Ž . Journal of Immunological Methods 256 2001 121–140 www.elsevier.comrlocaterjim Recombinant Technology Recombinant fusion proteins for haemagglutination-based rapid detection of antibodies to HIV in whole blood Amita Gupta, Sanjay Gupta, Vijay K. Chaudhary ) Department of Biochemistry, UniÕersity of Delhi South Campus, Benito Juarez Road, New Delhi-110 021, India Received 6 April 2001; received in revised form 30 May 2001; accepted 30 May 2001 Abstract Recombinant fusion proteins, consisting of a monovalent anti-human RBC monoclonal antibody B6, and conserved immunodominant peptide of HIV-1 envelope glycoprotein gp41 or HIV-2 envelope glycoprotein gp36, have been designed and purified after over-expression in E. coli. These fusion proteins are Fab-based and were obtained by assembling the light Ž . chain with Fd variable domain and the first constant domain of the heavy chain or Fd fusions containing HIV-derived peptide, and following a protocol of in vitro denaturation of inclusion bodies and subsequent renaturation to assemble functional Fab. Using a multistep column chromatographic procedure, monomeric Fab and Fab fusion proteins containing HIV-derived peptide were purified to high degree, free of aggregates. The yield of various proteins on the laboratory scale Ž . 1–2 l of shake flask culture was in the range of tens of milligram. Purified anti-human RBC Fab fusion proteins containing sequences derived from HIV-1 gp41 and HIV-2 gp36 were highly specific for detection of antibodies to HIV-1 and HIV-2, respectively. The described design, expression and purification protocols will make it possible to produce specific recombinant reagents in large quantities for agglutination-based rapid detection of antibodies to HIV in whole blood. q 2001 Elsevier Science B.V. All rights reserved. Keywords: Agglutination; Diagnosis; HIV AbbreÕiations: B6, an anti-human RBC monoclonal antibody; cmyc, decapeptide recognised by monoclonal antibody 9E10; env13, 31 Ž . Ž . Ž . Ž . amino acids 590–620 of HIV-1 envelope glycoprotein gp41 ; env24, 31 amino acids 581–611 of HIV-2 envelope glycoprotein gp36 ; Ž . Ž . E. coli , Escherichia coli; Fd, fragment consisting of variable domain V and the first constant domain C 1 of the heavy chain of H H antibody; Fab, antigen binding fragment; Fdenv, Fd fragment carrying 31 amino acid peptide from envelope glycoprotein of HIV-1 or HIV-2; Fabenv, Fab fusion protein carrying 31 amino acids from envelope glycoprotein of HIV-1 or HIV-2; IgG, immunoglobulin type G; Ž IPTG, isopropyl-b-D-thiogalactoside; LC, light chain of an antibody; MAb, monoclonal antibody; PBS, phosphate buffered saline 20 mM . phosphate buffer, pH 7.2 containing 145 mM NaCl ; RT, room temperature; RBC, human red blood cell. ) Corresponding author. Tel.: q 91-11-467-4157r58; fax: q 91-11-688-5270. Ž . E-mail address: [email protected]V.K. Chaudhary . 0022-1759r01r$ - see front matter q 2001 Elsevier Science B.V. All rights reserved. Ž . PII: S0022-1759 01 00435-5
Transcript
Ž .Journal of Immunological Methods 256 2001 121–140www.elsevier.comrlocaterjim
Recombinant Technology
Recombinant fusion proteins for haemagglutination-based rapiddetection of antibodies to HIV in whole blood
Amita Gupta, Sanjay Gupta, Vijay K. Chaudhary)
Department of Biochemistry, UniÕersity of Delhi South Campus, Benito Juarez Road, New Delhi-110 021, India
Received 6 April 2001; received in revised form 30 May 2001; accepted 30 May 2001
Abstract
Recombinant fusion proteins, consisting of a monovalent anti-human RBC monoclonal antibody B6, and conservedimmunodominant peptide of HIV-1 envelope glycoprotein gp41 or HIV-2 envelope glycoprotein gp36, have been designedand purified after over-expression in E. coli. These fusion proteins are Fab-based and were obtained by assembling the light
Ž .chain with Fd variable domain and the first constant domain of the heavy chain or Fd fusions containing HIV-derivedpeptide, and following a protocol of in vitro denaturation of inclusion bodies and subsequent renaturation to assemblefunctional Fab. Using a multistep column chromatographic procedure, monomeric Fab and Fab fusion proteins containingHIV-derived peptide were purified to high degree, free of aggregates. The yield of various proteins on the laboratory scaleŽ .1–2 l of shake flask culture was in the range of tens of milligram. Purified anti-human RBC Fab fusion proteins containingsequences derived from HIV-1 gp41 and HIV-2 gp36 were highly specific for detection of antibodies to HIV-1 and HIV-2,respectively. The described design, expression and purification protocols will make it possible to produce specificrecombinant reagents in large quantities for agglutination-based rapid detection of antibodies to HIV in whole blood. q 2001Elsevier Science B.V. All rights reserved.
Ž . Ž .E. coli, Escherichia coli; Fd, fragment consisting of variable domain V and the first constant domain C 1 of the heavy chain ofH H
antibody; Fab, antigen binding fragment; Fdenv, Fd fragment carrying 31 amino acid peptide from envelope glycoprotein of HIV-1 orHIV-2; Fabenv, Fab fusion protein carrying 31 amino acids from envelope glycoprotein of HIV-1 or HIV-2; IgG, immunoglobulin type G;
ŽIPTG, isopropyl-b-D-thiogalactoside; LC, light chain of an antibody; MAb, monoclonal antibody; PBS, phosphate buffered saline 20 mM.phosphate buffer, pH 7.2 containing 145 mM NaCl ; RT, room temperature; RBC, human red blood cell.
0022-1759r01r$ - see front matter q 2001 Elsevier Science B.V. All rights reserved.Ž .PII: S0022-1759 01 00435-5
( )A. Gupta et al.rJournal of Immunological Methods 256 2001 121–140122
1. Introduction
The high specificity of antibodies and antibodyfragments has been exploited for a variety of diag-
Žnostic and therapeutic applications Kemp et al.,1988; Pastan et al., 1992; Brinkmann and Pastan,
.1994 . For this, antibodies are used either directly orŽmodified to impart new functions Neuberger et al.,
.1984; Williams and Neuberger, 1986 . Initially,chemical conjugation methods were used for modifi-cations but these methods are now being replaced by
Žrecombinant methods Chaudhary et al., 1989; Wil-.son et al., 1991; Coia et al., 1996 . This recombinant
approach has been possible due to the availability ofsimple methods to obtain DNA encoding variable
Ždomains of antibodies Orlandi et al., 1989; Chaud-.hary et al., 1990a , and expression systems to pro-
duce antibody fragments such as single chain FvŽ . Ž .scFv , disulfide-linked Fv dsFv and Fab in E. coliand other procaryotic and eucaryotic hostsŽ .Pluckthun, 1992 . These fragments are modified bygene fusion to encode fusion proteins having neweffector functions. They include immunotoxins for
Žtargeted therapy of cancer and AIDS Brinkmann. Žand Pastan, 1994 , antibody conjugates Cho et al.,
.1997 in therapeutics, and antibody enzyme fusionsŽ .in diagnostics Muller et al., 1999 . In an interesting
Ž .application, Kemp et al. 1988 designed a novelbifunctional molecule for the detection of anti-HIVantibodies in the blood of an infected individual.This molecule was produced by chemical conjuga-tion of a non-agglutinating anti-human RBC MAbwith a peptide derived from HIV-1 envelope gp41.When added to whole blood from HIV-infected indi-vidual, this molecule resulted in agglutination ofRBCs within 2 min. In this assay, RBCs in the bloodof an HIV-infected person get coated with the bi-functional molecule and cross-linking of coated RBCsis effected by the naturally occurring bivalent anti-HIV antibodies in the blood resulting in agglutina-tion visible to naked eyes. Later, this technology wasdeveloped for the detection of a variety of antigens
Ž .and antibodies Rylatt et al., 1990 . Chemical conju-gation techniques were refined but remained cumber-
Žsome and resulted in low yields Wilson et al.,
.1991 . Recombinant methods were then employedfor expression of monovalent scFv fragment of anti-
ŽRBC antibody fused to HIV antigens Lilley et al.,.1994; Coia et al., 1996 . The fusion protein was
localised in the culture medium and was purified byaffinity chromatography. The yield of purified pro-tein was not reported although the culture super-natant was estimated to contain 1–5 mg of fusionprotein per liter. Later, the same group produced aFab fusion protein carrying immunodominant epi-topes of HIV-1 and HIV-2 fused to the C-terminus
Ž .of Fd and LC in Fab Dolezal et al., 1995 . However,Žthe yield of this protein was very low 50 mgrl
.culture . It was obvious that use of recombinantmolecules for such a novel approach of immunoas-say was hampered by the low yields of fusion pro-teins obtained.
An alternate approach to high level production offunctional monovalent antibody fragments relies uponcytosolic expression as inclusion bodies followed bydenaturation and in vitro renaturation procedureŽ .Buchner and Rudolph, 1991 . This procedure hasalso been effectively used for producing recombinant
Ž .immunotoxins Buchner et al., 1992a,b in largequantities. This method has been successful in theproduction of both Fv- and Fab-based proteinsŽ .Buchner et al., 1992a; Choe et al., 1994 .
The objective of the present study was to developrecombinant, monovalent, Fab-based bifunctional an-tibody molecules for detection of anti-HIV antibod-ies in whole blood. For this, HIV antigens derivedfrom immunodominant region of HIV-1 gp41 andHIV-2 gp36 have been separately fused at the C-terminus of Fd chain of an anti-RBC MAb, B6. MAbB6 was isolated by conventional hybridoma technol-ogy using spleen cells from mice immunised with ORhD negative human RBCs. The universal reactivityof this antibody was established by screening morethan 1000 random blood samples. The DNA encod-ing LC and Fd of B6 was cloned using PCR-based
Ž .methods our unpublished data .In the present paper, we describe designing, high
level expression in E. coli, purification and charac-terisation of B6Fab-based fusion proteins containingHIV antigens, for their use in the rapid detection ofanti-HIV antibodies in whole blood.
( )A. Gupta et al.rJournal of Immunological Methods 256 2001 121–140 123
2. Materials and methods
2.1. Materials
MAb B6 was isolated by conventional hybridomaŽtechnology in our laboratory our unpublished re-
.sults . The DNA encoding LC and Fd of B6 wereŽcloned using PCR-based methods our unpublished
.data . Q-Sepharose fast flow, SP-Sepharose fast flow,Ž .Sephacryl S-200 high resolution , Sephadex G-25
Ž .medium and chromatography columns, XK50r20,XK16r20, XK26r70, XK26r100, HR10r10 andHR16r10 were purchased from Amersham Pharma-cia Biotech, Buckinghamshire, England. Toyobutyl
Ž .gel M was obtained from Tosohaas, Mont-gomeryville, PA, USA. Human serum samples wereobtained from Post Graduate Institute of Medical
Ž .Education and Research PGI , Chandigarh, IndiaŽ .and Centre for Disease Control CDC , Atlanta, USA.
These were previously collected serum samples thathad been characterised for HIV infection using im-munoassays. For this study, they were provided ascoded samples and identity of the donor was notdisclosed.
