and bis-phosphates are formed in response to the activation ofsurface membrane receptors (9). At that time a rathersimple biochemical pathway was envisioned that involvedthe sequential dephosphorylation of (1,4,5)1P3 to (1,4)1P2to (1)IP, and finally to free inositol. Today, the pictureof inositol phosphate metabolism is exceedingly complex(Fig. 1) (10-12). Our knowledge of this complexity islargely attributable to high performance liquid chroma-tography (HPLC) analytical procedures, which canseparate inositol phosphates with only subtle structuraldifferences. (1,4,5)1P3 is dephosphorylated to (1,4)1P2, byan extremely active 5-phosphatase as originally suggested
(13), but it now seems that (1,4)1P2 is dephosphorylatedprimarily to (4)IP by a 1-phosphatase (14). In addition tothe 5-phosphatase, most cells contain a 3-kinase, whichtransfers a phosphate from ATP to the 3 position of(1,4,5)1P3 to form (1,3,4,5)1P4 (15). This latter molecule isthen dephosphorylated (presumably by the same 5-phos-phatase that degrades (1,4,5)1P3) to form an isomericinositol trisphosphate, (1,3,4)1P3. (1,3,4)1P3 is then de-phosphorylated by the same 1-phosphatase that degrades(1,4)1P2 to (3,4)1P2 (14); to a lesser extent, (1,3)1P2 is alsoformed (16) by an enzyme that has not been character-ized as well. These bisphosphates are dephosphorylatedprimarily to a mixture of (1)IP and (3)IP, which arestereoisomers, and are not resolvable by conventionalHPLC. In addition, in two instances it has been shownthat (1,3,4)1P3 can be phosphorylated, to a limited extent,
Agonist
R
by a 6-kinase to form (1,3,4,6)1P4 (17, 18). The functionof this pathway is not known, but there is evidence that(1,3,4,6)1P4 can be a precursor for the synthesis of thehigher inositol penta- and hexakisphosphates (19). Thecomplexity of this metabolic pathway suggests that somebiological function may be regulated by one of thesemetabolites.
An additional complication in the metabolism of theinositol phosphates is the demonstration that phospholi-pase C in vitro acts on phosphatidylinositol 4,5-bisphos-phate to produce a mixture of (1,4,5)1P3 and inositolcyclic-1:2,4,5-trisphosphate ([cl:2 ,4,5]1P3), a derivativewith the 1-phosphate cyclized between the 1- and 2-hydroxyls (20). In platelets, (cl:2,4,5)1P3 apparently canmobilize intracellular Ca2 with about the same potencyas its noncyclic analog (21); (cl:2,4,5)1P3 is not a substratefor the 3-kinase, and is dephosphorylated only slowly bythe 5-phosphatase (22, 23). Thus, if formed in cells, thiscompound might cause persistent activation of Ca2’-mo-bilization. In fact, reports of the production of (cl:2,4,5)1P3in cells are somewhat conflicting; it appears that with
brief stimulation, little of the cyclic derivative is formed(23), but it accumulates with prolonged stimulation (24),presumably because of its rather slow rate of metabolism.Estimates of kinetics of (1,4,5)1P3 and (cl:2,4,5)1P3 turn-over in the parotid gland suggest that as little as 1% ofthe phospholipase C product may be in cyclic form (25).
Inositol pentakisphosphate (believed to be primarily[(1,3,4,5,6)]1P5) and hexakisphosphate (1P6; phytic acid) arepresent in most mammalian cells. Their levels do notchange noticeably upon stimulation (26), despite the ex-istence, at least in some cells, of a pathway for the forma-tion of 1P5 from (1,3,4)1P3, a metabolite of (1,4,5)1P3 (19).Whether any functional relationship exists between theCa2 signaling pathway and higher inositol phosphatesis not known yet.
INOSITOL PHOSPHATES AND CALCIUM
MOBILIZATION
In virtually all systems examined, receptor-activated cal-cium mobilization involves an initial phase of calciumrelease from intracellular stores, followed by a moreprolonged phase of calcium entry (10, 27). The availableevidence strongly indicates that the internal release issignaled by (1,4,5)1P3 (6, 8). Ca2 is released from a dis-crete intracellular pool that does not include all of thenonmitochondrial sequestered Ca2 (28), and there is evi-dence that the size of this pool can be regulated in cellsby a GTP-dependent mechanism (29, 30). The organellefrom which (1,4,5)1P3 releases Ca2 was originally believedto be a component of the endoplasmic reticulum, butmore recently it has been suggested that the Ca2 comesfrom a novel (1,4,5)1P3 -responsive organelle, termed “cal-ciosome” (31). The mechanism by which (1,4,5)1P3releases Ca2 appears to involve its interaction with aspecific membrane receptor 32, 33). Binding of (1,4,5)1P3to its receptor increases Ca + efflux from the intracellu-lar pooi by opening a Ca2 channel, which may be closelyassociated with the receptor (34).
2
(cl:2)IP (1,3,4)1P3
(1 3
\A.6)IP4
(1 or 3)IP
(1,3)1P2 , (3,4)1P2 (1,4)1P2
______// I(4)XP
(1 ,3.4.5.6)1P5
‘P6
Inesitol
Figure 1. Major (known) pathways for metabolism of(1,4,5)1P3 and(cl:2,4,5)1P3. See text for details.
METABOLISM OF INOSITOL PHOSPHATES
1900 Vol. 3 June 1989 The FASEB Journal PUTNEY ET AL.
(cl:2,4)1P2
(1,3,4,5)1P4
CALCIUM AND INOSITOL PHOSPHATES 1901
As mentioned above, at least some cells release
(cl:2,4,5)1P3 and (1,4,5)1P3 upon receptor activation, andit has been suggested that the former may serve an intra-cellular messenger function (21). In vitro, (cl:2,4,5)1P3appears to be only somewhat less potent than (l,4,5)1P3in releasing intracellular Ca2 (21, 35). The slower inac-tivation of (cl:2,4,5)IP3 compared with that of(1,4,5)1P3suggests that if it is formed in cells, its effects on Ca2mobilization might be more sustained than those of(1,4,5)1P3. Thus, it is important to determine preciselywhich inositol trisphosphates are formed in vivo as wellas their role or roles in the Ca2 response. In this regard,several laboratories have reported receptor-stimulated(cl:2,4,5)1P3 formation in platelets (36, 37), pancreas (24,38), and parotid minilobules (39). In pancreatic minio-bules, Dixon and Hokin (38) determined that, upon mus-carinic receptor stimulation, the levels of (1,4,5)1P3 in-creased rapidly and then fell to a new elevated steadystate, whereas (cl:2,4,5)1P3 increased slowly. Accordingly,it has been suggested that (1,4,5)1P3 could be responsi-ble for intracellular Ca2 release during short periods ofstimulation, whereas (1,4,5)1P3 and (cl:2,4,5)1P3 couldcontribute equally to Ca2 release during prolongedstimulation (38, 40).
