Université Paris Descartes Ecole doctorale Bio-Sorbonne Paris Cité Biologie Cellulaire et Moléculaire, Physiologie, Physiopathologie INSERM U970, Centre de recherche cardiovasculaire de Paris, Equipe 1 Physiopathologie des évènements cardiovasculaires chez les malades atteints de syndrome myéloprolifératif Bcr/Abl-négatif Pathophysiology of cardiovascular events in Bcr/Abl- negative myeloproliferative neoplasms Par Johanne Poisson Thèse de doctorat de physiologie et physiopathologie Dirigée par Pr. Pierre-Emmanuel Rautou Présentée et soutenue publiquement le 27 Septembre 2018 Devant un jury composé de : Pr. Chloé James Rapporteur Pr. Jonel Trebicka Rapporteur Pr. Caroline Le Van Kim Examinateur Dr. Yacine Boulaftali Examinateur Pr. Pierre-Emmanuel Rautou Directeur de thèse
I. LISTOFABBREVIATIONS..........................................................................................................................................................6
II. INTRODUCTION............................................................................................................................................................................7
A. Myeloproliferativeneoplasms..........................................................................................................................................7
a) PolycythaemiaVera....................................................................................................................................................9
b) Essentialthrombocythemia..................................................................................................................................10
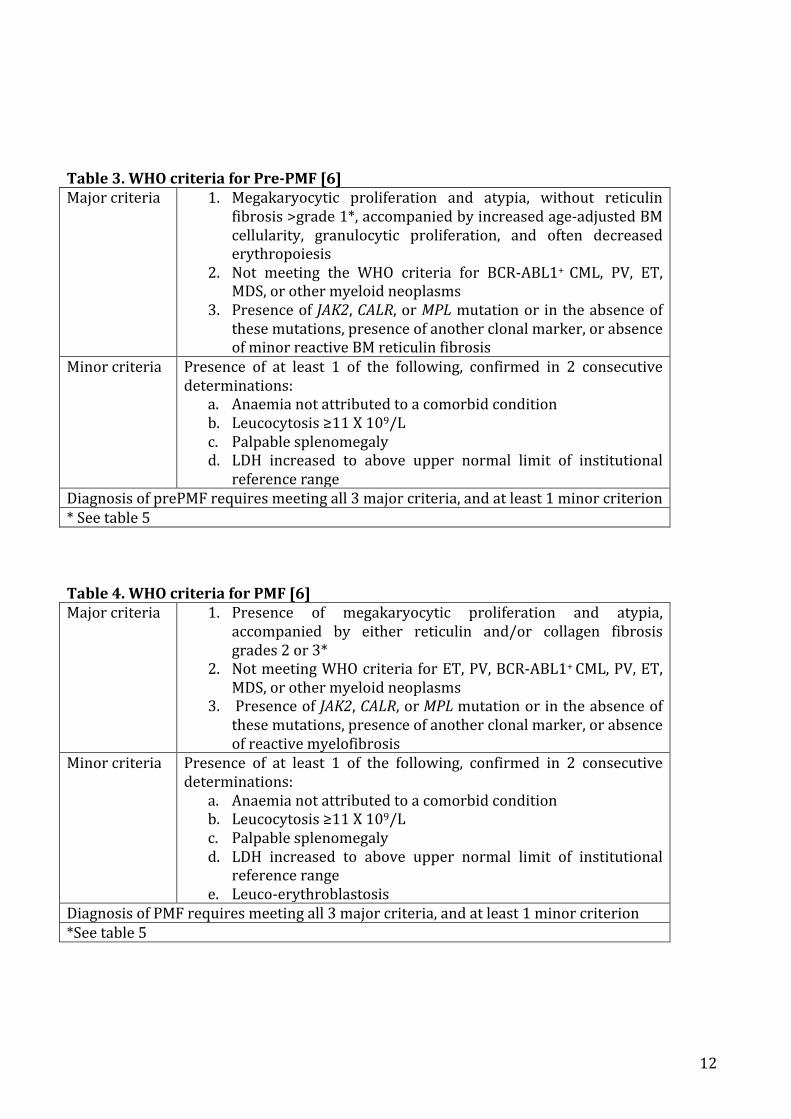
c) Preprimarymyelofibrosisandprimarymyelofibrosis.............................................................................11
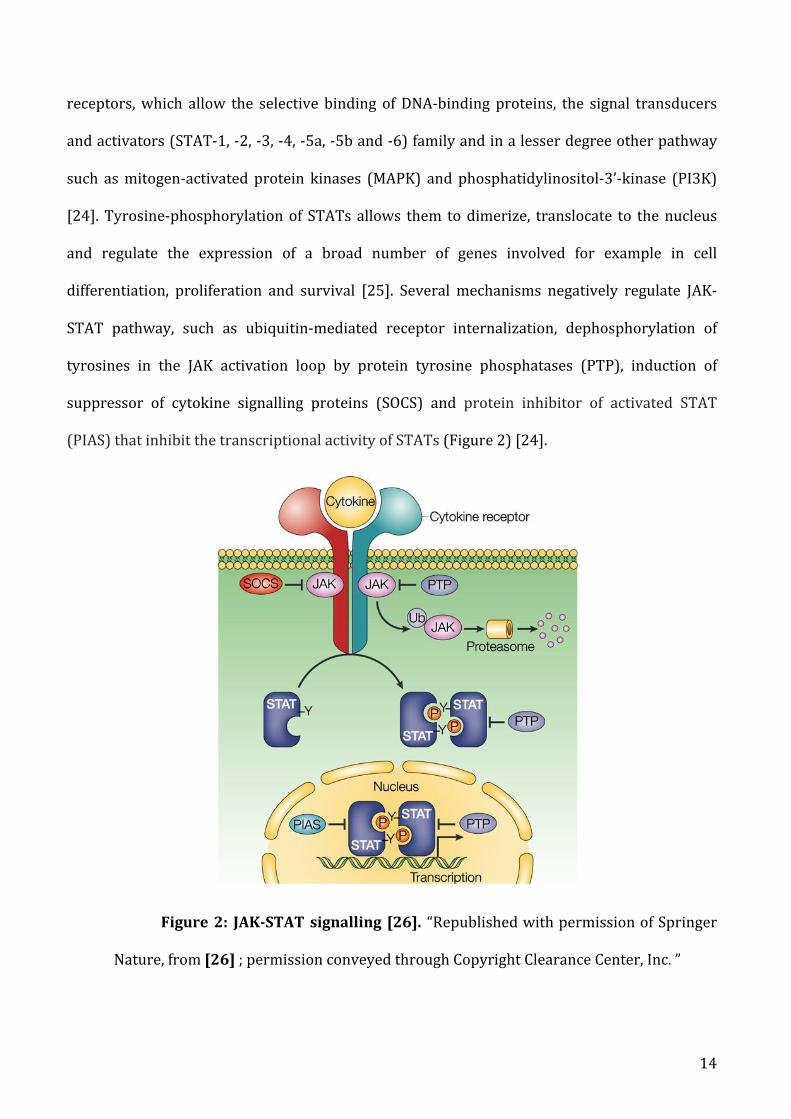
a) JAK/STATsignalling.................................................................................................................................................13
b) Mutationallandscape...............................................................................................................................................16
a) Secondarymyelofibrosis........................................................................................................................................24
b) Acutemyeloidleukaemia.......................................................................................................................................25
a) PolycythaemiaVera..................................................................................................................................................26
b) Essentialthrombocythemia..................................................................................................................................28
c) Preprimarymyelofibrosisandprimarymyelofibrosis.............................................................................31
B. Myeloproliferativeneoplasmsandcardiovascularcomplications..................................................................32
a) Drivermutations........................................................................................................................................................34
b) Leukocytes....................................................................................................................................................................35
a) Platelets..........................................................................................................................................................................36
b) Redbloodcells............................................................................................................................................................36
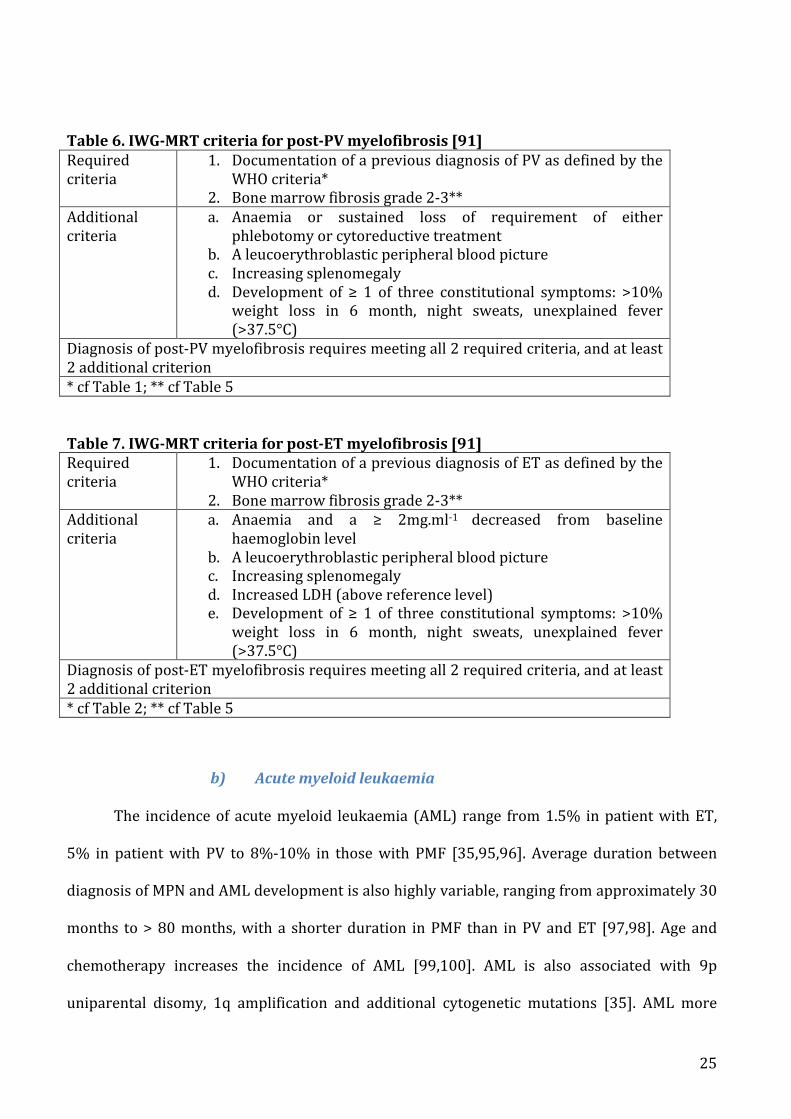
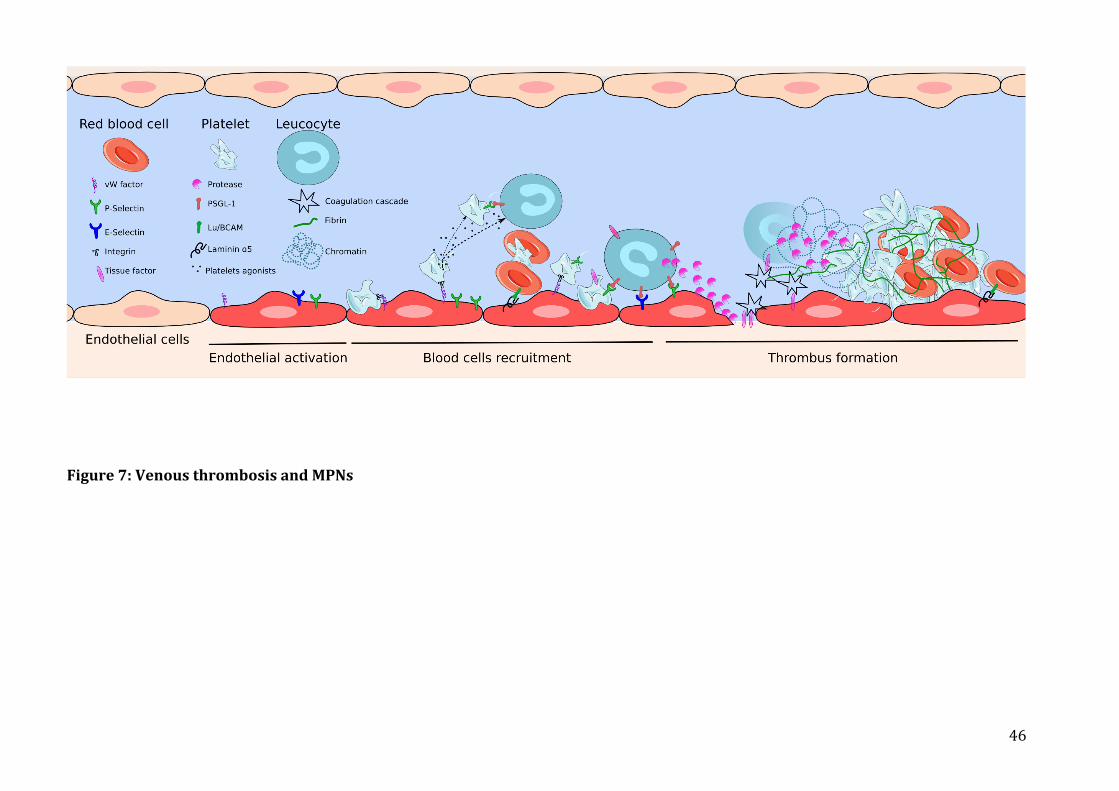
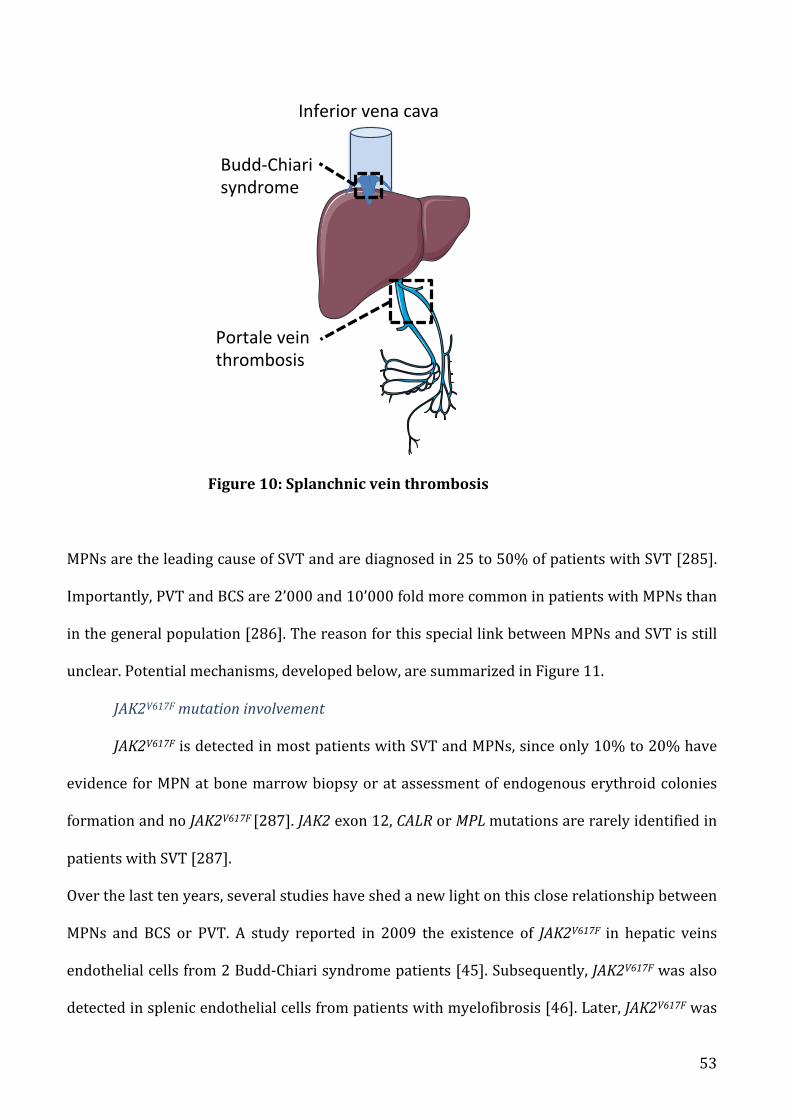
a) Pathophysiologyofvenousthrombosisinmyeloproliferativeneoplasms......................................37
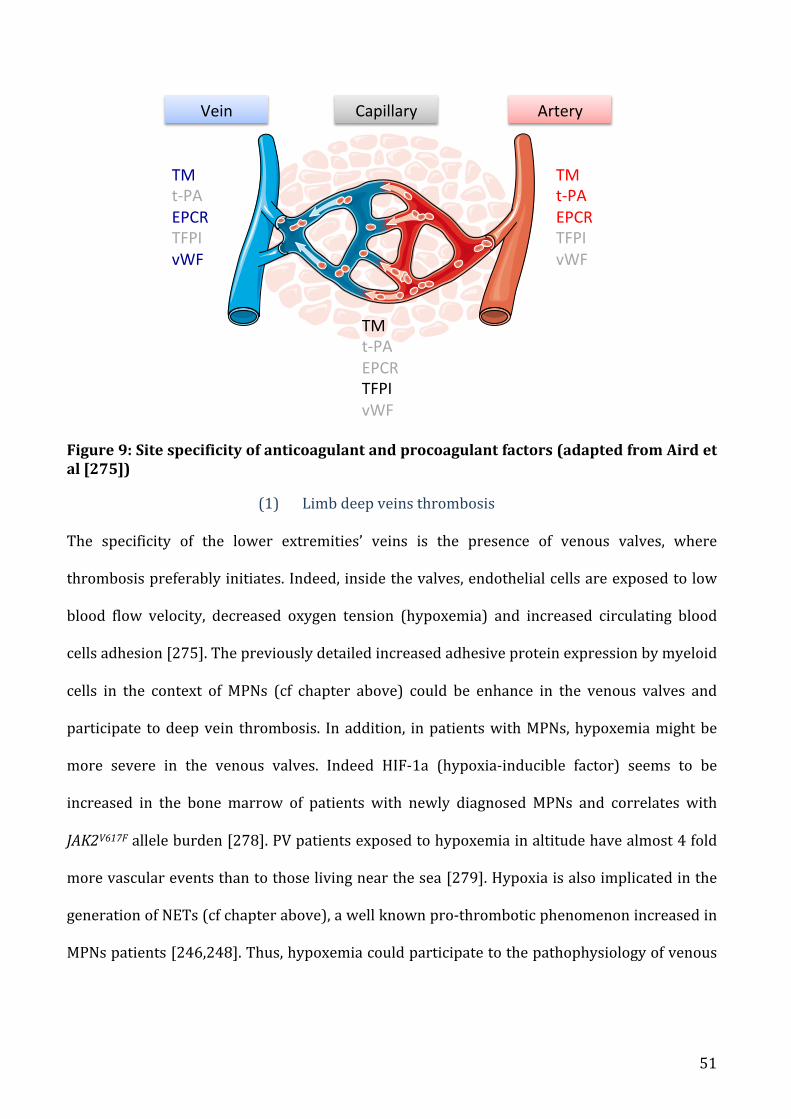
b) SitespecificityinMPNspatients.........................................................................................................................50
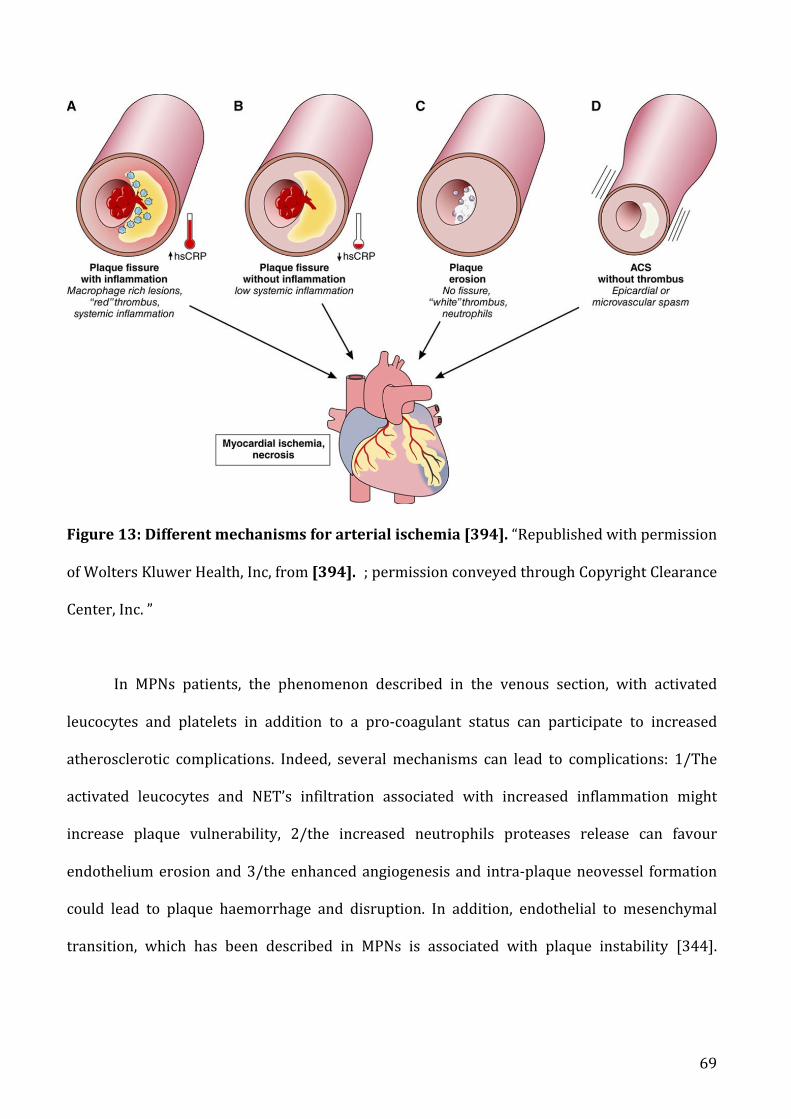
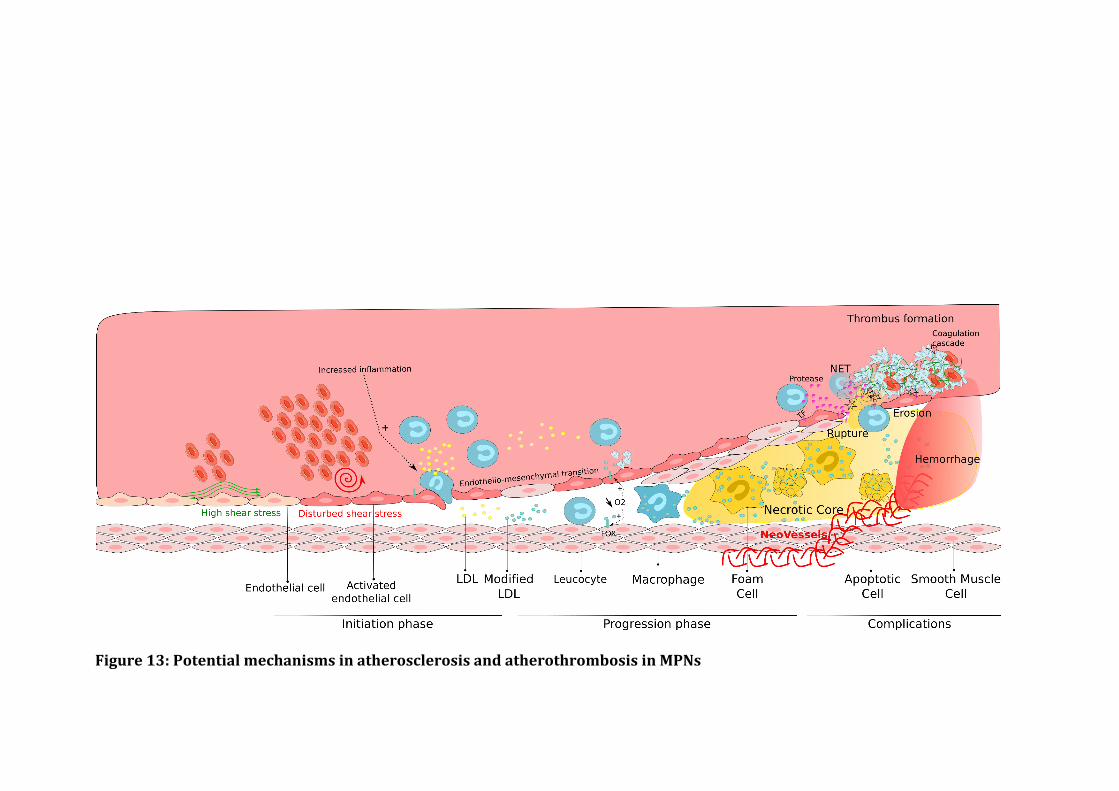
a) AtherosclerosisandMPNs.....................................................................................................................................58
b) Beyondatherosclerosis...........................................................................................................................................71
III. Thesiswork.................................................................................................................................................................................83
A. Aims............................................................................................................................................................................................83
B. JAK2V617Finarterialevents................................................................................................................................................841. Article1:ErythrocytemicrovesiclesincreasearterialcontractioninJAK2V617Fmyeloproliferativeneoplasms.....................................................................................................................................................................................84
C. JAK2V617Finsplanchnicveinthrombosis..................................................................................................................1201. EndothelialJAK2V617FandBudd-Chiarisyndrome..........................................................................................120a) Backgroundandaims............................................................................................................................................120
b) Materialsandmethods.........................................................................................................................................120
c) Article2:EndothelialJAK2V617FdoesnotenhanceliverlesionsinmicewithBudd-Chiari
IV. DISCUSSIONANDPROSPECTS.........................................................................................................................................137
A. JAK2V617Finarterialevents.............................................................................................................................................137B. JAK2V617FinBudd-Chiarisyndrome............................................................................................................................139C. CALRandsplanchnicveinthrombosis......................................................................................................................141
V. CONCLUSION.............................................................................................................................................................................142
VI. REFERENCES...........................................................................................................................................................................143
VII. APPENDIX...............................................................................................................................................................................170
A. Appendix1:Review:Liversinusoidalendothelialcells:physiologyandroleinliverdiseases......170
B. Appendix2:Replyto“CalreticulinmutationsandtheirimportanceinBudd-Chiarisyndrome”...187
C. Appendix3:Curriculumvitaeandlistofpublications.......................................................................................191
6
I. LISTOFABBREVIATIONS
AML:Acutemyeloidleukaemia
BCS:Budd-Chiarisyndrome
BM:Bonemarrow
CALR:Calreticulin
CML:Chronicmyelogenousleukaemia
COX:Cyclooxygenase
CV:Cardiovascular
CVT:Cerebralvenousthrombosis
EC:Endothelialcells
EPCR:ProteinCreceptor
EPO:erythropoietine
EPOR:Erythropoetinreceptor
ERK:Extracellularsignal-regulatedkinase
ET:Essentialthrombocythemia
FERM:Four-point-oneezrinradixinmoesin
G-CSF: Granulocyte colony-stimulating
factorreceptor
HU:Hydroxyurea
HUVECs:Humanumbilical vein endothelial
cells
IFN-α:Interferon-alpha
IL:Interleukin
IPSET: International prognostic score for
thrombosisinessentialthrombocythemia
IPSS: International prognostic scoring
system
JAK:JanusKinase
JH1:Jakhomology1
MAPK:Mitogen-activatedproteinkinases
MDS:myelodysplasticsyndrome
MPL: Myeloproliferative leukaemiavirus/
Thrombopoietinreceptor
MPNs:Myeloproliferativeneoplasms
NETs:Neutrophilextracellulartraps
NO:Nitricoxide
Pa:Pascal
PAD4:Petidyl-argininedeiminase
PAI-1: Type I plasminogen activator
inhibitor
Ph:Philadelphia
PI3K:Phosphatidylinositol-3’-kinase
PIAS:ProteininhibitorofactivatedSTAT
PMF:Primarymyelofibrosis
PrePMF:Prefibrotic/earlyprimary
PSGL-1:P-selectinglycoproteinligand-1
PTP:Proteintyrosinephosphatases
PV:Polycythaemiavera
PVT:Portalveinthrombosis
RBCs:Redbloodcells
SH2:Srchomology2
SOCS: Induction of suppressor of cytokine
signallingproteins
STAT:Signaltransducersandactivators
SVT:Splanchnicveinthrombosis
t-PA:tissueplasminogen
TAFI: Thrombin activated fibrinolysis
inhibitor
TF:Tissuefactor
TFPI:Tissuefactorpathwayinhibitor
TM:Thrombomodulin
TNFα:Tumornecrosisfactoralpha
TPO:Thrombopoietin
TYK:Tyrosinekinases
u-PA:urokinase
vWF:VonWillebrandfactor
WBC:Whitebloodcells
WHO:Worldhealthorganization
WT:Wild-type
7
II. INTRODUCTION
A. Myeloproliferativeneoplasms
1. Definitions
Myeloproliferative neoplasms (MPNs) are clonal hematopoietic diseases characterized
by an overproduction of differentiated hematopoietic cells. The first report of
«myeloproliferative disorders» in 1951 described a group of disease without a clear
separation between polycythaemia vera, thrombocythemia chronic granulocytic leukaemia,
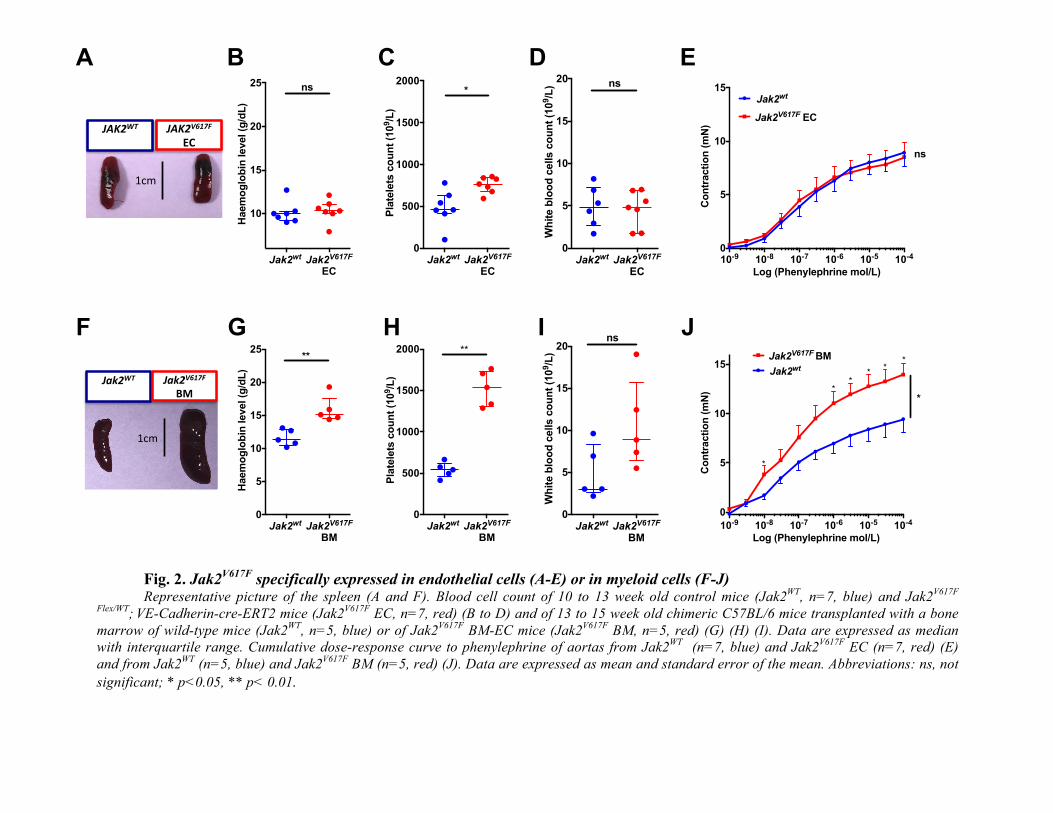
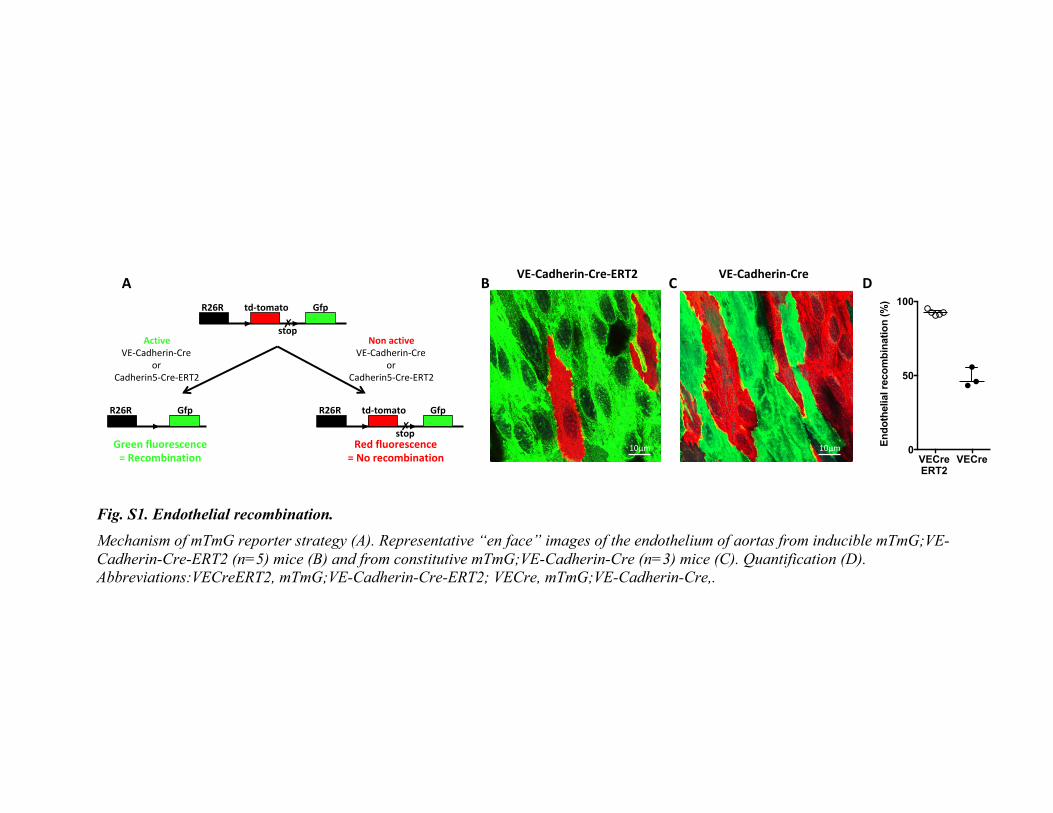
specifically expressed in endothelial cells (A-E) or in myeloid cells (F-J) Representative picture of the spleen (A and F). Blood cell count of 10 to 13 week old control mice (Jak2
WT, n=7, blue) and Jak2
V617F
Flex/WT;
VE-Cadherin-cre-ERT2 mice (Jak2
V617F EC, n=7, red) (B to D) and of 13 to 15 week old chimeric C57BL/6 mice transplanted with a bone
marrow of wild-type mice (Jak2WT
, n=5, blue) or of Jak2V617F
BM-EC mice (Jak2V617F
BM, n=5, red) (G) (H) (I). Data are expressed as median
with interquartile range. Cumulative dose-response curve to phenylephrine of aortas from Jak2WT
(n=7, blue) and Jak2V617F
EC (n=7, red) (E)
and from Jak2WT
(n=5, blue) and Jak2V617F
BM (n=5, red) (J). Data are expressed as mean and standard error of the mean. Abbreviations: ns, not
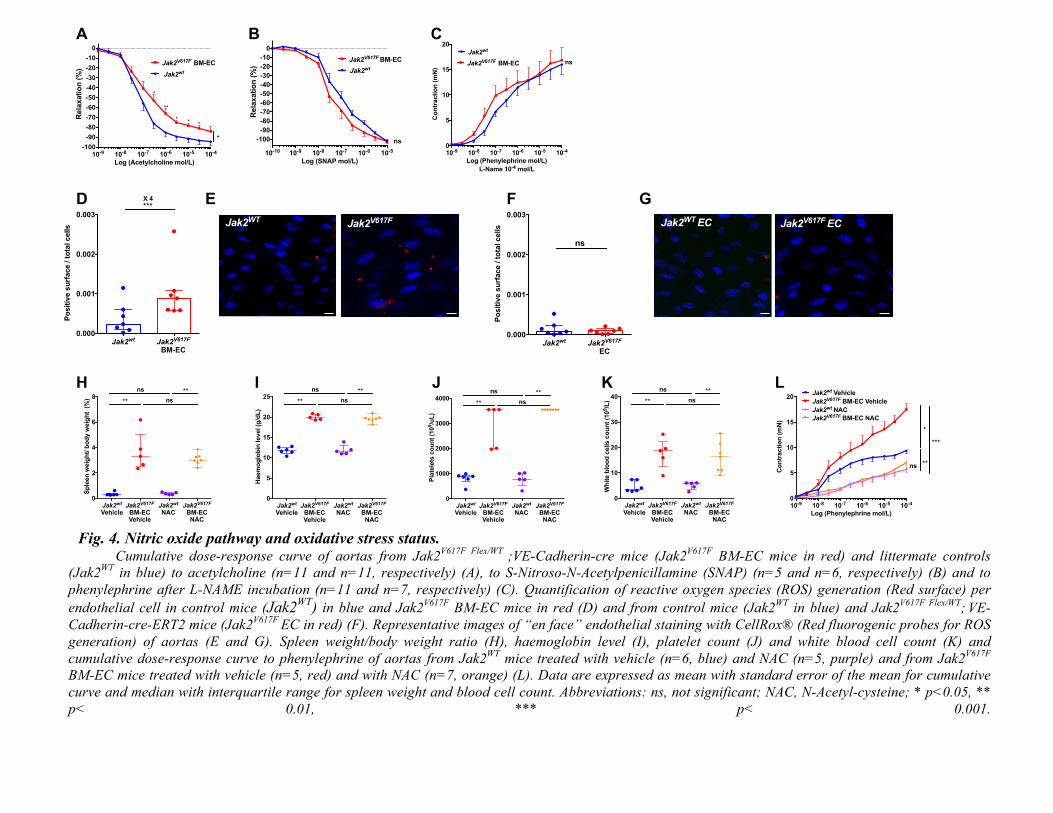
Altogether, these results show that the increased arterial contraction in Jak2V617F
BM-ECmice is induced by excessive oxidative stress in endothelial cells leading to a
decreasedavailabilityofNO.