2.2. Expression Õector
A high copy number T7 promoter-based vector,pVCCD41140, was used for the cloning and expres-sion of various fusion proteins. This vector wasconstructed by cloning a PCR product encoding thefirst 176 amino acids of human CD4 as Nde I–EcoR
Ž .I insert into pVC45fq t Chaudhary et al., 1990a,b .In this vector, the initiation codon is part of the Nde
Ž .I site CATATG . The CD4 encoding DNA is flankedby Nhe I and Mlu I site and is followed by codonsfor decapeptide tag, cmyc. The vector utilizes
Ž .BL21 lDE3 host that carries l-lysogen encodingT7 RNA polymerase gene under the control of
ŽlacUV5 promoter inducible by IPTG Studier et al.,.1990 . DNA encoding LC or Fd was cloned as Nhe
I–Mlu I fragment in this vector.pVCB6Fd1140 contains DNA sequence encoding
Fd region of anti-RBC monoclonal antibody, B6, asNhe I–Mlu I fragment followed by DNA encoding a10-amino acid tag, cmyc. This plasmid expresses Fd
fragment of B6 with decapeptide cmyc at the C-Ž .terminus B6Fd .
pVCB6Fdenv131240 is similar to pVCB6Fd1140but contains in its Mlu I site, a sequence encoding
Ž .31 amino acids 590–620 of HIV-1 gp41 envelopeŽ .Ratner et al., 1985 . This plasmid was constructedby cloning a synthetic oligonucleotide duplex havingMlu I compatible ends in a manner that upon cloningin the correct orientation Mlu I site between Fd andthe env sequence is destroyed, but the site betweenenv and cmyc sequence is regenerated. This plasmidexpresses a fusion protein of B6Fd carrying 31 aminoacids of HIV-1 gp41 with cmyc sequence at the
Ž .C-terminus B6Fdenv13 .pVCB6Fdenv241240 was constructed by cloning
into Mlu I site of pVCB6Fd1140, an oligonucleotideŽ .duplex encoding 31 amino acids 581–611 of HIV-2
Ž .envelope gp36 Clavel et al., 1986 . In this construct,the Mlu I site between Fd and env2 sequence isregenerated, but the site between env2 and cmyc isdestroyed. This plasmid expresses a fusion protein ofB6Fd carrying 31 amino acids of HIV-2 gp36 and
Ž .C-terminal cmyc tag B6Fdenv24 .pVCB6LC1140 is similar to pVCB6Fd1140 but
contains DNA sequence encoding light chain of B6Ž .B6LC as Nhe I–Mlu I fragment with a stop codonpreceding Mlu I site.
2.3. Expression and isolation of inclusion bodies
Ž .BL21 lDE3 cells were transformed with vari-ous plasmids and grown on LB plates containingampicillin for 16 h at 32 8C. Transformed cells from
Ž .six plates approximately 2000 colonies each werescrapped and inoculated in 1-l superbroth containingampicillin, and grown at 30 8C with vigorous shak-ing. At OD of 2.5–3.0, the culture was induced600 nm
Ž .with IPTG final concentration, 0.25 mM and grownat 30 8C. After 2 h of induction, the culture waschilled over ice. Cells were harvested by centrifuga-
Ž .tion at 4000=g GSA rotor, Sorvall RC5C for 10min at 4 8C.
Inclusion bodies were extracted following a proto-Ž .col as described earlier Buchner and Rudolph, 1991 .
ŽBriefly, the cell pellet from 1-l culture contained in.one 250-ml GSA bottle was suspended in 135 ml of
( )A. Gupta et al.rJournal of Immunological Methods 256 2001 121–140124
Ž .T E solution 50 mM Tris pH 8.0, 20 mM EDTA50 20Žusing tissuemiser with large probe Model T25, IKA,
.Germany at 20,500 rpm for 2 min. Forty-milligramlysozyme in 5 ml of T E was added and the50 20
suspension was incubated on an orbital shaker for 60min at room temperature. To this suspension, 15 mlof 5 M NaCl and 15 ml of 25% Triton X-100 wereadded. The bottles were shaken vigorously and kepton a reciprocating shaker for 1 h at 37 8C. The
Žsamples were centrifuged at 23,500=g GSA rotor,.Sorvall RC5C for 1 h at 4 8C. The supernatant was
discarded and the pellet was suspended in 100 ml1% Triton X-100 solution in T E , using tis-50 20
suemiser, as described above. The samples werecentrifuged at 23,500=g for 50 min at 4 8C. Thepellet was washed by suspending it in 100-ml T E50 20
using tissuemiser and centrifugation at 23,500=gfor 50 min at 4 8C. This step was repeated fourtimes. Finally, the pellet was suspended in 30-mlT E using tissuemiser, transferred to 50-ml Oak50 20
ridge tube and centrifuged at 27,000=g for 50 minŽ . Žat 4 8C SS34 rotor, Sorvall RC5C . An aliquot 30
.ml of the suspension was removed before centrifu-gation, mixed with 30 ml of 2= Laemmli bufferŽ .reducing , boiled at 110 8C for 3–5 min and anal-ysed on 0.1% SDS–12.5% PAGE. The pellet con-taining inclusion bodies in Oak ridge tubes wasstored at y70 8C until further use.
2.4. Assembly of Fab molecules
Inclusion bodies isolated from 1-l culture each ofB6Fd or B6Fdenv fusion protein and B6LC were
Žthawed and 10-ml solubilisation buffer 0.1 M Tris–.HCl, pH 8.0, 2 mM EDTA, 6 M guanidine HCl was
added to each tube. Inclusion bodies were solubilisedproperly using tissuemiser with small probe at 9500rpm for 4–5 min. Total protein of solubilised FdrFdfusion protein and LC was estimated by Bradfordmethod using BSA as standard. The final proteinconcentration was adjusted to 10 mgrml by addingsolubilisation buffer. Equimolar amounts of LC andFd or Fdenv fusion proteins were taken and solid
Ž .dithioerythritol DTE was added to a final concen-tration of 10 mgrml and the tubes were kept at roomtemperature for 2 h. After this, LC and Fd solutionswere mixed and centrifuged at 93,000=g, at 4 8C,
Žfor 30 min 70.1 Ti rotor, L7-65 Ultracentrifuge,.Beckman . The supernatant was collected and was
Ždiluted 100 fold in cold renaturation buffer 0.1 MTris–HCl, pH 8.0, 2 mM EDTA, 0.5 M arginine
.HCl, 0.9 mM oxidised glutathione by adding it dropwise to the renaturation buffer while stirring. Thismaterial was kept undisturbed at 10 8C for 70 h forrenaturation.
The renatured material was dialysed against 3–4volumes of 20 mM Tris, pH 8.0, containing 100 mMurea. Dialysis was done for 72 h with four bufferchanges. Conductivity of buffer was measured ateach change to monitor dialysis. The final dialysiswas done in buffer containing 50 mM urea. Thedialysed material was filtered through 2.3- and 0.4-mm filters and then used to purify renaturedmonomeric FabrFabenv fusion proteins.
2.5. Purification of monomeric FabenÕ fusion pro-teins
After renaturation, the assembled monomeric Fabfusion protein was separated from oligomerised Fabfusion protein, unassembled LC and Fd moleculesand other contaminants using a series of columnchromatographic steps. These steps were commonfor various fusion proteins. The procedure to purify
ŽB6Fabenv24 made by assembling B6LC and.B6Fdenv24 is described below in detail.
The pH of the dialysed and filtered solution con-taining Fab fusion protein was adjusted to ;5.0with 5% acetic acid and the samples were keptundisturbed at 4 8C for 12 h. After centrifugation at
Ž .23,500=g GSA, Sorvall , 4 8C for 10 min, thesupernatant was filtered through 0.4-mm filter. Using
Ž .FPLC Amersham Pharmacia Biotech, USA , theŽ .filtrate 3.5 l was applied at a flow rate of 12
Žmlrmin on a 50-ml SP-Sepharose column XK.50r20 pre-equilibrated with 20 mM acetate buffer,Ž .pH 5.0 Buffer A . All the chromatography steps
were carried out at 2–8 8C. The column was washedwith 100 ml of Buffer A containing 100 mM NaCl ata flow rate of 8 mlrmin and the protein was eluted
Žwith a 250-ml linear gradient of NaCl 100–600.mM in Buffer A. The elution was monitored at 280
Žnm and fractions were collected. The fractions 12.ml each were neutralized immediately with 120 ml
( )A. Gupta et al.rJournal of Immunological Methods 256 2001 121–140 125
1 M Tris solution. The fractions containing the de-Žsired protein analysed by SDS–PAGE under non-re-
ducing conditions and by RBC agglutination assay.described in Section 2.7 were pooled.
The pool of fractions obtained from SP-Sepharosecolumn was subjected to hydrophobic column chro-matography on Toyobutyl resin. Average salt con-centration of this pool was estimated from the chro-matogram and solid NaCl was added to the pool to afinal concentration of 2 M. The pH was made to;7.2 by adding 1 M Tris solution and the sample
Žwas loaded on a 25-ml Toyobutyl column XK.16r20 pre-equilibrated with 20 mM phosphate
Ž .buffer, pH 7.2 Buffer B containing 2 M NaCl. Thecolumn was washed with 50 ml of Buffer B contain-ing 2 M NaCl at a flow rate of 3 mlrmin, followedby 50 ml of Buffer B containing 1.0 M NaCl. Theprotein was eluted with a 100-ml step gradient ofBuffer B containing 0.6 M NaCl. The elution wasmonitored at 280 nm and 8-ml fractions were col-lected and analysed by agglutination assay. Fractionscontaining monomeric Fab fusion protein werepooled and iodoacetamide was added to a final con-centration of 50 mM. This protein sample was incu-bated at 25 8C for 15 min and then loaded on a
Ž325-ml Sephadex G-25 desalting column XK.26r70 pre-equilibrated with 20 mM Tris buffer, pH
Ž .8.5 Buffer C . After loading, the column was devel-oped with Buffer C at a flow rate of 5 mlrmin withmonitoring of absorbance at 280 nm and collectionof 12-ml fractions. Based on absorbance values,fractions containing protein were pooled. This pool
Žwas loaded on a 20-ml Q-Sepharose column HR.16r20 pre-equilibrated with Buffer C. The column
was washed with 40 ml of Buffer C at a flow rate of4 mlrmin, and the protein was eluted with a 120-ml
Ž .linear gradient of NaCl 0–350 mM in Buffer C.The elution was monitored at 280 nm and 4-mlfractions were collected. The fractions were analysedby SDS–PAGE under non-reducing conditions andby agglutination assay. The fractions containing de-
Ž .sired protein were pooled Q-Sepharose pool .The Q-Sepharose pool was loaded on 500-ml
Ž .Sephacryl S-200 column XK 26r100 . The columnwas developed with PBS at a flow rate of 1 mlrmin.The elution was monitored at 280 nm and 4-mlfractions were collected. The fractions were analysedby SDS–PAGE under non-reducing conditions and
by agglutination assay. Fractions containing active,monomeric Fab fusion protein were pooled.
2.6. Purification of B6 Fab protein
Ž .B6Fab protein without HIV-antigen peptide waspurified using protocol similar to that described inSection 2.5. The dialysed material was loaded onSP-Sepharose column, eluted and after analysis, rele-vant fractions were pooled and loaded on Toyobutylcolumn. However, Fab protein did not bind to Toy-obutyl column and was present in flow throughcollected during loading. The flow through wastreated with iodoacetamide, desalted on SephadexG-25 column and then purified on Q-Sepharose col-umn. Fractions eluted from Q-Sepharose were pooledafter analysis and this preparation was used forfurther work.
