Gene manipulation is an invaluable tool to investigate and understand the biology of an organism. Although this technology has been applied to both the human and rodent malarial parasites (RMP), Plasmodium berghei in particular offers a more robust system due to a higher and more ef fi cient transformation rate. Here, we describe a comprehensive transfection and selection protocol using P. berghei including a variant negative selection protocol administering 5- fl uorocytosine to the animals in drinking water. Additionally, we discuss and assess the latest advances in gene manipulation technologies developed in RMP to gain a better understanding of Plasmodium biology.
In the last decade, large-scale sequencing of Plasmodium genomes followed by global gene expression arrays, parallel RNA sequenc-ing, and proteomic studies have generated a wealth of information aiding a greater understanding of Plasmodium biology ( 1– 9 ) . Despite these advances, “hypotheticals” still encompass about 50% of the ~5,000 annotated genes. Consequently, genetic manipula-tion is necessary for assigning (and con fi rming) gene function in order to understand their role in various aspects of parasite biology, especially Plasmodium -speci fi c attributes. In this respect the rodent malaria parasites (RMP) have been particularly useful notably Plasmodium berghei where stable genetic manipulation was fi rst established ( 10 ) , although P. yoelii ( 11 ) and P. chabaudi ( 12 ) are also somewhat amenable to genetic manipulation. Apart from rela-tive ease of use, RMP offer a number of signi fi cant advantages:
1. Introduction
100 N. Philip et al.
their genomes are fundamentally homologous to those of the human parasites across the majority of the genome excluding the subtelomeric clusters of (typically) multigene families. Approximately 80% of the annotated genes in the genome, which typically con-tains ~5,500 genes, can be considered to be syntenic ( 13 ) , permit-ting gene function to be assessed in stages that are dif fi cult or impossible to access in human Plasmodium . They allow experi-mental procedures that cannot be considered with human subjects and offer experimental access to the entire life cycle. Different viru-lence patterns and pathologies might also be studied with RMP models. A range of reporter RMP are now available that, for example, express fl uorescent proteins either constitutively or stage speci fi cally and permit imaging and puri fi cation processes for further pheno-typic or biochemical analysis ( 14– 16 ) . Furthermore, both host and vector may now be genetically manipulated, allowing a consider-ation of all aspects of parasite interactions with those it infects. RMP are limited in a number of respects, notably the inability of the blood stages to be cultivated long term in vitro, restricting study of invasion of and egress from the erythrocyte, and the rela-tive lack of selectable markers with which to select manipulated parasites.
A further general consideration is that Plasmodium parasites are haploid organisms, which is both an experimental advantage and disadvantage when considering reverse genetics. The major advantage of haploidy is that only a single allele needs to be deleted/modi fi ed, which is a substantial bene fi t given the paucity of positive selectable markers available. However, the problem that arises from haploidy is that genes that are essential to the vegeta-tively growing phase of the parasite in blood, which are used to propagate schizonts for transfection, cannot be studied since dele-tion or disabling through modi fi cation would be a lethal event and such transfected parasites would perish prior to selection. As noted above, asexual blood-stage growth of Plasmodium includes a series of events (parasite egress, erythrocyte invasion, and cytoadherence) that are of intense interest from therapeutic considerations, and the inability to study these processes through gene deletions medi-ated by conventional reverse genetics is a signi fi cant handicap requiring other approaches, some of which are described below.
Nonetheless the haploid genome of the asexual blood stage of the parasite is amenable for genetic manipulation and is therefore used for transfection. However, introduction of exogenous DNA into the intracellular Plasmodium parasite is problematic because traversal of four membranes is required to reach the nucleus. In P. berghei , use of puri fi ed schizonts/merozoites partly avoids this problem, which when combined with advances in transfection technologies due to development of Amaxa nucleofection has resulted in much higher transfection frequencies (frequency of 10 −3 –10 −4 compared to 10 −6 –10 −9 using past methodology) than
1017 Transfection of Rodent Malaria Parasites
typically achieved with the human parasite, P. falciparum ( 17 ) . In addition to two detailed transfection and selection protocols, we discuss various gene manipulation strategies the technique can be applied to.
Gene inactivation or modi fi cation is the most reliable and useful approach to examine gene function in parasite biology. Given that Plasmodium exclusively incorporates DNA by homologous recom-bination, both single and double crossover strategies can be exploited for gene manipulation ( see Fig. 1 ). Insertion plasmids uti-lizing single crossover require minimally ~300 bp of homology on either side of the point of recombination (site of linearization in the plasmid) to integrate into the targeted genomic locus. Larger
1.1. Reverse Genetics
+ve -ve
-ve+ve
a b
c d
Fig. 1. Gene manipulation strategies: gene inactivation and modi fi cation can be achieved by both double and single cross-over homologous recombination. ( a ) Gene knockouts can be generated using plasmids containing a positive selection marker (SM) fl anked by arms homologous ( dashed lines ) to regions upstream and downstream of the gene of interest. The construct is linearized to generate a fragment with the SM and regions of homology; when transfected into the parasite, it undergoes double crossover leading to allelic exchange of the wild-type copy of the gene with the SM resulting in a gene knockout. ( b ) Gene disruption using insertion plasmids containing an internal fragment of the gene occurs by a single crossover event. The plasmid is linearized (gap) within the homology region, which recombines into the parasite genome to generate two truncated copies of the target gene. This process can be reversed with negative selection using 5-FC when the yFCU element is fused to the positive selectable marker. ( c ) Gene modi fi cation including N- and C-terminus tagging can be accomplished by constructs designed for double crossover events. The plasmid would contain an upstream homology region, followed by the SM, gene promoter, 5 ¢ UTR, the modi fi ed targeted gene, and fi nally the downstream homology region. ( d ) Insertion plasmids using single crossover recombination can be used to modify genes and introduce C-terminal tags. The construct contains a partial C-terminus of the modi fi ed gene of interest followed by a 3 ¢ UTR. The linearized plasmid recombines at the targeted locus to generate a promoter-less truncated gene and a full-length functional modi fi ed gene of interest. This process can be reversed with negative selection using 5-FC when the yFCU element is fused to the positive selectable marker.
102 N. Philip et al.
regions of homology tend to make the recombination event more ef fi cient/frequent. Such plasmids can be used for introducing subtle mutations and also for gene inactivation. Alternatively, linear replacement constructs containing a drug-selectable marker bor-dered by two arms of homology, which integrate into the genome by double homologous recombination, can be employed for gene modi fi cation and deletion. However, despite signi fi cant improve-ments in the technology ( 18 ) systematic functional genomic analy-sis is hampered by the relatively low ef fi ciency of homologous recombination in the parasite and dif fi culty of producing targeting plasmids containing A/T-rich Plasmodium DNA in Escherichia coli . A recent development from the Billker group attempted to address these issues ( see Chapter 8 ) employing a recombineering strategy that uses bacteriophage N15-derived library to stably maintain frag-ments of P. berghei gDNA up to 16 kb in size. Phage clones are manipulated to produce transfection-competent vectors, which through the use of long homology arms achieve over tenfold increased transfection ef fi ciency compared to conventional meth-ods. The approach was further scaled-up to generate P. berghei gene modi fi cation vectors in a 96-well format achieving 88% success (86/96 constructs). A genome-wide library of genetic modi fi cation vectors capable of gene deletion and tagging now makes it possible to perform large-scale functional studies of Plasmodium genes.
It was noted above that reverse genetics in P. berghei is hindered by the low number of selectable markers due to toxicity issues in the rodent host. Currently, dihydrofolate reductase-thymidylate syn-thase ( DHFR-TS ), an enzyme involved in folate metabolism, is com-monly used as a drug selectable marker. The P. berghei (Pb) and Toxoplasma gondii (Tg) DHFR-TS confer resistance to pyri-meth-amine, a drug now administered in drinking water ( 19 ) . The human DHFR gene, in addition to pyrimethamine, also confers resistance to WR99210 ( 20 ) . Combination of the two markers is possible only if Pb/Tg DHFR-TS ( fi rst) and human (second) DHFR are intro-duced sequentially due to resistance of human DHFR to both pyrimethamine and WR99210. The scarcity of selectable markers can be somewhat mitigated by recycling them using a positive–neg-ative selection strategy. A positive–negative selectable marker system can be applied for (1) complementing disrupted genes (generated using positive selection), followed by excision of the drug selection cassette by homologous recombination (negative selection); (2) counter-selecting episomal transformation and single crossover events to increase recovery of double crossover recombinants; (3) developing a hit-and-run strategy for generating mutations without introducing exogenous sequences into the targeted locus; and (4) recycling both the positive and negative selection markers to gener-ate additional sequential mutations (e.g., double, triple). A negative selection system based on a yeast bifunctional enzyme (yFCU)
consisting of cytosine deaminase and uracil phosphoribosyl trans-ferase has been developed for both P. falciparum and P. berghei ( 21, 22 ) . Negative selection pressure is applied by 5- fl uorocytosine (5-FC), which is converted by yFCU into a highly toxic compound 5- fl uorouracil that kills yFCU-expressing parasites within 48 h. In P. berghei , pure populations of negatively selected parasites are recov-ered within 5–6 days post 5-FC treatment. This system was success-fully utilized to restore gene function by homologous recombination and excision of the selectable marker at the disrupted gene locus. The strategy was further developed to recycle the positive selectable marker by fl anking the selection cassette by two 0.5-kb PbDHFR-T S 3 ¢ regions that under 5-FC pressure recombine and excise the select-able marker. Additionally, in P. falciparum the method was adapted to use site-speci fi c recombinases such as the Flp/ FRT system ( 23 ) . This chapter also describes a modi fi ed detailed protocol for negative selection using 5-FC in drinking water, which offers signi fi cant improvements in labor, drug use, animal welfare, reduction in ani-mal use, and reproducibility.