The metabolism of (cl:2,4,5)IP3 in parotid acinar cellshas been investigated in considerable detail, with attentiongiven to the possible relationship of its metabolism toCa2 signaling (25). The half-time of (1,4,5)1P3 in the aci-nar cells was estimated to be 7.6 s, whereas that of(cl:2,4,5)1P3 was almost 10 mm. Ca2” signaling in thesecells was found to inactivate in seconds, even under con-ditions where the levels of (cl:2,4,5)1P3 were comparableto those of (1,4,5)1P3. The simplest interpretation ofthese data is that, unlike those systems in which the Ca2-mobilizing activity of (cl:2,4,5)1P3 has been previouslydemonstrated, the cyclic compound may not be able toactivate the (1,4,5)1P3 receptor to promote Ca2 release inthe rat parotid gland. If this is so, it would be surprising,since there are other inositol phosphates with vicinal4 and 5 phosphates capable of releasing Ca2” from the(1,4,5)1P3-sensitive pool (41). Perhaps the cyclic 1:2 phos-phodiester bond imparts steric constraints on the mo-lecule, altering the conformations of the critical phos-phates at positions 4 and 5. It would not be surprising
if the receptor for (1,4,5)1P3 has evolved an ability to dis-tinguish between the rapidly metabolized (and thereforeeasily controlled) (1,4,5)1P3 and the much more slowlymetabolized (cl:2,4,5)1P3.
Recent evidence suggests that (1,3,4,5)1P4, together with(1,4,5)1P3, modulates Ca2” mobilization (42, 43). Morriset al. (42) found that neither (l,4,5)1P3 nor (1,3,4,5)1P4was capable of activating Ca2 mobilization when dia-lyzed into mouse lacrimal cells in patch pipettes. How-ever, when the two were added together, a biphasic re-sponse, which apparently resulted from Ca2 release andentry, was observed. In a more recent study, it was foundthat (1,4,5)1P3 was capable of inducing a transient mobili-zation of Ca2, but the response could be sustained only if(1,3,4,5)1P4 was also added (43). On the other hand,under certain conditions, AR42J exocrine pancreatomacells express undetectable (l,4,5)1P3 3-kinase activity and
make little or no (1,3,4,5)1P4 (44). However, these cellsare capable of mobilizing cellular Ca2” in response to sub-stance P, and the Ca2 signal generated appears to beattributable to Ca2 entry as well as to intracellular Ca2release (44). Thus, an obligatory role for (1,3,4,5)1P4 inCa2 signaling in all types of cells must be questioneduntil an explanation for this apparent paradox is found.
CALCIUM ENTRY
Despite the controversy regarding the roles of some of theinositol polyphosphates in Ca2 signaling, the function of(1,4,5)1P3 as the primary signal for intracellular releasephase of Ca2 mobilization is now firmly established. Bycomparison, the regulation of calcium entry, or the sec-ond phase of calcium mobilization, is poorly understood(45). In only one case is there convincing evidence forthe direct regulation of a plasma membrane Ca2 chan-nel by an activated receptor (46). Injection of (l,4,5)1P3into sea urchin eggs, lacrimal gland cells, and mast cellsproduces a response pattern that suggests an activation ofboth intracellular calcium release and an entry of calciumfrom the extracellular space (42, 43, 47-51). As discussedabove, in some (42, 43, 48, 49) but not all (47, 50, 51)cases, coinjection of (1,3,4,5)1P4 was necessary for fullexpression of the response. In most studies, the directapplication of (1,4,5)1P3 to plasma membranes did notincrease their permeability to calcium (52-54). However,in one study, (1,4,5)1P3 seemed to increase the permeabilityof plasma membrane vesicles to Ca2 (55), and (1,4,5)1P3was found to increase a Ca2 current in excised membranepatches of B lymphocytes (56). Injection of (1,4,5)1P3 intomast cells increased Ca2 by a mechanism that appeared
to involve Ca2 entry, but a (1,4,5)1P3-regulated Ca2
channel could not be identified (51). Collectively, theseobservations suggest that unlike intracellular Ca2” release,Ca2 entry may be regulated by a variety of mechanismsin different systems. There is evidence in a number ofcases that (l,4,5)1P3 may be responsible for the activa-
tion of calcium entry in cells, but not by acting directlyon a plasma membrane channel.
A mechanism that is consistent with this idea has been
proposed. According to this hypothesis, the emptying of a(1,4,5)1P3-sensitive pool secondarily signals calcium entry.A discussion of some of the evidence from which this ideaevolved has been presented elsewhere (45). Much of theoriginal evidence was derived from studies of the kineticsof emptying and refilling of a receptor-regulated calciumpool in the rat parotid gland. In the parotid gland underresting conditions, this intracellular calcium pool was re-sistant to depletion by extracellular chelating agents.However, when emptied by agonist stimulation, this poolcould be rapidly filled from outside the cell, even in theabsence of agonists and (presumably) second messengerssuch as inositol phosphates (57). These results suggestthat the loss of calcium from this pool activated a pathwayfor entry into the pool from the extracellular space. Thus,in the continued presence of agonist, when (1,4,5)1P3 iscontinuously maintained at an elevated level, the poolwould presumably be empty, the pathway from the extra-cellular space would be open, and calcium would enter the
C
+N
0U
100 &M Msthacheln.
300 1’1OdMrcpln.
:0 100 200 300 400
1902 Vol. 3 June 1989 The FASEBJournal PUTNEY ET AL.
pool and subsequently the cytosol through the (1,4,5)1P3-activated channels.
