

Cystic Fibrosis FRACP teaching
October 2007
Mark O’Carroll
Respiratory Physician

Introduction• Mode of inheritence• Genetic defect• Pathology• Pathophysiology• Clinical features• Diagnosis• Therapy• Survival

Cystic Fibrosis• The most common lethal inherited disease
affecting Caucasians• Autosomal recessive• Incidence 1:2500 Caucasian populations• Carrier frequency 4%• Single gene disease (CFTR 7q)• Gene discovered 1989
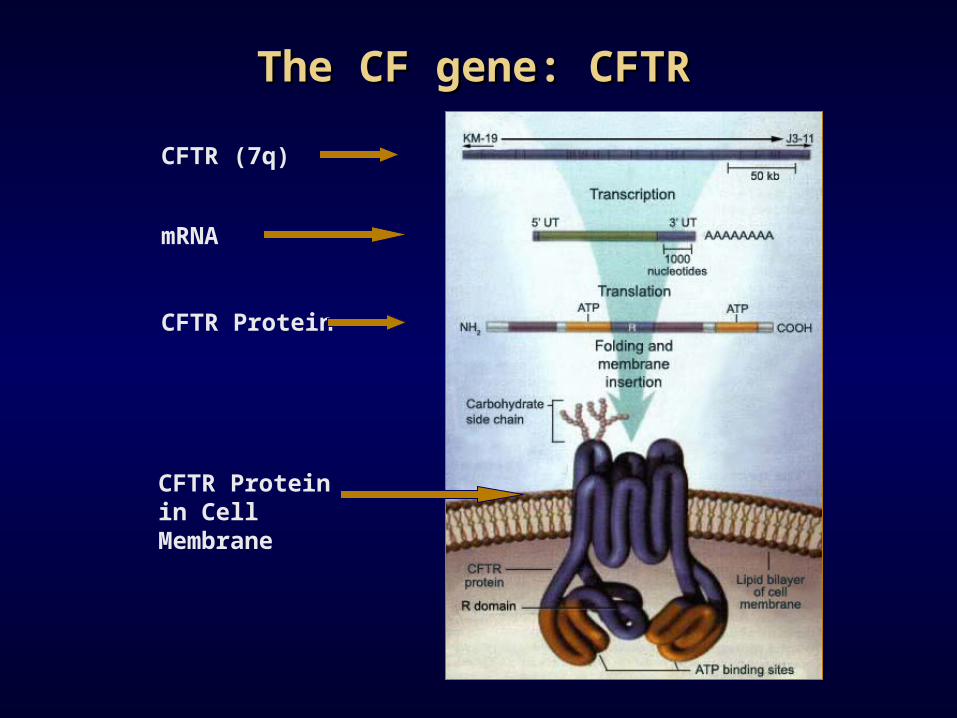
The CF gene: CFTRThe CF gene: CFTR
CFTR (7q)
mRNA
CFTR Protein
CFTR Protein in Cell Membrane

CFTR
• Gene on 7q
• 27 exons code for 1480 amino acid protein
• Member of the ATP-binding cassette (ABC) family of transporters
• Codes for a voltage gated chloride channel

CFTR mutations• > 1000 mutations described• ∆F508 accounts for 70%• Certain mutations occur more frequently in
particular ethnic groups• Commercially available genetic tests screen
for the 31 most common mutations found in NZ population (accounts for 85% of the mutations)