A common problem noted above is that genes that are essential in blood-stage asexual parasites cannot be deleted due to their essen-tial nature, and in the absence of robust conditional knockout (ko) strategies this remains an issue. However, the function of these essential genes can be examined in later stages of the parasite life cycle through an approach where the wild-type promoter, which might be active in multiple stages of the life cycle, is swapped for one that is active only in intraerythrocytic asexual stages. In this manner recombinant parasites that have undergone this type of promoter swap for an essential gene are viable in the asexual blood stages where their selection and cloning take place, yet gene expres-sion terminates once the parasites commit to sexual development and thereafter. Therefore, the function of the promoter-swapped gene can be established at its fi rst point of phenotype appearance after (or during) gametocyte development. This approach has been taken to study the FACT (facilitates chromatin transcription) pro-tein—four promoter swap constructs using four different promot-ers were introduced into the parasite and promoters from two genes (PBANKA_091420 and PBANKA_140060) gave viable parasites and were used to demonstrate the critical role of FACT in fertile male gamete formation ( 24 ) .
The tools mentioned earlier use either homologous recombination or insertion of foreign DNA into the genome to generate integra-tive transgenic lines. Alternatively, extrachromosomal or episomal vectors, which do not physically disrupt the genome, can be used for ectopic expression and analyzing gene function. Episomal transfection has been applied towards understanding transcrip-tional regulation, examining protein localization, performing
1.3. Promoter Swap
1.4. Episomal Transfection
104 N. Philip et al.
genetic rescue experiments, as well as dissecting molecular pathways. Although episomal vectors containing bacterial derived origin of replication do multiply in the parasite, they have a major drawback of not segregating evenly into daughter cells during cell division ( 25, 26 ) . Additionally, episomes are lost if not kept under constant drug pressure, which is possible only during the parasite asexual stages. Moreover, under selective drug pressure plasmids are main-tained in high copy numbers, which might result in elevated gene expression causing non-physiological effects.
Dif fi culties introduced by experimental artifacts and nonsegre-gation of plasmids can be overcome by designing vectors based on chromosomal elements derived from the parasite genome. The dis-covery of yeast centromeres, origins of replication, and telomeres led to the construction of the fi rst eukaryotic arti fi cial chromo-some ( 27, 28 ) . Arti fi cial chromosomes have the potential to carry large fragments of genomic DNA, persist in the nucleus without selection pressure, maintain low copy numbers, and segregate evenly during mitosis and meiosis. The recent identi fi cation of the P. berghei centromere and development of a Plasmodium arti fi cial chromosome (PAC) offers a signi fi cant advance in extrachromo-somal transfection technology ( 29 ) . Iwanaga et al. initially con-structed a P. berghei centromere-containing circular plasmid capable of ef fi ciently replicating and maintaining itself during cell division in the asexual blood stages, and zygote/ookinete and oocyst stages in the mosquito. The plasmid was further developed by addition of capped telomeres to generate a linear PAC (L-PAC). The L-PAC is stably maintained throughout the life cycle of the parasite in low copy number (2.1 per cell) and persists in 70% of the parasites after a complete cycle including the mosquito and liver stages. These advantages make the vector an excellent tool to (a) precisely exam-ine transcriptional regulation during any parasite life stage, (b) complement mutant phenotypes without the use of additional selectable markers, and (c) investigate Plasmodium chromosome function. Examining chromosome function, especially telomere biology, is key to understanding regulation of Plasmodium multi-gene families. These gene families are enriched in sub-telomeric regions and encode variable surface antigens (e.g. , var ), which are targeted to the erythrocytic surface. Expression of var genes is mutually exclusive with only a single member active at a given time ( 30, 31 ) . Additionally, switching of expression of the different var genes is used by the parasite to evade host immunity and promote parasite survival. Mechanisms controlling antigenic variation include histone modi fi cation, exchange of canonical histones with variant forms, and subnuclear compartmentalization ( 32– 34 ) . Given that L-PAC functions as natural parasite chromosome, the vector originally developed in P. berghei provides a powerful tool to better understand this complex mechanism of gene regulation in Plasmodium.
1057 Transfection of Rodent Malaria Parasites
The problems of studying essential genes in haploid organisms through reverse genetics have been outlined already. Therefore conditional mutagenesis, usually the interruption of gene/protein function in an inducible manner, would appear to be the ideal solution to these dif fi culties. To date the methods that have been typically employed for induction of gene inactivation have not been applied to RMP although they have been introduced with moder-ate success into the human parasite P. falciparum . Tetracycline-inducible expression of transgenes ( 35 ) and selective destruction of protein fusions to the ligand-regulatable FKBP protein destabiliza-tion domain (ddFKBP—regulated by shield) ( 36 ) or to a degradation domain derived from E. coli dihydrofolate reductase (regulated by trimethoprim) ( 37 ) have all been achieved in cultured P. falci-parum but not yet in RMP, in part due to the dif fi culties of long-term in vitro cultivation of blood stages.
However, transmission of RMP through Anopheles mosquitoes is well established as is the ability to create novel genetic crosses through the in vitro fertilization of gametes derived from distinct lines of P. berghei and their subsequent passage through mosqui-toes and recovery upon bite back on a mouse. This has been clev-erly exploited to develop FLP recombinase-mediated stage-speci fi c excision of blood stage-required genes in the mosquito with sub-sequent analysis downstream in the infected mouse. In principle, genes of interest (GoI) are fl anked fi rst by the FRT repeat sequences (substrates for the Saccharomyces cerevisiae FLP recombinase) in a fashion that upon excision brings GFP under control of the promoter of the GoI, making the parasites of interest identi fi able. In early forms of this approach the feasibility was established using a strategy where the recombinase was expressed in a stage-speci fi c manner (the late oocyst- and sporozoite-speci fi c promoter TRAP ) in an independent line, and gamete crossing performed to create a subpopulation that contained the correct combination of targeted locus and FLP recombinase ( 38 ) . Latterly and building on this proof of principle, P. berghei lines have been developed that express a form of FLP recombinase (FlpL) that is maximally active at the ambient temperature of the mosquito habitat. Furthermore FLP recombinase is placed under either a midgut or a salivary gland sporozoite-speci fi c promoter and GoI are FRT - fl anked in the same line ( 39 ) . The ef fi ciency has been shown to be as high as 95%, obvi-ating the need for the use of GFP expression to detect the experi-mental parasite subpopulation and allowing assignation of function downstream in liver stages. The essential and unknown role of a gene involved in merozoite formation in hepatocytes ( MSP1 ) has been demonstrated in this fashion, expanding our knowledge of the function of this protein that was previously shown to be essential for merozoite recognition of erythrocytes. It should be noted that for all gene disruption methods, the limitation remains that if the GoI is essential at any point in the life cycle then its role in
1.5. Conditional Mutagenesis: Roles of Essential Genes?
106 N. Philip et al.
Plasmodium life cycle events downstream of the fi rst critical point of function cannot be studied. Analysis of genes essential to blood stages can be facilitated by this FLP-mediated stage-speci fi c approach to excision of GoI. However, some genes that are essen-tial in blood stages are also critical at other stages as they play a conserved role in the invasive organelles shared by merozoite and sporozoite, for example AMA1 .
Although the recent developments described above and else-where have made it feasible to perform larger scale gene-by-gene disruption analysis, the large number of hypotheticals, some of which have already been shown to be responsible for parasite-speci fi c processes ( 40 ) , provides a challenge to making educated predictions of gene function. A complementary unbiased phenotype-driven approach might provide insights into critical aspects of parasite biology.
