

PH73CH07-Desvergne ARI 3 January 2011 16:15
Endocrine Disruptors:From Endocrine toMetabolic DisruptionCristina Casals-Casas and Beatrice DesvergneCenter for Integrative Genomics, Faculty of Biology and Medicine, University of Lausanne,Lausanne, 1015 Switzerland; email: [email protected]
Annu. Rev. Physiol. 2011. 73:135–62
First published online as a Review in Advance onNovember 5, 2010
The Annual Review of Physiology is online atphysiol.annualreviews.org
This article’s doi:10.1146/annurev-physiol-012110-142200
Copyright c© 2011 by Annual Reviews.All rights reserved
0066-4278/11/0315-0135$20.00
Keywords
persistent organic pollutants, phthalates, bisphenol A, obesity,diabetes, nuclear receptors
Abstract
Synthetic chemicals currently used in a variety of industrial and agri-cultural applications are leading to widespread contamination of theenvironment. Even though the intended uses of pesticides, plasticiz-ers, antimicrobials, and flame retardants are beneficial, effects on hu-man health are a global concern. These so-called endocrine-disruptingchemicals (EDCs) can disrupt hormonal balance and result in develop-mental and reproductive abnormalities. New in vitro, in vivo, and epi-demiological studies link human EDC exposure with obesity, metabolicsyndrome, and type 2 diabetes. Here we review the main chemical com-pounds that may contribute to metabolic disruption. We then presenttheir demonstrated or suggested mechanisms of action with respect tonuclear receptor signaling. Finally, we discuss the difficulties of fairlyassessing the risks linked to EDC exposure, including developmentalexposure, problems of high- and low-dose exposure, and the complex-ity of current chemical environments.
135
Ann
u. R
ev. P
hysi
ol. 2
011.
73:1
35-1
62. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by K
arol
insk
a In
stitu
tet o
n 07
/26/
11. F
or p
erso
nal u
se o
nly.

PH73CH07-Desvergne ARI 3 January 2011 16:15
EDCs: endocrine-disrupting chemicals
Metabolic syndrome:a combination ofdisorders includingimpaired glucosetolerance or insulinresistance,dyslipidemia, highblood pressure, andobesity
NRs: nuclearreceptors
1. INTRODUCTION
1.1. Endocrine Disruption
Endocrine disruptors are exogenous com-pounds with the potential to disturb hormonalregulation and the normal endocrine system,consequently affecting health and reproductionin animals and humans (1). Endocrine disrup-tors can interfere with the production, release,metabolism, and elimination of or can mimicthe occurrence of natural hormones (2). En-docrine disruptors may also be derived fromnatural animal, human, or plant (phytoestro-gen) sources; however, for the most part in-ternational concern is currently focused onsynthetic chemicals and endocrine-disruptingchemicals (EDCs). This concern is further am-plified by two factors, the expansion in chemicalproduction, which has now reached 400 milliontons globally, and the increased pollution fromthese chemicals. As such, the impact on humanhealth through known or unknown effects ofthese chemicals on hormonal systems is great.
The term endocrine disruptors was firstcoined by Ana Soto and collaborators, whoidentified a number of developmental effectsof EDCs in wildlife and humans (3). AlthoughEDCs can target various hormone systems, anumber of observations concerning reproduc-tive development and sex differentiation, to-gether with early embryonic development andpuberty, have focused on EDC interferencewith sex steroid hormones.
1.2. Metabolic Disruption:A Subdivision of Endocrine Disruption
In addition to the developmental and repro-ductive effects, there is also a growing con-cern that metabolic disorders may be linkedwith EDCs. Global obesity rates have risen dra-matically over the past three decades in adults,children, and adolescents, especially in devel-oped countries. Obesity is frequently associatedwith metabolic disorders (including type 2 dia-betes, metabolic syndrome, cardiovascular andpulmonary complications, and liver disease) aswell as other health issues such as psycholog-
ical/social problems, reproductive defects, andsome forms of cancer.
A combination of genetic, lifestyle, and en-vironmental factors likely account for the rapidand significant increase in obesity rates. Al-though genetic factors may explain a portionof obesity predisposition, they alone are unableto account for the sudden appearance and pro-gression of the current worldwide obesity epi-demic. Modern lifestyles that include excessiveenergy intake, lack of physical activity, sleepdeprivation, and more stable home tempera-tures appear to be major contributing factorsof obesity. However, the increased incidenceof metabolic diseases also correlates with sub-stantial changes in the chemical environmentresulting from new industrial and agriculturalprocedures initiated over the past 40 years. Thischange in the environment has led to the hy-pothesis that some of the numerous environ-mental pollutants are EDCs, interfering withvarious aspects of metabolism and adding an-other risk factor for obesity (4, 5). This hy-pothesis is supported by laboratory and ani-mal research as well as epidemiological studiesthat have shown that a variety of environmen-tal EDCs can influence adipogenesis and obe-sity (reviewed in References 5–10). Such EDCshave been referred to as environmental obeso-gens (11). However, because adverse effects byEDCs may also lead to other metabolic diseasessuch as metabolic syndrome and type 2 diabetes,this subclass of EDCs would be better referredto as metabolic disruptors (12).
1.3. A Common Molecular Mechanismfor Endocrine Disruptionand Metabolic Disruption
Hormones function mainly through interac-tions with their cognate receptors, which can beclassified into two large groups: (a) membrane-bound receptors, which respond primarilyto peptide hormones such as insulin, and(b) nuclear receptors (NRs), which are activatedby interaction with small lipophilic hormonessuch as sex steroid hormones. EDCs may pos-sess multiple mechanisms of action; however,
136 Casals-Casas · Desvergne
Ann
u. R
ev. P
hysi
ol. 2
011.
73:1
35-1
62. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by K
arol
insk
a In
stitu
tet o
n 07
/26/
11. F
or p
erso
nal u
se o
nly.