2.7. Agglutination assay of Õarious samples to checkfor the presence of actiÕe, monomeric Fab
Sixty microliters of column fraction or an appro-Žpriate dilution including sample before column and
.flow through obtained during loading was taken in aŽ .96-well round-bottom microtitre plate Wells 1–12 .
Two-fold serial dilution were made in PBS-BSAŽ . Ž .PBS containing 0.2% BSA Wells A–H . To each
Žwell, 30 ml of 2% human RBC suspension washed.RBC pellet reconstituted vrv to 2% in PBS–BSA
was added. After mixing, the plate was incubated at37 8C for 1 h. Agglutination was read visually and
Žalso using microplate reader Showscan software,.Cerus 900, Biotek Instruments . The maximum dilu-
Ž .tion of sample that gave agglutination end pointwas recorded. Agglutination in any well at this stageindicates the presence of aggregates in that sample.The plate was centrifuged at 800=g at RT for 5min. The supernatant was discarded by inversion andthe pellet was suspended in remaining buffer byvortexing. Sixty microliters of anti-cmyc MAbŽ .1:1000 dilution of 9E10 ascitic fluid in PBS–BSAwas added per well. After mixing, the plate wasincubated at 37 8C for 1 h. Agglutination was read asabove. Samples, which gave agglutination only afteraddition of 9E10 antibody, contain active monomericFabrFab fusion protein.
( )A. Gupta et al.rJournal of Immunological Methods 256 2001 121–140126
( )A. Gupta et al.rJournal of Immunological Methods 256 2001 121–140 127
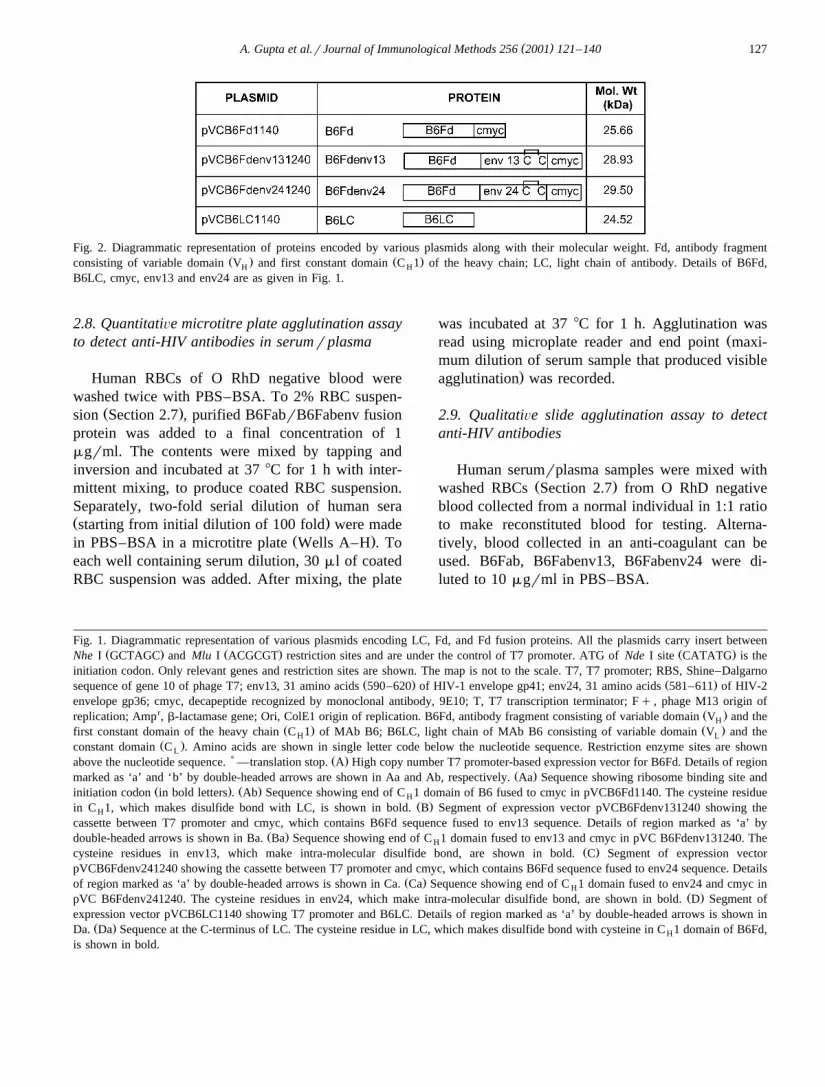
Fig. 2. Diagrammatic representation of proteins encoded by various plasmids along with their molecular weight. Fd, antibody fragmentŽ . Ž .consisting of variable domain V and first constant domain C 1 of the heavy chain; LC, light chain of antibody. Details of B6Fd,H H
B6LC, cmyc, env13 and env24 are as given in Fig. 1.
Human RBCs of O RhD negative blood werewashed twice with PBS–BSA. To 2% RBC suspen-
Ž .sion Section 2.7 , purified B6FabrB6Fabenv fusionprotein was added to a final concentration of 1mgrml. The contents were mixed by tapping andinversion and incubated at 37 8C for 1 h with inter-mittent mixing, to produce coated RBC suspension.Separately, two-fold serial dilution of human seraŽ .starting from initial dilution of 100 fold were made
Ž .in PBS–BSA in a microtitre plate Wells A–H . Toeach well containing serum dilution, 30 ml of coatedRBC suspension was added. After mixing, the plate
was incubated at 37 8C for 1 h. Agglutination wasŽread using microplate reader and end point maxi-
mum dilution of serum sample that produced visible.agglutination was recorded.
2.9. QualitatiÕe slide agglutination assay to detectanti-HIV antibodies
Human serumrplasma samples were mixed withŽ .washed RBCs Section 2.7 from O RhD negative
blood collected from a normal individual in 1:1 ratioto make reconstituted blood for testing. Alterna-tively, blood collected in an anti-coagulant can beused. B6Fab, B6Fabenv13, B6Fabenv24 were di-luted to 10 mgrml in PBS–BSA.
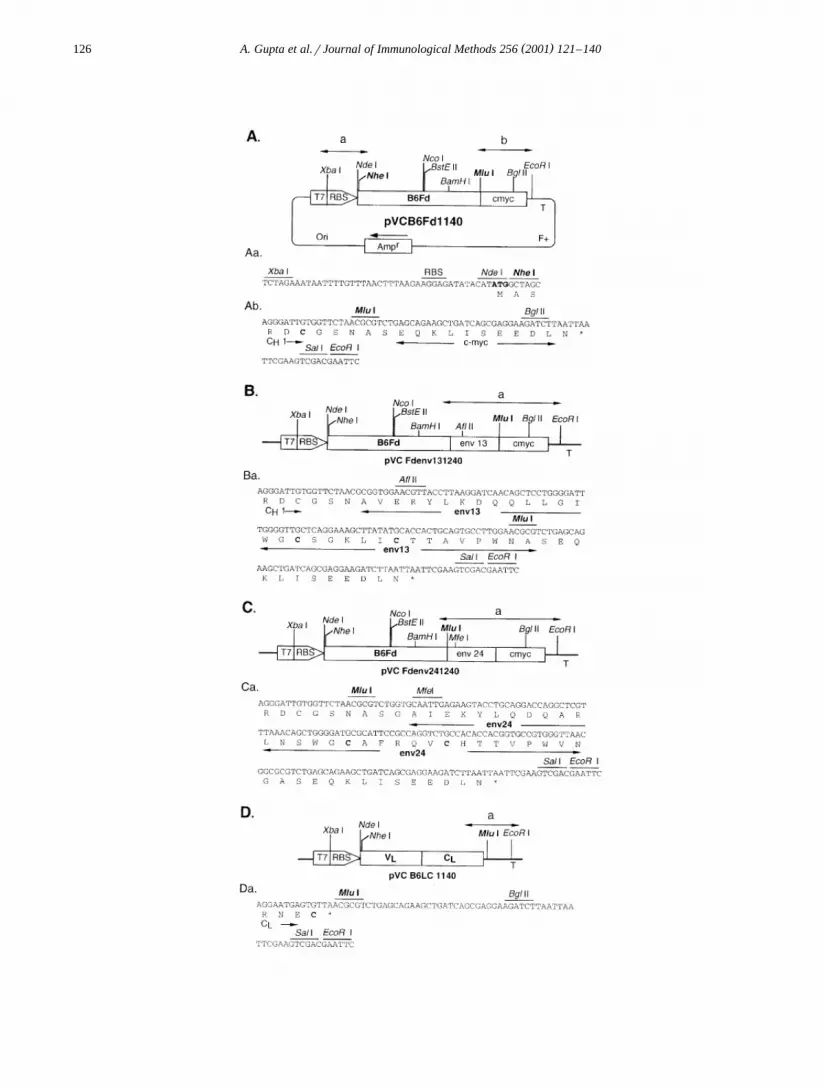
Fig. 1. Diagrammatic representation of various plasmids encoding LC, Fd, and Fd fusion proteins. All the plasmids carry insert betweenŽ . Ž . Ž .Nhe I GCTAGC and Mlu I ACGCGT restriction sites and are under the control of T7 promoter. ATG of Nde I site CATATG is the
initiation codon. Only relevant genes and restriction sites are shown. The map is not to the scale. T7, T7 promoter; RBS, Shine–DalgarnoŽ . Ž .sequence of gene 10 of phage T7; env13, 31 amino acids 590–620 of HIV-1 envelope gp41; env24, 31 amino acids 581–611 of HIV-2
envelope gp36; cmyc, decapeptide recognized by monoclonal antibody, 9E10; T, T7 transcription terminator; Fq , phage M13 origin ofr Ž .replication; Amp , b-lactamase gene; Ori, ColE1 origin of replication. B6Fd, antibody fragment consisting of variable domain V and theH
Ž . Ž .first constant domain of the heavy chain C 1 of MAb B6; B6LC, light chain of MAb B6 consisting of variable domain V and theH LŽ .constant domain C . Amino acids are shown in single letter code below the nucleotide sequence. Restriction enzyme sites are shownL
) Ž .above the nucleotide sequence. —translation stop. A High copy number T7 promoter-based expression vector for B6Fd. Details of regionŽ .marked as ‘a’ and ‘b’ by double-headed arrows are shown in Aa and Ab, respectively. Aa Sequence showing ribosome binding site and
Ž . Ž .initiation codon in bold letters . Ab Sequence showing end of C 1 domain of B6 fused to cmyc in pVCB6Fd1140. The cysteine residueHŽ .in C 1, which makes disulfide bond with LC, is shown in bold. B Segment of expression vector pVCB6Fdenv131240 showing theH
cassette between T7 promoter and cmyc, which contains B6Fd sequence fused to env13 sequence. Details of region marked as ‘a’ byŽ .double-headed arrows is shown in Ba. Ba Sequence showing end of C 1 domain fused to env13 and cmyc in pVC B6Fdenv131240. TheH
Ž .cysteine residues in env13, which make intra-molecular disulfide bond, are shown in bold. C Segment of expression vectorpVCB6Fdenv241240 showing the cassette between T7 promoter and cmyc, which contains B6Fd sequence fused to env24 sequence. Details
Ž .of region marked as ‘a’ by double-headed arrows is shown in Ca. Ca Sequence showing end of C 1 domain fused to env24 and cmyc inHŽ .pVC B6Fdenv241240. The cysteine residues in env24, which make intra-molecular disulfide bond, are shown in bold. D Segment of
expression vector pVCB6LC1140 showing T7 promoter and B6LC. Details of region marked as ‘a’ by double-headed arrows is shown inŽ .Da. Da Sequence at the C-terminus of LC. The cysteine residue in LC, which makes disulfide bond with cysteine in C 1 domain of B6Fd,H
is shown in bold.