In the experiments that led to this proposal, 86Rbeffiux was used to monitor changes in cytosolic Ca2 (i.e.,as an indicator of the activation of Ca2-dependent K4channels) (58). When muscarinic receptor activation wasterminated by the action of atropine, the intracellular Ca2pooi regulated by agonists rapidly refilled with Ca2” fromthe extracellular space (59). Because efflux of 86Rbwas not increased during reloading of the pool, it wasconcluded that the pathway for Ca2 entry from theextracellular space into the pool did not traverse thecytoplasm (57).
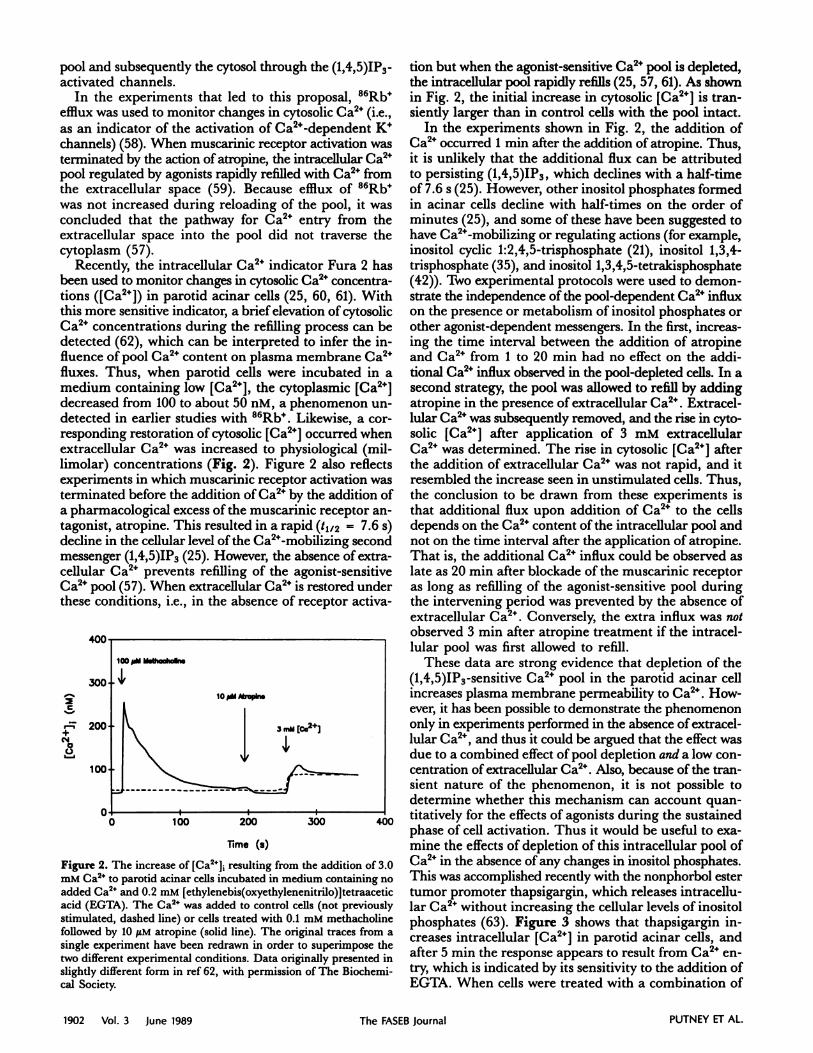
Recently, the intracellular Ca2 indicator Fura 2 hasbeen used to monitor changes in cytosolic Ca2 concentra-tions ([Ca2]) in parotid acinar cells (25, 60, 61). Withthis more sensitive indicator, a brief elevation of cytosolicCa2 concentrations during the refilling process can bedetected (62), which can be interpreted to infer the in-fluence of pool Ca2 content on plasma membrane Ca2fluxes. Thus, when parotid cells were incubated in amedium containing low [Ca2], the cytoplasmic [Ca2]decreased from 100 to about 50 nM, a phenomenon un-detected in earlier studies with 86Rb. Likewise, a cor-responding restoration of cytosolic [Ca2] occurred whenextracellular Ca2 was increased to physiological (mil-limolar) concentrations (Fig. 2). Figure 2 also reflectsexperiments in which muscarmnic receptor activation wasterminated before the addition of Ca2 by the addition ofa pharmacological excess of the muscarinic receptor an-tagonist, atropine. This resulted in a rapid (t112 = 7.6 s)decline in the cellular level of the Ca2-mobiizing secondmessenger (1,4,5)1P3 (25). However, the absence of extra-cellular Ca2 prevents refilling of the agonist-sensitiveCa2” pool (57). When extracellular Ca2 is restored underthese conditions, i.e., in the absence of receptor activa-
Time (s)
Figure 2. The increase of [Ca2], resulting from the addition of 3.0
mM Ca2 to parotid acinar cells incubated in medium containing no
added Ca2 and 0.2 mM [ethylenebis(oxyethylenenitrilo)]tetraaceticacid (EGTA). The Ca2 was added to control cells (not previouslystimulated, dashed line) or cells treated with 0.1 mM methacholinefollowed by 10 tM atropine (solid line). The original traces from a
single experiment have been redrawn in order to superimpose the
two different experimental conditions. Data originally presented in
slightly different form in ref 62, with permission of The Biochemi-cal Society.
tion but when the agonist-sensitive Ca2 pool is depleted,the intracellular pool rapidly refills (25, 57, 61). As shownin Fig. 2, the initial increase in cytosolic [Ca2] is tran-siently larger than in control cells with the pool intact.
