

Aneuploidy compensatory mechanisms and genome-wide regulation of gene expression in Drosophila melanogaster
Lina Lundberg
Department of Molecular Biology
Umeå University 2013, Sweden

This work is protected by the Swedish Copyright Legislation (Act 1960:729) ISBN: 978-91-7459-659-5 Cover photo by: Glenn Landgren
Back side photo: Section from an expression array (enlarged)
Electronic version is available at http://umu.diva-portal.org/
Printed by: Print & Media Umeå, Sweden 2013

“The bad news is time flies. The good news is you’re the pilot!”
~Michael Althsuler
Till Faster Kicki


Table of CONTENTS
i
TABLE OF CONTENTS
TABLE OF CONTENTS ....................................................................... i
LIST OF PUBLICATIONS ...................................................................iv
TERMINOLOGY AND ABBREVATIONS .............................................. v
ABSTRACT ..................................................................................... vii
SVENSK SAMMANFATTNING ........................................................ viii
INTRODUCTION ............................................................................. 1
EPIGENETICS ....................................................................................... 1 Cell differentiation ......................................................................................... 2
CHROMATIN ....................................................................................... 2 Histone acetylation ............................................................................................ 5
H4K16 acetylation .......................................................................................... 5 Histone and DNA methylation ........................................................................... 5
H3K9 methylation .......................................................................................... 6
DIFFERENT CHROMATIN STRUCTURES ................................................. 6 Euchromatin ....................................................................................................... 6 Heterochromatin ................................................................................................ 6 GREEN, BLUE, BLACK, RED, YELLOW chromatin ................................................. 8 Position-effect variegation ................................................................................. 9
HP1a (Su(var)2-5) ............................................................................... 10 Repressive or activating function of HP1a? ................................................. 11 Isoforms of HP1 ............................................................................................ 12
MEDIATION OF H3K9 METHYLATION MARKS ...................................... 12 G9a ................................................................................................................... 12 Su(var)3-9 ......................................................................................................... 13 SETDB1 ............................................................................................................. 14
HETEROCHROMATIN FORMATION ...................................................... 14 Heterochromatin formation and RNA interference ......................................... 15 Transposons ..................................................................................................... 16
WHY USE DROSOPHILA AS A MODEL ORGANISM? .............................. 17 General information about the fruit fly ........................................................... 17 Specific advantages of fruit fly in epigenetics .................................................. 19
Polytene chromosomes................................................................................ 19

Table of CONTENTS
ii
Two chromosome-wide regulatory systems ................................................. 19
DOSAGE COMPENSATION .................................................................. 19 General ............................................................................................................. 19 In mammals ...................................................................................................... 20 Up-regulation of mammalian X-chromosome .................................................. 21 In Drosophila ..................................................................................................... 22
MSL1 ............................................................................................................. 22 MSL2 ............................................................................................................. 23 MSL3 ............................................................................................................. 23 MLE ............................................................................................................... 23 MOF .............................................................................................................. 24 roX1 and roX2 ............................................................................................... 24 High affinity sites and spreading of DCC ....................................................... 25 Targeting mechanisms of the MSL complex ................................................. 25 Mechanism behind the X-chromosome up-regulation ................................. 26
Painting of Fourth (POF) ..................................................................... 27 Chromosome 4.............................................................................................. 28 Haplo-4
th lethality and POF ........................................................................... 29
Balanced regulation of chromosome 4 genes by POF and HP1a .................. 29 Evolutionary links between POF and the MSL complex ................................ 29
Do other compensating systems exist? ............................................................ 30
ANEUPLOIDY ..................................................................................... 31 Cancer, developmental diseases and aneuploidy ............................................. 31 Aneuploidy and evolution ................................................................................. 32 Aneuploidy in Drosophila .................................................................................. 33 Are there mechanisms for aneuploidy compensation? .................................... 33 Buffering ........................................................................................................... 33
Feedback regulation ..................................................................................... 36 Feedforward regulation ................................................................................ 36 Inverse dosage effect .................................................................................... 37
Challenges with genome-wide expression analysis .......................................... 37 Reference points ........................................................................................... 37 Skewness....................................................................................................... 37 Limitations in the arrays ............................................................................... 38
AIMS ............................................................................................ 39
RESULTS AND DISCUSSION ........................................................... 40
PAPER I AND II ................................................................................... 40

Table of CONTENTS
iii
General buffering levels ................................................................................... 40 Buffering of specific gene groups ..................................................................... 40
UEGs and NUEGs .......................................................................................... 40 Gene length and wildtype expression level affects buffering ...................... 41
Buffering mechanisms ...................................................................................... 41 POF compensates chromosome 4 ................................................................ 42 Buffering induces proteolysis ....................................................................... 43
Future perspectives .......................................................................................... 43 Conclusions ...................................................................................................... 44
PAPER III ............................................................................................ 45 HP1a has opposing functions on chromosome 4 and in pericentromeric
regions .............................................................................................................. 45 HP1a has different functions at the promoter and at the gene body .......... 45
SETDB1 and Su(var)3-9 are complementary to each other ............................. 46 HP1a displays a stronger repression of long genes .......................................... 47 HP1a effect in the pericentromeric regions depends on location ................... 47 Concluding remarks ......................................................................................... 48 Conclusions ...................................................................................................... 49
PAPER IV ............................................................................................ 50 POF targets roX proximal sites ......................................................................... 50 Connection with the MSL complex .................................................................. 50 Parts of PoX2 functions as POF high affinity target ......................................... 51 HP1a correlates with POF in the PoX sites ....................................................... 51 Conclusions ...................................................................................................... 52
FINAL CONCLUDING REMARKS ........................................................... 53
ACKNOWLEDGEMENTS ................................................................ 54
REFERENCES ................................................................................ 58
PAPER I-IV

LIST OF PUBLICATIONS
iv
LIST OF PUBLICATIONS
This thesis is based on the following papers, which in the text will be referred
to by their Roman numerals (I-IV).
I Per Stenberg, Lina E Lundberg, Anna-Mia Johansson, Patrik
Rydén, Malin J Svensson and Jan Larsson (2009). Buffering of
segmental and chromosomal aneuploidies in Drosophila
melanogaster, PLoS Genet 5:e1000465
II Lina E Lundberg, Margarida L A Figueiredo, Per Stenberg and Jan
Larsson (2012). Buffering and proteolysis are induced by segmental
monosomy in Drosophila melanogaster. Nucleic Acids Res 40:
5926-5937
III Lina E Lundberg, Per Stenberg and Jan Larsson (2013). HP1a,
Su(var)3-9, SETDB1 and POF stimulate or repress gene expression
depending on genomic position, gene length and expression pattern in
Drosophila melanogaster. Nucleic Acids Res doi:
10.1093/nar/gkt158
IV Lina E Lundberg, Maria Kim, Anna-Mia Johansson, Marie-Line
Faucillion, Rafael Josupeit and Jan Larsson (2013). Targeting of
Painting of fourth to roX1 and roX2 proximal sites links dosage
compensation to heterochromatin in Drosophila melanogaster.
Submitted manuscript
Paper I-III are reproduced with permission from the publishers.
The following paper is not included in this thesis;
Filip Crona, Olle Dahlberg, Lina E Lundberg, Jan Larsson and Mattias
Mannervik (2013). Gene regulation by the lysine demethylase KDM4A in
Drosophila. Dev Biol 373:453-463

TERMINOLOGY AND ABBREVATIONS
v
TERMINOLOGY AND ABBREVATIONS
Autosome All chromosomes which are not sex-chromosomes
CD Chromo domain
Centromere Part of the chromosome that links sister chromatids during
metaphase
Chromatin The DNA-protein structure, which all DNA in the nucleus
is present in
CSD Chromo-shadow domain
DCC Dosage Compensation Complex
dsRNA Double-stranded RNA, two RNA molecules that base-pair
with each other
Ectopic Occurring in an abnormal position
Exon The segments of a gene that will be present in the mRNA
when it is transcribed
Gene body The entire gene from the transcription start site to the end
of the transcript
Haplo-4th One copy of the 4th chromosome
Heterozygous Two different alleles for a single trait
Histone Small DNA binding proteins that forms the complex which
DNA is wrapped around in nucleosomes
HKMT Histone lysine methyltransferase
Homozygous Identical alleles of a single trait
HP1a Heterochromatin protein 1 a
H3K9me1, me2, me3 Histone H3 mono-, di-, trimethylated at lysine 9
(associated with inactive genes)
H3K36me3 Histone H3 tri-methylated at lysine 36 (associated with
active genes)
H4K16ac Histone H4 acetylated at lysine 16 (associated with active
genes)
Intron The segments in-between exons in a gene, they are not part
of the mRNA
Kb Kilo base pairs
Mb Mega bases
Mitosis When a cell separates all replicated chromosomes into two
identical groups, before dividing into two daughter cells
Meiosis Special type of cell division that forms eggs and sperm
cells. Four haploid cells are produced from one diploid cell
MOF Males absent on the first
Monosomic A chromosome region present in one copy
MRE MSL recognition element
mRNA messenger RNA, carrier of information from DNA

TERMINOLOGY AND ABBREVATIONS
vi
MSL Male specific lethal, the complex which mediates dosage
compensation of the Drosophila male X-chromosome
nm Nano meter
Nucleotide The building blocks of DNA: A, T, C, G
Nucleosome Basic DNA packaging unit consisting of a histone-DNA
complex
NUEG Non- Ubiquitously Expressed Gene, genes required for
tissue specific functions, not expressed in all tissues
Orthologue Genes in different species that evolved from a common
ancestral gene by speciation
Paralogue Related genes which have occurred due to duplication
within a species
Pericentromeric regions Heterochromatic regions near the centromere
Promoter A region of DNA that initiates transcription from the
nearby located gene
RNAi RNA interference
RNA polymerase II An enzyme that produces RNA from a gene
roX RNA on X, non-coding RNA which is part of the MSL
complex
S-phase The phase of the cell-cycle in which DNA is replicated,
occurs before mitosis
SXL Sex lethal (prevents MSL2 from forming in females)
Su(var) Suppressor of variegation
TE Transposable elements
Transcriptional elongation The process when RNA polymerase II reads a gene and
synthesizes RNA
Transgene A gene or genetic material that researchers have inserted in
a genome, or into another species
Trisomic A chromosome region present in three copies
UEG Ubiquitously Expressed Gene, genes required for
maintenance of basic cellular functions (housekeeping)
Xi Inactivated X chromosome, found in mammals
3’ end The end of a gene, is transcribed last
5’ end The beginning of a gene, is transcribed first

ABSTRACT
vii
ABSTRACT
Stimulation or repression of gene expression by genome-wide regulatory
mechanisms is an important epigenetic regulatory function which can act to
efficiently regulate larger regions or specific groups of genes, for example by
compensating for loss or gain of chromosome copy numbers. In Drosophila
melanogaster there are two known chromosome-wide regulatory systems;
the MSL complex, which mediates dosage compensation of the single male
X-chromosome and POF, which stimulates expression from the
heterochromatic 4th chromosome. POF also interacts with the
heterochromatin inducing protein HP1a, which represses expression from
the 4th chromosome but which also has been assigned stimulatory functions.
In addition to these two, there is another more elusive and less well-
characterized genome-wide mechanism called buffering, which can act to
balance transcriptional output of aneuploidy regions of the genome (i.e. copy
number variation).
In my thesis, I describe the presence of a novel physical link between dosage
compensation and heterochromatin; mediate by two female-specific POF
binding sites, proximal to roX1 and roX2 on the X chromosome (the two
non-coding RNAs in the MSL complex). These sites can also provide clues to
the mechanisms behind targeting of chromosome-specific proteins.
Furthermore, to clarify the conflicting reports about the function of HP1a, I
have suggested a mechanism in which HP1a has adopted its function to
different genomic locations and gene types. Different binding mechanisms to
the promoter vs. the exon of genes allows HP1a to adopt opposite functions;
at the promoter, HP1a binding opens up the chromatin structure and
stimulates gene expression, whereas the binding to exons condense the
chromatin and thus, represses expression. This also causes long genes to be
more bound and repressed by HP1a. Moreover, I show that buffering of
monosomic regions is a weak but significant response to loss of
chromosomal copy numbers, and that this is mediated via a general
mechanism which mainly acts on differentially expressed genes, where the
effect becomes stronger for long genes. I also show that POF is the factor
which compensates for copy number loss of chromosome 4.

SVENSK SAMMANFATTNING
viii
SVENSK SAMMANFATTNING
Alla celler i kroppen innehåller all vår arvsmassa, våra gener, i form av DNA
och funktionen för varje enskild cell styrs av vilka gener som är aktiva
(uttryckta) i just den cellen. Det är därför extremt viktigt att regleringen av
hur gener används fungerar som den ska. Denna reglering sker ofta på en
enskild gen-nivå, men förekommer också på en mer generell nivå på grupper
av gener eller på hela kromosomer. Fördelen med en generell genreglering är
att cellen på ett effektivt och synkroniserat sätt kan reglera grupper av gener
som är kopplade till liknande funktioner eller till samma region, till exempel
genom att kompensera uttrycket om delar av en kromosom av någon
anledning tappar eller får extra kopior, ett tillstånd som kallas aneuploidi.
Detta är vanligt förekommande i naturen och det är till och med troligt att
varje människa bär på hundratals små aneuploida regioner. Gravare
aneuploidi är starkt förknippat med tumörer och utvecklingsstörning, t.ex.
Downs syndrom är orsakat av en kopia för mycket av kromosom 21. Det
finns mekanismer som verkar i aneuploida regioner för att dämpa effekterna
av felaktig gen-dos, så kallad buffring, men hur de fungerar är fortfarande
mycket oklart. Jag visar i min avhandling att den buffring som motverkar
effekterna av en halverad gen-dos troligtvis är en generell mekanism som
känner igen regioner med felaktiga kopienummer, och som dessutom har
starkast effekt på långa gener. Gener som är viktiga för vävnadsspecifika
funktioner, och därmed bara aktiva i vissa celler, verkar också ha lättare för
att bli buffrade och bör alltså klara en halverad gen-dos bättre än gener som
är konstant aktiva och involverade i livsuppehållande processer i alla celler.
Utöver denna ännu ganska oklara buffringsmekanism finns det två mer
väldefinierade, kromosom-specifika system i bananflugan som stimulerar
genuttryck: proteinkomplexet MSL, som doskompenserar hanarnas enda X-
kromosom så att den får dubbelt så högt uttryck och blir likvärdig med
honornas två X-kromosomer, samt proteinet POF, som specifikt binder
kromosom 4 och stimulerar genuttrycket. POF är också starkt kopplat till
HP1a, ett proteins som är mycket viktigt för att cellen ska kunna bilda
heterokromatin, en tätt packad DNA struktur som tystar ner de flesta gener.
Jag visar att POF kan binda till två speciella ställen på honornas X-
kromosom, vilket dels kan vara mycket användbart för att förstå
mekanismen bakom hur POF känner igen kromosom 4, men som också
tyder på att POF har kopplingar till doskompensering. Jag visar också att
HP1a kan ha motsatta effekter på genreglering; om HP1a binder till en
promoter (en DNA-sekvens bredvid genen som hjälper till att reglera
uttrycket) uppstår en löst packad DNA struktur som leder till ökat
genuttryck, medan HP1a bindning till själva genen inhiberar genuttryck.

INTRODUCTION
1
INTRODUCTION
The processes behind gene expression are of course extremely complex, with
many different proteins involved in recognizing, binding to, reading, and
ultimately translating the messages within the genes, the DNA code, into
fully functional proteins that are required in different processes of the cell.
The cell nucleus is literally packed with proteins with the only purpose of
maintaining a balanced and functional flow of information from the genes.
All this would be far too complicated to describe in just one book, so this
thesis will be focused on one small part of the puzzle: genome-wide gene
regulation, which involves how the regulation of gene activation, or
repression, can be maintained on a larger scale. This means not only
regulation of individual genes but rather, the regulation of large genomic
regions, and even entire chromosomes. Most of this works falls under a
branch of genetics called epigenetics.
EPIGENETICS
The properties and characteristics of all life forms are said to depend on the
genetic factor, the DNA code containing all the heritable information, as well
as on the environmental factor which shapes us during life. Which one is the
actually determining factor for many of our traits is very often a subject of
dispute, but it has grown more and more evident that it is likely a
combination of both, and that they are linked by an intermediate factor: the
epigenetic factor.
Examples to illustrate the impact of epigenetics: compare one of your brain
cells with one of your skin cells or muscle cells; they contain the exact same
DNA, and yet they have such different features, or compare two adult
identical twins; they have identical DNA but will most likely differ in small
physical characteristics. This is caused by epigenetics.
Epigenetic literally means “above genetic” and is defined as changes and
patterns in gene expression, which can be inherited through mitosis and
sometimes also meiosis, but which are not caused by any changes in the
actual DNA sequence.
To explain in other words: The DNA code is (ideally) a fixed and non-
changing sequence of letters which, when put in different combinations (a
bit like the binary system in computers), can be interpreted into information

INTRODUCTION
2
that the cell uses to assemble proteins. But this does not mean that all parts
of this code are always read at all times (much like in a computer).
Depending on the cell and the function it has, different genes will be read,
and at different times.
So how can your body (or the bodies of identical twins), have emerged from
just one single cell and turned into all these different cell types that have
adopted so different fates. And an equally important question is: how do they
remember their fate throughout endless rounds of cell division?
Cell differentiation
The initiation of cell differentiation (i.e. establishing the fate of a cell) is
usually a complex orchestra of different signal molecules that permeate the
embryo during early development. In Drosophila (i.e. fruit fly), this process
is well studied and it all begins with a few number of signal molecules that
are deposited in opposite ends of the egg (oocyte) by the mother. These will
define the polarity of the embryo (i.e. determine the anterior- posterior axis)
and diffuse throughout the embryo, creating a gradient of the signal
molecules which will result in the activation or inhibition of various different
genes. These genes have evolved to respond differently (i.e. be active or
silent) to different concentrations of signal molecules, hence the position a
cell has within the embryo will determine its fate, “simply” by the different
gradients of the signal molecules that reaches the cell. This cell will then in
turn activate new genes which will submit new signal molecule gradients,
and thus the complex process of cell differentiation begins.
However, once the fate of a cell is established, it needs to maintain this
identity even in the absence of the initial signal. This is actually where
epigenetics comes into the picture; epigenetics is the memory by which
expression statuses assigned to various genes and/or genomic regions are
maintained through cell division. Essentially, this memory is mediated by
keeping active genes easily accessible to the transcription machinery,
whereas inactive genes are blocked from transcription. The key to this lies in
the organization of the DNA in the nucleus, and the various proteins which
are surrounding the DNA.
CHROMATIN
To really understand the concept of epigenetics, it is important to first
understand the organization of the chromosomes and the genetic material;

INTRODUCTION
3
since the DNA material within each individual cell measures a total of about
2 meters in humans, it is extremely important for the cell to keep the DNA
organized to uphold a smooth and efficient reading of the genetic material,
and a correct replication and distribution of the DNA into two daughter cells
during cell-division. This is done by packaging all DNA into a large,
condensed DNA-protein structure called chromatin. As a first step of
condensation, stretches of DNA are wrapped around a specific protein core
complex, forming a DNA-protein complex called a nucleosome (see figure
1A), which is found in all eukaryote genomes [1-3]. The protein core, also
called the histone core, is made up of an octamer of the different histone
proteins H3, H4, H2A and H2B, which are all present in two copies [4].
Around the histone octamer, 146 base pair (bp) of DNA are wrapped forming
1.65 turns [5] and the DNA is then “locked” in position on the histone core by
a linker histone protein 1 (H1) [6]. The nucleosome structure compacts the 2
nm thick DNA about a five- to ten-fold, into an 11 nm thick structure (see
figure 1B). Between each nucleosome is a short segment of linker DNA
which, depending on level of compaction, varies between about 10-80 bp in
length [7,8], giving the DNA a typical “beads on a string” like structure. This
structure is best visible if the linker histone 1 is removed [1-3].
This structure is then further condensed into an about 50-times folded, 30
nm thick fiber-structure [9] (see figure 1B), by either folding into a zigzag
structure or into a solenoid-like structure [1]. In a mitotic chromosome, a
partly unknown mechanism can condense this 30 nm structure even a few
hundred-fold more, to form the final compacted structure which makes the
chromosomes visible as separate units (see figure 1B), or the “X”-like shape
that we associate with a metaphase chromosome in a karyogram (the “X” is
formed when the cells are about to divide and each chromosome has
replicated into two copies, only attached by the centromere).
The histone core, on which the DNA resides, is the actual backbone for
almost all the epigenetic information that can control gene expression. This
is because each histone molecule has 15-38 amino acids protruding in the N-
terminal end, forming a “tail” domain, which is accessible outside of the
compact DNA-histone core [10]. These histone tails function as potential
modification site where a variety of different small tags can be attached by
different enzymes. These tags can affect the biological role of the underlying
DNA by modifying the level of chromatin compaction, often by functioning
as signals, or binding platforms, for specific chromatin-associated
proteins, which can bind to the nucleosome and affect the properties of the
local chromatin structure; either making it more condensed, and thus silent,

