

Discovery of berberine that targetedly induce autophagic degradation of both
BCR-ABL and BCR-ABL T315I through recruiting LRSAM1 for overcoming
imatinib-resistance
Zhao Yin1,2,3,4†
, Guiping Huang1,3,4†
, Chunming Gu1,2,3,4†
, Yanjun Liu1,3,4
, Juhua
Yang1,3,4
, Jia Fei1,2,3,4*
1 Department of Biochemistry and Molecular Biology, Medical College of Jinan
University, Guangzhou 510632, China
2 Institute of Chinese Integrative Medicine, Medical College of Jinan University,
Guangzhou 510632, China
3 Engineering Technology Research Center of Drug Development for Small Nucleic
Acids, Guangdong, China
4 Antisense Biopharmaceutical Technology Co., Ltd., Guangzhou, China
† Zhao Yin, Guiping Huang and Chunming Gu contributed equally to this work
Running title: Berberine induces autophagic BCR-ABL T315I degradation
Keywords: berberine, surface plasmon resonance (SPR)-LC-MS/MS, target
identification, imatinib resistance, BCR-ABL
Disclosure of potential conflicts of interest
The authors declare no potential conflicts of interest.
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

Author Information
Zhao Yin: 1 Department of Biochemistry and Molecular Biology, Medical College of Jinan
University, Guangzhou 510632. 2 Institute of Chinese Integrative Medicine, Medical College
of Jinan University, Guangzhou 510632, China. Email: [email protected]
Guiping Huang: 1 Department of Biochemistry and Molecular Biology, Medical College of
Jinan University, Guangzhou 510632, China. Email: [email protected]
Chunming Gu: 1 Department of Biochemistry and Molecular Biology, Medical College of
Jinan University, Guangzhou 510632, China. 2 Institute of Chinese Integrative Medicine,
Medical College of Jinan University, Guangzhou 510632, China. Email:
Yanjun Liu: 1 Department of Biochemistry and Molecular Biology, Medical College of Jinan
University, Guangzhou 510632, China. Email: [email protected]
Juhua Yang: 1 Department of Biochemistry and Molecular Biology, Medical College of Jinan
University, Guangzhou 510632, China. Email: [email protected]
Jia Fei: 1 Department of Biochemistry and Molecular Biology, Medical College of Jinan
University, Guangzhou 510632, China. 2 Institute of Chinese Integrative Medicine, Medical
College of Jinan University, Guangzhou 510632, China. Email: [email protected]
*Corresponding author:
Jia Fei, Department of Biochemistry and Molecular Biology, Medical College of Jinan
University, 601 Western Huangpu Avenue, 510632 Guangzhou, China; Tel: 86-20-85220256;
Fax: 86-20-85221343; Email: [email protected]
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

Translational Relevance
Acquired imatinib resistance is frequently characterized by BCR-ABL mutations that
affect imatinib binding and kinase inhibition in patients with CML. In this study, we
found that BBR, a Chinese traditional medicine, significantly inhibited the cell
viability and colony formation of CML cells and prolonged survival in CML mouse
models with imatinib sensitivity and resistance. Further studies demonstrated that
BBR not only inhibit BCR-ABL tyrosine kinase activity but also directly bind to
ABL1, which induce autophagic degradation of both BCR-ABL and BCR-ABL
T315I through recruiting LRSAM1 for overcoming imatinib-resistance. Our finding
would be of remarkable value for further therapy of CML with BCR-ABL-mutation.
Abstract
Purpose: Imatinib, the breakpoint cluster region protein (BCR)/Abelson murine
leukemia viral oncogene homolog (ABL) inhibitor, is widely used to treat chronic
myeloid leukemia (CML). However, imatinib resistance develops in many patients.
Therefore, new drugs with improved therapeutic effects are urgently needed.
Berberine (BBR) is a potent BCR-ABL inhibitor for imatinib-sensitive and -resistant
CML.
Experimental design: Protein structure analysis and virtual screening were used
to identify BBR targets in CML. Molecular docking analysis, surface plasmon
resonance imaging (SPRi), nuclear magnetic resonance (NMR) assays, and
thermoshift assays were performed to confirm the BBR target. The change in
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

BCR-ABL protein expression after BBR treatment was assessed by western blotting.
The effects of BBR were assessed in vitro in cell lines, in vivo in mice, and in human
CML bone marrow cells as a potential strategy to overcome imatinib resistance.
Results: We discovered that BBR bound to the protein tyrosine kinase (PTK)
domain of BCR-ABL. BBR inhibited the activity of BCR-ABL and BCR-ABL with
the T315I mutation, and it also degraded these proteins via the autophagic lysosome
pathway by recruiting E3 ubiquitin-protein ligase LRSAM1. BBR inhibited the cell
viability and colony formation of CML cells and prolonged survival in CML mouse
models with imatinib sensitivity and resistance.
Conclusions: The results show that BBR directly binds to and degrades BCR-ABL
and BCR-ABL T315I via the autophagic lysosome pathway by recruiting LRSAM1.
The use of BBR is a new strategy to improve the treatment of CML patients with
imatinib sensitivity or resistance.
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

Introduction
Chronic myeloid leukemia (CML) results from the transformation of primitive
hematopoietic cells by the breakpoint cluster region protein (BCR)/Abelson murine
leukemia viral oncogene homolog (ABL) oncogene. Imatinib, a tyrosine kinase
inhibitor that binds to the ATP-binding site of ABL, is remarkably effective at treating
CML. However, resistance, which develops in many patients, is the main barrier to
prolonged survival, and the use of a single tyrosine kinase inhibitor cannot cure CML.
Thus, the development of novel targeted therapeutic agents, or the use of imatinib in
combination with other drugs, is required to improve response rates and overcome
imatinib resistance (1-4).
Here, we describe the identification and characterization of the mechanism of action
of berberine (BBR) as an ABL-binding agent that can capable of degrading
BCR-ABL and overcome imatinib resistance both in vitro and in vivo. BBR is a
clinically important natural isoquinoline alkaloid derived from the plant Berberis
vulgaris that is known to have multiple pharmacological activities, including
anti-cancer effects (5-7). However, the mechanisms underlying the effects of BBR on
cancer cells have not been fully elucidated. In this study, BBR significantly inhibited
CML cell viability and colony formation and prolonged survival in both
imatinib-sensitive and -resistant CML mouse models. Hence, BBR is a promising new
inhibitor for use in CML treatment, particularly for imatinib-resistant CML.
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

Materials and Methods
Human samples and cell lines
Healthy peripheral blood mononuclear cells (PBMCs) and CML bone marrow (BM)
mononuclear cells were obtained from adult donors in Guangdong Provincial
Emergency Hospital/the Guangdong Second Provincial General Hospital after written
informed consent was obtained according to the institutional guidelines and the
Declaration of Helsinki. These cells were cultured in medium containing 100 ng/mL
stem cell factor (SCF), 100 ng/mL granulocyte-colony stimulating factor (G-CSF), 20
ng/mL FMS-like tyrosine kinase 3 (FLT3), 20 ng/mL interleukin (IL)-3, and 20
ng/mL IL-6. Preparation of CD34+ from umbilical cord blood was performed with the
EasySep human cord blood CD34+ selection kit (STEMCELL Technologies)
according to the manufacturer’s instructions and incubated in Iscove's modified
Dulbecco's medium supplemented with 10% FBS (21, 24). The studies were approved
by Institutional Review Board, Jinan University (Guangzhou, China)
The imatinib-sensitive CML cell line K562 was purchased from Shanghai Cell Bank
(Chinese Academy of Sciences, Shanghai). The imatinib-sensitive CML cell line
KCL-22 was kindly provided by Dr. Muschen (The Children’s Hospital, Los Angeles,
CA, USA). The imatinib-resistant CML cell line SFO2 (which does not express
BCR-ABL) was obtained from Dr. Muschen (The Children’s Hospital, Los Angeles,
CA, USA). BaF3-P210 and BaF3-P210-T315I cells were kindly presented by
Professor Wen li Feng (Chong qin Medical University, China). These cells were
grown in Roswell Park Memorial Institute (RPMI) 1640 medium containing 10%
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

fetal bovine serum (FBS) and 1% penicillin/streptomycin (all purchased from GIBCO,
Grand Island, NY, USA). Plat-E cells were generously provided by Dr. Jing Xuan Pan
(Pharmacy College Jinan University, Guangzhou, China)(8). Imatinib and BBR were
purchased from Sigma (Santa Clara, CA, USA).
Identification and quantification of BBR target proteins using surface plasmon
resonance (SPR) and high-performance liquid chromatography (HPLC) mass
spectrometry (MS)
To explore the direct cell target of BBR, we designed an SPR-HPLC-MS assay. BBR
at a concentration of 100 mM was formulated with 50% dimethyl sulfoxide (DMSO).
Consistent BBR samples were produced on a chip surface by auto-spotting three times
using a BioDot™-1520 array printer (California,USA). The chip surface was printed
with a 50×50 matrix of 18.75 µL (1.875 μmol) BBR sample in total, with 2.5 nL of
projected point of the solution.
Calibration of cell lysate: The protein concentration of K562 and KCL22 cell lysate,
including membrane proteins, was calibrated after extraction with certain SOPs, using
a Bicinchoninic Acid (BCA) Protein Assay Kit (Thermo Fisher), which led to a
measurement of 332.1 mg/mL. The concentration was adjusted using a 1X lysate
stock solution to a final concentration of 200 μg/mL.
Calibration of chip performance: Each chip was manufactured by Lumera Co. Ltd
Kaiserslautern, Germany)., with batch difference <0.5%. The resonance angle was
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