Pathology


Normal Airway CF Airway

Pathophysiology

Molecular biology of CFTR
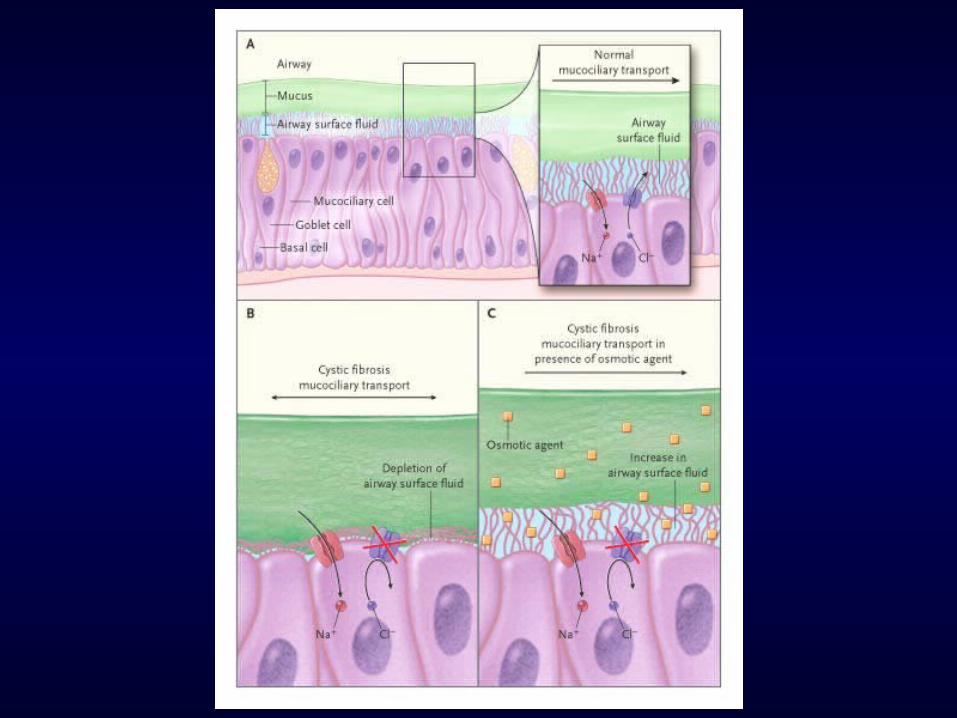
• There has been considerable debate about the mechanism by which defective CFTR impacts on airway physiology and mucociliary clearance (MCC)
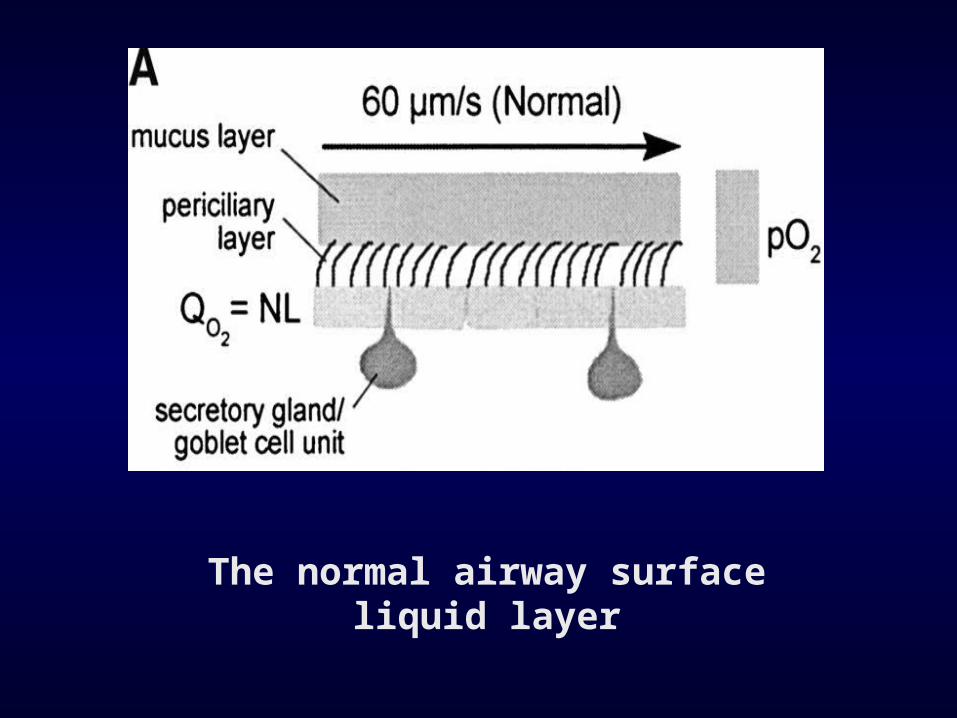
The normal airway surface liquid layer

Pathogenesis• Soon after birth there is intense neutrophilic
inflammation in the CF lung
• IL8 is the predominant cytokine and sentinel neutrophil chemo attractant
• IL8 is produced by stimulated epithelial cells, macrophages and neutrophils
• Various factors stimulate further IL8 production to sustain neutrophil influx (including IL1, TNF, LPS, Pseudomonas antigens and neutrophil elastase)

