HtrA2 Regulates �-Amyloid Precursor Protein (APP) Metabolismthrough Endoplasmic Reticulum-associated Degradation*□S
Received for publication, April 6, 2007, and in revised form, July 31, 2007 Published, JBC Papers in Press, August 6, 2007, DOI 10.1074/jbc.M702951200
Henri J. Huttunen‡§1, Suzanne Y. Guenette§1, Camilla Peach‡§, Christopher Greco‡§, Weiming Xia¶, Doo Yeon Kim‡§,Cory Barren‡§, Rudolph E. Tanzi§, and Dora M. Kovacs‡§2
From the ‡Neurobiology of Disease Laboratory and the §Genetics and Aging Research Unit, Massachusetts GeneralInstitute for Neurodegenerative Disease (MIND), Massachusetts General Hospital, Harvard Medical School,Charlestown, Massachusetts 02129 and the ¶Center for Neurologic Diseases, Brigham andWomen’s Hospital and Harvard Medical School, Boston, Massachusetts 02115
Alzheimer disease-associated �-amyloid peptide is generatedfrom its precursor protein APP. By using the yeast two-hybridassay, here we identified HtrA2/Omi, a stress-responsive chap-erone-protease as a protein binding to the N-terminal cysteine-rich region of APP. HtrA2 coimmunoprecipitates exclusivelywith immature APP from cell lysates as well as mouse brainextracts and degrades APP in vitro. A subpopulation of HtrA2localizes to the cytosolic side of the endoplasmic reticulum (ER)membranewhere it contributes to ER-associated degradation ofAPP together with the proteasome. Inhibition of the protea-some results in accumulation of retrotranslocated forms of APPand increased association of APP with HtrA2 and Derlin-1 inmicrosomal membranes. In cells lacking HtrA2, APP holopro-tein is stabilized and accumulates in the early secretory pathwaycorrelating with elevated levels of APP C-terminal fragmentsand increased A� secretion. Inhibition of ER-associated degra-dation (either HtrA2 or proteasome) promotes binding of APPto the COPII protein Sec23 suggesting enhanced trafficking ofAPP out of the ER. Based on these results we suggest a novelfunction for HtrA2 as a regulator of APP metabolism throughER-associated degradation.
A major neuropathological hallmark of Alzheimer disease(AD)3 is the accumulation of amyloid �-peptide (A�) in senileplaques (1). A� is generated from �-amyloid precursor protein(APP) by sequential proteolytic cleavages mediated by �- and�-secretases (2). An alternative nonamyloidogenic �-secretasecleavage cuts APP in the middle of the A� region (3). APP is aubiquitous type I transmembrane glycoprotein with many
putative functions related to cell adhesion and migration. Likemost plasma membrane proteins, APP is synthesized andN-glycosylated in the endoplasmic reticulum (ER) and trans-ported to the Golgi complex for maturation before transport tothe cell surface. Several molecular chaperones that interactwith APP have been identified including BiP/GRP78 (4) andcalreticulin (5).Genetic evidence shows that duplication of the APP gene
locus is sufficient to cause early-onset AD (6). APP gene dosageis doubled in Down syndrome (DS), and all adults over the ageof 40 years develop AD (7). Furthermore, analysis of the humangenome suggests that copy number variations play a significantrole in the etiology of complex diseases such as AD (8) high-lighting the importance of cellular regulatory mechanisms.Although proteolytic events resulting in the generation of A�fromAPP are well characterized, the regulation of APP proteinlevels in cells remains poorly understood. Altered maturation,processing, and degradation of APP holoprotein may regulatethe availability of APP for A� generation or affect the ratio ofA�42 to A�40, both of which appear to be important in thepathogenesis of AD. Several recent reports have suggested thatevents in the early secretory pathway strongly affect APPmetabolism and A� generation (9–11).HtrA2, a novel APP-interacting chaperone protease, was
recently reported to cleave APP in themitochondria (12). HtrAproteins are oligomeric, ATP-independent serine proteaseswidely found frombacteria tomammalswith important roles inprotein quality control (13). In general, HtrA proteins contain aprotease domain and one or two regulatory PDZ domainsthat recognize partially folded or misfolded proteins. Of thefour human HtrA family members HtrA2 is the only onewith an intracellular localization (13), and there is strongevidence showing that HtrA2 plays a role in the regulation ofapoptosis (14–16). However, the neurodegenerative pheno-type of the HtrA2 knock-out mice and increased susceptibil-ity of HtrA2�/� cells to ER stress suggest that HtrA2 mightplay an important role in protein quality control (17). HtrA2has been shown to bind through its PDZ domain to both A�(18) and the C termini of the presenilins (PS1 and PS2), keycomponents of the �-secretase complex (19).Our current results suggest that HtrA2 has a more general
role in the regulation of APPmetabolism. Partial localization ofHtrA2 to the cytosolic side of ERmembranes together with thefinding that HtrA2 exclusively binds to the immature form of
* This work was supported in part by grants from the NINDS, National Insti-tutes of Health (to D. M. K.) and the Helsingin Sanomat Centennial Foun-dation and Maud Kuistila Memorial Foundation (to H. J. H.). The costs ofpublication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked “advertisement” inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
□S The on-line version of this article (available at http://www.jbc.org) containssupplemental Figs. S1–S3.
1 These authors contributed equally to this work.2 To whom correspondence should be addressed: Neurobiology of Disease
Laboratory, Massachusetts General Hospital, Harvard Medical School, 11416th St., Charlestown, MA 02129. Tel.: 617-726-3668; Fax: 617-724-1823;E-mail: [email protected].
APP indicate that the HtrA2-APP interaction occurs in theearly secretory pathway. Lack of HtrA2 in cells results in stabi-lization of APP holoprotein, its accumulation inmicrosomes aswell as increased association of immature APP with the COPIIprotein Sec23. Furthermore, we show that APP is retrotranslo-cated from the ER to the cytosol and is a substrate for the ER-associated degradation (ERAD) pathway. Based on these data,we propose that HtrA2 collaborates with the proteasome inERAD to regulate cellular levels of APP.
EXPERIMENTAL PROCEDURES
Yeast Two-hybrid Assay—Identification of APP N-terminaldomain-binding proteins was performed as described previ-ously for the APP cytoplasmic domain (20). Briefly, the N-ter-minal cysteine-rich region (38–187 amino acids) of humanAPP was cloned in-frame in the LexA plasmid. The yeast strainEGY48 that contained LexAop-LEU2, LexAop-LacZ, andLexA-APP (38–187) was transformed with DNA from a galac-tose-inducible activation domain-cDNA fusion library con-structed from 22-week-old human fetal brain (a gift fromDimi-tri Krainc, Massachusetts General Hospital, Boston). Weidentified and confirmed 63 independent positives from thisscreen. Southern blot analysis of the cDNA inserts showed that61 of these clones were highly conserved sequences from sixclasses of overlapping cDNA clones. A full-length 458 aminoacid open reading frame for the newly identified human genewas assembled from the two hybrid library clone containingthe longest cDNA insert, a human adult brain lambda ZAPcDNA library (B616), a human kidney lambda phage cDNAlibrary (ClonTech) and a human cosmid library (a gift fromMarcyMacdonald, Massachusetts General Hospital, Boston).CellCultureandTransfection—ParentalCHO,H4,CHOAPP751,
H4APP751, andmouse embryonic fibroblasts (MEF; wild-type orHtrA2�/� (17)) were grown in Dulbecco’s modified Eagle’smedium supplemented with 10% (v/v) fetal bovine serum(Atlanta Biologicals), 2 mM L-glutamine (Lonza), 100 units/mlpenicillin, and 100 �g/ml streptomycin (Lonza). Stable celllineswere selected andmaintained inG418 (Calbiochem). Cellswere cultured at 37 °C in a water-saturated air/5% CO2 atmo-sphere.MEF cells were transiently transfected using theAmaxaNucleofector system according to the manufacturer’s instruc-tions (Amaxa Biosystems).Antibodies, Peptides, and Recombinant Proteins—Recombi-
nant human HtrA2 (rec-HtrA2), His-tagged recombinantBACE1 and HtrA2 antibody were from R&D Systems. The fol-lowing APP antibodies were used: 22C11 (N-terminal; Chemi-con), A8717 (C-terminal; Sigma), 6E10 (A� 1–17, Signet) andNAB228 (A� 1–11, Covance). C-66 is an affinity-purified rabbitpolyclonal antibody raised to the C-terminal 20 amino acids ofhuman APP (751–770) by the same protocol (BioSource) thatwas used for the widely used C7 antibody (21). Calreticulin andSmac/Diablo antibodies were from Calbiochem, V5 fromInvitrogen, His from Amersham Biosciences, BiP/GRP78 fromAffinity BioReagents, GAPDH from Chemicon, protein-disul-fide isomerase (PDI) and ubiquitin from Stressgen, Sec23 fromBethyl Laboratories, Derlin-1 from Novus Biologicals,GM130, EEA-1, cytochrome c, and syntaxin-6 from BD Bio-sciences. To construct C-terminally V5/His-tagged proteins,