automatically tuned to the optimum value using a bScreen LB 991 system (Berthold
Technologies, Germany).
Target protein capture process: During the SPR assays, BBR was immobilized on the
surface of the chip and K562 or KCL22 cell lysate was used as the liquid phase. The
H2 sample curve signal indicated target protein binding on the area spotted with BBR.
The background curve indicated the change in the signal in the non-spotted area.
The timing of the procedures was as follows. At 0–260 s, the system was pre-washed
to infiltrate the surface of the chip with running buffer. At this point, the resonance
intensity was about 0 resonance units (RU). At 260–520 s, binding began, with BBR
on the chip surface starting to capture the protein targets in the cell lysate. At 520–820
s, the chip was washed to gradually remove non-bound and non-specific molecules,
while the target protein that specifically bound to the BBR remained on the chip
surface. Resonance intensity decreased until it reached a plateau at ~542.65 RU. As
non-specific binding to the non-spotted areas gradually decreased, the background
resonance intensity gradually decreased to baseline (~37.72 RU), i.e., the chip
background noise returned to the normal level.
Molecular docking
The molecular docking assays between BBR and ABL1 were generated as described
previously(9). Using the Protein Preparation Wizard module of Schrödinger Maestro
9.3 software (Schrödinger, Cambridge, MA, USA), a model of BBR with ABL1 was
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

processed using default settings. A molecular docking analysis was implemented
using the Ligand Docking tab of the Glide module. Regarding the docking parameters,
precision was set as extra precision (XP), ligand sampling was set as flexible, the
number of poses per ligand was set at 5, and other parameters were set at the default
values. The residue compounds were further subjected to Prime molecular mechanics
(MM)-generalized Born surface area (GBSA) calculations and vision analysis.
Protein expression and purification
Protein expression and purification assays were performed according to our previous
report(10). Escherichia coli BL21 (DE3) was transformed with a
hexa-histidine-tagged recombinant human protein tyrosine kinase (PTK) domain of
ABL1 (isoform 2). After bacterial growth in Terrific Broth containing 30 mg/L
kanamycin at 37°C to an optical density (OD) at 600 nm of 0.4–0.6, induction was
carried out at 18°C using 0.5 mM isopropyl-β-D-thiogalactoside (IPTG), and growth
was then continued at 18°C overnight. Bacteria were collected by centrifugation. The
pellets were immediately resuspended in lysis buffer (20 mM PB, 150 mM NaCl, pH
7.4) containing a protease inhibitor cocktail. Cell lysis was performed in an ultrasonic
ice bath to generate crude protein samples. Extracted proteins were diluted five-fold
with balance buffer (500 mM NaCl, 20 mM tris, pH 8.0), incubated with Ni-agarose
beads (CWBio, Beijing, China), and washed to remove unbound proteins and
proteases. The proteins were eluted using different concentrations of imidazole (20,
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

50, 200, and 500 mM) to determine the absorption peak, and then purified samples of
the PTK domain of ABL1 were collected according to the identified absorption peak.
SPR imaging (SPRi)
SPRi assays were performed as previously described (9). BBR was bound to a sensor
chip, and protein samples were injected into the chip at a rate of 2 µL/s at 25℃. Oval
regions of interest (ROIs) in the imaging area were automatically set using the data
collection software. ROIs of rapamycin and DMSO were used as positive and
negative controls, respectively. The protein samples were diluted in
phosphate-buffered saline (PBS) containing Tween 20 (0.05%), pH 7.4, and used as
analytes with an association and dissociation flow rate of 2 µL/s at different
concentrations by serial dilution. A solution of glycine-HCl (pH 2.0) was used to
regenerate the surface of the sensor chip by removing bound proteins, enabling the
sensor chip to be reused for subsequent analyte injections.
Differential scanning fluorimetry (DSF) assay
Thermoshift assays were carried out in 96-well PCR plates with a real-time
thermo-cycler (CFX96, Bio-Rad California, USA) and the fluorescent dye SYPRO
Orange (1:1000). The fluorescence signal was initially measured at a temperature of
25°C, which was then increased to 100°C with a step size of 0.5°C/min. Interactions
of SYPRO Orange with hydrophobic surfaces increase the quantum yield of the dye
(11). Binding was assessed using a melting curve analysis.
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

Nuclear magnetic resonance (NMR) assay
BBR and the BBR-PTK complex were dissolved in heavy water. All 1H-NMR
experiments were performed on an AVANCE I-600 spectrometer (Bruker, Karlsruhe,
Germany) at 25°C (298.0 K), using a probe tuned at 600 MHz. The chemical shift of
BBR was assessed.
Extraction of BM cells from mice
BM cells were obtained from C57/BL6 mice by flushing the cavities of femurs and
tibias with PBS. After filtration through a 70-mm filter and depletion of erythrocytes
using lysis buffer (BD PharmLyse, BD Biosciences, New Jersey , USA), the cells
were washed with PBS and cultured with 10 ng/mL recombinant mouse IL-3, 25
ng/mL recombinant mouse IL-6, and 50 ng/mL recombinant mouse SCF medium
(12).
Cell viability assay
Cell viability was assessed using 3-(4,5-dimethylthiazol-2-yl)-2,5-
diphenyltetrazolium bromide (MTT) assays. Cells were seeded in 96-well plates at a
density of 5 × 103 cells/well, treated with either BBR or imatinib at the indicated
concentrations(0-20μM), and incubated at 37°C for 48 h. Thereafter, 20 μL MTT was
added to each well. After incubation for 4 h, OD at 540 nm was determined using a
microplate reader.
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

Colony formation assay
A colony formation assay for dispersed single cells was performed. Single cells
(BaF3-P210 or BaF3-P210-T315I) were seeded into a 24-well plate and mixed
completely with 0.9% methylcellulose solution in RPMI 1640 medium containing 20%
FBS and 5 μM BBR. Single cells were randomly and evenly distributed throughout
the wells. Colonies were formed during incubation for 1–2 weeks at 37°C with 5%
CO2. Light microscopy was used to observe and count the colonies (> 50 cells).
Real-time PCR
To assess the levels of BCR-ABL mRNA, total RNA was isolated from K562 cells
using a Trizol Total RNA Isolation kit (Tiangen, Beijing, China) and reverse
transcribed using a Fast Quant RT Kit (Tiangen). An SYBR Green kit (Tiangen) was
used for PCR. BCR-ABL primer sequences (forward,
AGCATTCCGCTGACCATCAA; reverse, GCCTAAGACCCGGAGCTTTT) were
designed using Primer-Basic Local Alignment Search Tool (BLAST). β-actin served
as an internal control, with β-actin primers being purchased from Sangon Biotech
(Shanghai, China). The cycling conditions were as follows: 95℃ for 15 min; 30
cycles of 95℃ for 10 s, 55℃ for 30 s, and 72℃ for 30 s. Data were processed using
CFX Manager 3.0 software (Bio-Rad).
PTK activity assay
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

CML Cells were diluted to 100/mL in PBS (pH 7.2–7.4) and lysed by repeated
freezing and thawing to release the intracellular components. After centrifugation for
20 min at 2,000–3,000g, the supernatants were carefully collected. If precipitates
appeared during collection, the samples were centrifuged again. PTK standards were
prepared according to the manufacturer’s instructions for the PTK activity assay. The
total volume in each well in the micro-enzyme-linked immunosorbent assay (ELISA)
strip plates was 50 μl. Ten wells were used for a 1:2 dilution series of standards (final
concentrations: 3600, 2400, 1200, 600, and 300 U/mL). One well was left empty as a
negative control. In the sample wells, 40 μl sample dilution buffer and 10 μL cell
lysate samples were added. Samples were loaded into the bottom of wells without
touching the well walls. The solutions were mixed well by gentle shaking and the
plates were sealed with plate membrane and incubated for 30 min at 37°C. The
solutions were then aspirated from the wells and the wells were washed for 30 s with
wash solution five times. Next, 50 μL horseradish peroxidase (HRP)-conjugated
reagent was added to each well, except the blank control, and then the plates were
incubated and washed as described above. Thereafter, 50 μL Chromogen Solution A
and 50 μL Chromogen Solution B were added to each well, mixed by gentle shaking,
and incubated at 37°C for 15 min. Finally, 50 μL stop solution was added to each well
to terminate the reaction and the OD at 450 nm was determined using a microplate
reader. The OD value of the blank control well was set at zero. Assays were
performed in triplicate, and two-sided paired t tests were used for statistical analyses.
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