Fig. 4. Nitric oxide pathway and oxidative stress status.
Cumulative dose-response curve of aortas from Jak2V617F Flex/WT
;VE-Cadherin-cre mice (Jak2V617F
BM-EC mice in red) and littermate controls
(Jak2WT
in blue) to acetylcholine (n=11 and n=11, respectively) (A), to S-Nitroso-N-Acetylpenicillamine (SNAP) (n=5 and n=6, respectively) (B) and to
phenylephrine after L-NAME incubation (n=11 and n=7, respectively) (C). Quantification of reactive oxygen species (ROS) generation (Red surface) per
endothelial cell in control mice (Jak2WT
) in blue and Jak2V617F
BM-EC mice in red (D) and from control mice (Jak2WT
in blue) and Jak2V617F Flex/WT
; VE-
Cadherin-cre-ERT2 mice (Jak2V617F
EC in red) (F). Representative images of “en face” endothelial staining with CellRox® (Red fluorogenic probes for ROS
generation) of aortas (E and G). Spleen weight/body weight ratio (H), haemoglobin level (I), platelet count (J) and white blood cell count (K) and
cumulative dose-response curve to phenylephrine of aortas from Jak2WT
mice treated with vehicle (n=6, blue) and NAC (n=5, purple) and from Jak2V617F
BM-EC mice treated with vehicle (n=5, red) and with NAC (n=7, orange) (L). Data are expressed as mean with standard error of the mean for cumulative
curve and median with interquartile range for spleen weight and blood cell count. Abbreviations: ns, not significant; NAC, N-Acetyl-cysteine; * p<0.05, **
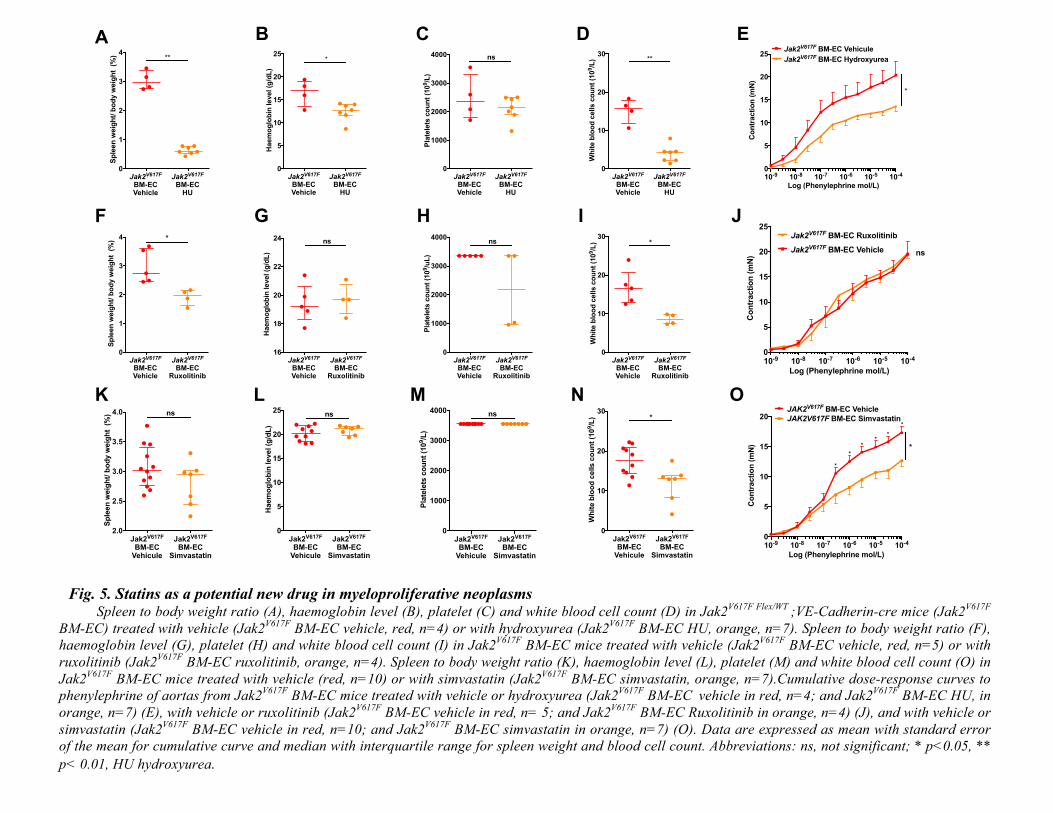
Fig. 5. Statins as a potential new drug in myeloproliferative neoplasms Spleen to body weight ratio (A), haemoglobin level (B), platelet (C) and white blood cell count (D) in Jak2
V617F Flex/WT ;VE-Cadherin-cre mice (Jak2
V617F
BM-EC) treated with vehicle (Jak2V617F
BM-EC vehicle, red, n=4) or with hydroxyurea (Jak2V617F
BM-EC HU, orange, n=7). Spleen to body weight ratio (F),
haemoglobin level (G), platelet (H) and white blood cell count (I) in Jak2V617F
BM-EC mice treated with vehicle (Jak2V617F
BM-EC vehicle, red, n=5) or with
ruxolitinib (Jak2V617F
BM-EC ruxolitinib, orange, n=4). Spleen to body weight ratio (K), haemoglobin level (L), platelet (M) and white blood cell count (O) in
Jak2V617F
BM-EC mice treated with vehicle (red, n=10) or with simvastatin (Jak2V617F
BM-EC simvastatin, orange, n=7).Cumulative dose-response curves to
phenylephrine of aortas from Jak2V617F
BM-EC mice treated with vehicle or hydroxyurea (Jak2V617F
BM-EC vehicle in red, n=4; and Jak2
V617F BM-EC HU, in
orange, n=7) (E), with vehicle or ruxolitinib (Jak2V617F
BM-EC vehicle in red, n= 5; and Jak2V617F
BM-EC Ruxolitinib in orange, n=4) (J), and with vehicle or
simvastatin (Jak2V617F
BM-EC vehicle in red, n=10; and Jak2V617F
BM-EC simvastatin in orange, n=7) (O). Data are expressed as mean with standard error
of the mean for cumulative curve and median with interquartile range for spleen weight and blood cell count. Abbreviations: ns, not significant; * p<0.05, **
p< 0.01, HU hydroxyurea.
10-9 10-8 10-7 10-6 10-5 10-40
5
10
15
20
25
Log (Phenylephrine mol/L)
Co
ntr
acti
on
(m
N)
Jak2V617F BM-EC Vehicule
Jak2V617F BM-EC Hydroxyurea
*
Jak2V617F
BM-ECVehicle
Jak2V617F
BM-ECHU
0
5
10
15
20
25
Haem
og
lob
in level (g
/dL
)
*
Jak2V617F
BM-ECVehicle
Jak2V617F
BM-ECHU
0
1000
2000
3000
4000
Pla
tele
ts c
ou
nt
(10
9/L
)
ns
Jak2V617F
BM-ECVehicle
Jak2V617F
BM-ECHU
0
10
20
30
Wh
ite b
loo
d c
ells c
ou
nt
(10
9/L
) **
10-9 10-8 10-7 10-6 10-5 10-40
5
10
15
20
25
Log (Phenylephrine mol/L)
Co
ntr
acti
on
(m
N)
Jak2V617F
BM-EC Vehicle ns
Jak2V617F
BM-EC Ruxolitinib
Jak2V617F
BM-ECVehicle
Jak2V617F
BM-ECRuxolitinib
0
1
2
3
4
Sp
leen
weig
ht/
bo
dy w
eig
ht
(%
) *
Jak2V617F
BM-ECVehicle
Jak2V617F
BM-ECRuxolitinib
16
18
20
22
24
Haem
og
lob
in level (g
/dL
)
ns
Jak2V617F
BM-ECVehicle
Jak2V617F
BM-ECRuxolitinib
0
1000
2000
3000
4000
Pla
tele
ts c
ou
nt
(10
9/u
L)
ns
Jak2V617F
BM-ECVehicle
Jak2V617F
BM-ECRuxolitinib
0
10
20
30
Wh
ite b
loo
d c
ells c
ou
nt
(10
9/L
) *
Jak2V617F
BM-ECVehicle
Jak2V617F
BM-ECHU
0
1
2
3
4
Sp
leen
weig
ht/
bo
dy w
eig
ht
(%
) **
A B C D E
F G H I J
10-9 10-8 10-7 10-6 10-5 10-40
5
10
15
20
Log (Phenylephrine mol/L)
Co
ntr
acti
on
(m
N) *
*
*
**
*
*
JAK2V617F BM-EC Vehicle
JAK2V617F BM-EC Simvastatin
Jak2V617F
BM-ECVehicule
Jak2V617F
BM-ECSimvastatin
0
5
10
15
20
25
Haem
og
lob
in level (g
/dL
)
ns
Jak2V617F
BM-ECVehicule
Jak2V617F
BM-ECSimvastatin
0
1000
2000
3000
4000
Pla
tele
ts c
ou
nt
(10
9/L
)ns
Jak2V617F
BM-ECVehicule
Jak2V617F
BM-ECSimvastatin
0
10
20
30
Wh
ite b
loo
d c
ells c
ou
nt
(10
9/L
) *
Jak2V617F
BM-ECVehicule
Jak2V617F
BM-ECSimvastatin
2.0
2.5
3.0
3.5
4.0
Sp
leen
weig
ht/
bo
dy w
eig
ht
(%
) ns
K L M N O
Discussion
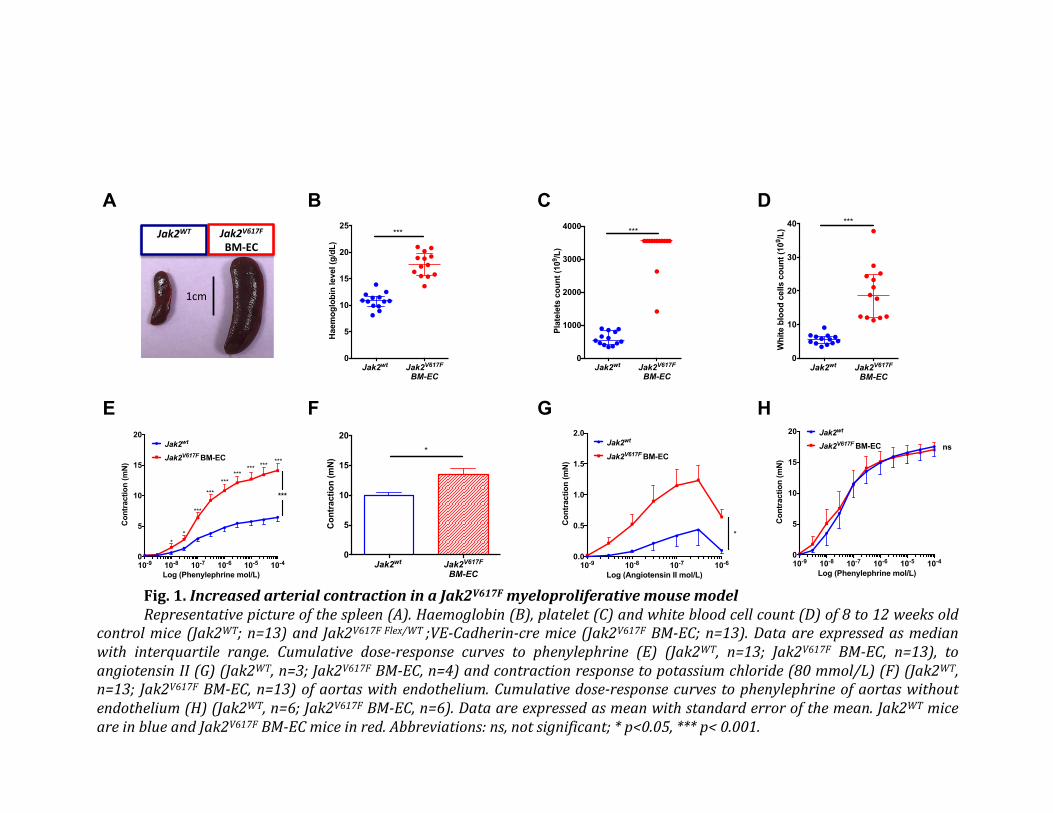
This study demonstrated that JAK2V617F
red blood cell derived microvesicles strongly
increased arterial contraction in response to vasoconstrictive agents, possibly accounting for
arterial events associated with MPNs. This effect was due to an augmented oxidative stress in
arterial endothelium and a decreased availability of NO. Simvastatin, a drug with anti-oxidant
properties, improved arterial contraction.
The first major finding of our study is the demonstration that JAK2V617F
MPN induces a
considerable increase in arterial contraction. This finding suggests a vasospastic phenomenon
associated with MPN and thus represents a paradigm shift in MPNs where arterial events were
only seen as a result of a thrombotic process (24). Our results obtained ex vivo would explain the
10 times higher incidence of arterial events in patients with polycythaemia vera than in the
general population (25, 26) and the high prevalence of myocardial infarction without significant
coronary stenosis by angiography in patients with MPN (14). Arterial spasm is an
underdiagnosed phenomenon that can happen in patients without atherosclerosis, but is also
favoured by underlying non-stenotic atherosclerotic plaques. This suggests that the effect we
observed might not only account for myocardial infarction without significant coronary stenosis
reported in patients with MPN, but might also more widely favour arterial events in patients with
atherosclerotic plaques and MPN (17, 18). Moreover, arterial spasm not only occurs in coronary
arteries, but also in brain arteries (16, 27–29). The mechanisms underlying arterial spasm are not
completely elucidated, but arterial contraction plays a central role (16, 28–30), which is
concordant with our findings, since we observed a pronounced increase in contraction in
response to different vasoconstrictive agents. We found only a mild impairment in arterial
dilatation, which is in line with the altered endothelial dependant flow mediated vasodilatation
reported in patients with polycythaemia vera, in the absence of overt arterial disease (31).
101
The second major finding of our work is the identification of JAK2V617F
red blood cell
derived microvesicles as responsible for the increased arterial contraction associated with MPN.
Importantly, we observed this effect with JAK2V617F
red blood cell microvesicles from mice, but
also with microvesicles isolated from patients carrying JAK2V617F
. We thus highlight here a
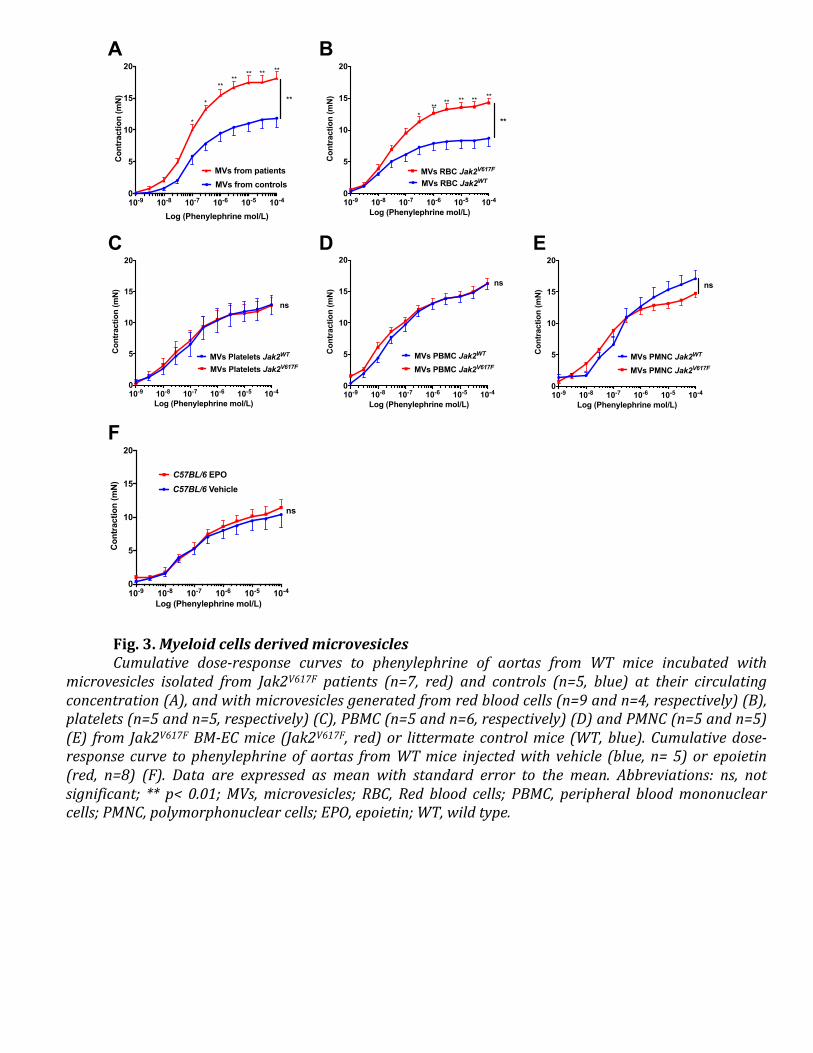
crucial vascular role of microvesicles in MPNs, beyond their so far described implication in
coagulation in this setting (32–36). Although patients with MPNs have higher circulating levels
of microvesicles than healthy individuals, we assessed vascular reactivity using the same
concentrations of microvesicles for both groups, suggesting that microvesicle composition, and
not concentration, accounts for the observed vascular effect (32, 33, 37–40). A specificity of
JAK2V617F
red blood cells derived microvesicles is also supported by our observation that
JAK2V617F
platelets or white blood cells derived microvesicles did not increase arterial
contraction, and that polycythaemia induced by epoietin without JAK2V617F
had no effect on
arterial contraction. These results are reminiscent of epidemiological studies showing that MPN
patients with JAK2V617F
have higher haematocrit level (41, 42) and a higher risk of
cardiovascular events than MPN patients without JAK2V617F
(42–46).
Finally, in our work we demonstrated that NO pathway inhibition and increased
endothelial oxidative stress are implicated in this increased arterial contraction in MPN. Several
groups reported high levels of circulating reactive oxygen species products (47–49) and low
antioxidant status in MPN (48) (50), but endothelial oxidative stress had never been investigated.
Besides explaining how MPN induce this increased arterial contraction, this observation opens
new potential therapeutic perspectives to prevent MPN. Indeed, statins being known to play a
protective role on endothelial function and on oxidative stress, we tested this drug and observed a
strong improvement in arterial response to vasocontricting agent in our MPN mouse model (22,
102
23). Simvastatin being a well-known and easily accessible drug, these results thus pave the way
for testing simvastatin to prevent arterial events in patients with MPN. We also tested available
treatments for MPN and observed that hydroxyurea, but not ruxolitinib, improved arterial
contraction. This difference might be explained by the fact that hydroxyurea decreased red blood
cell count in our mouse model whereas ruxolitinib did not (51, 52). Another explanation could be
that hydroxyurea has been shown to enhance NO release by endothelial cells while such an effect
has not been reported with ruxolitinib (53).
In conclusion, our study showed that red blood cell derived microvesicles are responsible
for an increased arterial contraction in Jak2V617F
MPNs. This effect implicated NO and
endothelial oxidative stress. Simvastatin appears as an original new approach to prevent arterial
1. J. L. Spivak, D. L. Longo, Ed. Myeloproliferative Neoplasms, N. Engl. J. Med. 376, 2168–2181 (2017).
2. W. Vainchenker, R. Kralovics, Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms, Blood 129, 667–679 (2017).
3. P. J. Campbell, A. R. Green, The myeloproliferative disorders, N. Engl. J. Med. 355, 2452–2466 (2006).
4. S. Sozer, M. I. Fiel, T. Schiano, M. Xu, J. Mascarenhas, R. Hoffman, The presence of JAK2V617F mutation in the liver endothelial cells of patients with Budd-Chiari syndrome, Blood 113, 5246–5249 (2009).
5. V. Rosti, L. Villani, R. Riboni, V. Poletto, E. Bonetti, L. Tozzi, G. Bergamaschi, P. Catarsi, E. Dallera, F. Novara, M. Massa, R. Campanelli, G. Fois, B. Peruzzi, M. Lucioni, P. Guglielmelli, A. Pancrazzi, G. Fiandrino, O. Zuffardi, U. Magrini, M. Paulli, A. M. Vannucchi, G. Barosi, Associazione Italiana per la Ricerca sul Cancro Gruppo Italiano Malattie Mieloproliferative (AGIMM) investigators, Spleen endothelial cells from patients with myelofibrosis harbor the JAK2V617F mutation, Blood 121, 360–368 (2013).
6. V. Rosti, E. Bonetti, G. Bergamaschi, R. Campanelli, P. Guglielmelli, M. Maestri, U. Magrini, M. Massa, C. Tinelli, G. Viarengo, L. Villani, M. Primignani, A. M. Vannucchi, F. Frassoni, G. Barosi, on behalf of the A. Investigators, High Frequency of Endothelial Colony Forming Cells Marks a Non-Active Myeloproliferative Neoplasm with High Risk of Splanchnic Vein Thrombosis, PLOS ONE 5, e15277 (2010).
7. Helman Ricardo, Pereira Welbert de O., Marti Luciana C., Campregher Paulo V., Puga Renato Davi, Hamerschlak Nelson, Chiattone Carlos S., Santos Fabio Pires de S., Granulocyte whole exome sequencing and endothelial JAK2V617F in patients with JAK2V617F positive Budd‐Chiari Syndrome without myeloproliferative neoplasm, Br. J. Haematol. 180, 443–445 (2018).
8. L. Teofili, M. Martini, M. G. Iachininoto, S. Capodimonti, E. R. Nuzzolo, L. Torti, T. Cenci, L. M. Larocca, G. Leone, Endothelial progenitor cells are clonal and exhibit the JAK2(V617F) mutation in a subset of thrombotic patients with Ph-negative myeloproliferative neoplasms, Blood 117, 2700–2707 (2011).
9. R. Marchioli, G. Finazzi, R. Landolfi, J. Kutti, H. Gisslinger, C. Patrono, R. Marilus, A. Villegas, G. Tognoni, T. Barbui, Vascular and Neoplastic Risk in a Large Cohort of Patients With Polycythemia Vera, J. Clin. Oncol. 23, 2224–2232 (2005).
10. P. J. Campbell, L. M. Scott, G. Buck, K. Wheatley, C. L. East, J. T. Marsden, A. Duffy, E. M. Boyd, A. J. Bench, M. A. Scott, G. S. Vassiliou, D. W. Milligan, S. R. Smith, W. N. Erber, D. Bareford, B. S. Wilkins, J. T. Reilly, C. N. Harrison, A. R. Green, Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: a prospective study, The Lancet 366, 1945–1953 (2005).
31
11. Buxhofer‐Ausch Veronika, Gisslinger Heinz, Thiele Jürgen, Gisslinger Bettina, Kvasnicka Hans‐Michael, Müllauer Leonhard, Frantal Sophie, Carobbio Alessandra, Passamonti Francesco, Rumi Elisa, Ruggeri Marco, Rodeghiero Francesco, Randi Maria L., Bertozzi Irene, Vannucchi Alessandro M., Antonioli Elisabetta, Finazzi Guido, Gangat Naseema, Tefferi Ayalew, Barbui Tiziano, Leukocytosis as an important risk factor for arterial thrombosis in WHO‐defined early/prefibrotic myelofibrosis: An international study of 264 patients, Am. J. Hematol. 87, 669–672 (2012).
12. Montanaro Marco, Latagliata Roberto, Cedrone Michele, Spadea Antonio, Rago Angela, Giandomenico Jonny, Spirito Francesca, Porrini Raffaele, Muro Marianna, Leonetti Sabrina Crescenzi, Villivà Nicoletta, Gregoris Cinzia, Breccia Massimo, Montefusco Enrico, Santoro Cristina, Cimino Giuseppe, Majolino Ignazio, Mazzucconi Maria Gabriella, Alimena Giuliana, Andriani Alesssandro, Thrombosis and survival in essential thrombocythemia: A regional study of 1,144 patients, Am. J. Hematol. 89, 542–546 (2014).
13. V. D. Stefano, I. Martinelli, Splanchnic vein thrombosis: clinical presentation, risk factors and treatment, Intern. Emerg. Med. 5, 487–494 (2010).
14. É. Pósfai, I. Marton, Z. Borbényi, A. Nemes, Myocardial infarction as a thrombotic complication of essential thrombocythemia and polycythemia vera, Anatol. J. Cardiol. 16, 397–402 (2016).
15. A. I. Larsen, P. D. Galbraith, W. A. Ghali, C. M. Norris, M. M. Graham, M. L. Knudtson, Characteristics and outcomes of patients with acute myocardial infarction and angiographically normal coronary arteries, Am. J. Cardiol. 95, 261–263 (2005).
16. S. Agewall, J. F. Beltrame, H. R. Reynolds, A. Niessner, G. Rosano, A. L. P. Caforio, R. De Caterina, M. Zimarino, M. Roffi, K. Kjeldsen, D. Atar, J. C. Kaski, U. Sechtem, P. Tornvall, ESC working group position paper on myocardial infarction with non-obstructive coronary arteries, Eur. Heart J. 38, 143–153 (2017).
17. M. J. Davies, The pathophysiology of acute coronary syndromes, Heart 83, 361–366 (2000).
18. F. Crea, P. Libby, Acute Coronary Syndromes: The Way Forward From Mechanisms to Precision Treatment, Circulation 136, 1155–1166 (2017).
19. E. Oberlin, B. El Hafny, L. Petit-Cocault, M. Souyri, Definitive human and mouse hematopoiesis originates from the embryonic endothelium: a new class of HSCs based on VE-cadherin expression, Int. J. Dev. Biol. 54, 1165–1173 (2010).
20. C. M. Boulanger, X. Loyer, P.-E. Rautou, N. Amabile, Extracellular vesicles in coronary artery disease, Nat. Rev. Cardiol. 14, 259–272 (2017).
21. P. M. Vanhoutte, Y. Zhao, A. Xu, S. W. S. Leung, Thirty Years of Saying NO: Sources, Fate, Actions, and Misfortunes of the Endothelium-Derived Vasodilator Mediator, Circ. Res. 119, 375–396 (2016).
22. S. Sen-Banerjee, S. Mir, Z. Lin, A. Hamik, G. B. Atkins, H. Das, P. Banerjee, A. Kumar, M. K. Jain, Kruppel-Like Factor 2 as a Novel Mediator of Statin Effects in Endothelial Cells, Circulation 112, 720–726 (2005).
32
23. R. P. Mason, M. F. Walter, R. F. Jacob, Effects of HMG-CoA reductase inhibitors on endothelial function: role of microdomains and oxidative stress, Circulation 109, II34-41 (2004).
24. T. Barbui, G. Finazzi, A. Falanga, Myeloproliferative neoplasms and thrombosis, Blood 122, 2176–2184 (2013).
25. R. Landolfi, L. Di Gennaro, T. Barbui, V. De Stefano, G. Finazzi, R. Marfisi, G. Tognoni, R. Marchioli, European Collaboration on Low-Dose Aspirin in Polycythemia Vera (ECLAP), Leukocytosis as a major thrombotic risk factor in patients with polycythemia vera, Blood 109, 2446–2452 (2007).
26. B. M. Kaess, S. R. Preis, A. Beiser, D. B. Sawyer, T. C. Chen, S. Seshadri, R. S. Vasan, Circulating vascular endothelial growth factor and the risk of cardiovascular events, Heart Br.
Card. Soc. 102, 1898–1901 (2016).
27. O. Y. Bang, K. Toyoda, J. F. Arenillas, L. Liu, J. S. Kim, Intracranial Large Artery Disease of Non-Atherosclerotic Origin: Recent Progress and Clinical Implications, J. Stroke 20, 208–217 (2018).
28. C. N. Bairey Merz, C. J. Pepine, M. N. Walsh, J. L. Fleg, Ischemia and No Obstructive Coronary Artery Disease (INOCA): Developing Evidence-Based Therapies and Research Agenda for the Next Decade, Circulation 135, 1075–1092 (2017).
29. J. F. Beltrame, F. Crea, J. C. Kaski, H. Ogawa, P. Ong, U. Sechtem, H. Shimokawa, C. N. Bairey Merz, Coronary Vasomotion Disorders International Study Group (COVADIS), The Who, What, Why, When, How and Where of Vasospastic Angina, Circ. J. Off. J. Jpn. Circ. Soc. 80, 289–298 (2016).
30. P. Ong, A. Aziz, H. S. Hansen, E. Prescott, A. Athanasiadis, U. Sechtem, Structural and Functional Coronary Artery Abnormalities in Patients With Vasospastic Angina Pectoris, Circ. J.
Off. J. Jpn. Circ. Soc. 79, 1431–1438 (2015).
31. T. Neunteufl, S. Heher, T. Stefenelli, I. Pabinger, H. Gisslinger, Endothelial dysfunction in patients with polycythaemia vera, Br. J. Haematol. 115, 354–359 (2001).
32. A. Charpentier, A. Lebreton, A. Rauch, A. Bauters, N. Trillot, O. Nibourel, V. Tintillier, M. Wemeau, J.-L. Demory, C. Preudhomme, B. Jude, T. Lecompte, N. Cambier, S. Susen, Microparticle phenotypes are associated with driver mutations and distinct thrombotic risks in essential thrombocythemia, Haematologica 101, e365-368 (2016).
33. X. Tan, J. Shi, Y. Fu, C. Gao, X. Yang, J. Li, W. Wang, J. Hou, H. Li, J. Zhou, Role of erythrocytes and platelets in the hypercoagulable status in polycythemia vera through phosphatidylserine exposure and microparticle generation, Thromb. Haemost. 109, 1025–1032 (2013).
34. H. Baccouche, M. Ben Jemaa, A. Chakroun, S. Chadi, S. Mahjoub, I. Sfar, Y. Gorgi, N. Ben Romdhane, The evaluation of the relevance of thrombin generation and procoagulant activity in thrombotic risk assessment in BCR-ABL-negative myeloproliferative neoplasm patients, Int. J.
Lab. Hematol. 39, 502–507 (2017).
33
35. J. Duchemin, V. Ugo, J.-C. Ianotto, L. Lecucq, B. Mercier, J.-F. Abgrall, Increased circulating procoagulant activity and thrombin generation in patients with myeloproliferative neoplasms, Thromb. Res. 126, 238–242 (2010).
36. M. Marchetti, C. J. Tartari, L. Russo, M. Panova-Noeva, A. Leuzzi, A. Rambaldi, G. Finazzi, B. Woodhams, A. Falanga, Phospholipid-dependent procoagulant activity is highly expressed by circulating microparticles in patients with essential thrombocythemia, Am. J. Hematol. 89, 68–73 (2014).
37. M. C. Trappenburg, M. van Schilfgaarde, M. Marchetti, H. M. Spronk, H. ten Cate, A. Leyte, W. E. Terpstra, A. Falanga, Elevated procoagulant microparticles expressing endothelial and platelet markers in essential thrombocythemia, Haematologica 94, 911–918 (2009).
38. W. Zhang, J. Qi, S. Zhao, W. Shen, L. Dai, W. Han, M. Huang, Z. Wang, C. Ruan, D. Wu, Y. Han, Clinical significance of circulating microparticles in Ph− myeloproliferative neoplasms, Oncol. Lett. 14, 2531–2536 (2017).
39. M.-P. Moles-Moreau, C. Ternisien, A. Tanguy-Schmidt, F. Boyer, M. Gardembas, M. Dib, A. Ponthieux, P. Guardiola, N. Ifrah, M. Hunault-Berger, Flow cytometry-evaluated platelet CD36 expression, reticulated platelets and platelet microparticles in essential thrombocythaemia and secondary thrombocytosis, Thromb. Res. 126, e394-396 (2010).
40. J. Kissova, P. Ovesna, A. Bulikova, J. Zavřelova, M. Penka, Increasing procoagulant activity of circulating microparticles in patients with Philadelphia-negative myeloproliferative neoplasms: a single-centre experience, Blood Coagul. Fibrinolysis Int. J. Haemost. Thromb. 26, 448–453 (2015).
41. E. Rumi, D. Pietra, V. Ferretti, T. Klampfl, A. S. Harutyunyan, J. D. Milosevic, N. C. Them, T. Berg, C. Elena, I. C. Casetti, JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes, Blood 123, 1544–1551 (2014).