APP751, APP695, and nectin-1 cDNAs were subcloned intothe pcDNA3.1-V5/His vector (Invitrogen). These constructswere stably transfected to CHO cells and V5/His-tagged pro-teins purified from detergent lysates using nickel-nitrilotri-acetic acid (Ni-NTA)-agarose columns (Qiagen).Immunoprecipitation, Western, and Lectin Blotting—Cells
were extracted on ice in a Triton-Nonidet P-40 buffer contain-ing 10 mM Tris-HCl, pH 6.8, 1 mM EDTA, 150 mMNaCl, 0.25%Nonidet P-40, 1% Triton X-100, and a protease inhibitor mix-ture (Roche Applied Science). Nuclei and unbroken cells wereremoved by centrifugation at 16,000 � g. The protein concen-trations were determined using the BCA protein assay kit(Pierce). For Western blots, 20–75 �g of total protein wasresolved in 4–12% gradient Bis-Tris gels (Novex/Invitrogen)under reducing conditions. For immunoprecipitation, 250–1000 �g of total protein was diluted to 1mg/ml with extractionbuffer and precleared with protein A or protein G-conjugatedagarose beads (Pierce). The precleared samples were incubatedovernight with excess amount of specific antibodies or nonim-mune IgG (Jackson Laboratories). Immunocomplexes werecaptured with Protein A/G agarose beads, washed five timeswith the extraction buffer and heated for 10 min at 70 °C in 1�LDS gel-loading buffer (Invitrogen) containing �-mercapto-ethanol. When needed, 20 �M UCF-101 (an HtrA2 inhibitor,Calbiochem) was included in the immunoprecipitation mixes.Band intensities on Western blot images were quantitated byusingQuantityOne software (Bio-Rad) and normalized as indi-cated in figure legends.Subcellular Fractionation—Cells were harvested in ice-cold
10mMHepes, pH 7.4, 0.25 M sucrose, 1mM EDTA, and homog-enized using a metal Dounce homogenizer. Postnuclear super-natants (1,000� g, S1) were centrifuged at 3,000� g for 10min(heavy mitochondria, P2), 12,000 � g for 10 min (light mito-chondria, P3) and 100,000 � g for 45 min (microsomes, P4).Each pellet waswashed once before proceeding to the next step.A portion of resuspended microsomal pellet (P4) was washedwith 100 mM Na2CO3, pH 11.5 for 30 min on ice and centri-fuged for 45min at 100,000� g. The pellet (P5) was the carbon-ate washedmicrosomal membrane fraction whereas the super-natant (S5) consistedmostly of proteins peripherally associatedwith microsomal membranes. Post-mitochondrial superna-tants of CHOAPP751 or parental CHO cells were fractionatedon a 7.5–30% continuous iodixanol gradients (OptiPrep;Axis-Shield) as described previously (22). Lipid rafts wereisolated as described previously (23) using 0.5% Lubrol WX(Lubrol 17A17; Serva) as a detergent. Because of variation indistribution of proteins between the two lipid raft fractionswithin a given gradient, combined APP signal from fractions3 and 4 was normalized to the sum of flotillin-1 signal fromthe same fractions.Confocal Fluorescence Microscopy—CHO cells were grown
on coverslips coated with poly-D-lysine/laminin mixture. Thecells were fixed with 3% paraformaldehyde/PBS, permeabilizedwith 0.1% Triton X-100/PBS for 10 min and blocked first with10 mM NH4Cl/PBS and then with 2% BSA/PBS. EndogenousHtrA2, PDI, syntaxin-6, and APP (rabbit C-terminal (Chemi-con) or 6E10 mAb (Signet)) were detected with correspondingantibodies and Alexa Fluor-conjugated secondary antibodies
HtrA2 Regulates APP Metabolism through ERAD
28286 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 282 • NUMBER 38 • SEPTEMBER 21, 2007
(Molecular Probes/Invitrogen). Mitochondria were stainedMitoTracker Red and nuclei with Hoechst 33342 (MolecularProbes/Invitrogen). Confocal fluorescence images were ob-tained using aOlympus DSU/IX70 spinning disc confocal micro-scope and processed using IPLab software (Scanalytics/BDBiosciences).In Vitro Cleavage Assay—Rec-HtrA2 was incubated with
purified recombinant APP751, APP695, nectin-1 or BACE1 in areaction buffer containing 50 mM Tris-HCl, pH 7.0, 0.5 mM
EDTA, 1 mM dithiothreitol. Depending on the assay, durationand temperature of incubation varied between 15 and 90 minand 37–42 °C. Reaction was stopped by adding 1� LDS gel-loading buffer (Invitrogen) and heating for 10 min at 70 °C.Occasionally, the 15 amino acid peptide corresponding to thepresenilin-1 C terminuswas included in the reactionmixture at10–100 �M (19). Alternatively, microsomal membranes reho-mogenized in 10mMHepes, pH7.4, 0.25M sucrose, 1mMEDTAwith or without 1% Triton X-100 were used as substratematerial.Retrotranslocation Assay—Retrotranslocation of proteins to
the cytosol was analyzed as described previously (24). Briefly,cells in monolayer were detached from tissue culture plateswith PBS containing 5 mM EDTA. After centrifugation at1,000 � g for 5 min, the cell pellet was resuspended in 100 �l ofDPB buffer (0.04% digitonin (Sigma), 50mMHepes, pH 7.5, 150mM NaCl, 2 mM CaCl2, protease inhibitor mixture (RocheApplied Sciences)), incubated on ice for 10min and centrifugedat 16,000 � g for 10 min at 4 °C. The supernatant was removed(cytosol), and the membrane pellet was washed with 500 �l ofcold PBS and extracted in 150�l ofDEBbuffer (1%digitonin, 25mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM MgCl2, 1 mM phen-ylmethylsulfonyl fluoride, protease inhibitormixture). Alterna-tively, the membrane pellet was homogenized in 10 mMHepes,pH 7.4, 0.25 M sucrose, 1 mM EDTAwith a glass-teflon homog-enizer. Postnuclear supernatant was centrifuged at 16,000 � gand carbonate washed as described above for microsomes.Where indicated, cells were treated with 1 �M epoxomicin(Biomol) for 5 h.Pulse Chase—Nearly confluent cells grown on 100-mm
plates were first preincubated in methionine/cysteine-freemedium for 1 h. 100 �Ci of [35S]methionine/cysteine (MP Bio-medicals) was then added per plate for 15 min (pulse). Then,cells were incubated in the presence of excess amounts of coldmethionine/cysteine (MP Biomedicals) and harvested at20-min intervals (chase). The cells were then washed, lysed inTriton-Nonidet P-40 buffer, and APPwas immunoprecipitated(A8717 antibody, Sigma). Fixed and dried gels were exposed toa phosphorimaging screen (Bio-Rad). Images were read andquantitated using a Personal Molecular Imager FX and Quan-tity One software (Bio-Rad).A� ELISA—For A� determination, conditioned media was
collected and cleared from debris. Secreted A� was immuno-precipitated with excess 6E10 antibody and quantitated bystandard sandwich ELISA (A� ELISA Core Facility, Center forNeurological Diseases, Harvard Institutes of Medicine, Har-vard Medical School).
Statistical Methods—Statistical comparisons were doneusing unpaired t tests or analysis of variance with significanceplaced at p � 0.05. Data are expressed as mean � S.D.
RESULTS
HtrA2 Binds and Degrades Immature APP—To identify pro-teins that interact with the cysteine-rich, N-terminal domain ofAPP, we performed a yeast two-hybrid assay with the APP cys-teine-rich domain as a bait sequence (depicted in Fig. 1A). Thesmallest APP-interacting cDNA contained 162 C-terminalamino acids of HtrA2/Omi chaperone-protease including theactive site motif, GNSGG, and the PDZ domain (data notshown). Furthermore, the HtrA2 two-hybrid fusion proteinsfailed to induce lacZ expression when the APLP2 cysteine-richregion (56–203 amino acids) was used in the yeast two-hybridassay, indicating that HtrA2 binding is specific for the APPCys-rich region.Next, we performed reciprocal immunoprecipitations with
HtrA2 and APP antibodies to verify the yeast two-hybrid find-ings. As shown in Fig. 1B, only immature form of APP interacts
FIGURE 1. HtrA2/Omi interacts with the ectodomain of immature APP.A, schematic diagram of APP751 domains and the yeast two-hybrid constructused in this study. The yeast two-hybrid construct was generated by fusingthe cysteine-rich region of APP-(38 –187) to LexA. SP, signal peptide; KPI,Kunitz-type protease inhibitor; TM, transmembrane; IC, intracellular domain.B, detergent extracts of CHO (750 �g) and CHOAPP751 (250 �g) cells wereimmunoprecipitated with HtrA2 or C-terminal APP antibodies. Nonimmunerabbit IgG and plain protein G beads were used as specificity controls. A rep-resentative Western blot of triplicate immunoprecipitations is shown for bothcell lines. C, reciprocal coimmunoprecipitation of detergent-extracted mousebrain hemispheres with HtrA2 or APP C-terminal antibodies.