In vitro tyrosine kinase assay
The in vitro tyrosine kinase assay was performed as previously described with some
modifications(13). The ABL kinase activity was tested using the Kinase-Glo
Luminescent Kinase Assay Platform (Promega Corporation, WI, USA, Cat# V6072),
which provides a homogeneous, High-throughput screening method for measuring
kinase activity by quantization the amount of ATP remaining in solution following a
kinase reaction. The assays are performed in a single well of a multi well plate by
adding a volume of Kinase Glo Reagent equal to the volume of a completed kinase
reaction and measuring luminescence. The luminescent signal is correlated with the
amount of ATP present and is inversely correlated with the amount of kinase activity.
Briefly, a 50 µL mixture containing 1 µL of BBR (0, 2, 4, 8, or 10 µmol/L), 2µL of
(1µg) ABL1(ABL protein tyrosine kinase:Pro137-Ser554, 65 kD, containing protein
tyrosine kinase activity, was supplied by Sino Biological Inc (Shanghai, China), 2µL
of (1µg) ABL1 kinase substrate (EAIYAAPFAKKK) (Substrate peptide of ABL1 was
synthesized by GL Biochem (Shanghai, China) and 5 µL of ATP (2 µmol/L) in 40 µL
kinase buffer [50 mmol/L HEPES (pH 7.3), 10 mmol/L MgCl2, 0.1% BSA, 2 mmol/L
DTT ] was added to the wells, and the reactions were incubated for 20 min at 30°C;
the control wells did not contain BBR. The reaction was stopped by addition of 50 µL
Kinase-Glo reagent, and the plate was read after a 10-min incubation time at
Cytation™5 Cell Imaging Multi-Mode Reader (Bio-Tek, USA)
Western blotting
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer in the presence of
protease inhibitors (Selleck Chemicals, Houston, TX, USA). Protein concentrations
were determined using the BCA method (Bioss, Beijing, China), and the proteins
were then denatured in Laemmli sample buffer (Bio-Rad) for 5 min at 100°C. Total
protein extracts (50 μg) were subjected to sodium dodecyl sulfate polyacrylamide gel
electrophoresis (SDS-PAGE) on 10% gels and transferred to nitrocellulose
membranes. The membranes were then blocked with 5% (w/v) fat-free milk powder
in tris-buffered saline with Tween 20 (TBST) for 1 h. The membranes were first
incubated with a primary anti-BCR antibody (1:1000; Abcam, Cambridge,UK) at
4°C overnight and then washed twice with TBST for 10 min and incubated with
HRP-conjugated secondary antibody for 1 h. BCR protein was detected using a
chemiluminescence kit (Millipore, MA, USA). Densitometric quantification was
performed using ImageJ software (National Institutes of Health, USA).
Plasmid Transfections
To analyze the effects of LRSAM1 in BBR mediated degradation of BCR-ABL,
K562 cells stably overexpressing LRSAM1 or control were established by
transfecting K562 cells with LRSAM1 expression vector or empty vector. The cells
were selected in medium containing puromycin (1μg/mL).
siRNA transfections
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

The sequences used in this study were: LRSAM1 siRNA target sequence
(CCACGATAATCAGCTGACA). The siRNA was synthesized and purified by
Ribobio company (Guang Zhou, China), and stored at −20°C. All siRNA (100 nM)
were transfected into K562 cells using Lipofectamine ™2000 according to the
manufacturer's instructions.
Immunoprecipitation
The immunoprecipitation assay was performed as described previously by us with
some modifications(14). K562 Cells were treated under the indicated conditions in
10-cm plates and then lysed with lysis buffer containing phosphatase and protease
inhibitor. Lysates were mixed with 4 μL primary anti-BCR antibody (Abcam) and
incubated overnight at 4°C with rocking. Protein A/G agarose beads (Santa Cruz, UK)
were added to the mixture and, after 4 h, the beads were pelleted, washed with lysis
buffer, resuspended in loading buffer, heated at 100°C for 10 min, and analyzed by
SDS-PAGE followed by western blotting. Antibodies against ABL1 (Santa Cruz) and
E3 ubiquitin-protein ligase LRSAM1 (Abcam) were used to detect the proteins in the
immunoprecipitates.
Autophagy assay
K562, KCL-22, BaF3-P210, and BaF3-P210-T315I cells were incubated for the
indicated times with different concentrations of BBR (0, 1, 3, and 5 μM). Thereafter,
the levels of the following autophagy-related proteins were assessed by western
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

blotting: light chain 3B (LC3B; CST, Boston,USA), Beclin1 (BECN1; CST, Boston,
USA), and P62 (Sigma, Louis, MO USA). Additionally, in experiments in which the
autophagy inhibitors chloroquine (CQ, 10 μM) and 3-methyladenine (MA, 25 μM;
Selleck Chemical ,Houston, TX, USA) was used, the cells were pre-treated for 4 h
with an autophagy inhibitor before the addition of BBR for 48h, or treated with the
autophagy inhibitor alone or BBR alone for 48h. After treatment, levels of BCR-ABL
protein were assessed by western blotting.
Colocalization immunofluorescence
K562 cells were treated with 5 μM BBR for 24 h, treated with lysosome dye
(KGMP006-1, Keygentec, Nanjing, China), smeared on slides, fixed for 10 min, and
incubated with anti-ABL or anti-P62 antibodies. Images were captured using a laser
scanning confocal microscope (TCS SP8, Leica, Weztlar, Germany). Additionally,
K562 cells were treated with 5 μM BBR for 24 h, smeared on slides, fixed for 10 min,
and incubated with anti-ABL or anti-LRSAM1 antibodies. Images were again
captured using the above microscope.
Imatinib-resistant CML T315I-luciferase mouse model
Experiments were performed on mice and these mice were sacrificed according to the
guidelines of the Jinan University Animal Research Committee. First, 106
BaF3-P210-T315I cells (BaF3 cells harboring the T315I-luciferase BCR-ABL
mutation, which express a mutant form of the BCR-ABL protein that is resistant to
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

imatinib) were injected into the tail veins of female BALB/c nu/nu mice (five mice
per group, 6 weeks of age). Before injection, the mice were irradiated with 2.5 Gy
X-rays. After 10 d, the mice were treated daily for 7 d via intraperitoneal (i.p.)
injection with BBR (15 mg/kg) or imatinib (50 mg/kg) and in vivo images were
assessed using an Xtreme system (Bruker).
To assess the effects of BBR in tumor xenograft models, 106 BaF3-P210-T315I cells
were implanted subcutaneously into female BALB/c nu/nu mice (6 weeks of age).
Tumors were allowed to reach 100 mm3 in size before the mice were randomly
assigned to treatment groups. Five mice per group were treated with BBR (15 mg/kg),
imatinib (50 mg/kg), BBR plus imatinib daily for 15 d or vehicle control (saline
solution) via i.p. injection twice daily for 15 d in two independent experiments.
Tumor volumes were assessed using vernier calipers, and the expression of ABL1 and
BCR-ABL in tumors was assessed by immunohistochemistry.
Retroviral construction
High-titer helper-free retroviruses were produced by transient transfection of Plat-E
cells with the retroviral construct murine stem cell virus (MSCV)-BCR-ABL-internal
ribosome entry site (IRES)-enhanced green fluorescent protein (EGFP), as described
previously (15).
Bone Marrow Transduction and Transplantation to Bulid CML-like Mice model
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

Bone marrow transduction was generated as described previously(8). Donor male
C57BL/6 mice (Jinan University Animal Research Center) were pretreated with
5-fluorouracil (5-FU, 200 mg/kg) and, 5d later, BM cells were harvested. These cells
were transduced for two rounds with the MSCV-BCR-ABL-IRES-EGFP retrovirus in
the presence of cytokines (SCF, IL3, and IL6). The cells were then transplanted into
sublethally irradiated (550 cGy) recipient female C57BL/6 mice. Following
transplantation, the mice were treated with vehicle, BBR (15 mg/kg/d, i.p.), or
imatinib (50 mg/kg/d, i.p.) for 14 d (16).
Immunohistochemistry
Tissue section staining was performed as described previously(10). Anti-ABL1
antibody (Santa Cruz) was used at a dilution of 1:50. Sections were processed and
developed using a Bond RX research stainer (Leica Biosystems). Images were
obtained using a Pannoramic 250 Flash Whole Slide Digital Scanner (Perkin Elmer)
and analyzed using ImageJ Plus software (National Institutes of Health).
Statistical analyses
Statistical analyses were carried out using GraphPad Prism 5 (Systat Software, San
Jose, CA, USA). Results are expressed as mean ± standard deviation. Paired analyses
were calculated using Student t test, and comparison of multiple groups by one-way
ANOVA, post hoc intergroup comparisons, Tukey test. Kaplan–Meier survival curves
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