42. A. Tefferi, E. A. Wassie, T. L. Lasho, C. Finke, A. A. Belachew, R. P. Ketterling, C. A. Hanson, A. Pardanani, N. Gangat, A. P. Wolanskyj, Calreticulin mutations and long-term survival in essential thrombocythemia, Leukemia 28, 2300–2303 (2014).
43. Y. C. Elala, T. L. Lasho, N. Gangat, C. Finke, D. Barraco, M. Haider, A. K. A. Hussein, C. A. Hanson, R. P. Ketterling, A. Pardanani, A. Tefferi, Calreticulin variant stratified driver mutational status and prognosis in essential thrombocythemia, Am. J. Hematol. 91, 503–506 (2016).
44. A. Carobbio, J. Thiele, F. Passamonti, E. Rumi, M. Ruggeri, F. Rodeghiero, M. L. Randi, I. Bertozzi, A. M. Vannucchi, E. Antonioli, H. Gisslinger, V. Buxhofer-Ausch, G. Finazzi, N. Gangat, A. Tefferi, T. Barbui, Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: an international study of 891 patients, Blood 117, 5857–5859 (2011).
45. G. Finazzi, A. Carobbio, P. Guglielmelli, C. Cavalloni, S. Salmoiraghi, A. M. Vannucchi, M. Cazzola, F. Passamonti, A. Rambaldi, T. Barbui, Calreticulin mutation does not modify the IPSET score for predicting the risk of thrombosis among 1150 patients with essential thrombocythemia, Blood 124, 2611–2612 (2014).
34
46. G. Rotunno, C. Mannarelli, P. Guglielmelli, A. Pacilli, A. Pancrazzi, L. Pieri, T. Fanelli, A. Bosi, A. M. Vannucchi, Impact of calreticulin mutations on clinical and hematological phenotype and outcome in essential thrombocythemia, Blood 123, 1552–1555 (2014).
47. C. Musolino, A. Allegra, A. Saija, A. Alonci, S. Russo, G. Spatari, G. Penna, D. Gerace, M. Cristani, A. David, S. Saitta, S. Gangemi, Changes in advanced oxidation protein products, advanced glycation end products, and s-nitrosylated proteins, in patients affected by polycythemia vera and essential thrombocythemia, Clin. Biochem. 45, 1439–1443 (2012).
48. A. Durmus, A. Mentese, M. Yilmaz, A. Sumer, I. Akalin, C. Topal, A. Alver, Increased oxidative stress in patients with essential thrombocythemia, Eur. Rev. Med. Pharmacol. Sci. 17, 2860–2866 (2013).
49. C. Vener, C. Novembrino, F. B. Catena, N. S. Fracchiolla, U. Gianelli, F. Savi, F. Radaelli, E. Fermo, A. Cortelezzi, S. Lonati, M. Menegatti, G. L. Deliliers, Oxidative stress is increased in primary and post-polycythemia vera myelofibrosis, Exp. Hematol. 38, 1058–1065 (2010).
50. H. C. Hasselbalch, M. Thomassen, C. H. Riley, L. Kjær, T. S. Larsen, M. K. Jensen, O. W. Bjerrum, T. A. Kruse, V. Skov, Whole blood transcriptional profiling reveals deregulation of oxidative and antioxidative defence genes in myelofibrosis and related neoplasms. Potential implications of downregulation of Nrf2 for genomic instability and disease progression, PloS
One 9, e112786 (2014).
51. A. Angona, A. Alvarez-Larrán, B. Bellosillo, R. Longarón, C. Fernández-Rodríguez, C. Besses, Dynamics of JAK2 V617F allele burden of CD34+ haematopoietic progenitor cells in patients treated with ruxolitinib, Br. J. Haematol. 172, 639–642 (2016).
52. W. Vainchenker, E. Leroy, L. Gilles, C. Marty, I. Plo, S. N. Constantinescu, JAK inhibitors for the treatment of myeloproliferative neoplasms and other disorders, F1000Research 7, 82 (2018).
53. V. P. Cokic, B. B. Beleslin-Cokic, M. Tomic, S. S. Stojilkovic, C. T. Noguchi, A. N. Schechter, Hydroxyurea induces the eNOS-cGMP pathway in endothelial cells, Blood 108, 184–191 (2006).
54. C. M. Boulanger, A. Scoazec, T. Ebrahimian, P. Henry, E. Mathieu, A. Tedgui, Z. Mallat, Circulating Microparticles From Patients With Myocardial Infarction Cause Endothelial Dysfunction, Circulation 104, 2649–2652 (2001).
55. N. Amabile, Circulating Endothelial Microparticles Are Associated with Vascular Dysfunction in Patients with End-Stage Renal Failure, J. Am. Soc. Nephrol. 16, 3381–3388 (2005).
56. P.-E. Rautou, J. Bresson, Y. Sainte-Marie, A.-C. Vion, V. Paradis, J.-M. Renard, C. Devue, C. Heymes, P. Letteron, L. Elkrief, D. Lebrec, D. Valla, A. Tedgui, R. Moreau, C. M. Boulanger, Abnormal plasma microparticles impair vasoconstrictor responses in patients with cirrhosis, Gastroenterology 143, 166-176.e6 (2012).
57. C. Marty, C. Lacout, N. Droin, J.-P. Le Couédic, V. Ribrag, E. Solary, W. Vainchenker, J.-L. Villeval, I. Plo, A role for reactive oxygen species in JAK2
58. Y. Wang, M. Nakayama, M. E. Pitulescu, T. S. Schmidt, M. L. Bochenek, A. Sakakibara, S. Adams, A. Davy, U. Deutsch, U. Lüthi, A. Barberis, L. E. Benjamin, T. Mäkinen, C. D. Nobes, R. H. Adams, Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis, Nature 465, 483–486 (2010).
59. C. Heymes, A. Habib, D. Yang, E. Mathieu, F. Marotte, J. Samuel, C. M. Boulanger, Cyclo-oxygenase-1 and -2 contribution to endothelial dysfunction in ageing, Br. J. Pharmacol. 131, 804–810 (2000).
60. A. Payancé, G. Silva-Junior, J. Bissonnette, M. Tanguy, B. Pasquet, C. Levi, O. Roux, O. Nekachtali, A. Baiges, V. Hernández-Gea, C. Laouénan, D. Lebrec, M. Albuquerque, V. Paradis, R. Moreau, D. Valla, F. Durand, C. M. Boulanger, J.-C. Garcia-Pagan, P.-E. Rautou, Hepatocyte microvesicle levels improve prediction of mortality in patients with cirrhosis, Hepatol. Baltim.
Md (2018), doi:10.1002/hep.29903.
61. L. Kubovcakova, P. Lundberg, J. Grisouard, H. Hao-Shen, V. Romanet, R. Andraos, M. Murakami, S. Dirnhofer, K.-U. Wagner, T. Radimerski, others, Differential effects of hydroxyurea and INC424 on mutant allele burden and myeloproliferative phenotype in a JAK2-V617F polycythemia vera mouse model, Blood 121, 1188–1199 (2013).
62. S. Maschalidi, F. E. Sepulveda, A. Garrigue, A. Fischer, G. de Saint Basile, Therapeutic effect of JAK1/2 blockade on the manifestations of hemophagocytic lymphohistiocytosis in mice, Blood 128, 60–71 (2016).
63. R. Kou, T. Shiroto, J. L. Sartoretto, T. Michel, Suppression of Gαs synthesis by simvastatin treatment of vascular endothelial cells, J. Biol. Chem. 287, 2643–2651 (2012).
64. L. Lamrani, C. Lacout, V. Ollivier, C. V. Denis, E. Gardiner, B. Ho Tin Noe, W. Vainchenker, J.-L. Villeval, M. Jandrot-Perrus, Hemostatic disorders in a JAK2V617F-driven mouse model of myeloproliferative neoplasm, Blood 124, 1136–1145 (2014).
36
Acknowledgment: We thank members of the INSERM UMR-970 animal facility (ERI) for
animal handling and breeding. We thank R. Adams for having provided Cadherin5Cre-ERT2
mice.
We also thank A. Payancé, K. Zekrini and D. Rezigue for their help in identifying the patients
and M. Salama for his help with myography experiments.
Author Contributions: J.P., and P-E.R. designed the experiments and wrote the manuscript.
J.P., H.D., F.C. performed myography experiments. J.P. performed oxidative stress experiments,
M.T. generated microvesicles. J-L.V., C.J. and M.S. provided transgenic mice. M-B.E-M., C.D.
and S.H. characterized the first mouse model. J.L. managed the mouse colony. M.K. performed
mTmG experiments. A.P. included the patients and healthy controls. P-E.R. obtained funding for
the project. All authors discussed and critically revised the manuscript.
Financial support: This work was supported by the Agence Nationale pour la Recherche (ANR
14 CE35 0022 03/ JAK-POT) and J.P by the “poste accueil INSERM”.
Conflict-of-interest disclosure: The authors declare no competing financial interest.
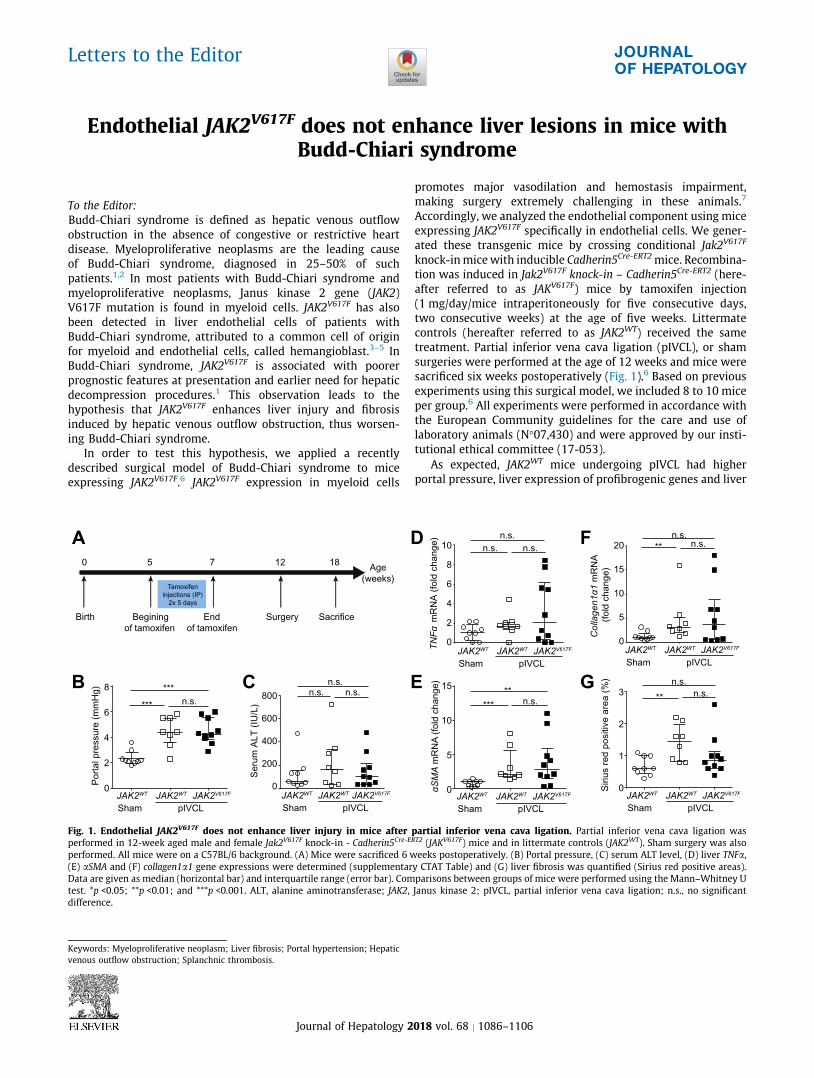
tion was induced in Jak2V617F knock-in – Cadherin5Cre-ERT2 (here-
after referred to as JAKV617F) mice by tamoxifen injection
(1 mg/day/mice intraperitoneously for five consecutive days,
two consecutive weeks) at the age of five weeks. Littermate
controls (hereafter referred to as JAK2WT) received the same
treatment. Partial inferior vena cava ligation (pIVCL), or sham
surgeries were performed at the age of 12 weeks and mice were
sacrificed six weeks postoperatively (Fig. 1).6 Based on previous
experiments using this surgical model, we included 8 to 10 mice
per group.6 All experiments were performed in accordance with
the European Community guidelines for the care and use of
laboratory animals (N!07,430) and were approved by our insti-
tutional ethical committee (17-053).
As expected, JAK2WT mice undergoing pIVCL had higher
portal pressure, liver expression of profibrogenic genes and liver
TN
Fα
mR
NA
(fo
ldch
an
ge
)
Co
llag
en
1α
1 m
RN
A
(fo
ldch
an
ge
)
Se
rum
AL
T(I
U/L
)
Siriu
sre
dp
ositiv
ea
rea
(%)
8
6
4
2
0
Port
al pre
ssure
(m
mH
g)
n.s.
JAK2WT
Sham pIVCL
JAK2WT JAK2V617F
***
***800
600
400
200
0
n.s.n.s. n.s.
JAK2WT
Sham pIVCL
JAK2WT JAK2V617F JAK2WT
Sham pIVCL
JAK2WT JAK2V617F
JAK2WT
Sham pIVCL
JAK2WT JAK2V617F JAK2WT
Sham pIVCL
JAK2WT JAK2V617F
JAK2WT
Sham pIVCL
JAK2WT JAK2V617F
n.s.
n.s.n.s.
n.s.n.s.
n.s.
n.s.n.s.
**
****
***
10
8
6
4
2
0
20
15
10
5
0
αS
MA
mR
NA
(fo
ld c
hange)
15
10
5
0
3
2
1
0
0 5 7 12 18Age
(weeks)
Birth Begining
of tamoxifen
End
of tamoxifen
Surgery Sacrifice
Tamoxifen
injections (IP)
2x 5 days
A
B C
D
E
F
G
Fig. 1. Endothelial JAK2V617F does not enhance liver injury in mice after partial inferior vena cava ligation. Partial inferior vena cava ligation was
performed in 12-week aged male and female Jak2V617F knock-in - Cadherin5Cre-ERT2 (JAKV617F) mice and in littermate controls (JAK2WT). Sham surgery was also
performed. All mice were on a C57BL/6 background. (A) Mice were sacrificed 6 weeks postoperatively. (B) Portal pressure, (C) serum ALT level, (D) liver TNFa,
(E) aSMA and (F) collagen1a1 gene expressions were determined (supplementary CTAT Table) and (G) liver fibrosis was quantified (Sirius red positive areas).
Data are given as median (horizontal bar) and interquartile range (error bar). Comparisons between groups of mice were performed using the Mann–Whitney U
1 Inserm,U970,ParisCardiovascularResearchCenter -PARCC,UniversitéParisDescartes,SorbonneParisCité,Paris,France;2DHUUnity,PôledesMaladiesdel’AppareilDigestif,Serviced’Hépatologie,CentredeRéférencedesMaladiesVasculairesduFoie,HôpitalBeaujon,AP-HP,Clichy,France;3Centred’InvestigationsCliniques,HôpitalStLouis,AP-HP,Clichy,France;4BarcelonaHepaticHemodynamicLaboratory,LiverUnit,HospitalClínic,IDIBAPS,Barcelona,Spain;5Serviced’HématologieBiologique,HôpitalBeaujon,AP-HP,Clichy,France;6Serviced’Hépato-gastroentérologie,CHURouen,Rouen,France;7Liver-GastroenterologyDepartment,UniversityHospitalandPaulSabatierUniversity,Toulouse,France;8 UPMC, Univ Paris 06, Groupe de Recherche Clinique sur les Myéloproliférations Aiguës et ChroniquesMYPAC,Paris,France;9Laboratoired’ImmunologieetHématologieBiologique,HôpitalSaint-Antoine,AP-HP,Paris,France;10InsermU1149,CentredeRecherche sur l’Inflammation (CRI), Paris, Université Paris 7-Denis-Diderot, Clichy, UFR de Médecine, Paris,France;11UniversitéParisDiderot,SorbonnePariscité,Paris,France;12CentrodeInvestigaciónBiomédicaenReddeEnfermedadesHepáticasyDigestivas(CIBERehd),Spain;13HematologyDepartment,HospitalClínic,IDIBAPS,UniversityofBarcelona,Spain;14HematopathologyUnit,HospitalClínic,IDIBAPS,CIBERONC,Spain
Lay summaryMutations of the CALR gene are detected
in 0 to 2% of patients with SVT, thus the
utility of systematic CALR mutation test-
ing to diagnose MPN is questionable. This
study demonstrates that CALR mutations
testing can be restricted to patients with
SVT, a spleen height #16 cm, a platelet
count >200"109/L, and no JAK2V617F. This
strategy avoids 96% of unnecessary CALR
mutations testing.
http://dx.doi.org/10.1016/j.jhep.2017.04.021! 2017 European Association for the Study of the Liver. Published by Elsevier B.V. All rights reserved. J. Hepatol. 2017, 67, 501–507
Research Article
Selective testing for calreticulin gene mutations in patientswith splanchnic vein thrombosis: A prospective cohort study
Johanne Poisson1, Aurélie Plessier2, Jean-Jacques Kiladjian3, Fanny Turon4, Bruno Cassinat3,Annalisa Andreoli3, Emmanuelle De Raucourt5, Odile Goria6, Kamal Zekrini3, Christophe Bureau7,
Florence Lorre8,9, Francisco Cervantes13, Dolors Colomer14, François Durand2,10,11,Juan-Carlos Garcia-Pagan4,12, Nicole Casadevall8,9, Dominique-Charles Valla2,10,11,Pierre-Emmanuel Rautou1,2,11,⇑,y, Christophe Marzac8,9,y, for the French national
network for vascular liver diseases
1Inserm, U970, Paris Cardiovascular Research Center - PARCC, Université Paris Descartes, Sorbonne Paris Cité, Paris, France; 2DHU Unity, Pôledes Maladies de l’Appareil Digestif, Service d’Hépatologie, Centre de Référence des Maladies Vasculaires du Foie, Hôpital Beaujon, AP-HP, Clichy,France; 3Centre d’Investigations Cliniques, Hôpital St Louis, AP-HP, Clichy, France; 4Barcelona Hepatic Hemodynamic Laboratory, Liver Unit,
Hospital Clínic, IDIBAPS, Barcelona, Spain; 5Service d’Hématologie Biologique, Hôpital Beaujon, AP-HP, Clichy, France; 6Serviced’Hépato-gastroentérologie, CHU Rouen, Rouen, France; 7Liver-Gastroenterology Department, University Hospital and Paul Sabatier University,Toulouse, France; 8UPMC, Univ Paris 06, GRC n!7, Groupe de Recherche Clinique sur les Myéloproliférations Aiguës et Chroniques MYPAC,
Paris, France; 9Laboratoire d’Immunologie et Hématologie Biologique, Hôpital Saint-Antoine, AP-HP, Paris, France; 10Inserm U1149, Centre deRecherche sur l’Inflammation (CRI), Paris, Université Paris 7-Denis-Diderot, Clichy, UFR de Médecine, Paris, France; 11Université Paris Diderot,Sorbonne Paris cité, Paris, France; 12Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBERehd), Spain;
13Hematology Department, Hospital Clínic, IDIBAPS, University of Barcelona, Spain; 14Hematopathology Unit, Hospital Clínic, IDIBAPS,CIBERONC, Spain
Background and Aims:Myeloproliferative neoplasms (MPN) arethe leading cause of splanchnic vein thrombosis (SVT). Januskinase 2 gene (JAK2)V617F mutations are found in 80 to 90% ofpatients with SVT and MPN. Mutations of the calreticulin (CALR)gene have also been reported. However, as their prevalenceranges from 0 to 2%, the utility of routine testing is questionable.This study aimed to identify a group of patients with SVT at highrisk of harboring CALR mutations and thus requiring this genetictesting.Methods: CALR, JAK2V617F and thrombopoietin receptor gene(MPL) mutations were analysed in a test cohort that included312 patients with SVT. Criteria to identify patients at high riskof CALR mutations in this test cohort was used and evaluated ina validation cohort that included 209 patients with SVT.Results: In the test cohort, 59 patients had JAK2V617F, five hadCALR and none had MPL mutations. Patients with CALR mutationshad higher spleen height and platelet count than patients with-out these mutations. All patients with CALR mutations had aspleen height P16 cm and platelet count [200!109/L. These
criteria had a positive predictive value of 56% (5/9) and a negativepredictive value of 100% (0/233) for the identification of CALR
mutations. In the validation cohort, these criteria had a positivepredictive value of 33% (2/6) and a negative predictive value of99% (1/96).Conclusion: CALR mutations should be tested in patients withSVT, a spleen height P16 cm, platelet count [200!109/L, andno JAK2V617F. This strategy avoids 96% of unnecessary CALR muta-tions testing.Lay summary:Mutations of the CALR gene are detected in 0 to 2%of patients with SVT, thus the utility of systematic CALR mutationtesting to diagnose MPN is questionable. This study demonstratesthat CALR mutations testing can be restricted to patients withSVT, a spleen height P16 cm, a platelet count[200!109/L, andno JAK2V617F. This strategy avoids 96% of unnecessary CALR
mutations testing." 2017 European Association for the Study of the Liver. Publishedby Elsevier B.V. All rights reserved.
Introduction
Splanchnic vein thrombosis (SVT) indicates Budd-Chiari syndrome(BCS) and portal venous system thrombosis (PVT). Primary BCS is arare disorder defined as a blocked hepatic venous outflow tract atvarious levels from small hepatic veins to the terminal portion ofthe inferior vena cava.1 Non-malignant non-cirrhotic extrahepaticPVT is characterized by thrombus development in the main portal
vein and/or its right or left branches and/or splenic or mesentericveins, or by the permanent obliteration that results from a priorthrombus.1 The pathogenesis of SVT is largely dependent on thepresence of systemic prothrombotic conditions that promotethrombus formation in the respective splanchnic veins.2,3
Myeloproliferative neoplasms (MPNs) are the leading cause ofSVT and are diagnosed in 25 to 50% of patients with SVT.4 In mostpatients with SVT and MPN, Janus kinase 2 gene (JAK2)V617F muta-tion is found. In 10% to 20% of patients with SVT this specificmutation is absent, whereas bone marrow biopsy or assessmentof endogenous erythroid colonies formation provide evidencefor MPN.5 Mutations across JAK2 exon 12 or the thrombopoietinreceptor gene (MPL) are rarely identified in patients with SVT.5
Two independent groups described heterozygous calreticulin(CALR) mutations as the second most prevalent acquired geneticalteration in essential thrombocythemia and primary myelofibro-sis.6,7 CALR mutations are mutually exclusive of JAK2 and MPL
mutations. Thereafter, CALR mutations have been found in 0 to2% of patients with SVT.8–17 Although CALR mutations appear tobe rare in patients with SVT and their detection not readily acces-sible to all centers, their identification influences patients’ clinicalmanagement. This prompted us to take advantage of a largeprospective cohort of SVT patients to identify the subgroup atthe highest risk of harboring CALR mutations and thus requiringthis genetic testing.
Patients and methods
Inclusion criteria
This study prospectively included patients with BCS or PVT seen between 2005
and 2013 at the French Reference Center for Vascular Disorders of the Liver
(Clichy, France) and for whom peripheral blood DNA was available for mutation
screening (Supplementary CTAT Table). The protocol was performed in accor-
dance with the ethical guidelines of the 1975 Declaration of Helsinki and was
approved by the institutional review board (CPP Ile de France IV, Paris; France).
Informed consent was obtained from all patients included in the study.
We looked for criteria characterizing patients at high risk of having CALR
mutations in this French cohort, thereafter referred to as ‘‘test cohort” and then
tested these criteria for validation in a previously reported cohort from Hospital
Clinic, Barcelona, thereafter referred to as ‘‘validation cohort”.8
Definitions
BCS was defined as hepatic outflow obstruction regardless of the cause or level of
obstruction, from the small hepatic veins to the entrance of the inferior vena cava
into the right atrium. BCS was confirmed by ultrasonography and/or multidetec-
tor computed tomography and/or magnetic resonance imaging, and/or
venography. Sinusoidal obstruction syndrome, as well as outflow obstruction
occurring in the setting of heart failure, orthotopic liver transplantation and hep-
atobiliary cancer were excluded from this definition. Diagnostic criteria for PVT
included recent portal, and/or splenic and/or mesenteric venous thrombosis or
portal cavernoma. PVT patients with cirrhosis or abdominal malignancies were
excluded.
Hematologic studies
JAK2V617F, MPL and CALR mutations were tested in all patients. JAK2V617F and MPL
mutation analyses were performed as previously described.5
For CALR mutations, we used DNA extracted by an automated standardized
procedure (Qiasymphony, Qiagen) from blood samples collected by venipuncture
in tubes containing 0.11 mol/L trisodium citrate and stored at "80 !C until anal-
ysis. The mutational status of CALR was determined using previously described
high-resolution sizing of fluorescent dye-labeled PCR amplification of exon 9,
with Sanger sequencing controls.7
Bone marrow biopsy and/or endogenous erythroid colonies formation were
performed when considered relevant by the physician according to French
recommendations.18
Investigations for other thrombotic risk factors
Patients were tested according to previously reported methods for the following
thrombotic risk factors:19 factor V R506Q mutation (factor V Leiden); G20210A
factor II gene mutation; deficiencies in protein C, protein S, or antithrombin
(regarded as primary deficiencies only in conjunction with a prothrombin index
P80%); paroxysmal nocturnal hemoglobinuria; and anti-phospholipid antibod-
ies.1 Oral contraceptive use was considered a thrombotic risk factor when taken
within the three months preceding diagnosis of SVT.20
Imaging analyses
All abdominal multidetector computed tomography or magnetic resonance imag-
ing performed within six months of SVT diagnosis were reviewed to measure the
greatest spleen height in coronal view.
Statistical analysis
Quantitative variables were expressed as median (interquartile range), and cate-
gorical variables as absolute and relative frequencies. Comparisons between
groups of quantitative and qualitative variables were performed using Mann
Whitney and the Fisher exact tests, respectively. All tests were two-sided and
used a significance level of 0.05. Data handling and analysis were performed with
SPSS 17.0 (SPSS Inc., Chicago, IL).
For further details regarding the materials used, please refer to the CTAT
table.
Results
Patient characteristics
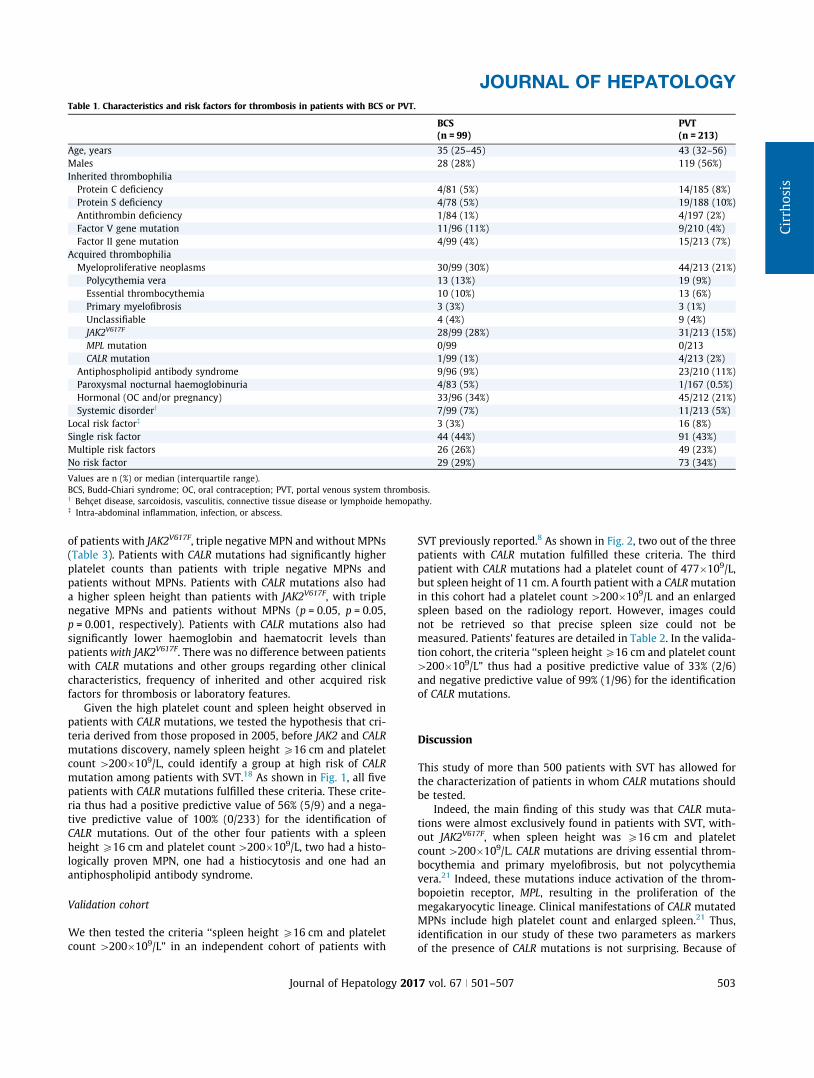
Three hundred twelve patients were enrolled, including 99 (32%)with BCS and 213 (68%) with PVT. Patients’ characteristics areshown in Table 1.
In patients with BCS, hepatic venous outflow obstruction wasdue to occlusion of one, two and three hepatic veins in 10, 19 and67 patients, respectively, and due to obstruction of the suprahep-atic segment of the inferior vena cava in two patients. The lastpatient had small hepatic veins BCS. Thirteen out of the 99patients with BCS also had a PVT. Out of the 213 patients withPVT without BCS, 39% had a portal cavernoma and 61% an acutePVT. Portal, splenic and mesenteric veins were involved in 199(93%), 79 (37%) and 118 (55%) of these 213 patients, respectively.
Risk factors for thrombosis are detailed in Table 1. The mostcommon cause was MPN. JAK2V617F was detected in 81% of theMPN patients. No MPL mutation was found.
CALR mutation
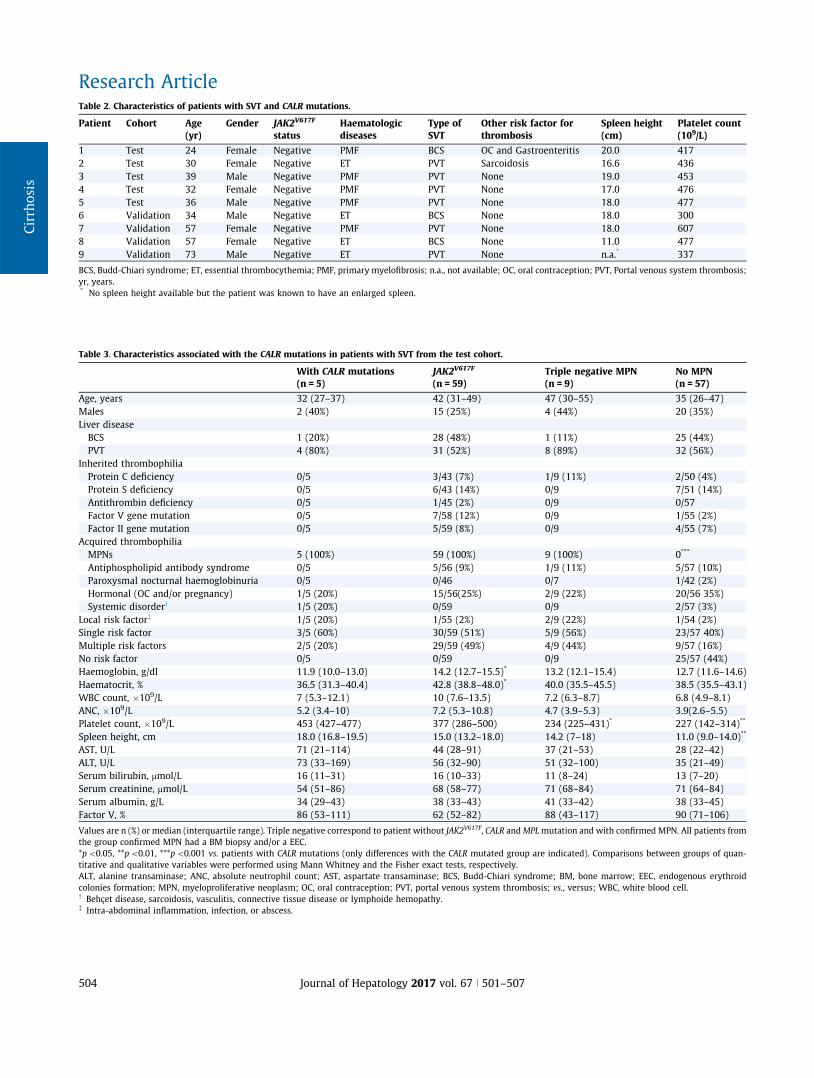
CALR mutations were detected in five patients (1.6%), a propor-tion in agreement with previous studies. Their individual charac-teristics are presented in Table 2. None of the patients with CALR
mutations had JAK2V617F or MPL mutations. Out of the patientswithout CALR mutations, 59 had JAK2V617F, nine had a triple neg-ative MPN (absence of JAK2V617F, CALR or MPL mutations but pos-itive bone marrow biopsy in eight patients, or endogenouserythroid colonies formation in one patient) and 57 did not haveMPN after bone marrow biopsy and/or endogenous erythroidcolonies formation examination. We compared the clinical andlaboratory features of patients with CALR mutations with those
Research Article
502 Journal of Hepatology 2017 vol. 67 j 501–507
of patients with JAK2V617F, triple negative MPN and without MPNs(Table 3). Patients with CALR mutations had significantly higherplatelet counts than patients with triple negative MPNs andpatients without MPNs. Patients with CALR mutations also hada higher spleen height than patients with JAK2V617F, with triplenegative MPNs and patients without MPNs (p = 0.05, p = 0.05,p = 0.001, respectively). Patients with CALR mutations also hadsignificantly lower haemoglobin and haematocrit levels thanpatients with JAK2V617F. There was no difference between patientswith CALR mutations and other groups regarding other clinicalcharacteristics, frequency of inherited and other acquired riskfactors for thrombosis or laboratory features.
Given the high platelet count and spleen height observed inpatients with CALR mutations, we tested the hypothesis that cri-teria derived from those proposed in 2005, before JAK2 and CALR
mutations discovery, namely spleen height P16 cm and plateletcount [200!109/L, could identify a group at high risk of CALR
mutation among patients with SVT.18 As shown in Fig. 1, all fivepatients with CALR mutations fulfilled these criteria. These crite-ria thus had a positive predictive value of 56% (5/9) and a nega-tive predictive value of 100% (0/233) for the identification ofCALR mutations. Out of the other four patients with a spleenheightP16 cm and platelet count[200!109/L, two had a histo-logically proven MPN, one had a histiocytosis and one had anantiphospholipid antibody syndrome.
Validation cohort
We then tested the criteria ‘‘spleen height P16 cm and plateletcount [200!109/L” in an independent cohort of patients with
SVT previously reported.8 As shown in Fig. 2, two out of the threepatients with CALR mutation fulfilled these criteria. The thirdpatient with CALR mutations had a platelet count of 477!109/L,but spleen height of 11 cm. A fourth patient with a CALRmutationin this cohort had a platelet count[200!109/L and an enlargedspleen based on the radiology report. However, images couldnot be retrieved so that precise spleen size could not bemeasured. Patients’ features are detailed in Table 2. In the valida-tion cohort, the criteria ‘‘spleen heightP16 cm and platelet count[200!109/L” thus had a positive predictive value of 33% (2/6)and negative predictive value of 99% (1/96) for the identificationof CALR mutations.