In the experiments shown in Fig. 2, the addition ofCa2 occurred 1 mm after the addition of atropine. Thus,it is unlikely that the additional flux can be attributedto persisting (1,4,5)1P3, which declines with a half-timeof 7.6 s (25). However, other inositol phosphates formedin acinar cells decline with half-times on the order ofminutes (25), and some of these have been suggested tohave Ca2”-mobiizing or regulating actions (for example,inositol cyclic 1:2,4,5-trisphosphate (21), inositol 1,3,4-trisphosphate (35), and inositol 1,3,4,5-tetrakisphosphate(42)). Two experimental protocols were used to demon-strate the independence of the pool-dependent Ca2 influxon the presence or metabolism of inositol phosphates orother agonist-dependent messengers. In the first, increas-ing the time interval between the addition of atropineand Ca24 from 1 to 20 mm had no effect on the addi-tional Ca2 influx observed in the pool-depleted cells. In asecond strategy the pool was allowed to refill by addingatropine in the presence of extracellular Ca2. Extracel-lular Ca2 was subsequently removed, and the rise in cyto-solic [Ca2] after application of 3 mM extracellularCa2 was determined. The rise in cytosolic [Ca2] afterthe addition of extracellular Ca2 was not rapid, and itresembled the increase seen in unstimulated cells. Thus,the conclusion to be drawn from these experiments isthat additional flux upon addition of Ca2 to the cellsdepends on the Ca2 content of the intracellular pool andnot on the time interval after the application of atropine.That is, the additional Ca2 influx could be observed aslate as 20 mm after blockade of the muscarinic receptoras long as refilling of the agonist-sensitive pool duringthe intervening period was prevented by the absence ofextracellular Ca2. Conversely, the extra influx was not
observed 3 mm after atropine treatment if the intracel-lular pool was first allowed to refill.
These data are strong evidence that depletion of the(1,4,5)1P3-sensitive Ca24 pool in the parotid acinar cellincreases plasma membrane permeability to Ca2. How-ever, it has been possible to demonstrate the phenomenononly in experiments performed in the absence of extracel-lular Ca24, and thus it could be argued that the effect wasdue to a combined effect of pool depletion and a low con-centration of extracellular Ca2. Also, because of the tran-sient nature of the phenomenon, it is not possible todetermine whether this mechanism can account quan-titatively for the effects of agonists during the sustainedphase of cell activation. Thus it would be useful to exa-mine the effects of depletion of this intracellular pool ofCa2 in the absence of any changes in inositol phosphates.This was accomplished recently with the nonphorbol estertumor promoter thapsigargin, which releases intracellu-lar Ca2 without increasing the cellular levels of inositolphosphates (63). Figure 3 shows that thapsigargin in-creases intracellular [Ca2] in parotid acinar cells, and
after 5 mm the response appears to result from Ca2 en-try, which is indicated by its sensitivity to the addition ofEGTA. When cells were treated with a combination of
Time (a)
CALCIUM AND INOSITOL PHOSPHATES 1903
C
+N
00
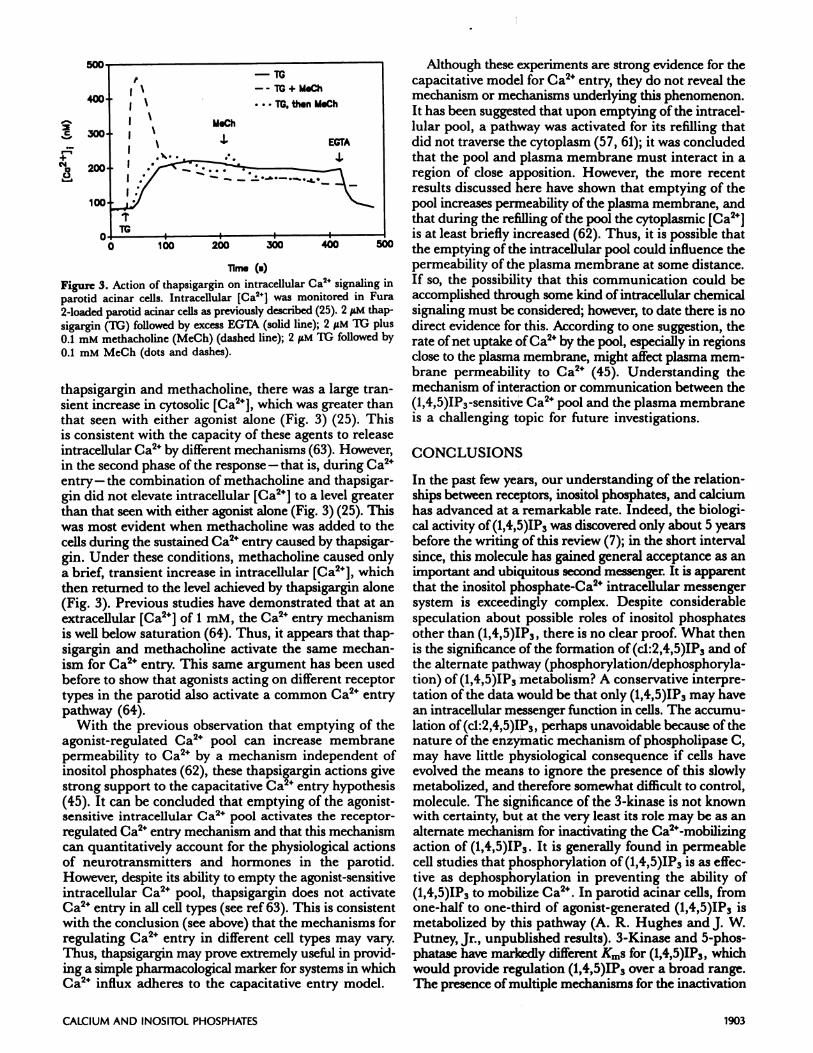
Figure 3. Action of thapsigargin on intracellular Ca2 signaling inparotid acinar cells. Intracellular [Ca2] was monitored in Fura
2-loaded parotid acinar cells as previously described (25). 2 M thap-
sigargin (it;) followed by excess EGTA (solid line); 2 iM TG plus
0.1 mM methacholine (MeCh) (dashed line); 2 iM It; followed by
0.1 mM MeCh (dots and dashes).
thapsigargin and methacholine, there was a large tran-sient increase in cytosolic [Ca2], which was greater thanthat seen with either agonist alone (Fig. 3) (25). Thisis consistent with the capacity of these agents to releaseintracellular Ca24 by different mechanisms (63). However,in the second phase of the response - that is, during Ca2entry-the combination of methacholine and thapsigar-gin did not elevate intracellular [Ca2] to a level greaterthan that seen with either agonist alone (Fig. 3) (25). Thiswas most evident when methacholine was added to thecells during the sustained Ca2 entry caused by thapsigar-gin. Under these conditions, methacholine caused onlya brief, transient increase in intracellular [Ca2], whichthen returned to the level achieved by thapsigargin alone(Fig. 3). Previous studies have demonstrated that at anextracellular [Ca2”] of 1 mM, the Ca2” entry mechanismis well below saturation (64). Thus, it appears that thap-sigargin and methacholine activate the same mechan-ism for Ca2 entry. This same argument has been usedbefore to show that agonists acting on different receptortypes in the parotid also activate a common Ca2 entrypathway (64).