PH73CH07-Desvergne ARI 3 January 2011 16:15
Estrogen receptors(ERs): ERα and ERβ
are members of thenuclear receptorsuperfamily. Theyform homodimers tobind to DNA
Retinoid X receptor(RXR): a member ofthe nuclear receptorsuperfamily and amajor partner of othernuclear receptors suchas PPAR, PXR, andCAR, with which itforms heterodimers
Persistent organicpollutants: chemicalsthat persist in theenvironment andbioaccumulate withrisks of adverse effectsto human health andenvironment
OCPs:organochlorinepesticides
because many EDCs are small lipophilic com-pounds, one privileged route is through theirdirect interaction with a given NR, which pre-sumably perturbs or modulates downstreamgene expression. For example, most EDC-associated reproductive and developmental de-fects are thought to result from EDCs interfer-ing with the function of the estrogen receptor(ER) and/or androgen receptor (AR), therebydisrupting the normal activity of estrogens andandrogens ligands.
In humans, the NR superfamily encom-passes 48 members that share a commonstructure and, once activated, bind as dimers tospecific response elements located near targetgene promoters. These dimers may be homo-dimers or heterodimers with retinoid X recep-tor (RXR), another member of the NR super-family. In addition to the sex steroid receptors,the NR superfamily includes transcription fac-tors that play pivotal roles in the integration ofthe complexities of metabolic homeostasis anddevelopment. The ability of EDCs to interactwith these NRs is supported by, and explains,the wide range of metabolic perturbations re-ported in both experimental and epidemiolog-ical studies. It also reinforces the concept of as-sociating endocrine and metabolic disruption.
The present review focuses on metabolicdisruptors and is organized into three sections.The first section discusses the chemical com-pounds that are presently considered to be ma-jor potential endocrine/metabolic disruptors.Also summarized is the impact of these chem-icals on human health and metabolism on thebasis of available epidemiological studies. Thesecond section highlights recent advances inestablished or proposed mechanisms of EDC-mediated metabolic disruption. The last sectionhighlights the main challenges that scientistsand regulators face in this field.
2. A MYRIAD OFENDOCRINE-DISRUPTINGCHEMICALS
EDCs encompass a variety of chemical classes,including pesticides, compounds used in the
plastic industry and in consumer products, andother industrial by-products and pollutants.They are often widely dispersed in the envi-ronment and, if persistent, can be transportedlong distances; EDCs are found in virtually allregions of the world (13–17). Persistent organicpollutants are prevalent among environmen-tal contaminants because they are resistant tocommon modes of chemical, biological, or pho-tolytic degradation. Moreover, many EDCs canbe stored for years in animal and human fatmass. However, other EDCs that are rapidlydegraded in the environment or the humanbody, or that may be present for only short pe-riods of time, can also have serious deleteriouseffects if exposure occurs during critical devel-opmental periods.
EDCs can be categorized according to theirintended use (e.g., pesticides) or their struc-tural properties (e.g., dioxins). The main cate-gories of chemicals with suspected metabolism-disrupting activity are presented below (seeTables 1 and 2). For more detailed informa-tion, the interested reader may refer to in-depthreviews focused on specific chemical categories,as discussed below.
2.1. Pesticides
Pesticides are any substance or mixture of sub-stances intended for preventing, destroying, re-pelling, or mitigating any pest (1, 18). Severalhundreds, if not thousands, of different chemi-cals are used as pesticides, and human exposureto these pesticides is widespread. Prominentchemical families include organochlorine pes-ticides (OCPs), organophosphates, carbamates,triazines, and pyrethroids. All OCPs are per-sistent. Even though OCPs such as the insec-ticide dichlorodiphenyltrichloroethane (DDT)are currently banned in most developed coun-tries and were subsequently replaced in 1975by organophosphates and carbamates, DDTcontamination still exists. OCPs are detectedin human breast milk and adipose tissueand may exhibit estrogenic, antiestrogenic,or antiandrogenic activity. Their associationwith breast cancer is suspected but not yet
www.annualreviews.org • Pollutants and Metabolic Disruption 137
Ann
u. R
ev. P
hysi
ol. 2
011.
73:1
35-1
62. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by K
arol
insk
a In
stitu
tet o
n 07
/26/
11. F
or p
erso
nal u
se o
nly.
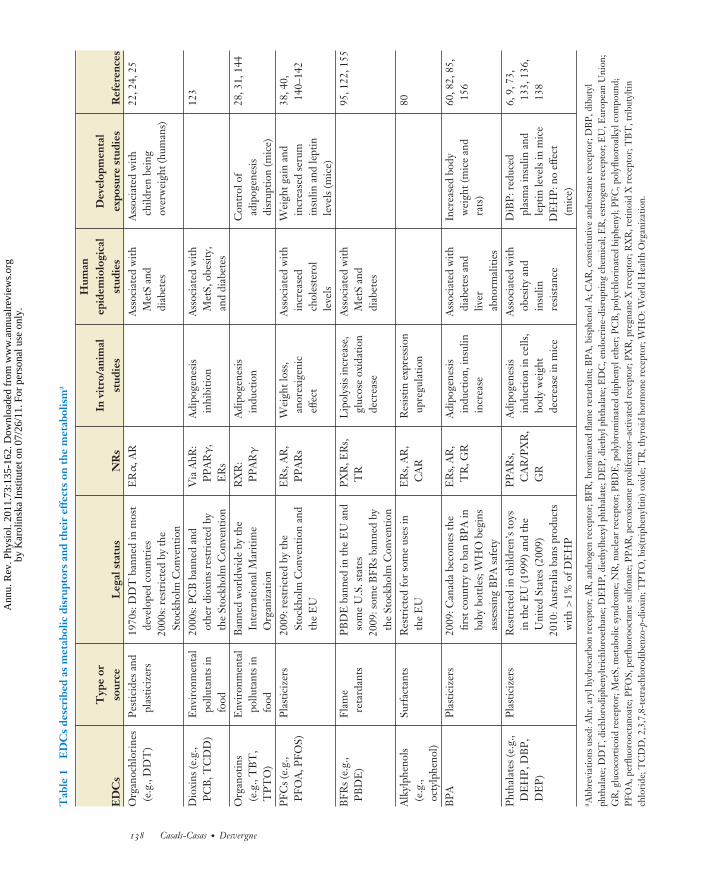
PH73CH07-Desvergne ARI 3 January 2011 16:15
Tab
le1
ED
Cs
desc
ribe
das
met
abol
icdi
srup
tors
and
thei
ref
fect
son
the
met
abol
ism
a
ED
Cs
Typ
eor
sour
ceL
egal
stat
usN
Rs
Invi
tro/
anim
alst
udie
s
Hum
anep
idem
iolo
gica
lst
udie
sD
evel
opm
enta
lex
posu
rest
udie
sR
efer
ence
sO
rgan
ochl
orin
es(e
.g.,
DD
T)
Pes
ticid
esan
dpl
astic
izer
s19
70s:
DD
Tba
nned
inm
ost
deve
lope
dco
untr
ies
2000
s:re
stri
cted
byth
eSt
ockh
olm
Con
vent
ion
ER
α,A
RA
ssoc
iate
dw
ithM
etS
and
diab
etes
Ass
ocia
ted
with
child
ren
bein
gov
erw
eigh
t(hu
man
s)
22,2
4,25
Dio
xins
(e.g
.,P
CB
,TC
DD
)E
nvir
onm
enta
lpo
lluta
nts
info
od
2000
s:P
CB
bann
edan
dot
her
diox
ins
rest
rict
edby
the
Stoc
khol
mC
onve
ntio
n
Via
AhR
:P
PA
Rγ
,E
Rs
Adi
poge
nesi
sin
hibi
tion
Ass
ocia
ted
with
Met
S,ob
esity
,an
ddi
abet
es
123
Org
anot
ins
(e.g
.,T
BT
,T
PT
O)
Env
iron
men
tal
pollu
tant
sin
food
Ban
ned
wor
ldw
ide
byth
eIn
tern
atio
nalM
ariti
me
Org
aniz
atio
n
RX
R:
PP
AR
γ
Adi
poge
nesi
sin
duct
ion
Con
trol
ofad
ipog
enes
isdi
srup
tion
(mic
e)
28,3
1,14
4
PFC
s(e
.g.,
PFO
A,P
FOS)
Pla
stic
izer
s20
09:r
estr
icte
dby
the
Stoc
khol
mC
onve
ntio
nan
dth
eE
U
ER
s,A
R,
PP
AR
sW
eigh
tlos
s,an
orex
igen
icef
fect
Ass
ocia
ted
with
incr
ease
dch
oles
tero
lle
vels
Wei
ghtg
ain
and
incr
ease
dse
rum
insu
linan
dle
ptin
leve
ls(m
ice)
38,4
0,14
0–14
2
BFR
s(e
.g.,
PB
DE
)Fl
ame
reta
rdan
tsP
BD
Eba
nned
inth
eE
Uan
dso
me
U.S
.sta
tes
2009
:som
eB
FRs
bann
edby
the
Stoc
khol
mC
onve
ntio
n
PX
R,E
Rs,
TR
Lip
olys
isin
crea
se,
gluc
ose
oxid
atio
nde
crea
se
Ass
ocia
ted
with
Met
San
ddi
abet
es
95,1
22,1
55
Alk
ylph
enol
s(e
.g.,
octy
lphe
nol)
Surf
acta
nts
Res
tric
ted
for
som
eus
esin
the
EU
ER
s,A
R,
CA
RR
esis
tinex
pres
sion
upre
gula
tion
80
BP
AP
last
iciz
ers
2009
:Can
ada
beco
mes
the
first
coun
try
toba
nB
PA
inba
bybo
ttle
s;W
HO
begi
nsas
sess
ing
BP
Asa
fety
ER
s,A
R,
TR
,GR
Adi
poge
nesi
sin
duct
ion,
insu
linin
crea
se
Ass
ocia
ted
with
diab
etes
and
liver
abno
rmal
ities
Incr
ease
dbo
dyw
eigh
t(m
ice
and
rats
)
60,8
2,85
,15
6
Pht
hala
tes
(e.g
.,D
EH
P,D
BP
,D
EP
)
Pla
stic
izer
sR
estr
icte
din
child
ren’
sto
ysin
the
EU
(199
9)an
dth
eU
nite
dSt
ates
(200
9)20
10:A
ustr
alia
bans
prod
ucts
with
>1%
ofD
EH
P
PP
AR
s,C
AR
/PX
R,
GR
Adi
poge
nesi
sin
duct
ion
ince
lls,
body
wei
ght
decr
ease
inm
ice
Ass
ocia
ted
with
obes
ityan
din
sulin
resi
stan
ce
DiB
P:r
educ
edpl
asm
ain
sulin
and
lept
inle
vels
inm
ice
DE
HP
:no
effe
ct(m
ice)
6,9,
73,
133,
136,
138
a Abb
revi
atio
nsus
ed:A
hr,a
rylh
ydro
carb
onre
cept
or;A
R,a
ndro
gen
rece
ptor
;BFR
,bro
min
ated
flam
ere
tard
ant;
BP
A,b
isph
enol
A;C
AR
,con
stitu
tive
andr
osta
nere
cept
or;D
BP
,dib
utyl
phth
alat
e;D
DT
,dic
hlor
odip
heny
ltric
hlor
oeth
ane;
DE
HP
,die
thyl
hexy
lpht
hala
te;D
EP
,die
thyl
phth
alat
e;E
DC
,end
ocri
ne-d
isru
ptin
gch
emic
al;E
R,e
stro
gen
rece
ptor
;EU
,Eur
opea
nU
nion
;G
R,g
luco
cort
icoi
dre
cept
or;M
etS,
met
abol
icsy
ndro
me;
NR
,nuc
lear
rece
ptor
;PB
DE
,pol
ybro
min
ated
diph
enyl
ethe
r;P
CB
,pol
ychl
orin
ated
biph
enyl
;PFC
,pol
yfluo
roal
kylc
ompo
und;
PFO
A,p
erflu
oroo
ctan
oate
;PFO
S,pe
rfluo
rooc
tane
sulfo
nate
;PP
AR
,per
oxis
ome
prol
ifera
tor–
activ
ated
rece
ptor
;PX
R,p
regn
ane
Xre
cept
or;R
XR
,ret
inoi
dX
rece
ptor
;TB
T,t
ribu
tylti
nch
lori
de;T
CD
D,2
,3,7
,8-t
etra
chlo
rodi
benz
o-p-
diox
in;T
PT
O,b
is(t
riph
enyl
tin)o
xide
;TR
,thy
roid
horm
one
rece
ptor
;WH
O:W
orld
Hea
lthO
rgan
izat
ion.
138 Casals-Casas · Desvergne
Ann
u. R
ev. P
hysi
ol. 2
011.
73:1
35-1
62. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by K
arol
insk
a In
stitu
tet o
n 07
/26/
11. F
or p
erso
nal u
se o
nly.
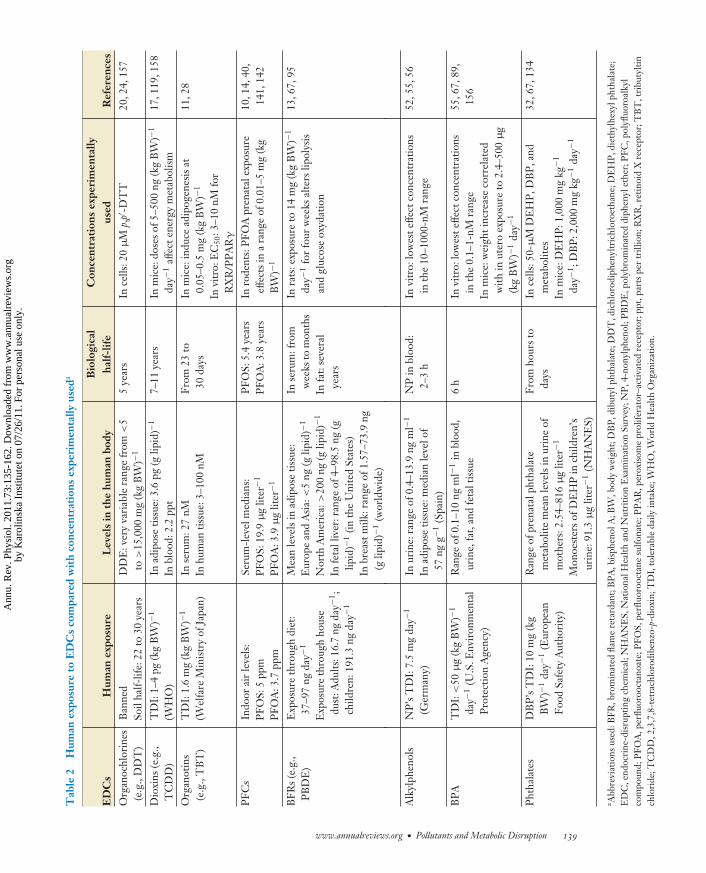
PH73CH07-Desvergne ARI 3 January 2011 16:15
Tab
le2
Hum
anex
posu
reto
ED
Cs
com
pare
dw
ith
conc
entr
atio
nsex
peri
men
tally
used
a
ED
Cs
Hum
anex
posu
reL
evel
sin
the
hum
anbo
dyB
iolo
gica
lha
lf-lif
eC
once
ntra
tion
sex
peri
men
tally
used
Ref
eren
ces
Org
anoc
hlor
ines
(e.g
.,D
DT
)B
anne
dSo
ilha
lf-lif
e:22
to30
year
sD
DE
:ver
yva
riab
lera
nge
from
<5
to>
15,0
00m
g(k
gB
W)−
15
year
sIn
cells
:20
μM
p,p′
-DT
T20
,24,
157
Dio
xins
(e.g
.,T
CD
D)
TD
I:1–
4pg
(kg
BW
)−1
(WH
O)
Inad
ipos
etis
sue:
3.6
pg(g
lipid
)−1
Inbl
ood:
2.2
ppt
7–11
year
sIn
mic
e:do
ses
of5–
500
ng(k
gB
W)−
1
day−
1af
fect
ener
gym
etab
olis
m17
,119
,158
Org
anot
ins
(e.g
.,T
BT
)T
DI:
1.6
mg
(kg
BW
)−1
(Wel
fare
Min
istr
yof
Japa
n)In
seru
m:2
7nM
Inhu
man
tissu
e:3–
100
nMFr
om23
to30
days
Inm
ice:
indu
cead
ipog
enes
isat
0.05
–0.5
mg
(kg
BW
)−1
Invi
tro:
EC
50:3
–10
nMfo
rR
XR
/PP
AR
γ
11,2
8
PFC
sIn
door
air
leve
ls:
PFO
S:5
ppm
PFO
A:3
.7pp
m
Seru
m-l
evel
med
ians
:P
FOS:
19.9
μg
liter
−1
PFO
A:3
.9μ
glit
er−1
PFO
S:5.
4ye
ars
PFO
A:3
.8ye
ars
Inro
dent
s:P
FOA
pren
atal
expo
sure
effe
cts
ina
rang
eof
0.01
–5m
g(k
gB
W)−
1
10,1
4,40
,14
1,14
2
BFR
s(e
.g.,
PB
DE
)E
xpos
ure
thro
ugh
diet
:37
–97
ngda
y−1
Exp
osur
eth
roug
hho
use
dust
:Adu
lts:1
6.7
ngda
y−1 ;
child
ren:
191.
3ng
day−
1
Mea
nle
vels
inad
ipos
etis
sue:
Eur
ope
and
Asi
a:<
5ng
(glip
id)−
1
Nor
thA
mer
ica:
>20
0ng
(glip
id)−
1
Infe
tall
iver
:ran
geof
4–98
.5ng
(glip
id)−
1(in
the
Uni
ted
Stat
es)
Inbr
east
milk
:ran
geof
1.57
–73.
9ng
(glip
id)−
1(w
orld
wid
e)
Inse
rum
:fro
mw
eeks
tom
onth
sIn
fat:
seve
ral
year
s
Inra
ts:e
xpos
ure
to14
mg
(kg
BW
)−1
day−
1fo
rfo
urw
eeks
alte
rslip
olys
isan
dgl
ucos
eox
ydat
ion
13,6
7,95
Alk
ylph
enol
sN
P’s
TD
I:7.
5m
gda
y−1
(Ger
man
y)In
urin
e:ra
nge
of0.
4–13
.9ng
ml−
1
Inad
ipos
etis
sue:
med
ian
leve
lof
57ng
g−1
(Spa
in)
NP
inbl
ood:
2–3
hIn
vitr
o:lo
wes
teffe
ctco
ncen
trat
ions
inth
e10
–100
0-nM
rang
e52
,55,
56
BP
AT
DI:
<50
μg
(kg
BW
)−1
day−
1(U
.S.E
nvir
onm
enta
lP
rote
ctio
nA
genc
y)
Ran
geof
0.1–
10ng
ml−
1in
bloo
d,ur
ine,
fat,
and
feta
ltis
sue
6h
Invi
tro:
low
este
ffect
conc
entr
atio
nsin
the
0.1–
1-nM
rang
eIn
mic
e:w
eigh
tinc
reas
eco
rrel
ated
with
inut
ero
expo
sure
to2.
4–50
0μ
g(k
gB
W)−
1da
y−1
55,6
7,89
,15
6
Pht
hala
tes
DB
P’s
TD
I:10
mg
(kg
BW
)−1
day−
1(E
urop
ean
Food
Safe
tyA
utho
rity
)
Ran
geof
pren
atal
phth
alat
em
etab
olite
mea
nle
vels
inur
ine
ofm
othe
rs:2
.54–
816
μg
liter
−1
Mon
oest
ers
ofD
EH
Pin
child
ren’
sur
ine:
91.3
μg
liter
−1(N
HA
NE
S)
From
hour
sto
days
Ince
lls:5
0-μ
MD
EH
P,D
BP
,and
met
abol
ites
Inm
ice:
DE
HP
:1,0
00m
gkg
−1
day−
1 ;D
BP
:2,0
00m
gkg
−1da
y−1
32,6
7,13
4
a Abb
revi
atio
nsus
ed:B
FR,b
rom
inat
edfla
me
reta
rdan
t;B
PA
,bis
phen
olA
;BW
,bod
yw
eigh
t;D
BP
,dib
utyl
phth
alat
e;D
DT
,dic
hlor
odip
heny
ltric
hlor
oeth
ane;
DE
HP
,die
thyl
hexy
lpht
hala
te;
ED
C,e
ndoc
rine
-dis
rupt
ing
chem
ical
;NH
AN
ES,
Nat
iona
lHea
lthan
dN
utri
tion
Exa
min
atio
nSu
rvey
;NP
,4-n
onyl
phen
ol;P
BD
E,p
olyb
rom
inat
eddi
phen
ylet
her;
PFC
,pol
yfluo
roal
kyl
com
poun
d;P
FOA
,per
fluor
ooct
anoa
te;P
FOS,
perfl
uoro
octa
nesu
lfona
te;P
PA
R,p
erox
isom
epr
olife
rato
r–ac
tivat
edre
cept
or;p
pt,p
arts
per
trill
ion;
RX
R,r
etin
oid
Xre
cept
or;T
BT
,tri
buty
ltin
chlo
ride
;TC
DD
,2,3
,7,8
-tet
rach
loro
dibe
nzo-
p-di
oxin
;TD
I,to
lera
ble
daily
inta
ke;W
HO
,Wor
ldH
ealth
Org
aniz
atio
n.
www.annualreviews.org • Pollutants and Metabolic Disruption 139
Ann
u. R
ev. P
hysi
ol. 2
011.
73:1
35-1
62. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by K
arol
insk
a In
stitu
tet o
n 07
/26/
11. F
or p
erso
nal u
se o
nly.