Discussion
This study of more than 500 patients with SVT has allowed forthe characterization of patients in whom CALR mutations shouldbe tested.
Indeed, the main finding of this study was that CALR muta-tions were almost exclusively found in patients with SVT, with-out JAK2V617F, when spleen height was P16 cm and plateletcount[200!109/L. CALR mutations are driving essential throm-bocythemia and primary myelofibrosis, but not polycythemiavera.21 Indeed, these mutations induce activation of the throm-bopoietin receptor, MPL, resulting in the proliferation of themegakaryocytic lineage. Clinical manifestations of CALR mutatedMPNs include high platelet count and enlarged spleen.21 Thus,identification in our study of these two parameters as markersof the presence of CALR mutations is not surprising. Because of
Table 1. Characteristics and risk factors for thrombosis in patients with BCS or PVT.
Values are n (%) or median (interquartile range). Triple negative correspond to patient without JAK2V617F, CALR andMPLmutation and with confirmed MPN. All patients from
the group confirmed MPN had a BM biopsy and/or a EEC.
*p\0.05, **p\0.01, ***p\0.001 vs. patients with CALR mutations (only differences with the CALR mutated group are indicated). Comparisons between groups of quan-
titative and qualitative variables were performed using Mann Whitney and the Fisher exact tests, respectively.
colonies formation; MPN, myeloproliferative neoplasm; OC, oral contraception; PVT, portal venous system thrombosis; vs., versus; WBC, white blood cell.y Behçet disease, sarcoidosis, vasculitis, connective tissue disease or lymphoide hemopathy.! Intra-abdominal inflammation, infection, or abscess.
Research Article
504 Journal of Hepatology 2017 vol. 67 j 501–507
hypersplenism and haemodilution related to portal hypertension,we chose a lower threshold for platelet count (200!109/L plate-lets) than in patients without SVT (450!109/L).18,21
When combining the test and validation cohorts together, thecriteria ‘‘spleen height P16 cm and platelet count[200!109/L”had a negative predictive value of 99.7%. Out of the 344 patientswithout JAK2V617F, these criteria would have avoided 329 unnec-essary CALR mutations tests with only one false negative result.Combining the two cohorts, four out of the eight patients withspleen height P16 cm and a platelet count[200!109/L and noCALR mutation had a triple negative MPN. These results suggestthat a bone marrow biopsy should be performed in patients withboth a spleen height P16 cm and a platelet count[200!109/L
when CALR mutations are absent. Six patients with SVT and CALR
mutations have been reported so far in the literature, in additionto those included in the present study.9,10,12,15 Spleen size andplatelet count were mentioned for three of them: all had enlargedspleens and a platelet count[200!109/L, which provide furtherevidence supporting the relevance of our findings.9,12
Due to the rarity of CALR mutations in patients with SVT, wewere not able to determine whether these mutations are associ-ated with a specific pattern of splanchnic vessels involvement ora particular outcome.
Our results have several implications. Firstly, identification ofCALR mutations allows the diagnosis of an underlying MPN and,in some cases, can remove the need for bone marrow biopsy,
312 patients with splanchnic vein thrombosis
(99 Budd-Chiari syndrome; 213 portal venous system thrombosis)
With JAK2V617F
(n = 59)
Without JAK2V617F
(n = 253)
CALR mutation
(n = 5)
Spleen height ≥16 cm and
platelet count >200x109/L
(n = 9)
Spleen height <16 cm or
platelet count ≤200x109/L
(n = 233)
Without JAK2V617F
or CALR
or MPL mutation
(n = 4)
Triple negative
MPN (n = 7)
No MPN
(n = 54)
Unavailable data
(n = 11)
Triple negative
MPN (n = 2)
No MPN
(n = 1)
BM
(n = 3)No BM
(n = 1)
BM and/or
EEC
(n = 61)
No BM
and/or
EEC
(n = 172)
No MPN
(n = 2)
BM and/or EEC
(n = 2)
No BM and/or
EEC (n = 9) CALR mutation
(n = 0)
Without JAK2V617F
or CALR or
MPL mutation
(n = 233)
Fig. 1. Flow chart for the test cohort. Unavailable data correspond to patients with platelet count[200!109/L but without spleen height available. Triple negative MPN
patients are patients without JAK2V617F, CALR and MPL mutation, but with a MPN proven by a BM biopsy and/or an EEC. BM, bone marrow; EEC, endogenous erythroid
(69 Budd-Chiari syndrome; 140 portal venous system thrombosis)
CALR mutation
(n = 2)
Spleen height ≥16 cm and
platelet count >200x109/L
(n = 6)
Spleen height <16 cm or
platelet count ≤200x109/L
(n = 96)
Without JAK2V617F
or CALR or
MPL mutation
(n = 4)
Unavailable data
(n = 46)
CALR mutation
(n = 1)
With JAK2V617F
(n = 61)
Without JAK2V617F
(n = 148)
CALR mutation
(n = 1)
Without JAK2V617F
or CALR or
MPL mutation
(n = 95)
Fig. 2. Flow chart for the validation cohort. Unavailable data correspond to patients with platelet count [200!109/L but without spleen height available. MPN,
Myeloproliferative neoplasm.
JOURNAL OF HEPATOLOGY
Journal of Hepatology 2017 vol. 67 j 501–507 505
an invasive procedure. Secondly, the criteria we proposed here(spleen height was P16 cm and platelet count[200!109/L) arereadily accessible and identify a patient population at high riskof having a MPN (with or without CALR mutations). Thesepatients should undergo rapid haematological investigations, toconsider cytoreductive therapy. Indeed, data from the Frenchnetwork on vascular liver diseases suggest that early introductionof cytoreductive therapy in patients with SVT and MPN reducessevere liver-related complications and improves event free sur-vival.22 Thirdly, by avoiding 96% of unnecessary CALR mutationstesting, this strategy will have economic consequences. Forinstance, in France, based on an incidence of primary SVT of 22per million inhabitants (around 2 per million for BCS and 2 per100, 000 for PVT23), on a population of 67 million inhabitantsand on a cost of CALR of 124 euros/test, this strategy would saveapproximately 200,000 euros per year.
In conclusion, this study provides the rationale for a new algo-rithm to diagnose MPNs in patients with SVT (Fig. 3). Given itshigh frequency in this setting, JAK2V617F must be tested first.Thereafter, patients without JAK2V617F but with spleen heightP16 cm and platelet count [200!109/L should be tested forCALR mutations. A bone marrow biopsy should be proposed inpatients without CALR mutations when the spleen is enlargedand platelets counts are normal or increased. In the remainingpatients, namely those without JAK2V617F, when spleen height is\16 cm and platelet count 6200!109/L, MPNs are extremelyuncommon and further studies are needed to identify thoserequiring a bone marrow biopsy.
Financial support
This work was supported by the Agence Nationale pour la
Recherche (ANR 14 CE35 0022 03/JAK-POT) and J.P by the ‘‘poste
accueil INSERM”.
Conflict of interest
The authors who have taken part in this study declared that theydo not have anything to disclose regarding funding or conflict ofinterest with respect to this manuscript.
Authors’ contributions
J.P., and P-E.R. wrote the paper. C.M., and N.C. performed themutational screening. A.P., O.G., C.B. and K.Z. collected the clinicaldata. F.T., F.C., D.C., and J.C.G.P., collected and analysed the datafrom the Spanish cohort. All authors discussed and criticallyrevised the manuscript.
Supplementary data
Supplementary data associated with this article can be found, inthe online version, at http://dx.doi.org/10.1016/j.jhep.2017.04.021.
References
Author names in bold designate shared co-first authorship
[1] Plessier A, Rautou P-E, Valla D-C. Management of hepatic vascular diseases. J
Hepatol 2012;56:S25–S38.
[2] Plessier A, Darwish-Murad S, Hernandez-Guerra M, Consigny Y, Fabris F,
Trebicka J, et al. Acute portal vein thrombosis unrelated to cirrhosis: a
[3] Darwish Murad S, Plessier A, Hernandez-Guerra M, Fabris F, Eapen CE, Bahr
MJ, et al. Etiology, management, and outcome of the Budd-Chiari syndrome.
Ann Intern Med 2009;151:167–175.
20-30%
55-65%
3%
Absent
CALR mutations
testing
2%
Spleen height ≥16 cm
and platelets >200x109/L
Absent
Bone marrow
biopsy
Spleen height <16 cm
or platelets ≤200x109/L
Consider bone marrow biopsy
and/or endogenous erythroid
colonies formation
JAK2V617F testing
Present
MPN
No
MPN
Present
Fig. 3. Proposed algorithm for the identification of myeloproliferative neoplasms (MPNs) in patients with primary splanchnic vein thrombosis (SVT). The first step
consists of JAK2V617F testing. Patients without JAK2V617F but with spleen height P16 cm and platelet count [200!109/L should be tested for CALR mutations; if CALR
mutations are absent, a bone marrow biopsy should be performed. In the remaining patients, MPNs are extremely uncommon and a bone marrow biopsy and/or
endogenous erythroid colonies formation can be considered on a case by case basis.
VI. REFERENCES[1] DameshekW.Editorial:SomeSpeculationsontheMyeloproliferativeSyndromes.Blood1951;6:372–5.[2] Thiele J, Kvasnicka HM. [Chronic myeloproliferative disorders. The new WHOclassification].Pathol2001;22:429–43.[3] Tefferi A, Vardiman JW. Classification and diagnosis ofmyeloproliferative neoplasms:the2008WorldHealthOrganizationcriteriaandpoint-of-carediagnosticalgorithms.Leukemia2008;22:14–22.[4] Nowell PC, Hungerford DA. Chromosome studies on normal and leukemic humanleukocytes.JNatlCancerInst1960;25:85–109.[5] Groffen J, Stephenson JR, Heisterkamp N, Klein A de, Bartram CR, Grosveld G.Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, onchromosome22.Cell1984;36:93–9.[6] Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Beau MML, et al. The 2016revision to the World Health Organization classification of myeloid neoplasms and acuteleukemia.Blood2016;127:2391–405.[7] MoulardOdile,MehtaJyotsna,FryzekJon,OlivaresRobert,IqbalUsman,MesaRubenA.Epidemiology of myelofibrosis, essential thrombocythemia, and polycythemia vera in theEuropeanUnion.EurJHaematol2014;92:289–97.[8] TitmarshGlenJ.,DuncombeAndrewS.,McMullinMaryFrances,O’RorkeMichael,MesaRuben, Vocht Frank, et al. How common are myeloproliferative neoplasms? A systematicreviewandmeta-analysis.AmJHematol2014;89:581–7.[9] VaquezH.Suruneformespécialedecyanoses’accompagnantd’hyperglobulieexcessiveetpersistante.CRSocBiolParis1892:vol.44,384-388.[10] OslerW.Osler,W. (1903).Chronic cyanosiswithpolycythemiaandenlargedspleen:anewclinicalentity.TransAmerPhycns1903:vol.18,299.[11] Epstein E, Goedel A. Hämorrhagische thrombocythämie bei vasculärer schrumpfmilz.VirchowsArchFürPatholAnatPhysiolFürKlinMed1934.[12] Heuck G. Zwei Fälle von Leukämie mit eigenthümlichem Blut- resp.Knochenmarksbefund.ArchFürPatholAnatPhysiolFürKlinMed1879;78:475–96.[13] TefferiA.MyelofibrosiswithMyeloidMetaplasia.NEnglJMed2000;342:1255–65.[14] Barbui T, Thiele J, Vannucchi AM, Tefferi A. Problems and pitfalls regarding WHO-defined diagnosis of early/prefibrotic primary myelofibrosis versus essentialthrombocythemia.Leukemia2013;27:1953–8.[15] Guglielmelli P, Pacilli A,RotunnoG,RumiE,Rosti V,Delaini F, et al. Presentation andoutcomeofpatientswith2016WHOdiagnosisofprefibroticandovertprimarymyelofibrosis.Blood2017;129:3227–36.[16] Mudireddy Mythri, Shah Sahrish, Lasho Terra, Barraco Daniela, Hanson Curtis A.,Ketterling Rhett P., et al. Prefibrotic versus overtly fibrotic primary myelofibrosis: clinical,cytogenetic,molecularandprognosticcomparisons.BrJHaematol2017;0.[17] BarbuiT,ThieleJ,PassamontiF,RumiE,BoveriE,RuggeriM,etal.SurvivalandDiseaseProgression in Essential Thrombocythemia Are Significantly Influenced by AccurateMorphologicDiagnosis:AnInternationalStudy.JClinOncol2011;29:3179–84.[18] Thiele J, Kvasnicka HM, Müllauer L, Buxhofer-Ausch V, Gisslinger B, Gisslinger H.Essentialthrombocythemiaversusearlyprimarymyelofibrosis:amulticenterstudytovalidatetheWHOclassification.Blood2011;117:5710–8.
144
[19] Barbui T, Thiele J, Carobbio A, Passamonti F, Rumi E, Randi ML, et al. Diseasecharacteristics and clinical outcome in young adults with essential thrombocythemia versusearly/prefibroticprimarymyelofibrosis.Blood2012;120:569–71.[20] RupoliS,GoteriG,PicardiP,MicucciG,CanafogliaL,ScortechiniAR,etal.Thrombosisinessential thrombocytemia and early/prefibrotic primarymyelofibrosis: the role of theWHOhistologicaldiagnosis.DiagnPathol2015;10.[21] KishimotoT,TagaT,AkiraS.Cytokinesignaltransduction.Cell1994;76:253–62.[22] O’SheaJJ,HollandSM,StaudtLM.JAKsandSTATsinImmunity,Immunodeficiency,andCancer.NEnglJMed2013;368:161–70.[23] VainchenkerW, Kralovics R. Genetic basis andmolecular pathophysiology of classicalmyeloproliferativeneoplasms.Blood2017;129:667–679.[24] SpringuelL,RenauldJ-C,KnoopsL.JAKkinasetargetinginhematologicmalignancies:asinuous pathway from identification of genetic alterations towards clinical indications.Haematologica2015;100:1240–53.[25] Carter-Su C, Schwartz J, Argetsinger LS. Growth hormone signaling pathways. GrowthHormIGFRes2016;28:11–5.[26] Shuai K, Liu B. Regulation of JAK–STAT signalling in the immune system. Nat RevImmunol2003;3:900–11.[27] Babon JJ, Lucet IS, Murphy JM, Nicola NA, Varghese LN. The molecular regulation ofJanuskinase(JAK)activation.BiochemJ2014;462:1–13.[28] CleyratCédric,DarehshouriAnza, SteinkampMaraP.,VilaineMathias,BoassaDaniela,Ellisman Mark H., et al. Mpl Traffics to the Cell Surface Through Conventional andUnconventionalRoutes.Traffic2014;15:961–82.[29] Walker JG, Smith MD. The Jak-STAT pathway in rheumatoid arthritis. J Rheumatol2005;32:1650–3.[30] AnanthakrishnanR,HallamK,LiQ,RamasamyR.JAK-STATpathwayincardiacischemicstress.VasculPharmacol2005;43:353–6.[31] KleinS,RickJ,LehmannJ,SchierwagenR,SchierwagenIG,VerbekeL,etal.Janus-kinase-2relatesdirectlytoportalhypertensionandtocomplications inrodentandhumancirrhosis.Gut2017;66:145–55.[32] Bose P, Gotlib J, Harrison CN, Verstovsek S. SOHO State-of-the-Art Update and NextQuestions:MPN.ClinLymphomaMyelomaLeuk2018;18:1–12.[33] LiJ,KentDG,GodfreyAL,ManningH,NangaliaJ,AzizA,etal.JAK2V617Fhomozygositydrives a phenotypic switch in myeloproliferative neoplasms, but is insufficient to sustaindisease.Blood2014;123:3139–3151.[34] Godfrey AL, Chen E, Pagano F, Ortmann CA, Silber Y, Bellosillo B, et al. JAK2V617Fhomozygosity arises commonly and recurrently in PV and ET, but PV is characterized byexpansionofadominanthomozygoussubclone.Blood2012;120:2704–7.[35] SpivakJL.MyeloproliferativeNeoplasms.NEnglJMed2017;376:2168–81.[36] JamesC,UgoV,LeCouédicJ-P,StaerkJ,DelhommeauF,LacoutC,etal.AuniqueclonalJAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature2005;434:1144–1148.[37] Kralovics R, Passamonti F, Buser AS, Teo S-S, Tiedt R, Passweg JR, et al. A Gain-of-FunctionMutationofJAK2inMyeloproliferativeDisorders.NEnglJMed2005;352:1779–90.[38] LevineRL,WadleighM,CoolsJ,EbertBL,WernigG,HuntlyBJP,etal.Activatingmutationin the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloidmetaplasiawithmyelofibrosis.CancerCell2005;7:387–97.[39] Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Acquiredmutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. The Lancet
145
2005;365:1054–61.[40] Silvennoinen O, Hubbard SR. Molecular insights into regulation of JAK2 inmyeloproliferativeneoplasms.Blood2015;125:3388–92.[41] LeroyE,DusaA,ColauD,MotamediA,CahuX,MoutonC,etal.UncouplingJAK2V617Factivation from cytokine-induced signalling by modulation of JH2 αC helix. Biochem J2016;473:1579–91.[42] Grinfeld J, Nangalia J, Green AR. Molecular determinants of pathogenesis and clinicalphenotypeinmyeloproliferativeneoplasms.Haematologica2017;102:7–17.[43] Hookham MB, Elliott J, Suessmuth Y, Staerk J, Ward AC, Vainchenker W, et al. Themyeloproliferative disorder–associated JAK2 V617F mutant escapes negative regulation bysuppressorofcytokinesignaling3.Blood2007;109:4924–9.[44] CampbellPJ,GreenAR.Themyeloproliferativedisorders.NEnglJMed2006;355:2452–66.[45] SozerS,FielMI,SchianoT,XuM,MascarenhasJ,HoffmanR.ThepresenceofJAK2V617Fmutation in the liver endothelial cells of patients with Budd-Chiari syndrome. Blood2009;113:5246–9.[46] RostiV,VillaniL,RiboniR,PolettoV,BonettiE,TozziL, et al. Spleenendothelial cellsfrompatientswithmyelofibrosisharbortheJAK2V617Fmutation.Blood2013;121:360–8.[47] Teofili L, Martini M, Iachininoto MG, Capodimonti S, Nuzzolo ER, Torti L, et al.Endothelial progenitor cells are clonal and exhibit the JAK2(V617F)mutation in a subset ofthromboticpatientswithPh-negativemyeloproliferativeneoplasms.Blood2011;117:2700–7.[48] Kralovics R, Guan Y, Prchal JT. Acquired uniparental disomy of chromosome 9p is afrequentstemcelldefectinpolycythemiavera.ExpHematol2002;30:229–36.[49] RumiE,PietraD,FerrettiV,KlampflT,HarutyunyanAS,MilosevicJD,etal.JAK2orCALRmutation status defines subtypes of essential thrombocythemia with substantially differentclinicalcourseandoutcomes.Blood2014;123:1544–1551.[50] Spivak JL, Considine M, Williams DM, Talbot CC, Rogers O, Moliterno AR, et al. TwoClinicalPhenotypesinPolycythemiaVera.NEnglJMed2014;371:808–17.[51] Dawson MA, Bannister AJ, Göttgens B, Foster SD, Bartke T, Green AR, et al. JAK2phosphorylateshistoneH3Y41andexcludesHP1αfromchromatin.Nature2009;461:819–22.[52] LiuF,ZhaoX,PernaF,WangL,KoppikarP,Abdel-WahabO,etal.JAK2V617F-MediatedPhosphorylation of PRMT5 Downregulates Its Methyltransferase Activity and PromotesMyeloproliferation.CancerCell2011;19:283–94.[53] Scott LM, Tong W, Levine RL, Scott MA, Beer PA, Stratton MR, et al. JAK2 Exon 12MutationsinPolycythemiaVeraandIdiopathicErythrocytosis.NEnglJMed2007;356:459–68.[54] PassamontiF,ElenaC,SchnittgerS,SkodaRC,GreenAR,GirodonF,etal.Molecularandclinical featuresof themyeloproliferativeneoplasmassociatedwith JAK2exon12mutations.Blood2011;117:2813–6.[55] Li S, Kralovics R, Libero GD, Theocharides A, Gisslinger H, Skoda RC. Clonalheterogeneity in polycythemia vera patients with JAK2 exon12 and JAK2-V617F mutations.Blood2008;111:3863–6.[56] Marty C, Pecquet C, NivarthiH, El-KhouryM, Chachoua I, TulliezM, et al. Calreticulinmutants in mice induce an MPL-dependent thrombocytosis with frequent progression tomyelofibrosis.Blood2016;127:1317–24.[57] Cazzola M. Mutant calreticulin: when a chaperone becomes intrusive. Blood2016;127:1219–21.[58] KlampflT,GisslingerH,HarutyunyanAS,NivarthiH,RumiE,MilosevicJD,etal.SomaticMutationsofCalreticulininMyeloproliferativeNeoplasms.NEnglJMed2013;369:2379–90.[59] Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G,Wedge DC, et al. Somatic CALR
146
Mutations in Myeloproliferative Neoplasms with Nonmutated JAK2. N Engl J Med2013;369:2391–405.[60] VainchenkerW, Delhommeau F, Constantinescu SN, Bernard OA. Newmutations andpathogenesisofmyeloproliferativeneoplasms.Blood2011;118:1723–35..[61] Pietra D, Rumi E, Ferretti VV, Buduo CAD, Milanesi C, Cavalloni C, et al. Differentialclinical effects of differentmutation subtypes in CALR-mutantmyeloproliferative neoplasms.Leukemia2016;30:431–8.[62] SzuberN,LamontagneB,BusqueL.Novelgermlinemutations in thecalreticulingene:implications for the diagnosis of myeloproliferative neoplasms. J Clin Pathol 2016:jclinpath-2016-203940.[63] ArakiM,YangY,MasubuchiN,HironakaY,TakeiH,MorishitaS,etal.ActivationofthethrombopoietinreceptorbymutantcalreticulininCALR-mutantmyeloproliferativeneoplasms.Blood2016;127:1307–1316.[64] ChachouaI,PecquetC,El-KhouryM,NivarthiH,AlbuR-I,MartyC,etal.Thrombopoietinreceptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood2016;127:1325–35.[65] ElfS,AbdelfattahNS,ChenE,Perales-PatónJ,RosenEA,KoA,etal.MutantCalreticulinRequires Both Its Mutant C-terminus and the Thrombopoietin Receptor for OncogenicTransformation.CancerDiscov2016;6:368–81.[66] Cabagnols X, Defour JP, Ugo V, Ianotto JC, Mossuz P, Mondet J, et al. Differentialassociation of calreticulin type 1 and type 2 mutations with myelofibrosis and essentialthrombocytemia:relevancefordiseaseevolution.Leukemia2015;29:249–52.[67] Cavalloni C, Rumi E, Ferretti VV, Pietra D, Roncoroni E, Bellini M, et al. SequentialevaluationofCALRmutantburdeninpatientswithmyeloproliferativeneoplasms.Oncotarget2017;8:33416–21.[68] TheocharidesAPA,LundbergP,LakkarajuAKK,LysenkoV,MyburghR,AguzziA,etal.Homozygous calreticulin mutations in patients with myelofibrosis lead to acquiredmyeloperoxidasedeficiency.Blood2016;127:3253–9.[69] TefferiA,WassieEA,LashoTL,FinkeC,BelachewAA,KetterlingRP,etal.Calreticulinmutationsandlong-termsurvivalinessentialthrombocythemia.Leukemia2014;28:2300–3.[70] AssafCA,ObberghFV,BillietJ,LiermanE,DevosT,GrauxC,etal.Analysisofphenotypeand outcome in essential thrombocythemia with CALR or JAK2 mutations. Haematologica2015;100:893–7.[71] Villeval J-L, Cohen-Solal K, TulliezM, Giraudier S, Guichard J, Burstein SA, et al. HighThrombopoietin Production by Hematopoietic Cells Induces a Fatal MyeloproliferativeSyndromeinMice.Blood1997;90:4369–83.[72] RumiE, PietraD,Guglielmelli P,BordoniR, Casetti I,Milanesi C, et al.Acquired copy-neutrallossofheterozygosityofchromosome1pasamoleculareventassociatedwithmarrowfibrosisinMPL-mutatedmyeloproliferativeneoplasms.Blood2013;121:4388–95.[73] ChalignéR,JamesC,TonettiC,BesancenotR,CouédicJPL,FavaF,etal.EvidenceforMPLW515L/K mutations in hematopoietic stem cells in primitive myelofibrosis. Blood2007;110:3735–43.[74] BeerPA,CampbellPJ,ScottLM,BenchAJ,ErberWN,BarefordD,etal.MPLmutationsinmyeloproliferativedisorders:analysisofthePT-1cohort.Blood2008;112:141–9.[75] PikmanY,LeeBH,MercherT,McDowellE,EbertBL,GozoM,etal.MPLW515LIsaNovelSomaticActivatingMutationinMyelofibrosiswithMyeloidMetaplasia.PLOSMed2006;3:e270.[76] VargheseLN,DefourJ-P,PecquetC,ConstantinescuSN.TheThrombopoietinReceptor:Structural Basis of Traffic and Activation by Ligand, Mutations, Agonists, and MutatedCalreticulin.FrontEndocrinol2017;8.
147
[77] Nangalia J, Green AR. Myeloproliferative neoplasms: from origins to outcomes. Blood2017;130:2475–83.[78] YaoH,MaY,HongZ,ZhaoL,MonaghanSA,HuM-C,etal.ActivatingJAK2mutantsrevealcytokine receptor couplingdifferences that impactoutcomes inmyeloproliferativeneoplasm.Leukemia2017;31:2122–31.[79] TiedtR,Hao-ShenH,SobasMA,LooserR,DirnhoferS,SchwallerJ,etal.RatioofmutantJAK2-V617F to wild-type Jak2 determines the MPD phenotypes in transgenic mice. Blood2008;111:3931–40.[80] AkadaH,AkadaS,HutchisonRE,MohiG.Lossofwild-typeJak2alleleenhancesmyeloidcell expansion and accelerates myelofibrosis in Jak2V617F knock-in mice. Leukemia2014;28:1627–35.[81] Rumi Elisa, Cazzola Mario. Advances in understanding the pathogenesis of familialmyeloproliferativeneoplasms.BrJHaematol2017;178:689–98.[82] Saliba J, Saint-Martin C, Di Stefano A, Lenglet G, Marty C, Keren B, et al. Germlineduplication of ATG2B and GSKIP predisposes to familial myeloid malignancies. Nat Genet2015;47:1131–40.[83] HarutyunyanAS,GiambrunoR,KrendlC,StukalovA,KlampflT,BergT,etal.GermlineRBBP6mutationsinfamilialmyeloproliferativeneoplasms.Blood2016;127:362–5.[84] Loscocco Giuseppe Gaetano, Mannarelli Carmela, Pacilli Annalisa, Fanelli Tiziana,Rotunno Giada, Gesullo Francesca, et al. Germline transmission of LNKE208Q variant in afamilywithmyeloproliferativeneoplasms.AmJHematol2016;91:E356–E356.[85] RumiE,HarutyunyanAS, PietraD, Feenstra JDM, Cavalloni C, Roncoroni E, et al. LNKmutationsinfamilialmyeloproliferativeneoplasms.Blood2016;128:144–5.[86] Tapper W, Jones AV, Kralovics R, Harutyunyan AS, Zoi K, Leung W, et al. GeneticvariationatMECOM,TERT,JAK2andHBS1L-MYBpredisposestomyeloproliferativeneoplasms.NatCommun2015;6:6691.[87] HindsDA,BarnholtKE,MesaRA,KieferAK,DoCB,ErikssonN,etal.GermlinevariantspredisposetobothJAK2V617Fclonalhematopoiesisandmyeloproliferativeneoplasms.Blood2016;128:1121–8.[88] Delic Sabit, Rose Dominic, Kern Wolfgang, Nadarajah Niroshan, Haferlach Claudia,Haferlach Torsten, et al. Application of an NGS-based 28-gene panel in myeloproliferativeneoplasms reveals distinct mutation patterns in essential thrombocythaemia, primarymyelofibrosisandpolycythaemiavera.BrJHaematol2016;175:419–26.[89] OrtmannCA,KentDG,NangaliaJ,SilberY,WedgeDC,GrinfeldJ,etal.EffectofMutationOrderonMyeloproliferativeNeoplasms.NEnglJMed2015;372:601–12.[90] TefferiA,GuglielmelliP,LarsonDR,FinkeC,WassieEA,PieriL,etal.Long-termsurvivaland blast transformation inmolecularly annotated essential thrombocythemia, polycythemiavera,andmyelofibrosis.Blood2014;124:2507–13.[91] Barosi G, Mesa RA, Thiele J, Cervantes F, Campbell PJ, Verstovsek S, et al. Proposedcriteria for the diagnosis of post-polycythemia vera and post-essential thrombocythemiamyelofibrosis:aconsensusstatement fromthe internationalworkinggroupformyelofibrosisresearchandtreatment.Leukemia2008;22:437–8.[92] CzaderM,OraziA.AcuteMyeloidLeukemiaandOtherTypesofDiseaseProgressioninMyeloproliferativeNeoplasms.AmJClinPathol2015;144:188–206.[93] BoiocchiL,MathewS,GianelliU,IurloA,RadiceT,Barouk-FoxS,etal.Morphologicandcytogeneticdifferencesbetweenpost-polycythemicmyelofibrosisandprimarymyelofibrosisinfibroticstage.ModPathol2013;26:1577–85.[94] LundbergP,KarowA,NienholdR,LooserR,Hao-ShenH,NissenI,etal.Clonalevolutionand clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood
148
2014;123:2220–8.[95] CervantesF,TassiesD,SalgadoC,RoviraM,PereiraA,RozmanC.AcuteTransformationin Nonleukemic Chronic Myeloproliferative Disorders: Actuarial Probability and MainCharacteristicsinaSeriesof218Patients.ActaHaematol1991;85:124–7.[96] Björkholm M, Derolf ÅR, Hultcrantz M, Kristinsson SY, Ekstrand C, Goldin LR, et al.Treatment-Related Risk Factors for Transformation to Acute Myeloid Leukemia andMyelodysplasticSyndromesinMyeloproliferativeNeoplasms.JClinOncol2011;29:2410–5.[97] AbdulkarimKhadija,GirodonFrançois,JohanssonPeter,MaynadiéMarc,KuttiJack,CarliPaule-Marie, et al. AML transformation in 56 patients with Ph− MPD in two well definedpopulations.EurJHaematol2008;82:106–11.[98] Heaney ML, Soriano G. Acute Myeloid Leukemia Following a MyeloproliferativeNeoplasm:ClinicalCharacteristics,GeneticFeaturesandEffectsofTherapy.CurrHematolMaligRep2013;8:116–22.d[99] BerkPD,GoldbergJD,SilversteinMN,WeinfeldA,DonovanPB,EllisJT,etal.Increasedincidenceofacuteleukemiainpolycythemiaveraassociatedwithchlorambuciltherapy.NEnglJMed1981;304:441–7.[100] Najean Y, Rain JD. Treatment of polycythemia vera: the use of hydroxyurea andpipobromanin292patientsundertheageof65years.Blood1997;90:3370–7.[101] TefferiAyalew,BarbuiTiziano.Polycythemiaveraandessentialthrombocythemia:2015updateondiagnosis,risk-stratificationandmanagement.AmJHematol2015;90:162–73.[102] TefferiA,VannucchiAM,BarbuiT.Polycythemiaveratreatmentalgorithm2018.BloodCancerJ2018;8:3.[103] VannucchiAM.HowItreatpolycythemiavera.Blood2014;124:3212–20.[104] Hernández-Boluda J-C, Arellano-Rodrigo E, Cervantes F, Alvarez-Larrán A, Gómez M,BarbaP,etal.Oralanticoagulationtopreventthrombosisrecurrenceinpolycythemiaveraandessentialthrombocythemia.AnnHematol2015;94:911–8.[105] FruchtmanSM,MackK,KaplanME,PetersonP,BerkPD,WassermanLR.Fromefficacyto safety: a Polycythemia Vera Study group report on hydroxyurea in patients withpolycythemiavera.SeminHematol1997;34:17–23.[106] FinazziG,CarusoV,MarchioliR,CapnistG,ChisesiT,FinelliC,etal.Acuteleukemiainpolycythemiavera:ananalysisof1638patientsenrolledinaprospectiveobservationalstudy.Blood2005;105:2664–70.[107] Gisslinger H, Zagrijtschuk O, Buxhofer-Ausch V, Thaler J, Schloegl E, Gastl GA, et al.Ropeginterferon alfa-2b, a novel IFNα-2b, induces high response rates with low toxicity inpatientswithpolycythemiavera.Blood2015;126:1762–9.[108] Larsen TS, Bjerrum OW, Pallisgaard N, Andersen MT, Møller MB, Hasselbalch HC.Sustainedmajormolecularresponseoninterferonalpha-2bintwopatientswithpolycythemiavera.AnnHematol2008;87:847–50.[109] Kiladjian J-J, Cassinat B, Chevret S, Turlure P, Cambier N, Roussel M, et al. Pegylatedinterferon-alfa-2ainducescompletehematologicandmolecularresponseswithlowtoxicityinpolycythemiavera.Blood2008;112:3065–72.[110] Mascarenhas JO,Prchal JT,RambaldiA,MesaRA,BerenzonD,YacoubA,etal. InterimAnalysisoftheMyeloproliferativeDisordersResearchConsortium(MPD-RC)112GlobalPhaseIII Trial of Front Line Pegylated Interferon Alpha-2a Vs. Hydroxyurea in High RiskPolycythemiaVeraandEssentialThrombocythemia.Blood2016;128:479–479.[111] KuriakoseET,GjoniS,WangYL,BaumannR,JonesAV,CrossNCP,etal.JAK2V617Falleleburden is reducedbybusulfan therapy:anewobservationusinganolddrug.Haematologica2013;98:e135–7.[112] DouglasG,HarrisonC,ForsythC,BennettM,StevensonW,HounsellJ,etal.Busulfanis
149
effectivesecond-line therapy forolderpatientswithPhiladelphia-negativemyeloproliferativeneoplasmsintolerantoforunresponsivetohydroxyurea.LeukLymphoma2017;58:89–95.[113] Begna K, Abdelatif A, Schwager S, Hanson C, Pardanani A, Tefferi A. Busulfan for thetreatment of myeloproliferative neoplasms: the Mayo Clinic experience. Blood Cancer J2016;6:e427.[114] Barosi G, BirgegardG, Finazzi G, GriesshammerM,Harrison C,HasselbalchH, et al. Aunifieddefinitionofclinicalresistanceandintolerancetohydroxycarbamideinpolycythaemiaveraandprimarymyelofibrosis:resultsofaEuropeanLeukemiaNet(ELN)consensusprocess.BrJHaematol2010;148:961–3.[115] VannucchiAM,Kiladjian JJ,GriesshammerM,MassziT,Durrant S, Passamonti F, et al.Ruxolitinib versus Standard Therapy for the Treatment of Polycythemia Vera. N Engl J Med2015;372:426–35.[116] Passamonti F, Griesshammer M, Palandri F, Egyed M, Benevolo G, Devos T, et al.Ruxolitinib for the treatment of inadequately controlled polycythaemia vera withoutsplenomegaly (RESPONSE-2): a randomised, open-label, phase 3b study. Lancet Oncol2017;18:88–99.[117] Vannucchi AM, Barbui T, Cervantes F, Harrison C, Kiladjian J-J, Kröger N, et al.Philadelphia chromosome-negative chronic myeloproliferative neoplasms: ESMO ClinicalPracticeGuidelinesfordiagnosis,treatmentandfollow-up.AnnOncol2015;26:v85–99.[118] Tefferi A, Vannucchi AM, Barbui T. Essential thrombocythemia treatment algorithm2018.BloodCancerJ2018;8:2.[119] Elala YC, Lasho TL, GangatN, Finke C, BarracoD,HaiderM, et al. Calreticulin variantstratifieddrivermutationalstatusandprognosisinessentialthrombocythemia.AmJHematol2016;91:503–6.[120] CarobbioA,ThieleJ,PassamontiF,RumiE,RuggeriM,RodeghieroF,etal.Riskfactorsfor arterial and venous thrombosis in WHO-defined essential thrombocythemia: aninternationalstudyof891patients.Blood2011;117:5857–9.[121] Finazzi G, Carobbio A, Guglielmelli P, Cavalloni C, Salmoiraghi S, Vannucchi AM, et al.Calreticulinmutation does notmodify the IPSET score for predicting the risk of thrombosisamong1150patientswithessentialthrombocythemia.Blood2014;124:2611–2.[122] RotunnoG,Mannarelli C,Guglielmelli P, PacilliA, PancrazziA, Pieri L, et al. Impact ofcalreticulin mutations on clinical and hematological phenotype and outcome in essentialthrombocythemia.Blood2014;123:1552–5.[123] Barbui T, Vannucchi AM, Buxhofer-Ausch V, De Stefano V, Betti S, Rambaldi A, et al.Practice-relevant revision of IPSET-thrombosis based on 1019 patients with WHO-definedessentialthrombocythemia.BloodCancerJ2015;5:e369–e369.[124] HaiderM,GangatN,LashoT,HusseinAKA,ElalaYC,HansonC,etal.Validationof therevised international prognostic score of thrombosis for essential thrombocythemia (IPSET-thrombosis)in585Mayoclinicpatients.AmJHematol2016;91:390–4.[125] BarbuiT,FinazziG,CarobbioA,ThieleJ,PassamontiF,RumiE,etal.Developmentandvalidation of an International Prognostic Score of thrombosis inWorldHealth Organization-essentialthrombocythemia(IPSET-thrombosis).Blood2012;120:5128–33;quiz5252.[126] CortelazzoS,VieroP,FinazziG,D’EmilioA,RodeghieroF,BarbuiT. Incidenceandriskfactors for thrombotic complications in a historical cohort of 100 patients with essentialthrombocythemia.JClinOncol1990;8:556–62.[127] LatagliataR,MontanaroM,CedroneM,VeroliAD,SpiritoF,SantoroC,etal.Highplateletcount at diagnosis is a protective factor for thrombosis in patients with essentialthrombocythemia.ThrombRes2017;156:168–71.[128] Collaborative overview of randomised trials of antiplatelet therapy--III: Reduction in
150
venous thrombosisandpulmonaryembolismbyantiplateletprophylaxis amongsurgical andmedicalpatients.AntiplateletTrialists’Collaboration.BMJ1994;308:235–46.[129] BecattiniC,AgnelliG,SchenoneA,EichingerS,BucheriniE,SilingardiM,etal.Aspirinforpreventingtherecurrenceofvenousthromboembolism.NEnglJMed2012;366:1959–67.[130] BarosiG,BessesC,BirgegardG,BriereJ,CervantesF,FinazziG,etal.Aunifieddefinitionof clinical resistance/intolerance to hydroxyurea in essential thrombocythemia: results of aconsensusprocessbyaninternationalworkinggroup.Leukemia2007;21:277–80.[131] VergerE,CassinatB,ChauveauA,DosquetC,GiraudierS,SchlageterM-H,etal.Clinicalandmolecular response to interferon-α therapy in essential thrombocythemia patientswithCALRmutations.Blood2015;126:2585–91.[132] Cassinat B, Verger E, Kiladjian J-J. Interferon alfa therapy in CALR-mutated essentialthrombocythemia.NEnglJMed2014;371:188–9.[133] Quintás-CardamaA,Abdel-WahabO,ManshouriT,KilpivaaraO,CortesJ,RoupieA-L,etal. Molecular analysis of patients with polycythemia vera or essential thrombocythemiareceivingpegylatedinterferonα-2a.Blood2013;122:893–901.[134] GisslingerH,GoticM,Holowiecki J,PenkaM,Thiele J,KvasnickaH-M,etal.Anagrelidecomparedwith hydroxyurea inWHO-classified essential thrombocythemia: the ANAHYDRETStudy,arandomizedcontrolledtrial.Blood2013;121:1720–8.[135] HarrisonCN,CampbellPJ,BuckG,WheatleyK,EastCL,BarefordD,etal.HydroxyureaCompared with Anagrelide in High-Risk Essential Thrombocythemia. N Engl J Med2005;353:33–45.[136] Birgegård G, Besses C, Griesshammer M, Gugliotta L, Harrison CN, Hamdani M, et al.Treatment of essential thrombocythemia in Europe: a prospective long-term observationalstudyof3649high-riskpatientsintheEvaluationofAnagrelideEfficacyandLong-termSafetystudy.Haematologica2018;103:51–60.[137] TefferiA,RumiE,FinazziG,GisslingerH,VannucchiAM,RodeghieroF,etal.Survivalandprognosisamong1545patientswithcontemporarypolycythemiavera:aninternationalstudy.Leukemia2013;27:1874–81.[138] VerstovsekS,PassamontiF,RambaldiA,BarosiG,RumiE,GattoniE,etal.Ruxolitinibforessentialthrombocythemiarefractorytoorintolerantofhydroxyurea:long-termphase2studyresults.Blood2017;130:1768–71.[139] Bose P, Verstovsek S. Ruxolitinib for essential thrombocythemia? Oncoscience2017;4:148–9.[140] Harrison CN, Mead AJ, Panchal A, Fox S, Yap C, Gbandi E, et al. Ruxolitinib vs bestavailable therapy forET intolerantor resistant tohydroxycarbamide.Blood2017;130:1889–97.[141] CervantesF,DupriezB,PereiraA,PassamontiF,ReillyJT,MorraE,etal.NewprognosticscoringsystemforprimarymyelofibrosisbasedonastudyoftheInternationalWorkingGroupforMyelofibrosisResearchandTreatment.Blood2009;113:2895–901.[142] Barbui T, Barosi G, Birgegard G, Cervantes F, Finazzi G, Griesshammer M, et al.Philadelphia-Negative Classical Myeloproliferative Neoplasms: Critical Concepts andManagementRecommendationsFromEuropeanLeukemiaNet.JClinOncol2011;29:761–70.[143] BallenKK,ShresthaS,SobocinskiKA,ZhangM-J,BasheyA,BolwellBJ,etal.Outcomeoftransplantation for myelofibrosis. Biol Blood Marrow Transplant J Am Soc Blood MarrowTransplant2010;16:358–67[144] Kröger N, Holler E, Kobbe G, Bornhäuser M, Schwerdtfeger R, Baurmann H, et al.Allogeneic stem cell transplantation after reduced-intensity conditioning in patients withmyelofibrosis:aprospective,multicenterstudyoftheChronicLeukemiaWorkingPartyoftheEuropeanGroupforBloodandMarrowTransplantation.Blood2009;114:5264–70.