Forward genetics has the potential to be a powerful medium/high-throughput approach to identify genes responsible for observ-able alterations in phenotype. The forward genetics approach, unlike reverse genetics, offers a valuable unbiased tool that is able to identify multiple genes as well as noncoding genomic regions responsible for a given phenotype. Variations in phenotype can be achieved either by random (chemical, radiation, and insertional) or site-speci fi c mutagenesis. The fi rst study using transposable ele-ments in Plasmodium followed a random mutagenesis approach and employed the Class II transposable element mariner, which moves in the genome using a cut and paste mechanism, in the pres-ence of a transposase enzyme. Unfortunately in P. falciparum mar-iner proved to be quite inef fi cient and its use has not been reported in other species of Plasmodium . Other transposable elements including P-element, Tc1, Tol2, and piggyBac have been exten-sively used to study gene function in model organisms ( 41– 45 ) . Site-speci fi c insertional mutagenesis employing Tn 5 shuttle muta-genesis has also been attempted ( 46 ) . However, this approach involved cloning every targeted gene into E. coli rendering it a long and cumbersome procedure.
Recently, the piggyBac transposable element, which inserts at TTAA sites in the genome, was adapted for both human and RMP ( 47, 48 ) . The piggyBac system was fi rst developed in P. falciparum and applied to a phenotypic screen to identify genes required for the intraerythrocytic development cycle of the parasite ( 49 ) . The system was adapted to a dual-plasmid system by separating the gene encoding the transposase enzyme and the integrating asym-metric repeats that normally fl ank the transposase. The asymmetric repeats fl anked a positive selectable marker, which allowed selec-tion for stably transformed parasites when integrated through the activity of the transposase, whose gene was transiently introduced
1.6. Forward Genetics
1077 Transfection of Rodent Malaria Parasites
on the co-transfected helper plasmid. Eighty-one independent transfections yielded about 180 unique insertion sites (all charac-terized by the AATT recognition site) randomly distributed across the 14 chromosomes but with a preferential bias towards 5 ¢ UTR regions. Although the strategy has a high ef fi ciency of random insertion, P. falciparum genome manipulation is however plagued by very low transfection ef fi ciency, making such mutagenesis unlikely to achieve saturation levels.
Taking advantage of the higher transfection ef fi ciency in P. ber-ghei , Fonager et al. further developed the piggyBac system in the RMP ( 48 ) . Compared to P. falciparum , the piggyBac system applied to P. berghei system yielded 16–18-fold more insertion sites at AATT repeats in the genome. Additionally no 5 ¢ UTR bias was observed and insertions were randomly spread throughout the genome. Stable integration of the transposase gene into the genome was also tested for persistent expression of the enzyme, anticipat-ing that this would promote further transposon mobility. This mobility can potentially be exploited for reversion of the mutant phenotype to establish a causal relationship between the mutation and the observed phenotype. Remobilization can also be used for transposon excision and reinsertion into other genomic loci, increasing the potential coverage of the transposon from a single transfection experiment. A further advance of the system was to identify promoters active during asexual stages. A “promoter trap” plasmid construct was designed consisting of a piggyBac 5 ¢ element followed by a promoter-less GFP cassette, where GFP expression was observed only when the construct inserted into transcription units downstream of active promoters. This approach can be fur-ther employed to identify promoters active during other develop-mental stages.
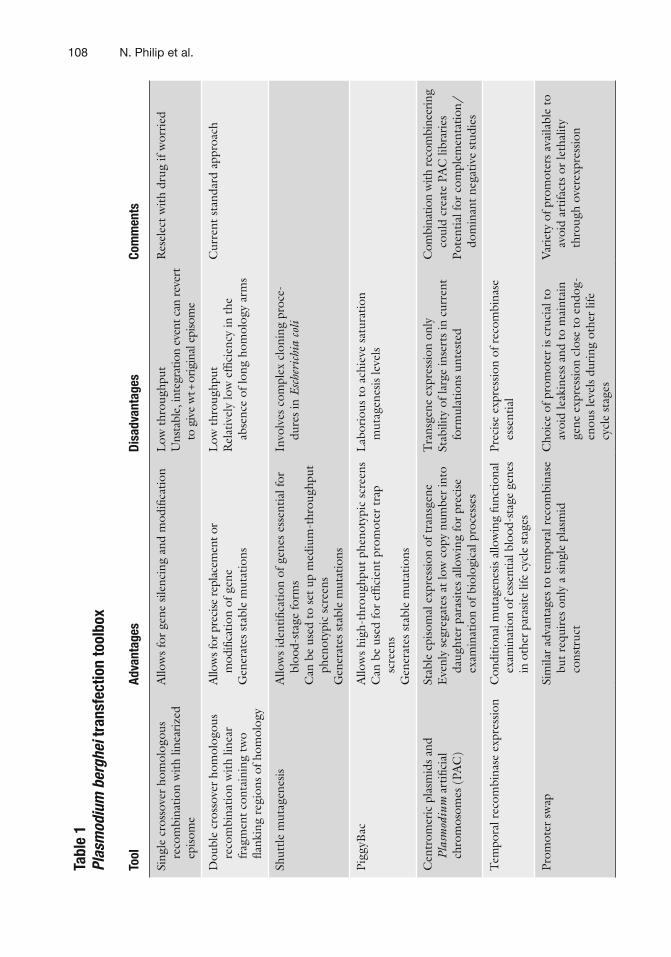
In the last decade the malaria research community has achieved signi fi cant technological advances in understanding gene function in the malarial parasite. Altogether, improved transfection fre-quency, increased site-speci fi c recombination ef fi ciency, conditional mutagenesis, transposon mutagenesis, and centromeric plasmids have provided the means to considerably facilitate examination of parasite biology. Table 1 summarizes the available gene manipula-tion tools and their uses. In spite of these technical advances, the Plasmodium molecular genetic toolbox misses some key pieces. Development of robust conditional knockout/inducible strategies is critical to understand functions of genes essential during blood-stage growth. The very essentiality of these genes makes them attractive targets for vaccine and drug development. Further devel-opment of strategies to regulate gene expression both post-tran-scriptionally and post-translationally would provide valuable tools to understand the druggable parasite genome and develop new intervention tactics.
108 N. Philip et al.
Tabl
e 1
Plas
mod
ium
ber
ghei
tran
sfec
tion
tool
box
Tool
Ad
vant
ages
Di
sadv
anta
ges
Com
men
ts
Sing
le c
ross
over
hom
olog
ous
reco
mbi
natio
n w
ith li
near
ized
ep
isom
e
Allo
ws
for
gene
sile
ncin
g an
d m
odi fi
catio
n L
ow t
hrou
ghpu
t U
nsta
ble,
inte
grat
ion
even
t can
reve
rt
to g
ive
wt +
orig
inal
epi
som
e
Res
elec
t w
ith d
rug
if w
orri
ed
Dou
ble
cros
sove
r ho
mol
ogou
s re
com
bina
tion
with
line
ar
frag
men
t co
ntai
ning
tw
o fl a
nkin
g re
gion
s of
hom
olog
y
Allo
ws f
or p
reci
se re
plac
emen
t or
mod
i fi ca
tion
of g
ene
Gen
erat
es s
tabl
e m
utat
ions
Low
thr
ough
put
Rel
ativ
ely
low
ef fi
cien
cy in
the
ab
senc
e of
long
hom
olog
y ar
ms
Cur
rent
sta
ndar
d ap
proa
ch
Shut
tle m
utag
enes
is
Allo
ws
iden
ti fi ca
tion
of g
enes
ess
entia
l for
bl
ood-
stag
e fo
rms
Can
be
used
to
set
up m
ediu
m-t
hrou
ghpu
t ph
enot
ypic
scr
eens
G
ener
ates
sta
ble
mut
atio
ns
Invo
lves
com
plex
clo
ning
pro
ce-
dure
s in
Esc
heri
chia
col
i
Pigg
yBac
A
llow
s hi
gh-t
hrou
ghpu
t ph
enot
ypic
scr
eens
C
an b
e us
ed fo
r ef
fi cie
nt p
rom
oter
tra
p sc
reen
s G
ener
ates
sta
ble
mut
atio
ns
Lab
orio
us t
o ac
hiev
e sa
tura
tion
mut
agen
esis
leve
ls
Cen
trom
eric
pla
smid
s an
d Pl
asm
odiu
m a
rti fi
cial
ch
rom
osom
es (
PAC
)
Stab
le e
piso
mal
exp
ress
ion
of t
rans
gene
E
venl
y se
greg
ates
at
low
cop
y nu
mbe
r in
to
daug
hter
par
asite
s al
low
ing
for
prec
ise
exam
inat
ion
of b
iolo
gica
l pro
cess
es
Tra
nsge
ne e
xpre
ssio
n on
ly
Stab
ility
of l
arge
inse
rts
in c
urre
nt
form
ulat
ions
unt
este
d
Com
bina
tion
with
reco
mbi
neer
ing
coul
d cr
eate
PA
C li
brar
ies
Pote
ntia
l for
com
plem
enta
tion/
dom
inan
t ne
gativ
e st
udie
s
Tem
pora
l rec
ombi
nase
exp
ress
ion
Con
ditio
nal m
utag
enes
is al
low
ing
func
tiona
l ex
amin
atio
n of
ess
entia
l blo
od-s
tage
gen
es
in o
ther
par
asite
life
cyc
le s
tage
s
Prec
ise
expr
essi
on o
f rec
ombi
nase
es
sent
ial
Prom
oter
sw
ap
Sim
ilar
adva
ntag
es t
o te
mpo
ral r
ecom
bina
se
but
requ
ires
onl
y a
sing
le p
lasm
id
cons
truc
t
Cho
ice
of p
rom
oter
is c
ruci
al t
o av
oid
leak
ines
s an
d to
mai
ntai
n ge
ne e
xpre
ssio
n cl
ose
to e
ndog
-en
ous
leve
ls d
urin
g ot
her
life
cycl
e st
ages
Var
iety
of p
rom
oter
s av
aila
ble
to
avoi
d ar
tifac
ts o
r le
thal
ity
thro
ugh
over
expr
essi
on
1097 Transfection of Rodent Malaria Parasites
All experimental procedures using animals should conform to the relevant local, national, and European legislations. Procedures must have been given ethical clearance. Consult your local animal welfare of fi cer for more information.