were analyzed by log-rank test. P-values <0.05 were considered statistically
significant.
Results
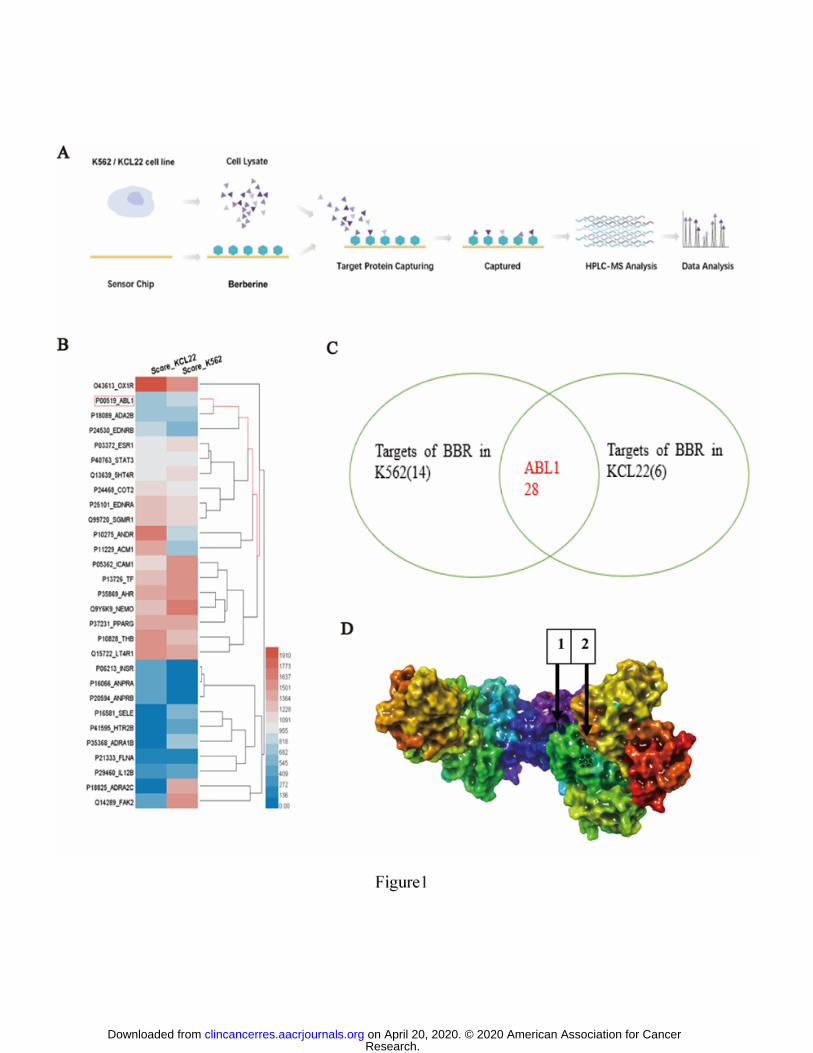
BBR directly bound to ABL1 according to SPR-HPLC-MS
To screen for the direct targets of BBR in K562 and KCL22 CML cells, SPR
combined with HPLC-MS was used, as shown in Fig. 1A. BBR was immobilized on a
chip, K562 and KCL22 cell lysates were incubated with the chip, and the possible
targets of BBR were identified by HPLC-MS. A total of 28 proteins from both K562
and KCL22 cells were identified, including ABL1 (Fig. 1B, C). SPRi assays of the
binding between the domains of ABL1 and BBR were executed, and the PTK domain
bound most strongly out of the tested ABL1 domains (Supplementary Fig. S3C).
BBR bound to the PTK domain of ABL1 according to the molecular docking
analysis
To reveal which domain is the direct binding site of BBR, we conducted a
computer-based molecular docking analysis using Schrödinger Maestro software. We
found that two sites of ABL1 (LWEIATYGMSP and NAVVLLYMATQ) bound to
BBR (Fig. 1D). These binding sites are in the PTK domain of ABL1 (Fig. 2E left, 2F
left). We then synthesized these peptides and conducted an SPRi assay, which
indicated that both sites bound to BBR (Fig. 2E right, 2F right)
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

BBR binding to the PTK domain of ABL1 was confirmed by NMR, SPRi, and
thermo shift assays
To confirm that BBR can directly bind to ABL1, we performed an NMR assay. BBR
and BBR with the PTK domain were dissolved in the same solvent (heavy water). By
comparing the chemical shift of BBR (blue line) with that of the BBR-PTK complex
(red line), we found that PTK markedly changed the chemical shift of BBR. The
NMR results indicated that BBR noncovalently bound to the PTK domain (Fig. 2C).
To confirm this finding, we performed SPRi. BBR was bound to a sensor chip, and
then the pure PTK domain protein (Supplementary Fig. S3B) was passed over the
chip surface, and a binding image was obtained. The mean equilibrium dissociation
constant (Kd) between the PTK domain and BBR was up to 10-7
, indicating that BBR
can directly interact with PTK (Fig. 2A, B).
To further examine whether BBR directly binds to ABL1, thermoshift assays were
performed. Different concentrations (0, 5, and 10 μM) of BBR were added to the PTK
domain protein and incubated at 4°C overnight. On the second day, SYPRO Orange
was added and the protein stability was assessed by observing the melting curve shifts,
with peaks at 91.5°C, 94.3°C, and 95.8°C for 0, 5, and 10 μM of BBR, respectively.
As the BBR concentration increased, the melting temperature (Tm) of PTK increased.
The results further confirmed that BBR directly binds to PTK (Fig. 2D)
BBR suppressed the viability of both imatinib-sensitive and -resistant cell lines
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

To determine whether BBR affected CML cell viability, imatinib-resistant
(BaF3-P210-T315I and SFO2) and imatinib-sensitive (K562, KCL22, and BaF3-P210)
CML cell lines were treated with different concentrations (0–10 μM) of BBR (Fig.
3B). The cell growth, in all cell lines, was significantly arrested by treatment with 5
μM BBR for 48 h (Fig. 3B). Imatinib plus BBR was more effective than imatinib
alone (Fig. 3D, E). The results clearly demonstrate that BBR can overcome resistance
to imatinib in CML cell lines. Resistance to imatinib remains a challenge in patients
with CML. Therefore, in this study, we focused on the function of BBR in
imatinib-resistant CML.
BBR inhibited the survival of human primary CML BM cells and CML-like
mouse BM cells in vitro
To assess the effect of BBR on human primary CML BM cells and CML-like mouse
BM cells, these cells were treated with different concentrations (0–20 μM) of BBR,
which significantly inhibited both human and mouse cell survival in vitro (Fig. 3C, F).
Imatinib plus BBR markedly decreased the human CML BM cell viability relative to
imatinib alone, showing that BBR increased the sensitivity to imatinib (Fig. 3F). BBR
was not cytotoxic for human PBMCs and normal CD34+ cells, indicating that BBR
specifically inhibited CML cell growth (Supplementary Fig. S6B).
BBR inhibited the colony formation of imatinib-sensitive and -resistant cell lines
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

Cell colony formation is closely associated with neoplastic capacity, so we
investigated the effects of BBR and/or imatinib on colony formation in
imatinib-sensitive and -resistant cell lines. Both BBR and imatinib significantly
reduced the colony formation of imatinib-sensitive cell lines (Fig. 3G, H). Importantly,
only BBR inhibited the colony formation ability of imatinib-resistant cell lines (Fig.
3I, J).
BBR inhibited PTK activity in CML cells
The effect of BBR on PTK activity was evaluated by treating CML cells with 5 μM
BBR for 12, 24, and 48 h and analyzing the PTK activity by ELISA. As shown in Fig.
3K–N, BBR significantly inhibited PTK activity in CML cells.
BBR inhibited ABL1 activity in vitro kinase assay
To explore whether BBR directly inhibit ABL kinase activity, 1 μg recombinant
ABL1 kinase proteins were mixed with different concentrations of BBR (0, 2 4 8
10μM), and kinase assays were performed as described in Materials and methods. As
shown in Supplementary Fig S8A, BBR directly inhibited ABL1 activity in vitro.
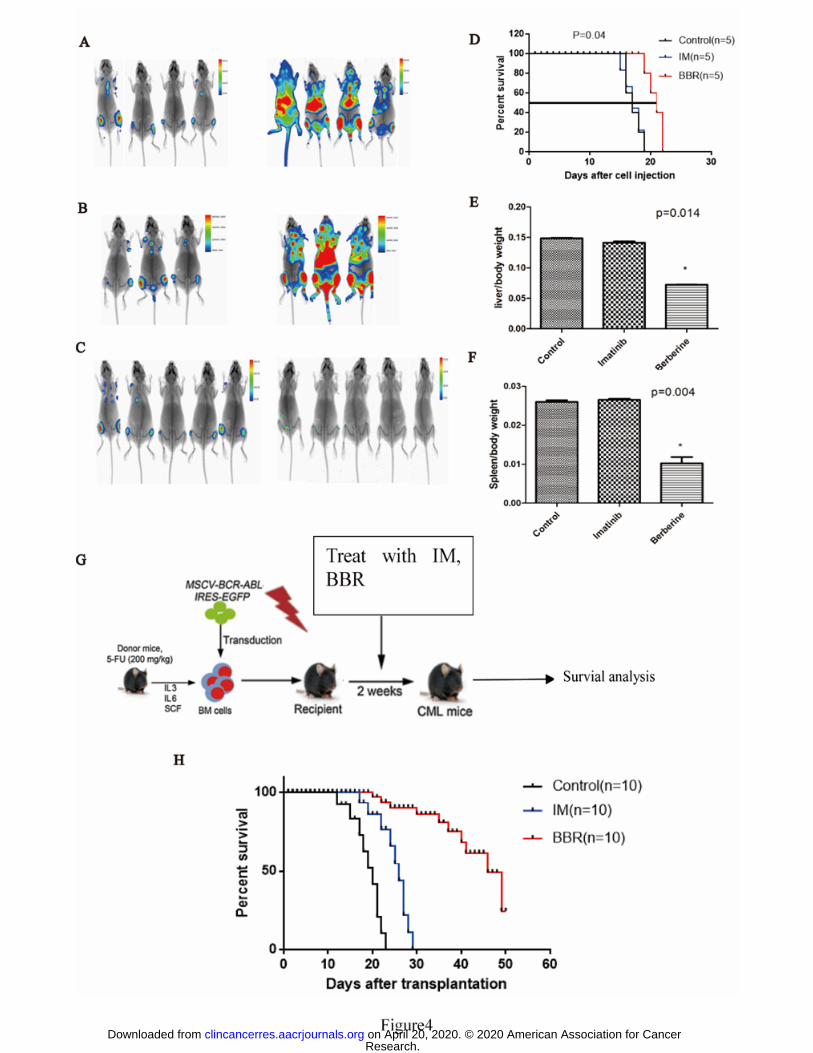
BBR therapeutic in CML mouse mode
To investigate the therapeutic potential of BBR, we intravenously injected BALB/c
nude mice with BaF3 cells harboring the T315I-luciferase BCR-ABL mutation, which
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