With the previous observation that emptying of theagonist-regulated Ca2 pool can increase membrane
permeability to Ca2” by a mechanism independent ofinositol phosphates (62), these thapsigargin actions givestrong support to the capacitative Ca2 entry hypothesis(45). It can be concluded that emptying of the agonist-sensitive intracellular Ca2 pool activates the receptor-regulated Ca2 entry mechanism and that this mechanismcan quantitatively account for the physiological actionsof neurotransmitters and hormones in the parotid.However, despite its ability to empty the agonist-sensitiveintracellular Ca2 pool, thapsigargin does not activateCa2” entry in all cell types (see ref 63). This is consistentwith the conclusion (see above) that the mechanisms forregulating Ca2” entry in different cell types may vary.Thus, thapsigargin may prove extremely useful in provid-ing a simple pharmacological marker for systems in whichCa2 influx adheres to the capacitative entry model.
Although these experiments are strong evidence for thecapacitative model for Ca2” entry, they do not reveal themechanism or mechanisms underlying this phenomenon.It has been suggested that upon emptying of the intracel-lular pooi, a pathway was activated for its refilling thatdid not traverse the cytoplasm (57, 61); it was concludedthat the pool and plasma membrane must interact in aregion of close apposition. However, the more recentresults discussed here have shown that emptying of thepool increases permeability of the plasma membrane, andthat during the refilling of the pool the cytoplasmic [Ca2”]is at least briefly increased (62). Thus, it is possible thatthe emptying of the intracellular pool could influence thepermeability of the plasma membrane at some distance.If so, the possibility that this communication could beaccomplished through some kind of intracellular chemicalsignaling must be considered; however, to date there is nodirect evidence for this. According to one suggestion, therate of net uptake of Ca2 by the pool, especially in regionsclose to the plasma membrane, might affect plasma mem-brane permeability to Ca2 (45). Understanding themechanism of interaction or communication between the(1,4,5)IP3-sensitive Ca2 pool and the plasma membraneis a challenging topic for future investigations.
CONCLUSIONS
In the past few years, our understanding of the relation-ships between receptors, inositol phosphates, and calciumhas advanced at a remarkable rate. Indeed, the biologi-cal activity of (1,4,5)1P3 was discovered only about 5 yearsbefore the writing of this review (7); in the short intervalsince, this molecule has gained general acceptance as animportant and ubiquitous second messenger. It is apparentthat the inositol phosphate-Ca2” intracellular messengersystem is exceedingly complex. Despite considerablespeculation about possible roles of inositol phosphatesother than (1,4,5)1P3, there is no clear proof. What thenis the significance of the formation of(cl:2,4,5)IP3 and ofthe alternate pathway (phosphorylation/dephosphoryla-tion) of (1,4,5)1P3 metabolism? A conservative interpre-tation of the data would be that only (l,4,5)1P3 may havean intracellular messenger function in cells. The accumu-lation of(cl:2,4,5)IP3, perhaps unavoidable because of thenature of the enzymatic mechanism of phospholipase C,may have little physiological consequence if cells haveevolved the means to ignore the presence of this slowlymetabolized, and therefore somewhat difficult to control,molecule. The significance of the 3-kinase is not knownwith certainty, but at the very least its role may be as analternate mechanism for inactivating the Ca24-mobiizingaction of(1,4,5)IP3. It is generally found in permeablecell studies that phosphorylation of(1,4,5)1P3 is as effec-tive as dephosphorylation in preventing the ability of(1,4,5)1P3 to mobilize Ca2. In parotid acinar cells, fromone-half to one-third of agonist-generated (1,4,5)1P3 ismetabolized by this pathway (A. R. Hughes and J. W.Putney, Jr., unpublished results). 3-Kinase and 5-phos-phatase have markedly different KmS for (1,4,5)1P3, whichwould provide regulation (1,4,5)IP3 over a broad range.The presence of multiple mechanisms for the inactivation
Ca 2+
Plasma
1904 Vol. 3 June 1989 The FASEB Journal PUTNEY ET AL.
Membrane
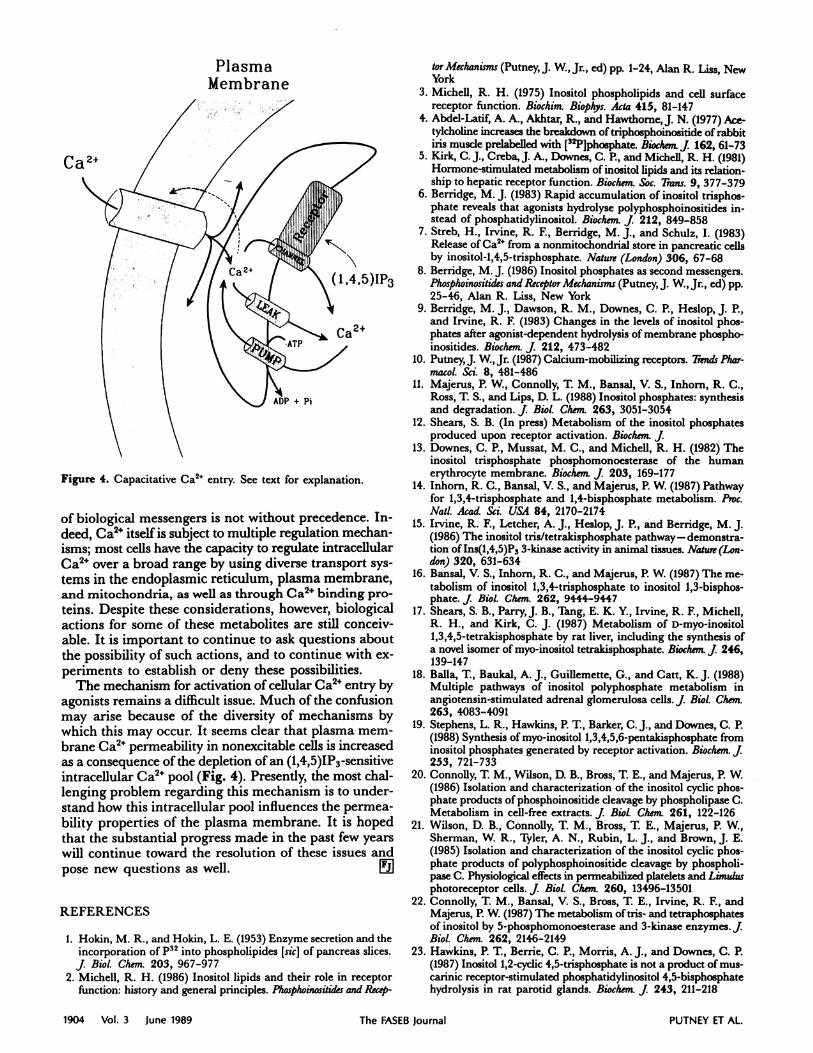
Figure 4. Capacitative Ca2 entry. See text for explanation.