HtrA2 Regulates APP Metabolism through ERAD
SEPTEMBER 21, 2007 • VOLUME 282 • NUMBER 38 JOURNAL OF BIOLOGICAL CHEMISTRY 28287
with HtrA2 in naıve and APP751-overexpressing CHO cells. Asimilar interaction of HtrA2 with immature APP was found inboth parental H4 human neuroglioma cells and H4 cells over-expressingAPP751 (data not shown). To confirm that theHtrA2interaction with APP occurs in vivo we immunoprecipitatedHtrA2 and APP frommouse brain extracts. Similar to our find-ings in cultured cells, the immature form of APP immunopre-cipitated with HtrA2 from detergent extracts of mouse brains(Fig. 1C).Although HtrA family proteases do not display strict cleav-
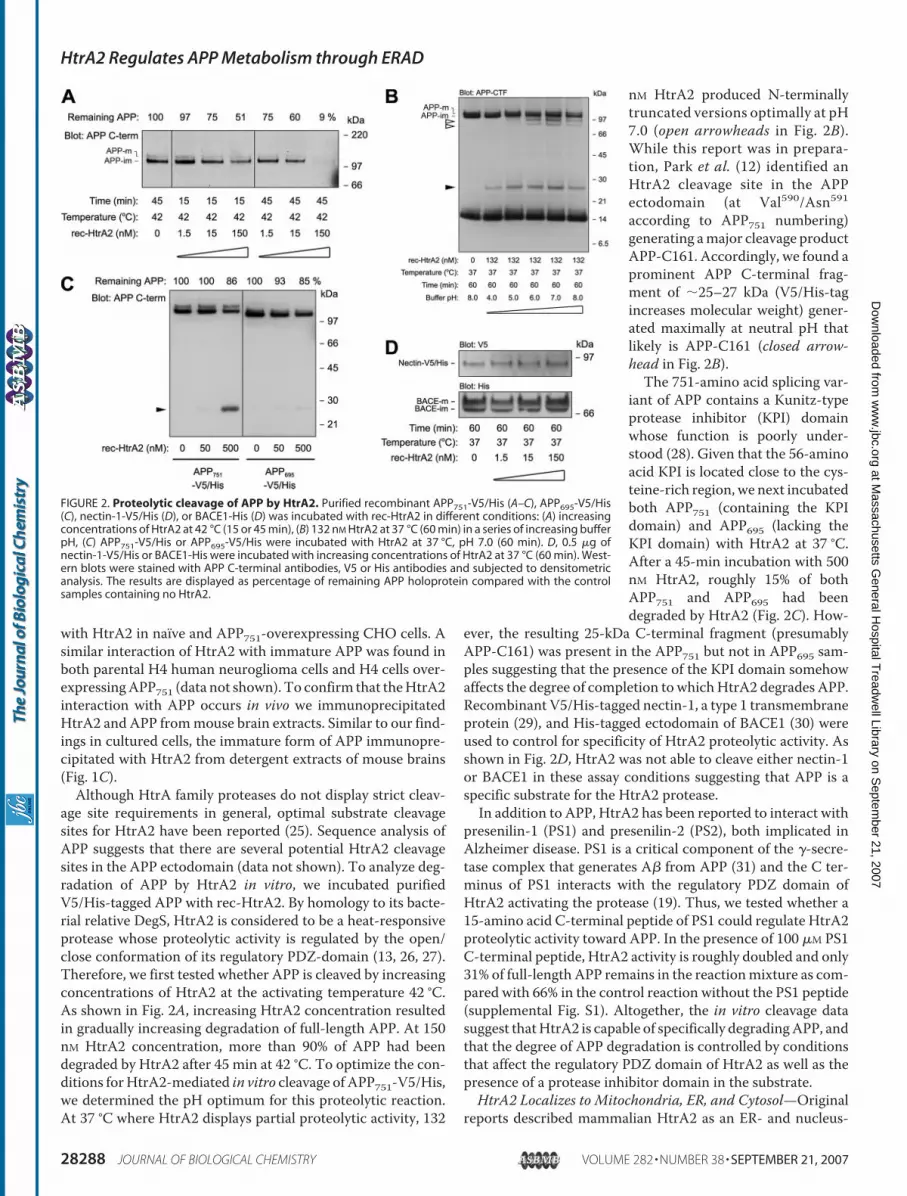
age site requirements in general, optimal substrate cleavagesites for HtrA2 have been reported (25). Sequence analysis ofAPP suggests that there are several potential HtrA2 cleavagesites in the APP ectodomain (data not shown). To analyze deg-radation of APP by HtrA2 in vitro, we incubated purifiedV5/His-tagged APP with rec-HtrA2. By homology to its bacte-rial relative DegS, HtrA2 is considered to be a heat-responsiveprotease whose proteolytic activity is regulated by the open/close conformation of its regulatory PDZ-domain (13, 26, 27).Therefore, we first tested whether APP is cleaved by increasingconcentrations of HtrA2 at the activating temperature 42 °C.As shown in Fig. 2A, increasing HtrA2 concentration resultedin gradually increasing degradation of full-length APP. At 150nM HtrA2 concentration, more than 90% of APP had beendegraded by HtrA2 after 45 min at 42 °C. To optimize the con-ditions forHtrA2-mediated in vitro cleavage of APP751-V5/His,we determined the pH optimum for this proteolytic reaction.At 37 °C where HtrA2 displays partial proteolytic activity, 132
nM HtrA2 produced N-terminallytruncated versions optimally at pH7.0 (open arrowheads in Fig. 2B).While this report was in prepara-tion, Park et al. (12) identified anHtrA2 cleavage site in the APPectodomain (at Val590/Asn591according to APP751 numbering)generating amajor cleavage productAPP-C161. Accordingly, we found aprominent APP C-terminal frag-ment of �25–27 kDa (V5/His-tagincreases molecular weight) gener-ated maximally at neutral pH thatlikely is APP-C161 (closed arrow-head in Fig. 2B).The 751-amino acid splicing var-
iant of APP contains a Kunitz-typeprotease inhibitor (KPI) domainwhose function is poorly under-stood (28). Given that the 56-aminoacid KPI is located close to the cys-teine-rich region, we next incubatedboth APP751 (containing the KPIdomain) and APP695 (lacking theKPI domain) with HtrA2 at 37 °C.After a 45-min incubation with 500nM HtrA2, roughly 15% of bothAPP751 and APP695 had beendegraded by HtrA2 (Fig. 2C). How-
ever, the resulting 25-kDa C-terminal fragment (presumablyAPP-C161) was present in the APP751 but not in APP695 sam-ples suggesting that the presence of the KPI domain somehowaffects the degree of completion to whichHtrA2 degrades APP.Recombinant V5/His-tagged nectin-1, a type 1 transmembraneprotein (29), and His-tagged ectodomain of BACE1 (30) wereused to control for specificity of HtrA2 proteolytic activity. Asshown in Fig. 2D, HtrA2 was not able to cleave either nectin-1or BACE1 in these assay conditions suggesting that APP is aspecific substrate for the HtrA2 protease.In addition to APP, HtrA2 has been reported to interact with
presenilin-1 (PS1) and presenilin-2 (PS2), both implicated inAlzheimer disease. PS1 is a critical component of the �-secre-tase complex that generates A� from APP (31) and the C ter-minus of PS1 interacts with the regulatory PDZ domain ofHtrA2 activating the protease (19). Thus, we tested whether a15-amino acid C-terminal peptide of PS1 could regulate HtrA2proteolytic activity toward APP. In the presence of 100 �M PS1C-terminal peptide, HtrA2 activity is roughly doubled and only31% of full-length APP remains in the reactionmixture as com-pared with 66% in the control reaction without the PS1 peptide(supplemental Fig. S1). Altogether, the in vitro cleavage datasuggest thatHtrA2 is capable of specifically degradingAPP, andthat the degree of APP degradation is controlled by conditionsthat affect the regulatory PDZ domain of HtrA2 as well as thepresence of a protease inhibitor domain in the substrate.HtrA2 Localizes to Mitochondria, ER, and Cytosol—Original
reports described mammalian HtrA2 as an ER- and nucleus-