( )A. Gupta et al.rJournal of Immunological Methods 256 2001 121–140128
Ž .On a glass slide having three marked spots , aŽdrop of reconstitutedrwhole blood sample ap-
.proximately 20 ml was dispensed on each spot.Twenty microliters of B6Fab was added on spotcontrol, 20 ml of B6Fabenv13 on spot ‘HIV-1’ and20 ml of B6Fabenv24 on spot ‘HIV-2’. The contentson each spot were mixed using plastic stick. The
Žslide was kept undisturbed for 3 min at RT 25–30. Ž .8C , and agglutination clumping of RBCs was
recorded visually while rotating the slide.
3. Results
3.1. Vector for expression of Õarious proteins
A T7 promoter-based expression vector,pVCCD41140, was employed for cloning DNA frag-
Žments as Nhe I–Mlu I inserts. pVCB6Fd1140 Fig..1A carries sequence encoding amino acids 1–219 of
Ž .the heavy chain Fd of an anti-RBC monoclonalantibody, B6, as Nhe I–Mlu I insert. Nhe I site in
Ž .the vector is preceded by Nde I site CATATGŽwhose ATG constitutes the initiation codon Fig.
.1Aa . Mlu I site is followed by sequence for a10-amino acid tag, cmyc that is recognised by MAb,
Ž .9E10 Fig. 1Ab . In pVCB6Fdenv131240, followingFd region, a sequence encoding 31 amino acidsŽ . Ž .590–620 of HIV-1 gp41 env13 has been insertedin a way that Mlu I site is regenerated between
Ž .env13 and cmyc sequence Fig. 1B,Ba . InpVCB6Fdenv241240, following Fd, a sequence en-
Ž .coding 31 amino acids 581–611 of HIV-2 gp36Ž .env24 has been inserted so that Mlu I site is
Ž .regenerated between Fd and env24 Fig. 1C,Ca .pVCB6LC1140 carries sequence encoding the light
Ž .chain amino acids 1–219 of B6 as Nhe I–Mlu IŽ .insert Fig. 1D . This plasmid contains a stop codon
after the coding sequence of B6LC and precedingŽ .Mlu I site Fig. 1Da . All the constructs encode a
Ž .protein with three additional amino acids MAS atŽ .their N-terminus Fig. 1Aa .
3.2. Expression of Õarious fusion proteins
Proteins encoded by different plasmids are shownŽ .diagrammatically in Fig. 2. BL21 lDE3 cells trans-
formed with various plasmids were grown and ex-pression of the desired protein was induced withIPTG. Cells from induced cultures were analysedfor expression by SDS–PAGE under reducing con-
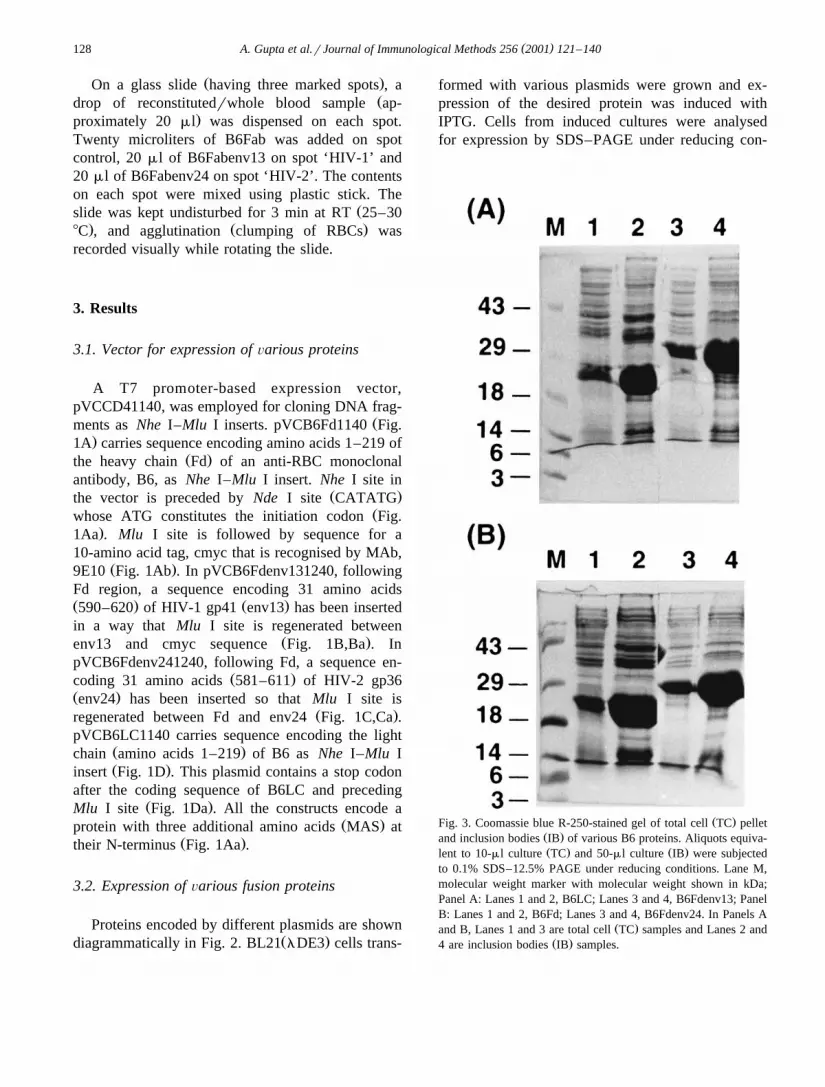
Ž .Fig. 3. Coomassie blue R-250-stained gel of total cell TC pelletŽ .and inclusion bodies IB of various B6 proteins. Aliquots equiva-Ž . Ž .lent to 10-ml culture TC and 50-ml culture IB were subjected
to 0.1% SDS–12.5% PAGE under reducing conditions. Lane M,molecular weight marker with molecular weight shown in kDa;Panel A: Lanes 1 and 2, B6LC; Lanes 3 and 4, B6Fdenv13; PanelB: Lanes 1 and 2, B6Fd; Lanes 3 and 4, B6Fdenv24. In Panels A
Ž .and B, Lanes 1 and 3 are total cell TC samples and Lanes 2 andŽ .4 are inclusion bodies IB samples.
( )A. Gupta et al.rJournal of Immunological Methods 256 2001 121–140 129
ditions. pVCB6Fd1140 expresses a protein ofapproximately 25 kDa, which constitutes ;20%
Ž .of total cellular protein Fig. 3B; Lane 1 .pVCB6Fdenv131240 and pVCB6Fdenv241240 ex-
Ž .press fusion proteins B6Fdenv13 Fig. 3A; Lane 3Ž .and B6Fdenv24 Fig. 3B; Lane 3 , respectively, of
approximately 29 kDa each. These two fusion pro-teins also constitute ;20% of total cellular protein.On Western Blot, B6Fd, B6Fdenv13 and B6Fdenv24reacted with MAb 9E10 showing the presence of
Ž .cmyc tag data not shown . pVCB6LC1140 ex-pressed a protein of approximately 24 kDa, but theexpression was very poor as compared to that of
Ž .B6Fd proteins data not shown . Analysis of B6LCsequence revealed that arginine residues at four posi-
Ž .tions 24, 58, 59 and 82 residues of LC were codedŽ .by minor codons AGA and AGG which might be
Ž .resulting in poor expression Kane, 1995 . Therefore,codons at positions 24, 58 and 59 were changed to
Ž .major codons CGT and CGG using two separateoligonucleotides and following the method for site-
Ž .directed mutagenesis as described by Kunkel 1985 .The expression of mutant LC increased several fold
Žand was similar to that of B6 Fd fusion proteins Fig..3A; Lane 1 . Mutant B6LC was used for further
work.
3.3. Preparation and purification of inclusion bodiesand assembly of Fab molecules
Localization studies showed that various fusionproteins produced insoluble mass of protein in the
cytosol, which could be isolated as inclusion bodiesŽ .data not shown . The recombinant fusion proteinconstituted approximately 70–80% of total protein in
Žthe purified inclusion body preparations Fig. 3A,B;.Lanes 2 and 4 .
For the assembly of various B6Fab derivatives, aprotocol comprising of in vitro denaturation ofFdrFdenv and LC, and their subsequent renaturationand association was optimised. B6Fd derivatives andB6LC were completely denatured and reduced andthen mixed in equimolar ratio. The assembly of Fabwas initiated by diluting the mixed Fd and LC in abuffer containing oxidised glutathione. This createdproper redox conditions for the refolding of Fd andLC, formation of intramolecular disulfide bonds, as-sociation of Fd with LC, and formation of inter-molecular disulfide bond to form Fab. The renaturedmaterial was dialysed to remove denaturants. Thiswas followed by a series of column chromatographysteps to purify monomeric Fab derivatives.
3.4. Purification of Õarious B6FabenÕ fusion pro-teins
A four-step protocol consisting of cation ex-change, hydrophobic, anion exchange and gel filtra-tion chromatography was developed to purifyB6Fabenv13 and B6Fabenv24. Fractions obtained atvarious chromatography steps were analysed for level
Ž .of purity by SDS–PAGE and for presence of func-Ž .tional and monomeric Fab by agglutination assay .