PH73CH07-Desvergne ARI 3 January 2011 16:15
NHANES
The National Health and Nutrition Examination Survey(NHANES) is the American food consumption database pro-gram conducted by the National Center for Health Statistics,Centers for Disease Control and Prevention. This program wasdesigned to collect data and to assess the health and nutritionalstatus of adults and children in the United States. The programbegan in 1960, but in 1999 NHANES was changed to include thetesting of blood and urine for an extensive number of chemicalsand to monitor roughly 5,000 to 10,000 nationally representativepersons per year. All NHANES surveys are cross-sectional andcontain a core set of physical examinations, clinical and labora-tory tests, and personal interviews. Information about disease andhealth status, diet, sociodemographics, occupation, and educationis collected. In addition, tests are conducted for a variety of mate-rials, such as micronutrients, disease markers, and environmentalpollutants. NHANES’s partnership with the EnvironmentalProtection Agency has also allowed for the implementation oflongitudinal studies of many important environmental influ-ences on health [NHANES I Epidemiologic Follow-Up Study(NHEFS)]. NHANES and NHEFS data are public and availablefor researchers at http://www.cdc.gov/nchs/nhanes.htm.
PVC: polyvinylchloride
demonstrated by epidemiological studies (19).A well-documented case study in the UnitedKingdom listed 127 pesticides identified ashaving endocrine-disrupting properties (16).Despite confounding issues stemming fromthe multifactorial causes of disease and thechallenges in monitoring pesticide exposure,this study underscores the link between med-ical problems and pesticide exposure (16).With respect to metabolic disorders, a largenumber of epidemiological studies have alsolinked pesticide exposure with obesity, dia-betes, insulin resistance, and metabolic syn-drome (9, 20). For example, an association wasdiscovered between prenatal exposure to theDDT breakdown product dichlorodiphenyl-dichloroethylene (DDE) and increased bodymass index in adult women (21). Similarly,cord blood levels of the OCP hexachloroben-zene correlated with a two- to threefold-higherrisk of an elevated body mass index and obe-sity in children (22). Another study used the
National Health and Nutrition ExaminationSurvey (NHANES) database and carried outa cross-sectional analysis of 1,721 adults (seeNHANES and Observational Studies side-bars); the study reported a positive associa-tion between diabetes and the levels of 19 dif-ferent persistent pollutants (including OCPs)measured in serum (21). Other epidemiolog-ical studies find a significant association be-tween pesticide exposure [mostly OCPs suchas heptachlore epoxide, oxychlordane, or β-hexachlorocyclohexane (β-HCH)] and higherincidences of metabolic syndrome, insulin re-sistance, and diabetes (23, 24). A higher preva-lence of diabetes is also associated with DDEexposure (25).
2.2. Dioxins
Dioxins consist of a group of organochlo-rines that include the polychlorinated dibenzo-dioxins (PCDDs), the polychlorinated diben-zofurans (PCDFs), and the polychlorinatedbiphenyls (PCBs) and other related com-pounds. Dioxins can be produced from nat-ural sources such as volcanoes and forestfires but are created mostly by human ac-tivity as by-products in organochloride pro-duction, in incineration of chlorine-containingsubstances such as polyvinyl chloride (PVC),and in bleached paper production. The PCDD2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)is the most toxic of all dioxins. This dioxin wasa major contaminant in the Seveso catastropheand in the Vietnam War (Agent Orange); it wasalso used as a poison in the attempted assassi-nation of Viktor Yushchenko (17).
Dioxins are fat soluble and readily climb thefood chain via their bioaccumulation in fat tis-sues. They are neither readily metabolized norexcreted, and TCDD has a half-life of approx-imately 8 years in humans. Several epidemio-logical studies have evaluated the toxic effects ofTCDD and others dioxins on the general popu-lation as well as heavily exposed subgroups suchas Vietnam War veterans (reviewed in Refer-ences 17 and 26). These studies demonstrate acause and effect between dioxin exposure, with
140 Casals-Casas · Desvergne
Ann
u. R
ev. P
hysi
ol. 2
011.
73:1
35-1
62. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by K
arol
insk
a In
stitu
tet o
n 07
/26/
11. F
or p
erso
nal u
se o
nly.

PH73CH07-Desvergne ARI 3 January 2011 16:15
PFCs: polyfluoroalkylcompounds
an increase in cancers, nervous system degen-eration, immune damage, thyroid disease, andreproductive and sexual development disorders.With respect to metabolism, exploration of theNHANES database indicated that PCDDs andPCDFs are weakly associated with metabolicdisorders, whereas PCBs are strongly associ-ated with type 2 diabetes (27). Several cross-sectional investigations further supported thesecorrelations (reviewed in Reference 25).
2.3. Organotins
Organotins, including tributyltin chloride(TBT) and bis(triphenyltin) oxide (TPTO), arepersistent organic pollutants that have beenwidely used as agricultural fungicides, as rodentrepellents, as molluscicides, and in antifoulingpaints for ships and fishing nets. Organotincompounds such as PVC are also used to stabi-lize plastics.
TBT and TPTO provide one of the clear-est examples of environmental endocrine dis-ruption: Exposure of marine gastropods to verylow concentrations of these compounds inducesan irreversible sexual abnormality in femalestermed imposex, resulting in impaired repro-ductive fitness and possibly sterility (28). Con-cerns over the toxicity of these compounds ledto a worldwide restriction and a ban on marineuses. Currently, human exposure may comefrom dietary sources, such as fish and shell-fish, or through contaminated drinking waterand food. However, no epidemiological data areavailable concerning human exposure, althoughTBT has been reported to have modest adverseeffects on mammalian male and female repro-ductive tracts (29, 30). Nonetheless, recent ex-perimental studies revealed proadipogenic ac-tivity of TBT and TPTO (11, 31).
2.4. Plastics
Owing to their variety, robustness, and ex-tremely low costs, plastics are fundamental inmodern life, public health, and medicine; plasticproduction exceeded 300 million tons in 2010(32). After more than five decades of debate and
OBSERVATIONAL STUDIES
Different types of observational studies are detailed athttp://www.vetmed.wsu.edu/courses-jmgay/glossclinstudy.htm. A summary is presented below.
Clinical studies aimed at evaluating the effects of EDC expo-sure rely mainly on observational studies, which interpret exper-iments of nature and are exposed to large confounding biases.However, such studies provide preliminary evidence to be usedas a basis for hypotheses.
Cross-sectional studies examine the relationship between dis-eases and other factors (such as EDC levels) at one point in timein a defined population. Cross-sectional studies lack any informa-tion on timing of exposure and include only prevalent cases. Theycan suggest a link between a disease and the factor of interest.
In longitudinal studies, or cohort studies, a panel of individu-als is interviewed repeatedly over a period of time. This allows aprospective and analytical study of a group in which some in-dividuals have had, currently have, or will have the exposureof interest to determine the association between that exposureand an outcome. Cohort studies are stronger than cross-sectionaland case-control studies when well executed, but they are moreexpensive.
Case-control studies are a retrospective comparison of theproportion of cases with a potential risk factor to the propor-tion of controls (individuals without the disease) with the samerisk factor. Due to the potential for many forms of bias in thisstudy type, case-control studies provide relatively weak empiricalevidence, even when properly executed.
research, controversy still surrounds the risksthat plastics may cause in humans, particularlywith respect to endocrine-disrupting proper-ties. Adverse effects can stem from the variouscomponents of plastics, the additives used, ora combination of both. Laboratory animal andepidemiological studies have studied the effectsof several of these substances on human health.A few examples are provided below.
2.4.1. Polyfluoroalkyl compounds. Polyflu-oroalkyl compounds (PFCs) are synthetic flu-orinated organic compounds used in a widerange of industrial applications and consumerproducts, including paper, leather, textile coat-ings, and fire-fighting foam, and in the polymer
www.annualreviews.org • Pollutants and Metabolic Disruption 141
Ann
u. R
ev. P
hysi
ol. 2
011.
73:1
35-1
62. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by K
arol
insk
a In
stitu
tet o
n 07
/26/
11. F
or p
erso
nal u
se o
nly.

PH73CH07-Desvergne ARI 3 January 2011 16:15
BFRs: brominatedflame retardants
industry. Among them, perfluorooctane sul-fonate (PFOS) and perfluorooctanoate (PFOA)are widely detected in the environment. PFCsare classified as persistent organic pollutants,even though they are not stored in fat tissue butinstead form chemical adducts with liver andserum proteins. Laboratory rodents exposed toPFCs exhibit developmental effects such as re-duced birth weight and increased neonatal mor-tality (33–35). In addition to hormonal pertur-bations with decreased testosterone levels andincreased estradiol levels in adult rats (36), re-ductions in serum cholesterol and/or triglyc-eride in mice and rats exposed to high doses ofPFOS and PFOA (37) suggest that these chem-icals disturb normal lipid metabolism.
Inverse relationships were observed be-tween PFOS and PFOA concentrations in cordblood and birth weight, ponderal index, andhead circumference in children (38; reviewedin Reference 15). Numerous studies have eval-uated the possible associations of blood PFClevels with metabolic parameters; the most con-sistent result is the positive association betweenPFC exposure and increased cholesterol, par-ticularly high-density lipoprotein (HDL) (39–44). The same reports discounted any associa-tion with metabolic diseases such as type 2 dia-betes and metabolic syndrome (39–44) and sug-gested a complex positive correlation of PFClevels with hyperglycemia but an inverse corre-lation with metabolic syndrome in adolescents(45). Most of these studies are cross-sectionaland as such fail to provide a causal link butrather draw attention to the potential effects ofPFCs on human physiology.
2.4.2. Brominated flame retardants. Bromi-nated flame retardants (BFRs), particularlypolybrominated diphenyl ethers (PBDEs), areadditives used as flame retardants in a greatnumber of consumer products such as houseelectronic equipment, clothing, and furniture.Although these compounds have decreased fireincidences, they are highly prevalent, ubiq-uitous, and persistent pollutants. The mainsources of human exposure come from indoorenvironments, diet, and occupational exposure
(13, 46, 47). Despite their beneficial effects,these chemicals are also thought to adverselyaffect human health through endocrine disrup-tion and developmental neurotoxicity (48). Inaddition, increased incidences of hepatocellu-lar carcinoma and thyroid adenoma have beenobserved in rodents, albeit with relatively highexposure doses (49). Taken together, these ob-servations have led to the recent ban of PBDEsin several American states and in the EuropeanUnion, where a ban on all BFR use is being con-templated. Meanwhile, PBDEs are still presentin environmental samples and are detected inmilk, serum, and adipose tissue in animals andhumans.
Published studies addressing the conse-quences of human BRF exposure remain scarce,and the effects of BFR exposure on sex steroidhormone systems are still poorly understood.One study demonstrated a correlation be-tween PBDEs in breast milk and congenitalcryptorchidism, although confounding factorsmay have been present (50). Only a few epi-demiological studies have addressed the pos-sible impacts of these persistent pollutants onmetabolic parameters in humans. The mosttelling study revealed that, among six dif-ferent BFRs, PBDE-153 and the polybromi-nated biphenyl PBB-153 showed an invertedU-shaped association with type 2 diabetes andmetabolic syndrome (51). Clearly, more stud-ies are needed to clarify the possible extent ofBFR-related damage.
2.4.3. Alkylphenols. Alkylphenols such as 4-n-nonylphenol and 4-n-octylphenol are sur-factants widely used in detergents, emulsifiers,antistatic agents, demulsifiers, and solubilizersand are found commonly in wastewater (52).They are also used as additives to plastics suchas PVC and polystyrene, from which they canleach. Alkylphenols are capable of initiatingproliferation in breast tumor cells in the lab-oratory (53), consistent with their capacities forestrogenic and antiandrogenic activity (54, 55,and references therein). However, given the lowenvironmental concentrations and the currentregulation of alkylphenol use in the European
142 Casals-Casas · Desvergne
Ann
u. R
ev. P
hysi
ol. 2
011.
73:1
35-1
62. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by K
arol
insk
a In
stitu
tet o
n 07
/26/
11. F
or p
erso
nal u
se o
nly.