151
[145] HarrisonC,KiladjianJ-J,Al-AliHK,GisslingerH,WaltzmanR,StalbovskayaV,etal. JAKInhibition with Ruxolitinib versus Best Available Therapy for Myelofibrosis. N Engl J Med2012;366:787–98.[146] Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. A Double-Blind,Placebo-ControlledTrialofRuxolitinibforMyelofibrosis.NEnglJMed2012;366:799–807.[147] HarrisonCN,VannucchiAM,KiladjianJ-J,Al-AliHK,GisslingerH,KnoopsL,etal.Long-term findings fromCOMFORT-II, a phase 3 study of ruxolitinib vs best available therapy formyelofibrosis.Leukemia2016;30:1701–7.[148] Verstovsek S, Mesa RA, Gotlib J, Gupta V, DiPersio JF, Catalano JV, et al. Long-termtreatmentwithruxolitinibforpatientswithmyelofibrosis:5-yearupdatefromtherandomized,double-blind, placebo-controlled, phase 3 COMFORT-I trial. J Hematol OncolJ Hematol Oncol2017;10:55.[149] VerstovsekS,GotlibJ,MesaRA,VannucchiAM,KiladjianJ-J,CervantesF,etal.Long-termsurvival in patients treated with ruxolitinib for myelofibrosis: COMFORT-I and -II pooledanalyses.JHematolOncolJHematolOncol2017;10:156[150] Marchioli R, Finazzi G, Landolfi R, Kutti J, Gisslinger H, Patrono C, et al. Vascular andNeoplastic Risk in a Large Cohort of Patients With Polycythemia Vera. J Clin Oncol2005;23:2224–32.[151] TefferiA,ElliottM.ThrombosisinMyeloproliferativeDisorders:Prevalence,PrognosticFactors,andtheRoleofLeukocytesandJAK2V617F.SeminThrombHemost2007;33:313–20.[152] Campbell PJ, Scott LM, Buck G, Wheatley K, East CL, Marsden JT, et al. Definition ofsubtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2V617Fmutationstatus:aprospectivestudy.TheLancet2005;366:1945–53.[153] Buxhofer-AuschVeronika,GisslingerHeinz,ThieleJürgen,GisslingerBettina,KvasnickaHans-Michael,Müllauer Leonhard, et al. Leukocytosis as an important risk factor for arterialthrombosis in WHO-defined early/prefibrotic myelofibrosis: An international study of 264patients.AmJHematol2012;87:669–72.[154] MontanaroMarco, LatagliataRoberto, CedroneMichele, SpadeaAntonio, RagoAngela,GiandomenicoJonny,etal.Thrombosisandsurvivalinessentialthrombocythemia:Aregionalstudyof1,144patients.AmJHematol2014;89:542–6.[155] Barbui T, Carobbio A, Cervantes F, Vannucchi AM, Guglielmelli P, Antonioli E, et al.Thrombosisinprimarymyelofibrosis:incidenceandriskfactors.Blood2010;115:778–82.[156] StefanoVD,Martinelli I. Splanchnic vein thrombosis: clinical presentation, risk factorsandtreatment.InternEmergMed2010;5:487–94.[157] LippiG,FranchiniM,TargherG.Arterialthrombusformationincardiovasculardisease.NatRevCardiol2011;8:502–12.[158] FranchiniM,MannucciPM.Venousandarterialthrombosis:Differentsidesofthesamecoin?EurJInternMed2008;19:476–81.[159] Jerjes-Sanchez C. Venous and arterial thrombosis: a continuous spectrumof the samedisease?EurHeartJ2005;26:3–4.[160] De Stefano Valerio, Rossi Elena, Za Tommaso, Ciminello Angela, Betti Silvia, LuzziClaudia,etal.JAK2V617Fmutationalfrequencyinessentialthrombocythemiaassociatedwithsplanchnicorcerebralveinthrombosis.AmJHematol2011;86:526–8.[161] Carobbio A, Finazzi G, Antonioli E, Guglielmelli P, Vannucchi AM, Dellacasa CM, et al.JAK2V617Falleleburdenandthrombosis:Adirectcomparison inessential thrombocythemiaandpolycythemiavera.ExpHematol2009;37:1016–21.[162] Ziakas PD. Effect of JAK2 V617F on thrombotic risk in patients with essentialthrombocythemia:measuringtheuncertain.Haematologica2008;93:1412–4.[163] Dahabreh IJ, Zoi K, Giannouli S, Zoi C, Loukopoulos D, Voulgarelis M. Is JAK2 V617F
152
mutationmorethanadiagnosticindex?LeukRes2009;33:67–73.[164] LussanaF,CaberlonS,PaganiC,KamphuisenPW,BüllerHR,CattaneoM.AssociationofV617F Jak2 mutation with the risk of thrombosis among patients with essentialthrombocythaemia or idiopathic myelofibrosis: A systematic review. Thromb Res2009;124:409–17.[165] Alvarez-Larran A, Pereira A, Guglielmelli P, Hernandez-Boluda JC, Arellano-Rodrigo E,Ferrer-Marin F, et al. Antiplatelet therapy versus observation in low-risk essentialthrombocythemiawithaCALRmutation.Haematologica2016;101:926–31.[166] PeiY-Q,WuY,WangF,CuiW.Prognostic valueofCALRvs. JAK2V617Fmutationsonsplenomegaly, leukemic transformation, thrombosis, and overall survival in patients withprimaryfibrosis:ameta-analysis.AnnHematol2016;95:1391–8.[167] LandolfiR,DiGennaroL,BarbuiT,DeStefanoV,FinazziG,MarfisiR,etal.Leukocytosisasamajorthromboticriskfactorinpatientswithpolycythemiavera.Blood2007;109:2446–52.[168] DeStefanoValerio,ZaTommaso,RossiElena,VannucchiAlessandroM.,RuggeriMarco,ElliElena,etal.Leukocytosisisariskfactorforrecurrentarterialthrombosisinyoungpatientswith polycythemia vera and essential thrombocythemia. Am J Hematol 2009;85:97–100.doi:10.1002/ajh.21593.[169] BarbuiT,MasciulliA,MarfisiMR,TognoniG,FinazziG,RambaldiA,etal.Whitebloodcell counts and thrombosis inpolycythemia vera: a subanalysis of theCYTO-PV study.Blood2015;126:560–1.[170] GangatNaseema, Strand Jacob,LiChin-Yang,WuWenting,PardananiAnimesh,TefferiAyalew. Leucocytosis in polycythaemia vera predicts both inferior survival and leukaemictransformation.BrJHaematol2007;138:354–8.[171] PassamontiF,RumiE,PascuttoC,CazzolaM,LazzarinoM.Increase in leukocytecountovertimepredictsthrombosisinpatientswithlow-riskessentialthrombocythemia.JThrombHaemostJTH2009;7:1587–9.[172] PassamontiF,Thiele J,GirodonF,RumiE,CarobbioA,GisslingerH,etal.Aprognosticmodel to predict survival in 867 World Health Organization–defined essentialthrombocythemiaatdiagnosis: a studyby the InternationalWorkingGrouponMyelofibrosisResearchandTreatment.Blood2012;120:1197–201.[173] Rumi E, Cazzola M. Diagnosis, risk stratification, and response evaluation in classicalmyeloproliferativeneoplasms.Blood2017;129:680–92.[174] TefferiA,BarbuiT.Polycythemiaveraandessentialthrombocythemia:2017updateondiagnosis,risk-stratification,andmanagement.AmJHematol2017;92:94–108.[175] Caramazza D, Caracciolo C, Barone R,Malato A, Saccullo G, Cigna V, et al. Correlationbetween leukocytosis and thrombosis in Philadelphia-negative chronic myeloproliferativeneoplasms.AnnHematol2009;88:967–71.[176] Buxhofer-AuschVeronika,GisslingerBettina,SchallingMartin,GleissAndreas,SchieferAna-Iris,MüllauerLeonhard, et al. Impactofwhiteblood cell counts atdiagnosis andduringfollow-upinpatientswithessentialthrombocythaemiaandprefibroticprimarymyelofibrosis.BrJHaematol2016;179:166–9.[177] CampbellPJ,MacLeanC,BeerPA,BuckG,WheatleyK,KiladjianJ-J,etal.Correlationofblood counts with vascular complications in essential thrombocythemia: analysis of theprospectivePT1cohort.Blood2012;120:1409–11.[178] Carobbio A, Antonioli E, Guglielmelli P, Vannucchi AM, Delaini F, Guerini V, et al.Leukocytosis and Risk Stratification Assessment in Essential Thrombocythemia. J Clin Oncol2008;26:2732–6.[179] TefferiA,GangatN,WolanskyjAP.Managementofextremethrombocytosisinotherwiselow-riskessentialthrombocythemia;doesnumbermatter?Blood2006;108:2493–4.
153
[180] Di Nisio Marcello, Barbui Tiziano, Di Gennaro Leonardo, Borrelli Giovanna, FinazziGuido,LandolfiRaffaele, et al.Thehaematocrit andplatelet target inpolycythemiavera.Br JHaematol2006;136:249–59.[181] Gugliotta L, Iurlo A, Gugliotta G, Tieghi A, Specchia G, GaidanoG, et al. Unbiased pro-thromboticfeaturesatdiagnosisin977thrombocythemicpatientswithPhiladelphia-negativechronicmyeloproliferativeneoplasms.LeukRes2016;46:18–25.[182] MichielsJJ,BernemanZ,SchroyensW,FinazziG,BuddeU,VlietHHDMvan.TheParadoxofPlateletActivationandImpairedFunction:Platelet-vonWillebrandFactorInteractions,andtheEtiologyofThromboticandHemorrhagicManifestationsinEssentialThrombocythemiaandPolycythemiaVera.SeminThrombHemost2006;32:589–604.[183] Regev Arie, Stark Pinhas, Blickstein Dorit, Lahav Meir. Thrombotic complications inessentialthrombocythemiawithrelativelylowplateletcounts.AmJHematol1998;56:168–72.[184] Falchi Lorenzo, Bose Prithviraj, Newberry Kate J., Verstovsek Srdan. Approach topatientswithessentialthrombocythaemiaandveryhighplateletcounts:whatistheevidencefortreatment?BrJHaematol2017;176:352–64.[185] CastamanG,LattuadaA,RuggeriM,TosettoA,MannucciPM,RodeghieroF.PlateletvonWillebrandfactorabnormalitiesinmyeloproliferativesyndromes.AmJHematol1995;49:289–93.[186] Turitto VT,Weiss HJ. Red blood cells: their dual role in thrombus formation. Science1980;207:541–3[187] Adams BD, Baker R, Lopez JA, Spencer S. Myeloproliferative disorders and thehyperviscositysyndrome.HematolOncolClinNorthAm2010;24:585–602.[188] ByrnesJR,WolbergAS.Redbloodcellsinthrombosis.Blood2017;130:1795–9.[189] MarchioliR,FinazziG,SpecchiaG,CacciolaR,CavazzinaR,CilloniD,etal.Cardiovasculareventsandintensityoftreatmentinpolycythemiavera.NEnglJMed2013;368:22–33.[190] FélétouM.Introduction.Morgan&ClaypoolLifeSciences;2011.[191] Verhamme P, Hoylaerts MF. The pivotal role of the endothelium in haemostasis andthrombosis.ActaClinBelg2006;61:213–9.[192] FélétouM.TheEndothelium.Morgan&ClaypoolLifeSciencesPublisher;2011.[193] JaffeEA,HoyerLW,NachmanRL.SynthesisofvonWillebrandfactorbyculturedhumanendothelialcells.ProcNatlAcadSciUSA1974;71:1906–9.[194] SavageB,SaldívarE,RuggeriZM.InitiationofplateletadhesionbyarrestontofibrinogenortranslocationonvonWillebrandfactor.Cell1996;84:289–97.[195] CarlosTM,HarlanJM.Leukocyte-endothelialadhesionmolecules.Blood1994;84:2068–101.[196] SpielAO,Gilbert JC, JilmaB. vonWillebrand factor in cardiovasculardisease: focusonacutecoronarysyndromes.Circulation2008;117:1449–59.[197] LipGY,BlannA.vonWillebrandfactor:amarkerofendothelialdysfunctioninvasculardisorders?CardiovascRes1997;34:255–65.[198] CellaG,MarchettiM,VianelloF,Panova-NoevaM,VignoliA,RussoL,etal.Nitricoxidederivatives and soluble plasma selectins in patients with myeloproliferative neoplasms.ThrombHaemost2010;104:151–6.[199] Piccin A, Steurer M, Feistritzer C, Murphy C, Eakins E, Van Schilfgaarde M, et al.Observationalretrospectivestudyofvascularmodulatorchangesduringtreatmentinessentialthrombocythemia.TranslRes2017;184:21–34.[200] KarakantzaM,GiannakoulasNC,ZikosP,SakellaropoulosG,KouraklisA,AktypiA,etal.Markers of Endothelial and In Vivo Platelet Activation in Patients with EssentialThrombocythemiaandPolycythemiaVera.IntJHematol2004;79:253–9.[201] BelottiAngelo,ElliElena,SperanzaTiziana,LanziEraldo,PioltelliPietro,PoglianiEnrico.
154
Circulatingendothelial cellsandendothelial activation inessential thrombocythemia:ResultsfromCD146+immunomagneticenrichment—flowcytometryandsolubleE-selectindetection.AmJHematol2012;87:319–20.[202] Jensen MK, de Nully Brown P, Thorsen S, Hasselbalch HC. Frequent occurrence ofanticardiolipin antibodies, Factor V Leiden mutation, and perturbed endothelial function inchronicmyeloproliferativedisorders.AmJHematol2002;69:185–91.[203] Falanga A, Marchetti M, Evangelista V, Vignoli A, Licini M, Balicco M, et al.Polymorphonuclear leukocyte activation and hemostasis in patients with essentialthrombocythemiaandpolycythemiavera.Blood2000;96:4261–6.[204] Santilli F, Romano M, Recchiuti A, Dragani A, Falco A, Lessiani G, et al. Circulatingendothelialprogenitorcellsandresidualinvivothromboxanebiosynthesisinlow-doseaspirin-treatedpolycythemiaverapatients.Blood2008;112:1085–90.[205] LancellottiS,DraganiA,RanalliP,PetrucciG,BassoM,TartaglioneR,etal.Qualitativeand quantitative modifications of von Willebrand factor in patients with essentialthrombocythemiaandcontrolledplateletcount.JThrombHaemostJTH2015;13:1226–37.[206] Prater DN, Case J, Ingram DA, YoderMC.Working hypothesis to redefine endothelialprogenitorcells.Leukemia2007;21:1141–9.[207] AlonciA,AllegraA,BellomoG,PennaG,D’AngeloA,QuartaroneE,etal.Evaluationofcirculating endothelial cells, VEGF and VEGFR2 serum levels in patients with chronicmyeloproliferativediseases.HematolOncol2008;26:235–9.[208] OppligerLeibundgutE,HornMP,BrunoldC,Pfanner-MeyerB,MartiD,HirsigerH,etal.Hematopoietic and endothelial progenitor cell trafficking in patientswithmyeloproliferativediseases.Haematologica2006;91:1465–72.[209] MassaM,RostiV,RamajoliI,CampanelliR,PecciA,ViarengoG,etal.CirculatingCD34+,CD133+, andVascularEndothelialGrowthFactorReceptor2–PositiveEndothelialProgenitorCellsinMyelofibrosisWithMyeloidMetaplasia.JClinOncol2005;23:5688–95.[210] Torres C, Fonseca AM, Leander M, Matos R, Morais S, Campos M, et al. CirculatingEndothelial Cells in Patients with Venous Thromboembolism and MyeloproliferativeNeoplasms.PLOSONE2013;8:e81574.[211] Rosti V, Bonetti E, BergamaschiG, Campanelli R, Guglielmelli P,MaestriM, et al.HighFrequency of Endothelial Colony Forming Cells Marks a Non-Active MyeloproliferativeNeoplasmwithHighRiskofSplanchnicVeinThrombosis.PLOSONE2010;5:e15277.[212] Helman Ricardo, PereiraWelbert de O., Marti Luciana C., Campregher Paulo V., PugaRenatoDavi,HamerschlakNelson,etal.GranulocytewholeexomesequencingandendothelialJAK2V617F in patients with JAK2V617F positive Budd-Chiari Syndrome withoutmyeloproliferativeneoplasm.BrJHaematol2018;180:443–5.[213] Guy A, Gourdou-Latyszenok V, Le Lay N, Vieira Dias J, Kilani B, Peghaire C, et al.Endothelial cells are important in the pathogenesis of thrombosis in myeloproliferativeneoplasms2017.[214] Etheridge SL, Roh ME, Cosgrove ME, Sangkhae V, Fox NE, Chen J, et al. JAK2V617F-positiveendothelialcellscontributetoclottingabnormalitiesinmyeloproliferativeneoplasms.ProcNatlAcadSciUSA2014;111:2295–300.[215] JensenMK,deNullyBrownP,LundBV,NielsenOJ,HasselbalchHC. Increasedplateletactivation and abnormal membrane glycoprotein content and redistribution inmyeloproliferativedisorders.BrJHaematol2000;110:116–24.[216] BlannA, CaineG, BarefordD. Abnormal vascular, platelet and coagulationmarkers inprimary thrombocythaemia are not reversed by treatments that reduce the platelet count.Platelets2004;15:447–9.[217] Arellano-RodrigoE,Alvarez-LarránA,ReverterJC,VillamorN,ColomerD,CervantesF.
155
Increased platelet and leukocyte activation as contributing mechanisms for thrombosis inessential thrombocythemia and correlation with the JAK2 mutational status. Haematologica2006;91:169–75.[218] RobertsonB,UrquhartC,FordI,TownendJ,WatsonHG,VickersMA,etal.Plateletandcoagulationactivationmarkersinmyeloproliferativediseases:relationshipswithJAK2V6I7Fstatus,clonality,andantiphospholipidantibodies.JThrombHaemostJTH2007;5:1679–85.[219] Panova-NoevaM,MarchettiM, SpronkHM, Russo L, Diani E, Finazzi G, et al. Platelet-inducedthrombingenerationbythecalibratedautomatedthrombogramassayisincreasedinpatientswithessentialthrombocythemiaandpolycythemiavera.AmJHematol2011;86:337–42.[220] Falanga A, Marchetti M, Vignoli A, Balducci D, Russo L, Guerini V, et al. V617F JAK-2mutation in patients with essential thrombocythemia: relation to platelet, granulocyte, andplasmahemostaticandinflammatorymolecules.ExpHematol2007;35:702–11.[221] VillmowT,Kemkes-MatthesB,MatzdorffAC.Markersofplateletactivationandplatelet-leukocyte interaction in patients with myeloproliferative syndromes. Thromb Res2002;108:139–45.[222] YaoH-X,HuangL,WuC-M,LinL-E,HuangZ-Q,WuJ-F,etal.[Effectsofsodiumozagrelinprimary thrombocytosis combined with thrombosis]. Zhongguo Shi Yan Xue Ye Xue Za Zhi2009;17:1360–2.[223] FalangaA,MarchettiM, Barbui T, Smith CW. Pathogenesis of Thrombosis in EssentialThrombocythemia and Polycythemia Vera: The Role of Neutrophils. Semin Hematol2005;42:239–47.[224] JensenMK,deNullyBrownP,LundBV,NielsenOJ,HasselbalchHC.Increasedcirculatingplatelet-leukocyte aggregates in myeloproliferative disorders is correlated to previousthrombosis,plateletactivationandplateletcount.EurJHaematol2001;66:143–51.[225] Arellano-RodrigoE,Alvarez-LarránA,ReverterJ-C,ColomerD,VillamorN,BellosilloB,et al. Platelet turnover, coagulation factors, and soluble markers of platelet and endothelialactivation in essential thrombocythemia: Relationshipwith thrombosis occurrence and JAK2V617Falleleburden.AmJHematol2009;84:102–8.[226] LandolfiR,RoccaB,PatronoC.Bleedingandthrombosisinmyeloproliferativedisorders:mechanismsandtreatment.CritRevOncolHematol1995;20:203–22.[227] WehmeierA, SüdhoffT,MeierkordF.Relationof platelet abnormalities to thrombosisandhemorrhageinchronicmyeloproliferativedisorders.SeminThrombHemost1997;23:391–402.[228] Le Blanc K, Berg A, Palmblad J, Samuelsson J. Stimulus-specific defect in plateletaggregationinpolycythemiavera.EurJHaematol1994;53:145–9.[229] Hosseini E, Ghasemzadeh M, Nassaji F, Jamaat ZP. GPVI modulation during plateletactivationandstorage:itsexpressionlevelsandectodomainsheddingcomparedtomarkersofplateletstoragelesion.Platelets2017;28:498–508.[230] Ghasemzadeh M, Hosseini E, Roudsari ZO, Zadkhak P. Intraplatelet reactive oxygenspecies(ROS)correlatewiththesheddingofadhesivereceptors,microvesiculationandplateletadhesion tocollagenduringstorage:DoesendogenousROSgenerationdownregulateplateletadhesivefunction?ThrombRes2018;163:153–61.[231] HobbsCM,ManningH,BennettC,VasquezL,SeverinS,BrainL,etal.JAK2V617Fleadstointrinsic changes inplatelet formation and reactivity in a knock-inmousemodel of essentialthrombocythemia.Blood2013;122:3787–97.[232] Lamrani L, Lacout C,OllivierV,Denis CV,GardinerE,HoTinNoeB, et al.Hemostaticdisorders in a JAK2V617F-driven mouse model of myeloproliferative neoplasm. Blood2014;124:1136–45.
156
[233] CalaminusSDJ,GuitartA,SinclairA,SchachtnerH,WatsonSP,HolyoakeTL,etal.LineageTracingofPf4-CreMarksHematopoieticStemCellsandTheirProgeny.PLoSONE2012;7.[234] PertuyF,AguilarA,StrasselC,EcklyA,FreundJ-N,DulucI,etal.Broaderexpressionofthe mouse platelet factor 4-cre transgene beyond the megakaryocyte lineage. J ThrombHaemostn.d.;13:115–25.[235] Strassel C, Kubovcakova L, Mangin PH, Ravanat C, Freund M, Skoda RC, et al.Haemorrhagic and thrombotic diatheses in mouse models with thrombocytosis. ThrombHaemost2015;113:414–25.[236] MusolinoC,AlonciA,BellomoG,TringaliO, SpatariG,QuartaroneC, et al.MarkersofEndothelialandPlateletStatusinPatientswithEssentialThrombocythemiaandPolycythemiaVera.Hematology1999;4:397–402.[237] GrignaniC,NorisP,TinelliC,BarosiG,BalduiniCL.Invitroplateletaggregationdefectsin patients with myeloproliferative disorders and high platelet counts: are they laboratoryartefacts?Platelets2009;20:131–4.[238] Mackman N. New insights into the mechanisms of venous thrombosis. J Clin Invest2012;122:2331–6.[239] von Brühl M-L, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al.Monocytes,neutrophils,andplateletscooperatetoinitiateandpropagatevenousthrombosisinmiceinvivo.JExpMed2012;209:819–35.[240] FalangaA,MarchettiM.Thromboticdisease in themyeloproliferativeneoplasms.ASHEducProgramBook2012;2012:571–81.[241] Marchetti M, Falanga A. Leukocytosis, JAK2V617F mutation, and hemostasis inmyeloproliferativedisorders.PathophysiolHaemostThromb2008;36:148–59.[242] Carulli G, Minnucci S, Gianfaldoni ML, Angiolini C, Azzarà A, Ambrogi F. Interactionsbetween platelets and neutrophils in essential thrombocythaemia. Effects on neutrophilchemiluminescenceandsuperoxideaniongeneration.EurJClinInvest1995;25:929–34.[243] Barbui T, Finazzi G, Falanga A. Myeloproliferative neoplasms and thrombosis. Blood2013;122:2176–84.[244] Augello C, Cattaneo D, Bucelli C, Terrasi A, Fermo E, Martinelli I, et al. <EmphasisType="Italic">CD18</Emphasis> promoter methylation is associated with a higher risk ofthromboticcomplicationsinprimarymyelofibrosis.AnnHematol2016;95:1965–9.[245] Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al.NeutrophilExtracellularTrapsKillBacteria.Science2004;303:1532–5.[246] MartinodK,WagnerDD.Thrombosis:tangledupinNETs.Blood2014;123:2768–76.[247] DemersM,KrauseDS,SchatzbergD,MartinodK,VoorheesJR,FuchsTA,etal.CancerspredisposeneutrophilstoreleaseextracellularDNAtrapsthatcontributetocancer-associatedthrombosis.ProcNatlAcadSci2012;109:13076–81[248] Wolach O, Sellar RS, Martinod K, Cherpokova D, McConkey M, Chappell RJ, et al.Increasedneutrophil extracellular trap formationpromotes thrombosis inmyeloproliferativeneoplasms.SciTranslMed2018;10.[249] MarinOyarzúnCP,CarestiaA,LevPR,GlembotskyAC,CastroRíosMA,MoiraghiB,etal.Neutrophil extracellular trap formationand circulatingnucleosomes inpatientswith chronicmyeloproliferativeneoplasms.SciRep2016;6:38738.[250] Colin Y, Van Kim CL, El Nemer W. Red cell adhesion in human diseases: Curr OpinHematol2014;21:186–92.[251] Cines DB, Lebedeva T, Nagaswami C, Hayes V, Massefski W, Litvinov RI, et al. Clotcontraction: compressionof erythrocytes into tightlypackedpolyhedra and redistributionofplateletsandfibrin.Blood2014;123:1596–603.[252] KraussS.Haptoglobinmetabolisminpolycythemiavera.Blood1969;33:865–76.