Fab molecules are monovalent in nature and, there-
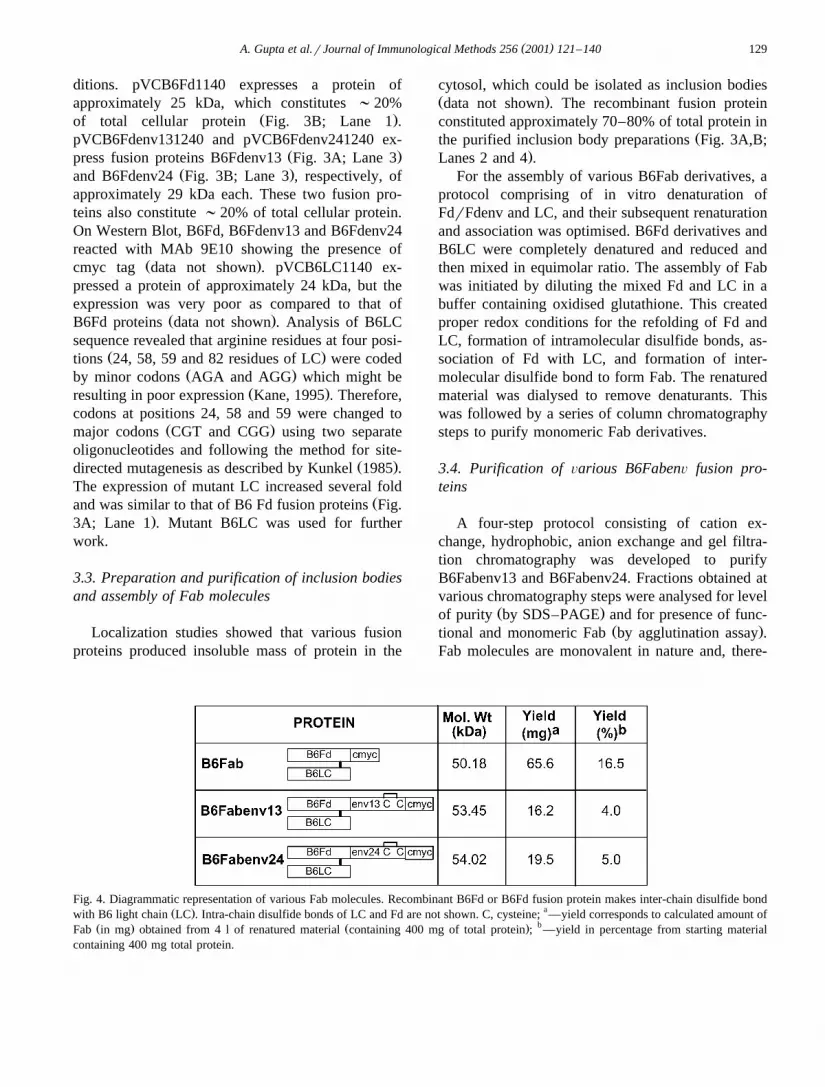
Fig. 4. Diagrammatic representation of various Fab molecules. Recombinant B6Fd or B6Fd fusion protein makes inter-chain disulfide bondŽ . awith B6 light chain LC . Intra-chain disulfide bonds of LC and Fd are not shown. C, cysteine; —yield corresponds to calculated amount of
Ž . Ž . bFab in mg obtained from 4 l of renatured material containing 400 mg of total protein ; —yield in percentage from starting materialcontaining 400 mg total protein.
( )A. Gupta et al.rJournal of Immunological Methods 256 2001 121–140130
Ž . Ž .Fig. 5. SP-Sepharose chromatography of B6Fabenv24. A Chromatogram showing elution profile of protein. Sample 3.4 l was loaded onŽ .50 ml SP-Sepharose packed in XK50r20 column at 12 mlrmin. Elution was carried out at 6 mlrmin with continuous monitoring of
Ž .absorbance at 280 nm, and 12-ml fractions were collected. B Coomassie blue-stained gel of the fractions eluted from SP-SepharoseŽ .column subjected to 0.1% SDS–10% PAGE. Samples were boiled in equal volume of 2= Laemmli sample buffer non-reducing . Each
lane contains sample equivalent to 10 ml of fraction. Molecular weight of Fab fusion protein is shown in kDa; Lane D, dialysed renaturedŽ .material; Lanes 19–33, fractions 19–33 from SP-Sepharose column; Lane C, a recombinant Fab protein purified by other protocol control ;
arrows indicate the position of B6Fabenv24 protein and B6LC. Aggn., end point of agglutination recorded as maximum dilution of eachŽ .sample, which gave visible agglutination. ‘0’ indicates no visible agglutination with undiluted sample neat . 9E10, a MAb which recognizes
a decapeptide tag, cmyc at the C-terminus of Fd in FabrFab fusion proteins.
( )A. Gupta et al.rJournal of Immunological Methods 256 2001 121–140 131
fore, can bind to RBCs but cannot cross-link RBCsto give visible agglutination. However, these Fab-coated RBCs can be cross-linked by MAb 9E10,which binds to cmyc tag present at the C-terminus ofall Fab molecules. Therefore, all samples containingfunctional Fab molecules will show agglutination ofRBCs on addition of 9E10, and the extent of aggluti-nation will be directly proportional to the amount ofactive Fab in that sample. However, if a sampleshows agglutination before addition of 9E10, it indi-cates that the Fab molecules in that sample are notmonovalent but have aggregated to become multiva-lent. Such samples are not processed further. Theyields obtained for B6Fabenv proteins are shown inFig. 4. The following section describes purificationof B6Fabenv24 in detail.
After renaturation of B6LC and B6Fdenv24 forassembly of B6Fabenv24, the renatured material wasdialysed and pH of the protein solution was adjustedto 5.0, which led to some precipitation. On analysisby SDS–PAGE, the precipitate was found to contain
Žmainly high molecular weight aggregates data not.shown . The precipitate was removed by centrifuga-
tion and filtration. The clear protein solution wasloaded on SP-Sepharose column and bound proteinswere eluted with a linear gradient of NaCl. Eachfraction was analysed by SDS–PAGE and for RBCagglutination. Absorbance profile shows that a largepeak eluted between 300 and 700 mM NaCl concen-tration with shoulder at 400 mM and also at 550 mM
Ž .NaCl concentration Fig. 5A . SDS–PAGE showedthat fractions 21–27 contained a band of ;53 kDacorresponding to calculated molecular weight of
Ž .B6Fabenv24 Figs. 4 and 5B . These fractions alsocontained band corresponding to B6LC and highmolecular weight contaminants. Fractions 18–20Ž .corresponding to the shoulder at 400 mM NaClcontained mainly free B6LC with very small quan-
Žtity of Fab Fig. 5B, data shown only for fraction. Ž19 . In agglutination assay, fractions 27–33 corre-
.sponding to the shoulder at 550 mM NaCl showedagglutination before the addition of MAb 9E10 indi-
Žcating the presence of aggregated B6Fabenv24 Fig..5B . Fractions 21–25 showed strong agglutination
only after addition of MAb 9E10 indicating thepresence of monomeric Fab; therefore, fractions 21–26 were pooled for further purification. The NaClconcentration of pool was estimated to be 0.4 M
Žcalculated from the chromatogram based on salt.concentration of fractions pooled and was made to 2
M by adding solid NaCl. The pH was adjusted to 7.2with 1 M Tris solution, and the pool was subjected tochromatography on Toyobutyl column. After loadingthe sample, the column was washed with 1.0 MNaCl, and the elution was effected with 0.6 M NaCl.
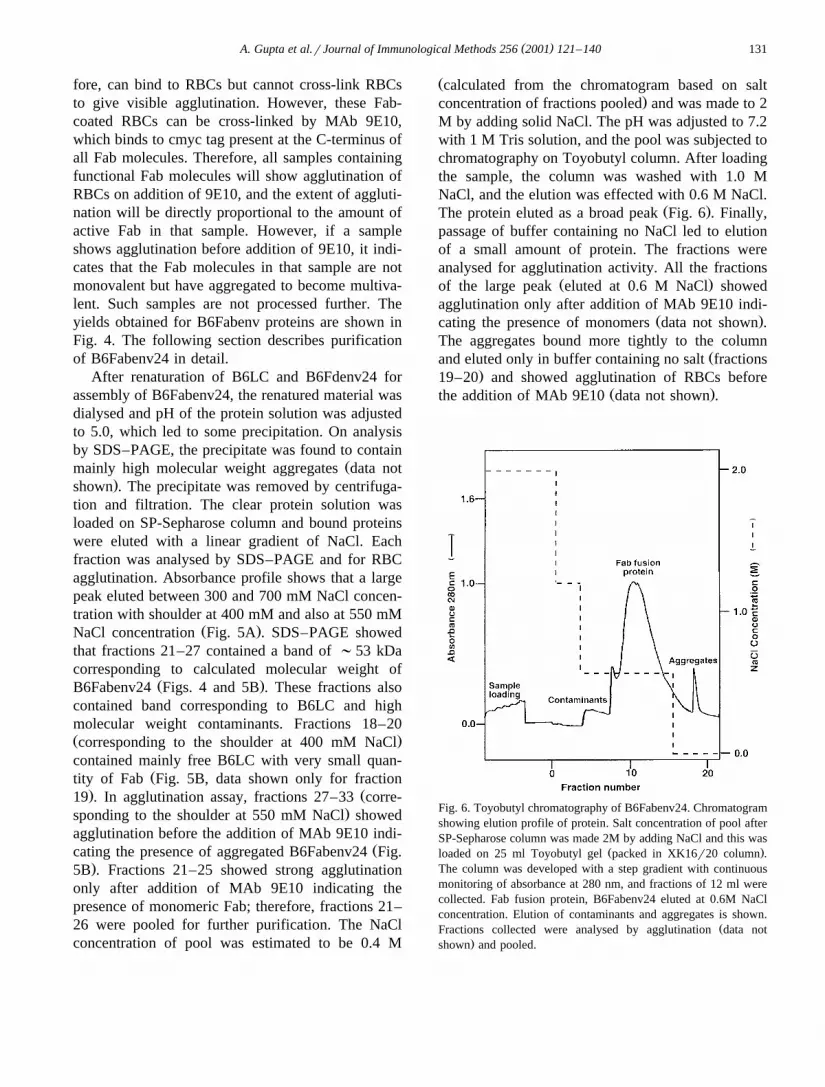
Ž .The protein eluted as a broad peak Fig. 6 . Finally,passage of buffer containing no NaCl led to elutionof a small amount of protein. The fractions wereanalysed for agglutination activity. All the fractions
Ž .of the large peak eluted at 0.6 M NaCl showedagglutination only after addition of MAb 9E10 indi-
Ž .cating the presence of monomers data not shown .The aggregates bound more tightly to the column
Žand eluted only in buffer containing no salt fractions.19–20 and showed agglutination of RBCs before
Ž .the addition of MAb 9E10 data not shown .
Fig. 6. Toyobutyl chromatography of B6Fabenv24. Chromatogramshowing elution profile of protein. Salt concentration of pool afterSP-Sepharose column was made 2M by adding NaCl and this was
Ž .loaded on 25 ml Toyobutyl gel packed in XK16r20 column .The column was developed with a step gradient with continuousmonitoring of absorbance at 280 nm, and fractions of 12 ml werecollected. Fab fusion protein, B6Fabenv24 eluted at 0.6M NaClconcentration. Elution of contaminants and aggregates is shown.
ŽFractions collected were analysed by agglutination data not.shown and pooled.
( )A. Gupta et al.rJournal of Immunological Methods 256 2001 121–140132
Ž .Fig. 7. Q-Sepharose chromatography of B6Fabenv24. A Chromatogram showing elution profile of protein. Sample after desalting onŽ . Ž .Sephadex G-25 ;130 ml was loaded on a 20-ml Q-Sepharose column packed in HR 16r10 at 4 mlrmin. Elution was done at 3 mlrmin
Ž .with continuous monitoring of absorbance at 280 nm, and 4-ml fractions were collected. B Coomassie blue-stained gel of the fractionseluted from Q-Sepharose column. Samples were processed and electrophoresed as in Fig. 5. Molecular weight of Fab fusion protein isshown in kDa; Lane P2, pool after Sephadex G-25 column; Lanes 26–37, fractions 26–37 from Q-Sepharose column. Arrows indicate theposition of B6Fabenv24 protein and aggregates. Aggn., as in Fig. 5.