INTRODUCTION
4
or making it more loose, which increases transcription activity. One example
of such a protein is HP1a, which will be mentioned later on in this thesis.
Several different types of tags have been found on the different histones, for
example: methylation, acetylation, phosphorylation, ubiquitylation and
sumoylation. Depending on which amino acid-residue they modify and at
which position on the histone tail, they will have different functions. These
modifications are dynamic and rapidly changing, they can be added or
disappear within minutes of stimuli on the cell surface. I will go into a
selection of these modifications that are relevant for this thesis:
Figure 1. DNA compaction. A) The nucleosome core, consisting of an octamer of two
copies each of four histone molecules. DNA (light blue) is wrapped around the core, and one
copy of the linker histone H1 secures the DNA binding. Each histone has an N-terminal tail
protruding out of the core, which can be labeled by different tags (for example a methyl
group). B) The different compaction levels of chromatin (i.e. the DNA-histone complex that
constitutes the chromosomes), from the 2 nm thick naked DNA helix to the 1400 nm thick
highly condensed metaphase chromosome.
A B

INTRODUCTION
5
Histone acetylation
Acetylation is generally associated with active chromatin and of the known
modifications, acetylation has the highest potential of unfolding chromatin,
since it can neutralize positive charges on the target residues and thus make
the tail of the histone less prone to interact with the negatively charged DNA.
This allows easier access of the transcriptional machinery to increase
transcription [11].
H4K16 acetylation
An example of acetylation is the covalent addition of an acetyl group onto the
16th amino acid, a lysine (K), on the tail of histone 4 (H4K16ac), which is
highly associated with the action of the dosage compensation system in male
Drosophila (this will be discussed in detail later). The acetylation of lysine 16
on histone 4 [12] has also been shown to inhibit the formation of the 30
nanometer fiber and higher-order chromatin structures [13] [14].
Histone and DNA methylation
Methyl groups can be covalently added to the histone tails of nucleosomes,
but also directly to the DNA. DNA methylation is a repressive mark highly
associated with epigenetic inheritance in mammals, however, the role of
DNA methylation in Drosophila is more elusive. Some traces of this has been
found in early stages of embryonic development [15], but in general, it is
believed not to have any functional significance in Drosophila and therefore;
I will focus on the methylation of histones. This modification can have both
positive and negative effects on the level of transcriptional activity and this
depends on the position of the target residue in the tail. Furthermore, this
modification adds even extra complexity because each histone tail binding
site can have between one and three methyl groups attached: Lysines (K) can
be mono- (me1), di- (me2) or tri (me3)- methylated and arginines (R) can be
mono- or di- methylated [16].
There are so far six well characterized methylation marks, three of which are
in general correlated to active transcription: H3K4 [17], H3K36 [18] and
H3K79 [19]. The other three are associated with transcriptional repression:
H3K9, H3K27 and H4K20 [20]. Most important for this thesis is the H3K9
methylation:

INTRODUCTION
6
H3K9 methylation
Methylation of lysine 9 on the tail of histone 3 results in repression of gene
expression and is highly associated with heterochromatin. In Drosophila, it
is primarily found on chromosome 4, in the centromeric and
pericentromeric regions. It is mediated by the proteins Su(var)3-9 and
SETDB1 and is essential for the binding of a protein named HP1a, these
will all be described in detail later.
DIFFERENT CHROMATIN STRUCTURES
Chromatin can be classified into different types depending on the different
levels of chromatin compaction, level of gene activity, and associated histone
modifications. Traditionally two different main types have been defined:
euchromatin and heterochromatin.
Euchromatin
The active parts of the genome essentially consists of euchromatin, a
structure in which the DNA is loosely packed around the histone cores (see
figure 2A) and this allows access of the transcriptional machinery to the
genes. Euchromatin is loosely packed throughout most part of the cell cycle
(interphase), and only becomes condensed during the relatively short mitotic
phase of the cell cycle when the duplicated DNA needs to be efficiently
distributed between the two daughter cells. Known histone modifications
associated with active chromatin are: H3K4 methylation, H3K9 acetylation
and H3S10 phosphorylation [21-23].
Heterochromatin
Heterochromatin is considered to be the “silent” chromatin, associated with
very little gene activity. It is a gene-poor, condensed structure (see figure
2A), which generally lacks mitotic recombination, is late replicating, and
remains condensed throughout the entire cell cycle. It is usually associated
with low levels of acetylation and high levels of some methylated sites such
as H3K27, H4K20 and most importantly, methylation of H3K9 [21-23].
Interestingly, the level of heterochromatinization can be affected by
environmental factors, for example temperature changes in Drosophila
during development; a decrease in temperature from 25˚C to 18˚C results in
more heterochromatic silencing of gene expression, whereas an increase in

INTRODUCTION
7
temperature to 29˚C results in less heterochromatic silencing [24]. Other
factors that affect the rate of development also show similar effects. The
highly heterochromatic Y-chromosome is known to affect the level of
heterochromatinization; flies carrying an extra copy of chromosome Y show
reduction in heterochromatic silencing in other parts of the genome. On the
other hand; male flies lacking the Y-chromosome (X0) display enhanced
heterochromatic silencing [25,26].
About one-third of the Drosophila genome consists of heterochromatin and
regions, which are located close to the centromeres, most parts of
chromosome 4, and the telomere regions [27] (see figure 3A). The
localization of heterochromatin might be important for protection of DNA
during replication and to separate sister chromatids in mitosis. The
pericentromeric regions and chromosome 4 are predominantly associated
with H3K9me2, whereas the centromeric regions are most enriched in
H3K9me3 [22,28]. The Y-chromosome, which corresponds to about 20-30%
of the male genome (~40 Mb) in size, is also considered to be
B
A
Figure 2. Heterochromatin vs. euchromatin. A) Schematic illustration of the
difference in compaction between heterochromatin and euchromatin, and the possible role
HP1a has in compacting chromatin into heterochromatin. HP1a binds to methyl groups on
lysine 9 on the N-terminal tail of histone H3 (H3K9me) and forms an HP1a dimer that links
together two adjacent nucleosomes. B) Schematic illustration of the protein-domains of
HP1a.

INTRODUCTION
8
heterochromatic and mostly contains repetitive elements and very few genes
[29]. In addition, small regions of heterochromatin are found dispersed in
the euchromatic parts of the genome.
Since the amount of genomic heterochromatinization is variable and can be
affected by factors such as temperature, it is possible that euchromatin and
heterochromatin are highly dynamic states that are sometimes more
transient, and sometimes more fixed, and that large heterochromatic regions
have the role of titrating the amount of heterochromatin associated proteins.
If the amount of heterochromatin proteins becomes too large, threatening
the expression of the active genome, it can be redirected to heterochromatic
regions, such as the Y-chromosome. On the opposite, if the Y-chromosome is
missing, the amount of heterochromatin proteins in the genome becomes
abundant, and can repress other regions.
GREEN, BLUE, BLACK, RED, YELLOW chromatin
The traditional view on chromatin is that heterochromatin and euchromatin
are the two main types of structure, but it has become more and more
evident that there are large variations also within these groups, and therefore
a more specific classification of chromatin has been proposed [30], in which
five principal chromatin types, defined by unique combinations of associated
chromatin binding proteins, are specified in Drosophila:
GREEN chromatin corresponds to “classical” heterochromatin and is
found primarily in pericentromeric regions and on chromosome 4. It is
defined by the presence of Su(var)3-9, HP1a (described later on in this
thesis), as well as a couple of HP1a interacting proteins: LHR and HP6. In
addition, GREEN chromatin is also highly and specifically enriched in
H3K9me2.
BLUE chromatin corresponds to PcG chromatin since it is associated with
the Polycomb-group (PcG) proteins PC, E(Z), PCL and SCE (these are
proteins important for maintaining silencing of specific genes during
Drosophila embryonic development). In addition, it is highly enriched with
the repressive histone modification H3K27me3, which is mediated by E(Z)
and recognized by PC. The Hox gene cluster (a well-known PcG target loci) is
found within BLUE chromatin.
BLACK chromatin is the most abundant chromatin type, covering 48% of
the probed genome of Drosophila. It is generally gene poor, and the enclosed

INTRODUCTION
9
genes (4162 genes) are either silent or expressed at very low levels. In fact, a
majority of the silent genes in the genome are found within the BLACK
chromatin definition. It lacks the active histone marks H3K4me2 and
H3K27me3 but is marked by a number of chromatin associated proteins, like
histone H1. BLACK chromatin appears to have an active role in
transcriptional silencing, since it has a higher ability to repress inserted
reporter genes than the rest of the genome.
RED chromatin and YELLOW chromatin both constitute the active
chromatin (euchromatin) and they share several marks for active chromatin.
Both are replicated early in S phase of the cell cycle, but characteristic for
RED chromatin is that it tends to be replicated even earlier in S-phase and is
strongly enriched in origin of replication complex (ORC). Apart from
replication timing, the YELLOW chromatin differs from RED chromatin by
being enriched in MRG15, a chromo domain-containing protein that has
been suggested to bind to H3K36me2 and me3, [31] and YELLOW
chromatin in general also contains more H3K36me3 than RED chromatin.
Genes that are ubiquitously expressed over many embryonic stages and
tissue types, such as ribosomal- and DNA repair genes and genes involved in
nucleic acid metabolic processes, are mostly found within YELLOW
chromatin whereas genes involve in more specific processes such as defense
responses, signal transduction etc. are more located in RED chromatin.
These five different types of chromatin are distributed in a mosaic around
the entire genome, with accumulation of repressive chromatin around the
centromeres. Although this classification is rough, it provides a more specific
definition of different chromatin types, which often differs extensively in
characteristics. A similar classification has been done by the modENCODE
project, in which they defined nine chromatin states [32]. Chromatin state 7
or the GREEN chromatin corresponds to the heterochromatin type that will
be in focus in this thesis.
Position-effect variegation
A chromatin connected mechanism, which has been particularly well studied
in Drosophila, is position-effect variegation, one of the first mutations that
was discovered in Drosophila was white, which changes the eye color of the
fly from the normal red into white. Muller experimented with X-ray as a
mutagen to induce mutations and discovered an unusual and interesting
phenotype; a fly with red and white patched eyes, i.e. variegated eyes. This
was highly intriguing since it meant that the white gene was indeed affected

INTRODUCTION
10
by the mutagenesis, but not destroyed. Had it been destroyed, every facet of
the eye would have been white. Moreover, this variegated phenotype could
be restored back into wildtype red eyed in some of the offspring of variegated
parents, by again using radiation. This meant that it was no ordinary
mutation, and to better understand this, they examined the chromosomes of
these variegated flies and found that a chromosome breakage and
subsequent inversion of a chromosomal segment had translocated (i.e.
moved) the white gene into close vicinity of heterochromatin. The gene was
thus still intact and could in principal be expressed, but the new
heterochromatic silencing surrounding could actually spread into the white
gene, causing varying degree of repression of the red eye pigmentation which
results in red and white patched eyes. This phenomenon was named
Position-effect variegation (PEV) (reviewed by [33]), since this
phenotype was a result of a change in position of the white gene. In
Drosophila, practically every gene examined has shown PEV when
translocated into the vicinity of heterochromatin. PEV of the white gene in
Drosophila has been used as a tool to identify genes involved in
heterochromatin formation; by combining PEV flies with various other
mutations, scientists could search for those mutations that result in an
increase of silencing of the reporter gene, termed Enhancer of variegation,
E(var) or in a decrease of silencing, termed Suppressor of variegation,
Su(var). Using this method, so far about 30 different genes involved in
modifying PEV have been isolated and characterized [33].
Two of these genes have particular importance for the formation of
heterochromatin and the silencing of genomic regions: Su(var)2-5, encoding
the protein HP1a and Su(var)3-9, encoding the protein Su(var)3-9, which
mediates a methylation mark required for HP1a targeting. I will describe
both in more detail.
HP1A (SU(VAR)2-5)
HP1a (Heterochromatin Protein 1a) is the most important factor involved in
establishing and maintaining heterochromatin by binding directly to and
affecting the compaction level of the chromatin. A complete removal of HP1a
using a homozygous mutant results in loss of heterochromatin and hence,
lethality [34]. HP1a essentially functions by binding to H3K9me2 and me3
marks and initiates a compaction of the nucleosomes. The protein is made
up of two functionally important parts: the N-terminal chromo domain (CD),
which mediates the interaction with H3K9me2 and me3 [35,36] [37], and
the C-terminal chromo-shadow domain (CSD), which can interact with the

INTRODUCTION
11
CSD of another HP1a molecule to create a HP1a dimer [38,39]. The two
functional domains are connected through a hinge domain, which gives the
protein flexibility and potentially aids in the recognition of H3K9
methylation [40] (see figure 2B).
HP1a binding sites in the Drosophila genome are highly associated with
GREEN chromatin [30] and are found primarily in the pericentromeric
region, on the 4th chromosome, and dispersed in a number of smaller
euchromatic sites, of which cytological region 31 on chromosome arm 2L is
the most distinct one [27,41-45]. On chromosome 4, HP1a is bound to the
gene body (the part of the gene that is transcribed) and the promoters (the
region which recruits transcription proteins and initiates transcription of the
gene) of active genes [30,43-47], whereas in the pericentromeric regions,
HP1a appears to bind in a more general manner and it spreads over tens of
kb rather than to individual genes [45,47].
Interestingly, HP1a binding to the gene body is dependent on H3K9me2 and
me3 whereas the binding peak at the promoters of the genes is methylation-
independent [45], this could indicate that HP1a binds to chromatin by two
different binding mechanisms.
Repressive or activating function of HP1a?
HP1a is, as already mentioned, essential for formation and maintenance of
the highly repressive heterochromatin, and has thus been assumed to have a
repressive function. This view was also supported by an RNAi mediated
knock-down of HP1a, which resulted in an increased expression of genes on
the 4th chromosome [48,49].
But more recently, several conflicting studies have emerged, providing
different evidence for an activating function of HP1a; knock-down
experiments using both RNAi [50,51], and mutation have discovered several
euchromatic genes that, as a result of the HP1a depletion, are down-
regulated in expression [42,52-54]. This is the opposite of the effect you
would expect given HP1a’s inhibiting properties. In addition, genes such as
light, rolled, RpL15 and Dbp80, which are all located within the
heterochromatic pericentromeric regions, have shown to actually depend on
HP1a and the heterochromatic surrounding to maintain proper expression
[55-60]. Furthermore, the existence of euchromatic binding sites of HP1a
also indicates an association with active gene transcription [41] [47,52],
although here, the binding seems mostly localized to the gene body rather

INTRODUCTION
12
than the promoter [52] and is mostly independent on Su(var)3-9 [42]. Other
sites with high gene activity, such as developmentally regulated genes and
heat-shock induced chromosomal puffs, have also been associated with HP1a
binding [52].
All these evidence has led to an unresolved debate whether HP1a has a
repressing or a stimulating function on gene expression, and this is an issue
explored and discussed in paper III in this thesis.
Isoforms of HP1
D. melanogaster encodes no less than 5 paralogues of HP1: HP1a, HP1b,
HP1c, HP1d/Rhino and HP1e. They are structurally similar, containing the
conserved chromo domain and chromo-shadow domain that are separated
by a variable hinge domain. Yet they appear to have different functions;
HP1a is the most studied isoform and usually the one that is referred to in
general studies of HP1 function. As described above, HP1a localizes mainly
with H3K9 methylation and the GREEN heterochromatic regions. HP1b is
found in both euchromatic and heterochromatic regions whereas HP1c is
primarily localized to euchromatic regions [61], and co-localizes with
H3K4me and RNA polymerase II (both marks for active gene transcription)
[62]. HP1a, HP1b and HP1c are all ubiquitously expressed whereas
HP1d/Rhino and HP1e are mainly functioning in the germline of males and
females [63].
MEDIATION OF H3K9 METHYLATION MARKS
As mentioned above, the binding of HP1a to the chromatin and the gene
bodies of active genes requires the presence of the histone modification
H3K9me2 and/or me3, i.e. the attachment of two or three methyl-groups to
lysine (K) 9 on the tail of histone H3. The exception is HP1a binding to
promoter regions, which is independent on H3K9 methylation. The proteins
that mediates these methylations are known as histone lysine methyl
transferases (HKMTs) and in Drosophila there are three known HKMTs;
G9a, Su(var)3-9 and SETDB1.
G9a
The Drosophila dG9a protein appears to be a functional orthologue of the
mammalian G9a, which mediates mono- and dimethylation in euchromatic
regions [64], however, in Drosophila G9a does not appear to affect the H3K9

INTRODUCTION
13
methylation patterns and is also not important for viability [65]. A role in
germ cell formation has been proposed [66], but in general, the function of
G9a in Drosophila remains elusive [45,65,67].
Su(var)3-9
Su(var)3-9 controls H3K9me2 and me3 [68] primarily in the centromeric
and pericentromeric regions, and is therefore important for gene silencing
through formation of heterochromatin [69-71]. Notably, in the centromeric
regions, Su(var)3-9 only seem to control the me3, but not the me2 [20,70].
It actually is a rather funny coincidence that this protein happens to
methylate H3K9, since the name 3-9 was given because the gene is situated
on chromosome 3, and 9 was just a serial number applied during the
screening experiment that identified it. The actual function was discovered
much later.
Su(var)3-9 mutants have depleted levels of HP1a and H3K9me2 in the
pericentromeric regions and, in contrast to HP1a mutants, are generally
viable, fertile and unaffected in germ-cell development [71]. A mutation that
gives the protein a hyperactive function results both in stronger PEV (i.e.
heterochromatic silencing of reporter genes), higher levels of H3K9me2 and
me3 at the chromocenter, and also ectopic H3K9 methylation in several
euchromatic sites. Su(var)3-9 contains a SET domain, which mediates the
methylations, and a chromo domain, commonly associated with chromatin
remodeling proteins [70,71]. Both the SET domain and the chromo domain
are required for the binding to heterochromatic sites [70].
The presence of Su(var)3-9 and its methylation of H3K9 in heterochromatic
regions is a prerequisite for HP1a binding to these regions, but in fact, these
two proteins are interdependent on each other; HP1a is essential for
restricted Su(var)3-9 binding to heterochromatin and the two proteins can
bind to each other through interactions between the chromo domain of
Su(var)3-9 and the chromo-shadow domain of HP1a [70]. Furthermore, in a
HP1a mutant background, the H3K9 methylation patterns are increased,
particularly in euchromatic sites [20].
In addition to the pericentromeric regions, Su(var)3-9 binding has been
detected on chromosome 4, but the H3K9 methylation pattern of the 4th
chromosome is unaffected in a Su(var)3-9 mutant, so the function of
Su(var)3-9 here is still unknown [20,45]. Furthermore, in contrast to
Su(var)3-9 binding to pericentromeric regions, it appears as if Su(var)3-9

INTRODUCTION
14
does not require the SET domain and the chromo domain to bind to
chromosome 4 [70].
SETDB1
The other HKMTs that mediates H3K9me2 and me3 in Drosophila is
SETDB1 [45,54,72-74], which is encoded by the gene Setdb1 (also named
eggless or egg) [64]. Su(var)3-9 and SETDB1 appear in principle to be
complementary and are responsible for H3K9 methylation in different
regions. So whereas Su(var)3-9 mediates H3K9 methylation in the
pericentromeric regions, SETDB1 is mainly responsible for mediating H3K9
methylation on chromosome 4 [45,72], although it has also been suggested
to mediate H3K9 methylation at some euchromatic sites [72]. In a Setdb1
mutant, H3K9 methylation and HP1a binding is impaired on chromosome 4,
but not in pericentromeric regions [73].
More specifically, SETDB1 methylates the gene body of active genes on
chromosome 4 [45], and in a Setdb1 mutant, HP1a binding to the gene body
of chromosome 4 active genes is impaired, while HP1a binding to the
promoters is still unaffected [45].
SETDB1 contains a methyl-CpG-binding domain (MDB), which seems able
to recruit deacetylase (HDAC) complexes [75] and a PreSET/SET domain,
which mediates the methylation function [64,73,74]. SETDB1 is essential for
female fertility [74,76,77] by mediating H3K9me3 in germ cells and somatic
cells of the germarium, and early stages of egg chamber require SETDB1 for
proper formation [74]. There are some results that indicate that in the germ
cells, SETDB1 is the only active HKMTs and is therefore responsible for
mediating the H3K9 methylation in the pericentromeric regions (instead of
Su(var)3-9), and then in a later stage of the oogenesis, this function is
transferred to Su(var)3-9 [76].
In addition to the pericentromeric regions and chromosome 4, H3K9
methylation is also detected at telomeres and at some euchromatic sites, but
neither Su(var)3-9 nor SETDB1 appear to be involved in establishing these
marks, indicating the presence of additional unknown HKMTs [20,70].
HETEROCHROMATIN FORMATION
Once H3K9me2 and me3 has been established by either SETDB1 or
Su(var)3-9, they function as docking sites that recruit HP1a [35,36,78,79]