1. Phenylhydrazine hydrochloride (Sigma-Aldrich): 12.5 mg/ml phenylhydrazine in 0.9% NaCl solution ( see Note 1 ).
2. 25 or 26G needle. 3. 1-ml Syringe. 4. Theiler’s original (TO) or NIH Swiss outbred mouse, female,
25 g, aged 6 weeks.
1. Microscope slides with frosted end. 2. Methanol. 3. Giemsa stain. 4. Sörensen staining buffer: KH 2 PO 4 2.541 g, Na 2 HPO 4 ·2 H 2
0.5507 g per 5 L of dH 2 O, pH to 7.2 with NaOH ( see Note 2 ).
5. Heparin: 200 U/ml prepared in PBS, fi ltered with a 0.22- m m fi lter device.
6. Fetal bovine serum (FBS) (heat inactivated). 7. Complete culture medium RPMI1640 with l -glutamine and
25 mM HEPES without NaHCO 3 : weigh 0.85 g NaHCO 3 , add 5 U/ml penicillin and 5 m g/ml streptomycin, pH to 7.3, fi lter with a 0.22- m m fi lter device, and supplement with 25% fetal calf serum.
8. Gas mixture: 5% CO 2 , 5% O 2 , and 90% N 2 . 9. Cell culture 150-cm 2 plug seal fl ask. 10. Orbital shaking incubator. 11. Light microscope with 100× lens plus immersion oil.
1. Nycodenz stock solution: 138 g of Nycodenz powder (Axis-shield) dissolved in 500 ml buffered medium, autoclaved at 120°C for 20 min ( see Note 3 ).
2. Amaxa Nucleofector II Device (Lonza). 3. Puri fi ed schizonts obtained from Subheading 3.3 . 4. Culture media from the top of Nycodenz centrifugation gra-
dient tube. 5. 5–10 m g of construct in 10 m l of dH 2 O or TE.
1. 0.3-ml Insulin syringe with 30 G needle (BD biosciences). 2. Hot box or infrared heat lamp. 3. TO or NIH Swiss outbred mouse, female, 25 g, aged 6 weeks,
one per transfection reaction.
1. Pyrimethamine (Sigma-Aldrich): stock (100×) is 7 mg/ml dissolved in DMSO ( see Note 4 ).
2. WR99210 in DMSO (gift from Jacobus Pharmaceuticals, Princeton, NJ. Drugs).
3. Darkened or opaque drinking bottles.
1. Microscope slides with frosted end. 2. Methanol. 3. Giemsa stain. 4. Sörensen staining buffer, as above in Subheading 2.2 , item 4 .
1. Cryotubes. 2. PBS. 3. Heparin: 200 U/ml prepared in PBS, fi lter with a 0.22- m m
fi lter device. 4. Plasmodipur fi lter (EuroProxima). 5. Zeba spin column, empty 10-ml (Fisher) ( see Note 5 ). 6. Whatman CF-11 cellulose powder (Fisher) ( see Note 5 ). 7. Erythrocyte lysis buffer: 10× stock, 1.5 M NH 4 Cl, 0.1 M
KHCO 3 , 0.01 M EDTA. This should be prepared to 1× working stock in prechilled dH 2 O.
8. 30% glycerol/PBS (v/v) with heparin 10 U/ml.
1. 50-Well disposable plug molds (BioRad). 2. TNE buffer: 50 mM Tris, pH 8.0, 100 mM NaCl, 5 mM EDTA. 3. Low-melting agarose (Sigma-Aldrich): 15 mg/ml TNE buffer. 4. Proteinase K: 20 mg/ml. 5. SE buffer: 0.5 M EDTA, 1% sarcosyl lauroyl sulphate, pH 8.0.
2.4. Electroporation of Schizonts
2.5. Intravenous Injection of Transfected Schizonts
2.6. Selection of Transfected Parasites
2.7. Monitoring the Growth of Transfected Parasites
2.8. Collection of Parasites for Cryopreservation and for Isolation of DNA for Analysis
2.9. Preparation of Agarose Blocks Containing Chromosomes
1. Pyrimethamine (Sigma-Aldrich): stock 100× is 7 mg/ml dis-solved in DMSO ( see Note 4 ).
1. 5-FC (Sigma-Aldrich): 1 mg/ml in water ( see Note 6 ).
A mouse is infected with P. berghei to serve as a source of blood-stage parasites for the culture and puri fi cation of schizonts. The schizont is the developmental stage that is used for introduction of foreign DNA by electroporation. Phenylhydrazine treatment results in the induction of reticulocytosis.
1. A TO outbred mouse of 25 g is intraperitoneally (i.p.) injected on day 0, typically a Wednesday, with 0.1 ml of 12.5 mg/ml phenylhydrazine hydrochloride ( see Note 1 ).
2. On day 2 the phenylhydrazine-treated mouse is i.p. infected with 0.2 ml of a thawed suspension of a cryopreserved stabilate ( see Note 7 ).
3. On day 4 (Monday) or 5 (Tuesday), Giemsa-stained thin blood fi lms are prepared from a droplet of tail blood to detect the level of parasitemia, generally between 0.5% and 10% at this stage ( see Note 2 ).
Blood stages of P. berghei are cultured in RPMI1640 medium (pH 7.3) containing FBS. In general, parasites are maintained in vitro for only one developmental cycle: ring forms/young trophozoites are allowed to develop into mature schizonts during a period of 16–23 h ( see Note 8 ).
2.10. Cloning of Transfected Parasites
2.11. In Vivo Maintenance and Production of Transgenic Parasites Containing Episomes
2.12. Negative Selection Using 5-Fluorocytosine In Vivo Provided in the Drinking Water
3. Methods
3.1. Phenylhydrazine Treatment of Schizont Donor (Day 0)
3.2. In Vitro Culture of Schizonts (Day 5)
112 N. Philip et al.
1. Collect 1–2 ml of infected blood by cardiac puncture using a 2-ml syringe from a mouse with a parasitemia of 1–10% ( see Note 9 ). Usually the blood is collected between 10 a.m. and 4 p.m. on day 4 or 5.
2. Transfer the blood to 40 ml complete culture medium per ml of blood collected, doubling this volume if infection is above 3%. It is important, however, to ensure that there are no multiply infected erythrocytes if parasitemia is above 3% ( see Note 10 ).
3. Cultures can be maintained in closed, plastic culture fl asks of 150 cm 2 that have been gassed at the start of the culture period using the gas mixture 5% CO 2 , 5% O 2 , and 90% N 2 . There should be no more than 80 ml of culture per fl ask; therefore any volume greater than this should be split equally between multiple fl asks ( see Note 11 ).
4. Put the fl ask on a shaker in a 37°C water bath, incubator, or climate room.
5. Switch on the orbital shaker after 5 min at a minimal speed (20–30 rpm) to keep the cells in suspension.
6. Leave the parasites in culture at 37°C until the next morning 9.00 a.m. ( see Note 12 ).
7. Take 0.5 ml from the culture in an Eppendorf tube at 9.00 a.m. to determine the “quality” of the parasites.
8. Pellet the cells (maximum speed, 5 s) and discard the supernatant. 9. Make a thin blood smear of the cells and Giemsa-stain the slide. 10. Examine schizont development using a light microscope
at a 1,000× magni fi cation (100× objective, immersion oil) ( see Note 13 ).
11. Start the puri fi cation of the schizonts.
Prior to electroporation, the schizonts (1–3% of the total cell pop-ulation) are separated from the uninfected erythrocytes that are present in the culture.
1. Prepare a 55% Nycodenz/PBS solution (v/v). In general a total volume of 30 ml, with 16.5 ml Nycodenz stock solution and 13.5 ml PBS, is required for a culture suspension of 80 ml ( see Note 14 ).