FIGURE 2. Proteolytic cleavage of APP by HtrA2. Purified recombinant APP751-V5/His (A–C), APP695-V5/His(C), nectin-1-V5/His (D), or BACE1-His (D) was incubated with rec-HtrA2 in different conditions: (A) increasingconcentrations of HtrA2 at 42 °C (15 or 45 min), (B) 132 nM HtrA2 at 37 °C (60 min) in a series of increasing bufferpH, (C) APP751-V5/His or APP695-V5/His were incubated with HtrA2 at 37 °C, pH 7.0 (60 min). D, 0.5 �g ofnectin-1-V5/His or BACE1-His were incubated with increasing concentrations of HtrA2 at 37 °C (60 min). West-ern blots were stained with APP C-terminal antibodies, V5 or His antibodies and subjected to densitometricanalysis. The results are displayed as percentage of remaining APP holoprotein compared with the controlsamples containing no HtrA2.
HtrA2 Regulates APP Metabolism through ERAD
28288 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 282 • NUMBER 38 • SEPTEMBER 21, 2007
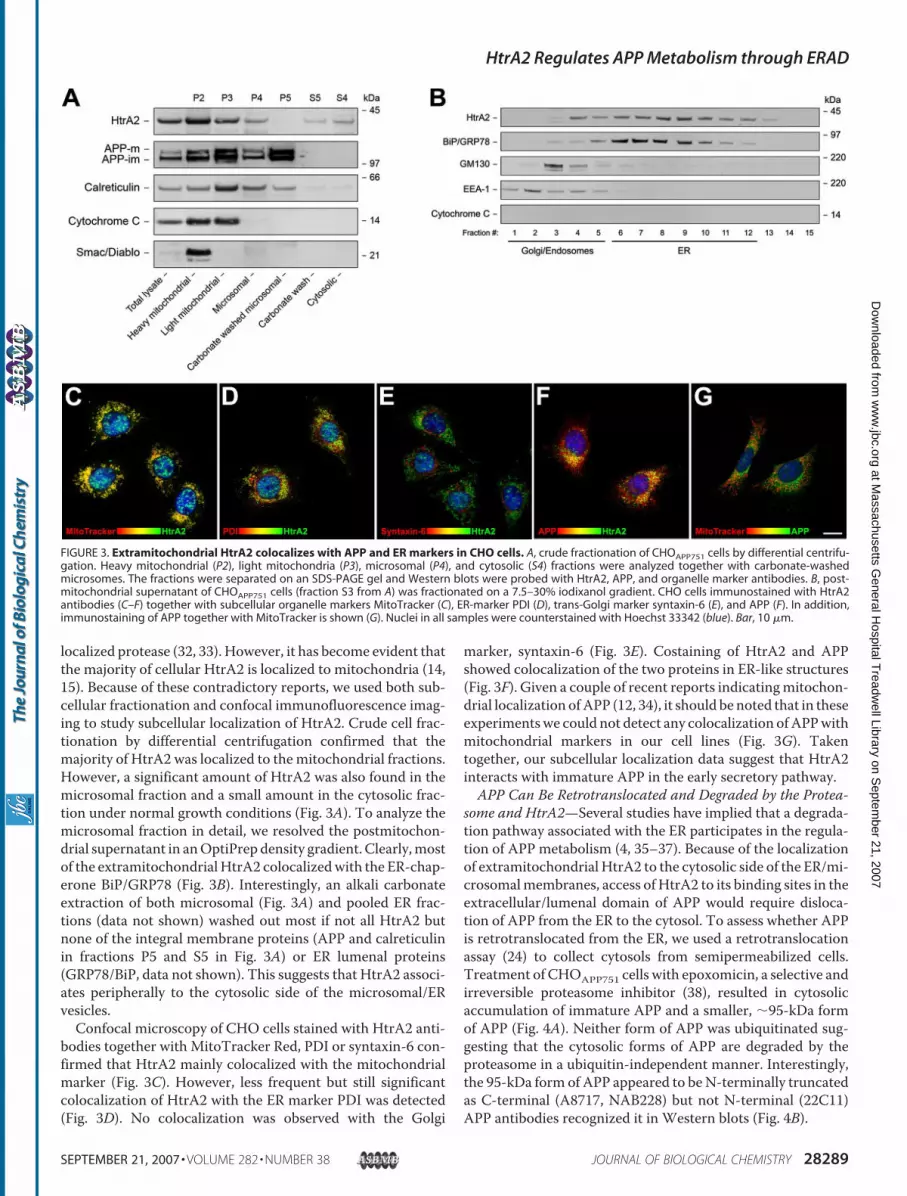
localized protease (32, 33). However, it has become evident thatthe majority of cellular HtrA2 is localized to mitochondria (14,15). Because of these contradictory reports, we used both sub-cellular fractionation and confocal immunofluorescence imag-ing to study subcellular localization of HtrA2. Crude cell frac-tionation by differential centrifugation confirmed that themajority of HtrA2 was localized to themitochondrial fractions.However, a significant amount of HtrA2 was also found in themicrosomal fraction and a small amount in the cytosolic frac-tion under normal growth conditions (Fig. 3A). To analyze themicrosomal fraction in detail, we resolved the postmitochon-drial supernatant in anOptiPrep density gradient. Clearly,mostof the extramitochondrial HtrA2 colocalizedwith the ER-chap-erone BiP/GRP78 (Fig. 3B). Interestingly, an alkali carbonateextraction of both microsomal (Fig. 3A) and pooled ER frac-tions (data not shown) washed out most if not all HtrA2 butnone of the integral membrane proteins (APP and calreticulinin fractions P5 and S5 in Fig. 3A) or ER lumenal proteins(GRP78/BiP, data not shown). This suggests that HtrA2 associ-ates peripherally to the cytosolic side of the microsomal/ERvesicles.Confocal microscopy of CHO cells stained with HtrA2 anti-
bodies together with MitoTracker Red, PDI or syntaxin-6 con-firmed that HtrA2 mainly colocalized with the mitochondrialmarker (Fig. 3C). However, less frequent but still significantcolocalization of HtrA2 with the ER marker PDI was detected(Fig. 3D). No colocalization was observed with the Golgi
marker, syntaxin-6 (Fig. 3E). Costaining of HtrA2 and APPshowed colocalization of the two proteins in ER-like structures(Fig. 3F). Given a couple of recent reports indicatingmitochon-drial localization ofAPP (12, 34), it should be noted that in theseexperimentswe could not detect any colocalization ofAPPwithmitochondrial markers in our cell lines (Fig. 3G). Takentogether, our subcellular localization data suggest that HtrA2interacts with immature APP in the early secretory pathway.APP Can Be Retrotranslocated and Degraded by the Protea-
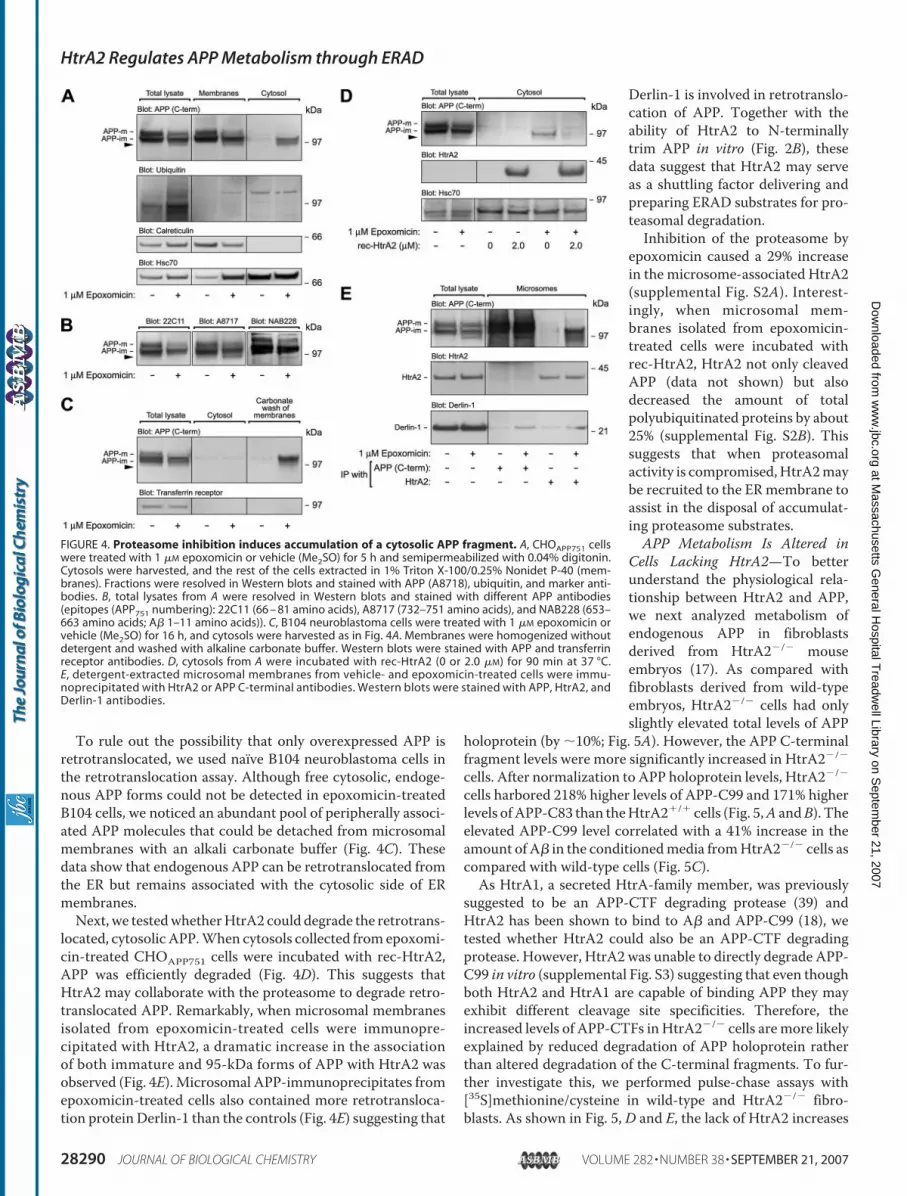
some and HtrA2—Several studies have implied that a degrada-tion pathway associated with the ER participates in the regula-tion of APP metabolism (4, 35–37). Because of the localizationof extramitochondrial HtrA2 to the cytosolic side of the ER/mi-crosomalmembranes, access of HtrA2 to its binding sites in theextracellular/lumenal domain of APP would require disloca-tion of APP from the ER to the cytosol. To assess whether APPis retrotranslocated from the ER, we used a retrotranslocationassay (24) to collect cytosols from semipermeabilized cells.Treatment of CHOAPP751 cells with epoxomicin, a selective andirreversible proteasome inhibitor (38), resulted in cytosolicaccumulation of immature APP and a smaller, �95-kDa formof APP (Fig. 4A). Neither form of APP was ubiquitinated sug-gesting that the cytosolic forms of APP are degraded by theproteasome in a ubiquitin-independent manner. Interestingly,the 95-kDa form of APP appeared to beN-terminally truncatedas C-terminal (A8717, NAB228) but not N-terminal (22C11)APP antibodies recognized it in Western blots (Fig. 4B).
FIGURE 3. Extramitochondrial HtrA2 colocalizes with APP and ER markers in CHO cells. A, crude fractionation of CHOAPP751 cells by differential centrifu-gation. Heavy mitochondrial (P2), light mitochondria (P3), microsomal (P4), and cytosolic (S4) fractions were analyzed together with carbonate-washedmicrosomes. The fractions were separated on an SDS-PAGE gel and Western blots were probed with HtrA2, APP, and organelle marker antibodies. B, post-mitochondrial supernatant of CHOAPP751 cells (fraction S3 from A) was fractionated on a 7.5–30% iodixanol gradient. CHO cells immunostained with HtrA2antibodies (C–F) together with subcellular organelle markers MitoTracker (C), ER-marker PDI (D), trans-Golgi marker syntaxin-6 (E), and APP (F). In addition,immunostaining of APP together with MitoTracker is shown (G). Nuclei in all samples were counterstained with Hoechst 33342 (blue). Bar, 10 �m.