PH73CH07-Desvergne ARI 3 January 2011 16:15
Bisphenol A (BPA):a high-volume-production plasticizerthat is an estrogenmimic and is detectedin 95% of humanurine samples
Union [Directive 2003/53/EC (2003)], someauthors argue that alkylphenols do not pose ahealth risk. Human exposure was recently eval-uated in a population of women in southernSpain; nonylphenol was detected in 100% ofthe adipose tissue samples tested. In this study,body mass index was associated with nonylphe-nol levels and emerged as a determinant of ex-posure (56). More studies with larger study pop-ulations are thus required to evaluate the risksstill posed by these chemicals.
2.4.4. Bisphenol A. Bisphenol A (BPA) is ahigh-volume-production monomer (>2.5 ×106 kg year−1) used in polycarbonated plastic,in polystyrene resins, and as dental sealants.It is also used as an additive to other plas-tics such as PVC, and halogenated derivativesof BPA are widely used as flame retardants(55). Because unbound monomers remain af-ter BPA polymerization, BPA molecules canbe released from beverage and food containers,for example, from plastic baby bottles or fromtin can liners. Human exposure to BPA is thuswidespread, and unconjugated BPA moleculesare detected in human blood, tissues, and milk.In a reference study in the United States, asmany as 95% of human urine samples containeddetectable levels of BPA in a range that is pre-dicted to be biologically active (38, 57). Estro-genic properties of BPA were first described in1936 (58). Since then, experiments performedin rodents have confirmed its hormonal activ-ity, although the models and the high doses re-ported do not allow direct transposition to hu-man risks. Thus, the potential human healthrisks caused by BPA exposure remain fiercelydebated. Experimental data have been usedto evaluate long-term exposure of mammalianmodel organisms during development and inadulthood to low doses of BPA [levels that fallbelow the regulatory safety standard (59)]. Inshort, these studies point to a number of ad-verse effects in mammals that include abnormalpenile/urethra development, decreased spermcount, early sexual maturation in females, andbrain and behavioral abnormalities. As such, the
potential impact of BPA on human health is noteasily dismissed.
A few epidemiological and preliminary stud-ies, based on small populations, have uncov-ered associations between BPA blood levels inwomen and various ailments, including obesity,recurrent miscarriages, and sterility (60–62).Additionally, higher urinary concentrations ofBPA are associated with an increased prevalenceof cardiovascular disease, diabetes, and liver en-zyme abnormalities (60). This last study high-lighted the need for regulatory action regardingBPA exposure, and Canada was the first countryto ban the use of BPA in baby bottles.
2.4.5. Phthalates. Phthalate esters have beenused worldwide as softeners to impart flexibil-ity, pliability, and elasticity to otherwise rigidpolymers such as PVC. Produced in large quan-tities since the 1930s, nearly all groups of indus-trial consumer products contain phthalates ortraces of phthalates. These molecules are foundmostly in industrial paints and solvents but alsoin toys, personal-care products, and medicaldevices such as intravenous tubing and bloodtransfusion bags. In such devices, they can makeup 80% of the product’s weight (32). UnlikeBPA, phthalates are not covalently bound to thepolymer matrix, making them highly suscepti-ble to leaching. As a result, phthalates contami-nate food, particularly meat and milk products,and are found nearly everywhere in interior en-vironments. In addition, important routes ofhuman exposure include dermal uptake frompersonal-care products and from plastic medi-cal devices that come into direct contact withbiological fluids. Exposure to phthalates canoccur in the developing fetus through theplacenta-blood barrier and in postnatal stagesduring breast feeding or from mouthing toysand baby-care products. Once incorporatedinto the human body, phthalates are short-lived and are rapidly metabolized in a two-phase process (63). In phase I, diester phtha-lates are hydrolyzed into monoester phthalates,whose dosage is used for biomonitoring humanexposure. The conjugation process in phase II
www.annualreviews.org • Pollutants and Metabolic Disruption 143
Ann
u. R
ev. P
hysi
ol. 2
011.
73:1
35-1
62. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by K
arol
insk
a In
stitu
tet o
n 07
/26/
11. F
or p
erso
nal u
se o
nly.

PH73CH07-Desvergne ARI 3 January 2011 16:15
Diethylhexylphthalate (DEHP):the most-usedphthalate; itsmetabolite, MEHP[mono(2-ethylhexyl)phthalate],interferes with severaltypes of nuclearreceptors
leads to the urinary excretion of the conjugatedmetabolites.
Among all the phthalates, diethylhexylphthalate (DEHP) elicits the most concern,with more than two million tons producedannually. This compound is widely used inmedical devices and in a variety of foodproducts. DEHP causes animal toxicity inmany physiological systems; however, many ofthe abnormalities that have been characterizedsince the 1940s have occurred at high DEHPdoses (32). In addition, DEHP promotes livertumor development in rodent models throughsevere peroxisomal proliferation. However,peroxisome proliferation has not been ob-served in humans, and according to a decisionof the International Agency for Research onCancer, DEHP cannot be classified as a humancarcinogen.
Experimental studies at low doses of DEHPexposure, which appear to be most pertinent tohuman health, have demonstrated subtle repro-ductive toxicity in male rodents (64, 65). Otherreproductive outcomes include testicular dys-genesis together with permanent feminizationand demasculinization, resulting in a reducedanogenital distance (66).
Some epidemiological studies reportedan association between cord blood levelsof mono(2-ethylhexyl)phthalate (MEHP), aDEHP metabolite, and shorter gestational ageof delivery. Indirect evidence also suggeststhat diethyl phthalate and dibutyl phthalatemay impart antiandrogenic effects in theperinatal period (reviewed in Reference 67).Maternal urine levels of metabolites of DEHP(benzylbutyl phthalate, diethyl phthalate, anddibutyl phthalate) are associated with a higherrisk of incomplete testicular descent for malehuman infants and are inversely correlatedwith the anogenital distance (68, 69). Otherdevelopmental effects of phthalate exposuremay cause damage to the pulmonary systemand may result in asthma (70).
More recently, several studies have demon-strated a correlation between phthalates andmetabolic disorders. In short- and long-termrodent studies, dose-related deregulation of
levels of serum insulin, blood glucose, liverglycogen, T3, T4, thyroid-stimulating hor-mone, and cortisol was observed (71, 72). Inhumans, the log-transformed concentrations ofseveral phthalate metabolites positively corre-lated with abdominal obesity and insulin resis-tance in adult males (73). These analyses sup-port the concept of environmental obesogensbut await further confirmation by longitudinalstudies.
At first glance, this presentation of so manytypes of chemicals suspected of generatingmetabolic disruptors may seem alarming (seeTables 1 and 2). However, because many stud-ies discussed here are cross-sectional, a defini-tive causal link between metabolic disorders andEDC exposure is still hypothetical. For that rea-son, parallel studies aimed at identifying themolecular mechanisms of EDC activity with re-gard to metabolism should provide greater in-sights into the real health risks posed by thesecompounds.
3. METABOLIC DISRUPTION:MECHANISTIC APPROACHESIn the context of endocrine disruption,metabolic disruption may result from threemain types of activity. First, hormones in gen-eral and sex steroid hormones in particular con-tribute to general body homeostasis throughdiverse metabolic regulations. Thus, a certainnumber of metabolic perturbations are simplythe result of hormonal disruption. Second, di-rect EDC activity through receptors respond-ing to xenobiotics and regulating xenobioticmetabolism may also contribute to a metabolicphenotype. Third, EDC interactions with spe-cialized metabolic receptors may serve as a pri-mary mechanism for metabolic disruption. Thisarticle presents and discusses experimental ob-servations linking EDCs with metabolic dis-ruption along these three types of activity (seeFigure 1).
3.1. Metabolic Disruption ThroughHormone Receptors
Hormone receptors belong to a class of clas-sic hormone receptors that recognize only one
144 Casals-Casas · Desvergne
Ann
u. R
ev. P
hysi
ol. 2
011.
73:1
35-1
62. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by K
arol
insk
a In
stitu
tet o
n 07
/26/
11. F
or p
erso
nal u
se o
nly.

PH73CH07-Desvergne ARI 3 January 2011 16:15
or a few molecules with high affinity. Thyroidhormone (TH), mineralocorticoid, glucocorti-coid, retinoic acid, estrogen, vitamin D, pro-gesterone, and androgen receptors belong tothis class. Initial studies identified ER and ARas the targets of many EDCs, which resulted indevelopmental and reproductive effects, as wellas metabolic alterations.
3.1.1. Metabolic disruption mediated by in-appropriate activation of the estrogen re-ceptor. ERα and ERβ are the main media-tors of the biological effects of estrogens. Uponestrogen binding, they form homodimers thatbind to the promoters of estrogen-responsivegenes. These molecules share a similar struc-ture and bind to the same response elementbut have varying relative binding affinities forsome steroid hormones. In addition to theirwell-established roles in reproduction, ERα
and ERβ are involved in brain development andfunction of many other organs, such as skin,bone, and liver.
Several lines of evidence link ERs tometabolism. For example, in postmenopausalwomen and ovarectomized rodents in whichestrogen is low, one observes an increase inwhite adipose tissue; estrogen replacementtherapy reverses these effects. ERα but notERβ appears to mediate these effects, asinferred from studies using mice in which ERα
is knocked out: Both male and female mutantmice show increased insulin resistance andimpaired glucose tolerance (74, 75). Althoughthe underlying mechanisms remain unclearfor these observed results, it seems likely thatERα activation modulates neural networkscontrolling food intake as well as acts directlyin adipose tissue (reviewed in Reference 76). Ata cellular level, preadipocytes also express ERα
and ERβ, and during development, estrogenscontribute to an increase in adipocyte number,with subsequent effects on adipocyte function(77). At the molecular level, ERs and estrogensregulate many aspects of metabolism, includingglucose transport, glycolysis, mitochondrialstructure and activity, and fatty acid oxidation(reviewed in Reference 8).
Bisphenol A
Adipogenesis Insulin levelsBody weight
Phthalates Organotins Dioxins
ARNTAhR
PFCs
?
CAR/PXR RXRPPARγ RXR PPARα RXRGRGR ER ER RXRTR
Figure 1Endocrine-disrupting chemicals (EDCs) interact with aryl hydrocarbonreceptor (AhR) and with diverse members of the nuclear receptor (NR)superfamily, which convey EDC-mediated metabolic disruption. Sensorreceptors like peroxisome proliferator–activated receptors (PPARs) play aprominent role: PPARγ activated by phthalates or organotins inducesadipogenesis in vitro, and this effect is inhibited by dioxins through AhRactivation. PPARα, which has a major role in fatty acid oxidation, limitsadipogenic activity and is activated in vivo by phthalates and polyfluoroalkylcompounds (PFCs). Estrogenic EDCs such as bisphenol A have also beeninvolved in metabolic disruption through complex interaction with hormonereceptors such as the estrogen receptors (ERs), thyroid hormone receptor(TR), and glucocorticoid receptor (GR). These receptors are important for thecontrol of adipogenesis, weight gain, and insulin levels, although theunderlying mechanisms are not yet well understood (dashed lines). Finally, eventhough the mechanisms have yet to be described (dashed lines and questionmark), the xenosensors pregnane X receptor (PXR), constitutive androstanereceptor (CAR), and AhR also play a crucial role in regulating metabolichomeostasis via direct or indirect interaction with the metabolic pathways.Other abbreviations used: ARNT, aryl hydrocarbon receptor nucleartranslocator; RXR, retinoid X receptor.
Experimental evidence showing the effectsof estrogen-mimicking EDCs such as BPAon metabolism remains scarce and has beenrestricted to cultured cell line models. Studiesusing 3T3-L1 cells suggested that early BPAexposure may enhance adipocyte differenti-ation in a dose-dependent manner and maypermanently disrupt adipocyte-specific geneexpression and leptin synthesis (78, 79). Forinstance, the estrogenic surfactant octylphenolelevates adipocyte production of resistinthrough activation of the ER and extracellularsignal–regulated kinase pathways in 3T3-L1
www.annualreviews.org • Pollutants and Metabolic Disruption 145
Ann
u. R
ev. P
hysi
ol. 2
011.
73:1
35-1
62. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by K
arol
insk
a In
stitu
tet o
n 07
/26/
11. F
or p
erso
nal u
se o
nly.

PH73CH07-Desvergne ARI 3 January 2011 16:15
Thyroid hormonereceptor (TR): TRα
and TRβ are nuclearreceptors that areactivated by thethyroid hormones andplay an important rolein development andmetabolism regulation
cells (80). Resistin is secreted by adipocytes andmay cause insulin resistance and predispositionto type 2 diabetes (81). These limited in vitrostudies suggest that octylphenol-induced se-cretion of resistin may contribute to metabolicdisorders. Finally, BPA may affect ERα activityin the pancreas and increase insulin secretion(82). According to this report, short exposure toBPA provokes chronic hyperinsulinemia, withperturbations of glucose and insulin tolerancetests. This activity has been related to ERα
expression in the pancreas, with 17β-estradiolshown to increase β-cell insulin content, insulingene expression, and insulin release (82).
There are two important aspects to con-sider with respect to estrogen-like activity andmetabolic changes. The first aspect concernsnongenomic responses to estrogen mediatedby the nonclassical transmembrane receptorGPR30. GPR30 deletion in mice revealed itsmajor role in many facets of estrogen metabolicactivity (83), with phenotypes including im-paired glucose tolerance and reduction of bonegrowth. This membrane receptor can also beactivated by BPA and nonylphenol, as assessedin an in vitro cell culture model (84). Furtherstudies are thus needed to evaluate the in vivorelevance of this activation.
The second major question concerns ex-posure to estrogenic EDCs during the criticalperiod of development. Indeed, embryos andfetuses are likely to be much more sensitiveto perturbation by endocrine-like activities.Protective mechanisms available in adultanimals, such as DNA repair mechanisms orliver detoxification and metabolism, are notfully functional in the fetus or neonate. Thus,exposure to EDCs during this period can causeadverse effects, some of which are not apparentuntil much later in life. This point is bestillustrated by prenatal exposure to the estrogenderivative diethylstilbestrol (DES), which waswidely used until the 1970s as an antimiscar-riage medication; this early exposure impairedreproduction later in life (85). Mice exposedto low DES doses during pregnancy producednormal-sized offspring but later showed anage-dependent increased body weight gain and
altered obesity-related gene expression. Prena-tal exposure to DES also led to elevated serumlevels of leptin, adiponectin, interleukin (IL)-6,and triglycerides in mice prior to their becom-ing overweight and obese (86). With regard toEDCs, the effects of prenatal exposure to BPAare well documented. In contrast to the reducedbody weight associated with BPA exposure inadult rodents, exposure to BPA during fetallife resulted in an increase in adult body weight(87). In rats, perinatal exposure to low BPAdoses increased adipogenesis and body weightin adult females, which exhibited adipocytehypertrophy and overexpression of lipogenicgenes (88). Accordingly, high- or low-doseexposure to BPA during gestation to pubertyleads to hyperlipidemia with increased bodyand adipose tissue weight in both sexes (89).
An epigenetic mechanism has been pro-posed to explain these transgenerational ef-fects. Epigenetic changes are inherited changesin phenotype or gene expression caused bymechanisms other than changes in the underly-ing DNA sequence. Epigenetic effects involvemodifications in the activation of certain genes.It is thus hypothesized that EDCs impact obe-sity via estrogen-driven epigenetic reprogram-ming of gene activity during development (90)(see Figure 1).
3.1.2. Metabolic disruption through inap-propriate activation of thyroid hormone re-ceptor and glucocorticoid receptor. EDCsmay also modulate other hormone nuclear re-ceptors, particularly thyroid hormone receptor(TR) and glucocorticoid receptor (GR). MostTH activity is mediated by the TRs TRα andTRβ, which form heterodimers with RXR tobind the promoter sequences of target genes.TR agonists relieve the repression that unli-ganded TRs may exert on some target genes,thus further inducing gene expression. In addi-tion to an important role in brain development,THs are tightly associated with metabolism.Elevated TH levels accelerate metabolism, in-crease lipolysis as well as hepatic cholesterolbiosynthesis and excretion, and provoke weight
146 Casals-Casas · Desvergne
Ann
u. R
ev. P
hysi
ol. 2
011.
73:1
35-1
62. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by K
arol
insk
a In
stitu
tet o
n 07
/26/
11. F
or p
erso
nal u
se o
nly.