INTRODUCTION
15
through an interaction between the H3K9me2 or me3, and a hydrophobic
pocket in the HP1a N-terminal chromo domain (CD) [35,36,80]. Two
adjacent nucleosome-bound HP1a molecules can interact with each other’s
C-terminal chromo-shadow domain (CSD) to form an HP1a dimer [39],
which causes two HP1a bound nucleosomes to be linked tightly together [38]
(see figure 2A). Depending on regions, Su(var)3-9 or SETDB1 is then
believed to interact with the CSD of the bound HP1 [70] to promote further
methylation of the neighboring nucleosome, forming a new binding site for
HP1a and thus, initiate a spreading mechanism that will cause the chromatin
to become condensed and inactive.
For euchromatin to be heterochromatinized, specific de-methylation, de-
acetylation and de-phosphorylation reactions need to take place within the
euchromatin, and this process seem to begin with the de-acetylation of H3K9
by the enzyme HDAC1 [81]. HDAC1 and Su(var)3-9 have been shown to
associate in vivo, and could thus provide extra complexity to the process of
heterochromatin formation [69].
Heterochromatin formation and RNA interference
In a number of eukaryotes, RNAi is involved in heterochromatin formation.
For example in S. pombe (fission yeast), RNAi is required for spreading of
H3K9 methylation into reporter genes that have been inserted into
heterochromatic regions [82]. RNA interference or RNAi was originally
identified as a mechanism involved in post-transcriptional regulation and
defense against for example retroviral infections. It is also believed to be
involved in post-transcriptional silencing of transposable elements. The
RNAi mechanism regulates the levels of RNA transcript in several steps; first
a group of proteins known as the dicer family targets and cleaves dsRNA into
short segments of 21-30 nucleotides, called small interfering RNAs (siRNA).
These siRNAs are then binding to argonaute proteins to form the RISC (RNA
Induced Silencing) complex. The siRNA will base pair with complementary
transcripts and hence, guide the RISC complex to degrade it.
Recently, an RNAi mediated guidance mechanism for targeting of chromatin
remodeling factors to heterochromatic sites has been proposed in
Drosophila, in which the protein Piwi (part of the argonaute protein family)
associates with, and utilizes various different piRNAs (Piwi-interacting
RNAs) to target piRNA complementary sites within heterochromatic regions,
and then recruiting HP1a and Su(var)3-9 to these sites to induce
heterochromatinization. The involvement of the RNAi machinery and small

INTRODUCTION
16
interfering RNAs may explain the lack of sequence recognition domains in
the chromatin remodeling proteins [83].
Transposons
Transposons or transposable elements (TE) are found in both eukaryotic and
prokaryotic genomes and are highly associated with heterochromatic
regions. They constitute a very special type of DNA sequences, also known as
“selfish” elements or as “jumping” genes [84]. Transposable elements can
have the ability to self-replicate and to change position within the genome,
some of them carry within them genes encoding proteins such as
transposases, which can cut and paste the transposon between genomic sites
(DNA transposons), other TE transcribe RNA which is converted into DNA
by a reverse transcriptase, and then inserted into new genomic sites
(retrotransposons). In active state, these elements can potentially cause
severe damage if the insertion site happens to be within an essential gene.
Fortunately, most TE are silent, either due to disruption of their important
genes, or by silencing mechanisms that have evolved in the host genome as a
response to the deleterious effects of active TE [85]. In humans for example,
RNA interference is important for silencing of transposons; small RNAs
from the transposon sequence are incorporated into the RISC complex,
which then can target and repress all the copies of that particular TE [86].
Chromatin modifications also appear to be involved in the repression of TE;
a big proportion of HP1a and H3K9me3 binding is reported to occur to
transposons and repeated elements, where they co-localize with each other.
Transcription activity marks (such as H3K9ac and RNA Polymerase II) on
the other hand, are more or less absent [44], showing that most transposons
in Drosophila, with few exceptions, are transcriptionally silent and that
HP1a is suggested to be involved in this silencing [87] by binding to
Su(var)3-9 mediated H3K9me3 marks [88].
Over the course of evolution, silenced TE seem to have accumulated within
heterochromatic regions, and about 77% of Drosophila heterochromatin (or
about 30% of the entire Drosophila genome) consists of transposable
elements or repeated regions [89], and organisms with larger genomes, for
example humans or maize consists of as much as 45% and 75% transposons
respectively, in the entire genome [82].
If these elements become active and change positions, they sometimes bring
part of the surrounding genome along for the ride. This of course can be very

INTRODUCTION
17
deleterious to a cell, but also, can be a driving force for evolution, and
Drosophila genetic researchers are often utilizing this feature to create
disruptions of genes or genomic segments for studies of gene function.
WHY USE DROSOPHILA AS A MODEL ORGANISM?
General information about the fruit fly
Drosophila melanogaster, or fruit fly, is a black and yellow fly with bright
red eyes, which is usually found around the fruit section in grocery stores, or
in the kitchen of your average student habitat. With a humble size of only 2.5
mm, it might appear very insignificant, or at most, slightly annoying. But in
fact, the fruit fly is very valuable for many researchers and it has been used
as a genetic model organism for more than 100 years (Thomas Morgan Hunt
began to use them for heredity studies around 1910). It became a popular
model organism for many different reasons; the most obvious is that they are
easy and cheap to cultivate and don’t require much space, and with a
generation time of about 10 days at room temperature, you can study several
generations within a few weeks. Genetically they also have several useful
traits; the complete genome of D. melanogaster was sequenced and first
published in 2000 [90]. They have nearly as many genes as humans, about
15 000 genes (compared to 20 000 in humans) and about 75% of human
disease genes have homologues in the genome of Drosophila [91], yet the
size of its genome is substantially smaller and easier to handle, about 260
Mb (2.6 x 108) (according to Drosophila annotation release 5) divided
between just four pairs of chromosomes (to compare with the human
genome which is about 6 x 109 bp divided between 23 pairs of
chromosomes). The genome consists of one pair of sex chromosomes: XX or
XY, and three pairs of autosomal chromosomes: chromosome 2, 3 and 4, of
which chromosome 2 and 3 are each divided into two chromosome arms, left
and right, separated approximately in the middle of the chromosome by the
centromere (they are more specifically named chromosome 2L, 2R, 3L and
3R) (See figure 3A).
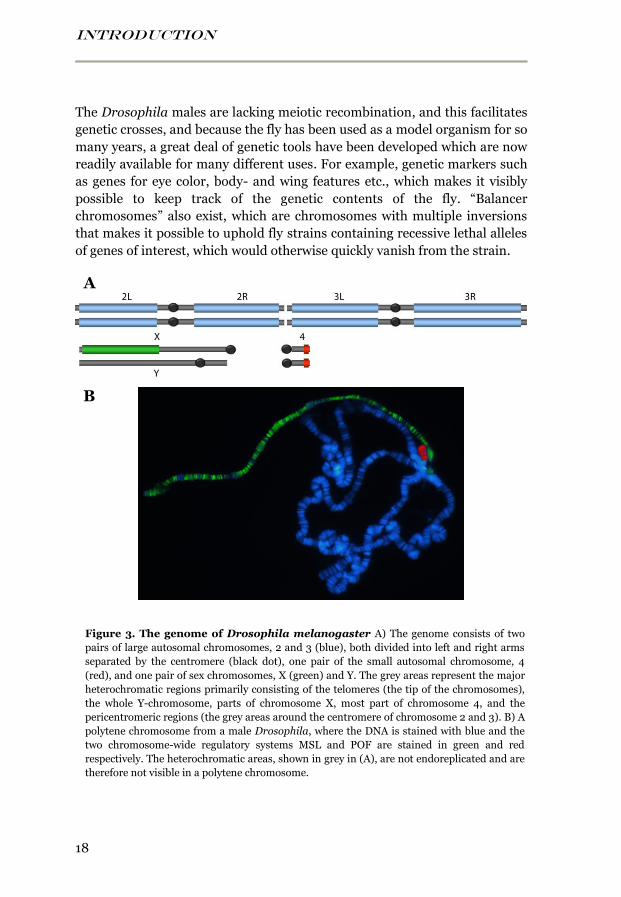
INTRODUCTION
18
The Drosophila males are lacking meiotic recombination, and this facilitates
genetic crosses, and because the fly has been used as a model organism for so
many years, a great deal of genetic tools have been developed which are now
readily available for many different uses. For example, genetic markers such
as genes for eye color, body- and wing features etc., which makes it visibly
possible to keep track of the genetic contents of the fly. “Balancer
chromosomes” also exist, which are chromosomes with multiple inversions
that makes it possible to uphold fly strains containing recessive lethal alleles
of genes of interest, which would otherwise quickly vanish from the strain.
Figure 3. The genome of Drosophila melanogaster A) The genome consists of two
pairs of large autosomal chromosomes, 2 and 3 (blue), both divided into left and right arms
separated by the centromere (black dot), one pair of the small autosomal chromosome, 4
(red), and one pair of sex chromosomes, X (green) and Y. The grey areas represent the major
heterochromatic regions primarily consisting of the telomeres (the tip of the chromosomes),
the whole Y-chromosome, parts of chromosome X, most part of chromosome 4, and the
pericentromeric regions (the grey areas around the centromere of chromosome 2 and 3). B) A
polytene chromosome from a male Drosophila, where the DNA is stained with blue and the
two chromosome-wide regulatory systems MSL and POF are stained in green and red
respectively. The heterochromatic areas, shown in grey in (A), are not endoreplicated and are
therefore not visible in a polytene chromosome.
B
A

INTRODUCTION
19
Specific advantages of fruit fly in epigenetics
However, there are two specific properties that are unique for the fruit fly,
which makes them specifically good as a model organism for my work:
Polytene chromosomes
One specifically useful feature of Drosophila, when it comes to studying
chromatin binding proteins, is their polytene chromosomes. These are
extremely large chromosomes found primarily in the salivary glands of third
instar larvae of Drosophila and other dipteran (two-winged) insects. The
purpose of these giant chromosomes is believed to be to increase expression
of proteins that are needed for pupation. The polytene chromosomes are
formed through repeated rounds of DNA replication, but instead of the
normal division into two cells, the cell grows in size and the sister
chromatids remain synapsed together, forming a chromosome consisting of
multiple copies (in Drosophila sometimes up to 1024 copies) adjacent to
each other (a process known as endoreplication). As a result, a very long and
thick chromosome (see figure 3B) is easily seen in microscopes and it is very
good for immunostaining and in situ hybridization, which allows
visualization of the binding sites of proteins and RNAs.
Two chromosome-wide regulatory systems
The second thing that makes Drosophila good for studies of chromatin
associated and chromosome-wide gene regulation, is that it has two known
chromosome-wide gene regulatory mechanisms; the MSL complex, which
decorates the male X-chromosome and mediates dosage compensation,
and POF, a chromosome 4 specific stimulating protein (see figure 3B). POF
is the first and only autosome-specific gene regulatory protein reported in
any organism to this day and therefore makes Drosophila unique compared
to other model organisms. I will go through these two mechanisms in detail:
DOSAGE COMPENSATION
General
In many organisms, sex is determined by the combination of the sex
chromosomes X and Y. Females have the combination XX and males have
the combination XY. These two chromosomes differ substantially in their
genetic content. The Y-chromosome mostly consists of heterochromatin and

INTRODUCTION
20
repetitive elements and very few genes, in mammals about 80 protein-
coding genes [92], out of which the most important one is the key gene in
male sex determination, SRY [93]. The Drosophila male Y-chromosome is
entirely heterochromatic and contains even fewer genes, between 12 to about
20 genes, of which several have suggested male related functions, but unlike
the mammalian sex determination, the Drosophila Y-chromosome is not the
key factor for male determination, but rather for fertility.
The Y-chromosome originally started out as a homologue to the X-
chromosome, the proto-Y, but over the course of evolution, the proto-Y-
chromosome has degenerated in the heterochromatic Y-chromosome,
leaving only a few genes and a small region of homology so that X and Y can
pair during cell division [29,94]. This means that an imbalance in X-linked
genes has occurred between the sexes (♂X: ♀XX) which need to be
compensated for. This is achieved by a dosage compensating mechanism,
which has the purpose of equalizing the gene expression from the X-
chromosome between males (XY) and females (XX) (see figure 4). Different
organisms have evolved different dosage compensating mechanism [95], but
I will focus on the mechanisms in mammals and in Drosophila.
In mammals
Mammals have evolved a mechanism in which one of the two female X-
chromosomes, Xi, in each cell is randomly inactivated after a few rounds of
Figure 4. Dosage compensation. The left part illustrates the problem with uneven X-
chromosome numbers between males (one copy) and females (two copies), and the uneven
ratio between the X-chromosome and the autosomes (two copies of each autosome) in
males. This is in mammals compensated by an inactivation of one of the female X-
chromosomes, by condensing it into a compact Barr body (small dot), but also by doubling
the expression of the X-chromosome in both sexes to equalize the X:A ratio. In Drosophila,
dosage compensation is mediated by restricting the doubling in expression of the X-
chromosome to males.

INTRODUCTION
21
cell division in the embryo [96], and condensed into a small, highly compact
element called the Barr body [97]. Females are thus mosaics for two cell
types that express one or the other X-chromosome. Only a few Xi-bound
genes escape the inactivation and are still expressed from the Barr body [96].
The X inactivation occurs early during embryogenesis and is initiated by
transcription of the 19 kb long non-coding RNA Xist, from an inactivation
center on the X-chromosome destined for inactivation. The Xist RNA
spreads in cis to cover the entire length of the X-chromosome [98,99] [100],
and initiates the silencing process, possibly by functioning as a platform for
recruiting repressive complexes (Polycomb complexes) [101]. Transgenes of
Xist placed on autosomes can induce spreading of Xist and different degrees
of gene silencing of autosomal genes surrounding the insertion site [102].
Evidence suggests that HP1 is involved in the process of inactivating the X-
chromosome in humans [103].
Up-regulation of mammalian X-chromosome
The inactivation of one of the female X-chromosomes solves the problem
with unbalanced sex chromosome ratio between males and female, however,
one additional problem remains with this model; the single active X-
chromosome in both males and females is out-numbered by the autosomal
chromosomes, which are all present in two copies. This leads to an
unbalanced X to autosomes ratio of 0.5-1, and the transcriptional output
from the single X-chromosome will thus be too small compared to the
transcriptional output from the autosomal chromosomes.
This problem appears to be solved by a doubling of the expression from the
X-chromosome in males and from the active X-chromosome in females
[102,104-106] (See figure 4). Up-regulation of mammalian X-chromosome
has been detected in several species including human, primates, rat and
mouse, and microarray studies have shown that the X:autosome
transcriptional output is close to 1 in most somatic tissues from both males
and females [104-107].
In fact, it is likely that up-regulation of the X-chromosome actually began to
evolve first, to balance the X to autosome ratio when the proto-Y in males
was gradually degenerated, and that the X-inactivation in females followed
as a response to the hyper-activation of the X-chromosome, which would be
unfavorable for females already possessing two X-chromosomes [108].

INTRODUCTION
22
Very recently, an additional long non-coding RNA, XACT, has been
discovered in a pluripotent human cell line. XACT coats the length of the
active X-chromosome in females as well as in males in a similar fashion to
the Xist covering of the inactive Xi-chromosome [109], indicating that this
non-coding RNA might be involved in the mechanism that up-regulates the
active X-chromosome in males as well as females. In absence of Xist, the
XACT RNA is expressed from, and covers the length of both X-
chromosomes.
However, it should be noted that mammalian X-chromosome up-regulation
is a controversial issue and some studies claims that up-regulation of the
mammalian X-chromosome does not exist at all [110].
In Drosophila
In Drosophila, dosage compensation is achieved by a two-fold up-regulation
of the male X-chromosome (see figure 4) [108], and this hyper-activation is
mediated at least to some extent by a male-specific ribonucleoprotein
complex, called the Male-Specific Lethal (or MSL) complex. The MSL
complex decorates the entire length of the male X-chromosome, by binding
to hundreds of distinct sites (shown by cytological studies on polytene male
X-chromosomes) (see figure 3B), and mediating H4K16 acetylation [111].
The proteins of this complex are, as implied by their name, essential for
viability in males but not in females and males lacking any one of the MSL
proteins will die during larval stage [95]. The complex consists of at least five
different proteins, MSL1, MSL2 and MSL3, MLE, and MOF and two non-
coding RNAs (ncRNAs), roX1 and roX2.
MSL1
MSL1 (Male-Specific Lethal 1), together with MSL2, appears to function as a
backbone for the assembly of the complex, and MSL1 can independently
interact with MSL2, MSL3 and MOF [112,113]. Evidence suggest that the C-
terminal domain (also called the PEHE domain) of MSL1 interacts with
MSL3 and MOF [114], and that a coiled-coil N-terminal domain of MSL1
interacts with a RING finger domain of MSL2 [115,116]. It has been
suggested that two MSL1 proteins initially form a dimer that is essential for
the MSL complex to assemble, recognize- and spread on the X-chromosome
[117].

INTRODUCTION
23
MSL2
MSL2 (Male-Specific Lethal 2) is the limiting component which stabilizes the
entire MSL complex; without MSL2 no complex is formed and in contrast to
the other components of the MSL complex, the MSL2 protein is only
expressed in males. The absence of MSL2 is also what prevents the MSL
complex from being formed in females, because the expression of MSL2 in
females is blocked at the level of translation by the female specific sex-lethal
(SXL) protein, thus, the remaining proteins expressed in females cannot be
assembled into a functional MSL complex. If ectopic expression of MSL2 is
induced in females, an MSL complex is formed and hyper-activates the
female X-chromosomes, leading to severely impaired viability [118-121].
MSL2 is a RING finger containing protein, meaning that it can potentially
bind to DNA [118,119,122]. An additional MSL2 regulating mechanism has
been proposed for male Drosophila, in which non-chromatin- associated
MSL complexes bind to msl2 mRNA in males. This blocks the translation
into MSL2 proteins and allows a fine tuning of the amount of functional MSL
complex, by feedback regulation of the rate-limiting component [123].
MSL3
MSL3 (Male-Specific Lethal 3) contains an N-terminal chromo domain, a
common domain within chromatin remodeling proteins [124], which is
needed for interacting with H3K36me3 on the nucleosomes of active genes.
MSL3 and MOF interact together and MSL3 must be acetylated by MOF at a
single lysine residue close to its chromo domain (at lysine 116) to be properly
included in the MSL complex [125].
MLE
MLE (Maleless) is an ATP-dependent RNA/DNA helicase, which means it
has the capacity to unwind DNA or double-stranded RNA by breaking the
hydrogen bonds between the two strands [126]. It is weakly associated with
the MSL complex [115,127], and the association of MLE with the X-
chromosome is sensitive to RNase treatment [128]. MLE interacts with the
roX RNAs, and this interaction appears to be necessary for both of them to
be incorporated into the MSL complex [129-131]. MLE has an ATPase
activity, which means it catalyzes the release of energy by de-
phosphorylating ATP into ADP, and this activity appears to be sufficient for
transcriptional activation, whereas the helicase activity is required for the
spreading of the MSL complex along the X-chromosome [132]. Additional

INTRODUCTION
24
function of MLE has also been proposed, in which it binds to newly
transcribed RNAs from the X-chromosome, and in this way it can direct the
MSL complex to active genes [128,133,134].
MOF
MOF (Males absent On the First) is a histone acetyltransferase (HAT) and it
specifically acetylates lysine 16 on histone H4 (H4K16), which is important
for the up-regulation of the X-chromosome [111,135,136]. The presence of
H4K16 acetylation does not appear to be directly involved in targeting or
spreading of the complex, since none of the components of the MSL complex
have any known domain for recognizing this modification. It thus seems
more likely that this modification facilitates spreading by opening the
chromatin structure, and thus, increasing the accessibility of the MSL
complex [137]. Unlike the other MSL proteins, MOF is encoded on the X-
chromosome.
MOF is the only MSL component that is also found associated with
autosomal chromosomes [137,138], and it appears to be part of a non-X-
specific complex called the NSL (Non-Specific Lethal) complex, which
targets promoters of constitutively expressed genes (housekeeping genes),
on both the X-chromosome and on autosomes in males and females. It
appears to be involved in the recruitment of RNA Pol II and the pre-
initiation complex to the promoters of the targeted genes [139,140].
However, in presence of the MSL complex, the catalytic activity of MOF is
mostly constrained to the X-chromosome [141].
roX1 and roX2
roX1 and roX2 (RNA on the X1 and 2) are two non-coding RNAs essential for
proper targeting of the MSL complex. They are both encoded on the X-
chromosome, at cytological section 3F3 and 10C7 respectively. These two
ncRNAs are very different in size and primary sequence, roX1 is
approximately 3.7 kb in length whereas roX2 is only 0.6 kb in length. They
only share one similar, 30 bp long sequence [142]. Despite these differences,
roX1 and roX2 have redundant functions, meaning that deleting one of these
two still creates a fully functional MSL complex, but deleting both is lethal to
males, with a few sterile escapers (in contrast to mutants of the other
components of the MSL complex which have no escapers). In these flies,
both the MSL complex and histone 4 acetylation are relocated to autosomal