of biological messengers is not without precedence. In-deed, Ca2 itself is subject to multiple regulation mechan-isms; most cells have the capacity to regulate intracellularCa2” over a broad range by using diverse transport sys-tems in the endoplasmic reticulum, plasma membrane,and mitochondria, as well as through Ca2’ binding pro-
teins. Despite these considerations, however, biologicalactions for some of these metabolites are still conceiv-able. It is important to continue to ask questions aboutthe possibility of such actions, and to continue with ex-periments to establish or deny these possibilities.
The mechanism for activation of cellular Ca2” entry byagonists remains a difficult issue. Much of the confusionmay arise because of the diversity of mechanisms by
which this may occur. It seems clear that plasma mem-brane Ca2 permeability in nonexcitable cells is increasedas a consequence of the depletion of an (1,4,5)1P3 -sensitiveintracellular Ca2 pool (Fig. 4). Presently, the most chal-lenging problem regarding this mechanism is to under-stand how this intracellular pool influences the permea-bility properties of the plasma membrane. It is hopedthat the substantial progress made in the past few yearswill continue toward the resolution of these issues andpose new questions as well. L!iI
REFERENCES
1. Hokin, M. R., and Hokin, L. E. (1953) Enzyme secretion and theincorporation of P32 into phospholipides [sic] of pancreas slices.J. BioL Chem. 203, 967-977
2. Michell, R. H. (1986) Inositol lipids and their role in receptorfunction: history and general principles. Phosphoinositides and Recep-
tor Mechanisms (Putney, J. W., Jr., ed) pp. 1-24, Alan R. Liss, NewYork
3. Michell, R. H. (1975) Inositol phospholipids and cell surfacereceptor function. Biochim. Biophys. Ada 415, 81-147
4. Abdel-Latif, A. A., Akhtar, R., and Hawthorne, J. N. (1977) Ace-tylcholine increases the breakdown of triphosphoinositide of rabbitiris muscle prelabelled with [32P]phosphate. Biochem. j 162, 61-73
5. Kirk, C. J., Creba, J. A., Downes, C. P., and Michell, R. H. (1981)Hormone-stimulated metabolism of inositol lipids and its relation-
ship to hepatic receptor function. Biochem. Soc. Trans. 9, 377-3796. Berridge, M. J. (1983) Rapid accumulation of inositol trisphos-
phate reveals that agonists hydrolyse polyphosphoinositides in-stead of phosphatidylinositol. Bioche,n. j 212, 849-858
7. Streb, H., Irvine, R. F, Berridge, M. J., and Schulz, I. (1983)Release of Ca2 from a nonmitochondrial store in pancreatic cells
by inositol-1,4,5-trisphosphate. Nature (London) 306, 67-68
8. Berridge, M. J. (1986) Inositol phosphates as second messengers.Phosphoinositides and Receptor Mechanisms (Putney, J. W., Jr., ed) pp.
25-46, Alan R. Liss, New York9. Berridge, M. J., Dawson, R. M., Downes, C. P., Heslop, J. P.,
and Irvine, R. F (1983) Changes in the levels of inositol phos-phates after agonist-dependent hydrolysis of membrane phospho-inositides. Bioc/zem. J. 212, 473-482
10. Putney, J. W, Jr. (1987) Calcium-mobilizing receptors. Trends Phar-macol. Sci. 8, 481-486
11. Majerus, P. W., Connolly, T M., Bansal, V. S., Inhorn, R. C.,Ross, T S., and Lips, D. L. (1988) Inositol phosphates: synthesisand degradation. j Biol. Chem. 263, 3051-3054
12. Shears, S. B. (In press) Metabolism of the inositol phosphatesproduced upon receptor activation. Biochem. j
13. Downes, C. P., Mussat, M. C., and Michell, R. H. (1982) Theinositol trisphosphate phosphomonoesterase of the human
erythrocyte membrane. Biochem. j 203, 169-17714. Inhorn, R. C., Bansal, V. S., and Majerus, P. W. (1987) Pathway
for 1,3,4-trisphosphate and 1,4-bisphosphate metabolism. Proc.NaIL Acad. Sci. USA 84, 2170-2174
15. Irvine, R. F, Letcher, A. J., Heslop, J. P., and Berridge, M. J.(1986) The inositol tris/tetrakisphosphate pathway-demonstra-tion of Ins(1,4,5)Ps 3-kinase activity in animal tissues. Nature (Lon-don) 320, 631-634
16. Bansal, V. S., Inhorn, R. C., and Majerus, P. W. (1987) The me-
tabolism of inositol 1,3,4-trisphosphate to inositol 1,3-bisphos-phate. J. Biol. Chem. 262, 9444-9447
17. Shears, S. B., ParryJ. B., Tang, E. K. Y., Irvine, R. F, Michell,R. H., and Kirk, C. J. (1987) Metabolism of D-myo-inositol1,3,4,5-tetrakisphosphate by rat liver, including the synthesis ofa novel isomer of myo-inositol tetrakisphosphate. Biochem. j 246,139-147
18. Balla, T., Baukal, A. J., Guillemette, G., and Catt, K. J. (1988)Multiple pathways of inositol polyphosphate metabolism inangiotensin-stimulated adrenal glomerulosa cells. j Biol. Chem.263, 4083-4091
19. Stephens, L. R., Hawkins, P. T, Barker, C. J., and Downes, C. P.(1988) Synthesis of myo-inositol l,3,4,5,6-pentakisphosphate from
inositol phosphates generated by receptor activation. Biochem. j253, 721-733
20. Connolly, T. M., Wilson, D. B., Bross, T. E., and Majerus, P. W.(1986) Isolation and characterization of the inositol cyclic phos-phate products of phosphoinositide cleavage by phospholipase C.Metabolism in cell-free extracts. j Biol. C/tern. 261, 122-126
21. Wilson, D. B., Connolly, T. M., Bross, T. E., Majerus, P. W.,Sherman, W. R., Tyler, A. N., Rubin, L. J., and Brown, J. E.(1985) Isolation and characterization of the inositol cyclic phos-phate products of polyphosphoinositide cleavage by phospholi-pase C. Physiological effects in permeabiized platelets and Limulusphotoreceptor cells. J. BioL Chem. 260, 13496-13501