( )A. Gupta et al.rJournal of Immunological Methods 256 2001 121–140 133
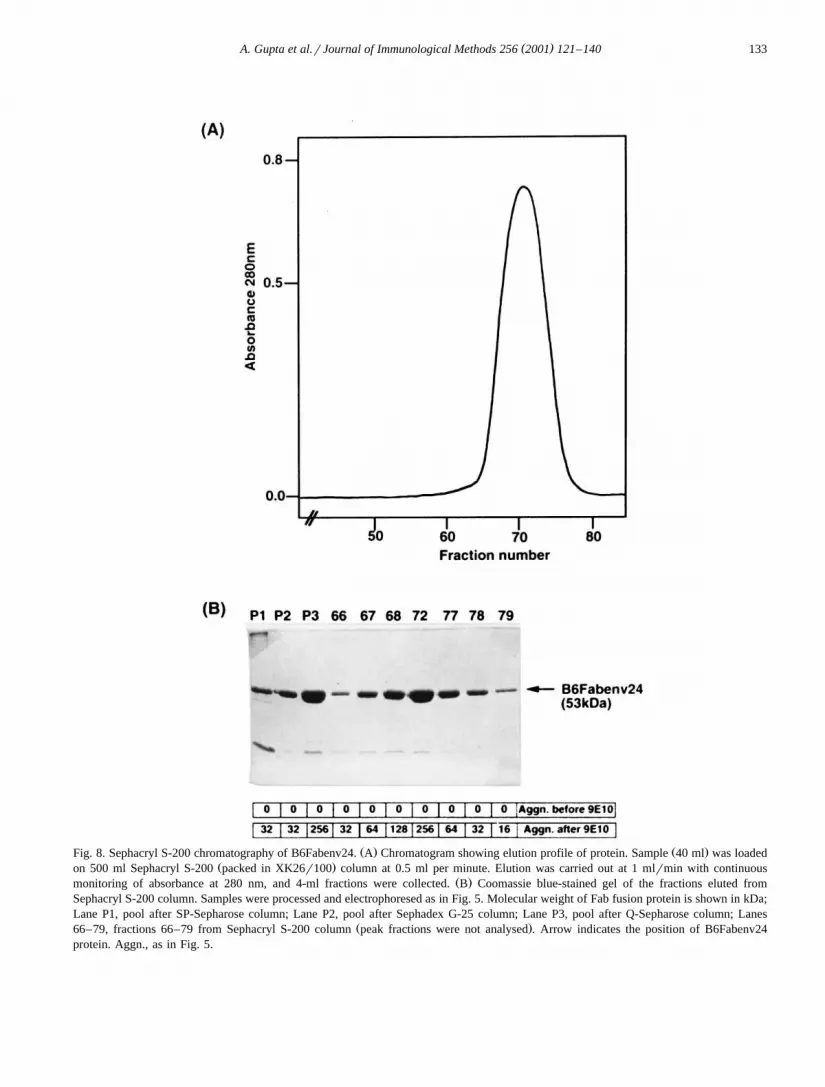
Ž . Ž .Fig. 8. Sephacryl S-200 chromatography of B6Fabenv24. A Chromatogram showing elution profile of protein. Sample 40 ml was loadedŽ .on 500 ml Sephacryl S-200 packed in XK26r100 column at 0.5 ml per minute. Elution was carried out at 1 mlrmin with continuous
Ž .monitoring of absorbance at 280 nm, and 4-ml fractions were collected. B Coomassie blue-stained gel of the fractions eluted fromSephacryl S-200 column. Samples were processed and electrophoresed as in Fig. 5. Molecular weight of Fab fusion protein is shown in kDa;Lane P1, pool after SP-Sepharose column; Lane P2, pool after Sephadex G-25 column; Lane P3, pool after Q-Sepharose column; Lanes
Ž .66–79, fractions 66–79 from Sephacryl S-200 column peak fractions were not analysed . Arrow indicates the position of B6Fabenv24protein. Aggn., as in Fig. 5.
( )A. Gupta et al.rJournal of Immunological Methods 256 2001 121–140134
The pool of fractions from hydrophobic column,free of most aggregated proteins, was treated withiodoacetamide to block free sulfhydryl groups pre-sent in any incorrectly folded Fab molecules andcontaminating free LC and Fd chains. The excessiodoacetamide and salt were then removed from thesample by desalting on Sephadex G-25 column be-fore loading on a 20-ml Q-Sepharose anion exchangecolumn. The Q-Sepharose column was developedwith a linear gradient of NaCl. B6Fabenv24 elutedbetween 100- and 250-mM gradient of NaCl as a
Ž .symmetrical peak Fig. 7A . SDS–PAGE of frac-
tions showed that fractions 26–32 contained a bandof 53 kDa corresponding to calculated molecularweight of B6Fabenv24 protein, while fractions 34–37contained some amount of Fab but also containedbands corresponding to high molecular weight aggre-
Ž .gates Fig. 7B . In agglutination assay, fractions34–37 showed agglutination before addition of 9E10antibody indicating the presence of aggregates. Afteraddition of 9E10, strong agglutination was observedin fractions 26–32. Fractions 26–33 were pooled andloaded on Sephacryl S-200 gel filtration column to
Ž .remove any remaining aggregates Fig. 8 . Fractions
Ž . Ž .Fig. 9. Purification of B6Fab. Coomassie blue-stained gel of the eluted fractions of B6Fabfrom A SP-Sepharose and B Q-Sepharosecolumn. Samples were processed and electrophoresed as in Fig. 5. Molecular weight of Fab fusion protein is shown in kDa; Panel A: LaneD, dialysed renatured material; Lanes 14–28, fractions 14–28 from SP-Sepharose column; Lane C, B6Fab protein purified by another
Ž .protocol control ; Panel B: Lane P1, pool after SP-Sepharose column; Lane P2, pool after Toyobutyl and Sephadex G-25 column; LanesŽ .26–33, fractions 26–33 from Q-Sepharose column; Lane E, BSA 10 mg control . Arrows indicate the position of B6Fab protein, free B6Fd
and free B6LC. Aggn., as in Fig. 5.
( )A. Gupta et al.rJournal of Immunological Methods 256 2001 121–140 135
66–79 of Sephacryl S-200 column showed bandcorresponding to B6Fabenv24 protein on SDS–
Ž .PAGE with purity of ;95% Fig. 8B . In agglutina-tion assay, none of these fractions showed agglutina-tion before addition of 9E10 antibody indicating theabsence of aggregates. After addition of 9E10, agglu-tination was observed in fractions 66–80. Fractions
Ž .68–80 were pooled. B6Fabenv24 protein 15.1 mgwas obtained from 3.4 l of renatured material con-taining 340 mg total protein.
B6Fabenv13 was purified following an identicalscheme and set of columns. The elution from hy-drophobic column was carried out using 0.7 M NaClconcentration. Elution profiles were also similar tothat of B6Fabenv24. Highly purified B6Fabenv13Ž .18.2 mg, ;95% purity was obtained from 4.5 l ofrenatured material containing 450 mg total protein.
3.5. Purification B6Fab
A three-step protocol consisting of cation ex-change, hydrophobic and anion-exchange column
chromatography was developed for purification ofFab protein containing no envelope fusion.
For the assembly of B6Fab, denatured inclusionbodies of B6LC and B6Fd were mixed in equimolarratio and renatured. The renatured material was dial-ysed to remove denaturants and B6Fab was purifiedfrom the dialysed material following column chro-matography procedures described below.
Ž .The pH of the dialysed material 2.7 l wasadjusted to ;5.0 and after filtration, it was loaded
Ž .on 50-ml SP-Sepharose column XK 50r20 . TheSP-Sepharose column was developed with a lineargradient of NaCl and protein eluted between 250-and 600-mM gradient of NaCl with elution profile
Ž .similar to that for B6Fabenv24 Section 3.4 . SDS–PAGE analysis of peak fractions showed that frac-tions 16–24 contained a 50-kDa band corresponding
Ž .to B6Fab protein Fig. 9A . In agglutination assay,none of the fractions showed agglutination beforeaddition of 9E10 antibody indicating the absence ofaggregates. After addition of 9E10, strong agglutina-
Ž .tion was observed in fractions 18–26 Fig. 9A ;these fractions were pooled. The concentration of
Table 1Activity of various B6Fab fusion proteinsIdentity of samples is based on data provided by the source of sample as described in Section 2.
a bSerum Source of sample Identity Plate agglutination Slide agglutinationsample B6Fab B6Fab B6 Fab B6Fab B6Fab B6 Fab
Ž .env13 env24 control env13 env24Ž . Ž .HIV-1 HIV-2
9E10, a monoclonal antibody which binds to decapeptide tag, cmyc; nd, not done.‘0’ indicates no agglutination with 1:100 dilution of sample.
a Numbers indicate the end point of agglutination recorded as maximum dilution of each sample that gave visible agglutination.b The agglutination was visibly recorded with ‘0’ as no agglutination and ‘4q ’ as the highest agglutination, as seen in Fig. 10.
( )A. Gupta et al.rJournal of Immunological Methods 256 2001 121–140136
NaCl in the pool was estimated to be approximatelyŽ .0.3 M calculated from the chromatogram and was
made to 2 M by adding NaCl. The pH was adjustedto 7.2 with 1 M Tris solution, and the sample wasloaded on 10-ml Toyobutyl column. B6Fab did notbind to the column and eluted in flow through during
Ž .loading data not shown . The flow through wastreated with iodoacetamide to block any freesulfhydryl groups. The excess iodoacetamide and saltwere then removed from the sample by desalting onSephadex G-25 column. The protein pool obtainedfrom G-25 column was loaded on a 20-ml Q-Sep-
Ž .harose column HR16r10 , and the column wasdeveloped with a linear gradient of NaCl. B6Fabeluted between 100- and 230-mM gradient of NaCl.SDS–PAGE analysis showed that fractions 26–31contained band corresponding to B6Fab with purity
Ž .of ;95% Fig. 9B . Faint bands of high molecularweight were also present. However, these bands didnot react with MAb 9E10 on Western blot, indicating
Žthat they were not multimers of B6Fab data not.shown . In agglutination assay, none of the fractions
showed agglutination before addition of 9E10 anti-body. After addition of 9E10, agglutination wasobserved in fractions 25–33. Fractions 25–32 werepooled. Forty-one milligrams of B6Fab protein wasobtained from 2.5 l of renatured material containing250 mg total protein, which corresponds to 16.5%
Žrecovery of total protein used for renaturation Fig..4 .
3.6. Characterization of purified B6 Fab andB6FabenÕ fusion proteins
Immunoreactivity of various purified proteins wasstudied using two normal, eight HIV-1 positive, twoHIV-2 positive and three HIV-1q2 positive serumsamples following a quantitative microtitre plate ag-glutination assay and a qualitative but simple slideagglutination assay as described in Section 2. Theidentity of the serum samples was based on severalimmunoassays, which are able to discriminate be-tween antibodies to HIV-1 and HIV-2. In microtitreplate assay, HIV-1 positive samples showed strongagglutination with B6Fabenv13-coated RBCs and no
Ž .reactivity with B6Fabenv24-coated RBCs Table 1 .Similarly, HIV-2 positive samples reacted stronglywith B6Fabenv24-coated RBCs and gave no reaction
with B6Fabenv13-coated RBCs. Samples positive forboth HIV-1 and HIV-2 showed agglutination withboth the proteins. The serum samples could be di-luted to very high degree indicating the sensitivity ofdetection. B6Fab-coated RBCs did not show aggluti-nation with any of the serum samples except forcontrol MAb 9E10 indicating the high specificity ofthe reagents.