INTRODUCTION
25
sites [143,144]. This creates interesting questions about the function of these
ncRNAs in the MSL complex.
The MSL complex binds to these two genes, and it has been suggested that
the MSL complex is incorporating the synthesizing roX RNAs at the site of
transcription, because autosomal transgenes of roX can induce spreading of
MSL binding at about a 1-2 Mb region surrounding sites of insertion
[145,146], and both roX RNAs are rapidly degraded if they do not associate
with the proteins of the complex [131]. Autosomal roX transgenes can also
rescue roX mutants which would otherwise die [144].
High affinity sites and spreading of DCC
It is known that the MSL complex binds to active genes on the male X-
chromosome, rather than over stretches of genomic regions (thus resulting
in the re-occurring banded pattern) (see figure 3B), and MSL1 and MSL2
forms the core of the complex that initially targets about 200 reproducible
sites distributed over the length of the chromosome. These sites have been
named High affinity sites (HAS) or chromatin entry sites (CESs)
[120,145,147-151] The MSL1- MSL2 core serves a platform on which the
additional proteins and ncRNAs assembles into a functional MSL complex,
which then can spread to cover the remaining non-CES targeting sites on the
X-chromosome. MSL3, MLE and MOF and at least one of roX1 or roX2 are
all required specifically for MSL spreading from the CES [144,149,151]. The
genes encoding roX1 and roX2 are furthermore two of the strongest CES for
the MSL complex and were the first CESs to be discovered [130]. CES
inserted into autosomal sites can induce short-range spreading to the 3’ end
of flanking genes surrounding the site of insertion [148].
Targeting mechanisms of the MSL complex
One very important question is how the dosage compensation mechanism is
able to distinguish the genes of the X-chromosome from the other
chromosome, and there are two main models for the targeting of MSL: the
transcription model and the sequence model.
The transcription model argues that MSL targeting depends on gene
activity, because the MSL complex is highly associated with active genes:
about 75% of all active genes on the male X-chromosome are bound by the
MSL complex; and about 80-90% of genes bound by the MSL complex are
associated with active chromatin marks, such as the transcription-associated

INTRODUCTION
26
H3K36me3 mark [146,152,153]. If the chromo domain of MSL3 is disrupted,
the MSL complex is not able to spread further than about 1 kb away from a
CES, which indicates that the recognition of H3K36me3 by MSL3 is involved
in spreading of the complex [154]. It is also known that the complex can
spread from an ectopic CES into active genes with no X-specific sequence
(i.e. translocation of CES to autosomes can induce spreading)
[145,146,154,155], and that autosomal genes inserted onto the X can be up-
regulated [156,157]. Also, non-dosage compensated X-bound genes can start
recruiting MSL upon activation [158]. Furthermore, it is interesting that even
though the dosage compensation seems to depend on H4K16 acetylation
mediated by MOF, many more genes (nearly all active genes) are acetylated
than are bound by the MSL complex [159] and more active genes also appear
to be dosage compensated than are bound by the MSL complex [105].
The sequence model on the other hand claims that the MSL complex
recognizes chromosome X specific sequences; sequence composition analysis
(i.e. searching for enrichment of specific types of sequences) of the X-
chromosome in Drosophila have revealed that the X is enriched for simple
repeat sequences, making it different from the autosomal chromosomes
[160,161]. Motif search in previously defined CES [162,163] has furthermore
identified a 21 bp long, GA-rich motif enriched in MSL binding, this motif is
named MSL recognition site (MRE) [148]. Sequence motif have also been
suggested by [153,164], although they do not perfectly predict the MSL
binding.
However, it is likely that the targeting mechanism is actually a combination
of these two models, so that the MSL complex first recognizes specific
sequences in the CES, and then spreads to nearby genes based on chromatin
marks for active transcription [146]. It has also been suggested that the
secondary spreading mechanism as well could depend on sequence-
specificity [165].
Mechanism behind the X-chromosome up-regulation
The MSL complex binds mainly to the gene body of active genes, with a
preference for the middle part and the 3’ end of the genes [152,153]. In
contrast to many transcriptional regulators, it does not bind to promoters.
The 3’ end binding bias is observed for many factors that regulate
transcription elongation, and this suggests that the MSL complex is
important for functions such as transcriptional elongation or recycling
of the polymerase back to the promoter [152,153] [166]. However, recently

INTRODUCTION
27
this 3’ bias was suggested to depend on the fact that the 3’ end usually
contains longer exons, and that the MSL complex rather binds with an exon
bias than a 3’ bias [147]. The H4K16 acetylation mediated by the MSL
complex on the X-chromosome is also said to be responsible for hyper-
activation by loosening up the chromatin structure and thus allowing easier
transcription initiation [167], or enhanced transcription elongation [168].
However, it is relevant to keep in mind that although the X-chromosome in
males is hyper-activated about two times, i.e. has an expression level in
range with that of the autosomes, the up-regulation mediated by the MSL
complex actually only counts for about a 1.4-fold hyper-activation [169-173].
This indicates that another compensating mechanism exists as a
complement to the MSL complex.
PAINTING OF FOURTH (POF)
For a long time, the concept of chromosome-specific gene regulation was
ascribed only to sex chromosomes and dosage compensation mechanisms;
however, with the discovery of a particular protein that seemed to only bind
to the small 4th chromosome in Drosophila, this view was challenged. This
protein was named Painting of Fourth, or POF [174]. POF binds to the
distal section of the fourth chromosome and is so far the only described
chromosome specific protein that binds to an autosome (see figure 3B). The
Pof gene is located in region 60E on chromosome 2R and it encodes a 495
amino acid long protein containing a predicted RNA-binding domain
(RRM1) in the central part of the protein [174].
Specifically, POF targets the gene body of active genes on the 4th
chromosome and has a strong preference for exons, with a tendency for a
stronger binding at the 3’ end [46,48,175], and it is responsible for
stimulating expression to similar levels as the MSL complex. A Pof mutant is
viable but has a significant general reduction of chromosome 4 gene
expression [48].
The binding of POF is quite specific for the 4th chromosome because partial
translocation of chromosome 4 onto the tip of other chromosomes cannot
recruit POF, nor can POF binding spread into regions of other autosomes
that are translocated onto the tip of the 4th chromosome [174].

INTRODUCTION
28
Chromosome 4
The 4th chromosome of Drosophila is unique and differs from the other
chromosomes in several ways; the most obvious difference is the size; the 4th
chromosome is very small, its total length is estimated to be 4.5-5.2 Mb or about 3.5% of the genome (see figure 3A) [176]. Secondly, even though it
contains about 92 genes, it is considered to be a mixture of both euchromatic
and highly heterochromatic regions. The entire 4th chromosome displays
heterochromatic features such as late replication [177] and no meiotic
recombination, and the proximal part of this chromosome, which also
constitutes the majority of the length (about 3-4 Mb), is a highly condensed,
completely heterochromatic structure. The proximal region is under-
replicated in polytene tissue (i.e. does not go through endoreplication and is
therefore not visible in polytene chromosome), mostly consists of simple
satellite repeats and does not contain any known genes [178].
The remaining 1.23 Mb of the 4th chromosome constitutes the polytenized
distal part, which roughly corresponds to the banded region visible in
polytene chromosomes (cytological sections 101E-102F). The 92 genes
associated with the 4th chromosome are all found within this banded section,
giving it a gene density close to that of the major chromosome arms.
However, this region also has several properties typical of heterochromatin:
the genes are interspersed with a mix of unique sequences, repetitive- and
transposable elements [179-182], and it is associated with the
heterochromatin inducing proteins HP1a, SETDB1 and the histone
modification H3K9me. It also has the capacity to induce variegated
repression on inserted reporter genes in a way that is typical of
heterochromatin position-effect variegation (PEV) [183].
So this gene-containing section of the chromosome is actually a mixture of
euchromatin and heterochromatin [180], and this means that the active
genes on the 4th chromosome have to be able to uphold expression within a,
in many aspects, repressive environment [184,185]. It is even so that the
ability of POF to bind chromosome 4 depends on this heterochromatic
background; increasing the compaction of heterochromatin, by decreasing
the temperature or removing the Y-chromosome, can results in binding of
POF to a translocated 4th chromosome [48]. This makes the 4th chromosome
a very interesting model for studying heterochromatin and its effects on gene
expression.

INTRODUCTION
29
Haplo-4th lethality and POF
One very interesting feature of the 4th chromosome is that it is the only
autosome in Drosophila that can exist in only one copy, without causing the
flies to die, i.e. it is haplo-viable [178]. But if you combine a haplo-4th
condition with a Pof mutation (which on its own does not cause lethality),
the flies apparently can no longer sustain sufficient chromosome 4
expression and die. Therefore, it seems as if POF is essential for maintaining
chromosome 4 gene expression in flies which are deficient in chromosome 4
dose [48].
Balanced regulation of chromosome 4 genes by POF and HP1a
Interestingly, the binding of the stimulating protein POF and the repressing
protein HP1a to the distal, euchromatic part of chromosome 4 genes overlap
almost perfectly and they both bind with a preference for exons of the active
genes [45,46,48]. Furthermore, the binding of the two proteins appears to be
interdependent on each other; if HP1a is absent, POF is released from the 4th
chromosome and if POF on the other hand is absent, the HP1a binding to the
4th chromosome is reduced; although, this reduction is less pronounced [48].
This was, however, contradicted by a recent study showing that POF binding
is essentially unaffected in an HP1a mutant [54], but either way, they still
bind to essentially the same genes, and the opposing effects on gene
transcription suggest a system in which the expression of chromosome 4
genes is fine-tuned by a balancing mechanism between HP1a and POF, in
which POF provides a stimulatory function and HP1a provides a repressing
function [48].
Importantly, there is an exception to the overlap between POF and HP1a
binding to the distal section of 4th chromosome, and that is the
uncharacteristic H3K9me-independent HP1a binding to the promoters,
which are not targeted by POF [45,46]. In line with the fact that POF only
correlates with H3K9me-dependent HP1a binding; POF is also suggested to
interact with SETDB1, and the H3K9 methylation pattern on chromosome 4
is impaired in a Pof mutant, suggesting that the proper function of SETDB1
depends on the presence of both HP1a and POF at the gene bodies [73].
Evolutionary links between POF and the MSL complex
The apparent requirement of POF to maintain sufficient chromosome 4 gene
transcription in a haplo-4th condition gives POF a whole-chromosome

INTRODUCTION
30
regulatory function, reminiscent of the dosage compensation system of the
male X-chromosome. The stimulating potential of POF and the MSL
complex are also comparable to each other. But there are also other
evolutionary aspects connecting POF and chromosome 4 to the MSL
complex and the X-chromosome: the binding of POF to chromosome 4 (also
known as the F element) is evolutionary conserved in several different
species of genus Drosophila (for example in D. virilis which diverged from
D. melanogaster about 39 million years ago [186]). And in some species, like
D. ananassae and D. malerkotliana, POF in fact decorates the entire male
X-chromosome and co-localizes perfectly with the MSL complex.
Furthermore, in D. busckii, where POF not only binds to the X-chromosome,
but where the entire F element is actually fused to the X-chromosome, there
is no apparent MSL complex (i.e. no complex that is detectable with a D.
melanogaster MSL-antibody), instead, POF is decorating the X-
chromosome in males, but not females, and this binding co-localizes with
H4K16 acetylation (which is characteristic for D. melanogaster dosage
compensation of the male X). This suggests that POF and the MSL complex
are evolutionary connected [187]. It is noteworthy that of the tested relatives
of D. melanogaster within genus Drosophila, D. busckii is also the most
distant relative that contained POF, and this suggests that POF might have
originated as an early form of dosage compensating mechanism for up-
regulating the male X-chromosome [46,48,172,174,187,188].
Moreover, in similarity with the X-chromosome, chromosome 4 seems to
have “female tendencies”, meaning that in intersexes with a 2X:3 autosome
ratio, an increase in chromosome 4 dose will lead to female development,
whereas a decrease in dose will lead to male development [189,190].
Furthermore, three copies of the 4th chromosome increase the frequency of
chromosome X nondisjunction, which indicates that chromosome 4
sometimes pairs with the X-chromosome during meiosis [191].
Do other compensating systems exist?
I have discussed the highly evolved mechanisms for compensation of sex
chromosomes that aim to equalize expressional output between the X-
chromosomes of XX:AA females and X:AA males, as well as the equalization
between the autosomes and the single X-chromosome ratio in mammals.
POF, and the up-regulation of chromosome, 4 presents the so far first
described autosome specific regulating protein, and this facts gives rise to
the perhaps not very far-fetched question: are there other similar

INTRODUCTION
31
mechanisms for compensating un-balanced autosomal regions in general?
Unbalance in genome copy number is generally referred to as aneuploidy,
and this is not at all an uncommon phenomenon in nature, so aneuploid
genomes can provide good models for investigating autosomal compensatory
mechanisms. I will first explain the concept of aneuploidy, and its general
effects and consequences:
ANEUPLOIDY
Aneuploidy and segmental aneuploidy refers to a state in which one or
more chromosomes or chromosome segments are present in abnormal copy
numbers. The normal chromosome copy number for many species, including
mammals and Drosophila, is two, meaning that all chromosomes (and
hence, all genes) are present in two essentially identical copies, one set is
inherited from the mother and one set is inherited from the father. The
exception is the sex chromosomes, which are unequally distributed between
the sexes. Aneuploidy is defined as loss or gain of one or more (but not all)
chromosomes, giving a chromosome number that is not an exact multiple of
the haploid number. This is slightly different from polyploidy, in which the
cell or organism carries three or more complete sets of chromosomes. This
phenomenon is frequently found in nature as the normal chromosome set up
of many plants and some animals, including a few specific human cell types.
Aneuploidy usually occurs during meiosis: Normally, when chromosomes
are being duplicated, the two copies of the chromosomes are equally
distributed to the two daughter cells, so that the total number of
chromosomes is maintained in the cell line. But if you have errors in the
chromosome segregation, chromosome copies can be lost or an extra copy
can be obtained (aneuploidy states which more specifically are termed
monosomy and trisomy, respectively). If the segregation problem is
combined with double strand breaks of the chromosome, it might lead to
segmental aneuploidy.
Cancer, developmental diseases and aneuploidy
Aneuploidy in humans in most cases lead to fetal abortion at early
developmental stages and is actually the leading cause of miscarriages in
humans [192,193]. It is predicted that over 10% of human oocytes are
aneuploid, although these oocytes rarely give rise to viable offspring [194].
However, in cases where the offspring is viable, the copy number defects
always give rise to developmental abnormalities and a reduction in fitness

INTRODUCTION
32
occurs in all species studied [193,195-200]. Most mental retardations in
humans are caused by aneuploidy [196,201], for example trisomy 21 (Down
syndrome) which is an extra copy of the relatively small chromosome 21 in
humans. As can be expected, the severity of the associated defects of
surviving individuals with aneuploidy can to some degree be correlated with
the length of the aneuploidy region, and in general, segmental trisomies are
better tolerated than segmental monosomies [195,197,200].
In addition to developmental abnormalities, aneuploidy is one of the
hallmarks of cancer, and tumors often contain extensive aneuploidy, with
gain or loss of multiple chromosome copies or segments of chromosomes
[201]. This is very contradictory with the nature of aneuploidy, since this in
normal cases is deleterious to a cell and to organisms as a whole. So the fact
that tumor cells survive these deleterious chromosomal rearrangements has
remained a mystery, but it also remains very complex and controversial
whether aneuploidy is a cause or a consequence of cancer [195,200]. Either
way, the aneuploid state, which would normally be deleterious, is clearly
accepted in tumor cells since these have gained proliferative advantages
compared to normal cells within their surroundings. This provides
interesting future questions to understand how or by which mechanisms
tumor cells are able to evade the deleterious effects of abnormal copy
numbers, and consequently the unbalanced levels of gene products.
Aneuploidy and evolution
One important aspect to consider in aneuploidy is that although an
aneuploidy state is usually deleterious to a cell, and in theory should be
selected against, variations in the number of chromosome or chromosomal
segment copies is important for genomic variance and hence in the long run,
evolution. With this in mind it is understandable that cells and organisms
need to be able to survive certain amount of copy number variation to
potentially obtain new traits, and will thus have evolved mechanisms for
this. These mechanisms need to be balanced between being efficient enough
so that the cells survive minor copy number variation, but without
compensating away the potential beneficial effects [172,199]. It turns out
that differences in copy number are relatively common in all individuals of
all species studied. About 500 individual regions or around in total 0.2% (6
Mb) of the genome may for example vary in copy number between two
normal human individuals [202,203], and recent studies have shown that up
to 13% may vary in copy number between two individuals [199], indicating
the importance of aneuploidy for creating genomic variation.

INTRODUCTION
33
Aneuploidy in Drosophila
The Drosophila genome has been divided into 102 numbered divisions based
on cytological analyses of the polytene chromosome [204], and each region
is about 800-1500 kb in length [205]. Induced monosomic regions (i.e one
copy number) longer than 800-1500kb, or more than 1% of the genome
reduces fertility and viability [197]. Although, there are a couple of
exceptions to this rule, where flies carrying a ~1.7 Mb or a ~2.8 Mb
monosomic region respectively, are still viable [206]. In general, Drosophila
are more sensitive to segmental monosomy than trisomy (in the whole
genome so far only one triplo-lethal allele and two alleles that give mutant
phenotype in triplo condition are described [197]). Still, only about 50 out of
all the approximately 15 000 genes in Drosophila are haplo-insufficient,
meaning that they are lethal when one of the copies (alleles) are mutated.
One large class of these haplo-lethal loci is the “Minute” genes. Monosomic
state of these loci results in flies with short, thin bristles, reduced
developmental rate, low viability and fertility and other abnormalities
[207,208]. This means that for the large majority of genes, one wildtype gene
copy is sufficient for proper function of the organism.
Are there mechanisms for aneuploidy compensation?
The various aspects of aneuploidy listed above hints that there should be
some mechanism involved, which can diminish the deleterious effects of
autosomal copy number variation and allow cells to survive minor
rearrangements. Also, that the MSL complex only accounts for about 1.4
times of the up-regulation of the two-fold hyper-acetylated male X-
chromosome [171] indicates the existence of another, more elusive
compensating mechanism.
Buffering
Indeed, several studies have shown that autosomal aneuploidy regions are
actually affected by an elusive mechanism which compensates the expression
level of these regions, both up or down, and this autosomal compensation
has been named buffering (see figure 5).
When considering aneuploidy, i.e. variations in the dose of a genomic region
and the effects on expression level, you would intuitively expect that if a
genomic region was present in either one copy (monosomy) or three copies
(trisomy), the transcriptional output from this region would drop to 0.5 or

INTRODUCTION
34
increase to 1.5 respectively, compared to a normal two copy region where the
transcriptional output would be 1. However, it has been shown in a number
of early studies in segmental trisomies, that duplicated genes had a lower
expression output than expected, indicating that something is reducing, or
buffering, the expression of the three autosomal gene copies, and this was
measured both on protein level [209-211] and RNA level [209,212]. Studies
using more genome-wide approaches, such as microarray, supported these
findings and showed that buffering appears to exist also on a wider scale
[105,213-216]. For example one of these studies performed in humans found
that triploid regions were expressed at a mere 1.1-fold, compared to the
expected 1.5-fold, indicated that the triploid regions are repressed to obtain
an transcriptional output level more similar to that of a wildtype [215]. Two
studies also showed that a three-fold gene dose difference (i.e. when
comparing the expression from three copies of an autosomal region to that of
one copy number of the same region) only results in a 1.4-fold change in
mRNA levels [105,216]. Thus, it is evident that autosomal compensation, i.e.
buffering exists, albeit elusive.
Buffering could explain why the male X-chromosome has a two-fold up-
regulation even though the MSL complex only accounts for a 1.35-fold up-
regulation [171]. It was speculated by [105,108,171,172] that if we assume
that buffering stimulates monosomic regions to an average of an 1.5-fold up-
regulation and that it can act on all chromosomes with a deviating copy
number, including the male X-chromosome, then buffering together with the
effects of the MSL complex can count for the two-fold hyper-activation of the
male X-chromosome, 1.50-fold x 1.35-fold=2.03-fold!
Even if this particular effect of buffering of the X-chromosome can be a bit
hard to measure exclusively, the conclusion is that autosomal buffering does
exist to allow cells and organisms to survive milder forms of chromosome
imbalance, and it appears to act on the transcript level. However, the
buffering effects do not fully restore transcript levels back to a wildtype
(diploid) level, and it is therefore possible that buffering occurs on more than
one level. A study in a number of yeast strains, carrying extra chromosome
copy numbers, showed that mutations leading to increased proteasomal
degradation had evolved to increase the fitness of several trisomic yeast
strains [217]. This suggests that buffering is something that also acts on a
protein level, and thus it is obvious that many aspects about buffering
remains to be explained.