HtrA2 Regulates APP Metabolism through ERAD
SEPTEMBER 21, 2007 • VOLUME 282 • NUMBER 38 JOURNAL OF BIOLOGICAL CHEMISTRY 28289
To rule out the possibility that only overexpressed APP isretrotranslocated, we used naıve B104 neuroblastoma cells inthe retrotranslocation assay. Although free cytosolic, endoge-nous APP forms could not be detected in epoxomicin-treatedB104 cells, we noticed an abundant pool of peripherally associ-ated APP molecules that could be detached from microsomalmembranes with an alkali carbonate buffer (Fig. 4C). Thesedata show that endogenous APP can be retrotranslocated fromthe ER but remains associated with the cytosolic side of ERmembranes.Next, we testedwhetherHtrA2 could degrade the retrotrans-
located, cytosolic APP.When cytosols collected from epoxomi-cin-treated CHOAPP751 cells were incubated with rec-HtrA2,APP was efficiently degraded (Fig. 4D). This suggests thatHtrA2 may collaborate with the proteasome to degrade retro-translocated APP. Remarkably, when microsomal membranesisolated from epoxomicin-treated cells were immunopre-cipitated with HtrA2, a dramatic increase in the associationof both immature and 95-kDa forms of APP with HtrA2 wasobserved (Fig. 4E). Microsomal APP-immunoprecipitates fromepoxomicin-treated cells also contained more retrotransloca-tion protein Derlin-1 than the controls (Fig. 4E) suggesting that
Derlin-1 is involved in retrotranslo-cation of APP. Together with theability of HtrA2 to N-terminallytrim APP in vitro (Fig. 2B), thesedata suggest that HtrA2 may serveas a shuttling factor delivering andpreparing ERAD substrates for pro-teasomal degradation.Inhibition of the proteasome by
epoxomicin caused a 29% increasein the microsome-associated HtrA2(supplemental Fig. S2A). Interest-ingly, when microsomal mem-branes isolated from epoxomicin-treated cells were incubated withrec-HtrA2, HtrA2 not only cleavedAPP (data not shown) but alsodecreased the amount of totalpolyubiquitinated proteins by about25% (supplemental Fig. S2B). Thissuggests that when proteasomalactivity is compromised,HtrA2maybe recruited to the ERmembrane toassist in the disposal of accumulat-ing proteasome substrates.APP Metabolism Is Altered in
Cells Lacking HtrA2—To betterunderstand the physiological rela-tionship between HtrA2 and APP,we next analyzed metabolism ofendogenous APP in fibroblastsderived from HtrA2�/� mouseembryos (17). As compared withfibroblasts derived from wild-typeembryos, HtrA2�/� cells had onlyslightly elevated total levels of APP
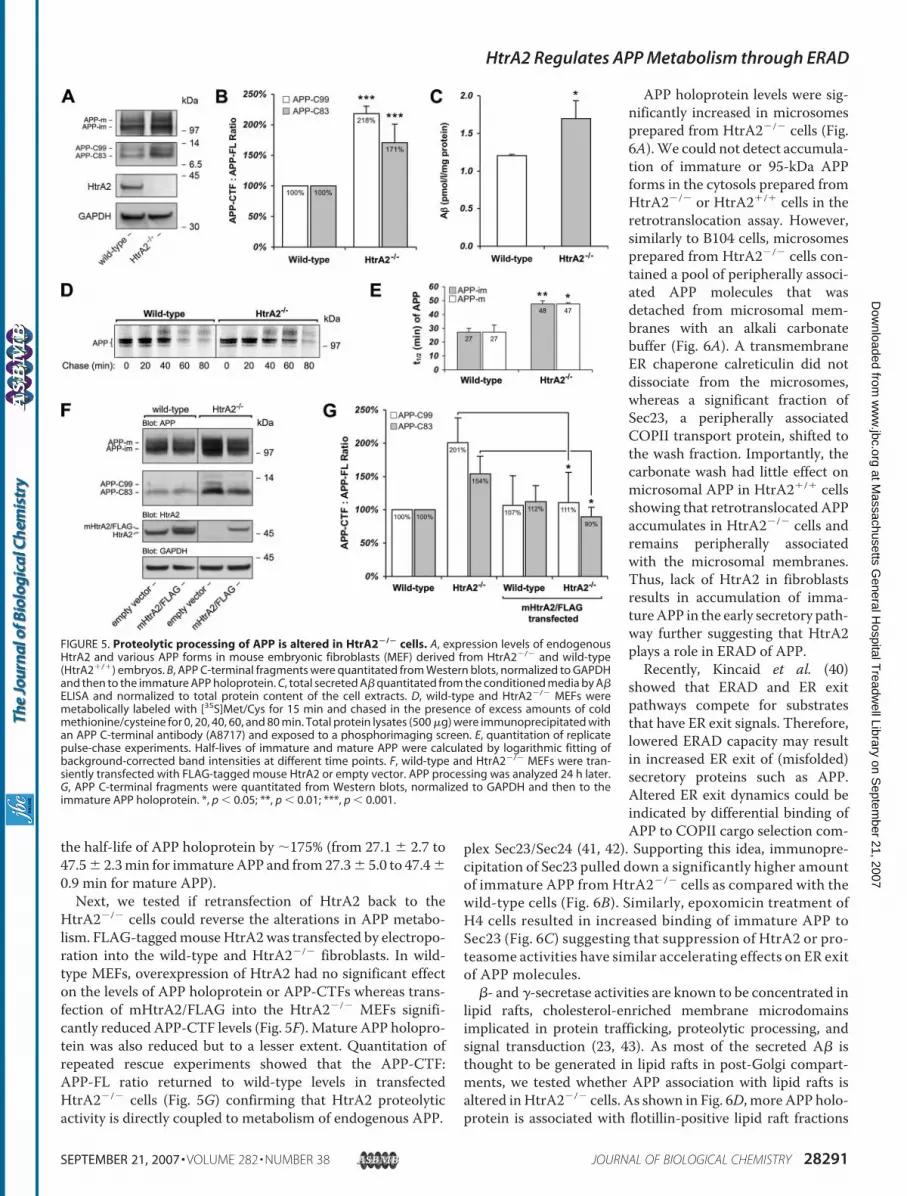
holoprotein (by �10%; Fig. 5A). However, the APP C-terminalfragment levels were more significantly increased in HtrA2�/�
cells. After normalization to APP holoprotein levels, HtrA2�/�
cells harbored 218% higher levels of APP-C99 and 171% higherlevels ofAPP-C83 than theHtrA2�/� cells (Fig. 5,A andB). Theelevated APP-C99 level correlated with a 41% increase in theamount of A� in the conditionedmedia fromHtrA2�/� cells ascompared with wild-type cells (Fig. 5C).As HtrA1, a secreted HtrA-family member, was previously
suggested to be an APP-CTF degrading protease (39) andHtrA2 has been shown to bind to A� and APP-C99 (18), wetested whether HtrA2 could also be an APP-CTF degradingprotease. However, HtrA2 was unable to directly degrade APP-C99 in vitro (supplemental Fig. S3) suggesting that even thoughboth HtrA2 and HtrA1 are capable of binding APP they mayexhibit different cleavage site specificities. Therefore, theincreased levels of APP-CTFs in HtrA2�/� cells aremore likelyexplained by reduced degradation of APP holoprotein ratherthan altered degradation of the C-terminal fragments. To fur-ther investigate this, we performed pulse-chase assays with[35S]methionine/cysteine in wild-type and HtrA2�/� fibro-blasts. As shown in Fig. 5, D and E, the lack of HtrA2 increases
FIGURE 4. Proteasome inhibition induces accumulation of a cytosolic APP fragment. A, CHOAPP751 cellswere treated with 1 �M epoxomicin or vehicle (Me2SO) for 5 h and semipermeabilized with 0.04% digitonin.Cytosols were harvested, and the rest of the cells extracted in 1% Triton X-100/0.25% Nonidet P-40 (mem-branes). Fractions were resolved in Western blots and stained with APP (A8718), ubiquitin, and marker anti-bodies. B, total lysates from A were resolved in Western blots and stained with different APP antibodies(epitopes (APP751 numbering): 22C11 (66 – 81 amino acids), A8717 (732–751 amino acids), and NAB228 (653–663 amino acids; A� 1–11 amino acids)). C, B104 neuroblastoma cells were treated with 1 �M epoxomicin orvehicle (Me2SO) for 16 h, and cytosols were harvested as in Fig. 4A. Membranes were homogenized withoutdetergent and washed with alkaline carbonate buffer. Western blots were stained with APP and transferrinreceptor antibodies. D, cytosols from A were incubated with rec-HtrA2 (0 or 2.0 �M) for 90 min at 37 °C.E, detergent-extracted microsomal membranes from vehicle- and epoxomicin-treated cells were immu-noprecipitated with HtrA2 or APP C-terminal antibodies. Western blots were stained with APP, HtrA2, andDerlin-1 antibodies.
HtrA2 Regulates APP Metabolism through ERAD
28290 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 282 • NUMBER 38 • SEPTEMBER 21, 2007
the half-life of APP holoprotein by �175% (from 27.1 � 2.7 to47.5� 2.3min for immatureAPP and from27.3� 5.0 to 47.4�0.9 min for mature APP).Next, we tested if retransfection of HtrA2 back to the
HtrA2�/� cells could reverse the alterations in APP metabo-lism. FLAG-taggedmouseHtrA2was transfected by electropo-ration into the wild-type and HtrA2�/� fibroblasts. In wild-type MEFs, overexpression of HtrA2 had no significant effecton the levels of APP holoprotein or APP-CTFs whereas trans-fection of mHtrA2/FLAG into the HtrA2�/� MEFs signifi-cantly reduced APP-CTF levels (Fig. 5F). Mature APP holopro-tein was also reduced but to a lesser extent. Quantitation ofrepeated rescue experiments showed that the APP-CTF:APP-FL ratio returned to wild-type levels in transfectedHtrA2�/� cells (Fig. 5G) confirming that HtrA2 proteolyticactivity is directly coupled to metabolism of endogenous APP.
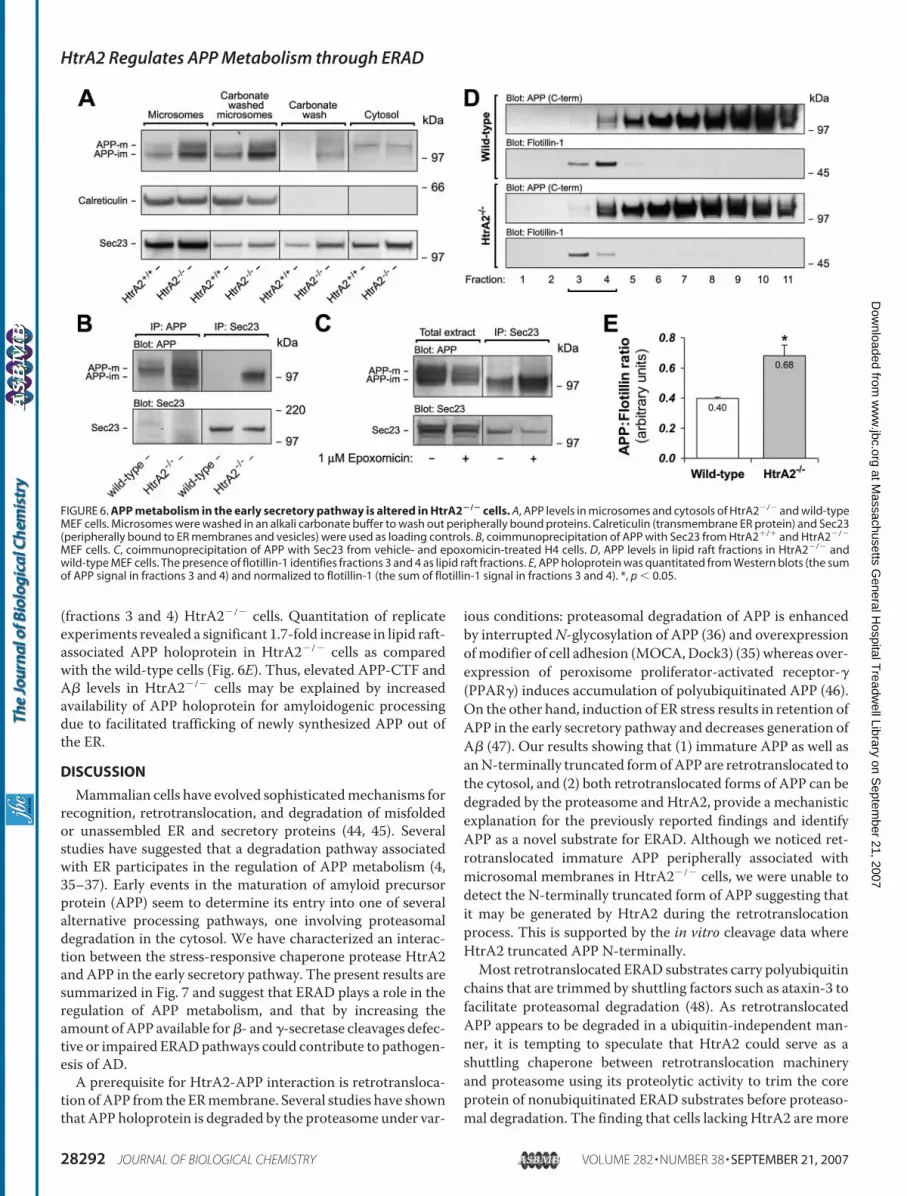
APP holoprotein levels were sig-nificantly increased in microsomesprepared from HtrA2�/� cells (Fig.6A).We could not detect accumula-tion of immature or 95-kDa APPforms in the cytosols prepared fromHtrA2�/� or HtrA2�/� cells in theretrotranslocation assay. However,similarly to B104 cells, microsomesprepared from HtrA2�/� cells con-tained a pool of peripherally associ-ated APP molecules that wasdetached from microsomal mem-branes with an alkali carbonatebuffer (Fig. 6A). A transmembraneER chaperone calreticulin did notdissociate from the microsomes,whereas a significant fraction ofSec23, a peripherally associatedCOPII transport protein, shifted tothe wash fraction. Importantly, thecarbonate wash had little effect onmicrosomal APP in HtrA2�/� cellsshowing that retrotranslocated APPaccumulates in HtrA2�/� cells andremains peripherally associatedwith the microsomal membranes.Thus, lack of HtrA2 in fibroblastsresults in accumulation of imma-tureAPP in the early secretory path-way further suggesting that HtrA2plays a role in ERAD of APP.Recently, Kincaid et al. (40)
showed that ERAD and ER exitpathways compete for substratesthat have ER exit signals. Therefore,lowered ERAD capacity may resultin increased ER exit of (misfolded)secretory proteins such as APP.Altered ER exit dynamics could beindicated by differential binding ofAPP to COPII cargo selection com-
plex Sec23/Sec24 (41, 42). Supporting this idea, immunopre-cipitation of Sec23 pulled down a significantly higher amountof immature APP from HtrA2�/� cells as compared with thewild-type cells (Fig. 6B). Similarly, epoxomicin treatment ofH4 cells resulted in increased binding of immature APP toSec23 (Fig. 6C) suggesting that suppression of HtrA2 or pro-teasome activities have similar accelerating effects on ER exitof APP molecules.