Pathogenesis• TNF stimulates neutrophil secretory and oxidative
processes
• TNF and IL1 prime neutrophils for a heightened response to chemo attractants
• Neutrophils then release massive amounts of elastase and other proteases which overwhelm local host defenses including 1AT and secretory leukocyte protease inhibitor (SLPI)

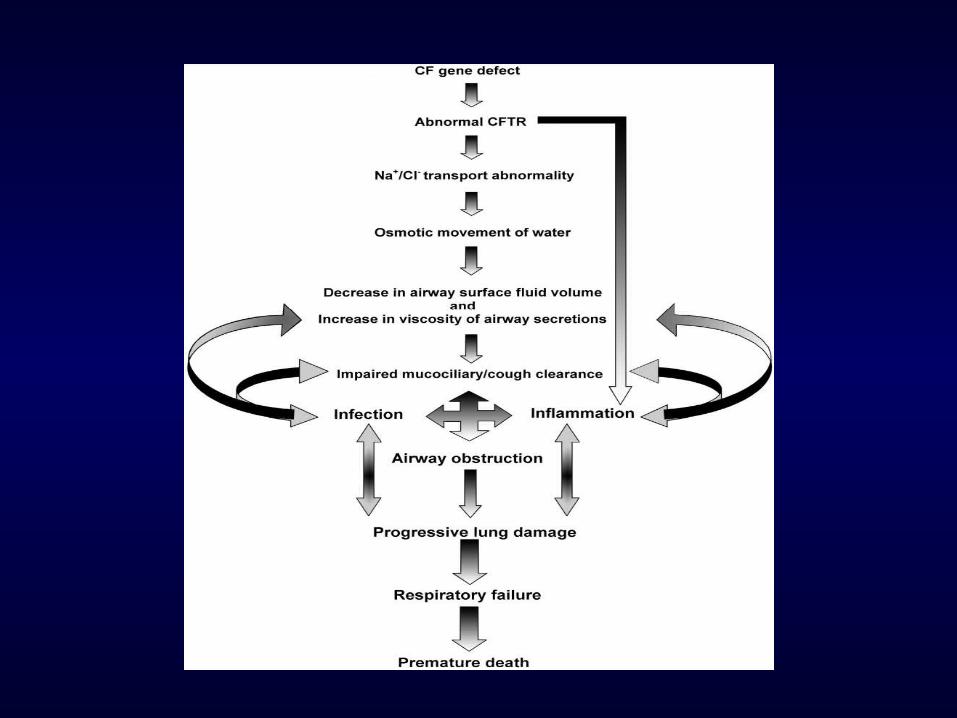
Pathogenesis
Neutrophil breakdown
Large amounts high MW DNA
viscosity of endobronchial secretions
mucociliary clearance

ENT: Chronic SinusitisNasal Polyps
GI: Pancreatic insufficiency (malnutrition)Pancreatitis (PS)
Meconium ileus and DIOSBiliary cirrhosis and portal hypertension
Adapted from Welsh and Smith. Sci Am. 1995;273:52-59.
Classical Clinical Features
Sex organs: Obstructive azoospermia (CBAVD)
Lungs: Cough and sputumAirflow obstructionRecurrent infection (Psa, S. aureus)

Diagnosis of CFConsensus Statement J Pediatr 1998
One or more typical phenotypic featuresoror
a history of CF in a siblinga history of CF in a siblingoror
a positive newborn screening testa positive newborn screening test
plusplus
Laboratory evidence of a CFTR abnormalityLaboratory evidence of a CFTR abnormality[Sweat test, 2 CFTR mutations or NPD]

CF Foundation Patient Registry 2003

Therapy

Approach to the Management of CF Lung Disease
Correction of Underlying DefectPharmacologic TherapyGene Therapy
Reduction in the Mucus BurdenAirway Clearance TechniquesPhysical TrainingDN’aseOther Mucolytic TherapyHyperosmolar Agents
Control of InfectionNebulised anti-pseudomonal ABsIV anti-pseudomonal ABsOral antibioticsVaccinationLong-term oral anti-staph ABs
Control of InflammationOral corticosteroidsICSNSAIDsMacrolides
Other ManagementBronchodilatorsTheophyllineLTRAsFlu vaccinationLTOTNIVLung transplantation