INTRODUCTION
35
One important question is how this buffering mechanism works; and also
what causes the haplo-insufficiency once the aneuploidy region becomes too
extended: either it could be a general mechanism that recognizes and targets
monosomic/duplicated regions and stimulates or inhibits gene expression
more or less evenly over these regions, and that the cause of haplo-
insufficiency is depleted levels of the buffering components. This model is
supported by the fact that when the size of an aneuploidy region reaches a
Figure 5. Schematic illustration of buffering. A normal disomic region in two copies
(one copy inherited from the mother and one copy inherited from the father) compared to a
monosomic region (i.e. a region that has lost one copy) with and without buffering, and a
trisomic region (i.e. a region with one extra copy) with buffering. In the absence of any
buffering, the expected transcriptional output of the monosomic region is 0.5 of that of the
disomic region. But if the monosomic region is buffered, an unknown mechanism induces a
higher expression of the remaining single copy genes so that the total amount of
transcriptional output is closer to that of the normal disomic level. In a trisomic region,
buffering works in the opposite way, repressing gene expression to reduce the amount of
transcriptional output.

INTRODUCTION
36
critical point, it is primarily the extent of the aneuploidy that causes the
lethality and not the uncovering of specific genes or regions [197]. The other
option is that buffering is the sum of regulation of individual genes, which
would mean that most genes within a deficient region are unaffected
whereas a few genes become fully compensated by feedback regulation. The
haplo-insufficiency would then primarily be caused by collapsing gene
networks. So as long as a deficiency is small enough, the probability is that
the affected genes are functionally unrelated to each other, and the
remaining components of the protein network can balance for the lack of one
network component. As the deficient region expands, you will eventually
reach a point in which more than one gene involved in the same protein
network is affected, and consequently, the other components of the network
can no longer cover up the losses, resulting in lethality [218].
It addition to the putative buffering mechanism or mechanisms, other less
general mechanisms could also be involved in gene dose regulation, such as
feedback regulation and feedforward regulation.
Feedback regulation
Feedback regulation is a mechanism for controlling gene expression by the
level of gene product present. If levels of a protein or RNA are too low, gene
product is continuously produced until sufficient levels are reached, and the
newly synthesized proteins will inhibit the formation of more proteins. This
makes feedback regulation a well-established error control mechanism
where the protein itself regulates the amount of new protein produced.
Feedforward regulation
Feedforward mechanism on the other hand functions by anticipating
possible effects of system errors rather than acting once the levels are out of
balance. For example the cells can detect copy number variation and adjust
transcription levels prior to major errors occur.
One specific example is the sex determination in male Drosophila embryos:
the cells detect the X copy number in relation to autosomal copy number and
assigns the MSL complex to the X before zygotic transcription is activated in
a feed-forward manner [219]. Although the MSL actually also displays
feedback properties by binding to transcribed genes [152,163,220,221].

INTRODUCTION
37
Inverse dosage effect
An alternative view on aneuploidy compensation is that since most gene
regulatory enzymes are repressing, loss of a chromosomal segment would
lead to a greater loss of negative regulators than of positive regulators, and
consequently the remaining genome will increase in transcription, rather
than the monosomic region being decreased in expression. According to this
model, the monosomic state of the X-chromosome in males causes a general
up-regulation of the entire genome and the MSL complex acts by attracting
and isolating the activating factor MOF to the X-chromosome, and hence
passively causing an up-regulating of the male X-chromosome [222]. This
model is however contradicted by the finding that an msl2 mutant causes a
reduction of chromosome X expression, but with very little effect on the
autosomes [169,223]. Yet, the true reasons behind these differences in X-
chromosome and autosomal expression levels are very hard to determine
and this issue presents one of the most problematic aspects of measuring
genome-wide effects of gene expression:
Challenges with genome-wide expression analysis
Reference points
In studies involving gene expression effects of chromatin-binding proteins,
chromosome copy number variation, or other conditions in general that
affect large parts of the genome, one key concern for accurate estimations is
to set valid reference points, and this is a general problem in these kinds of
studies. If you measure effects of a few genes, which most likely will have
very minor effects on the remaining genome, the whole genome or a few
reference genes can be used as the standard to which the effects of your
genes of interest can be compared to. But if you start applying severe
conditions to the cells, or study aneuploid regions that are big enough to
affect the rest of the genome, it becomes very difficult to set a reference
point, to what should you compare the effects that you are interested in?
Skewness
Another related issue is skewness since it is evident that different data set
will always be affected by both biological and technical variation. Two
replicates that are generated by genome-wide expression arrays (such as
next generation sequencing or microarrays) can never be directly compared
as raw data, instead they need to be normalized against each-other so that

INTRODUCTION
38
they obtain in principle the same array average value, and the same variance.
If gene expression of a large part of the genome is affected, for example by a
down-regulation of the X-chromosome, the normalization process will by
default try to equalize the overall average expression level of the whole data
set, and this will mean that the down-regulation of one large region will be
compensated by increasing the expression value of the remaining genes, thus
creating a bias [224]. This will have the result that it is very hard to
determine whether a change between the X and the autosomes is caused by
just a down-regulation of X or also by an up-regulation of the autosomes.
Thus, the challenge with genome-wide studies is keeping a balance between
measuring the effects of large genome rearrangements and still avoiding
biased data sets.
This is one of the reasons why the small 4th chromosome is such a good
model for studying chromosome-wide gene regulation; it has its own unique
regulatory mechanism (POF) and contains enough genes (92) to measure
significant effects, but it is still small enough not to affect the remaining
genome, and thus it is easy to set reference points. The same is true for
segmental aneuploid regions (such as those used in paper I and II in this
thesis). They are in approximately the same size range as chromosome 4, and
will thus be very useful in examining effects on copy number variation and
the potential autosomal buffering effect.
Limitations in the arrays
An additional problem with several genome-wide methods, especially
microarrays, is that there are limitations in the detection range. The
expression levels of genes that are expressed at very low levels (i.e. un-
expressed) will inevitably drown in background noise of the microarrays,
meaning that any differences in transcriptional output between genes in a
monosmic or trisomic state vs. a diploid (wildtype) state will not be detected,
and these genes will therefore appear as if they are fully compensated. This is
a general problem with many of the previous estimations of buffering level,
which have usually included all genes, both expressed and unexpressed.
Therefore, these studies may tend to overestimate the buffering effect, and
this is a problem we have addressed in paper I and II.

Aims
39
AIMS
The general aims of this thesis have been to:
Investigate the buffering effect of segmental monosomic regions and
find out if there are general responses in the genome that are
triggered by aneuploidy (Paper I and II).
Determine whether buffering is a general mechanism or a gene
specific mechanism, and potentially find features of genes and
regions which affect the level of buffering (Paper I and II).
Elucidate the conflicting reports about HP1as function on gene
expression; inhibiting or stimulating? (Paper III).
Determine which roles the two different HKMTs, SETDB1 and
Su(var)3-9 are playing on gene expression in different parts of the
genome (Paper III).
Use POF binding sites located on the female X-chromosome to
determine the relationship between POF and dosage compensation,
and furthermore, determine how POF binding is influenced by
chromatin environment (Paper IV).

RESULTS AND DISCUSSION
40
RESULTS AND DISCUSSION
PAPER I AND II
General buffering levels
In segmental monosomic regions, we found that the transcription output
was indeed enhanced in relation to the gene dose, i.e. buffered. The average
expression varied depending on region between 54%-58% of wildtype
expression in paper II, and up to 64% of wildtype expression level in paper I.
If no buffering effect existed, we would expect these regions to be expressed
at 50% of wildtype expression, corresponding to the 0.5 gene dose. Our
measured buffering effect is in general lower than previous studies have
shown [105,213,215], however, we postulate that this is caused by the more
stringent cutoff we applied, excluding all genes expressed below or above the
reliable detection range of the array. The excluded genes are in the risk of
being scored as fully compensated if the array, due to background noise or
over-saturation, is unable to detect differences in gene expression between
the aneuploid and the control samples. We conclude that buffering of
monosomic regions is a weak but significant effect, which acts to stimulate
expression of the remaining single copy genes to compensate for the reduced
gene dose. Furthermore, we find that the average buffering of entire
monosomic regions is weakly related to the number of genes that are in
single copy, the more genes affected, the lower the buffering is. Yet, buffering
is not clearly decreased if another monosomic region is added in the same
genome, nor could any other clear effects be seen when combining
monosomic regions.
Buffering of specific gene groups
On average, when studying compensation of whole regions, our estimated
buffering effect is small but significant, however, when we start looking at
specific groups of genes, the buffering effect becomes evident. We found that
buffering depends on expression pattern, wildtype expression level, and gene
length:
UEGs and NUEGs
We found that in monosomic regions, Non-Ubiquitously Expressed
Genes (NUEGs) (i.e. genes which are only expressed in some, but not all

Results and discussion
41
tissue types of the body) are more strongly buffered than Ubiquitously
Expressed Genes (UEGs) (i.e. genes which are expressed in all tissue
types). UEGs are likely important for maintaining general housekeeping
functions and need to be expressed at relatively high levels in all tissue types,
whereas NUEGs on the other hand are required for more tissue specific
functions in general, and are consequently only active in some tissue. We
therefore speculate that more regulatory mechanisms have evolved to fine
tune expression of NUEGs than UEGs. UEGs are already expressed close to
the maximum levels, and therefore, it is hard to further stimulate these
genes, whereas NUEGs still have the potential to have their transcription
enhanced in most tissues. One could even speculate whether UEGs are
buffered at all; it is possible that the weak effects measured for UEGs are
mainly due to misclassified NUEGs, which is likely to occur since
classifications such as the one used will never be perfect.
Gene length and wildtype expression level affects buffering
When looking at the buffering of individual genes, the strongest determinant
for buffering we found was gene length: long genes (>3 kb) display the
strongest buffering, and for these genes, the buffering effect is also
independent on wildtype expression level or expression pattern (i.e. NUEG
or UEG). Short genes (<3 kb) are less buffered on average, but interestingly,
these genes in contrast actually depend on both expression pattern and
expression level, with a low expression level resulting in nearly the same
buffering as long genes. The short genes with highest expression level are not
buffered at all, and virtually the same is true for short UEGs. Thus, gene
length appears to be the primary determinant for buffering (see figure 6).
Buffering mechanisms
One very interesting and important question concerning buffering is: what is
the underlying mechanism behind buffering? Is it the sum of feedback
regulation of a few individual genes, or in the form of protein network
effects? Or is it a more general mechanism that recognizes and acts on
regions present in only one copy?
If the buffering was caused by feedback regulation, we would most likely see
a strong buffering effect only in a few monosomic genes, whereas the
majority of genes are expressed at around the expected 50% of wildtype. In
contrast, we observe a normal distribution of the buffering, on average most
monosomic genes are up-regulated and the variation is likely caused by

RESULTS AND DISCUSSION
42
normal variance and array noise. This indicates that buffering is mediated by
a more general mechanism, although, there is also a possibility that the
normal distribution of the buffering is caused by feedback regulation of
specific genes and that the buffering effect spreads, since spreading is a well-
known mechanism in, e.g., dosage compensation [130,145,152,225].
However, we found that buffering does not spread to neither neighboring
genes within the monosomic regions, nor to neighboring diploid regions,
which argues against a spreading mechanism.
The finding that gene length is the primary determinant for buffering makes
it tempting to speculate that transcriptional elongation is involved in this
mechanism. Transcriptional elongation has been proposed as mechanism for
the MSL complex [168,226], and considering the hypothesis that total
compensation of the male X-chromosome is a combined effect of the MSL
complex and a more elusive general mechanism, such as the autosomal
buffering, makes this even more tempting. However, it remains to be
clarified whether transcription elongation is really a part of the buffering
mechanism. Buffering could also be mediated, or partially mediated, by
looping out effects, since the monosomic regions are unpaired, they might
loop-out from the normal nuclear position into more permissive
environments, or by increasing access of transcriptional machinery.
POF compensates chromosome 4
Buffering could potentially also be mediated by a specific factor, similar to
the chromosome 4 specific protein POF. Chromosome 4 is the only autosome
which is haplo-viable, meaning that flies can survive with only one copy of
chromosome 4 [178]. In paper I, we found that a haplo-4th chromosome is
buffered to 70% of wildtype expression, which is a more pronounced effect
than on the other autosomes. Since we know that haplo-4th flies dies in the
absence of POF [48], we speculate that POF is involved in the buffering of
chromosome 4 genes. In paper I, we further support this hypothesis by
showing that there is a negative correlation between gene expression in a
haplo-4th condition and a Pof mutant. Genes that are most tolerant to loss of
one copy of the 4th chromosome are also most down-regulated in a Pof
mutant, and reciprocally, the genes that are not compensated by POF are
also the genes which are most sensitive to loss of one copy of chromosome 4.
In general, it appeared as if the genes compensated by POF were
overrepresented by NUEGs. We conclude that POF is the mechanism that
compensates chromosome 4 genes and that it preferentially stimulates
expression of NUEGs.

Results and discussion
43
Buffering induces proteolysis
We were interested to find out if any general effects were induced elsewhere
in the genome (i.e. in diploid genes not uncovered by any copy number loss),
as a general response to deletions, and we discovered that genes involved in
peptidase and proteolytic activity where highly over-represented among the
100 most up-regulated genes in the genome. Furthermore, we found a
negative correlation between buffering level and expression of proteolytic
genes: the poorer buffering a monosomic region displayed, the more
expressed the proteolytic genes were. The correlation was stronger for short
genes, in line with the previous finding that buffering is lower for short
genes. Proteolytic proteins are involved in the degradation and digestion of a
vast number of proteins, and are responsible for degrading misfolded
proteins, thus giving them an important regulatory function. Other studies
performed on duplications, i.e. genome containing extra copy number, have
also shown induced proteotoxic stress [217,227,228]. It is possible that
proteolysis is a general response to unbalanced genomes and is induced to
compensate unbalanced genomes on a protein level, when the buffering on
transcription level has not been sufficient.
Future perspectives
The studies of buffering included in this thesis are all conducted on fly
strains in which the deletion of a genomic region was induced long ago,
meaning that many generations have passed since the induction, and this
inevitably includes the possibility that the effects we observed have occurred
over long time as an adaptation to these impairments. Therefore, one
relevant question to be addressed is: would the same effects be obtained if
these deletion where re-induced, and the effects monitored directly upon
induction? What would happen if the buffering effects where followed over
several generations?

RESULTS AND DISCUSSION
44
Conclusions
Buffering of monosomic regions is a weak but significant effect,
ranging between about 54-64% of wildtype expression level.
Buffering of entire regions partially depends on the number of genes
included in the monosomic region, where regions with many genes
are less buffered.
Buffering of one monosomic region does not affect the buffering of
another monosomic region, if they are combined in one genome.
Gene length is the primary determinant for buffering. Short genes
depend on expression patterns and expression level for proper
buffering, and short UEGs and short genes with a high expression
level are not buffered at all (see figure 6).
Buffering effect is normally distributed, indicating a general
mechanism rather than feedback regulation of individual genes, and
buffering does not display any spreading to nearby genes.
POF is responsible for the compensation of haplo-sensitive genes on
chromosome 4, and is also crucial for the survival of haplo-4th flies.
Aneuploidy in general induces expression of proteolytic genes, and
this effect is more pronounced in genomes with poor buffering.
Figure 6. Schematic illustration of genes in a monosomic region. Buffering is
strongest for long genes, irrespective of gene type, whereas buffering of short genes
depends on gene type. Short UEGs are weakly or not at all buffered.

Results and discussion
45
PAPER III
HP1a has opposing functions on chromosome 4 and in
pericentromeric regions
It has been debated whether HP1a has a repressive or a stimulating function
on gene expression, and to better understand the function of HP1a, as well as
that of the co-dependent proteins POF, SETDB1 and Su(var)3-9, we analyzed
mutants of each of these genes using microarray expression analysis. The
most striking finding was that HP1a inhibits gene expression on the 4th
chromosome and stimulates gene expression in the pericentromeric regions
(the heterochromatic regions adjacent to the highly heterochromatinized
centromeres). To add even extra complexity to this matter, we discovered
that these effects where different depending on gene type: An HP1a mutant
was primarily affecting NUEGs in chromosome 4, whereas in the
pericentromeric regions, only UEGs were significantly affected. These two
regions are very different in features and genomic environment which
indicates that HP1a have evolved to adapt to different chromatin
environments. This could also explain the contradictory reports about the
function of HP1a.
HP1a has different functions at the promoter and at the gene body
We know that HP1a binds to active genes on chromosome 4, and that it has a
stronger binding peak to the promoters than to the gene body of many of the
genes. When re-analyzing this HP1a binding data [45], we found that the
binding is elevated to both NUEGs and UEGs of chromosome 4 and the
pericentromeric regions, compared to a control region. Interestingly, we
found that there is a difference between the gene types; HP1a binds much
stronger to the promoters of UEGs than the promoters of NUEGs, and
furthermore, there was also a region specific difference; HP1a targets the
gene bodies of chromosome 4 UEGs stronger than the gene bodies of
pericentromeric UEGs. This is very intriguing considering that HP1a binding
to gene bodies of chromosome 4 is dependent on H3K9me2 and me3, (and
thus indirectly dependent on SETDB1) whereas the binding to promoters is
independent on H3K9me2 and me3. It has been suggested that HP1a
binding to the promoters results in an open chromatin conformation which
stimulates gene expression, whereas HP1a binding to gene bodies has a
repressive function. The H3K9me-independent binding to the promoter is
likely also more stable and less transient than the binding to H3K9
methylation at the gene body [45,229].

RESULTS AND DISCUSSION
46
We therefore propose a model in which HP1a has adopted its function to
different genomic environments and that this is mediated through different
binding mechanism (see figure 7): in UEGs, HP1a preferentially binds to the
promoters and stimulates expression, maybe by loosening up the chromatin
structure. In the pericentromeric regions, the strongest HP1a binding is
found at the promoters of UEGs and this would also explain both the average
stimulating effects of HP1a in these regions, and the fact that only UEGs are
significantly affected in an HP1a mutant. On chromosome 4, HP1a binding is
also enriched at the gene bodies, and this repressive binding balances the
stimulating promoter binding of UEGs, causing these genes to be less
affected in an HP1a mutant. The NUEGs on the other hand, which lack the
promoter peak, are dominated by the repressive effect of HP1a at the gene
bodies, and are thus up-regulated in an HP1a mutant. Interestingly, we show
in paper I that POF preferentially stimulate gene expression of NUEGs,
indicating that POF might have the role of balancing the repressive influence
of HP1a in those genes that lacks HP1a at the promoters. The hypothesis that
the repressive effect of HP1a correlates with H3K9me is also supported by
our finding that HP1a and Su(var)3-9 to a high extent appear to repress
expression of transposons in an overlapping manner.
SETDB1 and Su(var)3-9 are complementary to each other
SETDB1 mediates the H3K9me2 and me3 on chromosome 4, which repress
chromosome 4 gene expression, and in paper III we found that SETDB1 co-
localizes with the POF and HP1a binding regions on chromosome 4, and that
Setdb1 and HP1a mutants cause approximately the same up-regulation of
chromosome 4 gene expression, with a preference for NUEGs. This supports
the fact that SETDB1 is essential for the HP1a binding to gene bodies on
chromosome 4. The Su(var)3-9 mutant background resulted in down-
regulation of genes in the pericentromeric regions, in a similar manner as
the HP1a mutant. This is surprising since the function of Su(var)3-9 is
supposedly repressive in these region. However, the fact that HP1a
preferentially binds to the H3K9me-independent gene promoters in these
regions [45] indicates that Su(var)3-9 could have another role in the
pericentromeric regions than only preceding HP1a binding to gene bodies.
Su(var)3-9 is known to interact with the chromo-shadow domain of HP1a,
and maybe this is the primary binding mechanism of Su(var)3-9 in these
particular regions.
Furthermore, SETDB1 and Su(var)3-9 are known to be complementary,
mediating methylation in essentially different regions. However we found, a

Results and discussion
47
bit surprising, that a majority of the genes that are differentially up-or down-
regulated in Su(var)3-9 mutants are correspondingly up- or down-regulated
in a Setdb1 mutant, with an overrepresentation of down-regulated genes.
This indicates that SETDB1 and Su(var)3-9 have more redundant functions,
and they also appear to be more involved in active gene expression than
previously thought. Although, we cannot exclude the possibility that these
observations could be caused by indirect effects, for example by re-direction
of HP1a to normally unbound regions, it could also be that even if different
regions are primary targeted, the same genetic networks will in the end be
affected by the mutants.
In addition, we observed that Su(var)3-9 to our surprise was highly enriched
on chromosome 4, where it has no known function, and when looking at
expression level, we saw that Su(var)3-9 mutants have reduced repression of
chromosome 4 genes. We also noted that mutations in HP1a, Setdb1 or
Su(var)3-9 all individually lead to up-regulation of essentially the same
genes on chromosome 4, meaning that both SETDB1 and Su(var)3-9 appear
to inhibit expression of chromosome 4 genes in an overlapping manner. This
indicates that Su(var)3-9 either has a yet unknown repressive function on
chromosome 4 genes, or this up-regulation is indirectly caused by
redistribution of SETDB1 from the 4th chromosome to compensate for the
absence of HKMTs function in the pericentromeric regions.
HP1a displays a stronger repression of long genes
The repressive effects mediated by HP1a correlate with gene length in a
whole genome comparison. The longer the gene, the more up-regulated it is
in an HP1a mutant, whereas the average HP1a binding per length unit is
unaffected by gene length. This implies that HP1a binds along the gene body
with the same density irrespective of gene length, potentially acting as
“speed-bumps” that slows down the rate of the RNA polymerase II during
the elongation phase of gene transcription. This probably means that the de-
repression caused in an HP1a mutant is stronger for long genes simply
because these genes in total lose more HP1a molecules from the gene body.
HP1a effect in the pericentromeric regions depends on location
We found that the position of the genes can affect level of regulation by
HP1a: in the pericentromeric regions of chromosome arm 2L and 3L, HP1a
binds stronger to genes situated closer to the centromere than to genes
located more distally within the pericentromeric regions. The de-repressing

RESULTS AND DISCUSSION
48
effect in an HP1a mutant in these regions is also stronger the more
proximally located a gene is.
Concluding remarks
It appears that a balance between the amount of euchromatin and
heterochromatin exists in the genome, in which larger heterochromatic
regions have the role of titrating the amount of heterochromatin associated
proteins. If our hypothesis that HP1a has different functions depending on
binding mechanism is correct, it would mean an even more refined mean of
balancing the amount of heterochromatin in a genome, and perhaps also
provide a potentially fast switching mechanism between the two states.
Figure 7. Schematic illustration of the two different types of HP1a binding within
a gene. HP1a binding in the promoter is more stable and leads to activation of gene
expression, potentially by binding in nucleosome core and de-condensing the chromatin
structure. HP1a binding to exons of the gene body depends on H3K9me marks on the histone
tails and represses gene expression.