express a mutant form of the BCR-ABL protein that is resistant to imatinib. Prior to
injection, the mice were irradiated with 2.5 Gy X-rays. After the BaF3-P210-T315I
cells migrated to the BM, the mice were treated daily with 15 mg/kg BBR or 50
mg/kg imatinib alone for 7 d. Imatinib resulted in no significant inhibition of
BaF3-P210-T315I cell growth and did not affect the mouse survival rates (Fig. 4A, B).
In contrast, BBR led to significant inhibition of BaF3-P210-T315I cell growth and
improved the mouse survival rates (Fig. 4C, D).
BBR-induced inhibition of BCR-ABL and tumor growth were also evaluated in mice
T315I xenografts. BALB/c nude mice were inoculated subcutaneously with
BaF3-P210-T315I cells and treated i.p. with BBR (15 mg/kg), imatinib (50 mg/kg), or
BBR plus imatinib daily for 15 d. Thereafter, tumors were extracted and BCR-ABL
expression was examined by immunohistochemistry (Supplementary Fig. S1C, D).
BBR inhibited the growth of T315I tumors (Supplementary Fig. S1A, B), indicating
that it may be a promising drug for the treatment of CML with the T315I mutation.
However, preclinical and clinical testing of BBR in CML will be required to confirm
this hypothesis.
We employed a human BCR-ABL gene-driven CML mouse model to evaluate the in
vivo effect of BBR on CML. CML mice were randomized into three groups to be
treated with either vehicle, BBR, or imatinib for 14 d (Fig. 4G). BBR or imatinib
alone prolonged the survival of the mice (Fig. 4H). We found that BCR-ABL-T315I
protein was inhibited by BBR in vivo in the T315I xenograft models (Supplementary
Fig. S1C, D), indicates the different mechanism underlying the effect of BBR in the
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

treatment of imatinib-resistant CML compared to the mechanism underlying the effect
of imatinib.
BBR reduced BCR-ABL protein expression in imatinib-sensitive and -resistant
cell lines
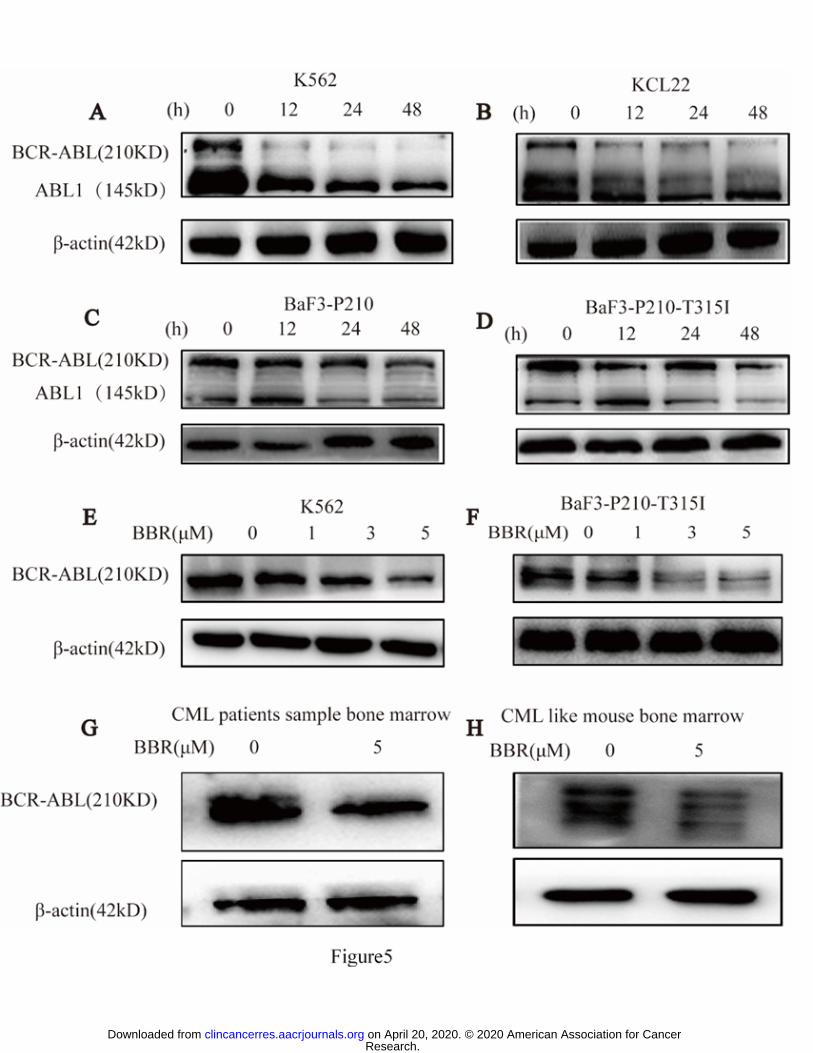
Levels of BCR-ABL protein in cell lines were determined after BBR treatment. BBR
reduced BCR-ABL protein expression in both imatinib-sensitive and -resistant CML
cells in a time- and concentration-dependent manner (Fig. 5A–F and Supplementary
Fig. S4A–F). In contrast, levels of BCR-ABL mRNA remained unchanged
(Supplementary Fig. S2). These results indicate that BBR may directly affect the level
of BCR-ABL protein.
BBR reduced BCR-ABL protein expression in human primary CML BM cells
and CML-like mouse BM cells in vitro
To investigate the effect of BBR on BCR-ABL in primary CML BM cells, we treated
CML BM mononuclear cells and CML-like mouse BM mononuclear cells with BBR
(5 μM) for 48 h, which decreased the BCR-ABL expression in vitro in these cells (Fig.
5G, H and Supplementary Fig. S4G, H).
BBR induced autophagy in CML cells
To investigate the mechanism of BBR-induced degradation of BCR-ABL, we
examined autophagy at the cellular level by assessing the levels of the autophagic
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

lysosome-related proteins LC3II, BECN1, and P62 in BBR-treated cells using western
blotting. BBR treatment of K562 and BaF3-P210-T315I cells induced autophagy,
which was confirmed by the upregulation of LC3II and BECN1 and downregulation
of P62 (Fig. 6B, C and Supplementary Fig. S5A–F). Furthermore, we assessed
whether BCR-ABL interacted with the autophagic lysosome-associated protein P62.
As expected, BBR induced this interaction (Fig. 6A).
Degradation of BCR-ABL by BBR can be reversed by autophagy inhibition
Pre-treatment with the autophagy inhibitors CQ and 3-MA counteracted the
BBR-mediated degradation of BCR-ABL (Fig. 6D–G and Supplementary Fig.
S7A–D). BBR mediated BCR-ABL protein degradation by autophagy, and inhibition
of autophagy by CQ and 3-MA significantly counteracted this degradation. Thus,
autophagy inhibition rescued CML cells from the cytotoxic effects of BBR
(Supplementary Fig. S7).
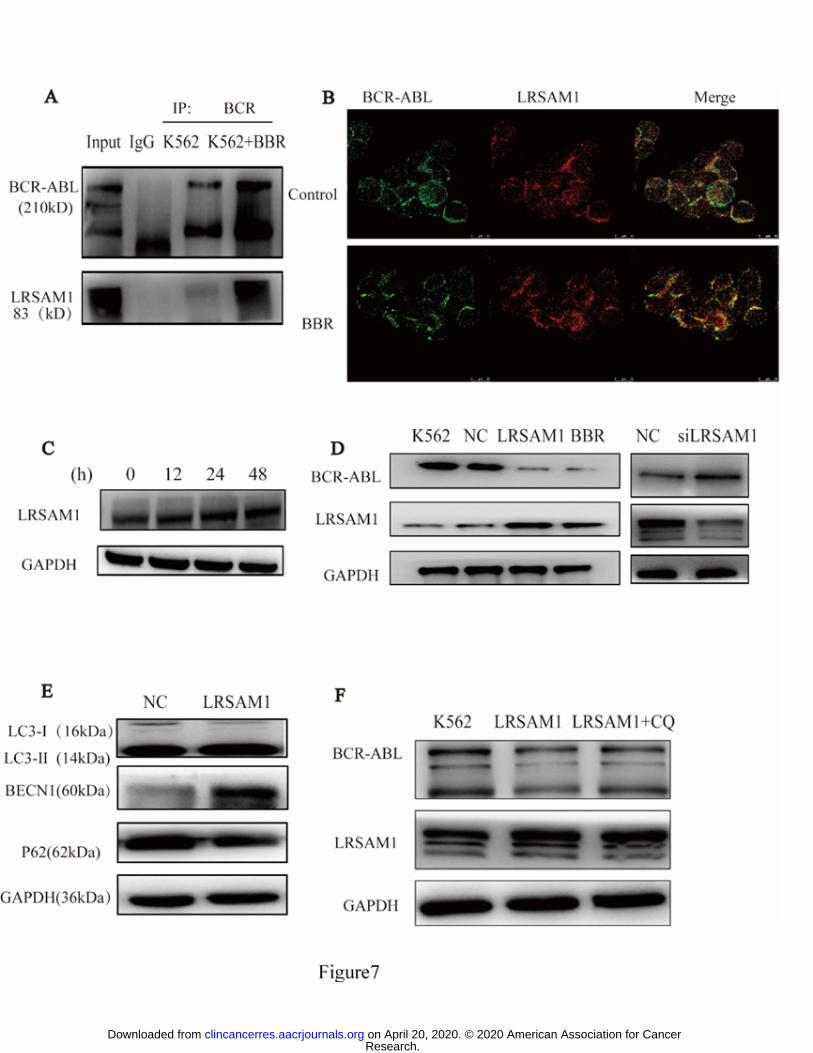
LRSAM1 was associated with BCR-ABL in a multiprotein complex
We used immunoprecipitation-2D and nano-HPLC-MS/MS on K562 cells and 293T
cells transfected with a FLAG-BCR-ABL construct to identify proteins that are
potentially associated with BBR-mediated BCR-ABL degradation. A number of
autophagic lysosome-related proteins were identified in the purified
immunoprecipitates (Supplementary TableS1 and S2) . Of these molecules, LRSAM1
attracted our attention because it is upregulated by BBR (Fig. 7C) and related to
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