�- and �-secretase activities are known to be concentrated inlipid rafts, cholesterol-enriched membrane microdomainsimplicated in protein trafficking, proteolytic processing, andsignal transduction (23, 43). As most of the secreted A� isthought to be generated in lipid rafts in post-Golgi compart-ments, we tested whether APP association with lipid rafts isaltered inHtrA2�/� cells. As shown in Fig. 6D, more APP holo-protein is associated with flotillin-positive lipid raft fractions
FIGURE 5. Proteolytic processing of APP is altered in HtrA2�/� cells. A, expression levels of endogenousHtrA2 and various APP forms in mouse embryonic fibroblasts (MEF) derived from HtrA2�/� and wild-type(HtrA2�/�) embryos. B, APP C-terminal fragments were quantitated from Western blots, normalized to GAPDHand then to the immature APP holoprotein. C, total secreted A� quantitated from the conditioned media by A�ELISA and normalized to total protein content of the cell extracts. D, wild-type and HtrA2�/� MEFs weremetabolically labeled with [35S]Met/Cys for 15 min and chased in the presence of excess amounts of coldmethionine/cysteine for 0, 20, 40, 60, and 80 min. Total protein lysates (500 �g) were immunoprecipitated withan APP C-terminal antibody (A8717) and exposed to a phosphorimaging screen. E, quantitation of replicatepulse-chase experiments. Half-lives of immature and mature APP were calculated by logarithmic fitting ofbackground-corrected band intensities at different time points. F, wild-type and HtrA2�/� MEFs were tran-siently transfected with FLAG-tagged mouse HtrA2 or empty vector. APP processing was analyzed 24 h later.G, APP C-terminal fragments were quantitated from Western blots, normalized to GAPDH and then to theimmature APP holoprotein. *, p � 0.05; **, p � 0.01; ***, p � 0.001.
HtrA2 Regulates APP Metabolism through ERAD
SEPTEMBER 21, 2007 • VOLUME 282 • NUMBER 38 JOURNAL OF BIOLOGICAL CHEMISTRY 28291
(fractions 3 and 4) HtrA2�/� cells. Quantitation of replicateexperiments revealed a significant 1.7-fold increase in lipid raft-associated APP holoprotein in HtrA2�/� cells as comparedwith the wild-type cells (Fig. 6E). Thus, elevated APP-CTF andA� levels in HtrA2�/� cells may be explained by increasedavailability of APP holoprotein for amyloidogenic processingdue to facilitated trafficking of newly synthesized APP out ofthe ER.
DISCUSSION
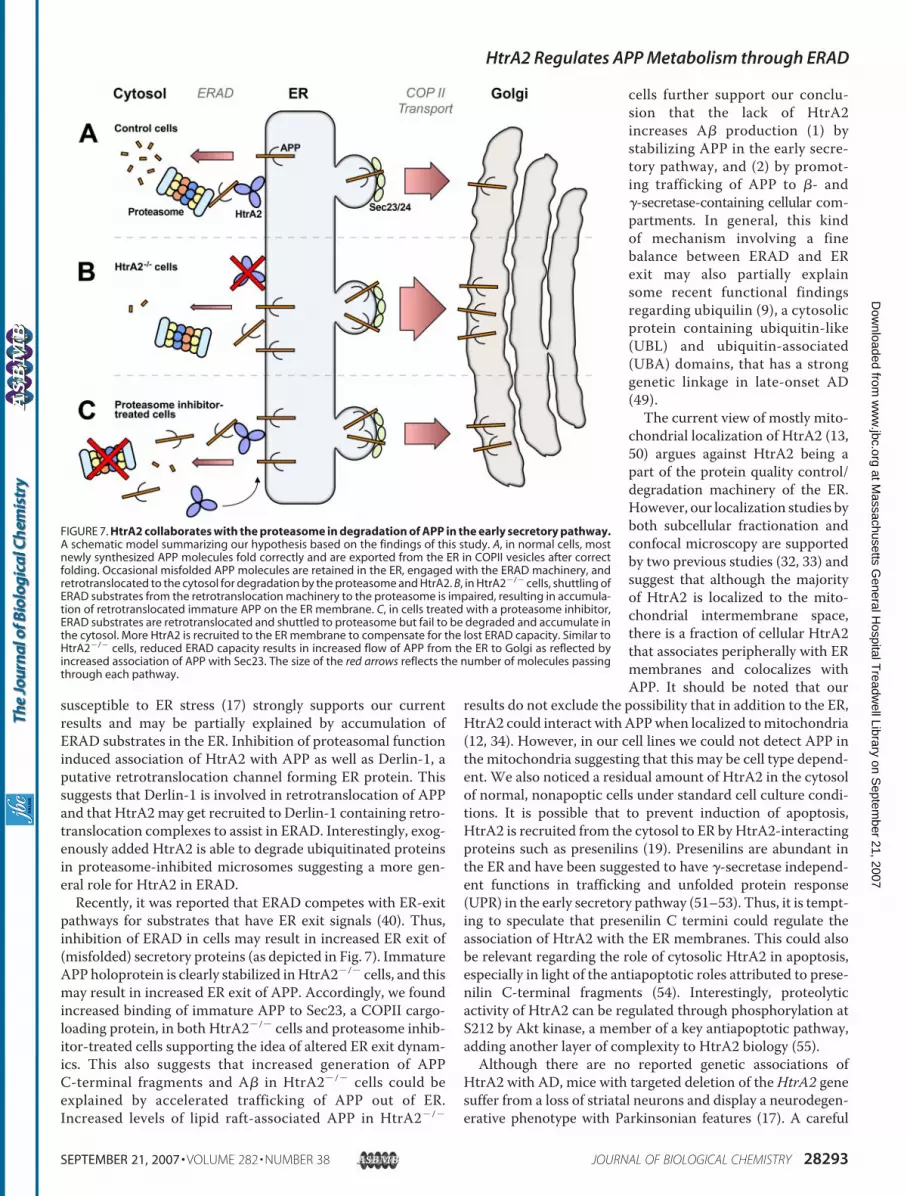
Mammalian cells have evolved sophisticatedmechanisms forrecognition, retrotranslocation, and degradation of misfoldedor unassembled ER and secretory proteins (44, 45). Severalstudies have suggested that a degradation pathway associatedwith ER participates in the regulation of APP metabolism (4,35–37). Early events in the maturation of amyloid precursorprotein (APP) seem to determine its entry into one of severalalternative processing pathways, one involving proteasomaldegradation in the cytosol. We have characterized an interac-tion between the stress-responsive chaperone protease HtrA2and APP in the early secretory pathway. The present results aresummarized in Fig. 7 and suggest that ERAD plays a role in theregulation of APP metabolism, and that by increasing theamount ofAPP available for�- and�-secretase cleavages defec-tive or impaired ERADpathways could contribute to pathogen-esis of AD.A prerequisite for HtrA2-APP interaction is retrotransloca-
tion ofAPP from the ERmembrane. Several studies have shownthat APP holoprotein is degraded by the proteasome under var-
ious conditions: proteasomal degradation of APP is enhancedby interruptedN-glycosylation of APP (36) and overexpressionofmodifier of cell adhesion (MOCA,Dock3) (35) whereas over-expression of peroxisome proliferator-activated receptor-�(PPAR�) induces accumulation of polyubiquitinated APP (46).On the other hand, induction of ER stress results in retention ofAPP in the early secretory pathway and decreases generation ofA� (47). Our results showing that (1) immature APP as well asanN-terminally truncated formofAPP are retrotranslocated tothe cytosol, and (2) both retrotranslocated forms of APP can bedegraded by the proteasome and HtrA2, provide a mechanisticexplanation for the previously reported findings and identifyAPP as a novel substrate for ERAD. Although we noticed ret-rotranslocated immature APP peripherally associated withmicrosomal membranes in HtrA2�/� cells, we were unable todetect the N-terminally truncated form of APP suggesting thatit may be generated by HtrA2 during the retrotranslocationprocess. This is supported by the in vitro cleavage data whereHtrA2 truncated APP N-terminally.Most retrotranslocated ERAD substrates carry polyubiquitin
chains that are trimmed by shuttling factors such as ataxin-3 tofacilitate proteasomal degradation (48). As retrotranslocatedAPP appears to be degraded in a ubiquitin-independent man-ner, it is tempting to speculate that HtrA2 could serve as ashuttling chaperone between retrotranslocation machineryand proteasome using its proteolytic activity to trim the coreprotein of nonubiquitinated ERAD substrates before proteaso-mal degradation. The finding that cells lacking HtrA2 are more
FIGURE 6. APP metabolism in the early secretory pathway is altered in HtrA2�/� cells. A, APP levels in microsomes and cytosols of HtrA2�/� and wild-typeMEF cells. Microsomes were washed in an alkali carbonate buffer to wash out peripherally bound proteins. Calreticulin (transmembrane ER protein) and Sec23(peripherally bound to ER membranes and vesicles) were used as loading controls. B, coimmunoprecipitation of APP with Sec23 from HtrA2�/� and HtrA2�/�
MEF cells. C, coimmunoprecipitation of APP with Sec23 from vehicle- and epoxomicin-treated H4 cells. D, APP levels in lipid raft fractions in HtrA2�/� andwild-type MEF cells. The presence of flotillin-1 identifies fractions 3 and 4 as lipid raft fractions. E, APP holoprotein was quantitated from Western blots (the sumof APP signal in fractions 3 and 4) and normalized to flotillin-1 (the sum of flotillin-1 signal in fractions 3 and 4). *, p � 0.05.