PH73CH07-Desvergne ARI 3 January 2011 16:15
Pregnane X receptor(PXR): a nuclearreceptor known as axenosensor and masterregulator ofdetoxificationpathways; known assteroid X receptor inhumans
Constitutiveandrostane receptor(CAR): a nuclearreceptor known as axenosensor and masterregulator ofdetoxificationpathways
AhR: arylhydrocarbon receptor
Cytochrome P450(CYP) family: a largeand diverse group ofenzymes playing animportant role in thedetoxificationpathways. Theirsubstrates includemetabolicintermediates andxenobiotic substances
loss. The exact opposite results are observedwith low TH levels.
In contrast to TR, GR forms homodimersand resides in the cytosol, forming complexeswith molecular chaperones. Ligand bindingreleases the chaperones, triggers GR nucleartranslocation, and influences gene expression.Glucocorticoids acting through GRs allow anorganism to adequately respond to physical oremotional stresses by promoting gluconeogen-esis, increasing blood glucose levels, and mo-bilizing the oxidation of fatty acids. The phar-macological uses of glucocorticoids, chiefly inthe context of controlling chronic inflamma-tion, have serious metabolic side effects such asdiabetes, muscle wasting, and growth retarda-tion in children.
EDCs also interact with these TR and GRreceptors. For instance, in differentiating 3T3-L1 cells, BPA and dicyclohexyl phthalate stim-ulate GR-mediated lipid accumulation and syn-ergize with a weak GR agonist to increaseexpression of adipocyte-specific markers (91).BPA may also act as an antagonist of the TRpathway by enhancing recruitment of the core-pressor NCoR to TR (92). In parallel, perinatalexposure of BPA increases levels of thyroxine(T4) (93). Given the important role of TH inenergy homeostasis, BPA effects on TR dur-ing development may be important in long-term body weight increase. BFRs also disruptthe TH pathway, and daily exposure of rats toPBDE over four weeks resulted in a significantincrease in lipolysis and a significant decreasein glucose oxidation, characteristics associatedwith obesity, insulin resistance, and type 2 di-abetes, although such exposure had no effecton body weight and adipocyte size. Althoughthe underlying molecular mechanisms remainto be experimentally addressed, these physio-logical effects are consistent with a change inER and TR pathways (94, 95).
3.2. Metabolic DisruptionThrough Xenosensors
The body is protected from the accumulationof toxic chemicals by a complex strategy that
in part takes place in the liver, regulating theexpression of drug-metabolizing enzymes andtransporters. This adaptive response incorpo-rates at least three xenosensors: pregnane X re-ceptor (PXR), constitutive androstane receptor(CAR), and aryl hydrocarbon receptor (AhR), aswell as xenobiotic metabolism and transportersystems.
3.2.1. Pregnane X receptor and constitutiveandrostane receptor. PXR and CAR aremembers of the NR superfamily of sensorreceptors, and although they were originallydefined as xenosensors involved in regulatingthe metabolism of xenobiotics, their contribu-tion to fatty acid, lipid, and glucose metabolismhas been only recently appreciated (96, 97).
PXR and CAR regulate gene expression byforming heterodimers with RXR that bind toxenobiotic response sequences present in thepromoters of their target genes. However, theirmechanisms of activation differ. PXR is locatedprimarily in the nucleus and is strongly acti-vated upon ligand binding. In contrast, in theabsence of ligand, CAR is retained in the cy-toplasm through association with the cytoplas-mic CAR-retention protein (CCRP) and heat-shock protein 90 (HSP90). In the presence ofactivators, CAR dissociates from its two chap-erones and translocates into the nucleus, whereit forms heterodimers with RXR (reviewed inReference 98).
PXR and CAR are highly expressed in theliver, where they act as master regulators ofdetoxification pathways through induction ofphase I to phase III enzymes. In the first phase,a polar group is added to hydrophobic sub-strates by hydroxylation and oxidation via thecytochrome P450 (CYP) mono-oxygenase sys-tem. CYP3A is responsible for the metabolismof up to 60% of the drugs presently on themarket (reviewed in Reference 94) and is amajor target gene of PXR, whereasphenobarbital-induced activation of CARtriggers the expression of CYP2B. Phase IIenzymes increase hydrophilicity of the com-pounds through various conjugation reactions,and phase III involves transporters that allow
www.annualreviews.org • Pollutants and Metabolic Disruption 147
Ann
u. R
ev. P
hysi
ol. 2
011.
73:1
35-1
62. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by K
arol
insk
a In
stitu
tet o
n 07
/26/
11. F
or p
erso
nal u
se o
nly.

PH73CH07-Desvergne ARI 3 January 2011 16:15
Peroxisomeproliferator–activated receptor(PPAR): PPARα,-β/δ, and -γ arenuclear receptors thatplay a prominent roleas lipid sensors
for removal of these compounds throughsecretion. Along these three phases, PXRand CAR have common target genes such asthose encoding glutathione-S-transferase andmultidrug resistance protein (MRP)2 and -3;these receptors also have distinct targets such asmultidrug resistance gene (MDR1) and MRP1,respectively. Thus, the combined activities ofPXR and CAR modify and eliminate nearly alltoxicants encountered by the living organism.
With the above in mind, ligands andactivators of PXR and CAR come from twomain sources. First, endogenous ligands forhuman PXR include some bile acid derivatives,pregnanes formed from cholesterol as imme-diate precursors of progesterone, and othermetabolic products of steroids. The ligandsof CAR are less promiscuous than those ofPXR, perhaps due to the smaller size of theCAR ligand-binding pocket. Examples of CARligands include the androstane metabolites andsteroid metabolites. This situation supports thehypothesis that PXR and CAR play an impor-tant role in endocrine system regulation. Theactivity of PXR is, however, defined primarilyby its interaction with exogenous compounds,including herbal medicines and pharmaceuticaldrugs (such as rifampicin), synthetic gluco-corticoids (such as dexamethasone), or steroidhormones (DES, 17β-estradiol). CAR alsoresponds to exogenous compounds such asphenobarbital, which induces CAR nucleartranslocation, or the well-characterized ligandTCPOBOP. A number of EDCs activate PXRand CAR; both may be activated by nonylphe-nol, DEHP, and MEHP. BPA and some PCBsactivate human PXR, whereas PFOA, PFOS,and the organochlorine methoxychlor canactivate CAR (99–102).
As mentioned above, PXR and CAR wereidentified chiefly as xenobiotic-metabolizingregulators; however, clinical observations re-vealed that many CAR and PXR activators af-fect lipid and glucose metabolism in patients.For instance, the known PXR activator ri-fampicin induced liver steatosis in tuberculosispatients (103), and long-term treatment withphenobarbital provoked significant changes in
hepatic and plasma metabolite profiles (104,105). Furthermore, laboratory animal and invitro studies show a similar trend: PXR acti-vation induced a steatogenic effect in rat andmouse liver (106–108), and CAR and PXR ac-tivators repressed hepatic gluconeogenic en-zymes and genes (109–111). CAR was recentlydescribed as an antiobesity NR that amelio-rates diabetes and fatty liver (112, 113). Inaddition to direct effects of PXR and CARon lipid and glucose metabolism, PXR and/orCAR indirectly affect these pathways by in-terfering with other regulatory pathways andNRs (114) (see Figure 2). CAR and PXR bindother transcription factors like forkhead boxesA2 and O1, inhibiting their DNA binding (96).PXR and CAR may also compete for the DR1-binding site recognized by the NR hepatocytenuclear factor 4α (HNF4α) and peroxisomeproliferator–activated receptor (PPAR)α. Fi-nally, PXR and CAR can also exert an inhibitoryeffect by targeting common coactivators likePPARγ coactivator 1α (PGC1α), which inter-acts with many transcription factors to regulatemetabolic homeostasis (96).
The activation of PXR and CAR by EDCsmay account for the metabolic responses notedafter exposure to these chemicals. For example,DEHP induces CAR-dependent activation ofthe nuclear receptor Rev-erbα pathway, whichin turn helps to control the cellular clock andfunctions in energy metabolism (101). BecausePXR and CAR regulate several CYP familymembers involved mainly in the metabolismof steroids and other endogenous compoundslike sex steroid hormones, their EDC-mediatedactivation may alter metabolism indirectly bychanging the effective concentrations of thesehormones (see Section 3.1.1) (98). Althoughthese hypotheses are appealing, to date no stud-ies have established a clear link between EDCsand metabolic disorders via PXR and CAR.
3.2.2. Aryl hydrocarbon receptor. AhR isa ligand-activated transcription factor thatbelongs to the basic helix-loop-helix Per-ARNT-SIM (bHLH-PAS—where Per denotesthe Drosophila melanogaster clock gene Period;
148 Casals-Casas · Desvergne
Ann
u. R
ev. P
hysi
ol. 2
011.
73:1
35-1
62. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by K
arol
insk
a In
stitu
tet o
n 07
/26/
11. F
or p
erso
nal u
se o
nly.

PH73CH07-Desvergne ARI 3 January 2011 16:15
ARNT denotes aryl hydrocarbon receptor nu-clear translocator; and SIM denotes a neurode-velopmental regulator in flies, single-minded )family of proteins. AhR is a xenosensor that me-diates the biological response to a wide spec-trum of xenobiotics; in particular, AhR is themajor factor sensing and mediating the toxiceffects of the dioxin TCDD.
The nonactivated AhR protein resides inthe cytosol and, upon ligand-mediated activa-tion, translocates into the nucleus, where it het-erodimerizes with the ubiquitously expressedARNT, a member of the same protein fam-ily. The AhR/ARNT complex binds to specificregulatory DNA sequences to regulate geneexpression. AhR activity may also be medi-ated by alternative ligands and by an ARNT-independent mechanism, although details ofthese mechanisms remain poorly understood(116).
Among the targets involved in detoxifica-tion, AhR target genes include the phase Ienzyme CYP1A1 and the phase II enzymesUGT1A1 and UGT1A6. In addition, AhR maycontribute to the coordinated regulation of hu-man drug-metabolizing enzymes and conjugatetransporters by inducing PXR and CAR expres-sion (117). Endogenous molecules that bindAhR and benefit from detoxification activity arelipoxin 4 and leukotriene derivatives, as wellas the heme metabolites biliverdin and biliru-bin. Xenobiotics that activate AhR include var-ious dietary phytochemicals, some PCBs, andTCDD. Because it is very poorly metabolized,TCDD triggers sustained activation of AhR,contributing to the toxic effects of dioxin. Thesetoxic effects thereby highlight the undesiredevents that may occur through inappropriateAhR activation and reveal a subset of AhR tar-get genes unrelated to detoxification. These tar-gets include the CDK inhibitors p21CIP1 andp27Kip1 (118), which may explain the broad roleof AhR in organogenesis, embryonic develop-ment, the cell cycle, immunosuppression, andcarcinogenicity.
Recently, AhR has been implicated as a reg-ulator of energy metabolism. Epidemiologicalstudies show an association between dioxin ex-
EDCe DNA-binding competition
NR2
NR1
f Proteasome activation
Proteasome
+
NRE1 +
a Activation or modulation
NR1coAct
–
NRE1
b Inhibition
NR1coRe
–
NRE1
c Squelching
NR1
NR2coAct
+
–
NRE1NRE2
d Synergism or inhibition
NR2 NR1
NRE1
–NR2NR1
Figure 2Endocrine-disrupting chemicals (EDCs) interfere with nuclear receptor (NR)signaling via multiple mechanisms. In this figure, NR1 and NR2 are genericnames for the NRs discussed in the text. EDCs can act as direct agonists orantagonists of NR1, taking the place of the endogenous ligand. They can directthe recruitment of coactivators (coAct) by the NR and trigger target genetranscription, although some EDCs act as modulators rather than as fullagonists by inducing the recruitment of only some of the coactivators. EDCsthat act as antagonists favor conformational changes that allow for therecruitment of corepressors (coRe) or inhibit DNA binding and target geneexpression (a,b). Moreover, EDCs can interfere by indirect mechanisms: EDCbinding to NR2 leads to disturbances of NR1 signaling via molecular cross-talksuch as competition for coactivators (c) or for DNA-binding sites (e). Otherindirect mechanisms are the binding of NR1 and NR2 to neighboringsequences, which may lead to either synergism or inhibition of the regulatoryactivity (d ) or to the EDC-mediated activation of NR2, which results in NR1degradation through proteasome activation ( f ). NRE1/NRE2, nuclearreceptor response element 1 and 2.
posure and type 2 diabetes (25). Other stud-ies also demonstrate that high and low dosesof dioxins affect genes in an AhR-dependentmanner linked with hepatic circadian rhythm,cholesterol biosynthesis, fatty acid synthesis,glucose metabolism, and adipocyte differenti-ation (119, 120). The mechanisms by whichAhR regulates energy metabolism are not yetwell described, but various direct and indirectmechanisms including cross-talk with ER maybe involved. AhR may disrupt the ER signal-ing pathways through increased ER proteaso-mal degradation, modulating estrogen levels via
www.annualreviews.org • Pollutants and Metabolic Disruption 149
Ann
u. R
ev. P
hysi
ol. 2
011.
73:1
35-1
62. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by K
arol
insk
a In
stitu
tet o
n 07
/26/
11. F
or p
erso
nal u
se o
nly.