autophagy. The interaction between LRSAM1 and BCR-ABL was confirmed in K562
cells (Fig. 7A, B). This observation prompted us to speculate that LRSAM1 might be
recruited by BCR-ABL and might be involved in the degradation induced by BBR.
To confirm the presumption that LRSAM1 mediates BBR-induced degradation of
BCR-ABL, we overexpressed LRSAM1 in K562 cells, and BCR-ABL
downregulation was observed (Fig. 7D left). We then designed siRNA sequences to
establish LRSAM1-knockdown K562 cell lines, which resulted in increased
BCR-ABL (Fig. 7D right), indicating the involvement of LRSAM1 in the regulation
of BCR-ABL turnover. To assess whether LRSAM1 was indeed involved in
autophagic degradation of BCR-ABL, we assessed the levels of LC3Ⅱ, BECN1, and
P62 autophagic lysosome-related proteins in LRSAM1-overexpressing K562 cells,
and we found that the autophagic pathway was activated by LRSAM1 (Fig. 7E). The
autophagic inhibitor CQ prevented BCR-ABL degradation mediated by LRSAM1
(Fig. 7F).
Discussion
BBR is an active agent in the treatment of various diseases, such as: Cardiovascular
and metabolic diseases (CVMD), depression and cancer (5,17-20). There is also
emerging evidence that BBR is a promising leukemia treatment (21). The
identification of druggable targets is extremely important for identifying therapeutic
drugs (22) (23). To explore the direct cell target of BBR, we designed an
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

SPR-HPLC-MS assay and found that it selectively targets ABL1. ABL1 contains a
critical PTK domain, which plays an important role in its tyrosine kinase activity, and
there has been a concerted effort to identify small-molecule inhibitors of ABL1, such
as imatinib and dasatinib. However, drug resistance is an important barrier in CML
treatment. The molecular docking results showed that BBR binds with the PTK
domain of ABL1, which was confirmed by the SPRi, NMR, and DSF assays. We
assume that BBR may have some effect in CML treatment.
The role of autophagic lysosomes in the specific BBR-dependent targeting
mechanisms in malignant cells was unclear. Notably, autophagy-modulating agents
have recently become the focus of clinical translational efforts to treat cancer (24), but
the underlying mechanisms connecting autophagy and cancer remained unclear. In
this study, we demonstrated that BBR can target the BCR-ABL oncoprotein in CML
and BBR induces degradation of BCR-ABL via the autophagic lysosome pathway.
According to our results, autophagy plays an important role in the BBR-mediated
degradation of BCR-ABL (Fig. 6). There are two mechanisms of cellular protein
degradation: the ubiquitin proteasome and autophagy-lysosome systems. The major
method for removal of bulky cellular material, including organelles and protein
complexes, is autophagy. This process involves the sequestration of cytosolic material
in membrane-bound vesicles, which eventually fuse with lysosomes, enabling the
degradation and recycling of components (25). Autophagy is required for the
degradation of the promyelocytic leukemia (PML)/retinoic acid receptor alpha (RAR)
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

oncogenic fusion protein (26,27), and arsenic trioxide-induced autophagy leads to
BCR-ABL degradation (28). Imatinib not only inhibits the tyrosine kinase activity of
BCR-ABL, but it also leads to BCR-ABL sequestration in autophagic vesicles (29).
BCR-ABL-expressing cells exhibit low basal levels of autophagy, and autophagy was
essential in a study that suppressed BCR-ABL-mediated leukaemogenesis (30).
Autophagy also actively suppresses hematopoietic stem cell metabolism (31), and the
promotion of autophagy may have therapeutic effects.
Selective autophagy shuttling proteins, such as P62, mediate degradation as they bind
to ubiquitinated substrates via their ubiquitin-binding domains and dock on
autophagosomes via the interaction of their LC3-interacting motif with LC3 (32).
However, in macroautophagy, several substrates can be targeted to autophagosomes
independently of ubiquitination. P62 has previously been implicated in the shuttling
of ubiquitinated proteins and functions as a “cargo receptor” for the autophagic
degradation of targeted proteins (33); P62 is degraded by the autophagic lysosomal
pathway (34).
LRSAM1 encodes a multidomain RING-type E3 ubiquitin ligase that covalently
ubiquitylates target proteins via its catalytic C-terminal zinc finger domain.
Posttranslational ubiquitylation directs cellular proteins to various fates and functions,
including proteasomal degradation, lysosomal targeting, modulation of
protein–protein interactions, transcriptional regulation, and cell signaling (35). We
found that LRSAM1 was recruited by BCR-ABL after BBR treatment (Fig. 7A, B),
and LRSAM1 overexpression increased the overall level of autophagy in CML cells.
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

BCR-ABL is associated with aberrant PTK activity and can phosphorylate a large
number of substrates, leading to activation of many downstream effectors, including
some that confer antiapoptotic and growth advantages to CML cells. BCR-ABL PTK
activity is selectively inhibited by current CML drugs, such as imatinib (36) and
dasatinib (37). We also found that the PTK domain is a direct target of BBR and its
activity is inhibited by BBR (Fig. 3K, L–N). As shown in Fig. 4 and Supplementary
Fig. S1, BBR significantly inhibited the growth of T315I tumors and extended the
lifespan of the T315I xenograft models. In the treatment of CML, imatinib is a safe
and effective first-line therapy for most patients with chronic-phase CML (38).
Although most patients attain a durable complete cytogenetic response, minimal
residual disease persists in nearly all patients, and active disease recurs if treatment is
discontinued. More importantly, discontinuation of imatinib due to intolerance or
resistance is necessary in up to 30% of patients within the first 5 years of therapy (39).
All current CML drugs act as ATP competitive inhibitors. Several PTK domain
mutations confer high-level resistance to one or more of these therapies, and the
BCR-ABL-T315I mutation confers resistance to all of them (40). The results of this
study showed that BCR-ABL-T315I protein was inhibited by BBR in vivo in the
T315I xenograft models (Supplementary Fig. S1C, D). The fact that the
BaF3-P210-T315I cells growth were inhibited in vivo indicates it maybe relay on the
different mechanism underlying the effect of BBR in the treatment of
imatinib-resistant CML compared to the mechanism underlying the effect of imatinib.
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

In summary, we discovered that the PTK domain of BCR-ABL is a binding site of
BBR. BBR can inhibit the cell viability and colony formation of CML cells and
prolong survival in CML mouse models with imatinib sensitivity or resistance. BBR
induces the degradation of BCR-ABL and BCR-ABL-T315I via the autophagic
lysosome pathway by recruiting LRSAM1, as shown in Supplementary Fig. S8B.
Therefore, BBR is a promising new inhibitor for the treatment of CML with imatinib
resistance.
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

Acknowledgements
This work was supported by grants from the Key Program for the National Natural
Science Foundation of China (no. 81830114), Research Project for Practice
Development of National TCM Clinical Research Bases (no. JDZX2015119), Science
and Technology Program of Guangdong Province (no. 2016A020226027, 2017B
030303001), Science and Technology Program of Guangzhou City (no.
201604020140), and Fundamental Research Funds for the Central Universities (no.
21617461).
Author contributions
JF conceived of and designed the experiments. ZY and CG performed the
experiments. YL, ZY, JY, GH, and CG analyzed the data. ZY and GH contributed
reagents, materials, and analytical tools. JF, ZY, and CG wrote the paper.
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