Results and discussion
49
Conclusions
HP1a represses expression of chromosome 4 active genes, with a
preference for NUEGs; and stimulate expression of pericentromeric
genes, with a slight preference for UEGs.
HP1a binds more predominantly to the promoters of UEGs than the
promoters of NUEGs in general, and on chromosome 4, HP1a
binding is also enhanced at the gene bodies.
HP1a and Su(var)3-9 repress transposon-derived transcripts.
Long genes are more repressed by HP1a, probably because HP1a
binds to gene bodies with the same density, irrespective of gene
length.
Within the pericentromeric regions of chromosome arms 2L and 3L,
HP1a binds to and stimulates expression stronger for genes situated
closer to the heterochromatic centromere.
We propose that HP1a, to better adapt to local chromatin
environment, has evolved opposite functions on gene expression,
and that this is mediated by different binding mechanisms. Either
via a more stable H3K9me-independent binding in the nucleosome
core of promoters which stimulates expression, or a more transient
binding to H3K9 methylation marks at the gene body, which inhibits
gene expression. This gene body binding depends on the presence of
HKMTs (SETDB1 or Su(var)3-9) and on chromosome 4 and it also
depends on the stimulating protein POF.

RESULTS AND DISCUSSION
50
PAPER IV
POF targets roX proximal sites
We made the exciting observation that POF, which displays chromosome 4
specific gene regulatory properties in both sexes, sometimes targets two
specific sites on chromosome X in females, but not in males. These loci were
named PoX1 and PoX2 (POF-on-X) and they contain in total at least seven
genes. The PoX1 site is primarily located in the 3’ end of the Mnt gene and in
a novel non-coding gene located just downstream of Mnt. The PoX2 site
involves the five genes Ck2β, Hsc70-3, CG1578, SelG and CG1840. These
non-4th specific binding sites provide a unique opportunity to study targeting
of POF specifically, but also targeting in general, since the specific locations
of these sites on the X-chromosome provides another very interesting aspect:
both PoX1 and PoX2 are located approximately 200 kb downstream of roX1
and of roX2, respectively. roX1 and roX2 are the two ncRNAs that are part of
the male MSL complex, and also function as high affinity sites for MSL
targeting. The relatively closeness between the roX and the PoX sites thus
provides an additional unique link between these two evolutionary
connected chromosome specific regulatory proteins.
Connection with the MSL complex
We find that POF binding to the PoX sites depends on, but does not require
roX transcription, since a deletion in one or both roX genes results in
decreased, but not entirely absent, PoX-binding. An overexpression of roX2
results in higher PoX targeting frequency. To find out if there are any other
connections between POF binding and the MSL complex binding to these
sites, we compared the POF binding profile in female to a MOF binding
profile in male and find that the MOF binding to the PoX sites is random,
and that no clear connection between POF and MSL complex binding exist
that differs these sites from other genes on the X-chromosome. We could
also not see that these sites differ in expression levels between males and
females, thus the female specific binding to these sites remains a mystery.
Another puzzling aspect is that, although roX transcription appears to
stabilize POF binding to the PoX sites, roX expression in wildtype females is
actually very low or absent, roX1 transcription is only detected in early
embryogenesis and then disappears [230].

Results and discussion
51
Parts of PoX2 functions as POF high affinity target
We used different transgenes or duplications of the PoX genes to investigate
the dependence of local chromatin environment for POF binding, and found
that ectopic autosomal localization of duplications containing either PoX1 or
two of the genes in the PoX2 site, SelG and CG1840, was sufficient to recruit
POF to the site of insertion. In addition, a 6 kb transgene covering SelG and
CG1840 was sufficient to recruit POF targeting.
The binding to the duplicated regions depended on roX transcription in a
similar way as the endogenous sites, whereas the binding to the 6 kb SelG
CG1840 transgene appeared less dependent on roX transcription levels, and
in contrast to the endogenous sites, POF targeted this transgene in both
males and females. We conclude that this 6 kb transgenic region of PoX2
works as a unique POF high-affinity recruitment site, which is independent
on genomic environment. This is a very important discovery, because up to
date, POF binding has been known to be chromosome 4 specific and to
depend on the chromatin environment surrounding the 4th chromosome;
POF cannot bind translocated chromosome 4 regions, nor can POF spread
into segments of other chromosomes translocated onto the tip of
chromosome 4. This is the first described high-affinity site for POF targeting,
and therefore provide a unique opportunity to study the mechanisms behind
POF targeting.
HP1a correlates with POF in the PoX sites
Similar to the situation on the 4th chromosome, HP1a binding co-localizes
with POF also at the PoX sites. In nuclei were no POF was detected at the
PoX sites, no HP1a binding was seen either, indicating that the binding of
HP1a to these sites depends on the presence of POF. Taken together, these
connections between the heterochromatin dependent protein POF, the MSL
complex, and also HP1a, support a model that links dosage compensation
with heterochromatin. The fact that HP1a has been proposed to bind to male
X-chromosome [231], and that in absence of the activating H3S10 kinase
JIL-1 (highly enriched on the male X-chromosome and potentially loosely
attached to the MSL-complex [232,233]), heterochromatic marks such as
H3K9me2 and HP1a are spreading on chromosome arms and most strongly
on chromosome X [234], supports the connection between dosage
compensation and heterochromatin.

RESULTS AND DISCUSSION
52
Conclusions
POF targets the two roX proximal sites, PoX1 and PoX2 on
chromosome X in females, which includes the genes Mnt, Ck2β,
Hsc70-3, CG1578, SelG, CG1840 and the novel, non-coding RNA
RE6469.
Gene expression is not a determinant for the female specific
targeting of POF to PoX1 and PoX2.
POF and HP1a co-localize on the PoX sites.
POF targeting to PoX sites is stabilized by roX activity.
POF targets ectopic PoX1 and partial PoX2 sites inserted on
chromosome 3L in both males and females.

Results and discussion
53
FINAL CONCLUDING REMARKS
Clearly, gene type and chromatin environment have great influence when it
comes to epigenetic regulation, since it appears as the number one
heterochromatin protein, HP1a, depending on context even can adopt
completely opposite functions. HP1a’s functions range from repressing
repetitive regions and potentially deleterious transposable elements, to
upholding expression (maybe by loosening up the chromatin structure at the
promoters) of the ubiquitously expressed UEGs. These are genes important
to maintain housekeeping functions in all cell types, and thus it should be
beneficial with a low energy consuming mechanism which upholds a
constitutive expression of these genes, maybe by a constantly open
chromatin structure. This could be especially important if the genes are
located within a repressive heterochromatic environment, such as the
pericentromeric regions and chromosome 4. HP1a even appears to be able to
alternate between the two opposing functions within the same gene, by
balancing the expression of NUEGs. These are differentially expressed genes
which might require a more fine-tuned expression to maintain correct
expression level in correct tissue.
This can probably also give some clues to the varying buffering capacity we
detected for NUEGs and UEGs, in cases of copy number variation. If, as
indicated by the action of HP1a, NUEGs highly depend on repressive
proteins at the gene body to correctly balance the expression, they should
also be flexible in case an up-regulation is required. UEGs in contrast, whose
actions are more or less constantly required for the maintenance of all cells,
cannot respond as efficiently as NUEGs to copy number reduction. So in
addition to regulating the expression of individual genes, it is also important
for a cell with more general regulatory systems which allows for fast and
simple means of regulating genes with similar functions, i.e. mechanisms
that can enhance or reduce gene expression as a response to different
cellular processes. These types of mechanisms are probably important for
some genes, whereas for other genes (UEGs) it is more essential to maintain
a stable and often high expression level.

ACKNOWLEDGEMENTS
54
ACKNOWLEDGEMENTS
I can honestly say that my years at Molbiol have been the best years of my
life so far! I have learned a lot about science and a lot about myself, and I had
much fun along the way. Mostly thanks to all smart, funny and inspirational
people I have met and come to know over the years, and especially thanks to
the one who personifies all these things, my supervisor Jan Larsson. I feel
truly privileged to have had you as my supervisor! You possess great wisdom,
about science but also about how to shed your knowledge, and simply about
life in general, and I will always be grateful that you saw enough potential in
me to take me on as a student and assigning me these fun projects, and for
supporting and encouraging me throughout these years, even on Monday
mornings . Other people who greatly helped in the formation of this thesis
are Per Stenberg, my almost co-supervisor, if not on paper at least in
practice. Thanks for all ideas and help with interpreting endless numbers of
diagrams and plots, all serious and all less serious discussions (which often
comes in a wonderful mix!), it’s never boring when you are around . And
thanks for introducing me to world of gourmet food (even though I will never
give up on the cheese doodles!). Karin Ekström, the iron lady of the flies,
for all the fly related work, tip on good fly pushing and all fun conversations
in the fika room, and for spreading a really good atmosphere around! I hope
to have your positive attitude, party spirit as well as physique when I retire!
Members of group Jan Larsson: Anna-Mia, “kilen” that holds up the group,
my years in the lab would not have been same without you! Thanks for
always spreading positivism and cheerfulness around you, no matter what,
you have a fantastic ability of making problems and setbacks suddenly
appear easy and trivial! And thanks for all fun conference experiences, late
nights on the dance floors (and tables) etc! Margarida, my partner in
science and in crime! You have brought spice to the group! What have we not
been through together? Conferences, publications, avalanches, awkward
saunas etc etc. Thanks for all our discussions, cooperation, and various
adventures in höga kusten and kittelfjäll, and of course, all beers and movies!
And above all, thanks for being a really good friend! Masha, it so nice that
you joined our group! Thanks for bringing a lot more knowledge and
wisdom, and for boosting dormant projects, Marie-Line, the next-
generation PhD, it’s fun that you have started and I am looking forward to
our up-coming conference trip!
Past members and students of the group: Anders, you are really missed in
our office, your knowledge about basically everything, your sense of humor

ACKNOWLEDGEMENTS
55
and your “bugg” enthusiasm, hope we sometime will get another opportunity
to dazzle the dancefloor! Malin, who was kind enough to leave both her
desk, lab bench and apartment to me when I started in this lab, it’s sad that I
only got to know you for such a short time before you left, it would have been
fun to work more with you, I think we would have gotten along well! Linnéa
and Rafael, for help with the HP1a expression project and the PoX project,
and Denise, really hope to see more of you in the future!
Grp PS: Philge, for your great knowledge in bioinformatics, your useful
scripts and for being kind enough to take the time to explain some of these
things to me as well , the rest of Grp PS: Somyadeep, Aman, Daniel
and Grp YS: Yuri, Tatiana, Sarina, Mischa, Isaac for discussions and
questions on group meetings and/or journal clubs.
The old genetics crew: Anssi, for your kindness and your never-ending
knowledge about genetics. Stefan E, thanks for teaching cooperation and all
the fun “befrielsefester”, but what do you think will become of my defense
party without our no.1 bordercollie? Anna B, thanks for organizing a great
genetics C-course, which really boosted my interest for genetics! And thanks
for movie making experience and “befrielsefester”. Bettan, for wanting to
sneak me in to “rex 27+”, too bad it never happened before the challenge was
gone ;) Also thanks to Magnus for interesting defense parties, and
Marianne R for inspiration!
Other people in the top sphere, the fly-floor: Fredrik, Yasuo, for nice talks,
afterworks, beers and Benjamin Buttons Grp DH: Dan, Jesper, Jens-
Ola, Inez, Martin, Sajna, Hairu and Silvia for creating a very nice
homey feeling in the fika room! And also great thanks to all other people on
this floor: Åsa, Ruth, Caroline, Camilla, Ingrid, Erik, John, Linn,
Sa, Murat, Barbara, Mahsa, Jesper, Behzad, Sofie and all past fly
floor/UCMP people; Anna L, Anna-Karin, Karin, Jana, Ulrika,
Dimitri, Margret, Mazen, Magdalena, Gaurav, Mattias, Therese N,
Olga, Maria W, Malin L-P, Anders O, and many more, for nice fika
room chats, for brightening up the corridors and for creating a genuine nice
and creative environment!
Friends from Molbiol (present or out on their own new adventures):
Therese E, I am so sad that you have left umeå, we have had a lot of fun
together! Thanks for all craft nights, you are a great source of inspiration,
and thanks for being a genuine good friend! Luckily, Sundsvall is not that far
away, and hope we will have more vacations together on Crete in the future!

ACKNOWLEDGEMENTS
56
With Erik N, Gunnar and Karin! Erik T, you are one of a kind, thanks for
being you, my years at molbiol would have been much less fun without you,
you’re always up for stuff and ready for adventures, your always kind and
funny, and take things in the right way! And thanks for your endless patients
in teaching beach-volleyboll! You are welcome to come and sink my canoe
again at any time you want in the future . Linn J, thanks for all fun events,
laughs, craft nights, spex movie making etc etc, if you ever resign from
science I think you should become an actress! John S-H, you are really fun
to be around, thanks for all lunches, fika, movie making etc, and thanks for
doing a great job as our fika over-lord, it would not have worked so well
without you! Chaz and Linus, I really miss you guys! You can light up every
occasion, and Chaz thanks for endless craft nights and for having the same
enthusiasm for costume parties as me! You should move back to umeå and
open “the drunken bead”, but maybe in the future our paths will cross again,
who knows Hande, thanks for all good times and nice discussions we
have had! Sa, for many pleasant dinners, craft nights and movie planning
nights in your home, you deserve the best! David G, probably the funniest
person I have met, it’s too bad you left umeå so early (in my perspective at
least) hope you survive the trip from Göteborg back to Umeå and my defense
party without any major mishaps ;) Sara R, for good times at the lunch
table, parties, spontaneous bonfires at parties ;) and for being an excellent
spex movie planner! Christina S, for crafts nights and BBQ at your balcony,
hope you are have a good time in Scotland! Anne-Laure, thanks for
movies, adventure on höga kusten and afterworks. The members of the lunch
and fika cluster (John, Linn, Erik, Margarida, Marie-Line, Caroline,
Christopher, Isabelle, Sajna, Victoria, Lisa, Jessica), thanks to all of you for
all great lunch discussions about small and big things, the lunches with you
guys can really make my day! Caroline, thanks for all nice chats and good
luck with the new family! Christopher, you have a talent for balancing
humor, bitterness and irony in a lovely mixture. Isabelle, for nice advices
about life, dissertations etc. Sajna, for all your sharp and funny stories.
Victoria, for teaching us pottery. Lisa, for lightening up my days by teasing
Andersson Jessica, our coffee-junkie, I think you’ve been gone too long!
Hasan, Tony, and previous presidents of Beer corner for maintaining a
good selection of beers! Hao, Hasan, Kristoffer L, Anais, Ayad,
Patrik, for nice teaching cooperation. Marek, for fixing and installing
computers, and Mediafolket, for always being helpful and nice! And thanks
to the fun people at beer corner, the molbiol crafting gang, and all other
people at the molbiol department that I have not mentioned but not
forgotten!

ACKNOWLEDGEMENTS
57
Människorna ute i den andra världen: Emma, det närmsta en syster jag har
kommit, kan inte minnas nån tid jag inte känt dig och jag tror mitt liv hade
varit väldigt annorlunda om jag inte hade känt dig, tack för allt vi har upplevt
ihop (det är omöjligt att lista upp) och för att du alltid har varit min vän!
Mogge, tack för att du tar hand om emma ;) Du är en hemskt roligt person
och jag är glad att ni har gift er! Vera, tack för att du varit min vän i alla
dessa år, jag saknar dig! Det är himla tur att det finns telefon och billiga
abonnemang! Hoppas vi nån dag kommer bo närmre varandra! Maria,
utan dig hade jag nog haft grava uppdämda psykologiska problem ;) tack för
din fina vänskap och för alla kloka råd och tankar om allt! Mats, ni två
passar väldigt bra ihop och jag ser fram emot att få lära känna dig ännu
bättre! Monika, tack för året som kombos och allt roligt vi har haft efter det,
jag hoppas på mycket mer i en snar framtid! Jag och umeå har saknat dig,
även om du inte saknar umeå ;) och tack för att du betrodde mig med dina
mysiga katter!
Slutligen min familj, den bästa man skulle kunna fått: Det är väldigt svårt att
beskriva i ord vad ni betyder för mig, Mamma och Pappa, tack för livets
gåva och all stöttning ni ger mig, för att ni alltid finns där! Mamma, jag
hoppas jag blir som du, jag tror inte det finns nån klokare kvinna, Pappa, för
att jag fick ärva ditt intresse för naturvetenskap, och tack för att du alltid,
alltid ställt upp och funnit till hands när jag behövt! Från att lösa
matteproblem och pussla ihop vattenlås till att köra 100 mil för att hjälpa
mig flytta, Arvid och Markus, mina älskade mallis, småbröder, vad vore väl
nånting utan er? Markus, minns mig när du blir rik och berömd
spelutvecklare Arvid, jag saknar att ha dig i närheten, varför måste ni bo
så långt bort! Mary, välkommen in i familjen, hoppas verkligen vi får fler
resor till solen tillsammas, eller åtminstone fler mysdagar i stugan så länge
Farfar, som tydligt visat att det är annat virke i den generationen och att
man kan klara det mesta på egen hand, bara envisheten och viljan finns där,
med eller utan brutna lårbenshalsar.
“Nya möjligheter är ofta förklädda till smärtsamma slut.” ~ Lao Zi

REFERENCES
58
REFERENCES
1. Khorasanizadeh S (2004) The nucleosome: from genomic organization to
genomic regulation. Cell 116: 259-272. 2. Olins AL, Olins DE (1974) Spheroid chromatin units (v bodies). Science
183: 330-332. 3. Oudet P, Gross-Bellard M, Chambon P (1975) Electron microscopic and
biochemical evidence that chromatin structure is a repeating unit. Cell 4: 281-300.
4. Thomas JO, Kornberg RD (1975) An octamer of histones in chromatin and free in solution. Proc Natl Acad Sci U S A 72: 2626-2630.
5. Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ (1997) Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389: 251-260.
6. Happel N, Doenecke D (2009) Histone H1 and its isoforms: contribution to chromatin structure and function. Gene 431: 1-12.
7. Kornberg RD (1974) Chromatin structure: a repeating unit of histones and DNA. Science 184: 868-871.
8. Wong H, Victor JM, Mozziconacci J (2007) An all-atom model of the chromatin fiber containing linker histones reveals a versatile structure tuned by the nucleosomal repeat length. PLoS One 2: e877.
9. Robinson PJ, Fairall L, Huynh VA, Rhodes D (2006) EM measurements define the dimensions of the "30-nm" chromatin fiber: evidence for a compact, interdigitated structure. Proc Natl Acad Sci U S A 103: 6506-6511.
10. Zheng C, Hayes JJ (2003) Structures and interactions of the core histone tail domains. Biopolymers 68: 539-546.
11. Gräff J, Tsai LH (2013) Histone acetylation: molecular mnemonics on the chromatin. Nat Rev Neurosci 14: 97-111.
12. Smith CM, Gafken PR, Zhang Z, Gottschling DE, Smith JB, et al. (2003) Mass spectrometric quantification of acetylation at specific lysines within the amino-terminal tail of histone H4. Anal Biochem 316: 23-33.
13. Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, et al. (2006) Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311: 844-847.
14. Tse C, Sera T, Wolffe AP, Hansen JC (1998) Disruption of higher-order folding by core histone acetylation dramatically enhances transcription of nucleosomal arrays by RNA polymerase III. Mol Cell Biol 18: 4629-4638.
15. Lyko F, Ramsahoye BH, Jaenisch R (2000) DNA methylation in Drosophila melanogaster. Nature 408: 538-540.