PH73CH07-Desvergne ARI 3 January 2011 16:15
CYP expression, altering ER transcriptional ac-tivity via coactivator squelching, or promot-ing DNA-binding competition (121, 122) (seeFigure 2). In addition, AhR also indirectlyaffects adipogenesis through inhibition ofPPARγ expression (123).
Additional experimental and epidemiologi-cal studies are still required to assess whetherAhR-mediated responses affect metabolism inaddition to the well-known roles of Ahr in im-munity, development, and cancer.
3.3. Metabolic Disruption ThroughPeroxisome Proliferator–ActivatedReceptors
Metabolic homeostasis requires a controlledbalance between energy storage and consump-tion; several NRs and their coregulators are in-strumental in these processes. Among these, thePPARs act as lipid sensors that cooperate indifferent organs to adapt gene expression to agiven metabolic status. PPARs are sensor recep-tors with a rather large ligand-binding domain,which can accommodate a variety of ligands,primarily lipid derivatives. In the presence ofligand, PPARs heterodimerize with RXR andbind to the PPAR response elements localizedin the promoter regions of their target genes(124).
The PPAR family is composed of threeisotypes: PPARα, -β/δ, and -γ. PPARα is ex-pressed predominantly in tissues characterizedby a high rate of fatty acid catabolism such asliver, kidney, heart, and muscle. PPARα wasfirst identified as the protein responsible forthe induction of peroxisome proliferation inrodents exposed to a variety of compoundscollectively termed peroxisome proliferators.However, humans do not undergo peroxisomeproliferation and are thereby protected fromthe consequent liver tumors observed insensitive species. PPARα plays a major role infatty acid oxidation in all species, controllinglipoprotein metabolism and limiting inflamma-tion. PPARβ is ubiquitously expressed, sharespartially overlapping functions with PPARα,and also plays a role in cell differentiation and
survival (125, 126). Finally, PPARγ functionsin adipogenesis, lipid storage, and the controlof insulin sensitivity; it also participates ininflammatory responses (127).
Plasticizers, surfactants, pesticides, anddioxins can modulate PPAR activity, althoughfairly little is known about the molecular mech-anisms and the physiological outputs involved.The specificity of this PPAR-mediated re-sponse is highlighted in a study in which200 pesticides were systematically screened fortheir peroxisome proliferation activity. Onlythree compounds were identified as havingPPARα transcriptional activity, and none pos-sessed PPARγ transcriptional activity (127).Among these pesticides, diclofop-methyl andpyrethrins induced PPARα target gene expres-sion at levels similar to those induced by classicagonists in mice (128, 129).
The phthalates are another group of well-characterized peroxisome proliferators (130).In vitro transactivation assays and intact cellu-lar systems were used to reveal that phthalatesand their metabolites bind and activate the threePPARs, among other NRs (131–134). Thesestudies also determined the range of potencyand efficacy of phthalate monoesters, show-ing differences between isotypes and species.Modeling the DEHP metabolite MEHP in thePPARγ ligand-binding pocket indicates thatMEHP may contact residues similar to thosedefined for the classic PPARγ agonist rosigli-tazone (135). MEHP induces adipogenesis in aPPARγ-dependent manner, albeit with lowerefficiency than rosiglitazone in 3T3-L1 cells(134). Accordingly, gene expression microarrayanalyses indicate that 70% of the genes are reg-ulated by either ligand, some of them to differ-ing degrees. However, 30% of the genes are ex-clusively regulated by rosiglitazone and not byMEHP, suggesting that MEHP acts as a selec-tive modulator of PPARγ rather than as a fullagonist. This differential activity results fromthe different abilities of MEHP and rosiglita-zone to induce the release of corepressors suchas NCoR and the recruitment of coactivatorssuch as p300 or PGC1α (133). Taken together,these in vitro data demonstrate that MEHP is
150 Casals-Casas · Desvergne
Ann
u. R
ev. P
hysi
ol. 2
011.
73:1
35-1
62. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by K
arol
insk
a In
stitu
tet o
n 07
/26/
11. F
or p
erso
nal u
se o
nly.

PH73CH07-Desvergne ARI 3 January 2011 16:15
proadipogenic in a cell culture model, suggest-ing that it may act as a metabolic disruptor andmay promote obesity in vivo.
Paradoxically, in vivo experiments partiallycontradict these results (136). Adult micetreated with high or low doses of DEHP areprotected from weight gain, gaining 30% lessweight than controls. These mice possess a re-duced fat mass and a metabolic improvement,with lower levels of triglycerides in the liver andthe blood, smaller adipocytes, and enhancedglucose tolerance. These effects were not ob-served in PPARα-null mice, confirming thatin vivo DEHP activity is mediated primarilyby PPARα in the liver, leading to increasedfatty acid catabolism and induced expressionof PPARα target genes such as that encodingfibroblast growth factor 21 (FGF21) (137) orgenes controlling fatty acid β-oxidation. Sur-prisingly, this phenotype is also completelyabolished in PPARα-humanized mice (mice inwhich the mouse PPARα alleles are replaced bythe human PPARα gene). These mice, whenexposed to a DEHP-containing diet, tend tobe more sensitive to diet-induced obesity thanare untreated controls (136). These observa-tions point to the possibility of species-specificEDC activity, due at least in part to evolution-ary differences in the receptors interacting withthem.
Studies of phthalate exposure in utero haveyielded dissimilar results. Male and female off-spring of rats exposed to diisobutyl phthalateand butylparaben exhibit reduced plasma lep-tin and insulin levels, similar to the modifi-cations observed upon in utero exposure torosiglitazone (138). In contrast, a study evalu-ating the impact of in utero exposure to DEHPcould not identify parameters indicating adultmetabolic disorders (6). These differences maybe attributable to the compounds tested as wellas the specific experimental protocols. In anycase, these studies highlight the necessity to in-vestigate the risks engendered by fetal exposureto phthalates.
The PFCs, particularly PFOA and PFOS,can also activate mouse and human PPARs intransactivation assays (139), although the in
vivo consequences of such activity remain quitecontroversial. Adult mice exposed to high dosesof PFOA exhibit weight loss, which is abro-gated in PPARα-null mice (140). The pro-posed mechanism involves PPARα-dependentanorexigenic activity in the hypothalamus ofadult rodents (141). In contrast, Hines et al.(142) reported that PFOA has no effect on bodyweight gain when exposure occurs at the adultstage. However, developmental exposure to lowPFC levels results in increased body weightand increased serum insulin and leptin levelsat midlife (142). Again, species-specific PPARα
activity was proposed because low doses ofPFOA significantly activate the function ofPPARα in wild-type mice but not in PPARα-humanized mice. Human PPARα may there-fore be less responsive to PFOA, increasingthe possibility of species-specific EDC activ-ity. More specifically, the extent to which thesePPAR activators influence metabolic home-ostasis in humans deserves more study (143).
Several EDCs also specifically targetPPARγ. Using a high-throughput method,Kanayama et al. (31) showed that among 40EDCs, organotins such as TBT and TPTOare activators of human PPARγ and RXR.TBT binds to and activates the three humansubtypes of RXR as well as many permissiveheterodimeric partners such as liver X receptor(LXR), nuclear receptor–related 1 protein(NURR1), PPARβ, and PPARγ, but notPPARα. Organotins bind and activate, pri-marily through RXR and not through PPARγ,the PPARγ:RXR heterodimer at nanomolarconcentrations. The crystal structure of theRXRα ligand-binding domain bound to TBTindicates that TBT binds with high affinity toRXR, even though TBT is structurally distinctfrom above-described ligands and only partlyoccupies the RXRα ligand-binding pocket(144). Consistent with the critical role playedby PPARγ:RXR signaling in mammalianadipogenesis, TBT promotes adipogenesisin 3T3-L1 cells by direct transcriptionaleffects on the PPARγ target genes. In uteroexposure to TBT in rodents led to alter-ations in fat structure and metabolism, with a
www.annualreviews.org • Pollutants and Metabolic Disruption 151
Ann
u. R
ev. P
hysi
ol. 2
011.
73:1
35-1
62. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by K
arol
insk
a In
stitu
tet o
n 07
/26/
11. F
or p
erso
nal u
se o
nly.

PH73CH07-Desvergne ARI 3 January 2011 16:15
disorganization of hepatic and gonadal archi-tecture, steatosis in the liver, and an increase inlipid accumulation and mature adipocytes. Thefat mass—but not the total body weight—of inutero TBT-treated mice significantly increasesin adulthood, supporting the conclusion thatembryonic and chronic lifetime organotinexposure may contribute to the incidence ofobesity through disruption of the PPARγ:RXRpathway (28).
Altogether, these many examples of EDCinteraction with receptors highlight the factthat a given compound can interfere withdifferent NRs and different pathways (seeTable 1). For example, depending on the com-pound, BFRs interface with AR, ER, and pro-gesterone receptor to elicit both agonist- andantagonist-like effects (94). PBDEs bind but donot activate AhR (145); in contrast, they in-duce the expression of various CYP enzymes,in part through the activation of PXR (146,147). PBDEs are also active in TH regula-tion by disrupting peripheral TH transport andmetabolism/deactivation or by binding and ac-tivating TRs (148, 149). The final consequencesof EDCs exposure are thus due to cross-talk be-tween these pathways, rather than to a linearcausation chain (96, 114, 122, 123, 150), andare much more complex to decipher in vivo.
4. METABOLIC DISRUPTORS:TOWARD MANY CHALLENGES
This review emphasizes the remarkable emer-gence of EDC-related research, which hasshifted focus from endocrine disruption tometabolic disruption. Epidemiological studiesthat underscore the parallels between EDC ex-posure and obesity incidence, as well as animallaboratory studies that demonstrate the abilityof EDCs to act on metabolic transcription fac-tors, are lines of investigation that cannot becasually discounted. However, there are manydifficulties to overcome before one can fairly as-sess the risks that past, present, and future envi-ronmental chemicals engender on human andwildlife health. In this last section, we highlight
some of the important questions that remain forresearchers and regulators.
4.1. Monitoring Exposure Levels
Ambient monitoring is performed by samplingair, dust, water, etc., and by measuring the lev-els of the pollutants of interest in these samples.This method is often reasonably easy and reli-able. However, ambient monitoring provides avalue valid only at the time of sampling, whichmay not reflect levels of chronic exposure; it alsodoes not take into account the efficiency withwhich living organisms breathe, ingest, or ab-sorb these compounds. In that respect, biomon-itoring is a more appropriate evaluation of thepresence of compounds or their metabolites inbiological samples, particularly in blood andurine, and ideally in tissue samples. Biomoni-toring does not identify the source of the con-tamination but provides the individual expo-sure level at a given time point. Biomonitoringassays also reflect past exposure to persistentpollutants. However, biomonitoring is invasiveand costly and cannot be proposed as a standardroutine evaluation, except for occupational ex-posure. Alternatively, biomonitoring of wildlifesamples can be more easily performed and mayserve as a good indicator of exposure to somepollutants widely found in the environment.
4.2. Identifying the MetabolicEffective Dose
One current issue in identifying the metaboliceffective dose concerns the so-called U-shapedor inverted U-shaped dose-response curve. Adose-response curve of this form is reportedfor BPA: Effects are observed at very low doses(from 10−12 M) and at high doses (10−8 M), butno effects are observed at intermediate doses(10−9 M). These U-shaped curves suggest theexistence of two independent mechanisms forlow doses and high doses (151, 152). However,mechanistic studies are still lacking, and evi-dence for low-dose activity is not available inepidemiological studies.
152 Casals-Casas · Desvergne
Ann
u. R
ev. P
hysi
ol. 2
011.
73:1
35-1
62. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by K
arol
insk
a In
stitu
tet o
n 07
/26/
11. F
or p
erso
nal u
se o
nly.

PH73CH07-Desvergne ARI 3 January 2011 16:15
A second complication is exposure to a mix-ture of EDCs rather than to a single EDC. Hu-mans and wildlife are exposed daily to a varietyof compounds, and it is thus likely that even ifnone of the compounds reach an effective level,the combination or mixture of chemicals maybecome effective. This scenario is supported bythe observation that various EDCs share re-ceptors, and thus additive effects should be ob-served (see Figure 1; Tables 1 and 2). Fewexperimental studies addressing this issue ex-ist, and given the vast number of EDCs, it isunclear how to monitor exposure and how touse or develop assays that capture the effects ofthese mixtures. At a practical level it is also un-clear how regulatory decisions will be amendedto account for exposure to mixtures rather thanto single compounds.
4.3. Establishing the Link BetweenExposure and Metabolic Effects
Metabolic alterations such as metabolic syn-drome and type 2 diabetes are complex andmultifactorial. Elevated body mass index andobesity are not diseases but rather contributeto various alterations that lead to the diseasedstate. EDC exposure is one more factor thatincreases an already long list of predisposingfactors that act in combination to increase therisk of obesity or other metabolic alterations.Such contributions can be revealed only bycomprehensive studies of large and well-characterized cohorts, such as the cohort usedfor the NHANES project (see sidebar entitledNHANES). However, these studies are cross-sectional (see sidebar entitled ObservationalStudies), due to the difficulties inherent in theevaluation of exposure, and may be subjectto unidentified confounding factors. At best,these studies can demonstrate correlations butnot causal links between exposure and effects.
4.4. Experimental Exploration of theMetabolic Disruption Properties ofEndocrine-Disrupting Chemicals
How can we reach the most appropriate un-derstanding of EDC biology that will help to
define appropriate actions? Although cellularmodels are flexible and allow large numbersof conditions and doses to be tested, they arelimited and unable to factor in bioavailabil-ity of the compounds and their metabolites orthe route of exposure. Reductionist molecu-lar and cellular studies may not take into ac-count the knowledge that several EDCs inter-fere with a variety of NRs, not all of whichare expressed in the same tissue or at the sametime. Additionally, EDCs can affect several or-gans in the body with different intensities, lead-ing to a global response that may be oppositeof that observed in cell cultures. Experimen-tation in animals is therefore unavoidable, asanimals are threatened by environmental con-taminants and provide a reasonable approxi-mation of human metabolism for most com-pounds. However, two main issues must beconsidered. First, EDC-interacting receptorsdisplay species-specific activity that is well de-scribed for PPARα, PXR, and CAR (98, 136).The development of humanized mice, in whichthe gene of the receptor of interest is exchangedfor the human allele, provides new tools for thistype of investigation (136, 143). Second, issuesof dose and exposure time complicate exper-imental design (see Table 2). Mice may livefor two years, and a three-month exposure canbe considered chronic. Does this experimentalsetup accurately reflect the decades over whichhumans might be exposed?
4.5. Exposure During Critical Periodsof Development
The dramatic effect of DES exposure on femalebabies illustrates the critical issue of exposureduring development (see Section 3.1.1). Con-sistent with the well-known functions of clas-sic hormones in developing reproductive or-gans, the effects are easily understood whenthese hormones are disrupted or mimicked.Even disregarding transgenerational effects, themetabolic consequences of prenatal exposurethat manifest in adulthood are difficult to as-sess and to understand. As discussed above (seeSection 3.1.1), controversy surrounds many of
www.annualreviews.org • Pollutants and Metabolic Disruption 153
Ann
u. R
ev. P
hysi
ol. 2
011.
73:1
35-1
62. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by K
arol
insk
a In
stitu
tet o
n 07
/26/
11. F
or p
erso
nal u
se o
nly.