Macrolides• Most significant recent advance in CF therapy• 3 RCTs (n=300 pts) + Cochrane review• All used azithromycin but probable class effect• Observed improvements in;
lung function hospitalisation rate intravenous antibiotic use quality of life weight

Macrolides• Mechanism of action uncertain• Potential mechanisms;
Anti-inflammatory Up-regulation of CFTR Antibacterial effects
• Seem to work in patients without PsA infection • Appropriate use remains unclear
Biofilm formation
Quorum sensing
Bacterial adherence

A Controlled Trial of Long-Term Inhaled Hypertonic Saline in Patients with Cystic Fibrosis
Mark R. Elkins, Michael Robinson, Barbara R. Rose, Colin Harbour, Carmel P. Moriarty, Guy B. Marks, Elena G. Belousova, Wei Xuan,
and Peter T.P. Bye.
NEJM 2006; 354(3): 229-240

Study Overview
• Patients with cystic fibrosis have inspissated mucus that is thought to contribute to the pulmonary exacerbations characteristic of the disease
• As compared with treatment with normal saline, twice-daily treatment with inhaled hypertonic saline after the inhalation of a bronchodilator did not affect the linear rate of change in the forced expiratory volume in one second (FEV
1) but was associated with improved
FEV1 values and with fewer and shorter pulmonary
exacerbations

Absolute Change from Baseline in FVC (Panel A) and the FEV1 (Panel B)
Elkins, M. et al. N Engl J Med 2006;354:229-240

Effect of Hypertonic Saline on Lung Function
Elkins, M. et al. N Engl J Med 2006;354:229-240

Percentage of Participants in Each Group Remaining Free of Exacerbations during the Trial
Elkins, M. et al. N Engl J Med 2006;354:229-240

Conclusion
Hypertonic saline preceded by a bronchodilator is an inexpensive, safe, and effective additional therapy
for patients with cystic fibrosis