157
[253] Strati P, Masarova L, Bose P, Daver N, Pemmaraju N, Verstovsek S. Haptoglobin isfrequentlylowinpatientswithmyelofibrosis:Clinicalrelevance.LeukRes2017;57:85–8.[254] RusakT,MisztalT,PiszczJ,TomasiakM.Nitricoxidescavengingbycell-freehemoglobinmay be a primary factor determining hypertension in polycythemic patients. FreeRadicRes2014;48:230–8.[255] WautierM-P,NemerWE,GaneP,RainJ-D,CartronJ-P,ColinY,etal.Increasedadhesionto endothelial cells of erythrocytes from patients with polycythemia vera is mediated bylamininα5chainandLu/BCAM.Blood2007;110:894–901.[256] GrandisMD,CambotM,WautierM-P,CassinatB,ChomienneC,ColinY,etal.JAK2V617Factivates Lu/BCAM-mediated red cell adhesion in polycythemia vera through an EpoR-independentRap1/Aktpathway.Blood2013;121:658–65.[257] Rautou P-E, Vion A-C, Amabile N, Chironi G, Simon A, Tedgui A, et al. Microparticles,VascularFunction,andAtherothrombosis.CircRes2011;109:593–606.[258] BoulangerCM,LoyerX,RautouP-E,AmabileN.Extracellularvesiclesincoronaryarterydisease.NatRevCardiol2017;14:259–72.[259] Wood JP, Ellery PER, Maroney SA, Mast AE. Protein S is a cofactor for platelet andendothelial tissue factor pathway inhibitor-α but not for cell surface-associated tissue factorpathwayinhibitor.ArteriosclerThrombVascBiol2014;34:169–76.[260] TongD,YuM,GuoL,LiT,Li J,NovakovicVA,etal.Phosphatidylserine-exposingbloodand endothelial cells contribute to the hypercoagulable state in essential thrombocythemiapatients.AnnHematol2018;97:605–16.[261] Duchemin J, Ugo V, Ianotto J-C, Lecucq L,Mercier B, Abgrall J-F. Increased circulatingprocoagulantactivityandthrombingenerationinpatientswithmyeloproliferativeneoplasms.ThrombRes2010;126:238–42.[262] MarchettiM,CastoldiE,SpronkHMH,vanOerleR,BalducciD,BarbuiT,etal.ThrombingenerationandactivatedproteinCresistanceinpatientswithessentialthrombocythemiaandpolycythemiavera.Blood2008;112:4061–8.[263] TripodiA, ChantarangkulV, Gianniello F, ClericiM, LemmaL, PadovanL, et al. Globalcoagulationinmyeloproliferativeneoplasms.AnnHematol2013;92:1633–9.[264] TrappenburgMC,vanSchilfgaardeM,MarchettiM,SpronkHM,tenCateH,LeyteA,etal.Elevatedprocoagulantmicroparticlesexpressingendothelialandplateletmarkersinessentialthrombocythemia.Haematologica2009;94:911–8.[265] Kogan I, Chap D, Hoffman R, Axelman E, Brenner B, Nadir Y. JAK-2 V617F mutationincreasesheparanaseprocoagulantactivity.ThrombHaemost2016;115:73–80.[266] GroverSP,MackmanN.TissueFactor:AnEssentialMediatorofHemostasisandTriggerofThrombosis.ArteriosclerThrombVascBiol2018;38:709–25.[267] Carmeliet P, Stassen JM, Schoonjans L, Ream B, van den Oord JJ, De Mol M, et al.Plasminogen activator inhibitor-1 gene-deficient mice. II. Effects on hemostasis, thrombosis,andthrombolysis.JClinInvest1993;92:2756–60.[268] Friedenberg WR, Roberts RC, David DE. Relationship of thrombohemorrhagiccomplicationstoendothelialcellfunctioninpatientswithchronicmyeloproliferativedisorders.AmJHematol1992;40:283–9.[269] SonmezM,SaglamF,KarahanSC,ErkutN,MenteseA,SonmezB,etal.Treatmentrelatedchanges in antifibrinolytic activity in patients with polycythemia vera. Hematology2010;15:391–6.[270] KaftanO,BalcikOS,CipilH,OzetG,BavbekN,KoşarA,etal.Plasmalevelsofthrombin-activatablefibrinolysis inhibitor inprimaryandsecondarythrombocytosis.ClinApplThrombOffJIntAcadClinApplThromb2005;11:449–54.[271] BirdaneA,HaznedaroğluIC,BavbekN,KoşarA,BüyükaşikY,OzcebeO,etal.Theplasma
158
levels of prostanoids and plasminogen activator inhibitor-1 in primary and secondarythrombocytosis.ClinApplThrombOffJIntAcadClinApplThromb2005;11:197–201.[272] PósánE,UjjG,KissA,TelekB,RákK,UdvardyM.ReducedinvitroclotlysisandreleaseofmoreactiveplateletPAI-1inpolycythemiaveraandessentialthrombocythemia.ThrombRes1998;90:51–6.[273] MałeckiR,GackaM,Kuliszkiewicz-JanusM,Jakobsche-PolichtU,KwiatkowskiJ,AdamiecR, et al. Altered plasma fibrin clot properties in essential thrombocythemia. Platelets2016;27:110–6.[274] Rusak T, Piszcz J, Misztal T, Brańska-Januszewska J, Tomasiak M. Platelet-relatedfibrinolysis resistance inpatients suffering fromPV. Impactof clot retractionand isovolemicerythrocytapheresis.ThrombRes2014;134:192–8.[275] AirdWC.Vascularbed-specificthrombosis.JThrombHaemostJTH2007;5Suppl1:283–91.[276] RosenbergRD,AirdWC.Vascular-bed--specifichemostasisandhypercoagulablestates.NEnglJMed1999;340:1555–64.[277] Yuan L, Chan GC, Beeler D, Janes L, Spokes KC, Dharaneeswaran H, et al. A role ofstochastic phenotype switching in generating mosaic endothelial cell heterogeneity. NatCommun2016;7:10160.[278] Cheng Z, Fu J, Liu G, Zhang L, Xu Q, Wang S. Angiogenesis in JAK2 V617F positivemyeloproliferative neoplasms and ruxolitinib decrease VEGF, HIF-1 enesis in JAK2 V617Fpositivecells.LeukLymphoma2018;59:196–203.[279] Zangari M, Fink L, Tolomelli G, Lee JCH, Stein BL, Hickman K, et al. Could hypoxiaincrease the prevalence of thrombotic complications in polycythemia vera? Blood CoagulFibrinolysisIntJHaemostThromb2013;24:311–6.[280] Plessier A, Rautou P-E, Valla D-C.Management of hepatic vascular diseases. J Hepatol2012;56:S25–S38.[281] AgenoW,DentaliF,PomeroF,FenoglioL,SquizzatoA,PaganiG,etal. IncidenceratesandcasefatalityratesofportalveinthrombosisandBudd-ChiariSyndrome.ThrombHaemost2017;117:794–800.[282] Ollivier-HourmandI,AllaireM,GoutteN,MorelloR,Chagneau-DerrodeC,GoriaO,etal.TheepidemiologyofBudd-ChiarisyndromeinFrance.DigLiverDisOffJItalSocGastroenterolItalAssocStudyLiver2018.[283] DarwishMuradS, PlessierA,Hernandez-GuerraM, FabrisF, EapenCE,BahrMJ, et al.Etiology, management, and outcome of the Budd-Chiari syndrome. Ann Intern Med2009;151:167–75.[284] HeitJA.Epidemiologyofvenousthromboembolism.NatRevCardiol2015;12:464–74.[285] Smalberg JH, Arends LR, Valla DC, Kiladjian J-J, Janssen HL, Leebeek FW.Myeloproliferative neoplasms in Budd-Chiari syndrome and portal vein thrombosis: ameta-analysis.Blood2012;120:4921–4928.[286] How J, Zhou A, Oh ST. Splanchnic vein thrombosis in myeloproliferative neoplasms:pathophysiologyandmolecularmechanismsofdisease.TherAdvHematol2017;8:107–18.[287] KiladjianJ-J,CervantesF,LeebeekFWG,MarzacC,CassinatB,ChevretS,etal.TheimpactofJAK2andMPLmutationsondiagnosisandprognosisofsplanchnicveinthrombosis:areporton241cases.Blood2008;111:4922–9.[288] DeStefanoV,FioriniA,RossiE,ZaT,FarinaG,ChiusoloP,etal. Incidenceof the JAK2V617Fmutationamongpatientswith splanchnicor cerebralvenous thrombosisandwithoutovertchronicmyeloproliferativedisorders.JThrombHaemostJTH2007;5:708–14.[289] PrimignaniM,BarosiG, BergamaschiG, GianelliU, Fabris F,ReatiR, et al. Role of theJAK2 mutation in the diagnosis of chronic myeloproliferative disorders in splanchnic vein
159
thrombosis.HepatolBaltimMd2006;44:1528–34.[290] Barosi G, Buratti A, Costa A, Liberato LN, Balduini C, Cazzola M, et al. An atypicalmyeloproliferative disorderwith high thrombotic risk and slow disease progression. Cancer1991;68:2310–8.[291] ChaitY,CondatB,Cazals-HatemD,RufatP,AtmaniS,ChaouiD,etal.Relevanceof thecriteria commonly used to diagnosemyeloproliferative disorder in patients with splanchnicveinthrombosis.BrJHaematol2005;129:553–60.[292] VallaD,CasadevallN,HuisseMG,TulliezM,GrangeJD,MullerO,etal.Etiologyofportalvein thrombosis in adults. A prospective evaluation of primarymyeloproliferative disorders.Gastroenterology1988;94:1063–9.[293] Lamy T, Devillers A, Bernard M, Moisan A, Grulois I, Drenou B, et al. Inapparentpolycythemiavera:anunrecognizeddiagnosis.AmJMed1997;102:14–20.[294] DeStefanoV,ZaT,RossiE,VannucchiAM,RuggeriM,ElliE,etal.Recurrentthrombosisinpatientswithpolycythemiaveraandessentialthrombocythemia:incidence,riskfactors,andeffectoftreatments.Haematologica2008;93:372–80.[295] Colaizzo D, Amitrano L, Guardascione MA, Tiscia GL, D’Andrea G, Longo VAC, et al.OutcomeofpatientswithsplanchnicvenousthrombosispresentingwithoutovertMPN:arolefortheJAK2V617Fmutationre-evaluation.ThrombRes2013;132:e99–104.[296] Aird WC. Phenotypic Heterogeneity of the Endothelium: I. Structure, Function, andMechanisms.CircRes2007;100:158–73.[297] PoissonJ,LemoinneS,BoulangerC,DurandF,MoreauR,VallaD,etal.Liversinusoidalendothelialcells:Physiologyandroleinliverdiseases.JHepatol2017;66:212–27.[298] Smalberg JH, Kruip MJHA, Janssen HLA, Rijken DC, Leebeek FWG, de Maat MPM.Hypercoagulabilityandhypofibrinolysisandriskofdeepveinthrombosisandsplanchnicveinthrombosis:similaritiesanddifferences.ArteriosclerThrombVascBiol2011;31:485–93.[299] Mathew R, Huang J, Wu JM, Fallon JT, Gewitz MH. Hematological disorders andpulmonaryhypertension.WorldJCardiol2016;8:703–18.[300] García-ManeroG,SchusterSJ,PatrickH,MartinezJ.Pulmonaryhypertensioninpatientswithmyelofibrosissecondarytomyeloproliferativediseases.AmJHematol1999;60:130–5.[301] TabarrokiA,LindnerDJ,VisconteV,ZhangL,RogersHJ,ParkerY,etal.Ruxolitinibleadsto improvement of pulmonary hypertension in patients with myelofibrosis. Leukemia2014;28:1486–93.[302] Masri FA, XuW, Comhair SAA,AsosinghK,KooM,Vasanji A, et al.Hyperproliferativeapoptosis-resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am JPhysiol-LungCellMolPhysiol2007;293:L548–54.[303] InraCN,ZhouBO,AcarM,MurphyMM,RichardsonJ,ZhaoZ,etal.Aperisinusoidalnicheforextramedullaryhaematopoiesisinthespleen.Nature2015;527:466–71.[304] Kvasnicka HM, Thiele J. Bone marrow angiogenesis: methods of quantification andchangesevolvinginchronicmyeloproliferativedisorders.HistolHistopathol2004;19:1245–60.[305] Ni H, Barosi G, Hoffman R. Quantitative evaluation of bone marrow angiogenesis inidiopathicmyelofibrosis.AmJClinPathol2006;126:241–7.[306] MedingerM,SkodaR,GratwohlA,TheocharidesA,BuserA,HeimD,etal.Angiogenesisandvascularendothelialgrowthfactor-/receptorexpressioninmyeloproliferativeneoplasms:correlation with clinical parameters and JAK2-V617F mutational status. Br J Haematol2009;146:150–7.[307] Gianelli U, Vener C, Raviele PR, Savi F, Somalvico F, Calori R, et al. VEGF ExpressionCorrelates With Microvessel Density in Philadelphia Chromosome–Negative ChronicMyeloproliferativeDisorders.AmJClinPathol2007;128:966–73.[308] Panteli K, Bai M, Hatzimichael E, Zagorianakou N, Agnantis NJ, Bourantas K. Serum
160
levels, and bone marrow immunohistochemical expression of, vascular endothelial growthfactorinpatientswithchronicmyeloproliferativediseases.Hematology2007;12:481–6.[309] BoiocchiL,VenerC,SaviF,BonoldiE,MoroA,FracchiollaNS,etal.Increasedexpressionofvascularendothelialgrowthfactorreceptor1correlateswithVEGFandmicrovesseldensityin Philadelphia chromosome-negative myeloproliferative neoplasms. J Clin Pathol2011;64:226–31.[310] Subotički T, Mitrović Ajtić O, Beleslin-Čokić BB, Nienhold R, Diklić M, Djikić D, et al.Angiogenic factors are increased in circulating granulocytes and CD34+ cells ofmyeloproliferativeneoplasms.MolCarcinog2017;56:567–79.[311] Bogdanova A, Mihov D, Lutz H, Saam B, Gassmann M, Vogel J. Enhanced erythro-phagocytosisinpolycythemicmiceoverexpressingerythropoietin.Blood2007;110:762–9.[312] StamJ.Thrombosisofthecerebralveinsandsinuses.NEnglJMed2005;352:1791–8.[313] MartinelliI,DeStefanoV.Rarethrombosesofcerebral,splanchnicandupper-extremityveins.Anarrativereview.ThrombHaemost2010;103:1136–44.[314] Passamonti SM, Biguzzi E, CazzolaM, Franchi F, Gianniello F, Bucciarelli P, et al. TheJAK2V617F mutation in patients with cerebral venous thrombosis. J Thromb Haemost JTH2012;10:998–1003.[315] LamyM,PalazzoP,AgiusP,ChomelJC,CironJ,BerthometA,etal.ShouldWeScreenforJanusKinase2V617FMutationinCerebralVenousThrombosis?CerebrovascDisBaselSwitz2017;44:97–104.[316] DeT,PrabhakarP,NagarajaD,ChristopherR. Januskinase(JAK)2V617Fmutation inAsianIndianswithcerebralvenousthrombosisandwithoutovertmyeloproliferativedisorders.JNeurolSci2012;323:178–82.[317] DentaliF,AgenoW,RumiE,CasettiI,PoliD,ScodittiU,etal.Cerebralvenousthrombosisand myeloproliferative neoplasms: results from two large databases. Thromb Res2014;134:41–3.[318] HasselbalchHC.PerspectivesontheimpactofJAK-inhibitortherapyuponinflammation-mediated comorbidities in myelofibrosis and related neoplasms. Expert Rev Hematol2014;7:203–16.[319] FinazziG,DeStefanoV,BarbuiT.AreMPNsvasculardiseases?CurrHematolMaligRep2013;8:307–16.[320] Vrtovec M, Anzic A, Zupan IP, Zaletel K, Blinc A. Carotid Artery Stiffness, DigitalEndothelialFunction,andCoronaryCalciuminPatientswithEssentialThrombocytosis,FreeofOvertAtheroscleroticDisease.RadiolOncol2017;51:203–10.[321] JaiswalS,FontanillasP,Flannick J,ManningA,GraumanPV,MarBG,etal.Age-relatedclonalhematopoiesisassociatedwithadverseoutcomes.NEnglJMed2014;371:2488–98.[322] Fuster JJ,WalshK.SomaticMutationsandClonalHematopoiesis:UnexpectedPotentialNewDriversofAge-RelatedCardiovascularDisease.CircRes2018;122:523–32.[323] JaiswalS,NatarajanP,SilverAJ,GibsonCJ,BickAG,ShvartzE,etal.ClonalHematopoiesisandRiskofAtheroscleroticCardiovascularDisease.NEnglJMed2017;377:111–21.[324] CassinatB,HarrisonC,KiladjianJ-J.ClonalHematopoiesisandAtherosclerosis.NEnglJMed2017;377:1400–1.[325] JaiswalS,NatarajanP,EbertBL.ClonalHematopoiesisandAtherosclerosis.NEnglJMed2017;377:1401–2.[326] Muendlein A, Gasser K, Kinz E, Stark N, Leiherer A, Rein P, et al. Evaluation of theprevalenceandprospectiveclinical impactof the JAK2V617Fmutation in coronarypatients.AmJHematol2014;89:295–301.[327] MuendleinA,KinzE,GasserK,LeihererA,ReinP,SaelyCH,etal.OccurrenceoftheJAK2V617Fmutationinpatientswithperipheralarterialdisease.AmJHematol2015;90:E17-21.
161
[328] Fuster JJ,MacLauchlan S, ZuriagaMA, PolackalMN, Ostriker AC, Chakraborty R, et al.ClonalhematopoiesisassociatedwithTET2deficiencyacceleratesatherosclerosisdevelopmentinmice.Science2017;355:842–7.[329] RidkerPM,MacFadyenJG,EverettBM,LibbyP,ThurenT,GlynnRJ,etal.RelationshipofC-reactive protein reduction to cardiovascular event reduction following treatment withcanakinumab:asecondaryanalysisfromtheCANTOSrandomisedcontrolledtrial.LancetLondEngl2018;391:319–28.[330] DaviesPF.Flow-mediatedendothelialmechanotransduction.PhysiolRev1995;75:519–60.[331] MalekAM,AlperSL,IzumoS.Hemodynamicshearstressanditsroleinatherosclerosis.JAMA1999;282:2035–42.[332] ParmarKM,LarmanHB,DaiG,ZhangY,WangET,MoorthySN,etal.Integrationofflow-dependentendothelialphenotypesbyKruppel-likefactor2.JClinInvest2006;116:49–58.[333] Wade JP, Pearson TC, Russell RW,Wetherley-Mein G. Cerebral blood flow and bloodviscosityinpatientswithpolycythaemiasecondarytohypoxiclungdisease.BrMedJClinResEd1981;283:689–92.[334] WellsRE,MerrillEW.INFLUENCEOFFLOWPROPERTIESOFBLOODUPONVISCOSITY-HEMATOCRITRELATIONSHIPS*.JClinInvest1962;41:1591–8.[335] Pearson TC, Wetherley-Mein G. VASCULAR OCCLUSIVE EPISODES AND VENOUSHÆMATOCRIT IN PRIMARY PROLIFERATIVE POLYCYTHÆMLX. The Lancet 1978;312:1219–22.[336] GolseN,BucurPO,AdamR,CastaingD,SaCunhaA,VibertE.Newparadigms inpost-hepatectomyliverfailure.JGastrointestSurgOffJSocSurgAlimentTract2013;17:593–605.[337] Yamanaka K, Hatano E, Narita M, Kitamura K, Yanagida A, Asechi H, et al. Olprinoneattenuatesexcessiveshearstressthroughup-regulationofendothelialnitricoxidesynthaseina rat excessive hepatectomymodel. Liver Transplant Off Publ AmAssoc Study Liver Dis IntLiverTransplantSoc2011;17:60–9.[338] Martini J,CarpentierB,NegreteAC,Frangos JA, IntagliettaM.Paradoxicalhypotensionfollowing increased hematocrit and blood viscosity. Am J Physiol-Heart Circ Physiol2005;289:H2136–43.[339] Dekker RJ, van Soest S, Fontijn RD, Salamanca S, de Groot PG, VanBavel E, et al.Prolonged fluid shear stress induces adistinct set of endothelial cell genes,most specificallylungKrüppel-likefactor(KLF2).Blood2002;100:1689–98.[340] LusisAJ.Atherosclerosis.Nature2000;407:233–41.[341] Knowles JW, Reddick RL, Jennette JC, Shesely EG, Smithies O, Maeda N. Enhancedatherosclerosis and kidney dysfunction in eNOS(-/-)Apoe(-/-) mice are ameliorated byenalapriltreatment.JClinInvest2000;105:451–8.[342] Moss JWE, Ramji DP. Nutraceutical therapies for atherosclerosis. Nat Rev Cardiol2016;13:513–32.[343] Neunteufl T, Heher S, Stefenelli T, Pabinger I, GisslingerH. Endothelial dysfunction inpatientswithpolycythaemiavera.BrJHaematol2001;115:354–9.[344] Li Y, Lui KO, Zhou B. Reassessing endothelial-to-mesenchymal transition incardiovasculardiseases.NatRevCardiol2018.[345] Cho JG, Lee A, Chang W, Lee M-S, Kim J. Endothelial to Mesenchymal TransitionRepresentsaKeyLink in the Interactionbetween InflammationandEndothelialDysfunction.FrontImmunol2018;9:294.[346] ChenP-Y,QinL,BaeyensN,LiG,AfolabiT,BudathaM,etal.Endothelial-to-mesenchymaltransitiondrivesatherosclerosisprogression.JClinInvest2015;125:4514–28.[347] Erba BG, Gruppi C, Corada M, Pisati F, Rosti V, Bartalucci N, et al. Endothelial-to-
162
Mesenchymal Transition in BoneMarrow and Spleen of PrimaryMyelofibrosis. Am J Pathol2017;187:1879–92.[348] ZahrAA,SalamaME,CarreauN,TremblayD,VerstovsekS,MesaR,etal.Bonemarrowfibrosis in myelofibrosis: pathogenesis, prognosis and targeted strategies. Haematologica2016;101:660–71.[349] Chou JM, Li C-Y, Tefferi A. Bone marrow immunohistochemical studies of angiogeniccytokines and their receptors in myelofibrosis with myeloid metaplasia. Leuk Res2003;27:499–504.[350] CampanelliR,RostiV,VillaniL,CastagnoM,MorettiE,BonettiE,etal.Evaluationofthebioactive and total transforming growth factor β1 levels in primarymyelofibrosis. Cytokine2011;53:100–6.[351] Simon SI, Green CE. Molecular Mechanics and Dynamics of Leukocyte RecruitmentDuringInflammation.AnnuRevBiomedEng2005;7:151–85.[352] Carman CV, Springer TA. A transmigratory cup in leukocyte diapedesis both throughindividualvascularendothelialcellsandbetweenthem.JCellBiol2004;167:377–88.[353] LibbyP,RidkerPM,HanssonGK,LeducqTransatlanticNetworkonAtherothrombosis.Inflammation in atherosclerosis: from pathophysiology to practice. J Am Coll Cardiol2009;54:2129–38.[354] Tedgui A,Mallat Z. Cytokines in atherosclerosis: pathogenic and regulatory pathways.PhysiolRev2006;86:515–81.[355] RamjiDP,DaviesTS. Cytokines in atherosclerosis:Keyplayers in all stages of diseaseandpromisingtherapeutictargets.CytokineGrowthFactorRev2015;26:673–85.[356] BarbuiT,CarobbioA,FinazziG,VannucchiAM,BarosiG,AntonioliE,etal.Inflammationand thrombosis in essential thrombocythemia and polycythemia vera: different role of C-reactiveproteinandpentraxin3.Haematologica2011;96:315–8.[357] BarosiG.AnimmunedysregulationinMPN.CurrHematolMaligRep2014;9:331–9.[358] Hasselbalch HC. Chronic inflammation as a promotor of mutagenesis in essentialthrombocythemia, polycythemia vera and myelofibrosis. A human inflammation model forcancerdevelopment?LeukRes2013;37:214–20.[359] RossR.Atherosclerosis--aninflammatorydisease.NEnglJMed1999;340:115–26.[360] Hasselbalch HC. Perspectives on chronic inflammation in essential thrombocythemia,polycythemia vera, andmyelofibrosis: is chronic inflammation a trigger and driver of clonalevolution and development of accelerated atherosclerosis and second cancer? Blood2012;119:3219–25.[361] TabasI.Macrophagedeathanddefectiveinflammationresolutioninatherosclerosis.NatRevImmunol2010;10:36–46.[362] FebbraioM,PodrezEA,SmithJD,HajjarDP,HazenSL,HoffHF,etal.TargeteddisruptionoftheclassBscavengerreceptorCD36protectsagainstatheroscleroticlesiondevelopmentinmice.JClinInvest2000;105:1049–56.[363] Silverstein RL. Inflammation, atherosclerosis, and arterial thrombosis: role of thescavengerreceptorCD36.CleveClinJMed2009;76Suppl2:S27-30.[364] GinsbergH,GilbertHS,GibsonJC,LeNA,BrownWV.Increasedlow-density-lipoproteincatabolisminmyeloproliferativedisorders.AnnInternMed1982;96:311–6.[365] Ginsberg H, Goldberg IJ,Wang-Iverson P, Gitler E, Le NA, Gilbert HS, et al. Increasedcatabolismofnativeandcyclohexanedione-modified lowdensity lipoprotein in subjectswithmyeloproliferativediseases.ArteriosclerDallasTex1983;3:233–41.[366] Vallabhajosula S, Gilbert HS, Goldsmith SJ, Paidi M, Hanna MM, Ginsberg HN. Low-densitylipoprotein(LDL)distributionshownby99mtechnetium-LDLimaginginpatientswithmyeloproliferativediseases.AnnInternMed1989;110:208–13.
163
[367] Gardai SJ,McPhillips KA, Frasch SC, JanssenWJ, Starefeldt A,Murphy-Ullrich JE, et al.Cell-surfacecalreticulininitiatesclearanceofviableorapoptoticcellsthroughtrans-activationofLRPonthephagocyte.Cell2005;123:321–34.[368] Daitoku S, Takenaka K, Yamauchi T, Yurino A, Jinnouchi F, Nunomura T, et al.Calreticulinmutationdoesnotcontributetodiseaseprogressioninessentialthrombocythemiabyinhibitingphagocytosis.ExpHematol2016;44:817-825.e3.[369] Getz GS, Reardon CA. Natural killer T cells in atherosclerosis. Nat Rev Cardiol2017;14:304–14.[370] Ait-OufellaH, SalomonBL, Potteaux S,RobertsonA-KL,GourdyP, Zoll J, et al.NaturalregulatoryTcellscontrol thedevelopmentofatherosclerosis inmice.NatMed2006;12:178–80.[371] Pastrana JL, Sha X, Virtue A, Mai J, Cueto R, Lee IA, et al. Regulatory T cells andAtherosclerosis.JClinExpCardiol2012;2012:002.[372] LiuzzoG,GoronzyJJ,YangH,KopeckySL,HolmesDR,FryeRL,etal.MonoclonalT-cellproliferationandplaqueinstabilityinacutecoronarysyndromes.Circulation2000;101:2883–8.[373] Cheng X, Yu X, Ding Y-J, Fu Q-Q, Xie J-J, Tang T-T, et al. The Th17/Treg imbalance inpatientswithacutecoronarysyndrome.ClinImmunolOrlandoFla2008;127:89–97.[374] LimK-H,ChenCG-S,ChangY-C,ChiangY-H,KaoC-W,WangW-T,etal.IncreasedBcellactivation is present in JAK2V617F-mutated, CALR-mutated and triple-negative essentialthrombocythemia.Oncotarget2017;8:32476–91.[375] Sanchez C, Baier C, Colle JG, Chelbi R, Rihet P, Le Treut T, et al. Natural killer cells inpatientswithpolycythemiavera.HumImmunol2015;76:644–50.[376] ChoiDC,TremblayD,Iancu-RubinC,MascarenhasJ.Programmedcelldeath-1pathwayinhibition in myeloid malignancies: implications for myeloproliferative neoplasms. AnnHematol2017;96:919–27.[377] ZhaoW,LiY,LiuX,ZhangL,WangX.InvolvementofCD4+CD25+regulatoryTcells inthepathogenesisofpolycythaemiavera.ChinMedJ(Engl)2008;121:1781–6.[378] Keohane C, Kordasti S, Seidl T, Abellan PP, Thomas NSB, Harrison CN, et al. JAKinhibition induces silencing of T Helper cytokine secretion and a profound reduction in Tregulatorycells.BrJHaematol2015;171:60–73.[379] Shoeibi S, Mozdziak P, Mohammadi S. Important signals regulating coronary arteryangiogenesis.MicrovascRes2018;117:1–9.[380] Moreno PR, Purushothaman K-R, Sirol M, Levy AP, Fuster V. Neovascularization inhumanatherosclerosis.Circulation2006;113:2245–52.[381] VirmaniR,KolodgieFD,BurkeAP,FinnAV,GoldHK,TulenkoTN,etal.Atheroscleroticplaque progression and vulnerability to rupture: angiogenesis as a source of intraplaquehemorrhage.ArteriosclerThrombVascBiol2005;25:2054–61.[382] Rohde E, Malischnik C, Thaler D, Maierhofer T, Linkesch W, Lanzer G, et al. Bloodmonocytesmimicendothelialprogenitorcells.StemCellsDaytOhio2006;24:357–67.[383] Case J, Mead LE, Bessler WK, Prater D, White HA, Saadatzadeh MR, et al. HumanCD34+AC133+VEGFR-2+ cells are not endothelial progenitor cells but distinct, primitivehematopoieticprogenitors.ExpHematol2007;35:1109–18.[384] Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, et al. Isolation ofputativeprogenitorendothelialcellsforangiogenesis.Science1997;275:964–7.[385] IngramDA,MeadLE,TanakaH,MeadeV,FenoglioA,MortellK,etal.Identificationofanovel hierarchy of endothelial progenitor cells using human peripheral and umbilical cordblood.Blood2004;104:2752–60.[386] YoderMC,MeadLE,PraterD,KrierTR,MrouehKN,LiF, et al.Redefiningendothelial
164
progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood2007;109:1801–9.[387] Eelen G, de Zeeuw P, Treps L, Harjes U, Wong BW, Carmeliet P. Endothelial CellMetabolism.PhysiolRev2017;98:3–58.[388] Silvestre J-S, Smadja DM, Lévy BI. Postischemic Revascularization: From Cellular andMolecularMechanismstoClinicalApplications.PhysiolRev2013;93:1743–802.[389] Hill JM,ZalosG,HalcoxJPJ,SchenkeWH,WaclawiwMA,QuyyumiAA,etal.CirculatingEndothelial Progenitor Cells, Vascular Function, and Cardiovascular Risk.Http://DxDoiOrgGate2InistFr/101056/NEJMoa0222872009.[390] WernerN,KosiolS,SchieglT,AhlersP,WalentaK,LinkA,etal.Circulatingendothelialprogenitorcellsandcardiovascularoutcomes.NEnglJMed2005;353:999–1007.[391] Piaggio G, Rosti V, Corselli M, Bertolotti F, Bergamaschi G, Pozzi S, et al. Endothelialcolony-formingcellsfrompatientswithchronicmyeloproliferativedisorderslackthedisease-specificmolecularclonalitymarker.Blood2009;114:3127–30.[392] Medinger M, Mross K. Clinical trials with anti-angiogenic agents in hematologicalmalignancies.JAngiogenesisRes2010;2:10.[393] DaviesMJ.Thepathophysiologyofacutecoronarysyndromes.Heart2000;83:361–6.[394] Crea F, Libby P. Acute Coronary Syndromes: TheWay Forward FromMechanisms toPrecisionTreatment.Circulation2017;136:1155–66.[395] CasscellsW, Naghavi M,Willerson JT. Vulnerable atherosclerotic plaque: a multifocaldisease.Circulation2003;107:2072–5.[396] Newby AC. Matrix metalloproteinases regulate migration, proliferation, and death ofvascularsmoothmusclecellsbydegradingmatrixandnon-matrixsubstrates.CardiovascRes2006;69:614–24.[397] Michiels JJ, Berneman ZN, Schroyens W, Van Vliet HHDM. Pathophysiology andtreatment of platelet-mediated microvascular disturbances, major thrombosis and bleedingcomplications in essential thrombocythaemia and polycythaemia vera. Platelets 2004;15:67–84.[398] Petrides PE, Siegel F. Thrombotic complications in essential thrombocythemia (ET):clinicalfactsandbiochemicalriddles.BloodCellsMolDis2006;36:379–84.[399] Ozben B, Ekmekci A, Bugra Z, Umman S, MericM. Multiple coronary thrombosis andstent implantation to the subtotally occluded right renal artery in a patient with essentialthrombocytosis:acasereportwithreview.JThrombThrombolysis2006;22:79–84.[400] Agewall S, Beltrame JF, Reynolds HR, Niessner A, Rosano G, Caforio ALP, et al. ESCworkinggrouppositionpaperonmyocardialinfarctionwithnon-obstructivecoronaryarteries.EurHeartJ2017;38:143–53.[401] Beltrame JF,CreaF,Kaski JC,OgawaH,OngP,SechtemU,etal.TheWho,What,Why,When,HowandWhereofVasospasticAngina.CircJOffJJpnCircSoc2016;80:289–98.[402] LandolfiR,DiGennaroL,BarbuiT,DeStefanoV,FinazziG,MarfisiR,etal.Leukocytosisasamajorthromboticriskfactorinpatientswithpolycythemiavera.Blood2007;109:2446–52.[403] KaessBM,PreisSR,BeiserA,SawyerDB,ChenTC,SeshadriS,etal.Circulatingvascularendothelial growth factor and the risk of cardiovascular events. Heart 2016:heartjnl-2015-309155.[404] Pósfai É, Marton I, Borbényi Z, Nemes A. Myocardial infarction as a thromboticcomplication of essential thrombocythemia and polycythemia vera. Anatol J Cardiol2016;16:397–402.[405] LarsenAI,GalbraithPD,GhaliWA,NorrisCM,GrahamMM,KnudtsonML.Characteristicsand outcomes of patients with acute myocardial infarction and angiographically normalcoronaryarteries.AmJCardiol2005;95:261–3.