2. Transfer the culture suspension containing the schizonts to two 50-ml tubes (35–40 ml per tube).
3. Using a 10-ml pipette, gently add 10 ml of the Nycodenz solu-tion in each tube under the culture suspension so that a sharp contrasting division is visible between the two suspensions ( see Note 15 ).
4. Centrifuge for 20–30 min 450 ́ g at 1,500 rpm in a swing out rotor at room temperature (RT) with no brake.
3.3. Puri fi cation of Mature Schizonts (Day 6)
1137 Transfection of Rodent Malaria Parasites
5. Collect carefully the “brown” (grey) layer at the interface between the two suspensions using a plastic Pasteur pipette.
6. In general a total volume of about 15–30 ml is collected from the two tubes ( see Note 16 ). Do not discard the gradient tube as the media from the top is reused later in this protocol.
7. Pellet the schizonts by centrifugation at 1,500 rpm for 8 min. For this “washing” step, add to the schizont suspension ~20 ml culture medium obtained from the top of the gradients.
8. Discard the supernatant from the puri fi ed schizonts and retain the media from step 6 .
The Amaxa Nucleofector Device II, sourced from Lonza, is used in this protocol.
1. The schizont pellet obtained from one mouse is used for fi ve to eight transfections ( see Note 17 ).
2. Resuspend the schizont pellet in 5–8 ml of culture medium. 3. Transfer the suspension to fi ve to eight Eppendorf tubes (1 ml/
tube). The parasites, 1–3 × 10 7 schizonts, from one tube are used for one transfection.
4. Pellet the cells by centrifugation (5 s, maximum speed). 5. Discard the supernatant. 6. Add 100 m l of the Human T-cell Nucleofector solution 2 with
supplement 3 ¢ -buffer to 5–10 m l of the DNA solution (5–10 m g DNA construct in water or TE buffer), and resuspend the parasites.
7. Transfer the parasite/DNA/buffer solution to a cuvette pro-vided, dispersing any air bubbles with a gentle tap.
8. Transfect using the Amaxa Nucleofector Device with protocol U-033 to electroporate.
9. Add 50–100 m l of culture medium (retained from the earlier gradient tube) to the cuvette immediately after transfection.
10. The 150- m l suspension containing parasites transfected with the Amaxa Nucleofector Device is injected immediately into one mouse.
1. Leave the mice at 37 °C for 15–20 min before electroporation of the parasites. The tail veins swell at 37°C, simplifying the intravenous injection procedure with 0.3-ml insulin syringes with 30G needles.
2. Inject the transfected parasites as quickly as possible into the tail veins of mice that have been anesthetized with iso fl uorane or restrained using a tunnel (consult local guidelines). It is best to carry out this procedure in a timely manner since the veins constrict rapidly after the mouse exits the hot box. Usually the animal is injected between 10.30 and 11.00 am.
3.4. Electroporation of Schizonts (Day 6)
3.5. Intravenous Injection of Transfected Schizonts (Day 6)
114 N. Philip et al.
Selection of transfected parasites can be carried out using either pyrimethamine for the PbDHFR/TS selectable marker or WR99210 for both the hDHFR and PbDHFR/TS . Where possible, it is pref-erable to use pyrimethamine due to its ease of application and ef fi ciency in drinking water ( see Note 18 ).
1. Provide the animals with drinking water containing pyrimethamine ( see Note 4 ) one day after transfected parasites have been injected.
2. Provide the drug for a period of 4–7 days.
1. Inject a single dose of 0.1 ml WR99210 solution (ranging from 6 to 20 mg/kg bodyweight) subcutaneously (in the neck) into mice (20 g) ( see Note 19 ).
2. Repeat the treatment on the following 2–3 days (for a total of three to four treatments).
1. Parasitemia is checked in Giemsa-stained blood fi lms from day 7 after electroporated parasites have been injected ( see Note 20 ).
2. Parasites are collected from mice when parasitemia is 1–5% and are stored in liquid nitrogen (cryopreservation) or used for genotype analysis.
1. Cryopreservation is carried out by collecting 0.5 ml of heart blood by cardiac puncture in 0.5 ml of a 30% (v/v) glycerol/PBS solution with 10 U Heparin/ml. This suspension is trans-ferred to 2 cryotubes, 0.5 ml per tube, kept for 5 min at 4 °C or in an isopropanol cryobox stored at −80°C, and then frozen directly in liquid nitrogen for storage.
2. The rest of the heart blood is collected in 5 ml of PBS to extract DNA for genotype analysis.
3. “Prewash” a Plasmodipur fi lter (EuroProxima) with 10 ml of culture medium or PBS using a 20-ml syringe placed on top of the fi lter. Always remove the fi lter before withdrawing the plunger.
4. The infected blood suspension is passed through the fi lter using a 20-ml syringe to remove leukocytes ( see Note 5 ).
5. Elute with 15–20 ml of culture medium or PBS. 6. Pellet the infected erythrocytes by centrifugation at 1,500 rpm
for 8 min. 7. Remove the supernatant. 8. Lyse the uninfected red blood cell (RBC) by resuspending the
RBC pellet in 50 ml of cold (4°C) 1× erythrocyte lysis buffer. 9. Incubate the suspension on ice for 3–5 min. The clarity of the
suspension is an indicator of the state of the lysis procedure, i.e., when fully clear the lysis is complete.
3.6. Selection of Transfected Parasites (Day 7)
3.6.1. Selection with Pyrimethamine, Provided in the Drinking Water
3.6.2. Selection with WR99210 in DMSO, Injected Subcutaneously
3.7. Monitoring the Growth of Transfected Parasites (Day 14)
3.8. Collection of Parasites for Cryopreservation and for Isolation of DNA for Analysis (Days 14–18)
1157 Transfection of Rodent Malaria Parasites
10. Pellet the parasites by centrifugation for 8 min at 1,500 rpm. 11. Remove the supernatant. 12. Resuspend the small parasite pellet in PBS to wash once more
by pelleting and remove the supernatant. 13. Remove 15–20 m l of the pellet to mix with 20 m l low-melting
agarose for the preparation of an agarose block ( see Subheading 3.9 ) for pulsed- fi eld gel electrophoresis.
14. Store the rest of the pellet in an Eppendorf tube at −20°C for further genomic DNA analysis by PCR, Southern analysis of restricted DNA, and plasmid rescue ( see Notes 21 and 22 ).
1. Pellet the parasites by centrifugation at 1,500 rpm for 8 min after erythrocyte lysis as described in Subheading 3.8 .
2. Mix part of the parasite pellet 1:1 with 1.5–2% low-melting agarose stored at 37°C in a water bath or a hot block.
3. Prepare small blocks using a plug mold or by carefully pouring the agarose suspension on a microscope slide in the area that is marked with waterproof marker-pen.
4. Let the agarose set at RT for a few minutes. 5. Cut the blocks (5 mm/5 mm/2 mm; length/width/height). 6. Place the blocks in 5 ml of SE buffer in a 15-ml tube. 7. Add 50 m l of 20 mg/ml proteinase K to the SE buffer and
incubate overnight at 37°C. 8. Store the blocks in SE buffer at 4°C. Blocks can be stored for
several years without loss of quality. 9. Southern analysis of pulse- fi eld gel electrophoresis or fi eld
inversion gel electrophoresis performed with the agarose chro-mosome should con fi rm correct integration of the transfected DNA construct into the target chromosome or the presence of circular plasmid and wild-type parasites ( see Note 22 ).
Start the cloning procedure after con fi rmation of correct integra-tion of the constructs by Southern blot analysis of separated chro-mosomes or of restricted genomic DNA ( see Note 23 ).
1. Inject a mouse i.p. with phenylhydrazine ( see Subheading 3.1 ) on day 0, Friday.
2. Infect a mouse i.p. with 0.3 ml of a blood suspension from cryopreserved parasites on day 3 (Monday). Take one cryo-tube from the liquid nitrogen stocks to thaw at RT.
3. Check the parasitemia on day 4 (Tuesday) by examining Giemsa-stained blood fi lms ( see Note 24 ).
4. Use a mouse with 0.3–1% parasitemia calculated accurately by counting ³ 6,000 total cells (with no multiply infected erythro-cytes, which is essential).
3.9. Preparation of Agarose Blocks Containing Chromosomes (Days 14–18)
3.10. Cloning of Transfected Parasites
116 N. Philip et al.
5. Collect 5 m l of infected blood in a heparinized capillary tube. 6. Suspend the blood in 1 ml of complete culture medium or PBS
in an Eppendorf tube. 7. Take a small sample (10 m l) and count the RBC in a
hemocytometer. 8. Calculate the number of RBC per m l of blood cell suspension
(usually one grid containing 16 of the smallest squares on the hemocytometer holds a 4-nl volume of cell suspension). By combining the parasitemia and number of RBC per m l, the number of infected RBC per m l can be calculated.