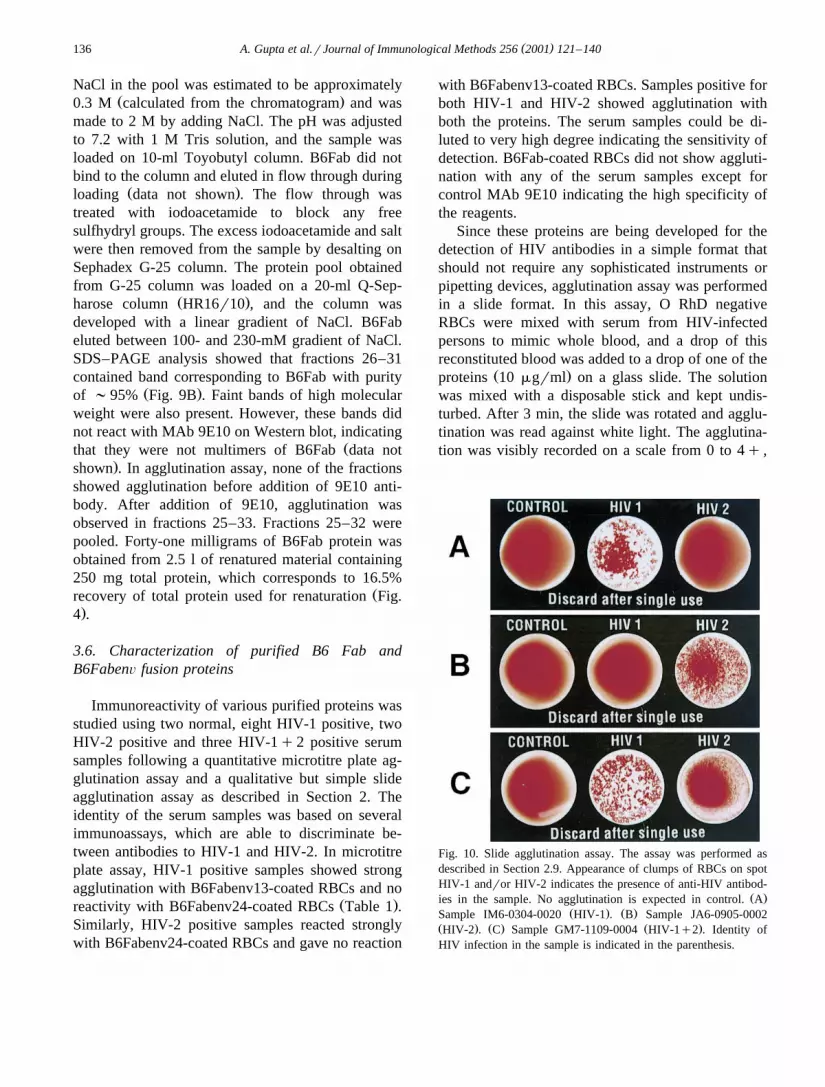
Since these proteins are being developed for thedetection of HIV antibodies in a simple format thatshould not require any sophisticated instruments orpipetting devices, agglutination assay was performedin a slide format. In this assay, O RhD negativeRBCs were mixed with serum from HIV-infectedpersons to mimic whole blood, and a drop of thisreconstituted blood was added to a drop of one of the
Ž .proteins 10 mgrml on a glass slide. The solutionwas mixed with a disposable stick and kept undis-turbed. After 3 min, the slide was rotated and agglu-tination was read against white light. The agglutina-tion was visibly recorded on a scale from 0 to 4q ,
Fig. 10. Slide agglutination assay. The assay was performed asdescribed in Section 2.9. Appearance of clumps of RBCs on spotHIV-1 andror HIV-2 indicates the presence of anti-HIV antibod-
Ž .ies in the sample. No agglutination is expected in control. AŽ . Ž .Sample IM6-0304-0020 HIV-1 . B Sample JA6-0905-0002
Ž . Ž . Ž .HIV-2 . C Sample GM7-1109-0004 HIV-1q2 . Identity ofHIV infection in the sample is indicated in the parenthesis.
( )A. Gupta et al.rJournal of Immunological Methods 256 2001 121–140 137
where ‘0’ indicates no agglutination and ‘4q ’ indi-Ž .cates the highest agglutination. B6Fab control did
Žnot show any agglutination of RBCs Table 1; Fig.. Ž .10 with any serum sample. B6Fabenv13 HIV-1
showed agglutination with HIV-1 and HIV-1q2Ž .positive samples Fig. 10A,C; Table 1 while
.1 . These results highlight the specificity of theproteins on their use in a simple slide assay format.
4. Discussion
The data presented here clearly demonstrates thatrecombinant fusion proteins can be designed andobtained in purified form in large quantities for usein haemagglutination-based detection of antibodiesto HIV in whole blood.
There were many important considerations whiledeveloping these recombinant proteins. Since theproteins comprise of chimeric molecules consistingof anti-RBC monovalent fragment fused to immun-odominant epitopes of HIV-1 and HIV-2, attentionwas given to designing of both the components.Selection of anti-human RBC antibody was based onthe premise that they should possess high affinity,bind to all types of human RBCs irrespective ofvariation in major and minor blood group antigens,and the molecule recognised by them should bepresent in high density on RBC surface. Similarly,the immunodominant antigens of HIV should beconserved and not be subtype-specific. A chimericprotein made from these two components shouldgive high yield using simple purification proceduresshould not aggregate, should be stable for long dura-tion at 4 8C and ambient temperatures, and should beable to detect antibodies against HIV in serum fromindividuals infected with any subtype of HIV, withhigh sensitivity and specificity. Finally, the format oftesting should be easy and result should be readablewithout requirement of any sophisticated instrument.The following discussion highlights the developmentof these molecules in line with the above considera-tions.
The present study employs Fab fragment of ananti-RBC MAb, B6, which was isolated in our labo-ratory by immunizing mice with a pool of O RhD
negative human RBCs, and following conventionalhybridoma technology. The evaluation of its bindingcharacteristics to human RBC was based on initialscreening of clones on 20 different blood samplesfollowed by final screening of a selected clone, B6,for reactivity to approximately 1000 randomly col-
Ž .lected blood samples our unpublished results . TheDNA sequence encoding Fd and LC was obtained by
ŽPCR-based techniques sequence of B6Fd and B6LC.will be published elsewhere . Selection of HIV epi-
topes was based on the alignment of available HIVgenome sequence of different subtypes to identifyconserved regions and information available regard-
Žing their immunodominant nature Gnann et al.,.1987a,b; Broliden et al., 1991 . A 31-amino acid
sequence in envelope glycoprotein gp41 of HIV-1Ž .amino acids 590–620 is highly conserved in mostsubtypes of HIV-1 and is known to be immunodomi-nant. Antibodies to this region have been reported to
Žbe present in most cases of HIV-1 infection Gnann.et al., 1987a,b; Broliden et al., 1991 . Similarly, a
31-amino acid sequence in HIV-2 envelope glyco-Ž .protein, gp36 amino acids 581–611 of gp36 , analo-
gous to gp41 sequence is highly conserved and hasbeen shown to be immunodominant. These regionsof gp41 and gp36 were therefore selected for makingB6Fdenv fusion proteins.
The method of preparing Fab and Fabenv fusionproteins was based on denaturation of FdrFdenv andLC, their mixing in equimolar ratio followed by invitro renaturation by dilution in a buffer creatingappropriate redox conditions. Since assembly of Fabrequires formation of many intra-molecular and in-ter-molecular disulfide bonds, after refolding, it wasnecessary to evolve a strategy to isolate monomeric,correctly folded Fab free from aggregates andunassembled Fd and LC chains. The B6Fabenv fu-sion proteins are likely to make more intra- andinter-molecular interactions due to hydrophobic na-ture of the envelope sequences and the two cysteineresidues present in env sequence, which make adisulfide bond. Keeping the above factors in consid-eration, a multistep protocol involving cation ex-change, hydrophobic, anion-exchange and gel filtra-tion chromatography was developed to isolate highlypure, monomeric Fab and Fabenv fusion proteins.
All the three proteins, B6Fab, B6Fabenv13 andB6Fabenv24, bound tightly to SP-Sepharose and
( )A. Gupta et al.rJournal of Immunological Methods 256 2001 121–140138
Ž .eluted with NaCl 400–500 mM NaCl . The pres-ence of monomeric protein was tested in an RBCagglutination assay where monomeric proteins showagglutination only after the addition of MAb 9E10which binds to a 10-amino acid tag, cmyc present atthe C-terminus of each protein. The aggregated pro-teins show agglutination of RBCs before the additionof MAb 9E10. For further processing, fractions con-taining monomeric protein were pooled. This step ofcation exchange chromatography primarily concen-trates the large volume of renatured protein and alsoseparates monomers from majority of free LC andaggregates. However, the eluate contains high mole-cular aggregates and other contaminants visible as a
Ž .smear on the gel Fig. 8B, Lane P1 . When thiseluate was loaded on hydrophobic Toyobutyl col-umn, both B6Fabenv13 and B6Fabenv24 bound at 2M NaCl concentration and the desired protein waseluted with lower salt concentration, 0.7 M NaCl forB6Fabenv13 and 0.6 M NaCl for B6Fabenv24. Thisstep removed remaining aggregates, which elutedfrom the column at zero salt concentration. SinceB6Fab does not carry the HIV-envelope sequences, itis less hydrophobic and, therefore, did not bind tothe hydrophobic matrix and came in the flow through.Aggregates and most of the contaminants eluted at
Ž .zero salt concentration data not shown , indicatingthat this step was essential to remove hydrophobicimpurities and other aggregates.
Fd and LC molecules contain cysteine residues,which make intrachain and interchain disulfide bondsto assemble functional Fab molecule. Besides, thesedisulfide-forming cysteine residues, B6Fabenv13 andB6Fabenv24, also contain cysteine residues in enve-lope sequence that make a disulfide bond. There isalways a possibility that any uncoupled cysteine canmake disulfide bonds with cysteine of anothermolecule or within the same improperly foldedmolecule that may lead to incorrect inter- or intra-molecular disulfide bond formation. Also, unassoci-ated LC and Fd contain free sulfhydryl groups thatcould interact with Fab thereby disrupting its struc-ture. To eliminate such interactions, free sulfhydrylgroups were blocked irreversibly with iodoacetamideduring purification. During optimisation of this pro-tocol, it was observed that B6Fabenv13 andB6Fabenv24 purified without iodoacetamide treat-ment aggregated upon storage, even though the final
purified protein prior to storage was monomeric.This indicated that this preparation had incorrectlyfolded molecules with reactive groups, which re-
Žsulted in aggregation of stored protein data not.shown . This aggregation was not observed after
pre-treatment with iodoacetamide.After iodoacetamide treatment, anion exchange
chromatography was performed. On Q-Sepharosecolumn, high molecular weight proteins were foundto elute at higher salt concentration in the case of
Ž .Fabenv proteins Fig. 7B; Lanes 34–37 . No suchaggregates were observed in Fab proteins, whichcontained no envelope sequence. Therefore, Fab pro-tein was used without any further purification, butthe Fabenv fusion proteins were further purified on agel-filtration column for complete removal of allhigh molecular weight species.