22. Connolly, T. M., Bansal, V. S., Bross, T E., Irvine, R. F, andMajerus, P. W. (1987) The metabolism of tris- and tetraphosphatesof inositol by 5-phosphomonoesterase and 3-kinase enzymes. jBioL Che,n. 262, 2146-2149
23. Hawkins, P. T., Berrie, C. P.,Morris, A. J., and Downes, C. P.(1987) Inositol 1,2-cyclic 4,5-trisphosphate is not a product of mus-carinic receptor-stimulated phosphatidylinositol 4,5-bisphosphatehydrolysis in rat parotid glands. Biochern. j 243, 211-218
CALCIUM AND INOSITOL PHOSPHATES 1905
24. Sekar, M. C., Dixon, J. F, and Hokin, L. E. (1987) The forma-
tion of inositol 1,2-cyclic 4,5-trisphosphate and inositol 1,2-cyclic4-bisphosphate on stimulation of mouse pancreatic minilobuleswith carbamylcholine. J. Bid. C/tern. 262, 340-344
25. Hughes, A. R., Takemura, H., and Putney,J. W.,Jr. (1988) Ki-netics of inositol 1,4,5-trisphosphate and inositol cyclic1:2,4,5-trisphosphate metabolism in intact rat parotid acinar cells:relationship to calcium signalling.j BioL C/tern. 263, 10314-10319
26. Heslop,J. P., Irvine, R. F, Tashjian, A. H., and Berridge, M. J.(1985) Inositol tetrakis- and pentakisphosphates in GH4 cells. jExp. Biol. 119, 395-401
27. Putney, J. W., Jr., Poggioli, J., and Weiss, S. J. (1981) Receptorregulation of calcium release and calcium permeability in parotidgland cells. Phi/os. Trans. R. Soc. Lond. B. Biol. Sci. 296, 3 7-45
28. Taylor, C. W., and Putney, J. W., Jr. (1985) Size of the inositol1,4,5-trisphosphate-sensitive calcium pool in guinea-pig hepato-cytes. Biochem. j 232, 435-438
29. Mullaney, J. M., Chueh, S-H., Ghosh, T. K., and Gill, D. L.(1987) Intracellular calcium uptake activated by GTP: evidence
for a possible guanine nucleotide-induced transmembrane con-veyance of intracellular calcium.j B:ol. C/tern. 262, 13865-13872
30. Mullaney, J. M., Yu, M., Ghosh, T. K., and Gill, D. L. (1988)Calcium entry into the inositol 1,4,5-trisphosphate-releasable cal-cium pool is mediated by a GTP-regulatory mechanism. Proc.Nail. Acad. Sci. USA 85, 2499-2503
31. Volpe, P., Krause, K. -H., Hashimoto, S., Zorzato, F, Pozzan, T.,Meldolesi, J., and Lew, D. P. (1988) “Calciosome,” a cytoplasmic
organelle: the inositol 1,4,5-trisphosphate-sensitive Ca2 store ofnonmuscle cells? Proc. NaIL Acad. Sci. USA 85, 1091-1095
32. Spat, A., Bradford, P. G., McKinney, J. S., Rubin, R. P., andPutney, J. W., Jr. (1986) A saturable receptor for 32P-inositol-1,4,5-triphosphate in hepatocytes and neutrophils. Nature (Lon-don) 319, 514-516
33. Supattapone, S., Worley, P. F., Baraban,J. M., and Snyder, S. H.(1988) Solubilization, purification, and characterization of an in-ositol trisphosphate receptor. J. Biol. Chern. 263, 1530-1534
34. Ehrlich, B. E., and Watras, J. (1988) Inositol 1,4,5-trisphosphate
activates a channel from smooth muscle sarcoplasmic reticulum.Nature (London) 336, 583-586
35. Irvine, R. F., Letcher, A. J., Lander, D. J., and Berridge, M. J.(1986) Specificity of inositol phosphate-stimulated Ca2 mobili-zation from Swiss-mouse 3T3 cells. Biochern. j 240, 301-304
36. Ishii, H., Connolly, T M., Bross, T E., and Majet-us, P. W. (1986)
Inositol cyclic trisphosphate (inositol 1:2-cyclic 4,5-trisphosphate)is formed upon thrombin stimulation of human platelets. Proc.NaIL Acad. Sci. USA 83, 6397-6401
37. Tarver, A. P., King, W. G., and Rittenhouse, S. E. (1987) In-ositol l,4,5-trisphosphate and inositol 1,2-cyclic 4,5-trisphosphate
are minor components of total mass of inositol trisphosphate in
thrombin-stimulated platelets. J. Biol. Chem. 262, 17268-1727138. Dixon, J. F., and Hokin, L. E. (1987) Inositol 1,2-cyclic 4,5-
trisphosphate concentration relative to inositol 1,4,5-trisphosphatein pancreatic minilobules on stimulation with carbamyicholinein the absence of lithium. Possible role as a second messengerin long- but not short-term responses. J. BioL Chem. 262,13892-13895
39. Dixon, J. F., and Hokin, L. E. (1987) Inositol 1,2-cyclic 4,5-trisphosphate is formed in the rat parotid gland on muscarinicstimulation. Biochem. Biophys. Res. Commun. 149, 1208-1213
40. Muallem, S., Pandol, S. J., and Becker, T. G. (1988) Two com-ponents of hormone-evoked calcium release from intracellularstores of pancreatic acinar cells. Biochem. j 255, 301-307
41. Aub, D. L., and Putney,J. W.,Jr. (1987) Mobilization of intracel-lular calcium by methacholine and inositol 1,4,5-trisphosphatein rat parotid acinar cells. j Dent. Res. 66, 547-551
42. Morris, A. P., Gallacher, D. V., Irvine, R. F, and Petersen, 0. H.(1987) Synergism of inositol trisphosphate and tetrakisphosphatein activating Ca2-dependent K channels. Nature (London) 330,653-655
43. Changya, L, Gallacher, D. V., Irvine, R. F, Potter, B. V. L., andPetersen, 0. H. (In press) Inositol 1,3,4,5-trisphosphate is essen-tial for sustained activation of the Ca2”-dependent K current in
single internally perfused lacrimal cells. j Mernbr. Biol.44. Horstman. D. A., Takemura, H., and Putney, J. W., Jr. (1988)
Formation and metabolism of [3Hlinositol phosphates in AR42Jpancreatoma cells: substance P-induced Ca2 mobilization in ap-parent absence of inositol 1,4,5-trisphosphate 3-kinase activity.