Effect of Aerosolized Recombinant Human DN’ase on Exacerbations of Respiratory Symptoms and on Pulmonary Function in Patients with Cystic Fibrosis
Henry J. Fuchs, Drucy S. Borowitz, David H. Christiansen, Edward M. Morris, Martha L. Nash, Bonnie W. Ramsey, Beryl J.
Rosenstein, Arnold L. Smith, Mary Ellen Wohl,
for The Pulmozyme Study Group
NEJM 1994; 331: 637-642
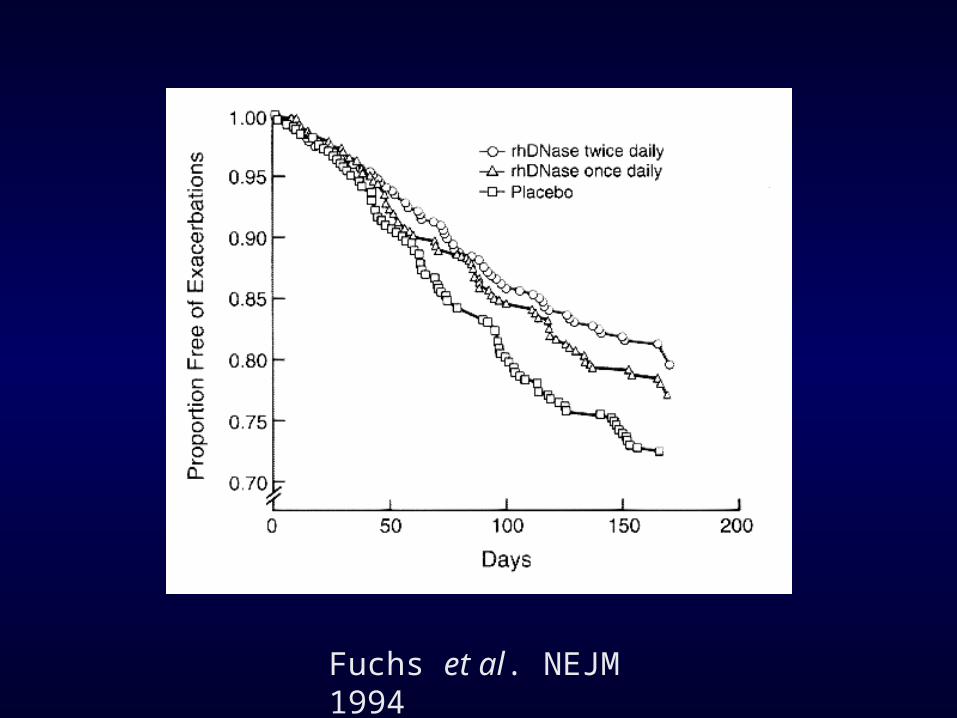
Fuchs et al. NEJM 1994
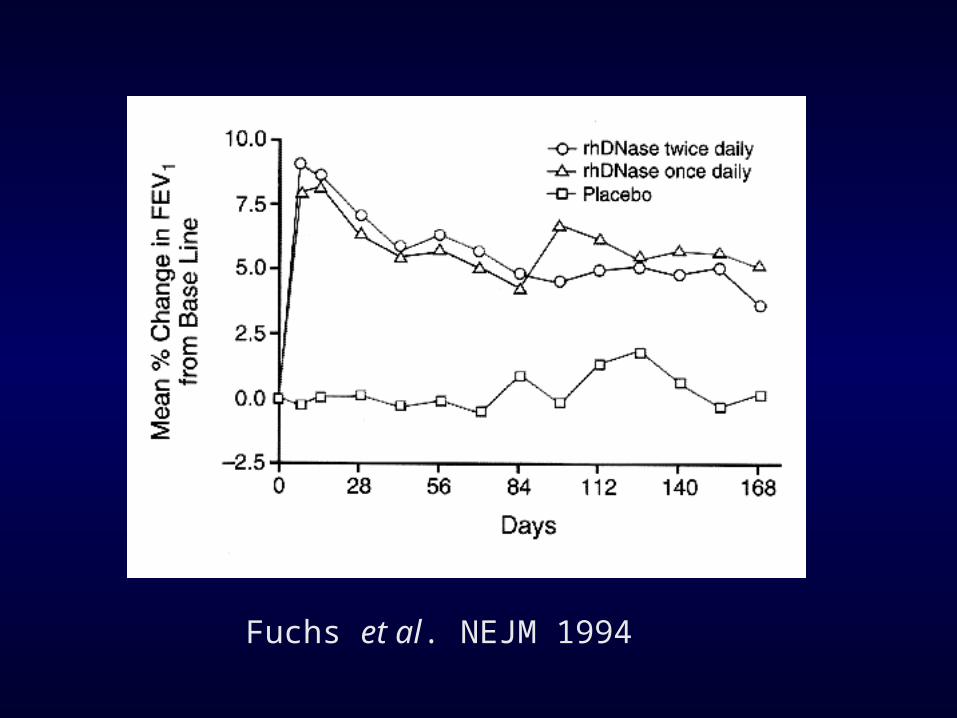
Fuchs et al. NEJM 1994

Intermittent Administration of Inhaled Tobramycin in Patients with Cystic Fibrosis
Bonnie W. Ramsey, Margaret S. Pepe, Joanne M. Quan, Kelly L. Otto, A. Bruce Montgomery, Judy Williams-Warren, Michael Vasiljev-K, Drucy Borowitz, C. Michael Bowman, Bruce C.
Marshall, Susan Marshall, Arnold L. Smith,
for The Cystic Fibrosis Inhaled Tobramycin Study Group
NEJM Ramsey et al. 1999; 340 (1): 23

NEJM Ramsey et al. 1999; 340 (1): 23

Survival

Median survival age in cystic fibrosis, 1985–2001. Data from the U.S. Cystic Fibrosis Foundation Patient Registry. Median survival in 2001 was 33.4 years.

Improved survival• Screening (early diagnosis)• Multi-disciplinary team based care• Nutritional supplementation• Better management of meconium ileus• Enhanced measures for sputum clearance• Improved antibiotics• Better management of respiratory failure• Transplantation

Impact of FEV1 on survival
Thorax 2001

Impact of nutrition on survival
Thorax 2001