The envelope sequences influence the folding ofFd and its association with LC to form Fab. Also,these sequences, probably due to their hydrophobicnature and presence of cysteine residues, are moreprone to incorrect intermolecular interactions, result-ing in an increased proportion of molecules undergo-ing aggregation. This is also reflected in the yields
Žobtained for various proteins. While 66 mg ;16.5%.of the starting protein of B6Fab can be obtained
from a 4-l renaturation mix containing 400 mg ofdenatured LC and Fd isolated from 1-l culture each,
Ž .approximately 15–18 mg ;5% of B6Fabenv fu-sion proteins were obtained from same amount of
Ž .starting material Fig. 4 .The purified proteins were stable for 7–10 days at
37 8C. However, for use in agglutination assay, amixture of sugars and protease inhibitors was addedto these proteins to enhance their stability. Underthese conditions, the proteins were stable for more
Žthan 30 days at 37 8C and for 6 months at 4 8C our.unpublished results .
The original description of rapid whole bloodagglutination assay for HIV antibodies employedchemical conjugation procedures to produce amonomeric bifunctional molecule, which was mar-keted as Simpli-Red whole blood immunoassay sys-
Ž .tem AGEN Biomedical Limited, Australia . Later,efforts were made to produce these reagents in E.coli either as scFv or Fab fusion proteins. However,the yields of these proteins were only in hundreds ofmicrogram and were not enough for practical use.
( )A. Gupta et al.rJournal of Immunological Methods 256 2001 121–140 139
The present work breaks that barrier and yields tensof milligrams of fusion proteins on the laboratoryscale using simple column chromatography steps.The good yield is probably due to the intracellularover-expression followed by in vitro renaturationstrategy for Fab assembly while earlier workers de-pended upon a secretion-based strategy where hy-drophobic envelope sequences might have affectedthe secretion of the molecules. The in vitro renatura-tion protocol to assemble Fab molecules is veryefficient with reported recovery of up to 40%Ž .Buchner and Rudolph, 1991 . The recovery of re-folded and purified B6Fab was approximately 17%.
ŽThe lower recovery of envelope fusion protein ap-.proximately 5% could be due to the presence of
hydrophobic envelope sequences, as mentionedabove. The recovery would also depend upon thefolding characteristics of the Fab molecule, which inturn depend on the primary sequence of the antibody.In case of B6, we have identified a mutation in theframework region of B6V , which results in approx-H
imately two-fold improvement in the yield of B6FabŽ .protein our unpublished results . Further optimisa-
tion by varying the concentration of L-arginine andoxidised glutathione might also improve refoldingŽ .Buchner et al., 1992a . Moreover, it may be possi-ble to select a variant B6 with improved binding andfolding characteristics using phage display technol-
Ž .tive slide agglutination format of evaluation ofthese proteins shows that envelope fusion proteinsproduce strong agglutination reaction with highspecificity, and can detect very low levels of anti-body in human serum as revealed by high dilution ofserum sample, which could produce visible aggluti-nation in microtitre plate assay. The qualitative slideagglutination format is for use on whole blood withno need of dilution.
Detection of individuals infected with HIV is avital key in attempts to curtail the spread of thevirus, particularly by way of contaminated bloodproducts. Simple, rapid, inexpensive tests, which canbe performed without the need of any expensive andsophisticated instruments, are desired not only forthe developing and under-developed countries butalso for developed countries in clinical settings suchas emergency rooms where obtaining immediate re-
sults could be beneficial. The above mentioned datademonstrates the potential of the described fusionproteins for the development of sensitive and spe-cific whole blood-based agglutination assay for de-tection of antibodies to HIV-1 and HIV-2.
A HIV diagnostic kit for antibody detection maynot possess desired sensitivity if the reagents containonly one immunodominant region of HIV antigens,as the antibodies to this immunodominant regionmay not be present in all samples from differentstages of disease. Moreover, with the emergence ofnew subtypes having differences in sequence, identi-fication and inclusion of other immunodominant epi-topes becomes necessary. For this, fusion proteinswith other immunodominant epitopes of HIV havebeen prepared in our laboratory and are under evalu-ation as cocktail with the fusion proteins describedabove.
The technology described in this paper can alsobe used for development of similar molecules fordetecting various infections, such as hepatitis B,hepatitis C and syphilis, which are relevant in bloodtransfusion.
Acknowledgements
AG and SG were recipients of Research Fellow-ship from the Council of Scientific and IndustrialResearch, Government of India. We are thankful to
Ž .Dr. Shobha Sehgal PGI, Chandigarh, India and Dr.Ž .R. Lal CDC, Atlanta, USA for serum samples. Mr.
Rajiv Chawla is acknowledged for help in prepara-tion of manuscript. This work was funded by theDepartment of Biotechnology, Government of India.
References
Brinkmann, U., Pastan, I., 1994. Immunotoxins against cancer.Biochim. Biophys. Acta 1198, 27–45.
Broliden, P.A., Ruden, U., Ouattara, A.S., de The, G., Solver, E.,Trojnar, J., Wahren, B., 1991. Specific synthetic peptides fordetection of and discrimination between HIV-1 and HIV-2infection. J. Acquired Immune Defic. Syndr. 4, 952–958.
Buchner, J., Rudolph, R., 1991. Renaturation, purification andcharacterization of recombinant Fab-fragments produced inEscherichia coli. BiorTechnology 9, 158–162.
Buchner, J., Pastan, I., Brinkmann, U., 1992a. A method forincreasing the yield of properly folded recombinant fusion
( )A. Gupta et al.rJournal of Immunological Methods 256 2001 121–140140
proteins: single-chain immunotoxins from renaturation of bac-terial inclusion bodies. Anal. Biochem. 205, 263–270.
Buchner, J., Brinkmann, U., Pastan, I., 1992b. Renaturation of asingle-chain immunotoxin facilitated by chaperones and pro-tein disulfide isomerase. BiorTechnology 10, 682–685.
Chaudhary, V.K., Queen, C., Junghans, R.P., Waldmann, T.A.,FitzGerald, D.J., Pastan, I., 1989. A recombinant immunotoxinconsisting of two antibody variable domains fused to Pseu-domonas exotoxin. Nature 339, 394–397.
Chaudhary, V.K., Batra, J.K., Gallo, M.G., Willingham, M.C.,FitzGerald, D.J., Pastan, I., 1990a. A rapid method of cloningfunctional variable-region antibody genes in Escherichia colias single-chain immunotoxins. Proc. Natl. Acad. Sci. U. S. A.87, 1066–1070.
Chaudhary, V.K., Jinno, Y., Gallo, M.G., FitzGerald, D., Pastan,I., 1990b. Mutagenesis of Pseudomonas exotoxin in identifi-cation of sequences responsible for the animal toxicity. J. Biol.Chem. 265, 16306–16310.
Ž .Choe, M., Webber, K.O., Pastan, I., 1994. B3 Fab -PE38M: arecombinant immunotoxin in which a mutant form of Pseu-domonas exotoxin is fused to the Fab fragment of monoclonalantibody B3. Cancer Res. 54, 3460–3467.
Clavel, F., Guetard, D., Brun-Vezinet, F., Chamaret, S., Rey,M.A., Santos-Ferreira, M.O., Laurent, A.G., Dauguet, C.,Katlama, C., Rouzioux, C. et al., 1986. Isolation of a newhuman retrovirus from West African patients with AIDS.Science 233, 343–346.
Coia, G., Hudson, P.J., Lilley, G.G., 1996. Construction of recom-binant extended single-chain antibody peptide conjugates foruse in the diagnosis of HIV-1 and HIV-2. J. Immunol. Meth-ods 192, 13–23.
Dolezal, O., Coia, G., Guthrie, R.E., Lilley, G.G., Hudson, P.J.,1995. Escherichia coli expression of a bifunctional Fab-peptideepitope reagent for the rapid diagnosis of HIV-1 and HIV-2.Immunotechnology 1, 197–209.
Gnann Jr., J.W., Nelson, J.A., Oldstone, M.B., 1987a. Fine map-ping of an immunodominant domain in the transmembraneglycoprotein of human immunodeficiency virus. J. Virol. 61,2639–2641.
Gnann Jr., J.W., Schwimmbeck, P.L., Nelson, J.A., Truax, A.B.,Oldstone, M.B., 1987b. Diagnosis of AIDS by using a 12-amino acid peptide representing an immunodominant epitopeof the human immunodeficiency virus. J. Infect. Dis. 156,261–267.
Hoogenboom, H.R., 1997. Designing and optimising library selec-tion strategies for generating high-affinity antibodies. Tibtech15, 62–70.
Kane, J.F., 1995. Effects of rare codon clusters on high-levelexpression of heterologous proteins in Escherichia coli. Curr.Opin. Biotechnol. 6, 494–500.
Kemp, B.E., Rylatt, D.B., Bundesen, P.G., Doherty, R.R., McPhee,D.A., Stapleton, D., Cottis, L.E., Wilson, K., John, M.A.,Khan, J.M. et al., 1988. Autologous red cell agglutinationassay for HIV-1 antibodies: simplified test with whole blood.Science 241, 1352–1354.
Kunkel, T.A., 1985. Rapid and efficient site-specific mutagenesiswithout phenotypic selection. Proc. Natl. Acad. Sci. U. S. A.82, 488–492.
Lilley, G.G., Dolezal, O., Hillyard, C.J., Bernard, C., Hudson,P.J., 1994. Recombinant single-chain antibody peptide conju-gates expressed in Escherichia coli for the rapid diagnosis ofHIV. J. Immunol. Methods 171, 211–226.
Muller, B.H., Chevrier, D., Boulain, J.C., Guesdon, J.L., 1999.Recombinant single-chain Fv antibody fragment-alkaline phos-phatase conjugate for one-step immunodetection in molecularhybridization. J. Immunol. Methods 227, 177–185.
Orlandi, R., Gussow, D.H., Jones, P.T., Winter, G., 1989. Cloningimmunoglobulin variable domains for expression by the poly-merase chain reaction. Proc. Natl. Acad. Sci. U. S. A. 86,3833–3837.
Pastan, I., Chaudhary, V., FitzGerald, D.J., 1992. Recombinanttoxins as novel therapeutic agents. Annu. Rev. Biochem. 61,331–354.
Pluckthun, A., 1992. Mono and bivalent antibody fragments pro-duced in Escherichia coli: engineering, folding and antigenbinding. Immunol. Rev. 130, 151–188.
Ratner, L., Haseltine, W., Patarca, R., Livak, K.J., Starcich, B.,Josephs, S.F., Doran, E.R., Rafalski, J.A., Whitehorn, E.A.,Baumeister, K. et al., 1985. Complete nucleotide sequence ofthe AIDS virus, HTLV-III. Nature 313, 277–284.
Rylatt, D.B., Kemp, B.E., Bundesen, P.G., John, M.A., O’Reilly,E.J., Cottis, L.E., Miles, S.J., Khan, J.M., Dinh, D.P., Staple-ton, D., Hillyard, C.J., 1990. A rapid whole-blood immunoas-say system. Med. J. Aust. 152, 75–77.
Studier, F.W., Rosenberg, A.H., Dunn, J.J., Dubendorff, J.W.,1990. Use of T7 RNA polymerase to direct expression ofcloned genes. Methods Enzymol. 185, 60–89.
Williams, G.T., Neuberger, M.S., 1986. Production of antibody-tagged enzymes by myeloma cells: application to DNA poly-merase I Klenow fragment. Gene 43, 319–324.
Wilson, K.M., Gerometta, M., Rylatt, D.B., Bundesen, P.G.,McPhee, D.A., Hillyard, C.J., Kemp, B.E., 1991. Rapid wholeblood assay for HIV-1 seropositivity using an Fab-peptideconjugate. J. Immunol. Methods 138, 111–119.