REFERENCES
59
16. Peters AH, Kubicek S, Mechtler K, O'Sullivan RJ, Derijck AA, et al. (2003) Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol Cell 12: 1577-1589.
17. Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, et al. (2002) Active genes are tri-methylated at K4 of histone H3. Nature 419: 407-411.
18. Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, et al. (2007) Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448: 553-560.
19. Martin C, Zhang Y (2005) The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol 6: 838-849.
20. Ebert A, Schotta G, Lein S, Kubicek S, Krauss V, et al. (2004) Su(var) genes regulate the balance between euchromatin and heterochromatin in Drosophila. Genes Dev 18: 2973-2983.
21. Grewal SI, Elgin SC (2002) Heterochromatin: new possibilities for the inheritance of structure. Curr Opin Genet Dev 12: 178-187.
22. Eissenberg JC, Reuter G (2009) Cellular mechanism for targeting heterochromatin formation in Drosophila. Int Rev Cell Mol Biol 273: 1-47.
23. Richards EJ, Elgin SC (2002) Epigenetic codes for heterochromatin formation and silencing: rounding up the usual suspects. Cell 108: 489-500.
24. Gowen JW, Gay EH (1933) Effect of temperature on eversporting eye color in Drosophila melanogaster. Science 77: 312.
25. Dimitri P, Pisano C (1989) Position effect variegation in Drosophila melanogaster: relationship between suppression effect and the amount of Y chromosome. Genetics 122: 793-800.
26. Gowen JW, Gay EH (1934) Chromosome constitution and behavior in eversporting and mottling in Drosophila melanogaster. Genetics 19: 189-208.
27. James TC, Eissenberg JC, Craig C, Dietrich V, Hobson A, et al. (1989) Distribution patterns of HP1, a heterochromatin-associated nonhistone chromosomal protein of Drosophila. Eur J Cell Biol 50: 170-180.
28. Ebert A, Lein S, Schotta G, Reuter G (2006) Histone modification and the control of heterochromatic gene silencing in Drosophila. Chromosome Res 14: 377-392.
29. Carvalho A, Koerich L, Clark A (2009) Origin and evolution of Y chromosomes: Drosophila tales. Trends Genet: 270-277.
30. Filion GJ, van Bemmel JG, Braunschweig U, Talhout W, Kind J, et al. (2010) Systematic protein location mapping reveals five principal chromatin types in Drosophila cells. Cell 143: 212-224.

REFERENCES
60
31. Zhang P, Du J, Sun B, Dong X, Xu G, et al. (2006) Structure of human MRG15 chromo domain and its binding to Lys36-methylated histone H3. Nucleic Acids Res 34: 6621-6628.
32. Kharchenko PV, Alekseyenko AA, Schwartz YB, Minoda A, Riddle NC, et al. (2011) Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature 471: 480-485.
33. Wallrath LL (1998) Unfolding the mysteries of heterochromatin. Curr Opin Genet Dev 8: 147-153.
34. Eissenberg JC, James TC, Foster-Hartnett DM, Hartnett T, Ngan V, et al. (1990) Mutation in a heterochromatin-specific chromosomal protein is associated with suppression of position-effect variegation in Drosophila melanogaster. Proc Natl Acad Sci U S A 87: 9923-9927.
35. Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, et al. (2001) Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410: 120-124.
36. Lachner M, O'Carroll D, Rea S, Mechtler K, Jenuwein T (2001) Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410: 116-120.
37. Vermaak D, Malik HS (2009) Multiple roles for heterochromatin protein 1 genes in Drosophila. Annu Rev Genet 43: 467-492.
38. Aasland R, Stewart AF (1995) The chromo shadow domain, a second chromo domain in heterochromatin-binding protein 1, HP1. Nucleic Acids Res 23: 3168-3173.
39. Cowieson NP, Partridge JF, Allshire RC, McLaughlin PJ (2000) Dimerisation of a chromo shadow domain and distinctions from the chromodomain as revealed by structural analysis. Curr Biol 10: 517-525.
40. Mishima Y, Watanabe M, Kawakami T, Jayasinghe CD, Otani J, et al. (2013) Hinge and chromoshadow of HP1α participate in recognition of K9 methylated histone H3 in nucleosomes. J Mol Biol 425: 54-70.
41. Fanti L, Berloco M, Piacentini L, Pimpinelli S (2003) Chromosomal distribution of heterochromatin protein 1 (HP1) in Drosophila: a cytological map of euchromatic HP1 binding sites. Genetica 117: 135-147.
42. Cryderman DE, Grade SK, Li Y, Fanti L, Pimpinelli S, et al. (2005) Role of Drosophila HP1 in euchromatic gene expression. Dev Dyn 232: 767-774.
43. Riddle NC, Minoda A, Kharchenko PV, Alekseyenko AA, Schwartz YB, et al. (2011) Plasticity in patterns of histone modifications and chromosomal proteins in Drosophila heterochromatin. Genome Res 21: 147-163.
44. Yin H, Sweeney S, Raha D, Snyder M, Lin H (2011) A high-resolution whole-genome map of key chromatin modifications in the adult Drosophila melanogaster. PLoS Genet 7: e1002380.

REFERENCES
61
45. Figueiredo MLA, Philip P, Stenberg P, Larsson J (2012) HP1a recruitment to promoters is independent of H3K9 methylation in Drosophila melanogaster. PLoS Genet 8: e1003061.
46. Johansson AM, Stenberg P, Pettersson F, Larsson J (2007) POF and HP1 bind expressed exons, suggesting a balancing mechanism for gene regulation. PLoS Genet 3: e209.
47. de Wit E, Greil F, van Steensel B (2007) High-resolution mapping reveals links of HP1 with active and inactive chromatin components. PLoS Genet 3: e38.
48. Johansson AM, Stenberg P, Bernhardsson C, Larsson J (2007) Painting of fourth and chromosome-wide regulation of the 4th chromosome in Drosophila melanogaster. EMBO J 26: 2307-2316.
49. Liu LP, Ni JQ, Shi YD, Oakeley EJ, Sun FL (2005) Sex-specific role of Drosophila melanogaster HP1 in regulating chromatin structure and gene transcription. Nat Genet 37: 1361-1366.
50. De Lucia F, Ni JQ, Vaillant C, Sun FL (2005) HP1 modulates the transcription of cell-cycle regulators in Drosophila melanogaster. Nucleic Acids Res 33: 2852-2858.
51. Schwaiger M, Kohler H, Oakeley EJ, Stadler MB, Schübeler D (2010) Heterochromatin protein 1 (HP1) modulates replication timing of the Drosophila genome. Genome Res 20: 771-780.
52. Piacentini L, Fanti L, Berloco M, Perrini B, Pimpinelli S (2003) Heterochromatin protein 1 (HP1) is associated with induced gene expression in Drosophila euchromatin. J Cell Biol 161: 707-714.
53. Piacentini L, Fanti L, Negri R, Del Vescovo V, Fatica A, et al. (2009) Heterochromatin protein 1 (HP1a) positively regulates euchromatic gene expression through RNA transcript association and interaction with hnRNPs in Drosophila. PLoS Genet 5: e1000670.
54. Riddle NC, Jung YL, Gu T, Alekseyenko AA, Asker D, et al. (2012) Enrichment of HP1a on Drosophila chromosome 4 genes creates an alternate chromatin structure critical for regulation in this heterochromatic domain. PLoS Genet 8: e1002954.
55. Hearn MG, Hedrick A, Grigliatti TA, Wakimoto BT (1991) The effect of modifiers of position-effect variegation on the variegation of heterochromatic genes of Drosophila melanogaster. Genetics 128: 785-797.
56. Wakimoto BT, Hearn MG (1990) The effects of chromosome rearrangements on the expression of heterochromatic genes in chromosome 2L of Drosophila melanogaster. Genetics 125: 141-154.
57. Clegg NJ, Honda BM, Whitehead IP, Grigliatti TA, Wakimoto B, et al. (1998) Suppressors of position-effect variegation in Drosophila melanogaster affect expression of the heterochromatic gene light in the absence of a chromosome rearrangement. Genome 41: 495-503.

REFERENCES
62
58. Lu BY, Emtage PC, Duyf BJ, Hilliker AJ, Eissenberg JC (2000) Heterochromatin protein 1 is required for the normal expression of two heterochromatin genes in Drosophila. Genetics 155: 699-708.
59. Schulze SR, Sinclair DA, Fitzpatrick KA, Honda BM (2005) A genetic and molecular characterization of two proximal heterochromatic genes on chromosome 3 of Drosophila melanogaster. Genetics 169: 2165-2177.
60. Yasuhara JC, Wakimoto BT (2006) Oxymoron no more: the expanding world of heterochromatic genes. Trends Genet 22: 330-338.
61. Smothers JF, Henikoff S (2001) The hinge and chromo shadow domain impart distinct targeting of HP1-like proteins. Mol Cell Biol 21: 2555-2569.
62. Font-Burgada J, Rossell D, Auer H, Azorín F (2008) Drosophila HP1c isoform interacts with the zinc-finger proteins WOC and Relative-of-WOC to regulate gene expression. Genes Dev 22: 3007-3023.
63. Vermaak D, Henikoff S, Malik HS (2005) Positive selection drives the evolution of rhino, a member of the heterochromatin protein 1 family in Drosophila. PLoS Genet 1: 96-108.
64. Mis J, Ner SS, Grigliatti TA (2006) Identification of three histone methyltransferases in Drosophila: dG9a is a suppressor of PEV and is required for gene silencing. Mol Genet Genomics 275: 513-526.
65. Seum C, Bontron S, Reo E, Delattre M, Spierer P (2007) Drosophila G9a is a nonessential gene. Genetics 177: 1955-1957.
66. Stabell M, Eskeland R, Bjørkmo M, Larsson J, Aalen RB, et al. (2006) The Drosophila G9a gene encodes a multi-catalytic histone methyltransferase required for normal development. Nucleic Acids Res 34: 4609-4621.
67. Brower-Toland B, Riddle NC, Jiang H, Huisinga KL, Elgin SC (2009) Multiple SET methyltransferases are required to maintain normal heterochromatin domains in the genome of Drosophila melanogaster. Genetics 181: 1303-1319.
68. Rea S, Eisenhaber F, O'Carroll D, Strahl BD, Sun ZW, et al. (2000) Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406: 593-599.
69. Czermin B, Schotta G, Hülsmann BB, Brehm A, Becker PB, et al. (2001) Physical and functional association of SU(VAR)3-9 and HDAC1 in Drosophila. EMBO Rep 2: 915-919.
70. Schotta G, Ebert A, Krauss V, Fischer A, Hoffmann J, et al. (2002) Central role of Drosophila SU(VAR)3-9 in histone H3-K9 methylation and heterochromatic gene silencing. EMBO J 21: 1121-1131.
71. Tschiersch B, Hofmann A, Krauss V, Dorn R, Korge G, et al. (1994) The protein encoded by the Drosophila position-effect variegation

REFERENCES
63
suppressor gene Su(var)3-9 combines domains of antagonistic regulators of homeotic gene complexes. EMBO J 13: 3822-3831.
72. Seum C, Reo E, Peng H, Rauscher FJ, Spierer P, et al. (2007) Drosophila SETDB1 is required for chromosome 4 silencing. PLoS Genet 3: e76.
73. Tzeng TY, Lee CH, Chan LW, Shen CK (2007) Epigenetic regulation of the Drosophila chromosome 4 by the histone H3K9 methyltransferase dSETDB1. Proc Natl Acad Sci U S A 104: 12691-12696.
74. Clough E, Moon W, Wang S, Smith K, Hazelrigg T (2007) Histone methylation is required for oogenesis in Drosophila. Development 134: 157-165.
75. Stabell M, Bjørkmo M, Aalen RB, Lambertsson A (2006) The Drosophila SET domain encoding gene dEset is essential for proper development. Hereditas 143: 177-188.
76. Yoon J, Lee KS, Park JS, Yu K, Paik SG, et al. (2008) dSETDB1 and SU(VAR)3-9 sequentially function during germline-stem cell differentiation in Drosophila melanogaster. PLoS One 3: e2234.
77. Koch CM, Honemann-Capito M, Egger-Adam D, Wodarz A (2009) Windei, the Drosophila homolog of mAM/MCAF1, is an essential cofactor of the H3K9 methyl transferase dSETDB1/Eggless in germ line development. PLoS Genet 5: e1000644.
78. Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI (2001) Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 292: 110-113.
79. Jacobs SA, Khorasanizadeh S (2002) Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail. Science 295: 2080-2083.
80. Jacobs SA, Taverna SD, Zhang Y, Briggs SD, Li J, et al. (2001) Specificity of the HP1 chromo domain for the methylated N-terminus of histone H3. EMBO J 20: 5232-5241.
81. Mottus R, Sobel RE, Grigliatti TA (2000) Mutational analysis of a histone deacetylase in Drosophila melanogaster: missense mutations suppress gene silencing associated with position effect variegation. Genetics 154: 657-668.
82. Slotkin RK, Martienssen R (2007) Transposable elements and the epigenetic regulation of the genome. Nat Rev Genet 8: 272-285.
83. Huang XA, Yin H, Sweeney S, Raha D, Snyder M, et al. (2013) A major epigenetic programming mechanism guided by piRNAs. Dev Cell 24: 502-516.
84. McClintock B (1950) The origin and behavior of mutable loci in maize. Proc Natl Acad Sci U S A 36: 344-355.
85. Craig NL (1995) Unity in transposition reactions. Science 270: 253-254.

REFERENCES
64
86. Rao M, Sockanathan S (2005) Molecular mechanisms of RNAi: implications for development and disease. Birth Defects Res C Embryo Today 75: 28-42.
87. Fanti L, Dorer DR, Berloco M, Henikoff S, Pimpinelli S (1998) Heterochromatin protein 1 binds transgene arrays. Chromosoma 107: 286-292.
88. Greil F, van der Kraan I, Delrow J, Smothers JF, de Wit E, et al. (2003) Distinct HP1 and Su(var)3-9 complexes bind to sets of developmentally coexpressed genes depending on chromosomal location. Genes Dev 17: 2825-2838.
89. Smith CD, Shu S, Mungall CJ, Karpen GH (2007) The Release 5.1 annotation of Drosophila melanogaster heterochromatin. Science 316: 1586-1591.
90. Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, et al. (2000) The genome sequence of Drosophila melanogaster. Science 287: 2185-2195.
91. Reiter LT, Potocki L, Chien S, Gribskov M, Bier E (2001) A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res 11: 1114-1125.
92. Skaletsky H, Kuroda-Kawaguchi T, Minx PJ, Cordum HS, Hillier L, et al. (2003) The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 423: 825-837.
93. Koopman P, Gubbay J, Vivian N, Goodfellow P, Lovell-Badge R (1991) Male development of chromosomally female mice transgenic for Sry. Nature 351: 117-121.
94. Carvalho AB (2002) Origin and evolution of the Drosophila Y chromosome. Curr Opin Genet Dev 12: 664-668.
95. Lucchesi JC, Kelly WG, Panning B (2005) Chromatin remodeling in dosage compensation. Annu Rev Genet 39: 615-651.
96. Lyon MF (1961) Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature 190: 372-373.
97. Barr ML, Carr DH (1962) Correlations between sex chromatin and sex chromosomes. Acta Cytol 6: 34-45.
98. Lee JT, Strauss WM, Dausman JA, Jaenisch R (1996) A 450 kb transgene displays properties of the mammalian X-inactivation center. Cell 86: 83-94.
99. Clemson CM, McNeil JA, Willard HF, Lawrence JB (1996) XIST RNA paints the inactive X chromosome at interphase: evidence for a novel RNA involved in nuclear/chromosome structure. J Cell Biol 132: 259-275.
100. Herzing LB, Romer JT, Horn JM, Ashworth A (1997) Xist has properties of the X-chromosome inactivation centre. Nature 386: 272-275.

REFERENCES
65
101. Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT (2008) Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 322: 750-756.
102. Heard E, Disteche CM (2006) Dosage compensation in mammals: fine-tuning the expression of the X chromosome. Genes Dev 20: 1848-1867.
103. Chadwick BP, Willard HF (2003) Chromatin of the Barr body: histone and non-histone proteins associated with or excluded from the inactive X chromosome. Hum Mol Genet 12: 2167-2178.
104. Deng X, Hiatt JB, Nguyen DK, Ercan S, Sturgill D, et al. (2011) Evidence for compensatory upregulation of expressed X-linked genes in mammals, Caenorhabditis elegans and Drosophila melanogaster. Nat Genet 43: 1043‐1047.
105. Gupta V, Parisi M, Sturgill D, Nuttall R, Doctolero M, et al. (2006) Global analysis of X-chromosome dosage compensation. J Biol 5: 3.
106. Nguyen DK, Disteche CM (2006) Dosage compensation of the active X chromosome in mammals. Nat Genet 38: 47-53.
107. Adler DA, Rugarli EI, Lingenfelter PA, Tsuchiya K, Poslinski D, et al. (1997) Evidence of evolutionary up-regulation of the single active X chromosome in mammals based on Clc4 expression levels in Mus spretus and Mus musculus. Proc Natl Acad Sci U S A 94: 9244-9248.
108. Prestel M, Feller C, Becker PB (2010) Dosage compensation and the global re-balancing of aneuploid genomes. Genome Biol 11: 216.
109. Vallot C, Huret C, Lesecque Y, Resch A, Oudrhiri N, et al. (2013) XACT, a long noncoding transcript coating the active X chromosome in human pluripotent cells. Nat Genet 45: 239-241.
110. Xiong Y, Chen X, Chen Z, Wang X, Shi S, et al. (2010) RNA sequencing shows no dosage compensation of the active X-chromosome. Nat Genet 42: 1043-1047.
111. Smith ER, Pannuti A, Gu W, Steurnagel A, Cook RG, et al. (2000) The drosophila MSL complex acetylates histone H4 at lysine 16, a chromatin modification linked to dosage compensation. Mol Cell Biol 20: 312-318.
112. Kadlec J, Hallacli E, Lipp M, Holz H, Sanchez-Weatherby J, et al. (2011) Structural basis for MOF and MSL3 recruitment into the dosage compensation complex by MSL1. Nat Struct Mol Biol 18: 142-149.
113. Chang KA, Kuroda MI (1998) Modulation of MSL1 abundance in female Drosophila contributes to the sex specificity of dosage compensation. Genetics 150: 699-709.
114. Morales V, Straub T, Neumann MF, Mengus G, Akhtar A, et al. (2004) Functional integration of the histone acetyltransferase MOF into the dosage compensation complex. EMBO J 23: 2258-2268.
115. Copps K, Richman R, Lyman LM, Chang KA, Rampersad-Ammons J, et al. (1998) Complex formation by the Drosophila MSL proteins: role

REFERENCES
66
of the MSL2 RING finger in protein complex assembly. EMBO J 17: 5409-5417.
116. Scott MJ, Pan LL, Cleland SB, Knox AL, Heinrich J (2000) MSL1 plays a central role in assembly of the MSL complex, essential for dosage compensation in Drosophila. EMBO J 19: 144-155.
117. Hallacli E, Lipp M, Georgiev P, Spielman C, Cusack S, et al. (2012) Msl1-mediated dimerization of the dosage compensation complex is essential for male X-chromosome regulation in Drosophila. Mol Cell 48: 587-600.
118. Bashaw GJ, Baker BS (1995) The msl-2 dosage compensation gene of Drosophila encodes a putative DNA-binding protein whose expression is sex specifically regulated by Sex-lethal. Development 121: 3245-3258.
119. Zhou S, Yang Y, Scott MJ, Pannuti A, Fehr KC, et al. (1995) Male-specific lethal 2, a dosage compensation gene of Drosophila, undergoes sex-specific regulation and encodes a protein with a RING finger and a metallothionein-like cysteine cluster. EMBO J 14: 2884-2895.
120. Kelley RL, Wang J, Bell L, Kuroda MI (1997) Sex lethal controls dosage compensation in Drosophila by a non-splicing mechanism. Nature 387: 195-199.
121. Kelley RL, Solovyeva I, Lyman LM, Richman R, Solovyev V, et al. (1995) Expression of msl-2 causes assembly of dosage compensation regulators on the X chromosomes and female lethality in Drosophila. Cell 81: 867-877.
122. Bashaw GJ, Baker BS (1997) The regulation of the Drosophila msl-2 gene reveals a function for Sex-lethal in translational control. Cell 89: 789-798.
123. Johansson AM, Allgardsson A, Stenberg P, Larsson J (2011) msl2 mRNA is bound by free nuclear MSL complex in Drosophila melanogaster. Nucleic Acids Res 39: 6428-6439.
124. Koonin EV, Zhou S, Lucchesi JC (1995) The chromo superfamily: new members, duplication of the chromo domain and possible role in delivering transcription regulators to chromatin. Nucleic Acids Res 23: 4229-4233.
125. Buscaino A, Köcher T, Kind JH, Holz H, Taipale M, et al. (2003) MOF-regulated acetylation of MSL-3 in the Drosophila dosage compensation complex. Mol Cell 11: 1265-1277.
126. Lee CG, Chang KA, Kuroda MI, Hurwitz J (1997) The NTPase/helicase activities of Drosophila maleless, an essential factor in dosage compensation. EMBO J 16: 2671-2681.
127. Wang CI, Alekseyenko AA, Leroy G, Elia AE, Gorchakov AA, et al. (2013) Chromatin proteins captured by ChIP-mass spectrometry are linked