PH73CH07-Desvergne ARI 3 January 2011 16:15
these consequences, and only systematic animalstudies may lead to a fair risk evaluation. Futureefforts should be aimed at elucidating whetherand how epigenetic imprinting is involved inthese pathologies (153).
4.6. How Should New ChemicalsBe Regulated?
As described in Table 1, several chemical prod-ucts are either totally banned or authorized foruse under strict conditions. Banning a persistentorganic pollutant will immediately limit expo-sure, but a number of years will pass before thechemical is entirely suppressed, and such sup-pression will occur only if large transnationalterritories ban the chemical of interest (seeTable 2). Inevitably, banned products are re-placed by others that bring other unwanted ef-fects. How do we establish the most pertinentand effective modes of risk evaluation not onlyfor the present but also for any future com-pound released to consumers?
From the research point of view, recent yearshave witnessed many efforts to set up new tech-nologies for EDC detection in human tissues,including scenarios of low doses, nonpersistentEDCs, and the developmental period (27, 56,67). New integrative approaches combining ge-nomics, proteomics, metabolomics, systems bi-ology, and computational modeling should helpto understand the complexity of the cocktail ef-fect and its consequences when exposure occursat various life stages (101, 150, 154). In addi-tion, these approaches should help to evaluate
the global effects of EDC interference with dif-ferent NRs and the activation of a complex bi-ological network. Before reaching this level ofunderstanding, these integrative strategies mayhelp define a signature: a composite signal ofno explanatory value but reflecting exposure tometabolic disruptors. Such a signature may helpin experimentally establishing a predictive valueto new compounds.
From the regulatory point of view, govern-ments face the challenge of making appropri-ate decisions by balancing potential risks againstdemonstrated advantages. Help for future deci-sions will come from two complementary pro-cesses. The first process is illustrated by theEuropean Registration, Evaluation, Authorisa-tion, and Restriction of Chemicals (REACH)program, aimed at protection of human healthand the environment through systematic reg-istration, assessment, and annotation of an ex-haustive list of industry compounds. The sec-ond process encourages research through grantsubsidiaries, as illustrated by the recent financ-ing of BPA research by the National Insti-tutes of Health (NIH). These initiatives includeincentives for transparency and collaboration,both at the level of government and betweenscientists. Metabolic perturbations are only onesmall aspect of the EDC-related problems tobe solved, but what we know now may be onlythe tip of the iceberg. In the present context ofendemic metabolic disorders, with severe eco-nomic, social, and professional consequences,every action first to understand and then to con-trol risk factors is beneficial on all counts.
SUMMARY POINTS
1. Increasing human exposure to endocrine-disrupting chemicals (EDCs) has been asso-ciated with the development of some of the main ailments of the industrialized world,particularly metabolic disorders like obesity, diabetes, and metabolic syndrome.
2. Among different mechanisms of action, lipophilic EDCs compounds can bind specificallyto nuclear receptors and can displace the corresponding endogenous ligands to modulatehormone-responsive pathways.
3. Persistent organic pollutants such as organochlorine pesticides, dioxins, and polyfluo-roalkyl compounds and nonpersistent pollutants such as bisphenol A and several phtha-lates are suspected of metabolic disruption activity.
154 Casals-Casas · Desvergne
Ann
u. R
ev. P
hysi
ol. 2
011.
73:1
35-1
62. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by K
arol
insk
a In
stitu
tet o
n 07
/26/
11. F
or p
erso
nal u
se o
nly.

PH73CH07-Desvergne ARI 3 January 2011 16:15
4. A major mechanism of EDC-mediated metabolic disruption is through EDC interactionwith nuclear receptors, including (a) sex steroid hormone receptors, (b) receptors actingas xenobiotic sensors, and (c) receptors specialized in metabolic regulations.
5. This field is littered by controversies, in part due to the difficulties in proving or disprovingEDC activity. The major issues are the monitoring of exposure levels, the identificationof the metabolic effective dose, and the establishment of a link between (a) either expo-sure during critical periods of development or chronic exposure at very low doses and(b) metabolic effects. Finally, this area of research would benefit tremendously if com-mon methodologies of experimental EDC exposure were established. All these issuesneed further work to create a common and effective regulatory policy for environmentalchemical pollutants.
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings thatmight be perceived as affecting the objectivity of this review.
LITERATURE CITED
1. U.S. Environ. Prot. Agency. 2000. Endocrine Disruptor Screening and Testing Advisory Com-mittee (EDSTAC). Washington, DC: U.S. Environ. Prot. Agency. http://www.epa.gov/scipoly/oscpendo/edspoverview/edstac.htm (accessed September 14, 2006)
2. Tabb M, Blumberg B. 2006. New modes of action for endocrine-disrupting chemicals. Mol. Endocrinol.20:475–82
3. Colborn T, vom Saal FS, Soto AM. 1993. Developmental effects of endocrine-disrupting chemicals inwildlife and humans. Environ. Health Perspect. 101:378–84
4. McAllister EJ, Dhurandhar NV, Keith SW, Aronne LJ, Barger J, et al. 2009. Ten putative contributorsto the obesity epidemic. Crit. Rev. Food Sci. Nutr. 49:868–913
5. Baillie-Hamilton PF. 2002. Chemical toxins: a hypothesis to explain the global obesity epidemic.J. Altern. Complement Med. 8:185–92
6. Casals-Casas C, Feige JN, Desvergne B. 2008. Interference of pollutants with PPARs: Endocrine dis-ruption meets metabolism. Int. J. Obes. 32(Suppl. 6):53–61
7. Elobeid MA, Allison DB. 2008. Putative environmental-endocrine disruptors and obesity: a review. Curr.Opin. Endocrinol. Diabetes Obes. 15:403–8
8. Chen JQ, Brown TR, Russo J. 2009. Regulation of energy metabolism pathways by estrogens andestrogenic chemicals and potential implications in obesity associated with increased exposure to endocrinedisruptors. Biochim. Biophys. Acta 1793:1128–43
9. Hatch EE, Nelson JW, Stahlhut RW, Webster TF. 2010. Association of endocrine disruptors andobesity: perspectives from epidemiological studies. Int. J. Androl. 33:324–32
10. Grun F, Blumberg B. 2009. Endocrine disrupters as obesogens. Mol. Cell. Endocrinol. 304:19–2911. Grun F, Blumberg B. 2006. Environmental obesogens: organotins and endocrine disruption via nuclear
receptor signaling. Endocrinology 147:S50–5512. Ben-Jonathan N, Hugo ER, Brandebourg TD. 2009. Effects of bisphenol A on adipokine release from
human adipose tissue: implications for the metabolic syndrome. Mol. Cell. Endocrinol. 304:49–5413. Costa LG, Giordano G, Tagliaferri S, Caglieri A, Mutti A. 2008. Polybrominated diphenyl ether (PBDE)
flame retardants: environmental contamination, human body burden and potential adverse health effects.Acta Biomed. 79:172–83
14. Jensen AA, Leffers H. 2008. Emerging endocrine disrupters: perfluoroalkylated substances. Int. J. Androl.31:161–69
www.annualreviews.org • Pollutants and Metabolic Disruption 155
Ann
u. R
ev. P
hysi
ol. 2
011.
73:1
35-1
62. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by K
arol
insk
a In
stitu
tet o
n 07
/26/
11. F
or p
erso
nal u
se o
nly.

PH73CH07-Desvergne ARI 3 January 2011 16:15
15. Kovarova J, Svobodova Z. 2008. Perfluorinated compounds: occurrence and risk profile. Neuro. En-docrinol. Lett. 29:599–608
16. McKinlay R, Plant JA, Bell JN, Voulvoulis N. 2008. Endocrine disrupting pesticides: implications forrisk assessment. Environ. Int. 34:168–83
17. Schecter A, Birnbaum L, Ryan JJ, Constable JD. 2006. Dioxins: an overview. Environ. Res. 101:419–2818. U.S. Environ. Prot. Agency. 2007. What is a pesticide? http://www.epa.gov/pesticides/about/. Re-
trieved on September 15, 200719. Salehi F, Turner MC, Phillips KP, Wigle DT, Krewski D, Aronson KJ. 2008. Review of the etiology
of breast cancer with special attention to organochlorines as potential endocrine disruptors. J. Toxicol.Environ. Health B 11:276–300
20. Porta M, Lee DH, Puigdomenech E. 2009. Transgenerational inheritance of environmental obesogens.Occup. Environ. Med. 66:141–42
21. Karmaus W, Osuch JR, Eneli I, Mudd LM, Zhang J, et al. 2009. Maternal levels of dichlorodiphenyl-dichloroethylene (DDE) may increase weight and body mass index in adult female offspring. Occup.Environ. Med. 66:143–49
22. Smink A, Ribas-Fito N, Garcia R, Torrent M, Mendez MA, et al. 2008. Exposure to hexachlorobenzeneduring pregnancy increases the risk of overweight in children aged 6 years. Acta Paediatr. 97:1465–69
23. Lee DH, Lee IK, Porta M, Steffes M, Jacobs DR Jr. 2007. Relationship between serum concentrationsof persistent organic pollutants and the prevalence of metabolic syndrome among nondiabetic adults:results from the National Health and Nutrition Examination Survey 1999–2002. Diabetologia 50:1841–51
24. Park SK, Son HK, Lee SK, Kang JH, Chang YS, et al. 2010. Relationship between serum concentrationsof organochlorine pesticides and metabolic syndrome among non-diabetic adults. J. Prev. Med. PublicHealth 43:1–8
25. Turyk M, Anderson H, Knobeloch L, Imm P, Persky V. 2009. Organochlorine exposure and incidenceof diabetes in a cohort of Great Lakes sport fish consumers. Environ. Health Perspect. 117:1076–82
26. Pelclova D, Urban P, Preiss J, Lukas E, Fenclova Z, et al. 2006. Adverse health effects in humans exposedto 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Rev. Environ. Health 21:119–38
27. Lee DH, Lee IK, Steffes M, Jacobs DR Jr. 2007. Extended analyses of the association between serumconcentrations of persistent organic pollutants and diabetes. Diabetes Care 30:1596–98
28. Grun F, Watanabe H, Zamanian Z, Maeda L, Arima K, et al. 2006. Endocrine-disrupting organotincompounds are potent inducers of adipogenesis in vertebrates. Mol. Endocrinol. 20:2141–55
29. Omura M, Ogata R, Kubo K, Shimasaki Y, Aou S, et al. 2001. Two-generation reproductive toxicitystudy of tributyltin chloride in male rats. Toxicol. Sci. 64:224–32
30. Ogata R, Omura M, Shimasaki Y, Kubo K, Oshima Y, et al. 2001. Two-generation reproductive toxicitystudy of tributyltin chloride in female rats. J. Toxicol. Environ. Health A 63:127–44
31. Kanayama T, Kobayashi N, Mamiya S, Nakanishi T, Nishikawa J. 2005. Organotin compounds promoteadipocyte differentiation as agonists of the peroxisome proliferator-activated receptor gamma/retinoidX receptor pathway. Mol. Pharmacol. 67:766–74
32. Halden RU. 2010. Plastics and health risks. Annu. Rev. Public Health 31:179–9433. Fuentes S, Colomina MT, Rodriguez J, Vicens P, Domingo JL. 2006. Interactions in developmental
toxicology: concurrent exposure to perfluorooctane sulfonate (PFOS) and stress in pregnant mice. Toxicol.Lett. 164:81–89
34. Lau C, Butenhoff JL, Rogers JM. 2004. The developmental toxicity of perfluoroalkyl acids and theirderivatives. Toxicol. Appl. Pharmacol. 198:231–41
35. Luebker DJ, York RG, Hansen KJ, Moore JA, Butenhoff JL. 2005. Neonatal mortality from in uteroexposure to perfluorooctanesulfonate (PFOS) in Sprague-Dawley rats: dose-response, and biochemicaland pharamacokinetic parameters. Toxicology 215:149–69
36. Shi Z, Zhang H, Liu Y, Xu M, Dai J. 2007. Alterations in gene expression and testosterone synthesis inthe testes of male rats exposed to perfluorododecanoic acid. Toxicol. Sci. 98:206–15
37. Thibodeaux JR, Hanson RG, Rogers JM, Grey BE, Barbee BD, et al. 2003. Exposure to perfluorooctanesulfonate during pregnancy in rat and mouse. I. Maternal and prenatal evaluations. Toxicol. Sci. 74:369–81
156 Casals-Casas · Desvergne
Ann
u. R
ev. P
hysi
ol. 2
011.
73:1
35-1
62. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by K
arol
insk
a In
stitu
tet o
n 07
/26/
11. F
or p
erso
nal u
se o
nly.