165
[406] BaireyMerzCN,PepineCJ,WalshMN, Fleg JL. Ischemia andNoObstructiveCoronaryArteryDisease (INOCA):DevelopingEvidence-BasedTherapies andResearchAgenda for theNextDecade.Circulation2017;135:1075–92.[407] Ong P, Aziz A, Hansen HS, Prescott E, Athanasiadis A, Sechtem U. Structural andFunctionalCoronaryArteryAbnormalitiesinPatientsWithVasospasticAnginaPectoris.CircJOffJJpnCircSoc2015;79:1431–8.[408] BangOY,ToyodaK,ArenillasJF,LiuL,KimJS.IntracranialLargeArteryDiseaseofNon-AtheroscleroticOrigin:RecentProgressandClinicalImplications.JStroke2018;20:208–17.[409] FurchgottRF,Zawadzki JV.Theobligatory roleofendothelial cells in the relaxationofarterialsmoothmusclebyacetylcholine.Nature1980;288:373–6.[410] Rapoport RM, Murad F. Agonist-induced endothelium-dependent relaxation in ratthoracicaortamaybemediatedthroughcGMP.CircRes1983;52:352–7.[411] Ignarro LJ, Buga GM,WoodKS, Byrns RE, Chaudhuri G. Endothelium-derived relaxingfactor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci U S A1987;84:9265–9.[412] Palmer RM, Ashton DS, Moncada S. Vascular endothelial cells synthesize nitric oxidefromL-arginine.Nature1988;333:664–6.[413] SessaWC.eNOSataglance.JCellSci2004;117:2427–9.[414] DimmelerS,Haendeler J,NehlsM,ZeiherAM.Suppressionofapoptosisbynitricoxideviainhibitionofinterleukin-1beta-convertingenzyme(ICE)-likeandcysteineproteaseprotein(CPP)-32-likeproteases.JExpMed1997;185:601–7.[415] Kang-DeckerN,CaoS,ChatterjeeS,YaoJ,EganLJ,SemelaD,etal.NitricoxidepromotesendothelialcellsurvivalsignalingthroughS-nitrosylationandactivationofdynamin-2.JCellSci2007;120:492–501.[416] Xu L, Eu JP,Meissner G, Stamler JS. Activation of the cardiac calcium release channel(ryanodinereceptor)bypoly-S-nitrosylation.Science1998;279:234–7.[417] Shiva S. Nitrite: A physiological store of nitric oxide andmodulator of mitochondrialfunction.RedoxBiol2013;1:40–4.[418] Hogg N. Detection of Nitric Oxide by Electron Paramagnetic Resonance Spectroscopy.FreeRadicBiolMed2010;49:122–9.[419] DimmelerS,FlemingI,FisslthalerB,HermannC,BusseR,ZeiherAM.ActivationofnitricoxidesynthaseinendothelialcellsbyAkt-dependentphosphorylation.Nature1999;399:601–5.[420] Vanhoutte PM, Zhao Y, Xu A, Leung SWS. Thirty Years of Saying NO: Sources, Fate,Actions, and Misfortunes of the Endothelium-Derived Vasodilator Mediator. Circ Res2016;119:375–96.[421] Cortelazzo S,VieroP,Morelli C,BarbuiT, deGaetanoG.Normal vascularprostacyclingenerationinpatientswithchronicmyeloproliferativedisorders.ThrombRes1985;39:139–41.[422] Cavalca V, Rocca B, Squellerio I, Dragani A, Veglia F, Pagliaccia F, et al. In vivoprostacyclin biosynthesis and effects of different aspirin regimens in patientswith essentialthrombocythaemia.ThrombHaemost2014;112:118–27.[423] CortelazzoS,GalliM,CastagnaD,VieroP,GaetanoGde,BarbuiT.IncreasedResponsetoArachidonic Acid and U-46619 and Resistance to Inhibitory Prostaglandins in Patients withChronicMyeloproliferativeDisorders.ThrombHaemost1988;59:73–6.[424] CooperB,AhernD.CharacterizationofthePlateletProstaglandinD2Receptor:LOSSOFPROSTAGLANDIN D2 RECEPTORS IN PLATELETS OF PATIENTS WITH MYELOPROLIFERATIVE
DISORDERS.JClinInvest1979;64:586–90.[425] CooperB,SchaferAI,PuchalskyD,HandinRI.PlateletresistancetoprostaglandinD2inpatientswithmyeloproliferativedisorders.Blood1978;52:618–26.[426] Nimmo GR,McInnes G, Turner AR, Evan-Wong A. Severemesenteric ischemia due to
166
primarythrombocythemiatreatedwithprostacyclin.CritCareMed1990;18:889–90.[427] ZeidmanA,DickerD,MittelmanM. Interferon-InducedVasospasm inChronicMyeloidLeukaemia.ActaHaematol1998;100:94–6.[428] RubanyiGM,PolokoffMA.Endothelins:molecularbiology,biochemistry,pharmacology,physiology,andpathophysiology.PharmacolRev1994;46:325–415.[429] Thorin E, ClozelM. The cardiovascular physiology and pharmacology of endothelin-1.AdvPharmacolSanDiegoCalif2010;60:1–26.[430] Yachoui R, Kristianto J, Sitwala K, Blank RD. Role of Endothelin-1 in a Syndrome ofMyelofibrosisandOsteosclerosis.JClinEndocrinolMetab2015;100:3971–4.[431] NakahataN.ThromboxaneA2:physiology/pathophysiology,cellularsignaltransductionandpharmacology.PharmacolTher2008;118:18–35.[432] YaoH-X,HuangL,LinL-E,WuC-M,WangH,ChenW-T,etal.[Thestudyofrelationshipbetween platelet function and thrombus in patients with essential thrombocythaemia].ZhonghuaYiXueZaZhi2010;90:253–5.[433] SchaferAI.Deficiencyofplateletlipoxygenaseactivityinmyeloproliferativedisorders.NEnglJMed1982;306:381–6.[434] Smith IL, Martin TJ. Platelet thromboxane synthesis and release reactions inmyeloproliferativedisorders.Haemostasis1982;11:119–27.[435] Okuma M, Uchino H. Altered arachidonate metabolism by platelets in patients withmyeloproliferativedisorders.Blood1979;54:1258–71.[436] RoccaB,CiabattoniG,TartaglioneR,CortelazzoS,BarbuiT,PatronoC,etal. Increasedthromboxanebiosynthesisinessentialthrombocythemia.ThrombHaemost1995;74:1225–30.[437] Dragani A, Pascale S, Recchiuti A, Mattoscio D, Lattanzio S, Petrucci G, et al. Thecontribution of cyclooxygenase-1 and -2 to persistent thromboxane biosynthesis in aspirin-treated essential thrombocythemia: implications for antiplatelet therapy. Blood2010;115:1054–61.[438] LandolfiR,CiabattoniG,PatrignaniP,CastellanaMA,PoglianiE,BizziB,etal.Increasedthromboxane biosynthesis in patients with polycythemia vera: evidence for aspirin-suppressibleplateletactivationinvivo.Blood1992;80:1965–71.[439] PatronoC,CiabattoniG,PatrignaniP,RoccaB,LandolfiR.Eicosanoidbiosynthesisandmetabolisminmyeloproliferativedisorders.AnnNYAcadSci1994;744:229–36.[440] ZahaviM, Zahavi J. Platelet function and thromboxane synthesis inmyeloproliferativedisorders.Blood1993;82:1377–8.[441] ZahaviJ,ZahaviM,FirsteterE,FrishB,TurleanuR,RachmaniR.Anabnormalpatternofmultiple platelet function abnormalities and increased thromboxane generation in patientswithprimarythrombocytosisandthromboticcomplications.EurJHaematol1991;47:326–32.[442] van Genderen PJ, Prins FJ, Michiels JJ, Schrör K. Thromboxane-dependent plateletactivationinvivoprecedesarterialthrombosisinthrombocythaemia:arationalefortheuseoflow-doseaspirinasanantithromboticagent.BrJHaematol1999;104:438–41.[443] Pascale S, Petrucci G, Dragani A, Habib A, Zaccardi F, Pagliaccia F, et al. Aspirin-insensitive thromboxane biosynthesis in essential thrombocythemia is explained byacceleratedrenewalofthedrugtarget.Blood2012;119:3595–603.[444] TefferiA.Overcoming“aspirinresistance”inMPN.Blood2012;119:3377–8.[445] Karantanos T, Moliterno AR. The roles of JAK2 in DNA damage and repair in themyeloproliferativeneoplasms:Opportunitiesfortargetedtherapy.BloodRev2018.[446] BjørnME, Hasselbalch HC. The Role of Reactive Oxygen Species inMyelofibrosis andRelatedNeoplasms.MediatorsInflamm2015;2015:648090.[447] Kagoya Y, Yoshimi A, Tsuruta-Kishino T, Arai S, Satoh T, Akira S, et al. JAK2V617F+myeloproliferative neoplasm clones evoke paracrine DNA damage to adjacent normal cells
167
throughsecretionoflipocalin-2.Blood2014;124:2996–3006.[448] MartyC,LacoutC,DroinN,LeCouédic J-P,RibragV,SolaryE,etal.Arole forreactiveoxygen species in JAK2V617F myeloproliferative neoplasm progression. Leukemia2013;27:2187–95.[449] Plo I, Nakatake M, Malivert L, de Villartay J-P, Giraudier S, Villeval J-L, et al. JAK2stimulates homologous recombination and genetic instability: potential implication in theheterogeneityofmyeloproliferativedisorders.Blood2008;112:1402–12.[450] AhnJS,LiJ,ChenE,KentDG,ParkHJ,GreenAR.JAK2V617FmediatesresistancetoDNAdamage-induced apoptosis by modulating FOXO3A localization and Bcl-xL deamidation.Oncogene2016;35:2235–46.[451] Hurtado-NedelecM,Csillag-GrangeM-J,BoussettaT,BelambriSA,FayM,CassinatB,etal.Increasedreactiveoxygenspeciesproductionandp47phoxphosphorylationinneutrophilsfrom myeloproliferative disorders patients with JAK2 (V617F) mutation. Haematologica2013;98:1517–24.[452] MusolinoC,AllegraA, SaijaA,AlonciA,RussoS, SpatariG, et al.Changes inadvancedoxidation protein products, advanced glycation end products, and s-nitrosylated proteins, inpatients affected by polycythemia vera and essential thrombocythemia. Clin Biochem2012;45:1439–43.[453] DurmusA,MenteseA,YilmazM, SumerA,Akalin I,TopalC, et al. Increasedoxidativestressinpatientswithessentialthrombocythemia.EurRevMedPharmacolSci2013;17:2860[454] Vener C, Novembrino C, Catena FB, Fracchiolla NS, Gianelli U, Savi F, et al. Oxidativestress is increased in primary and post-polycythemia vera myelofibrosis. Exp Hematol2010;38:1058–65.[455] Hasselbalch HC, Thomassen M, Riley CH, Kjær L, Larsen TS, Jensen MK, et al. Wholeblood transcriptional profiling reveals deregulation of oxidative and antioxidative defencegenesinmyelofibrosisandrelatedneoplasms.PotentialimplicationsofdownregulationofNrf2forgenomicinstabilityanddiseaseprogression.PloSOne2014;9:e112786.[456] IurloA,DeGiuseppeR,SciumèM,CattaneoD,FermoE,DeVitaC,etal.Oxidativestatusintreatment-naïveessentialthrombocythemia:apilotstudyinasinglecenter.HematolOncol2017;35:335–40.[457] BarbuiT,MasciulliA,GhirardiA,CarobbioA.ACEinhibitorsandcytoreductivetherapyinpolycythemiavera.Blood2017;129:1226–7.[458] AksuS,BeyazitY,HaznedarogluIC,KekilliM,CanpinarH,MisirlioğluM,etal.Enhancedexpressionofthelocalhaematopoieticbonemarrowrenin-angiotensinsysteminpolycythemiarubravera.JIntMedRes2005;33:661–7.[459] Haznedaroğlu IC, Büyükaşik Y. Current evidence for the existence of a local renin-angiotensinsystemaffectingphysiologicalandpathologicalhaemopoiesisinthebonemarrow.BrJHaematol1997;99:471.[460] VrsalovicMM, Pejsa V, Veic TS, Kolonic SO, Ajdukovic R, Haris V, et al. Bonemarrowrenin-angiotensin system expression in polycythemia vera and essential thrombocythemiadependsonJAK2mutationalstatus.CancerBiolTher2007;6:1434–6.[461] Haznedaroglu IC, Malkan UY. Local bone marrow renin-angiotensin system in thegenesisofleukemiaandothermalignancies.EurRevMedPharmacolSci2016;20:4089–111.[462] Rodrigues-Ferreira S, Abdelkarim M, Dillenburg-Pilla P, Luissint A-C, di-Tommaso A,DeshayesF,etal.AngiotensinIIfacilitatesbreastcancercellmigrationandmetastasis.PloSOne2012;7:e35667.[463] Shi K, ZhaoW, Chen Y,HoWT, Yang P, Zhao ZJ. Cardiac hypertrophy associatedwithmyeloproliferativeneoplasmsinJAK2V617Ftransgenicmice.JHematolOncolJHematolOncol2014;7:25.
168
[464] Tarach JS, Nowicka-Tarach BM, Matuszek B, Nowakowski A. Erythromelalgia--athromboticcomplicationinchronicmyeloproliferativedisorders.MedSciMonitIntMedJExpClinRes2000;6:204–8.[465] VallaD-C.PrimaryBudd-Chiarisyndrome.JHepatol2009;50:195–203.[466] Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, et al.Ephrin-B2controlsVEGF-inducedangiogenesisandlymphangiogenesis.Nature2010;465:483–6.[467] Marty C, Lacout C,Martin A, Hasan S, Jacquot S, BirlingM-C, et al.Myeloproliferativeneoplasm induced by constitutive expression of JAK2V617F in knock-in mice. Blood2010;116:783–7.[468] Simonetto DA, Yang H, Yin M, de Assuncao TM, Kwon JH, Hilscher M, et al. Chronicpassive venous congestion drives hepatic fibrogenesis via sinusoidal thrombosis andmechanicalforces.HepatolBaltimMd2015;61:648–59.[469] Schaefer A, Jung M, Miller K, Lein M, Kristiansen G, Erbersdobler A, et al. Suitablereference genes for relative quantification ofmiRNA expression in prostate cancer. ExpMolMed2010;42:749–58.[470] KaessBM,PreisSR,BeiserA,SawyerDB,ChenTC,SeshadriS,etal.Circulatingvascularendothelial growth factor and the risk of cardiovascular events. Heart Br Card Soc2016;102:1898–901.[471] Zhao B, Mei Y, Cao L, Zhang J, Sumagin R, Yang J, et al. Loss of pleckstrin-2 revertslethality and vascular occlusions in JAK2V617F-positivemyeloproliferative neoplasms. J ClinInvest2017.[472] Sen-BanerjeeS,MirS,LinZ,HamikA,AtkinsGB,DasH,etal.Kruppel-LikeFactor2asaNovelMediatorofStatinEffectsinEndothelialCells.Circulation2005;112:720–6.[473] MasonRP,WalterMF,JacobRF.EffectsofHMG-CoAreductaseinhibitorsonendothelialfunction:roleofmicrodomainsandoxidativestress.Circulation2004;109:II34-41.[474] AngonaA,Alvarez-LarránA,BellosilloB,LongarónR,Fernández-RodríguezC,BessesC.Dynamics of JAK2V617F allele burdenof CD34+haematopoietic progenitor cells in patientstreatedwithruxolitinib.BrJHaematol2016;172:639–42.[475] VainchenkerW,LeroyE,GillesL,MartyC,PloI,ConstantinescuSN.JAKinhibitorsforthetreatmentofmyeloproliferativeneoplasmsandotherdisorders.F1000Research2018;7:82.[476] Cokic VP, Beleslin-Cokic BB, Tomic M, Stojilkovic SS, Noguchi CT, Schechter AN.HydroxyureainducestheeNOS-cGMPpathwayinendothelialcells.Blood2006;108:184–91.[477] Miki T, SuzukiM, Shibasaki T, UemuraH, SatoT, YamaguchiK, et al.Mousemodel ofPrinzmetalanginabydisruptionoftheinwardrectifierKir6.1.NatMed2002;8:466–72.[478] ZorovDB,JuhaszovaM,SollottSJ.Mitochondrialreactiveoxygenspecies(ROS)andROS-inducedROSrelease.PhysiolRev2014;94:909–50.[479] HeitzerT,SchlinzigT,KrohnK,MeinertzT,MünzelT.Endothelialdysfunction,oxidativestress,andriskof cardiovascularevents inpatientswithcoronaryarterydisease.Circulation2001;104:2673–8.[480] PonomarevaAA,NevzorovaTA,MordakhanovaER,Andrianova IA,RauovaL, LitvinovRI, et al. Intracellular origin and ultrastructure of platelet-derived microparticles. J ThrombHaemostJTH2017;15:1655–67.[481] CamusSM,GausserèsB,BonninP,LoufraniL,GrimaudL,CharueD, et al.Erythrocytemicroparticles can induce kidney vaso-occlusions in a murine model of sickle cell disease.Blood2012;120:5050–8.[482] CamusSM,DeMoraesJA,BonninP,AbbyadP,LeJeuneS,LionnetF,etal.Circulatingcellmembrane microparticles transfer heme to endothelial cells and trigger vasoocclusions insicklecelldisease.Blood2015;125:3805–14.
169
[483] Merle NS, Grunenwald A, Rajaratnam H, Gnemmi V, Frimat M, Figueres M-L, et al.Intravascular hemolysis activates complement via cell-free heme and heme-loadedmicrovesicles.JCIInsight2018;
170
VII. APPENDIX
A. Appendix1:Review:Liversinusoidalendothelialcells:physiologyandrole
Liver sinusoidal endothelial cells: Physiology and role inliver diseases
Johanne Poisson1,2,y, Sara Lemoinne3,4,y, Chantal Boulanger1,2, François Durand5,6,7,Richard Moreau5,6,7, Dominique Valla5,6,7, Pierre-Emmanuel Rautou1,2,5,6,7,⇑
Summary
Liver sinusoidal endothelial cells (LSECs) are highly specialized endothelial cells representingthe interface between blood cells on the one side and hepatocytes and hepatic stellate cells onthe other side. LSECs represent a permeable barrier. Indeed, the association of ‘fenestrae’,absence of diaphragm and lack of basement membrane make them the most permeableendothelial cells of the mammalian body. They also have the highest endocytosis capacityof human cells. In physiological conditions, LSECs regulate hepatic vascular tone contributingto the maintenance of a low portal pressure despite the major changes in hepatic blood flowoccurring during digestion. LSECs maintain hepatic stellate cell quiescence, thus inhibitingintrahepatic vasoconstriction and fibrosis development. In pathological conditions, LSECs playa key role in the initiation and progression of chronic liver diseases. Indeed, they become cap-illarized and lose their protective properties, and they promote angiogenesis and vasoconstric-tion. LSECs are implicated in liver regeneration following acute liver injury or partialhepatectomy since they renew from LSECs and/or LSEC progenitors, they sense changes inshear stress resulting from surgery, and they interact with platelets and inflammatory cells.LSECs also play a role in hepatocellular carcinoma development and progression, in ageing,and in liver lesions related to inflammation and infection. This review also presents a detailedanalysis of the technical aspects relevant for LSEC analysis including the markers these cellsexpress, the available cell lines and the transgenic mouse models. Finally, this review providesan overview of the strategies available for a specific targeting of LSECs.! 2016 European Association for the Study of the Liver. Published by Elsevier B.V. All rightsreserved.
Introduction
The vascular endothelium, representing the inter-face between blood and other tissues, is not onlya physical barrier, but contributes to different phys-iological and pathological processes, includinghemostasis/thrombosis, metabolites transporta-tion, inflammation, angiogenesis and vascular tone[1]. Liver sinusoidal endothelial cells (LSECs) formthe wall of the liver sinusoids and representapproximately 15 to 20% of liver cells but only 3%of the total liver volume [2]. LSECs are highly spe-cialized endothelial cells. They have a discontinu-ous architecture meaning that fusion of theluminal and abluminal plasma membrane occursat other sites than cell junctions, in areas called‘fenestrae’. This review focuses on the role of LSECsin physiological conditions and their involvementin liver diseases.
LSECs in the normal liver
Formation of sinusoids during embryogenesis
As illustrated in Fig. 1, an early structural differentia-tion of hepatic sinusoids occurs between gestationalweeks 5 and 12 in human embryos [3]. During thatperiod, LSECs gradually loose cellmarkers of continu-ous endothelial cells including platelet endothelialadhesion molecule-1 (PECAM-1, also called clusterof differentiation (CD)31), CD34 and 1F10 antigen,and acquire markers of adult sinusoidal cells includ-ing CD4, CD32 and the intracellular adhesionmolecule-1 (ICAM-1). This differentiation of LSECsis regulated by hepatoblasts, both via the vascularendothelial growth factor (VEGF) they release andvia direct intercellular interactions [4,5].
Journal of Hepatology 2017 vol. 66 j 212–227
Review
Review
Keywords: Liver sinusoidal endo-
thelial cells; Capillarization; End-
othelial dysfunction; Cirrhosis;
Liver regeneration; Angiogenesis;
Drug delivery system;
Endothelium.
Received 24 May 2016; received in
revised form 5 July 2016; accepted 7
July 2016
1INSERM, UMR-970, Paris Cardio-
vascular Research Center – PARCC,
Paris, France;2Université Paris Descartes, Sor-
bonne Paris Cité, Paris, France;3INSERM, UMRS 938, Centre de
The embryological origin of LSECs is still a mat-ter of debate. Initial observational studies describedcapillaries progressively surrounded by growingcords of hepatoblasts in the septum transversum,suggesting that LSECs derive from the septumtransversum mesenchyme, a part of the mesoderm[3,6,7]. However, recent cell lineage experimentsperformed in mice showed that the septumtransversum gives rise to mesothelial cells, hepaticstellate cells, portal fibroblasts, and perivascularmesenchymal cells, but not to LSECs [8]. A part ofLSECs rather derives from a common progenitorto endothelial and blood cells, called the ‘‘heman-gioblast”, as attested by overlapping expression ofhematopoietic and endothelial cell markers byLSECs and by fate tracing experiments [9–14].These progenitor cells form veins crossing the sep-tum transversum, i.e., vitelin veins [15], umbilicalveins or cardinal veins and then LSECs [16,17].Another part of LSECs derives from the endo-cardium of the sinus venosus, a compartment ofthe primitive cardiac tube [18]. These two embry-ological origins might explain the heterogeneity ofthe markers expressed by LSECs in adults.
LSECs renewal
Although specific data are lacking, we can specu-late that in a physiological state LSECs are quies-cent, i.e., with a low proliferation rate and a longlife span, similar to endothelial cells from large ves-sels [19]. LSECs renewal differs in physiological andin pathological conditions. Three cell types con-tribute to LSEC renewal, namely mature LSECs,intrahepatic or resident sinusoidal endothelial cellprogenitors, and bone marrow derived sinusoidalendothelial cell progenitors [20]. Mature LSECscan self-proliferate in normal conditions, whenstimulated with growth factors such as VEGF andFGF (fibroblast growth factor) [20,21]. Residentsinusoidal endothelial cell progenitors represent 1to 7% of the LSECs of a normal rodent liver andprobably contribute to LSECs regeneration [20].Bone marrow derived sinusoidal endothelial cellprogenitors do not participate in LSEC turnover ina normal liver [22]. By contrast, after liver injury,these cells are the main drivers of liver regenera-tion [20,22]. Indeed, a subtoxic dose of monocro-talin, a toxic agent for LSECs, elicits liver injuryonly when bone marrow is suppressed. In addition,infusion of bone marrow cells after a toxic dose ofmonocrotalin almost fully corrects liver lesions[23].
Hepatic blood flow regulation
Liver sinusoids have a dual blood supply, receivingblood flow from the portal vein (70%) and the hep-atic artery (30%) [24]. Blood pressure equalizes inthe sinusoid and blood is then drained into the hep-atic veins and the inferior vena cava. Despite major
circadian changes in hepatic blood flow due todigestion, hepatic venous pressure gradient remainsat 4 mmHg or less in a normal individual, attesting afine regulation of hepatic vascular tone [25]. Intra-hepatic shear stress is recognized as a main driverof hepatic blood flow regulation [26]. Shear stressis a frictional force applied by blood flow onendothelial surface [26]. It is proportional to flowintensity and to blood viscosity and inversely pro-portional to the cubic radius of the vessel [26]. Intra-hepatic shear stress has never been directlymeasured in human or animal. Its evaluation isindeed difficult since the radius of sinusoids is verysmall and varies within the liver. Moreover, viscos-ity is hard to estimate in this specific area and alsovaries with hemodilution. In normal conditions, inthe liver like in other vascular beds, the endothe-lium is able to generate vasodilator agents inresponse to increased shear stress in order to atten-uate the increase in blood pressure. The loss of thisproperty is called endothelial dysfunction. Anendothelial specific transcription factor induced byprolonged shear stress, called Kruppel-like factor 2(KLF2) mediates this effect of shear stress [27].KLF2 induces the endothelial upregulation ofvasodilating agents including nitric oxide (NO) [28](Fig. 2). Shah and colleagues previously demon-strated that LSECs are the main source of NO inthe normal liver through endothelial nitric oxidesynthase (eNOS) activation by shear stress [29].KLF2 also induces the downregulation of vasocon-strictive molecules including endothelin-1 [28].Other molecules released by LSECs regulating bloodflow include the vasodilating agent carbon monox-ide (CO) and the metabolites of the cyclooxygenase(COX) pathway (thromboxane A2, Prostacyclin)[30]. All these molecules act in a paracrine manneron hepatic stellate cells localized in the space ofDisse [31]. Healthy LSECs maintain hepatic stellatecell quiescence, thus inhibiting their vasoconstric-tive effect [34]. The concept that hepatic stellate cellactivation induces sinusoid constriction is based ontheir expression of molecules found in smooth mus-cle cells including aSMA, on their position wrappedaround the exterior of LSECs and on the ex vivo
observation of their ability to contract [32,33].Although still controversial, LSEC could also regulateblood flow by swelling, thus creating an inlet and anoutlet sphincter [32]. Kupffer cells possess contrac-tile proteins as well, but their role in the regulationof hepatic blood flow remains controversial [32]. Incontrast to most vascular beds where blood flow ismostly regulated by smooth muscle cells, in theliver, smooth muscle cells play a limited role since,although present in hepatic arterioles, they are onlyfound in limited numbers in portal venules [32].
LSECs, a selective barrier
LSECs are positioned at an interface. On theirsinusoidal side, they are exposed to the highly
Key point
In a normal liver, differenti-ated LSECs are gatekeepersof fibrogenesis by maintain-ing hepatic stellate cells intheir inactivated state. LSECsregulate sinusoidal bloodflow through their actionon hepatic stellate cells andthus maintain a low portalpressure.
Review
JOURNAL OF HEPATOLOGY
Journal of Hepatology 2017 vol. 66 j 212–227 213
oxygenated arterial blood mixed with the portalblood derived from the gut and the pancreas con-taining nutrients, bile acids, and hormones includ-ing insulin and glucagon. On the abluminal side,they interact with hepatic stellate cells and hepato-cytes that are crucial for protein, lipid and glucosemetabolism. LSECs thus represent a permeable bar-rier allowing exchanges but also active uptake anddegradation of molecules [35].
Fluid exchange through fenestrae
Like endothelial cells located in other exchange ter-ritories, such as the glomeruli, the spleen and thebone marrow, LSECs are highly permeable [36].The association of fenestrae, absence of diaphragmand lack of basement membrane make them themost permeable endothelial cells of the mam-malian body [24]. These fenestrae are organizedin clusters termed sieve plates [37]. LSEC fenestraehave a diameter ranging from 50 to 150 nm[2,37,38]. Their size and number varies dependingon their localization in the liver, with larger butfewer fenestrae per sieve plate in the periportalregion and smaller but more numerous fenestraeper sieve plate in the centrilobular region [37,39].
This distribution could be related to the progressivedecrease in oxygen tension along the lobule accom-panied with an increasing need for oxygen exchange[36]. Alternatively, this distribution could be a mar-ker of LSEC maturation as they spread along the lob-ule [39]. Fenestrae are not static structures. Theirnumber and size varies in physiological conditionslike fasting that decreases the number but increasesthe size of the fenestrae [40] and in pathologicalconditions [36,39,41–43]. Using super-resolutionoptical microscopy, Mönkemöller and colleaguesrecently showed that sieve plates are surroundedand separated by microtubuli and that each fenes-trae within a sieve plate is surround by actin fila-ments [38]. Cytoskeleton is thus of greatimportance for the LSECS fenestrations. Fifteen yearsago, LSEC fenestrations were thought to be sort ofcaveolae [44]. Caveolae are uncoated plasma mem-brane invaginations found in lipid-ordered domainsof cell membranes called lipid rafts. Caveolin is amajor structural protein of caveolae. Althoughcaveolin-1 has been observed in LSECs fenestrations[44], caveolin-1 knockout mice have normal fenes-trations [45]. In addition, Svistounov and colleagues[46,47] described the ‘‘sieve-raft crosstalk”, wherefenestrations are formed in reduced lipid-raftregions of endothelial cells. Thus, fenestrations arenot dependent on caveolin-1 and are different struc-tures from caveolae.
In a normal liver, LSECs retain blood cells in thevessels, while molecules, such as metabolites,plasma proteins, pharmaceutical drugs, lipoproteinsand small chylomicron remnants, viruses (<200 nm)and exosomes can access the space of Disse to betaken up by hepatocytes and hepatic stellate cells[2,38,48]. There is no significant osmotic and hydro-static pressure gradient across the normal liver sinu-soids [41,49]. Small molecules and gasses freelydiffuse through the fenestrae, so that the space ofDisse contains a para-vascular part of the plasmavolume. In addition, as blood cells squeeze into thesinusoids, they massage the endothelial cells andfurther mix plasma and space of Disse fluids [49].Larger molecules, may also cross LSEC by a processcalled permselectivity or ‘‘sieving”, namely therestricted transport of large molecules due to theirdeformation capacity through membrane pores[41]. The fluid present in the space of Disse isdrained into hepatic lymphatics, then into hepatichilum lymphatics, cisterna chili, thoracic duct andeventually the central venous circulation, succes-sively [50]. The fluid formed in excess gains freeaccess to the Glisson’s capsule on the liver surface[49]. Contrary to the mesentery, the liver is thusleaky to large molecules including proteins. Thisexplains why ascites related to post-sinusoidalobstruction, such as cardiac failure or Budd-Chiarisyndrome, is protein rich while ascites resultingfrom cirrhosis is not [50].
Septum transversum
Foregut
Blood vesselsD
Liver primordium
V
A
B
Foregut
Hepatoblasts(liver primordium)
Future LSEC
Differentiated LSEC
(Weeks of gestation)
5 8 12
Markers expressed by LSECs
CD4; CD32; ICAM-1; Fenestrae
CD31; CD34; 1F10
Time
Fig. 1. Formation of sinusoids during human embryogenesis. (A) Frontal section of an embryo
showing the formation of an outgrowth of the foregut (endoderm), called the liver primordium, which
extends into the septum transversum (mesoderm), in which blood vessels are developing. V, ventral; D,
dorsal. (B) Transversal section of the embryo showing the liver primordium (i.e., hepatoblasts arranged
in thick cords separated by vascular spaces) growing into the septum transversum. The hepatic
sinusoids are progressively established. First, the endothelial lining is continuous with a basement
membrane (pink region) and no fenestrations. Around gestation week 12, fenestrations appear initially
with diaphragms. These diaphragms disappear during development [15,170,171].
Key point
LSECs act as a selective bar-rier, since exchanges occurthrough fenestrae as well asby transcytosis and LSECscavenging functions.
Review
Review
214 Journal of Hepatology 2017 vol. 66 j 212–227
Endocytic capacity
LSECs have one of the highest endocytic capacity inthe human body [51]. This property combined witha strong lysosomal activity give LSECs the ability toclear waste from the blood, as part of the ‘‘dual-cellprinciple” of waste clearance. This principle statesthat the mononuclear system represents the pro-fessional phagocyte, eliminating large particles,and that the scavenger endothelial cells, includingLSECs, represents the professional pinocyte, clear-ing soluble macromolecules and small particlesthrough endocytic receptors [52]. This propertycan be used to specifically target LSECs. LSEC endo-cytosis also contributes to the transfer of moleculesfrom the sinusoids to the space of Disse, a processcalled transcytosis [35]. Endocytosis by LSECsimplies different high affinity endocytosis recep-tors, including scavenger receptors (SR-A, SR-Band SR-H), mannose receptor and Fc gamma-receptor IIb2 [51,52]. The SRs mediate endocytosisof polyanionic molecules, such as oxidized andacetylate low-density lipoproteins (oxLDL and
acLDL), advanced glycation end products and wasteproducts (hyaluronan, chondroitin sulfate or N-terminal propeptides of procollagen (I, III)). Themain SRs of LSECs are SR-H/stabilin-1 and SR-H/stabilin-2. Stabilin1/2 double-knockout mice showonly a mild liver fibrosis without liver dysfunctionbut a severe renal glomerular fibrosis [53], suggest-ing that stabilin-1 and 2 are major liver endocyticreceptors implicated in the clearance of moleculestoxic mainly for the kidney. The mannose receptorsare not specific of LSECs and bind a wide range ofglycoproteins and microbial glycans, such as colla-gen alpha chains (I, II, III, IV, V, XI), tissue plasmino-gen activator regulating fibrinolytic activity, andlysosomal enzymes that are recruited for furtheruse in LSEC [54]. Thus, they have a role both inimmunity and in glycoprotein homeostasis [52].The Fc gamma-receptor IIb2 is the only Fc gamma-receptor expressed by LSECs and mediates the clear-ance of small circulating immune complexes; LSECplay a role in vascular immunity through this recep-tor [51,52].
E
EE
E
Shear stress
LSEC
↑KLF2
Fenestrae sieve plate
Space of Disse
Hepatocyte
VASODILATION VASOCONSTRICTION
HSC
QuiescentCollagen
LSEC
Hepatocyte
Space of Disse
HSC
Activated
NO
NO
NO
NO
NO NOStatins
Endothelial dysfunction
↑KLF2
BM
↑Shear stress
↑Resistance
NORMAL LIVER CIRRHOTIC LIVER
x
Fig. 2. Role of liver sinusoidal cells (LSECs) in chronic liver diseases. In normal conditions, LSECs maintain hepatic stellate cell
quiescence through a NO-dependent pathway as long as they are differentiated [101]. Exposure of LSECs to a physiological shear
stress activates the transcriptor factor KLF2 leading to the release of vasodilating agents including nitric oxide (NO) and to the
downregulation of vasoconstrictive molecules including endothelin-1. In a cirrhotic liver, LSECs become capillarized, meaning that
they lose their fenestrae and a basement membrane appears. Capillarized LSECs permit hepatic stellate cell activation and thus
production of collagen and of fibrosis. This change is associated with an endothelial dysfunction meaning that increased shear stress
no longer leads to vasodilation but rather to vasoconstriction and thus to increased intrahepatic resistance. Simvastatin restores the
vasoprotective effect of KLF2 and improves HSC phenotype through a NO-dependent pathway (the effect of simvastatin appears in
Identification and isolation of LSECs is a majorchallenge for the understanding of liver physiologyand diseases. However, technical barriers as well asa lack of consensual specific LSEC markers explainthat LSECs populations differ between researchgroups, which limits the interpretation and thecomparison of the results.
Features used to identify LSECs include: (a) theirhigh and rapid endocytic capacity, using labeledformaldehyde-treated serum albumin, collagenalpha chains or acLDL. As other cells, includingKupffer cells, also have endocytic capacities,labeled molecules have to be incubated in smallamount and for a short period of time to be specificfor LSECs [55]. (b) Fenestrae without diaphragmand organized in sieve plates, using electron micro-scopy. Although this feature is the only one specificof LSECs, it has some limitations. First, the distribu-tion of the fenestration varies along the lobule [37].Second, LSEC isolation methods, including liver per-fusion and cell preparation for electron microscopy,dilate fenestrae and might even create holes in cellsurface [55]. Third, fenestrae rapidly disappearwhen LSEC are cultured as a monolayer of cells,out of their environment [56]. This loss of fenestra-tion associated with basement membrane synthe-sis and modification of the expression of surfacemarkers is called capillarization. Capillarizationnot only happens in cultured LSEC but also in vivo
in most liver diseases [56]. (c) surface markers[24] (Table 1). Some markers are common to otherendothelial cells and some to hematopoietic cells.No single marker is specific for LSECs and a combi-nation is required. For instance, Ding and col-leagues considered that LSEC are VEGFR3+ CD34!
VEGFR2+ VE-Cadherin+ FactorVIII+ CD45! [57],while Lalor and colleagues selected CD31+, LYVE-1+, L-SIGN+, Stabilin-1+, CD34!, PROX-1! cells[56]. CD31, CD45 and CD33 deserve specific com-ments. CD31 (PECAM-1) is an intercellular adhe-sion molecule classically expressed at the surfaceof endothelial cells, but also of several leukocytes[58]. The expression of CD31 by LSECs is controver-sial. Several studies reported CD31 positivity ofLSECs in liver slices analyzed by immunohisto-chemistry or in cultured cells permeabilized beforestaining [55]. Conversely, for the isolation of LSECsusing flow cytometry, LSECs are considered asCD31 negative, CD31 positive cells being arterialand venous endothelial cells as well as capillarizedLSECs. An electron microscopic analysis reconciledthese results by showing that CD31 is located intra-cellularly shortly after establishing LSEC cultures,but, when fenestrae disappear few days later,CD31 becomes expressed at the cell surface likein other endothelial cells [59]. CD45 is ahematopoietic cell marker, expressed by leuco-
cytes. LSECs are usually described as CD45!, andliver CD45+ cells are often considered as Kupffercells. However, the reality may be more complex,as LSEC CD45 positivity appears to depend on thelocalization and the differentiation of LSECs[24,39]: bright CD45 positivity is found in periportalarea where LSECs have less fenestration, while CD45negativity appears to predominate in centrilobularareas where LSECs are more differentiated withmore fenestrae.
Knowledge of LSEC markers helps understandingsome drug adverse effects. For instance, Mylotarg"
(gemtuzumab ozogamicin), a drug used for acutemyeloid leukemia, consists of a humanized antibodyanti-CD33, linked to a potent antitumor antibiotic(calicheamicin). CD33 is expressed on the surfaceof acute myeloid leukemia cells, but also of LSECslikely explaining the high prevalence of hepaticsinusoidal obstruction syndrome following thistreatment [60].