REFERENCES
67
to dosage compensation in Drosophila. Nat Struct Mol Biol 20: 202-209.
128. Richter L, Bone JR, Kuroda MI (1996) RNA-dependent association of the Drosophila maleless protein with the male X chromosome. Genes to Cells 1: 325-336.
129. Morra R, Yokoyama R, Ling H, Lucchesi JC (2011) Role of the ATPase/helicase maleless (MLE) in the assembly, targeting, spreading and function of the male-specific lethal (MSL) complex of Drosophila. Epigenetics Chromatin 4: 6.
130. Oh H, Park Y, Kuroda MI (2003) Local spreading of MSL complexes from roX genes on the Drosophila X chromosome. Genes Dev 17: 1334-1339.
131. Meller VH, Gordadze PR, Park Y, Chu X, Stuckenholz C, et al. (2000) Ordered assembly of roX RNAs into MSL complexes on the dosage-compensated X chromosome in Drosophila. Curr Biol 10: 136-143.
132. Morra R, Smith ER, Yokoyama R, Lucchesi JC (2008) The MLE subunit of the Drosophila MSL complex uses its ATPase activity for dosage compensation and its helicase activity for targeting. Mol Cell Biol 28: 958-966.
133. Kotlikova IV, Demakova OV, Semeshin VF, Shloma VV, Boldyreva LV, et al. (2006) The Drosophila dosage compensation complex binds to polytene chromosomes independently of developmental changes in transcription. Genetics 172: 963-974.
134. Stuckenholz C, Kageyama Y, Kuroda MI (1999) Guilt by association, non-coding RNAs, chromosome-specific proteins and dosage compensation in Drosophila. Trends Genet 15: 454-458.
135. Akhtar A, Becker PB (2000) Activation of transcription through histone H4 acetylation by MOF, an acetyltransferase essential for dosage compensation in Drosophila. Mol Cell 5: 367-375.
136. Hilfiker A, Hilfiker-Kleiner D, Pannuti A, Lucchesi JC (1997) mof, a putative acetyl transferase gene related to the Tip60 and MOZ human genes and to the SAS genes of yeast, is required for dosage compensation in Drosophila. EMBO J 16: 2054-2060.
137. Kind J, Vaquerizas JM, Gebhardt P, Gentzel M, Luscombe NM, et al. (2008) Genome-wide analysis reveals MOF as a key regulator of dosage compensation and gene expression in Drosophila. Cell 133: 813-828.
138. Bhadra U, Pal-Bhadra M, Birchler JA (1999) Role of the male specific lethal (msl) genes in modifying the effects of sex chromosomal dosage in Drosophila. Genetics 152: 249-268.
139. Lam KC, Mühlpfordt F, Vaquerizas JM, Raja SJ, Holz H, et al. (2012) The NSL complex regulates housekeeping genes in Drosophila. PLoS Genet 8: e1002736.

REFERENCES
68
140. Raja SJ, Charapitsa I, Conrad T, Vaquerizas JM, Gebhardt P, et al. (2010) The nonspecific lethal complex is a transcriptional regulator in Drosophila. Mol Cell 38: 827-841.
141. Prestel M, Feller C, Straub T, Mitlöhner H, Becker PB (2010) The activation potential of MOF is constrained for dosage compensation. Mol Cell 38: 815-826.
142. Franke A, Baker BS (1999) The rox1 and rox2 RNAs are essential components of the Compensasome, which mediates dosage compensation in Drosophila. Mol Cell 4: 117-122.
143. Meller VH, Wu KH, Roman G, Kuroda MI, Davis RL (1997) roX1 RNA paints the X chromosome of male Drosophila and is regulated by the dosage compensation system. Cell 88: 445-457.
144. Meller VH, Rattner BP (2002) The roX genes encode redundant male-specific lethal transcripts required for targeting of the MSL complex. EMBO J 21: 1084-1091.
145. Kelley RL, Meller VH, Gordadze PR, Roman G, Davis RL, et al. (1999) Epigenetic spreading of the Drosophila dosage compensation complex from roX RNA genes into flanking chromatin. Cell 98: 513-522.
146. Larschan E, Alekseyenko AA, Gortchakov AA, Peng S, Li B, et al. (2007) MSL complex is attracted to genes marked by H3K36 trimethylation using a sequence-independent mechanism. Mol Cell 28: 121-133.
147. Straub T, Zabel A, Gilfillan GD, Feller C, Becker PB (2013) Different chromatin interfaces of the Drosophila dosage compensation complex revealed by high-shear ChIP-seq. Genome Res 23: 473–485.
148. Alekseyenko AA, Peng S, Larschan E, Gorchakov AA, Lee OK, et al. (2008) A sequence motif within chromatin entry sites directs MSL establishment on the Drosophila X chromosome. Cell 134: 599-609.
149. Palmer MJ, Richman R, Richter L, Kuroda MI (1994) Sex-specific regulation of the male-specific lethal-1 dosage compensation gene in Drosophila. Genes Dev 8: 698-706.
150. Demakova OV, Kotlikova IV, Gordadze PR, Alekseyenko AA, Kuroda MI, et al. (2003) The MSL complex levels are critical for its correct targeting to the chromosomes in Drosophila melanogaster. Chromosoma 112: 103-115.
151. Lyman LM, Copps K, Rastelli L, Kelley RL, Kuroda MI (1997) Drosophila male-specific lethal-2 protein: structure/function analysis and dependence on MSL-1 for chromosome association. Genetics 147: 1743-1753.
152. Alekseyenko AA, Larschan E, Lai WR, Park PJ, Kuroda MI (2006) High-resolution ChIP-chip analysis reveals that the Drosophila MSL complex selectively identifies active genes on the male X chromosome. Genes Dev 20: 848-857.

REFERENCES
69
153. Gilfillan GD, Straub T, de Wit E, Greil F, Lamm R, et al. (2006) Chromosome-wide gene-specific targeting of the Drosophila dosage compensation complex. Genes Dev 20: 858-870.
154. Sural TH, Peng S, Li B, Workman JL, Park PJ, et al. (2008) The MSL3 chromodomain directs a key targeting step for dosage compensation of the Drosophila melanogaster X chromosome. Nat Struct Mol Biol 15: 1318-1325.
155. Bell O, Conrad T, Kind J, Wirbelauer C, Akhtar A, et al. (2008) Transcription-coupled methylation of histone H3 at lysine 36 regulates dosage compensation by enhancing recruitment of the MSL complex in Drosophila melanogaster. Mol Cell Biol 28: 3401-3409.
156. Scholnick SB, Morgan BA, Hirsh J (1983) The cloned dopa decarboxylase gene is developmentally regulated when reintegrated into the Drosophila genome. Cell 34: 37-45.
157. Spradling AC, Rubin GM (1983) The effect of chromosomal position on the expression of the Drosophila xanthine dehydrogenase gene. Cell 34: 47-57.
158. Sass GL, Pannuti A, Lucchesi JC (2003) Male-specific lethal complex of Drosophila targets activated regions of the X chromosome for chromatin remodeling. Proc Natl Acad Sci U S A 100: 8287-8291.
159. Gelbart ME, Larschan E, Peng S, Park PJ, Kuroda MI (2009) Drosophila MSL complex globally acetylates H4K16 on the male X chromosome for dosage compensation. Nat Struct Mol Biol 16: 825-832.
160. Stenberg P, Pettersson F, Saura AO, Berglund A, Larsson J (2005) Sequence analysis of chromosome identity in three Drosophila species. BMC Bioinformatics 6: 1-17.
161. Gallach M, Arnau V, Marín I (2007) Global patterns of sequence evolution in Drosophila. BMC Genomics 8: 408.
162. Park Y, Mengus G, Bai X, Kageyama Y, Meller VH, et al. (2003) Sequence-specific targeting of Drosophila roX genes by the MSL dosage compensation complex. Mol Cell 11: 977-986.
163. Gilfillan GD, König C, Dahlsveen IK, Prakoura N, Straub T, et al. (2007) Cumulative contributions of weak DNA determinants to targeting the Drosophila dosage compensation complex. Nucleic Acids Res 35: 3561-3572.
164. Legube G, McWeeney SK, Lercher MJ, Akhtar A (2006) X-chromosome-wide profiling of MSL-1 distribution and dosage compensation in Drosophila. Genes Dev 20: 871-883.
165. Philip P, Pettersson F, Stenberg P (2012) Sequence signatures involved in targeting the Male-Specific Lethal complex to X-chromosomal genes in Drosophila melanogaster. BMC Genomics 13: 97.

REFERENCES
70
166. Smith ER, Allis CD, Lucchesi JC (2001) Linking global histone acetylation to the transcription enhancement of X-chromosomal genes in Drosophila males. J Biol Chem 276: 31483-31486.
167. Conrad T, Cavalli FM, Vaquerizas JM, Luscombe NM, Akhtar A (2012) Drosophila dosage compensation involves enhanced Pol II recruitment to male X-linked promoters. Science 337: 742-746.
168. Larschan E, Bishop EP, Kharchenko PV, Core LJ, Lis JT, et al. (2011) X chromosome dosage compensation via enhanced transcriptional elongation in Drosophila. Nature 471: 115-118.
169. Hamada FN, Park PJ, Gordadze PR, Kuroda MI (2005) Global regulation of X chromosomal genes by the MSL complex in Drosophila melanogaster. Genes Dev 19: 2289-2294.
170. Deng X, Koya SK, Kong Y, Meller VH (2009) Coordinated regulation of heterochromatic genes in Drosophila melanogaster males. Genetics 182: 481-491.
171. Zhang Y, Malone JH, Powell SK, Periwal V, Spana E, et al. (2010) Expression in aneuploid Drosophila S2 cells. PLoS Biol 8: e1000320.
172. Stenberg P, Larsson J (2011) Buffering and the evolution of chromosome-wide gene regulation. Chromosoma 120: 213-225.
173. Zhang Y, Oliver B (2010) An evolutionary consequence of dosage compensation on Drosophila melanogaster female X-chromatin structure? BMC Genomics 11: 6.
174. Larsson J, Chen JD, Rasheva V, Rasmuson Lestander A, Pirrotta V (2001) Painting of fourth, a chromosome-specific protein in Drosophila. Proc Natl Acad Sci U S A 98: 6273-6278.
175. Johansson AM, Stenberg P, Allgardsson A, Larsson J (2012) POF regulates the expression of genes on the fourth chromosome in Drosophila melanogaster by binding to nascent RNA. Mol Cell Biol 32: 2121-2134.
176. Locke J, McDermid H (1993) Analysis of Drosophila chromosome four by pulse field electrophoresis. Chromosoma 102: 718-723.
177. Barigozzi C, Dolfini S, Fraccaro M, Raimondi GR, Tiepolo L (1966) In vitro study of the DNA replication patterns of somatic chromosomes of Drosophila melanogaster. Exp Cell Res 43: 231-234.
178. Hochman B (1976) The fourth chromosome of Drosophila melanogaster. In: Ashburner M, Novitski E, editors. The Genetics and biology of Drosophila: Academic Press. pp. 903-928.
179. Miklos GLG, Yamamoto MT, Davies J, Pirrotta V (1988) Microcloning reveals a high frequency of repetitive sequences characteristic of chromosome four and the β-heterochromatin of Drosophila melanogaster. Proc Natl Acad Sci U S A 85: 2051-2055.
180. Sun FL, Cuaycong MH, Craig CA, Wallrath LL, Locke J, et al. (2000) The fourth chromosome of Drosophila melanogaster: interspersed

REFERENCES
71
euchromatic and heterochromatic domains. Proc Natl Acad Sci U S A 97: 5340-5345.
181. Pimpinelli S, Berloco M, Fanti L, Dimitri P, Bonaccorsi S, et al. (1995) Transposable elements are stable structural components of Drosophila melanogaster heterochromatin. Proc Natl Acad Sci U S A 92: 3804-3808.
182. Locke J, Podemski L, Roy K, Pilgrim D, Hodgetts R (1999) Analysis of two cosmid clones from chromosome 4 of Drosophila melanogaster reveals two new genes amid an unusual arrangement of repeated sequences. Genome Res 9: 137-149.
183. Wallrath LL, Elgin SC (1995) Position effect variegation in Drosophila is associated with an altered chromatin structure. Genes Dev 9: 1263-1277.
184. Riddle NC, Elgin SC (2006) The dot chromosome of Drosophila: insights into chromatin states and their change over evolutionary time. Chromosome Res 14: 405-416.
185. Riddle NC, Shaffer CD, Elgin SC (2009) A lot about a little dot - lessons learned from Drosophila melanogaster chromosome 4. Biochem Cell Biol 87: 229-241.
186. Russo CA, Takezaki N, Nei M (1995) Molecular phylogeny and divergence times of drosophilid species. Mol Biol Evol 12: 391-404.
187. Larsson J, Svensson MJ, Stenberg P, Mäkitalo M (2004) Painting of fourth in genus Drosophila suggests autosome-specific gene regulation. Proc Natl Acad Sci U S A 101: 9728-9733.
188. Larsson J, Meller VH (2006) Dosage compensation, the origin and the afterlife of sex chromosomes. Chromosome Res 14: 417-431.
189. Bridges CB (1925) Sex in relation to chromsomes and genes. Am Nat 59: 127-137.
190. Fung STC, Gowen JW (1960) Role of autosome-IV in Drosophila melanogaster sex balance. Genetics 45: 988-989.
191. Sandler L, Novitski E (1956) Evidence for genetic homology between chromosomes I and IV in Drosophila melanogaster, with a proposed explanation for the crowding effect in triploids. Genetics 41: 189-193.
192. FitzPatrick DR (2005) Transcriptional consequences of autosomal trisomy: primary gene dosage with complex downstream effects. Trends Genet 21: 249-253.
193. Brown S (2008) Miscarriage and its associations. Semin Reprod Med 26: 391-400.
194. Rosenbusch B (2004) The incidence of aneuploidy in human oocytes assessed by conventional cytogenetic analysis. Hereditas 141: 97-105.
195. Torres EM, Williams BR, Amon A (2008) Aneuploidy: cells losing their balance. Genetics 179: 737-746.

REFERENCES
72
196. Hassold TJ, Jacobs PA (1984) Trisomy in man. Annu Rev Genet 18: 69-97.
197. Lindsley DL, Sandler L, Baker BS, Carpenter AT, Denell RE, et al. (1972) Segmental aneuploidy and the genetic gross structure of the Drosophila genome. Genetics 71: 157-184.
198. Hodgkin J, Horvitz HR, Brenner S (1979) Nondisjunction mutants of the nematode Caenorhabditis elegans. Genetics 91: 67-94.
199. Stankiewicz P, Lupski JR (2010) Structural variation in the human genome and its role in disease. Annu Rev Med 61: 437-455.
200. Williams BR, Prabhu VR, Hunter KE, Glazier CM, Whittaker CA, et al. (2008) Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science 322: 703-709.
201. Holland AJ, Cleveland DW (2009) Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol 10: 478-487.
202. Schrider DR, Hahn MW (2010) Gene copy-number polymorphism in nature. Proc Biol Sci 277: 3213-3221.
203. McCarroll SA, Hadnott TN, Perry GH, Sabeti PC, Zody MC, et al. (2006) Common deletion polymorphisms in the human genome. Nat Genet 38: 86-92.
204. Bridges CB (1935) Salivary chromosome maps with a key to the banding of the chromosomes of Drosophila melanogaster. J Hered 26: 60-64.
205. Sorsa V (1988) Chromosome maps of Drosophila. Inc: CRC press. 206. Ashburner M, Golic KG, Hawley RS (2005) Drosophila A laboratory
handbook. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press.
207. Marygold SJ, Roote J, Reuter G, Lambertsson A, Ashburner M, et al. (2007) The ribosomal protein genes and Minute loci of Drosophila melanogaster. Genome Biol 8: R216.
208. Lambertsson A (1998) The Minute genes in Drosophila and their molecular functions. Adv Genet 38: 69-134.
209. Birchler JA, Hiebert JC, Paigen K (1990) Analysis of autosomal dosage compensation involving the alcohol dehydrogenase locus in Drosophila melanogaster. Genetics 124: 679-686.
210. Devlin RH, Holm DG, Grigliatti TA (1982) Autosomal dosage compensation Drosophila melanogaster strains trisomic for the left arm of chromosome 2. Proc Natl Acad Sci U S A 79: 1200-1204.
211. Devlin RH, Holm DG, Grigliatti TA (1988) The influence of whole-arm trisomy on gene expression in Drosophila. Genetics 118: 87-101.
212. Guo M, Birchler JA (1994) Trans-acting dosage effects on the expression of model gene systems in maize aneuploids. Science 266: 1999-2002.

REFERENCES
73
213. Makarevitch I, Phillips RL, Springer NM (2008) Profiling expression changes caused by a segmental aneuploid in maize. BMC Genomics 9: 7.
214. McAnally AA, Yampolsky LY (2010) Widespread transcriptional autosomal dosage compensation in Drosophila correlates with gene expression level. Genome Biol Evol 2: 44-52.
215. FitzPatrick DR, Ramsay J, McGill NI, Shade M, Carothers AD, et al. (2002) Transcriptome analysis of human autosomal trisomy. Hum Mol Genet 11: 3249-3256.
216. Zhang Y, Oliver B (2007) Dosage compensation goes global. Curr Opin Genet Dev 17: 113-120.
217. Torres EM, Dephoure N, Panneerselvam A, Tucker CM, Whittaker CA, et al. (2010) Identification of aneuploidy-tolerating mutations. Cell 143: 71-83.
218. Malone JH, Cho DY, Mattiuzzo NR, Artieri CG, Jiang L, et al. (2012) Mediation of Drosophila autosomal dosage effects and compensation by network interactions. Genome Biol 13: r28.
219. Franke A, Dernburg A, Bashaw GJ, Baker BS (1996) Evidence that MSL-mediated dosage compensation in Drosophila begins at blastoderm. Development 122: 2751-2760.
220. Kind J, Akhtar A (2007) Cotranscriptional recruitment of the dosage compensation complex to X-linked target genes. Genes Dev 21: 2030-2040.
221. Straub T, Grimaud C, Gilfillan GD, Mitterweger A, Becker PB (2008) The chromosomal high-affinity binding sites for the Drosophila dosage compensation complex. PLoS Genet 4: e1000302.
222. Birchler JA, Pal-Bhadra M, Bhadra U (2003) Dosage dependent gene regulation and the compensation of the X chromosome in Drosophila males. Genetica 117: 179-190.
223. Straub T, Gilfillan GD, Maier VK, Becker PB (2005) The Drosophila MSL complex activates the transcription of target genes. Genes Dev 19: 2284-2288.
224. Landfors M, Philip P, Rydén P, Stenberg P (2011) Normalization of high dimensional genomics data where the distribution of the altered variables is skewed. PLoS One 6: e27942.
225. Gorchakov AA, Alekseyenko AA, Kharchenko P, Park PJ, Kuroda MI (2009) Long-range spreading of dosage compensation in Drosophila captures transcribed autosomal genes inserted on X. Genes Dev 23: 2266-2271.
226. Lucchesi JC (1998) Dosage compensation in flies and worms: the ups and downs of X-chromosome regulation. Curr Opin Genet Dev 8: 179-184.

REFERENCES
74
227. Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, et al. (2007) Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 317: 916-924.
228. Tang YC, Williams BR, Siegel JJ, Amon A (2011) Identification of aneuploidy-selective antiproliferation compounds. Cell 144: 499-512.
229. Cryderman DE, Vitalini MW, Wallrath LL (2011) Heterochromatin protein 1a is required for an open chromatin structure. Transcription 2: 95-99.
230. Meller VH (2003) Initiation of dosage compensation in Drosophila embryos depends on expression of the roX RNAs. Mech Dev 120: 759-767.
231. de Wit E, Greil F, van Steensel B (2005) Genome-wide HP1 binding in Drosophila: developmental plasticity and genomic targeting signals. Genome Res 15: 1265-1273.
232. Jin Y, Wang Y, Johansen J, Johansen KM (2000) JIL-1, a chromosomal kinase implicated in regulation of chromatin structure, associates with the male specific lethal (MSL) dosage compensation complex. J Cell Biol 149: 1005-1010.
233. Jin Y, Wang Y, Walker DL, Dong H, Conley C, et al. (1999) JIL-1: a novel chromosomal tandem kinase implicated in transcriptional regulation in Drosophila. Mol Cell 4: 129-135.
234. Zhang W, Deng H, Bao X, Lerach S, Girton J, et al. (2006) The JIL-1 histone H3S10 kinase regulates dimethyl H3K9 modifications and heterochromatic spreading in Drosophila. Development 133: 229-235.