9. Dilute the suspension with culture medium or PBS by serial dilution in such a way that 0.5–2 parasites are present per 0.2 ml of culture medium or PBS ( see Notes 24 and 25 ).
10. Place mice at 37°C for 10–20 min before the injection of the infected RBC.
11. Inject 0.2 ml of the suspension intravenously into the tail veins of ten mice using 0.3-ml insulin syringes with 30G needles.
12. Check the parasitemia of these mice in Giemsa-stained smears at day 8 after infection. In a successful experiment 20–50% of the mice become positive and typically have a parasitemia of 0.3–1% at day 8. Collect the blood from the positive mouse for cryopreservation and for collection of DNA for PCR analysis ( see Subheading 3.8 ).
The growth characteristics of blood stages from transgenic parasites containing episomes are different from transgenic parasites con-taining DNA integrated into their genome ( see Notes 26 and 27 ). The initial infection to maintain production of transgenic parasites can be established by two methods: via passage from an existing infection, or i.p. from a cryopreserved parasite stock.
1. Collect on day 0, usually Thursday, 15 m l of tail blood (three droplets from a heparinized capillary) in 0.2 ml PBS of a mouse with a parasitemia of 5–15% (collect this tail blood from a mouse that has been infected the previous week by mechanical passage, either from mouse to mouse on a Thursday or directly from liquid nitrogen storage on Friday).
2. Inject the 0.2 ml blood/PBS suspension i.p. immediately into a mouse.
3. Proceed with the drug treatment.
1. Infect a mouse i.p. at day 0 (usually a Friday) with 0.3 ml of a blood suspension from cryopreserved parasites (take one cryo-tube from the liquid nitrogen, thaw at RT).
2. Proceed with the drug treatment.
3.11. In Vivo Maintenance and Production of Transgenic Parasites Containing Episomes
3.11.1. Infection of Mice via Mechanical Passage
3.11.2. Infection of Mice from a Cryopreserved Parasite Stock
1177 Transfection of Rodent Malaria Parasites
Mice are treated 1 or 2 days after infection. Selection with pyrimethamine in vivo, provided in the drinking
water:
1. Provide the animals with drinking water containing pyrimethamine 1 day after parasites have been injected and for a period of 3–4 days.
2. Parasites are collected at a parasitemia of 3–15%, not later than 2 days after the fi nal treatment of pyrimethamine to prevent the accumulation of parasites in the population that have lost the episomes.
Selection with WR99210 in DMSO injected subcutaneously:
1. Inject a single dose of 0.1 ml WR99210 solution as in Subheading 3.6.2 .
2. Repeat this treatment on the following 2–3 days for a total of three to four treatments.
3. Parasites are collected at a parasitemia of 3–15%, not later than 2 days after the fi nal treatment of pyrimethamine to prevent the accumulation of parasites in the population that have lost the episomes.
This protocol is based upon the use of outbred mice weighing 20–30 g, presuming that the animal consumes 5–10 ml of drinking water daily, to receive an equivalent dose of 5–10 mg of 5-FC per day ( see Notes 28 and 29 ).
1. A TO or an NIH Swiss outbred mouse at 25 g is infected i.p. on day 0, typically a Friday, with 0.1 ml of a thawed suspension of cryopreserved infected erythrocytes.
2. Monitor the parasitemia daily from day 3 until the negative selection process has reached a close.
3. Collect the blood by cardiac puncture when the parasitemia is 0.5–5%, typically a Monday, day 3, enabling passage of parasitized blood into two mice in such a fashion that parasitemia is observed on day 5 ( see Notes 30 and 31 ).
4. Drinking water with 1 mg/ml 5-FC pro-drug should be applied once a parasitemia of £ 0.1% is achieved; this can be from day 4 to 6 ( see Note 32 ). The drug water should be replenished after 4 days of treatment if the experiment is still running.
5. The infection levels should drop off to zero after 48 h of 5-FC treatment. Parasite clearance is generally around days 6–9, with the parasitemia rising again at days 8–10.
6. Once the parasitemia is greater than 3%, typically a Monday (day 10), collection of the parasites should be carried out with a cardiac puncture. Prepare cryopreserved stabilates and a parasite
3.11.3. Drug Treatment of the Mice
3.12. Negative Selection Using 5-Fluorocytosine In Vivo Provided in the Drinking Water
118 N. Philip et al.
pellet following Subheading 3.8 to prepare DNA and an agarose chromosome block.
7. Cloning is recommended prior to further manipulation of the line.
1. Phenylhydrazine hydrochloride is a suspected carcinogen. A mask must be worn when handling the powder form and gloves used at all times as it can be absorbed through the skin. Take care when disposing of this substance to fl ush with copious amounts of water due to its toxic effect on aquatic organisms.
2. Giemsa solution is prepared at 12% (v/v) with Sörensen staining buffer. Once the thin blood fi lm smears have dried, they are fi xed with methanol for 2 s, air-dried, and left to stain for a minimum of 10 min before rinsing with tap water.
3. Because Nycodenz powder becomes very viscose when dissolv-ing, it is not necessary to fully dissolve the powder into the buffered medium before autoclaving as this assists the process. Autoclave at 120°C for 20 min.
4. Pyrimethamine stock needs to be vortexed to dissolve into DMSO. Working stock should be diluted from 100× stock with tap water and then pH adjusted to pH 3.5–5 with 1 M HCl before a clear solution is obtained. Pyrimethamine should then be dispensed from an opaque or darkened bottle due to it being light sensitive.
5. Where Plasmodipur fi lters are not available or a cheaper alter-native is required, an empty 10-ml Zeba spin column can be fi lled with 3–4 cm of Whatman CF-11 powder. Continue to equilibrate with a wash as per the method in Subheading 3.8 .
6. Drinking water containing 5-FC should be prepared using tap water and requires vigorous shaking to dissolve. Additionally, vortexing and the use of warmed water can help to force the crystals into solution. 5-FC can be dissolved up to 15 mg/ml solution. The solution is light sensitive; therefore any transpar-ent drinking bottles should be covered to prevent exposure to light. As with any other drug in water it should be replenished after 4 days.
7. Blood is collected at 1–10% parasitemia to be mixed 1:1 with 30% glycerol/PBS (v/v) with heparin 10 U/ml. The blood/glycerol suspension is aliquoted into 500 m l volumes to be stored in 1-ml cryotubes at −80°C initially, before being trans-ferred to liquid nitrogen for long-term storage.
4. Notes
1197 Transfection of Rodent Malaria Parasites
8. Blood is collected from the positive mouse (1–2 ml) or rat (5–7 ml) (infected at day 2) at a parasitemia of 1–3% at day 5 (Monday) or 6 (Tuesday) between 10.00 a.m. and 4.00 p.m. Higher parasitemia is suboptimal since many erythrocytes will become multiply infected. In animals that are kept under the normal day/night light regime, the development of the para-sites is relatively synchronous. In these animals rupturing of the schizonts and invasion by merozoites occur in the early morning between 4.00 and 6.00 a.m. Therefore, most para-sites are in the ring form/young trophozoite stage when the infected blood is collected. The infected blood is then cultured overnight at 36.5–37°C. By 9.00 am the next day, all parasites have developed into mature schizonts, which do not rupture under in vitro conditions. Schizonts of P. berghei containing mature merozoites can survive for several hours and can be manipulated without rupturing and loss of viability. There is no need to remove the leucocytes from the blood when the schizonts are used for transfection.
9. Alternatively when requiring large-scale culture to accommo-date over ten transfections a Wistar rat (180–220 g) is injected i.p. with 0.5 ml of phenylhydrazine 12.5 mg/ml.
10. RBC should be singly infected in order to produce healthy mature schizonts. If multiple infected cells are present a pas-sage to another mouse should be performed to continue the experiment with optimal infection conditions. Larger scale cul-ture of schizonts can be carried out using a rat to provide infected erythrocytes, which usually generates enough sch-izonts for up to 30 transfections. Follow the method in Subheading 3.2 collecting 5–7 ml of blood using a 10-ml syringe at step 1 of the protocol.
11. Continuous gassing systems are also available to incubate cul-tures, but are not necessary. If gassing once at the start of cul-turing, ensure that the fl ask has a non-vented, plug seal cap to prevent gas escaping. This is crucial since it has been observed that without gas, as long as the fl ask is sealed and not vented, the culture will still develop viable schizonts.
12. The culture temperature is critical since the parasite develop-mental rate is dependent on the temperature. Above 38.5°C, parasites will degenerate, and at temperatures lower than 37°C, the parasites will develop into healthy parasites but the devel-opmental time is extended. Even at a temperature of 30°C, the parasites will reach the mature schizont stage, but the develop-ment of ring forms into schizonts will take longer than 48 h.