LSECs culture
As mentioned above, obtaining a pure culture of pri-mary LSECs is challenging because of the lack ofspecific markers of these cells. LSEC isolation proto-cols are detailed elsewhere [61,62]. The culture ofLSECs has at least four particularities. First, culturedLSEC tend to lose their typical phenotype. In order toprevent this dedifferentiation, several methods havebeen developed. Co-culture with hepatocytes andfibroblasts rather than with hepatocytes aloneallows LSECs to maintain their phenotype for up to2 weeks [63]. Extracellular matrix coating mimick-ing the space of Disse and its modifications inpathology can also be used, e.g., low-density base-ment membrane-like matrix imitating normal con-ditions, and interstitial type matrix (fibril-formingcollagen) imitating cirrhosis [63]. The addition ofVEGF to the medium or the use of hepatocyte-conditioned medium can also prevent LSECs dedif-ferentiation [56,64,65]. Second, when culturedalone, LSECs undergo apoptosis within 2 days [63];methods preventing dedifferentiation also preventcell death. Third, serum supplementation is toxicfor LSECs [55]. Fourth, in the normal liver, LSECsare exposed to an oxygen pressure decreasing alongthe liver lobule from 90 to 30 mmHg [66]; oxygenlevel is thus lower than in atmospheric conditionswhere oxygen pressure is 160 mmHg; actually, LSECare particularly sensitive to hyperoxia and to theresulting oxidative stress [67]; survival of primaryLSECs is improved under 5% oxygen instead of thecommonly used 20% [51,66].
To overcome the difficulties of culturing primaryLSECs, several teams have developed human andmurine immortalized LSECs lines. However, the firstimmortalized lines, obtained by viral transfectionsuch as M1LEC, had no fenestrae [68–72]. Subse-quently, several humans and murine immortalizedLSEC lines have been developed. As summarized in
Key point
There is no unique specificmarker of LSECs, apart fromtheir fenestrae devoid of dia-phragm in the absence ofbasement membrane. Acombination of markers isthus mandatory for theiridentification.
Key point
When cultured, primaryLSECs rapidly lose theirspecific phenotype. How-ever, human and murineimmortalized LSECs lineshave been successfullydeveloped.
Review
Review
216 Journal of Hepatology 2017 vol. 66 j 212–227
Table 2, these cell lines display many characteris-tics of LSEC. Each cell line has particular advantagesmaking it more appropriate for specific studies. Forinstance, TSECs are adequate for angiogenesis anal-yses and Sk Hep1 for fenestration. However, thefact that these cell lines are immortalized impliesthat they may react differently from primary cellsin response to stress. Therefore, a confirmation ofthe findings using primary cells is useful.
Mouse models
Transgenic mice using the Cre/Lox system can bevery useful to study the properties of LSECs in vivo.Briefly, Cre-recombinase, which can be regulatedby a tissue-specific promoter, excises essentialloxP-flanked (‘‘floxed’’) genes via intrachromosomalrecombination to generate so called conditionalknockouts, i.e., knockouts specifically affecting
Common with EC markers Endocytic markers Antigen presentation Common with leucocytes Common with lymphatic EC
CD34$
CD105*
CD146
Cytoplasmic CD31
ICAM-1
Ulex Lectin binding
vWf (Factor VIII)$
CD36
DC-SIGN
L-SIGN
Lectins
LYVE-1
SR-A/SR-B
Stabilin-1
Stabilin-2
Uptake of acLDL or denatured
alpha-collagen chain
CD40$
CD80$
CD86$
Fc Gamma R (CD32b**)
Mannose R
MHC I/MHC II$
CD4
CD11b
CD11c$
CD33
CD45$
Cytoplasmic CD31
VAP-1
⁄Also expressed by hepatic stellate cells and myofibroblasts; ⁄⁄correlates with fenestration and corresponds to SE-1 in rats [63]; $controversial [55].
tissues where the promoter is expressed. Severalmodels with an endothelial cell expression of theCre-recombinase have been developed and are sum-marized in Table 3. Mice with a constitutive expres-sion of the Cre-recombinase appeared first.However, the expression of the Cre-recombinase isnot restricted to endothelial cells, especially in adultmice, as recombination also occurs in hematopoieticcells. Indeed, early embryonic endothelial andhematopoietic cells arise froma commonembryonicprecursor called the hemangioblast [14]. This limita-tion can be overcome by performing a transplanta-tion of wild-type bone marrow together with aclodronate mediated Kupffer cell depletion [73].Indeed, in the absence of clodronate treatment,2 months after bone marrow transplantation, 85%of the Kupffer cells are still derived from the recipi-ent [74]. Myeloablation conditionings required forbone marrow transplantation might however alterLSEC function. Another way to overcome the con-comitant expression of the Cre-recombinase inendothelial and hematopoietic cells is to use trans-genic lines where Cre expression is induced in adultendothelial cells after tamoxifen administration. Inthat case, there is no expression of the transgene inhematopoietic cells.
LSECs in liver diseases
Chronic liver diseases
LSECs play a key role in chronic liver disease initi-ation and progression, through four processes:
sinusoid capillarization, angiogenesis, angiocrinesignals and vasoconstriction.
Capillarization of LSECs, also called dedifferentia-tion, occurs following liver injury in animal modelsas well as in patients [75–80]. Capillarization is anearly event since it precedes the activation of hep-atic stellate cells and macrophages and the onsetof liver fibrosis, suggesting that it could be a prelim-inary step necessary for fibrogenesis [76,81,82]. Themechanisms of capillarization and the cross talkbetween LSECs and hepatic stellate cells has beenreviewed elsewhere [83]. Briefly, LSECs are able tomaintain hepatic stellate cells quiescent as long asthey are differentiated so that differentiated LSECsare gatekeepers of fibrosis [34,84]. VEGF contributesto the maintenance of LSEC differentiation (Fig. 3).The role of LSECs in fibrosis regression is less clear.Indeed, in experimental models, restoration of LSECdifferentiation in vivo promotes regression of mildfibrosis [34,85]. However, immunohistochemicalanalysis of paired liver biopsies from 38 hepatitis Cvirus patients with cirrhosis, before and after antivi-ral treatment, revealed that sinusoid capillarizationpersists despite the regression of cirrhosis. LSEC dif-ferentiation is thus not crucial for fibrosis regressionin this setting [86].
Angiogenesis is defined by the development ofnew vessels from preexistent vessels [87]. Hepaticangiogenesis occurs during liver fibrogenesis andthese two processes are closely linked [88,89]. Liverfibrosis enhances angiogenesis and, in turn, liverangiogenesis aggravates liver fibrosis, as attestedby the anti-fibrotic effect of most anti-angiogenic
Key point
The loss of the specific phe-notype of LSECs, includingthe disappearance of the fen-estrae, the development of abasement membrane, andthe appearance of specificmarkers is called capillariza-tion and is an early even inchronic liver injury. Whencapillarized, LSECs lose theircapacity to inactivate hepaticstellate cells, thus promotingfibrogenesis and intrahepaticvasoconstriction.
Table 3. Characteristics of transgenic mice available to study the properties of liver endothelial cells in vivo.
Transgenic mice [Ref.] Constitutive/
inducible
Liver endothelial expression in adults Expression by hematopoietic
cells in adults
Limitation
Portal vein Sinusoids Centri lobular
vein
PECAM1-Cre [149] Constitutive n.a. n.a. n.a. Likely Poorly described
Tie1-Cre [150] Constitutive n.a. Good Good Yes (20%) Hematopoietic cell
expression
Tie2-Cre [151] Constitutive Good Good Good Yes (90%) Strong hematopoietic cell
Cdh5-Cre [12,14] Constitutive Good Good Good Yes (50%) Moderate hematopoietic
cell expression
Tie2-CreERT2 [153] Inducible Good Absent n.a. No No expression in LSEC
Endothelial-SCL-CreERT2 [154] Inducible Good Absent Absent No No expression in LSEC
Cdh5-CreERT2 [155] Inducible n.a. Mild Mild No Less penetrant than
Cdh5(PAC)-CreERT2
Cdh5(PAC)-CreERT2 [156] Inducible Good Good Good No
Pdgfb-iCreERT2 [157] Inducible n.a. Absent Good No during the first month
after induction of Cre-
mediated recombination
No expression in LSEC
Bmx cre [158] Constitutive Absent Absent Absent n.a. Artery specific
Recombination was classified as good (>66%), moderate (33–66%), mild (5–33%); absent (<5%) based on data provided in the articles describing each model for all mouse
lines but Tie2-Cre, Pdgfb-iCreERT2 and Cdh5 (PAC)-CreERT2. Indeed, these last 3 lines were independently and thoroughly analyzed and compared back to back using mT/
mG reporter mice by the group of C James, Pessac, France (Kilani et al., unpublished). Cdh5-CreERT2 mice were also analyzed using mT/mG reporter by our group
(unpublished data). Regarding LSEC expression, caution is needed since in all cases LacZ staining was performed without immunohistochemistry. Cells considered as LSEC
were thus sinusoidal cells. They may be LSEC but also may be Kupffer cells.
LSEC, liver sinusoidal endothelial cell; n.a., information not available.
Review
Review
218 Journal of Hepatology 2017 vol. 66 j 212–227
agents in animal models of liver fibrosis [90,91].However, analysis of the relationships betweenangiogenesis and fibrogenesis is not straightfor-ward since most tools used to inhibit angiogenesisalso act on fibrogenesis. For instance, VEGF, themaster regulator of angiogenesis, is also implicatedin fibrogenesis (Fig. 3) [87,92–95]. Besides LSECs,endothelial progenitor cell (EPC), i.e., endothelialcells derived from bone marrow, also contributeto liver angiogenesis, as reviewed elsewhere[96,97].
LSECs also regulate fibrosis by releasing angio-crine signals. This latter term refers to the paracrinefactors produced by endothelial cells that maintainorgan homeostasis, balance the self-renewal anddifferentiation of stem cells and orchestrate organregeneration and tumor growth. A recent landmarkstudy demonstrated that LSECs release divergentangiocrine signals balancing liver regenerationand fibrosis. After acute liver injury, activation ofCXCR7-Id1 pathway in LSECs stimulates productionof hepatic-active angiocrine factors leading to liverregeneration. By contrast, chronic injury causespersistent FGFR1 activation in LSECs that perturbsCXCR7-Id1 pathway and favors a CXCR4-drivenpro-fibrotic angiocrine response, thereby provokingliver fibrosis. Therefore, in response to injury, dif-ferentially primed LSECs deploy divergent angio-crine signals to balance liver regeneration andfibrosis [98].
Endothelial dysfunction occurs early in chronicliver disease, even before fibrosis and inflammationtake place, and persists in advanced cirrhosis[84,99,100] (Fig. 2). The mechanisms of endothelialdysfunction have been reviewed elsewhere and aresummarized in Fig. 2 [83,84]. Importantly, pharma-cologic strategies improving LSECs in chronic liverdiseases, including statins, decrease liver fibrosis,endothelial dysfunction and portal pressure[101,103,104].
Role of LSECs in hepatocellular carcinoma
Hepatocellular carcinoma (HCC) most often emergesin the context of chronic liver disease. The develop-ment of HCC is thought to be a multistep processfrom precancerous lesions (low then high grade dys-plastic nodule) to early and advanced HCC [105].Dysplastic nodules receive blood supply preferen-tially via the portal vein similarly to regenerativenodules of cirrhosis. A switch to prominent arterialblood supply occurs at the stage of early HCC[106]. Then, angiogenesis results in a highly vascu-larized tumor and promotes tumorigenesis and thedevelopment of metastasis. HCC is associated withchanges in endothelial cells within and around thetumor.
Endothelial cells present within HCC sequentiallylose during tumor progression LSECs markers,including stabilin-1, stabilin-2, LYVE-1 and CD32b,
Fenestrae sieve plate
Space of Disse
HepatocyteHepatocyte
HSC
Activated
Capillarized LSEC
BM
NORMAL LIVER CIRRHOTIC LIVER
Differentiated LSEC
Space of Disse
TGF-β1
Fibrosis Angiogenesis
Cholangiocyte
↑VEGF
HSC
Quiescent
VEGF
Fig. 3. A dual role of VEGF in chronic liver disease progression. In physiological conditions, VEGF released by hepatocytes,
cholangiocytes and HSC, maintains LSEC differentiation (blue arrow) and consequently HSC quiescence. VEGF is thus anti-fibrogenic.
During fibrogenesis, liver expression of VEGF increases. These high VEGF levels have a pro-fibrogenic action (red arrows) by inducing
liver angiogenesis and by activating HSC. The activation of HSC results from a direct action of VEGF on HSC and from the release of
as observed both in murine HCC models and inhuman HCC [107]. Moreover, as compared to LSECsfrom a healthy human liver, endothelial cellsderived from human HCC have a higher expressionof integrins, lower expression of ICAM-1, and exhi-bit higher angiogenic, procoagulant and fibrinolyticcapacities [108].
LSECs in the peritumoral tissue also undergochanges as HCC progresses including the loss ofthe LSEC markers stabilin-2 and CD32b [107]. In amouse tumor xenograft model, peritumoral livertissue displays a higher microvascular density andexpression of the proangiogenic genes,interleukin-6 (IL-6) and interleukin-6 receptor (IL-6R) than the model tumoral tissue [109]. In thesame line, peritumoral endothelial cells isolatedfrom patients with HCC proliferate more when cul-tured with IL-6 and soluble IL-6R than tumoralendothelial cells. IL-6 is secreted by peritumoralendothelial cells in response to hypoxia while IL-6R is secreted by macrophages, present in largenumber in the peritumoral liver tissue duringtumoral progression. These data suggesting a majorrole of peritumoral endothelial cells in HCC pro-gression echo the previous observation that geneexpression in the nontumoral liver from patientswith HCC has a higher prognostic value of thangene expression in HCC [110].
LSEC and liver regeneration following acute liver
injury or partial hepatectomy
Liver regeneration following acute liver injuryor partial hepatectomy is a complex process whereLSECs play a key role. LSECs sense the majorchanges in shear stress resulting from resection.They proliferate, and orchestrate the harmoniousregeneration of the different cell types by interact-ing with sinusoidal progenitor cells, platelets andinflammatory cells (Fig. 4).
After an acute liver injury or a partial hepatec-tomy, LSECs play a central role in liver regenerationthrough a dynamic regulation of the balancebetween hepatocytes proliferation and vascularproliferation. There is an asynchronism betweenhepatocyte and LSEC proliferation. In the earlyphase (at day 2), non-proliferative LSECs activatehepatocytes proliferation by two complementarymechanisms: (a) the downregulation of the hepato-cyte growth inhibitor TGF-b, through the downreg-ulation of the Tie2 receptor antagonist,angiopoiteine-2 [111]; and (b) the secretion of hep-atotropic cytokines, Wnt and hepatocyte growthfactor (HGF), through the upregulation of the tran-scription factor Id1 via the VEGFR2/VEGFA path-ways [57]. Following liver resection, the portalflow per gram of tissue immediately increases,enhancing the shear stress on LSECs [112,113]. Inresponse to this increased shear stress, LSECsrelease NO that sensitizes hepatocytes to HGF
[112,114]. Shear stress is thus a key inducer of liverregeneration. However, when resection is excessive,exaggerated shear stress can damage LSECs and leadto hemorrhagic necrosis [112]. Limiting shear stresscould be a potential strategy to prevent post-hepatectomy liver failure as suggested by the bene-ficial effect of portosystemic shunts, splenectomy orsplenic artery embolization in murine models and inpatients with large liver resections [112,115–121]. Aless invasive surgical intervention is being tested ina prospective trial (NCT02390713), using a pneu-matic ring to modulate the diameter of the portalvein and thus the post-hepatectomy shear stress.New promising molecules decreasing shear stressto prevent post-hepatectomy liver failure andsmall-for-size-syndrome have been proposedincluding the vasodilator olprinone, a phosphodi-esterase III inhibitor [122,123] currently tested in aprospective trial (NCT00966745).
In the second phase following hepatectomy (atday 4), LSECs begin to proliferate, via the upregula-tion of angiopoietin-2 and VEGFR2/VEGFA pathways[111]. VEGFR2 is a classical mediator of the mito-genic and the angiogenic effect of VEGFA. The roleof VEFGA/VEGFR1 pathway is more controversialthan that of the VEGFR2 pathway. Le Couteur et al.
described that VEGFR1 activation in LSECs after liverinjury, can paracrinally induce hepatocyte prolifera-tion, without LSEC proliferation and protectsparenchymal cells from the injury [124].
Liver regeneration not only implicates liver cellsbut also circulating cells including sinusoidal pro-genitor cells, platelets and inflammatory cells. Therole of sinusoidal progenitor cells in liver regenera-tion has been recently reviewed elsewhere and issummarized in Fig. 4 [20]. Briefly, liver injuryinduces increased hepatic VEGF expression, whichdrives recruitment of hepatocyte growth factor-rich bone marrow sinusoidal progenitor cells andpromotes expression of HGF by resident sinusoidalprogenitor cells and LSECs. HGF in turn stimulatesthe proliferation of hepatocytes in liver regenera-tion. In addition, sinusoidal progenitor cells replaceLSECs that were lost during injury. The role of theinteraction between LSECs and platelets in liverregeneration is summarized in Fig. 4 [125]. Follow-ing liver injury, platelets are recruited to andtrapped within the liver, where they adhere to LSEC.Subsequent platelet activation results in the releaseof platelet granules, which stimulate hepatocyteproliferation. Platelets activate LSECs, leading tothe secretion of growth factors, such as IL-6 [125].Finally, LSECs and hepatocytes can also internalizeplatelets, but the effects of this alternate processon liver regeneration remain to be explored. Theimprovement in survival following subtotal liverresection in rats and mice obtained by the inductionof thrombocytosis by thrombopoietin injection,splenectomy or platelet-rich plasma transfusionillustrates importance of platelets in liver regenera-tion [126–128]. The endothelial-monocyte interac-
Key point
LSECs are implicated in liverregeneration following acuteliver injury or partial hepa-tectomy since they renewfrom LSECs and/or LSEC pro-genitors, they sense theshear stress changes result-ing from surgery and inter-act with platelets andinflammatory cells.
Review
Review
220 Journal of Hepatology 2017 vol. 66 j 212–227
tion is also implicated in liver regeneration. Indeed,circulating monocytes are recruited in the injuredliver and stimulate parenchymal but also endothe-lial regeneration. LSECs regulate the infiltration ofmonocyte in the liver through a destabilization ofVE-Cadherin junction and through adhesive mole-cule expression [129].
Lastly, liver regeneration also depends on theexistence of lesion related to ischemia reperfusion.Mechanisms of ischemia reperfusion injury havebeen reviewed previously and will not be detailedhere [130].
Inflammation and infection
LSECs regulate liver inflammation in two manners.First, LSECs are a barrier separating the blood fromthe rest of liver, and thus restrict or enable theentry of circulating leucocytes into the liver tissue.The detailed mechanisms of the interactionsbetween leucocytes and LSECs have been previ-ously reviewed [131]. Briefly, LSECs expressICAM-1 and vascular adhesion protein-1 (VAP-1),allowing adhesion of leucocytes to the endothe-lium. During inflammation, expression of ICAM-1
increases and expression of vascular cell adhesionmolecule-1 (VCAM-1) and CD31 are induced, lead-ing to the transendothelial migration of leucocytes.Stabilin-1 has also been reported to promotetransendothelial migration of leucocytes, preferen-tially regulatory T cells [132]. Second, LSECs canmodulate lymphocytes behavior. In physiologicalconditions, antigen presentation by LSECs leads totolerance induction in CD8+ cells [133]. LSECs canalso induce differentiation of T cells into immuno-suppressive regulatory T cells (Treg) that are func-tional in vitro and in vivo [134]. As an application,the selective delivery of autoantigen peptides toLSECs in vivo using a polymeric nanoparticle carriercan efficiently prevent and treat an animal model ofautoimmunity, by increasing the number of Treg[135]. In inflammatory conditions, LSECs also tendto have an anti-inflammatory action since theyincrease the expression of the anti-inflammatorycytokine IL-10 in Th1 cells via the Notch pathway[136].
But LSECs can also be targeted by pathogens. Dueto their scavenging ability, LSECs can capture circu-lating viruses via the expression of lectin at theirsurface and in turn induce infection of hepatocytes,
Space of Disse
Hepatocytes
LSEC
Engraftment Differentiation
HGF
Shear stressPlateletsVEGF A
NO
Space of Disse
++
+
+
Liver injury
Mobilization
ProliferationVEGF
ProliferationReleaseStimulation
+
SPCs
Fig. 4. Liver sinusoidal cell (LSECs) and liver regeneration following acute liver injury or partial hepatectomy. Following liver
injury, liver expression of VEGF increases, leading to the proliferation of bone marrow sinusoidal progenitor cells (SPC) (1), to their
mobilization to the circulation (2), their engraftment in the sinusoids (3) and their differentiation in mature LSECs (4). VEGF A
stimulates liver regeneration trough LSECs (a) leading to HGF production (b), hepatocyte proliferation (c) and LSECs proliferation (d)
[20]. Increased shear stress associated with liver resection induces LSECs derived nitric oxide (NO) (I), which increase the effect of
HGF on hepatocytes proliferation (II). Platelets are rapidly recruited in the liver after liver surgery (A). They adhere to LSECs and
stimulate secretion of key molecules involved in hepatocytes (F) and LSECs (B) proliferation and survival. Platelets can also be
endocytosed by LSECs (C), or trapped in the space of Disse (E), a migration facilitated by the increased size of fenestration associated
with liver surgery (D). Abbreviations: HSC, hepatic stellate cell; NO, nitric oxide; LSEC, liver sinusoidal cell; HGF, hepatocyte growth
as observed for hepatitis B and hepatitis C viruses[137,138]. Lectin expressed by LSECs is not onlyinvolved in regulation of the entry of viruses butalso in the regulation of their clearance by modu-lating functions of T cells as it has been shown foradenovirus [139]. LSECs can also be infected withCMV (cytomegalovirus) which upregulates ICAM-1 and CXCL10 expression, thus favoring CD4 T celltransendothelial migration. Migration of effectormemory T cells through CMV-infected LSECs isassociated with a change in memory T cells pheno-type towards an activated phenotype facilitatinghepatic inflammation, while regulatory T cells
transmigrating retain a suppressive phenotype,favoring virus persistence [140]. This change inendothelial cells towards a proinflammatory pheno-type induced by CMV might explain why acute CMVinfection can trigger portal vein thrombosis [141].The effect of CMV on LSECs and lymphocytes mayalso be of particular interest in the setting of livertransplantation where CMV infection may favoracute rejection, a disease characterized byendotheliitis.
LSECs can also be infected with bacteria as elec-tron microscopy studies revealed Bartonella bacilli inLSECs associated with angiomatosis and peliosis
Table 4. Drug delivery system to LSEC in vivo.
Carrier distribution in vivo
Reference Carrier Size (nm) LSEC* KC* Hep* Other organs Experimental strategies and results Toxicity
Sano et al.
[159]
HA n.a. Yes n.a. n.a. n.a. Delivering of sphingosine-1-
phosphate
Hepatic I/R injury model in rats
→↓ALT level and hepatocytes and
LSECs apoptosis
n.a.
Fraser et al.
[160]
HA n.a. 90% 0 4% Low in spleen Bio-distribution in rats n.a.
Toriyabe et al.
[161]
HA-SA-liposome 254 ± 19 Yes Yes (NQ) No (NQ) Low expression in
lungs
Bio-distribution in mice n.a.
Takei et al.
[162]
PLL-g-HA 100 to 200 Yes No (NQ) No (NQ) >93% liver
2.5-1% spleen,
intestine and kidney
<1% heart, thymus,
lung and blood$
Delivering of DNA complexes
Bio-distribution in rats
n.a.
Kren et al.
[163]
HA-nanocapsule
(polyethyleneimine)
<50 Yes n.a. No (NQ) Not in lung, kidney,
spleen, heart and
gonads
Delivering of transposon vectors
expressing FVIII
Hemophilia A mice model
→Normalization of plasmatic
FVIII expression and activity
up to 11 mo
No
toxicity
in 72 h
and 3
mo
Carambia et
al. [135]
Iron oxide
nanocrystals
<50 Yes n.a. No (NQ) 90% liver
10% spleen and
kidney$
Delivering of auto-antigen peptide
Autoimmune encephalomyelitis mice
model
→Controlled disease progression by
Treg induction by LSECs
No
toxicity
(9 wk)
Tanoi et al.
[164]
STR-KLGR
modified YSK05-
MEND
80-120 Yes n.a. Lower (NQ) n.a. Delivering of BAX siRNA
Acute liver damage (anti-FAS Ab)
in mice
→↓Hepatocytes apoptosis. Preserve
sinusoidal structure
100%
alive at
24 h
Akhter et al.
[165]
STR-KLGR
modified YSK05-
MEND
80-120 Yes n.a. Low High in liver
Lower expression in
lung and kidney
Delivering of Tie2 siRNA
Bio-distribution in mice
→80% knockdown in LSECs
No liver
toxicity
(24 h)
Bartsch et al.
[166]
Aco-HSA-CCLs 154 ± 12 60% 40% 1.30% 60% liver
4% spleen
<1% lungs, heart,
kidneys£
Delivering of ODN
Bio-distribution in rats
n.a.
Kamps et al.
[167]
Aco-HSA
liposomes
92.1 ± 10 65% 25% 10% 80% liver
5% spleen£
Bio-distribution in rats n.a.
Bartsch et al.
[168]
Aco-HSA-PEG-
SAPLs
164 ± 45 75% 25% n.a. 80 % liver
5% spleen£
Delivering of anti-ICAM-1-ODN
No efficiency analyze in vivo
n.a.
⁄ % of expression in liver cells; $Express as % of body distribution; £Express as % of injected dose.
Ab, antibody; Aco-HAS, cis-aconitylated human serum albumin; ALT, alanine aminotransferase; CCLs, lipid-coated cationic lipoplexes; Hep, hepatocytes; HA, hyaluronic
hepatitis [142]. The fact that LSECs can be targetsfor pathogens with an impact on the local environ-ment might explain why nodular regenerativehyperplasia develops in patients with primaryhypogammaglobulinemia, a condition frequentlyassociated with intra-sinusoidal lymphocytic infil-tration. Immunodeficiency might favor infectionof LSECs with pathogens, leading to a change intheir phenotype towards a proinflammatory andprothrombotic phenotype and eventually to sinu-soid obstruction [143].
LSEC and ageing
Pseudocapillarization refers to changes in the liversinusoidal endothelium related with ageing. Elec-tron microscopy analyses showed that ageing isassociated with a 50% increase in the thickness ofLSECs, a 50% reduction in the number of LSEC fen-estrae and the formation of a basement membranewith perisinusoidal fibrosis and central vein fibrosis[42,82]. These changes decrease porosity and endo-cytic capacity of LSECs. Consequently clearance ofchylomicron remnants is impaired leading to postprandial triglyceridemia, which could participateto atherosclerosis development in older individuals[42,51]. Moreover, these LSEC changes can inducehepatocytes hypoxemia decreasing oxidative drugmetabolism and possibly promoting adverse drugreactions [42,51,144].
Conclusion
In conclusion, LSECs have a unique highly perme-able phenotype allowing the passage of certainbut not all molecules and cells. They also have avery special localization at the interface betweenblood cells on the one side and hepatocytes andhepatic stellate cells on the other side. LSECs arein constant interaction with other liver cells [83].
LSECs are implicated in most liver diseases includingchronic liver disease initiation and progression, hep-atocellular carcinoma development and progression,liver regeneration following acute liver injury orpartial hepatectomy, liver ageing and liver lesionsrelated to inflammation and infection. This role inmost liver diseases makes them an attractive thera-peutic target. Data summarized in Table 4 suggest apromising place to specific LSECs targeting. Suchcell-specific approaches may limit the adverseeffects associated with systemic drug delivery.
Financial support
This work was supported by the Agence Nationalepour la Recherche (ANR-14-CE12-0011 and ANR-14-CE35-0022) and by the Association Francaisepour l’Etude du foie (AFEF 2014) and J.P by the‘‘poste d’accueil INSERM”.
Conflict of interest
The authors declared that they do not have anythingto disclose regarding funding or conflict of interestwith respect to this manuscript.
Authors’ contributions
JP, SL and PER drafted the manuscript. FD, CMB, RMand DV discussed and critically revised themanuscript.
Acknowledgments
We thank Servier medical art for providing someimages included in the figures.
References
Author names in bold designate shared co-first authorship
[1] Chiu J-J, Chien S. Effects of disturbed flow on vascular endothelium:
pathophysiological basis and clinical perspectives. Physiol Rev
2011;91:327–387.
[2] Maslak E, Gregorius A, Chlopicki S. Liver sinusoidal endothelial cells (LSECs)
function and NAFLD; NO-based therapy targeted to the liver. Pharmacol Rep
2015;67:689–694.
[3] Couvelard A, Scoazec JY, Dauge MC, Bringuier AF, Potet F, Feldmann G.
Structural and functional differentiation of sinusoidal endothelial cells
during liver organogenesis in humans. Blood 1996;87:4568–4580.
[4] Walter TJ, Cast AE, Huppert KA, Huppert SS. Epithelial VEGF signaling is
required in the mouse liver for proper sinusoid endothelial cell identity and
hepatocyte zonation in vivo. Am J Physiol Gastrointest Liver Physiol
2014;306:G849–G862.
[5] Takabe Y, Yagi S, Koike T, Shiojiri N. Immunomagnetic exclusion of E-
cadherin-positive hepatoblasts in fetal mouse liver cell cultures impairs
morphogenesis and gene expression of sinusoidal endothelial cells. J Anat
2012;221:229–239.
[6] Gouysse G, Couvelard A, Frachon S, Bouvier R, Nejjari M, Dauge MC, et al.
Relationship between vascular development and vascular differentiation
during liver organogenesis in humans. J Hepatol 2002;37:730–740.
[7] Shiojiri N, Sugiyama Y. Immunolocalization of extracellular matrix compo-
nents and integrins during mouse liver development. Hepatology
2004;40:346–355.
[8] Asahina K, Zhou B, Pu WT, Tsukamoto H. Septum transversum-derived
mesothelium gives rise to hepatic stellate cells and perivascular mesenchy-
mal cells in developing mouse liver. Hepatology 2011;53:983–995.
[9] Jaffredo T, Nottingham W, Liddiard K, Bollerot K, Pouget C, de Bruijn M. From
hemangioblast to hematopoietic stem cell: an endothelial connection? Exp
Hematol 2005;33:1029–1040.
[10] Bollerot K, Pouget C, Jaffredo T. The embryonic origins of hematopoietic stem
cells: a tale of hemangioblast and hemogenic endothelium. APMIS
2005;113:790–803.
[11] Bailey AS, Fleming WH. Converging roads: evidence for an adult heman-
9 Validation 73 Male ET PVT NA* 337 52-bp deletion (1)
BCS, Budd-Chiari syndrome; ET, essential thrombocythemia; PMF, primary myelofibrosis; NA, not available; PVT, Portal venous system thrombosis; yrs, years.* No spleen height available but the patient was known to have an enlarged spleen.
JOURNAL OF HEPATOLOGY
Journal of Hepatology 2017 vol. 67 j 1106–1121 1113
RésuméLessyndromesmyéloprolifératifsBcr/Abl-negatifs(SMP)sontdesmaladieshématopoïétiquesclonales,secondairesdans80%descasàlamutationsporadiqueJAK2V617F.JAK2V617Faétérécemmentmiseen évidence dans les cellules endothéliales. Les deuxièmesmutations en ordre de fréquence sont lesmutationsdeCalréticulin(CALR).LesévènementscardiovasculairessontlapremièrecausedemortalitédesmaladesatteintsdeSMP(2/3artérielset1/3veineux).Lesévènementsveineuxsontcaractérisésparunefréquenceparticulièrementélevéedethrombosessurvenantdansdessitesinhabituels,commeles veines splanchniques (veines hépatiques (syndrome de Budd-Chiari) ou veine porte). Lesmécanismesresponsablesdecesévènementscardiovasculaireschez lesmaladesatteintsdeSMPsontmal compris. Les conséquences phénotypiques et fonctionnelles de la mutation JAK2V617F
Sur le plan artériel, j'ai montré, grâce à des expériences de myographie, que les aortes desourisportant JAK2V617Fà la foisdans leurs celluleshématopoïétiqueset endothélialesontune trèsforte augmentation de la réponse aux agents vasoconstricteurs alors que cet effet n’est pas observélorsquelamutationestuniquementendothéliale.J’aiensuiteisolédesmicrovésiculesplasmatiquesdemalades porteurs de JAK2V617F, non traités pour leur SMP, et ai observé que ces microvésiculesreproduisentl’effetd’hyperréponseartérielleauxagentsvasoconstricteurs.J'aiparlasuitemontréqueseuleslesmicrovésiculesdeglobulesrougesportantlamutationJAK2V617Fétaientresponsablesdeceteffet.J’aiensuiteanalysélesmécanismesimpliquésetaidéterminéquecettehyperréactivitévasculaireest dépendante de l’endothélium et des NO synthases. De plus, j'ai aussi mis en évidence une forteaugmentationdustressoxydantdansl'endothéliumdesaortesdesourisportantlamutationJAK2V617Fencomparaisonauxsourissauvages,suggérantquelesperturbationsdelavoieduNOrésultentd’unegénérationdestressoxydant induitepar lesmicrovésiculesdeglobulesrouges.Cesdonnéesnousontpoussés à évaluer de nouvelles thérapeutiques, comme les statines, qui sont des molécules anti-cholestérolémiques ayant indépendamment du cholestérol un effet anti-oxydant bénéfique sur lafonction endothélial. J'aimontré que l'utilisation de simvastatine chez les souris portant lamutationJAK2V617Fdiminuesignificativecettehyperréactivitévasculaireencomparaisonauxsouriscontrôles.Mes résultats suggèrent que chez les malades atteints de SMP une hyperréponse aux agents
vasoconstricteurs induite par les microvésicules circulantes d'origine érythrocytaire pourrait
participer aux accidents artériels et que de nouvelles thérapeutiques, comme les statines
pourraientêtreprometteuses.Sur le plan veineux, en analysant une cohorte prospective de 312 patients atteints de
thrombosessplanchniques,j’aipudéterminerlaprévalencedesmutationsCALRdanscettepopulation(2%)etidentifierungroupedemalades(ceuxsansJAK2V617Fetayantunetaillederate≥16cmetdesplaquettes>200G/L)chez lesquels larecherchedesmutationsCALRdoitêtreeffectuée.Cescritèresontuneexcellentevaleurprédictivenégative(100%)etpermettentd’éviter96%detestsinutiles.J’aiconfirmécescritèresgrâceàunecollaborationeuropéennedansunecohortedevalidationespagnolecomprenant209patients.Jemesuisaussiattachéeàdéterminer le rôlede JAK2V617Fendothélial dans les conséquences hépatiques des thromboses des veines hépatiques. Nousavons montré que la présence de la mutation JAK2V617F dans l'endothélium n'aggrave pas ledéveloppement des lésions hépatiques secondaires à un syndrome de Budd-Chiari, ni en termes defibrosehépatiquenientermesd’hypertensionportale.