References
1. Nash I. Chronic myeloid leukemia. N Engl J Med 1999;341(10):765 doi
10.1056/NEJM199909023411016.
2. O'Hare T, Deininger MW, Eide CA, Clackson T, Druker BJ. Targeting the
BCR-ABL signaling pathway in therapy-resistant Philadelphia
chromosome-positive leukemia. Clin Cancer Res 2011;17(2):212-21 doi
10.1158/1078-0432.CCR-09-3314.
3. Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, et al.
Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in
chronic myeloid leukemia. N Engl J Med 2001;344(14):1031-7 doi
10.1056/NEJM200104053441401.
4. Baccarani M, Saglio G, Goldman J, Hochhaus A, Simonsson B, Appelbaum F,
et al. Evolving concepts in the management of chronic myeloid leukemia:
recommendations from an expert panel on behalf of the European
LeukemiaNet. Blood 2006;108(6):1809-20 doi
10.1182/blood-2006-02-005686.
5. Kong W, Wei J, Abidi P, Lin M, Inaba S, Li C, et al. Berberine is a novel
cholesterol-lowering drug working through a unique mechanism distinct from
statins. Nat Med 2004;10(12):1344-51 doi 10.1038/nm1135.
6. Fonseca R, Barlogie B, Bataille R, Bastard C, Bergsagel PL, Chesi M, et al.
Genetics and cytogenetics of multiple myeloma: a workshop report. Cancer
Res 2004;64(4):1546-58.
7. Zhang X, Gu L, Li J, Shah N, He J, Yang L, et al. Degradation of MDM2 by
the interaction between berberine and DAXX leads to potent apoptosis in
MDM2-overexpressing cancer cells. Cancer Res 2010;70(23):9895-904 doi
10.1158/0008-5472.CAN-10-1546.
8. Liu C, Nie D, Li J, Du X, Lu Y, Li Y, et al. Antitumor Effects of Blocking
Protein Neddylation in T315I-BCR-ABL Leukemia Cells and Leukemia Stem
Cells. Cancer Res 2018;78(6):1522-36 doi 10.1158/0008-5472.CAN-17-1733.
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

9. Wang L, Cheng J, Wang S, Zhang X, Cai X. Screening of inhibitors of Taenia
solium glycogen synthase Kinase-3 beta. Rsc Advances 2017;7(68):43319-26
doi 10.1039/c7ra05873j.
10. Gu C, Liu Y, Yin Z, Yang J, Huang G, Zhu X, et al. Discovery of the
Oncogenic Parp1, a Target of bcr-abl and a Potential Therapeutic, in
mir-181a/PPFIA1 Signaling Pathway. Mol Ther Nucleic Acids 2019;16:1-14
doi 10.1016/j.omtn.2019.01.015.
11. Niesen FH, Berglund H, Vedadi M. The use of differential scanning
fluorimetry to detect ligand interactions that promote protein stability. Nat
Protoc 2007;2(9):2212-21 doi 10.1038/nprot.2007.321.
12. Duy C, Hurtz C, Shojaee S, Cerchietti L, Geng H, Swaminathan S, et al.
BCL6 enables Ph+ acute lymphoblastic leukaemia cells to survive BCR-ABL1
kinase inhibition. Nature 2011;473(7347):384-8 doi 10.1038/nature09883.
13. Wu LX, Wu Y, Chen RJ, Liu Y, Huang LS, Lou LG, et al. Curcumin derivative
C817 inhibits proliferation of imatinib-resistant chronic myeloid leukemia
cells with wild-type or mutant Bcr-Abl in vitro. Acta Pharmacol Sin
2014;35(3):401-9 doi 10.1038/aps.2013.180.
14. Yang J, Cao D, Zhang Y, Ou R, Yin Z, Liu Y, et al. The role of
phosphorylation of MLF2 at serine 24 in BCR-ABL leukemogenesis. Cancer
Gene Ther 2020;27(1-2):98-107 doi 10.1038/s41417-019-0152-4.
15. Li S, Ilaria RL, Jr., Million RP, Daley GQ, Van Etten RA. The P190, P210, and
P230 forms of the BCR/ABL oncogene induce a similar chronic myeloid
leukemia-like syndrome in mice but have different lymphoid leukemogenic
activity. J Exp Med 1999;189(9):1399-412.
16. Hurtz C, Hatzi K, Cerchietti L, Braig M, Park E, Kim YM, et al.
BCL6-mediated repression of p53 is critical for leukemia stem cell survival in
chronic myeloid leukemia. J Exp Med 2011;208(11):2163-74 doi
10.1084/jem.20110304.
17. Huang M, Liang Y, Liu Q, Chang X, Guo Y. Berberine attenuates
Abeta25-35-induced apoptosis in primary cultured hippocampal neurons.
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

Biochem Biophys Res Commun 2016 doi 10.1016/j.bbrc.2016.12.166.
18. Kong WJ, Zhang H, Song DQ, Xue R, Zhao W, Wei J, et al. Berberine reduces
insulin resistance through protein kinase C-dependent up-regulation of insulin
receptor expression. Metabolism 2009;58(1):109-19 doi
10.1016/j.metabol.2008.08.013.
19. Fan J, Li B, Ge T, Zhang Z, Lv J, Zhao J, et al. Berberine produces
antidepressant-like effects in ovariectomized mice. Sci Rep 2017;7(1):1310
doi 10.1038/s41598-017-01035-5.
20. Feng X, Sureda A, Jafari S, Memariani Z, Tewari D, Annunziata G, et al.
Berberine in Cardiovascular and Metabolic Diseases: From Mechanisms to
Therapeutics. Theranostics 2019;9(7):1923-51 doi 10.7150/thno.30787.
21. Yu FS, Yang JS, Lin HJ, Yu CS, Tan TW, Lin YT, et al. Berberine inhibits
WEHI-3 leukemia cells in vivo. In Vivo 2007;21(2):407-12.
22. Fetz V, Prochnow H, Bronstrup M, Sasse F. Target identification by image
analysis. Nat Prod Rep 2016;33(5):655-67 doi 10.1039/c5np00113g.
23. Wang J, Gao L, Lee YM, Kalesh KA, Ong YS, Lim J, et al. Target
identification of natural and traditional medicines with quantitative chemical
proteomics approaches. Pharmacol Ther 2016;162:10-22 doi
10.1016/j.pharmthera.2016.01.010.
24. Garber K. Inducing indigestion: companies embrace autophagy inhibitors. J
Natl Cancer Inst 2011;103(9):708-10 doi 10.1093/jnci/djr168.
25. Klionsky DJ. Autophagy: from phenomenology to molecular understanding in
less than a decade. Nat Rev Mol Cell Biol 2007;8(11):931-7 doi
10.1038/nrm2245.
26. Isakson P, Bjoras M, Boe SO, Simonsen A. Autophagy contributes to
therapy-induced degradation of the PML/RARA oncoprotein. Blood
2010;116(13):2324-31 doi 10.1182/blood-2010-01-261040.
27. Wang Z, Cao L, Kang R, Yang M, Liu L, Zhao Y, et al. Autophagy regulates
myeloid cell differentiation by p62/SQSTM1-mediated degradation of
PML-RARalpha oncoprotein. Autophagy 2011;7(4):401-11.
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

28. Goussetis DJ, Gounaris E, Wu EJ, Vakana E, Sharma B, Bogyo M, et al.
Autophagic degradation of the BCR-ABL oncoprotein and generation of
antileukemic responses by arsenic trioxide. Blood 2012;120(17):3555-62 doi
10.1182/blood-2012-01-402578.
29. Elzinga BM, Nyhan MJ, Crowley LC, O'Donovan TR, Cahill MR, McKenna
SL. Induction of autophagy by Imatinib sequesters Bcr-Abl in
autophagosomes and down-regulates Bcr-Abl protein. Am J Hematol
2013;88(6):455-62 doi 10.1002/ajh.23428.
30. Altman BJ, Jacobs SR, Mason EF, Michalek RD, MacIntyre AN, Coloff JL, et
al. Autophagy is essential to suppress cell stress and to allow
BCR-Abl-mediated leukemogenesis. Oncogene 2011;30(16):1855-67 doi
10.1038/onc.2010.561.
31. Ho TT, Warr MR, Adelman ER, Lansinger OM, Flach J, Verovskaya EV, et al.
Autophagy maintains the metabolism and function of young and old stem cells.
Nature 2017;543(7644):205-10 doi 10.1038/nature21388.
32. Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective
autophagy. Mol Cell 2009;34(3):259-69 doi 10.1016/j.molcel.2009.04.026.
33. Lamark T, Kirkin V, Dikic I, Johansen T. NBR1 and p62 as cargo receptors for
selective autophagy of ubiquitinated targets. Cell Cycle 2009;8(13):1986-90
doi 10.4161/cc.8.13.8892.
34. Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, et al. Homeostatic
levels of p62 control cytoplasmic inclusion body formation in
autophagy-deficient mice. Cell 2007;131(6):1149-63 doi
10.1016/j.cell.2007.10.035.
35. Metzger MB, Hristova VA, Weissman AM. HECT and RING finger families
of E3 ubiquitin ligases at a glance. J Cell Sci 2012;125(Pt 3):531-7 doi
10.1242/jcs.091777.
36. Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J.
Structural mechanism for STI-571 inhibition of abelson tyrosine kinase.
Science 2000;289(5486):1938-42.
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