13. Healthy, viable schizonts are distinguished by the presence of 12–24 “free” merozoites within one RBC and one cluster of pigment (hemozoin). Smearing the cells on the microscope
120 N. Philip et al.
slides often damages the red cell membrane and the merozoites are visible as more or less clustered yet free parasites. A purple (red) de fi ned compact nucleus and a dot of blue cytoplasm are characteristics of viable merozoites. About 15–25% of the para-sites in these smears are single-nucleated (young) gametocytes. Degenerate schizonts often show a compact morphology in which the separate merozoites are dif fi cult to recognize. Be careful not to mistake developing schizonts (which are still in the process of nuclear division prior to budding off of the merozoites) for degenerated schizonts ( 17 ) .
14. Start the puri fi cation procedure between 9.00 am and 10.00 am. Starting later in the morning results in a higher per-centage of degenerated schizonts. For the density gradients, use Nycodenz instead of Percoll. Percoll is used by many work-ers to separate parasite stages. However, in contrast to Percoll, Nycodenz does not affect the viability of parasites. Collect about 0.6 × 10 8 to 0.16 × 10 9 schizonts (i.e., 0.6 × 10 9 to 1.6 × 10 9 merozoites) from 1 to 2 ml of heart blood from a mouse and from a rat about 3 × 10 8 to 1 × 10 9 schizonts (i.e., 3 × 10 9 to 1 × 10 10 merozoites) from 5 to 7 ml of heart blood.
15. When performing a density gradient, it is possible to practice using colored water or RPMI1640 to determine the speed and pressure to release the Nycodenz solution to create a sharp line. It is useful to note that when loading a 10-ml pipette the Nycodenz should be taken to 14 ml to more easily visualize when 10 ml have been dispensed into the falcon. If a sharp line is not seen, the culture can be pelleted at 1,800 rpm for 8 min and resuspended in medium to start the gradient process again.
16. A total volume of about 30–40 ml is collected from the four tubes aliquoted from 160 ml of culture when using a rat . The schizonts (and leucocytes, gametocytes, and old trophozoites if present) will collect at the interface of the two suspensions, while the uninfected cells will pellet on the bottom of the tubes.
17. Thirty transfections can be set up from the schizont pellet of one rat, resuspending in 1 ml of culture medium per transfec-tion intended.
18. To date three selectable markers exist for the transformation of Plasmodium : the pyrimethamine-resistant form of the DHFR/TS gene of Plasmodium and of T. gondii , and the human DHFR gene. The latter gene confers not only resistance to pyrimethamine but also to the antimalarial drug WR99210. Introduction of all three genes into pyrimethamine-sensitive P. berghei parasites gives rise to a large increase (~1,000×) in pyrimethamine resistance that allows for a relatively simple in vivo selection procedure. The Tg DHFR/TS gene is preferable to the Plasmodium gene as a selectable marker because it
1217 Transfection of Rodent Malaria Parasites
reduces the likelihood of unwanted recombination with the endogenous Pb DHFR/TS gene and it may confer higher levels of resistance to pyrimethamine. Pyrimethamine selection in vivo is preferred over WR99210 selection, since the latter drug generates more side effects in rodents. The WR99210/ hDHFR selection system can be used in conjunction with the pyrimethamine/Tg DHFR-TS selection system, allowing for multiple manipulation of the genome such as the knockout of two genes in the same parasite clone or for complementation of knockout parasites. Since h DHFR also confers resistance to pyrimethamine, the h DHFR marker can only be used as the second selectable marker when both selectable markers are required.
Treatment of the animals starts 1 day after transfected para-sites have been injected into the mice (to allow the parasites to complete one full developmental cycle in the absence of drug pressure) and treatment is performed on 3–4 consecutive days. Since inoculation of the transfected schizonts occurs between 10.30 and 11.00 a.m. and one cycle takes 22–24 h, start treat-ing the animals in the afternoon. Treatment with pyrimethamine is now performed by providing the pyrimethamine in the drink-ing water and not by drug injection.
19. The concentration of WR99210 is dependent on the selectable markers present in the parasite line and the episomal/inte-grated nature of the constructs. If only one copy of the h DHFR gene is present, select with lower concentrations of 6–8 mg; if both a resistant DHFR-TS and the h DHFR genes are present, select with 12–16 mg.
20. One day after injection of the transfected parasites, parasitemia is usually between 0.05 and 3%. After the fi rst two drug treat-ments parasitemia rapidly becomes undetectable, indicating that most of the parasites do not contain the constructs. In unsuccessful experiments, parasites are often detected between day 13 and 15 after the injection of transfected parasites. These parasites are usually wild-type parasites that survived the drug treatment protocol.
21. Any material that is intended for use as a chromosome agarose block must be processed as such on that day and not stored at −20 °C since freezing fractures the chromosomes, therefore damaging the block.
22. Characterize the genotype of the uncloned, transfected para-sites as follows: (a) PCR to show the presence of the selectable marker (for
example Tg DHFR-TS , h DHFR ) and correct integration. These PCR are performed to rapidly have an indication whether the transfection was successful.
122 N. Philip et al.
(b) Separation of the chromosomes using Pulsed-Field Gel Electrophoresis (FIGE or CHEF) followed by hybridiza-tion of the separated chromosomes using the 3 ¢ UTR of the Pb DHFR-TS gene. This DNA fragment is present in most available constructs and is also present at chromosome 7 as endogenous gene in P. berghei . This is a standard analysis performed in all transfection experiments. The hybridiza-tion pattern and intensity give information on (1) the inte-gration event in the target chromosome, (2) the presence of episomes (that migrate in a different pattern compared to the chromosomes), and (3) the ratio between wild-type and transfected parasites in the population.
(c) Southern analysis of restricted genomic DNA. This is only done if the PCR and the chromosome hybridizations give confusing results and con fi rmation of correct integration is needed. Usually Southern analysis is performed on genomic DNA from cloned parasites. Southern analysis is also performed as a means to determine the ratio of wild-type to recombinant parasites in primary populations of transfected parasites.
(d) Plasmid rescue: plasmid rescue provides a means to con fi rm the integrity of introduced episomes and to recover genomic DNA fl anking the insertion site (single crossover only).
23. Do not clone parasites that are transfected with a single, circular construct unless more than one construct is present in the pop-ulation. The demonstration of the presence of the unaltered construct by southern analysis and plasmid rescue is normally suf fi cient for these parasite populations.
24. The limiting dilution cloning procedure should be started within 1–2 h after preparation of the Giemsa-stained slides. If there is a subsequent delay longer than 2 h, parasitemia might increase as a result of invasion of new parasites. A higher parasitemia in the starting material will of course affect the cloning procedure.
25. Parasites are cloned using the method of limiting dilution of blood-stage parasites, i.e., injection of a single parasite into animals. The method described here is empirically adapted to speci fi c laboratory conditions and it is possible that other labo-ratories have to make small changes in order to get reliable results with this cloning procedure. In the Leiden laboratory the inoculum is 2 parasites per mouse, whereas in the Glasgow’s laboratory we observe that 0.5 parasite per mouse is optimal resulting in an infection rate of 20–50% of the mice. It is con-sidered that a cloning experiment is successful if less than 50% of the mice become positive.
26. Under drug pressure transformed parasites that contain epi-somes grow slower than transgenic parasites containing DNA
1237 Transfection of Rodent Malaria Parasites
integrated into their genome. The slower growth rate is due to the unstable segregation of the episomes during schizogony, resulting in the production of merozoites in each cycle, which do not contain episomes. These latter parasites are therefore sensitive to pyrimethamine during their development into the next schizont generation. Up to 50–60% of merozoites pro-duced from episome-containing parasites lack the episomes. The episome-positive parasites may contain up to 20–40 cop-ies of the episome per nucleus.
27. It is important to maintain episomally transfected parasites under drug pressure during in vivo multiplication to prevent the loss of the episomes from the parasite population. Wild-type parasites show about a ten times multiplication rate per 24 h in mice, up to a parasitemia of 3–5%. At higher para-sitemia the multiplication rate is slower as a result of a shortage of suitable host cells and multiple infected cells, which does not support optimal growth of the parasites. Transgenic para-sites containing episomes grow much more slowly under drug pressure. It is preferable to start infections in mice with rela-tively high numbers of these transgenic parasites to obtain the required parasites in a relatively short period (4–8 days) reduc-ing the period of pyrimethamine treatment of the mice to 3–4 days.
28. 5-FC has dissolving capabilities of up to 15 mg/ml solution and should be dissolved in tap water when used for negative selection provided in drinking water. Dissolving it requires vig-orous shaking, although vortexing and the use of warmed water can help force the crystals into solution. The solution is light sensitive; therefore any transparent drinking bottles should be covered to darken them. As with any other drug water it should be replenished after 4 days if required.