HtrA2 Regulates APP Metabolism through ERAD
28292 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 282 • NUMBER 38 • SEPTEMBER 21, 2007
susceptible to ER stress (17) strongly supports our currentresults and may be partially explained by accumulation ofERAD substrates in the ER. Inhibition of proteasomal functioninduced association of HtrA2 with APP as well as Derlin-1, aputative retrotranslocation channel forming ER protein. Thissuggests that Derlin-1 is involved in retrotranslocation of APPand that HtrA2may get recruited to Derlin-1 containing retro-translocation complexes to assist in ERAD. Interestingly, exog-enously added HtrA2 is able to degrade ubiquitinated proteinsin proteasome-inhibited microsomes suggesting a more gen-eral role for HtrA2 in ERAD.Recently, it was reported that ERAD competes with ER-exit
pathways for substrates that have ER exit signals (40). Thus,inhibition of ERAD in cells may result in increased ER exit of(misfolded) secretory proteins (as depicted in Fig. 7). ImmatureAPP holoprotein is clearly stabilized inHtrA2�/� cells, and thismay result in increased ER exit of APP. Accordingly, we foundincreased binding of immature APP to Sec23, a COPII cargo-loading protein, in both HtrA2�/� cells and proteasome inhib-itor-treated cells supporting the idea of altered ER exit dynam-ics. This also suggests that increased generation of APPC-terminal fragments and A� in HtrA2�/� cells could beexplained by accelerated trafficking of APP out of ER.Increased levels of lipid raft-associated APP in HtrA2�/�
cells further support our conclu-sion that the lack of HtrA2increases A� production (1) bystabilizing APP in the early secre-tory pathway, and (2) by promot-ing trafficking of APP to �- and�-secretase-containing cellular com-partments. In general, this kindof mechanism involving a finebalance between ERAD and ERexit may also partially explainsome recent functional findingsregarding ubiquilin (9), a cytosolicprotein containing ubiquitin-like(UBL) and ubiquitin-associated(UBA) domains, that has a stronggenetic linkage in late-onset AD(49).The current view of mostly mito-
chondrial localization of HtrA2 (13,50) argues against HtrA2 being apart of the protein quality control/degradation machinery of the ER.However, our localization studies byboth subcellular fractionation andconfocal microscopy are supportedby two previous studies (32, 33) andsuggest that although the majorityof HtrA2 is localized to the mito-chondrial intermembrane space,there is a fraction of cellular HtrA2that associates peripherally with ERmembranes and colocalizes withAPP. It should be noted that our
results do not exclude the possibility that in addition to the ER,HtrA2 could interact with APPwhen localized tomitochondria(12, 34). However, in our cell lines we could not detect APP inthe mitochondria suggesting that this may be cell type depend-ent. We also noticed a residual amount of HtrA2 in the cytosolof normal, nonapoptic cells under standard cell culture condi-tions. It is possible that to prevent induction of apoptosis,HtrA2 is recruited from the cytosol to ER by HtrA2-interactingproteins such as presenilins (19). Presenilins are abundant inthe ER and have been suggested to have �-secretase independ-ent functions in trafficking and unfolded protein response(UPR) in the early secretory pathway (51–53). Thus, it is tempt-ing to speculate that presenilin C termini could regulate theassociation of HtrA2 with the ER membranes. This could alsobe relevant regarding the role of cytosolic HtrA2 in apoptosis,especially in light of the antiapoptotic roles attributed to prese-nilin C-terminal fragments (54). Interestingly, proteolyticactivity of HtrA2 can be regulated through phosphorylation atS212 by Akt kinase, a member of a key antiapoptotic pathway,adding another layer of complexity to HtrA2 biology (55).Although there are no reported genetic associations of
HtrA2 with AD, mice with targeted deletion of theHtrA2 genesuffer from a loss of striatal neurons and display a neurodegen-erative phenotype with Parkinsonian features (17). A careful
FIGURE 7. HtrA2 collaborates with the proteasome in degradation of APP in the early secretory pathway.A schematic model summarizing our hypothesis based on the findings of this study. A, in normal cells, mostnewly synthesized APP molecules fold correctly and are exported from the ER in COPII vesicles after correctfolding. Occasional misfolded APP molecules are retained in the ER, engaged with the ERAD machinery, andretrotranslocated to the cytosol for degradation by the proteasome and HtrA2. B, in HtrA2�/� cells, shuttling ofERAD substrates from the retrotranslocation machinery to the proteasome is impaired, resulting in accumula-tion of retrotranslocated immature APP on the ER membrane. C, in cells treated with a proteasome inhibitor,ERAD substrates are retrotranslocated and shuttled to proteasome but fail to be degraded and accumulate inthe cytosol. More HtrA2 is recruited to the ER membrane to compensate for the lost ERAD capacity. Similar toHtrA2�/� cells, reduced ERAD capacity results in increased flow of APP from the ER to Golgi as reflected byincreased association of APP with Sec23. The size of the red arrows reflects the number of molecules passingthrough each pathway.
HtrA2 Regulates APP Metabolism through ERAD
SEPTEMBER 21, 2007 • VOLUME 282 • NUMBER 38 JOURNAL OF BIOLOGICAL CHEMISTRY 28293
analysis of these HtrA2�/� mice resulted in a conclusion thatdespite of the suggested role of HtrA2 as a mitochondrial reg-ulator of apoptosis, mammalianHtrA2 is likely involved in pro-tection against cell stresses that involve protein misfolding.Notably, loss of function mutations in HtrA2 gene result inneurodegenerative phenotype with Parkinsonian features inboth humans and inmice. Furthermore, S276C loss of functionmutation in HtrA2 is responsible for the neurodegenerativephenotype in mnd2 (motor neuron degeneration 2) mice (56).Recently, two mutations in HtrA2 gene (G399S and A141S)were reported in German Parkinson disease (PD) patients (57).Both mutations resulted in defective induction of the proteo-lytic activity of HtrA2. In this regard, our findings identify yetanother PD-associated protein as a component of the proteinquality control and proteasome-related disposal machinery.Our results raise interesting new questions on the neuronalroles of HtrA2, especially in the light of the neurodegenerativephenotypes ofmnd2 andHtrA2 knock-outmice. Being a part ofprotein quality control systems in both mitochondria and ERputs HtrA2 in a central position in terms of pathophysiology ofneurodegeneration.
Acknowledgments—The MEF cells from HtrA2�/� mice and themouse HtrA2 cDNAwere kind gifts from Dr. Julian Downward (Can-cer Research UK, London) and Dr. Emad Alnemri (Thomas JeffersonUniversity, Philadelphia), respectively. We thank Dr. Dennis Selkoe(Brigham and Women’s Hospital, Boston) for the CHOAPP-C99 cellline.