j Biol. Chern. 263, 15297-1530345. Putney, J. W., Jr. (1986) A model for receptor-regulated calcium
entry. Cell Calcium 7, 1-1246. Benham, C. D., and Tsien, R. W. (1987) A novel receptor-
operated Ca2”-permeable channel activated by ATP in smoothmuscle. Nature (London) 328, 275-278
47. Slack, B. E., Bell, J. E., and Benos, D. J. (1986) Inositol1,4,5-trisphosphate injection mimics fertilization potentials in sea
urchin eggs. Am. j Physiol. 250, C340-C34448. Irvine, R. F., and Moor, R. M. (1986) Micro-injection of inositol
1,3,4,5-tetrakisphosphate activates sea urchin eggs by a mechan-ism dependent on external Ca2. Btochem. j 240, 917-920
49. Irvine, R. F., and Moor, R. M. (1987) Inositol (l,3,4,5)tetrakis-phosphate-induced activation of sea urchin eggs requires thepresence of inositol trisphosphate. Biochem. Biophys. lies. Commun.146, 284-290
50. Llano, I., Marty, A., and Tanguy,J. (1987) Dependence of intra-cellular effects of GTP gamma S and inositoltrisphosphate on cellmembrane potential and on external Ca ions. Pftuegers Arch. 409,
499-50651. Penner, R., Matthews, G., and Neher, E. (1988) Regulation of
calcium influx by second messengers in rat mast cells. Nature (Lon-don) 334, 499-504
52. Delfert, D. M., Hill, S., Pershadsingh, H. A., and Sherman,W. R. (1986) myo-Inositol 1,4,5-trisphosphate mobilizes Ca2 fromisolated adipocyte endoplasmic reticulum but not from plasmamembranes. Biochern. j 236, 3 7-44
53. Ueda, T., Church, S. H., Noel, M. W., and Gill, D. L. (1986)Influence of inositol 1,4,5-trisphosphate and guanine nucleotideson intracellular calcium release within the N1E-1l5 neuronal cellline. j BioL C/tern. 261, 3184-3192
54. Dargemont, C., Hilly, M., Claret, M., and Mauger,J.-P. (1988)
Characterization of Ca2 fluxes in rat liver plasma-membrane vesi-des. Biochern. J. 256, 117-124
55. Zschauer, A., Van Breemen, C., Buhler, F R., and Nelson, M. T.(1988) Calcium channels in thrombin-activated human plateletmembrane. Nature (London) 334, 703-705
56. Kuno, M., and Gardner, P. (1987) Ion channels activated by in-
ositol 1,4,5-trisphosphate in plasma membrane of human T-lymphocytes. Nature (London) 326, 301-304
57. Aub, D. L., McKinney, J. S., and Putney, J. W., Jr. (1982) Na-
ture of the receptor-regulated calcium pool in the rat parotidgland. J. PhysioL (London) 331, 557-565
58. Putney,J. W.,Jr. (1976) Biphasic modulation of potassium releasein rat parotid gland by carbachol and phenylephrine.j PharrnacoLExp. They 198, 375-384
59. Leslie, B. A., Burgess, G. M., and Putney, J. W., Jr. (1988) Per-sistent inhibition by inositol 1,4,5-trisphosphate of oxalate-
dependent 45calcium uptake in permeable guinea-pig hepatocytes.Cell Calcium 9, 9-16
60. Merritt, J. E., and Rink, T. J. (1987) The effects of substanceP and carbachol on inositoltris-and titrakisphosphate forma-
tion and cytosolic free calcium in rat parotid acinar cells. A corre-lation between inositol phosphate levels and calcium entry.] BioLC/tern. 262, 14912-14916
61. Merritt,J. E., and Rink, T.J. (1987) Regulation of cytosolic freecalcium in fura-2-loaded rat parotid acinar cells. j Biol. Chern.262, 17362-17369
62. Takemura, H., and Putney,J. W., Jr. (1989) Capacitative calcium
entry in parotid acinar cells. Biochem. j 258, 409-412
63. Jackson, T. R., Patterson, S. I., Thastrup, 0., and Hanley, M. R.(1988) A novel tumour promoter, thapsigargin, transiently in-creases cytoplasmic free Ca2 without generation of inositol phos-phates in NG1I5-401L neuronal cells. Bioc/tem.j 253, 81-86
64. Marier, S. H., Putney, J. W., Jr.,and van de Walle, C. M. (1978)Control of calcium channels by membrane receptors in the ratparotid gland. j P/tysiol. (London) 279, 141-151