37. Hochhaus A. Dasatinib for the treatment of Philadelphia chromosome-positive
chronic myelogenous leukaemia after imatinib failure. Expert Opin
Pharmacother 2007;8(18):3257-64 doi 10.1517/14656566.8.18.3257.
38. Druker BJ, Guilhot F, O'Brien SG, Gathmann I, Kantarjian H, Gattermann N,
et al. Five-year follow-up of patients receiving imatinib for chronic myeloid
leukemia. N Engl J Med 2006;355(23):2408-17 doi 10.1056/NEJMoa062867.
39. O'Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F, et al.
AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently
inhibits the T315I mutant and overcomes mutation-based resistance. Cancer
Cell 2009;16(5):401-12 doi 10.1016/j.ccr.2009.09.028.
40. O'Hare T, Eide CA, Tyner JW, Corbin AS, Wong MJ, Buchanan S, et al.
SGX393 inhibits the CML mutant Bcr-AblT315I and preempts in vitro
resistance when combined with nilotinib or dasatinib. Proc Natl Acad Sci U S
A 2008;105(14):5507-12 doi 10.1073/pnas.0800587105.
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

Figure legends
Figure 1. BBR directly bound to ABL1 in CML cells
(A) BBR target identification in K562 and KCL22 cells. (B) Heatmap of BBR target
proteins in K562 and KCL22 cells. (C) Venn diagram of BBR target proteins in K562
and KCL22 cells, showing that 28 proteins (including ABL1) from both K562 and
KCL22 cells were identified. (D) Molecular docking model of BBR with ABL1.
Figure 2. BBR directly bound to the PTK domain of ABL1 in CML cells
(A) Surface plasmon resonance imaging (SPRi) binding assay. Sensor chips with
BBR (molecular weight [MW]: 371) immobilized on the surface were first mock
pre-treated (Mock), and then treated with pure PTK domain protein, mouse IgG, or
rabbit IgG. SPR signals are expressed in resonance units (RU). (B) Mean equilibrium
dissociation constant (Kd) between PTK domain and BBR. (C) Chemical shift of
BBR based on 1H-NMR assays before and after adding PTK proteins. (D)
Thermoshift assays assessing the binding between BBR and the PTK domain of
ABL1. (E) Left: predicted binding site of BBR (LWEIATYGMSP) that targets the
PTK domain. Right: SPRi assay involving LWEIATYGMSP peptide and BBR. (F)
Left: predicted binding site of BBR (NAVVLLYMATQ) that targets the PTK domain.
Right: SPRi assay involving NAVVLLYMATQ peptide and BBR.
Figure 3. Growth inhibition of CML cell lines, CML-like mouse cells, and human
CML BM cells by BBR.
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

Cell viability was determined using MTT assays after treatment with imatinib and/or
BBR for 48 h in (A) SFO2 and (B) K562, KCL22, Baf3-P210, and Baf3-P210-T315I
cells. BBR increased the sensitivity to imatinib in (C) CML-like mouse bone marrow
(BM) (D) BaF3-P210-T315I, (E) BaF3-P210, and (F) human CML BM cells.
Histograms and images showing the number of cell colonies after treatment with
imatinib or BBR for 7 d in (G, H) BaF3-P210 cells and (I, J) BaF3-P210-T315I cells.
PTK activity based on ELISAs after BBR treatment (5 μM for 12, 24, and 48 h) in
CML cell lines: (K) K562, (L) KCL-22, (M) BaF3-P210, and (N) BaF3-P210-T315I
cells.
Figure 4. Efficacy of BBR in a CML mouse model.
BaF3 cells (106) harboring the T315I-luciferase BCR-ABL mutation were injected
into the tail vein of female BALB/c nude mice (five mice per group, 6 weeks of age)
and the mice were treated once daily (i.p.) for 7 d with BBR (15 mg/kg) or imatinib
(50 mg/kg). Images showing the tumor burden of the mice after treatment with (A)
saline control, (B) Imatinib (C) BBR. (D) Survival analysis of mice. (E) Liver/body
weight ratios of mice. (F) Spleen/body weight ratios of mice. (G) Schematic diagram
of BCR-ABL-driven CML mouse model and drug treatment. (H) Kaplan–Meier
survival curves of mice treated with BBR or imatinib, P < 0.01; P < 0.001, log-rank
test.
Figure 5. BBR-mediated downregulation of BCR-ABL protein in CML cell lines,
human CML BM cells, and CML-like mouse BM cells.
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

CML cells were treated with BBR (5 μM) for 12, 24, and 48 h, which downregulated
BCR-ABL protein levels, based on western blotting, in (A) K562, (B) KCL-22, (C)
BaF3-P210, and (D) BaF3-P210-T315I cells. CML cells were treated with different
concentrations of BBR (0, 1, 3, and 5 μM) and BCR-ABL protein levels were
determined by western blotting in (E) K562 and (F) BaF3-P210-T315I cells. Primary
CML cells were treated with BBR (5 μM) for 48 h and the BCR-ABL protein levels
were determined by western blotting in (G) human CML bone marrow (BM)
mononuclear cells and (H) CML-like mouse BM mononuclear cells.
Figure 6. Autophagic degradation of BCR-ABL induced by BBR in CML cells.
(A) Lysosomal colocalization of BCR-ABL and P62. K562 cells were treated with
control or BBR (5 μM) for 24 h. Before collection, cells were stained with lysosome
probe, and after collection they were stained with either anti-ABL (green) or anti-P62
(red) antibodies and signals were detected by confocal microscopy. Merged panels
indicate overlapping images of the three fluorescent signals. Additionally, the overall
levels of autophagy at the cellular level were assessed by using western blotting to
assess the protein levels of LC3Ⅱ, BECN1, and P62 after treatment with different
concentrations of BBR (1, 3, and 5 μM) in (B) K562 and (C) BaF3-P210-T315I cells.
β-actin served as a loading control. Next, (D) K562 and (E) BaF3-P210-T315I cells
were treated with the autophagy inhibitor CQ (10 μM) or BBR for 48 h, or
pre-exposed to CQ (10 μM) for 4 h and then treated with BBR for 48 h, and
BCR-ABL protein levels were determined by western blotting, which showed that CQ
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

inhibited the BBR-induced degradation of BCR-ABL. Lastly, (F) K562 and (G)
BaF3-P210-T315I cells were treated with the autophagy inhibitor 3-MA (25 μM) or
BBR for 48 h, or pre-exposed to 3-MA (25 μM) for 4 h and then treated with BBR (5
μM) for 48 h, and BCR-ABL protein levels were determined by western blotting,
which showed that 3-MA inhibited the BBR-induced degradation of BCR-ABL.
Figure 7. BBR-induced autophagic degradation of BCR-ABL via LRSAM1
recruitment in CML cells.
(A) K562 cells were treated with BBR (5 μM) for 24 h and immunoprecipitation was
performed using anti-BCR antibodies. To detect the proteins in the
immunoprecipitates, SDS-PAGE followed by western blotting with anti-ABL1 and
anti-LRSAM1 antibodies was conducted. (B) After BBR treatment, LRSAM1 protein
was recruited by BCR-ABL in K562 cells treated with BBR (5 μM) for 24 h. After
collection, cells were stained with either anti-ABL (green) or anti-LRSAM1 (red)
antibodies and signals were detected by confocal microscopy. Merged panels indicate
overlapping images of the two fluorescent signals. (C) K562 cells were treated with
BBR (5 μM) for 12, 24, and 48 h and the protein level of LRSAM1 was measured by
western blotting, showing that LRSAM1 protein was upregulated by BBR. (D) Left:
LRSAM1 was upregulated (using an LRSAM1 plasmid) in K562 cells and the level
of BCR-ABL was assessed by western blotting. Right: LRSAM1 was downregulated
(using LRSAM1 siRNA) in K562 cells and the level of BCR-ABL was assessed by
western blotting. There were negative correlations between LRSAM1 and BCR-ABL
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

in both experiments. (E) LRSAM1 was upregulated (using an LRSAM1 plasmid) in
K562 cells and the levels of autophagy signaling-related proteins were assessed by
western blotting. (F) LRSAM1 was upregulated (using an LRSAM1 plasmid) and the
lysosome inhibitor CQ (10 μM) was added, and the level of BCR-ABL was assessed
by western blotting.
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460

Published OnlineFirst February 25, 2020.Clin Cancer Res Zhao Yin, Guiping Huang, Chunming Gu, et al. recruiting LRSAM1 for overcoming imatinib-resistancedegradation of both BCR-ABL and BCR-ABL T315I through Discovery of berberine that targetedly induce autophagic
Updated version
10.1158/1078-0432.CCR-19-2460doi:
Access the most recent version of this article at:
Material
Supplementary
http://clincancerres.aacrjournals.org/content/suppl/2020/02/25/1078-0432.CCR-19-2460.DC1
Access the most recent supplemental material at:
Manuscript
Authorbeen edited. Author manuscripts have been peer reviewed and accepted for publication but have not yet
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://clincancerres.aacrjournals.org/content/early/2020/02/25/1078-0432.CCR-19-2460To request permission to re-use all or part of this article, use this link
Research. on April 20, 2020. © 2020 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 25, 2020; DOI: 10.1158/1078-0432.CCR-19-2460