REFERENCES1. Selkoe, D. J. (2001) Physiol. Rev. 81, 741–7662. Haass, C. (2004) EMBO J. 23, 483–4883. Kojro, E., and Fahrenholz, F. (2005) Subcell. Biochem. 38, 105–1274. Yang, Y., Turner, R. S., and Gaut, J. R. (1998) J. Biol. Chem. 273,
25552–255555. Johnson, R. J., Xiao, G., Shanmugaratnam, J., and Fine, R. E. (2001) Neu-
robiol. Aging 22, 387–3956. Rovelet-Lecrux, A., Hannequin, D., Raux, G., LeMeur, N., Laquerriere, A.,
Vital, A., Dumanchin, C., Feuillette, S., Brice, A., Vercelletto,M., Dubas, F.,Frebourg, T., and Campion, D. (2006) Nat. Genet. 38, 24–26
7. Lott, I. T., and Head, E. (2005) Neurobiol. Aging 26, 383–3898. Redon, R., Ishikawa, S., Fitch, K. R., Feuk, L., Perry, G. H., Andrews, T. D.,
Fiegler, H., Shapero,M.H., Carson, A. R., Chen,W., Cho, E. K., Dallaire, S.,Freeman, J. L., Gonzalez, J. R., Gratacos, M., Huang, J., Kalaitzopoulos, D.,Komura, D., MacDonald, J. R., Marshall, C. R., Mei, R., Montgomery, L.,Nishimura, K., Okamura, K., Shen, F., Somerville, M. J., Tchinda, J., Vals-esia, A.,Woodwark, C., Yang, F., Zhang, J., Zerjal, T., Zhang, J., Armengol,L., Conrad, D. F., Estivill, X., Tyler-Smith, C., Carter, N. P., Aburatani, H.,Lee, C., Jones, K.W., Scherer, S. W., and Hurles, M. E. (2006)Nature 444,444–454
9. Hiltunen, M., Lu, A., Thomas, A. V., Romano, D.M., Kim,M., Jones, P. B.,Xie, Z., Kounnas, M. Z., Wagner, S. L., Berezovska, O., Hyman, B. T.,Tesco, G., Bertram, L., and Tanzi, R. E. (2006) J. Biol. Chem. 281,32240–32253
11. Vetrivel, K. S., Gong, P., Bowen, J. W., Cheng, H., Chen, Y., Carter, M.,Nguyen, P. D., Placanica, L., Wieland, F. T., Li, Y. M., Kounnas, M. Z., andThinakaran, G. (2007)Mol. Neurodegener. 2, 4
12. Park, H. J., Kim, S. S., Seong, Y.M., Kim, K. H., Goo, H. G., Yoon, E. J., Mindo, S., Kang, S., and Rhim, H. (2006) J. Biol. Chem. 281, 34277–34287
13. Clausen, T., Southan, C., and Ehrmann, M. (2002)Mol. Cell 10, 443–455
14. Suzuki, Y., Imai, Y., Nakayama, H., Takahashi, K., Takio, K., and Taka-hashi, R. (2001)Mol. Cell 8, 613–621
15. Hegde, R., Srinivasula, S.M., Zhang, Z.,Wassell, R.,Mukattash, R., Cilenti,L., DuBois, G., Lazebnik, Y., Zervos, A. S., Fernandes-Alnemri, T., andAlnemri, E. S. (2002) J. Biol. Chem. 277, 432–438
16. Yang, Q. H., Church-Hajduk, R., Ren, J., Newton, M. L., and Du, C. (2003)Genes Dev. 17, 1487–1496
17. Martins, L. M., Morrison, A., Klupsch, K., Fedele, V., Moisoi, N., Teis-mann, P., Abuin, A., Grau, E., Geppert, M., Livi, G. P., Creasy, C. L., Mar-tin, A., Hargreaves, I., Heales, S. J., Okada, H., Brandner, S., Schulz, J. B.,Mak, T., and Downward, J. (2004)Mol. Cell. Biol. 24, 9848–9862
18. Park, H. J., Seong, Y.M., Choi, J. Y., Kang, S., and Rhim,H. (2004)Neurosci.Lett. 357, 63–67
19. Gupta, S., Singh, R., Datta, P., Zhang, Z., Orr, C., Lu, Z., Dubois, G., Zervos,A. S., Meisler, M. H., Srinivasula, S. M., Fernandes-Alnemri, T., and Al-nemri, E. S. (2004) J. Biol. Chem. 279, 45844–45854
20. Guenette, S. Y., Chen, J., Jondro, P. D., and Tanzi, R. E. (1996) Proc. Natl.Acad. Sci. U. S. A. 93, 10832–10837
21. Podlisny, M. B., Tolan, D. R., and Selkoe, D. J. (1991) Am. J. Pathol. 138,1423–1435
22. Puglielli, L., Konopka, G., Pack-Chung, E., Ingano, L. A., Berezovska, O.,Hyman, B. T., Chang, T. Y., Tanzi, R. E., andKovacs, D.M. (2001)Nat. CellBiol. 3, 905–912
23. Vetrivel, K. S., Cheng, H., Lin, W., Sakurai, T., Li, T., Nukina, N., Wong,P. C., Xu, H., and Thinakaran, G. (2004) J. Biol. Chem. 279, 44945–44954
24. Forster, M. L., Sivick, K., Park, Y. N., Arvan, P., Lencer, W. I., and Tsai, B.(2006) J. Cell Biol. 173, 853–859
25. Martins, L. M., Turk, B. E., Cowling, V., Borg, A., Jarrell, E. T., Cantley,L. C., and Downward, J. (2003) J. Biol. Chem. 278, 49417–49427
26. Wilken, C., Kitzing, K., Kurzbauer, R., Ehrmann, M., and Clausen, T.(2004) Cell 117, 483–494
27. Li, W., Srinivasula, S. M., Chai, J., Li, P., Wu, J. W., Zhang, Z., Alnemri,E. S., and Shi, Y. (2002) Nat. Struct. Biol. 9, 436–441
28. Tanzi, R. E.,McClatchey, A. I., Lamperti, E. D., Villa-Komaroff, L., Gusella,J. F., and Neve, R. L. (1988) Nature 331, 528–530
29. Kim, D. Y., Ingano, L. A., and Kovacs, D. M. (2002) J. Biol. Chem. 277,49976–49981
30. Vassar, R., Bennett, B. D., Babu-Khan, S., Kahn, S., Mendiaz, E. A., Denis,P., Teplow, D. B., Ross, S., Amarante, P., Loeloff, R., Luo, Y., Fisher, S.,Fuller, J., Edenson, S., Lile, J., Jarosinski, M. A., Biere, A. L., Curran, E.,Burgess, T., Louis, J. C., Collins, F., Treanor, J., Rogers, G., and Citron, M.(1999) Science 286, 735–741
31. De Strooper, B. (2003) Neuron 38, 9–1232. Faccio, L., Fusco, C., Chen, A., Martinotti, S., Bonventre, J. V., and Zervos,
A. S. (2000) J. Biol. Chem. 275, 2581–258833. Gray, C.W.,Ward, R. V., Karran, E., Turconi, S., Rowles, A., Viglienghi,
D., Southan, C., Barton, A., Fantom, K. G., West, A., Savopoulos, J.,Hassan, N. J., Clinkenbeard, H., Hanning, C., Amegadzie, B., Davis,J. B., Dingwall, C., Livi, G. P., and Creasy, C. L. (2000) Eur. J. Biochem.267, 5699–5710
34. Anandatheerthavarada, H. K., Biswas, G., Robin, M. A., and Avadhani,N. G. (2003) J. Cell Biol. 161, 41–54
35. Chen, Q., Kimura, H., and Schubert, D. (2002) J. Cell Biol. 158, 79–8936. Hare, J. F. (2006) Arch. Biochem. Biophys. 451, 79–9037. Bunnell, W. L., Pham, H. V., and Glabe, C. G. (1998) J. Biol. Chem. 273,
31947–3195538. Meng, L., Mohan, R., Kwok, B. H., Elofsson, M., Sin, N., and Crews, C. M.
(1999) Proc. Natl. Acad. Sci. U. S. A. 96, 10403–1040839. Grau, S., Baldi, A., Bussani, R., Tian, X., Stefanescu, R., Przybylski, M.,
Richards, P., Jones, S. A., Shridhar, V., Clausen, T., and Ehrmann, M.(2005) Proc. Natl. Acad. Sci. U. S. A. 102, 6021–6026
40. Kincaid, M. M., and Cooper, A. A. (2007)Mol. Biol. Cell 18, 455–46341. Wang, X., Matteson, J., An, Y., Moyer, B., Yoo, J. S., Bannykh, S., Wilson,
I. A., Riordan, J. R., and Balch, W. E. (2004) J. Cell Biol. 167, 65–7442. Kim, J., Hamamoto, S., Ravazzola, M., Orci, L., and Schekman, R. (2005)
J. Biol. Chem. 280, 7758–776843. Riddell, D. R., Christie, G., Hussain, I., and Dingwall, C. (2001) Curr. Biol.
11, 1288–1293
HtrA2 Regulates APP Metabolism through ERAD
28294 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 282 • NUMBER 38 • SEPTEMBER 21, 2007
44. Tsai, B., Ye, Y., and Rapoport, T. A. (2002) Nat. Rev. Mol. Cell. Biol. 3,246–255
45. Meusser, B., Hirsch, C., Jarosch, E., and Sommer, T. (2005) Nat. Cell Biol.7, 766–772
46. d’Abramo, C., Massone, S., Zingg, J. M., Pizzuti, A., Marambaud, P., DallaPiccola, B., Azzi, A., Marinari, U. M., Pronzato, M. A., and Ricciarelli, R.(2005) Biochem. J. 391, 693–698
47. Kudo, T., Okumura, M., Imaizumi, K., Araki, W., Morihara, T., Tanimu-kai, H., Kamagata, E., Tabuchi, N., Kimura, R., Kanayama, D., Fukumori,A., Tagami, S., Okochi, M., Kubo, M., Tanii, H., Tohyama, M., Tabira, T.,and Takeda, M. (2006) Biochem. Biophys. Res. Commun. 344, 525–530
48. Wang, Q., Li, L., and Ye, Y. (2006) J. Cell Biol. 174, 963–97149. Bertram, L., Hiltunen, M., Parkinson, M., Ingelsson, M., Lange, C., Ra-
masamy, K., Mullin, K., Menon, R., Sampson, A. J., Hsiao, M. Y., Elliott,K. J., Velicelebi, G., Moscarillo, T., Hyman, B. T., Wagner, S. L., Becker,K. D., Blacker, D., and Tanzi, R. E. (2005) N. Engl. J. Med. 352, 884–894
50. Vaux, D. L., and Silke, J. (2003) Biochem. Biophys. Res. Commun. 304,499–504
51. Kovacs, D. M., Fausett, H. J., Page, K. J., Kim, T.W., Moir, R. D., Merriam,
D. E., Hollister, R. D., Hallmark, O. G., Mancini, R., Felsenstein, K. M.,Hyman, B. T., Tanzi, R. E., and Wasco, W. (1996) Nat. Med. 2, 224–229
52. Naruse, S., Thinakaran, G., Luo, J. J., Kusiak, J. W., Tomita, T., Iwatsubo,T., Qian, X., Ginty, D. D., Price, D. L., Borchelt, D. R., Wong, P. C., andSisodia, S. S. (1998) Neuron 21, 1213–1221
53. Katayama, T., Imaizumi, K., Sato, N., Miyoshi, K., Kudo, T., Hitomi, J.,Morihara, T., Yoneda, T., Gomi, F.,Mori, Y., Nakano, Y., Takeda, J., Tsuda,T., Itoyama, Y., Murayama, O., Takashima, A., St George-Hyslop, P.,Takeda, M., and Tohyama, M. (1999) Nat. Cell Biol. 1, 479–485
54. McCarthy, J. V. (2005) Biochem. Soc. Trans. 33, 568–57255. Yang, L., Sun,M., Sun, X.M., Cheng, G. Z., Nicosia, S. V., and Cheng, J. Q.
E., Hajnoczky, G., Saunders, T. L., Van Keuren,M. L., Fernandes-Alnemri,T., Meisler, M. H., and Alnemri, E. S. (2003) Nature 425, 721–727
57. Strauss, K. M., Martins, L. M., Plun-Favreau, H., Marx, F. P., Kautzmann,S., Berg, D., Gasser, T., Wszolek, Z., Muller, T., Bornemann, A., Wolburg,H., Downward, J., Riess, O., Schulz, J. B., and Kruger, R. (2005)Hum. Mol.Genet. 14, 2099–2111
HtrA2 Regulates APP Metabolism through ERAD
SEPTEMBER 21, 2007 • VOLUME 282 • NUMBER 38 JOURNAL OF BIOLOGICAL CHEMISTRY 28295