29. Previously, negative selection was carried out over 4 days during which 10 mg of 5-FC was i.p. injected every day. The 5-FC stock was at 10 mg/ml in 0.9% NaCl solution, a concentration at the limit of the solubility of the compound. Negative selec-tion via drinking water is less laborious, using a lower dose of 5-FC with cage mates using the same drinking water, thereby allowing for a reduced and re fi ned animal usage.
30. If the parasitemia is between 0.2 and 1% then 50 m l of blood should be mixed with 0.2 ml of PBS to be injected ip, produc-ing a parasitemia between 0.2 and 1.5% after 48 h, typically a Wednesday.
31. Alternatively, two mice can be infected directly with 0.1–0.25 ml of a cryopreserved stabilate on a Monday, reducing animal numbers required.
32. If clearance of parasites is not visible after 48 h the concentration can be increased to 1.5 mg/ml.
124 N. Philip et al.
References
1. Bártfai R et al (2010) H2A.Z demarcates inter-genic regions of the Plasmodium falciparum epigenome that are dynamically marked by H3K9ac and H3K4me3. PLoS Pathog 6:e1001223
2. Bozdech Z et al (2003) The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum . PLoS Biol 1:e5
3. Florens L et al (2002) A proteomic view of the Plasmodium falciparum life cycle. Nature 419:520–526
4. Gardner MJ et al (2002) Genome sequence of the human malaria parasite Plasmodium falci-parum . Nature 419:498–511
5. Khan SM et al (2005) Proteome analysis of separated male and female gametocytes reveals novel sex-speci fi c Plasmodium biology. Cell 121:675–687
6. Lasonder E et al (2002) Analysis of the Plasmodium falciparum proteome by high-accuracy mass spectrometry. Nature 419:537–542
7. Le Roch KG et al (2003) Discovery of gene function by expression pro fi ling of the malaria parasite life cycle. Science 301:1503–1508
8. Otto TD et al (2010) New insights into the blood-stage transcriptome of Plasmodium falciparum using RNA-Seq. Mol Microbiol 76:12–24
9. Salcedo-Amaya AM et al (2009) Dynamic his-tone H3 epigenome marking during the intra-erythrocytic cycle of Plasmodium falciparum . Proc Natl Acad Sci USA 106:9655–9660
10. van Dijk MR et al (1995) Stable transfection of malaria parasite blood stages. Science 268:1358–1362
11. Mota MM et al (2001) Gene targeting in the rodent malaria parasite Plasmodium yoelii . Mol Biochem Parasitol 113:271–278
12. Reece SE, Thompson J (2008) Transformation of the rodent malaria parasite Plasmodium cha-baudi and generation of a stable fl uorescent line PcGFPCON. Malar J 7:183
13. Kooij TW et al (2005) A Plasmodium whole-genome synteny map: indels and synteny breakpoints as foci for species-speci fi c genes. PLoS Pathog 1:e44
14. Franke-Fayard B et al (2004) A Plasmodium ber-ghei reference line that constitutively expresses GFP at a high level throughout the complete life cycle. Mol Biochem Parasitol 137:23–33
15. Mair GR et al (2010) Universal features of post-transcriptional gene regulation are critical for Plasmodium zygote development. PLoS Pathog 6:e1000767
16. Ponzi M et al (2009) Egress of Plasmodium berghei gametes from their host erythrocyte is mediated by the MDV-1/PEG3 protein. Cell Microbiol 11:1272–1288
17. Janse CJ et al (2006) High-ef fi ciency transfec-tion and drug selection of genetically trans-formed blood stages of the rodent malaria parasite Plasmodium berghei . Nat Protoc 1:346–356
18. Janse CJ et al (2006) High ef fi ciency transfec-tion of Plasmodium berghei facilitates novel selection procedures. Mol Biochem Parasitol 145:60–70
19. van Dijk MR et al (1994) Mechanisms of pyrimethamine resistance in two different strains of Plasmodium berghei . Mol Biochem Parasitol 68:167–171
20. Fidock DA, Wellems TE (1997) Transformation with human dihydrofolate reductase renders malaria parasites insensitive to WR99210 but does not affect the intrinsic activity of proguanil. Proc Natl Acad Sci USA 94:10931–10936
21. Braks JA et al (2006) Development and appli-cation of a positive-negative selectable marker system for use in reverse genetics in Plasmodium . Nucleic Acids Res 34:e39
22. Maier AG et al (2006) Negative selection using yeast cytosine deaminase/uracil phosphoribosyl transferase in Plasmodium falciparum for tar-geted gene deletion by double crossover recom-bination. Mol Biochem Parasitol 150:118–121
23. van Schaijk BC et al (2010) Removal of heter-ologous sequences from Plasmodium falci-parum mutants using FLPe-recombinase. PLoS One 5:e15121
24. Laurentino EC et al (2011) Experimentally controlled downregulation of the histone chaperone FACT in Plasmodium berghei reveals that it is critical to male gamete fertility. Cell Microbiol 13:1956–1974
25. O’Donnell RA et al (2001) An alteration in concatameric structure is associated with ef fi cient segregation of plasmids in transfected Plasmodium falciparum parasites. Nucleic Acids Res 29:716–724
26. van Dijk MR et al (1997) Replication, expres-sion and segregation of plasmid-borne DNA in genetically transformed malaria parasites. Mol Biochem Parasitol 86:155–162
27. Murray AW, Szostak JW (1983) Construction of arti fi cial chromosomes in yeast. Nature 305:189–193
28. Stinchcomb DT et al (1979) Isolation and characterisation of a yeast chromosomal repli-cator. Nature 282:39–43
1257 Transfection of Rodent Malaria Parasites
29. Iwanaga S et al (2010) Functional identi fi cation of the Plasmodium centromere and generation of a Plasmodium arti fi cial chromosome. Cell Host Microbe 7:245–255
30. Chookajorn T et al (2007) Epigenetic memory at malaria virulence genes. Proc Natl Acad Sci USA 104:899–902
31. Freitas-Junior LH et al (2005) Telomeric het-erochromatin propagation and histone acetyla-tion control mutually exclusive expression of antigenic variation genes in malaria parasites. Cell 121:25–36
32. Hernandez-Rivas R et al (2010) Telomeric heterochromatin in Plasmodium falciparum . J Biomed Biotechnol. doi: 10.1155/2010/290501
33. Lopez-Rubio JJ et al (2007) 5 ¢ fl anking region of var genes nucleate histone modi fi cation pat-terns linked to phenotypic inheritance of viru-lence traits in malaria parasites. Mol Microbiol 66:1296–1305
34. Petter M et al (2011) Expression of P. falci-parum var genes involves exchange of the histone variant H2A.Z at the promoter. PLoS Pathog 7:e1001292
35. Meissner M et al (2005) Tetracycline analogue-regulated transgene expression in Plasmodium falciparum blood stages using Toxoplasma gondii transactivators. Proc Natl Acad Sci USA 102:2980–2985
36. Armstrong CM, Goldberg DE (2007) An FKBP destabilization domain modulates pro-tein levels in Plasmodium falciparum . Nat Methods 4:1007–1009
37. Muralidharan V et al (2011) Asparagine repeat function in a Plasmodium falciparum protein assessed via a regulatable fl uorescent af fi nity tag. Proc Natl Acad Sci USA 108:4411–4416
38. Carvalho TG et al (2004) Conditional muta-genesis using site-speci fi c recombination in
Plasmodium berghei . Proc Natl Acad Sci USA 101:14931–14936
39. Combe A et al (2009) Clonal conditional mutagenesis in malaria parasites. Cell Host Microbe 5:386–396
40. de Koning-Ward TF et al (2009) A newly dis-covered protein export machine in malaria parasites. Nature 459:945–949
41. Ivics Z et al (2009) Transposon-mediated genome manipulation in vertebrates. Nat Methods 6:415–422
42. Mátés L et al (2009) Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in verte-brates. Nat Genet 41:753–761
43. Patton EE, Zon LI (2001) The art and design of genetic screens: zebra fi sh. Nat Rev Genet 2:956–966
44. Schneider A, Leister D (2006) Forward genetic screening of insertional mutants. Methods Mol Biol 323:147–161
45. Yergeau DA, Mead PE (2007) Manipulating the Xenopus genome with transposable ele-ments. Genome Biol 8(Suppl 1):S11
46. Sakamoto H et al (2005) Towards systematic identi fi cation of Plasmodium essential genes by transposon shuttle mutagenesis. Nucleic Acids Res 33:e174
47. Balu B et al (2005) High-ef fi ciency transfor-mation of Plasmodium falciparum by the lepi-dopteran transposable element piggyBac. Proc Natl Acad Sci USA 102:16391–16396
48. Fonager J et al (2011) Development of the pig-gyBac transposable system for Plasmodium ber-ghei and its application for random mutagenesis in malaria parasites. BMC Genomics 12:155
49. Balu B, Adams JH (2006) Functional genom-ics of Plasmodium falciparum through trans-poson-mediated mutagenesis. Cell Microbiol 8:1529–1536