PH73CH07-Desvergne ARI 3 January 2011 16:15
38. Apelberg BJ, Witter FR, Herbstman JB, Calafat AM, Halden RU, et al. 2007. Cord serum concentrationsof perfluorooctane sulfonate (PFOS) and perfluorooctanoate (PFOA) in relation to weight and size atbirth. Environ. Health Perspect. 115:1670–76
39. Gilliland FD, Mandel JS. 1996. Serum perfluorooctanoic acid and hepatic enzymes, lipoproteins, andcholesterol: a study of occupationally exposed men. Am. J. Ind. Med. 29:560–68
40. Nelson JW, Hatch EE, Webster TF. 2010. Exposure to polyfluoroalkyl chemicals and cholesterol, bodyweight, and insulin resistance in the general U.S. population. Environ. Health Perspect. 118:197–202
41. Costa G, Sartori S, Consonni D. 2009. Thirty years of medical surveillance in perfluooctanoic acidproduction workers. J. Occup. Environ. Med. 51:364–72
42. MacNeil J, Steenland NK, Shankar A, Ducatman A. 2009. A cross-sectional analysis of type II diabetesin a community with exposure to perfluorooctanoic acid (PFOA). Environ. Res. 109:997–1003
43. Olsen GW, Zobel LR. 2007. Assessment of lipid, hepatic, and thyroid parameters with serum perflu-orooctanoate (PFOA) concentrations in fluorochemical production workers. Int. Arch. Occup. Environ.Health 81:231–46
44. Lin CY, Lin LY, Chiang CK, Wang WJ, Su YN, et al. 2010. Investigation of the associations between low-dose serum perfluorinated chemicals and liver enzymes in US adults. Am. J. Gastroenterol. 105(6):1354–63
45. Lin CY, Chen PC, Lin YC, Lin LY. 2009. Association among serum perfluoroalkyl chemicals, glucosehomeostasis, and metabolic syndrome in adolescents and adults. Diabetes Care 32:702–7
46. Sjodin A, Hagmar L, Klasson-Wehler E, Kronholm-Diab K, Jakobsson E, Bergman A. 1999. Flameretardant exposure: polybrominated diphenyl ethers in blood from Swedish workers. Environ. HealthPerspect. 107:643–48
47. Jakobsson K, Thuresson K, Rylander L, Sjodin A, Hagmar L, Bergman A. 2002. Exposure to polybromi-nated diphenyl ethers and tetrabromobisphenol A among computer technicians. Chemosphere 46:709–16
48. Hamers T, Kamstra JH, Sonneveld E, Murk AJ, Kester MH, et al. 2006. In vitro profiling of theendocrine-disrupting potency of brominated flame retardants. Toxicol. Sci. 92:157–73
49. Darnerud PO. 2003. Toxic effects of brominated flame retardants in man and in wildlife. Environ. Int.29:841–53
50. Main KM, Kiviranta H, Virtanen HE, Sundqvist E, Tuomisto JT, et al. 2007. Flame retardants inplacenta and breast milk and cryptorchidism in newborn boys. Environ. Health Perspect. 115:1519–26
51. Lim JS, Lee DH, Jacobs DR Jr. 2008. Association of brominated flame retardants with diabetes andmetabolic syndrome in the U.S. population, 2003–2004. Diabetes Care 31:1802–7
52. Soares A, Guieysse B, Jefferson B, Cartmell E, Lester JN. 2008. Nonylphenol in the environment: acritical review on occurrence, fate, toxicity and treatment in wastewaters. Environ. Int. 34:1033–49
53. Soto AM, Justicia H, Wray JW, Sonnenschein C. 1991. p-Nonyl-phenol: an estrogenic xenobiotic re-leased from “modified” polystyrene. Environ. Health Perspect. 92:167–73
54. Lee HJ, Chattopadhyay S, Gong EY, Ahn RS, Lee K. 2003. Antiandrogenic effects of bisphenol A andnonylphenol on the function of androgen receptor. Toxicol. Sci. 75:40–46
55. Bonefeld-Jorgensen EC, Long M, Hofmeister MV, Vinggaard AM. 2007. Endocrine-disrupting poten-tial of bisphenol A, bisphenol A dimethacrylate, 4-n-nonylphenol, and 4-n-octylphenol in vitro: new dataand a brief review. Environ. Health Perspect. 115(Suppl. 1):69–76
56. Lopez-Espinosa MJ, Freire C, Arrebola JP, Navea N, Taoufiki J, et al. 2009. Nonylphenol and octylphe-nol in adipose tissue of women in Southern Spain. Chemosphere 76:847–52
57. Calafat AM, Kuklenyik Z, Reidy JA, Caudill SP, Ekong J, Needham LL. 2005. Urinary concentrationsof bisphenol A and 4-nonylphenol in a human reference population. Environ. Health Perspect. 113:391–95
58. Dodds EC. 1936. The pharmacological action and clinical use of drugs with a camphor- and coramine-like action. Proc. R. Soc. Med. 29:655–57
59. Richter CA, Birnbaum LS, Farabollini F, Newbold RR, Rubin BS, et al. 2007. In vivo effects of bisphenolA in laboratory rodent studies. Reprod. Toxicol. 24:199–224
60. Lang IA, Galloway TS, Scarlett A, Henley WE, Depledge M, et al. 2008. Association of urinary bisphenolA concentration with medical disorders and laboratory abnormalities in adults. JAMA 300:1303–10
61. Sugiura-Ogasawara M, Ozaki Y, Sonta S, Makino T, Suzumori K. 2005. Exposure to bisphenol A isassociated with recurrent miscarriage. Hum. Reprod. 20:2325–29
www.annualreviews.org • Pollutants and Metabolic Disruption 157
Ann
u. R
ev. P
hysi
ol. 2
011.
73:1
35-1
62. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by K
arol
insk
a In
stitu
tet o
n 07
/26/
11. F
or p
erso
nal u
se o
nly.

PH73CH07-Desvergne ARI 3 January 2011 16:15
62. Takeuchi T, Tsutsumi O, Ikezuki Y, Takai Y, Taketani Y. 2004. Positive relationship between androgenand the endocrine disruptor, bisphenol A, in normal women and women with ovarian dysfunction. Endocr.J. 51:165–69
63. Frederiksen H, Skakkebaek N, Andersson A. 2007. Metabolism of phthalates in humans. Mol. Nutr. FoodRes. 51:899–911
64. Arcadi F, Costa C, Imperatore C, Marchese A, Rapisarda A, et al. 1998. Oral toxicity of bis(2-ethylhexyl)phthalate during pregnancy and suckling in the Long-Evans rat. Food Chem. Toxicol. 36:963–70
65. Li L, Jester WJ, Orth J. 1998. Effects of relatively low levels of mono-(2-ethylhexyl) phthalate oncocultured Sertoli cells and gonocytes from neonatal rats. Toxicol. Appl. Pharmacol. 153:258–65
66. Foster P. 2006. Disruption of reproductive development in male rat offspring following in utero exposureto phthalate esters. Int. J. Androl. 29:140–47
67. Meeker J, Sathyanarayana S, Swan S. 2009. Phthalates and other additives in plastics: human exposureand associated health outcomes. Philos. Trans. R. Soc. Lond. Ser. B 364:2097–113
68. Swan S, Main K, Liu F, Stewart S, Kruse R, et al. 2005. Decrease in anogenital distance among maleinfants with prenatal phthalate exposure. Environ. Health Perspect. 113:1056–61
69. Sharpe R. 2005. Phthalate exposure during pregnancy and lower anogenital index in boys: wider impli-cations for the general population? Environ. Health Perspect. 113:A504–5
70. Bornehag C, Sundell J, Weschler C, Sigsgaard T, Lundgren B, et al. 2004. The association betweenasthma and allergic symptoms in children and phthalates in house dust: a nested case-control study.Environ. Health Perspect. 112:1393–97
71. Gayathri N, Dhanya C, Indu A, Kurup P. 2004. Changes in some hormones by low doses of di (2-ethylhexyl) phthalate (DEHP), a commonly used plasticizer in PVC blood storage bags & medical tubing.Indian J. Med. Res. 119:139–44
72. Heudorf U, Mersch-Sundermann V, Angerer J. 2007. Phthalates: toxicology and exposure. Int. J. Hyg.Environ. Health 210:623–34
73. Stahlhut R, van Wijngaarden E, Dye T, Cook S, Swan S. 2007. Concentrations of urinary phthalatemetabolites are associated with increased waist circumference and insulin resistance in adult U.S. males.Environ. Health Perspect. 115:876–82
74. Tchernof A, Calles-Escandon J, Sites CK, Poehlman ET. 1998. Menopause, central body fatness, andinsulin resistance: effects of hormone-replacement therapy. Coron. Artery Dis. 9:503–11
75. Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS. 2000. Increased adipose tissue in male andfemale estrogen receptor-α knockout mice. Proc. Natl. Acad. Sci. USA 97:12729–34
76. Brown LM, Gent L, Davis K, Clegg DJ. 2010. Metabolic impact of sex hormones on obesity. Brain Res.1350:77–85
77. Cooke PS, Naaz A. 2004. Role of estrogens in adipocyte development and function. Exp. Biol. Med.229:1127–35
78. Wada K, Sakamoto H, Nishikawa K, Sakuma S, Nakajima A, et al. 2007. Lifestyle-related diseases of thedigestive system: Endocrine disruptors stimulate lipid accumulation in target cells related to metabolicsyndrome. J. Pharmacol. Sci. 105:133–37
79. Phrakonkham P, Viengchareun S, Belloir C, Lombes M, Artur Y, Canivenc-Lavier MC. 2008. Dietaryxenoestrogens differentially impair 3T3-L1 preadipocyte differentiation and persistently affect leptinsynthesis. J. Steroid Biochem. Mol. Biol. 110:95–103
80. Lee MJ, Lin H, Liu CW, Wu MH, Liao WJ, et al. 2008. Octylphenol stimulates resistin gene expressionin 3T3-L1 adipocytes via the estrogen receptor and extracellular signal-regulated kinase pathways. Am.J. Physiol. Cell Physiol. 294:1542–51
81. Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, et al. 2001. The hormone resistin links obesityto diabetes. Nature 409:307–12
82. Alonso-Magdalena P, Ropero AB, Carrera MP, Cederroth CR, Baquie M, et al. 2008. Pancreatic insulincontent regulation by the estrogen receptor ERα. PLoS ONE 3:e2069
83. Martensson UE, Salehi SA, Windahl S, Gomez MF, Sward K, et al. 2009. Deletion of the G protein-coupled receptor 30 impairs glucose tolerance, reduces bone growth, increases blood pressure, andeliminates estradiol-stimulated insulin release in female mice. Endocrinology 150:687–98
158 Casals-Casas · Desvergne
Ann
u. R
ev. P
hysi
ol. 2
011.
73:1
35-1
62. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by K
arol
insk
a In
stitu
tet o
n 07
/26/
11. F
or p
erso
nal u
se o
nly.

PH73CH07-Desvergne ARI 3 January 2011 16:15
84. Thomas P, Dong J. 2006. Binding and activation of the seven-transmembrane estrogen receptor GPR30by environmental estrogens: a potential novel mechanism of endocrine disruption. J. Steroid Biochem.Mol. Biol. 102:175–79
85. Rubin MM. 2007. Antenatal exposure to DES: lessons learned. . .future concerns. Obstet. Gynecol. Surv.62:548–55
86. Newbold RR, Padilla-Banks E, Snyder RJ, Jefferson WN. 2007. Perinatal exposure to environmentalestrogens and the development of obesity. Mol. Nutr. Food Res. 51:912–17
87. Rubin BS, Soto AM. 2009. Bisphenol A: perinatal exposure and body weight. Mol. Cell. Endocrinol.304:55–62
88. Somm E, Schwitzgebel VM, Toulotte A, Cederroth CR, Combescure C, et al. 2009. Perinatal exposureto bisphenol A alters early adipogenesis in the rat. Environ. Health Perspect. 117:1549–55
89. Miyawaki J, Sakayama K, Kato H, Yamamoto H, Masuno H. 2007. Perinatal and postnatal exposure tobisphenol A increases adipose tissue mass and serum cholesterol level in mice. J. Atheroscler. Thromb.14:245–52
90. Heindel JJ, vom Saal FS. 2009. Role of nutrition and environmental endocrine disrupting chemicalsduring the perinatal period on the aetiology of obesity. Mol. Cell. Endocrinol. 304:90–96
91. Sargis RM, Johnson DN, Choudhury RA, Brady MJ. 2010. Environmental endocrine disruptors promoteadipogenesis in the 3T3-L1 cell line through glucocorticoid receptor activation. Obesity 18:1283–88
92. Moriyama K, Tagami T, Akamizu T, Usui T, Saijo M, et al. 2002. Thyroid hormone action is disruptedby bisphenol A as an antagonist. J. Clin. Endocrinol. Metab. 87:5185–90
93. Zoeller RT, Bansal R, Parris C. 2005. Bisphenol-A, an environmental contaminant that acts as a thyroidhormone receptor antagonist in vitro, increases serum thyroxine, and alters RC3/neurogranin expressionin the developing rat brain. Endocrinology 146:607–12
94. Legler J. 2008. New insights into the endocrine disrupting effects of brominated flame retardants.Chemosphere 73:216–22
95. Hoppe AA, Carey GB. 2007. Polybrominated diphenyl ethers as endocrine disruptors of adipocytemetabolism. Obesity 15:2942–50
96. Moreau A, Vilarem MJ, Maurel P, Pascussi JM. 2008. Xenoreceptors CAR and PXR activation andconsequences on lipid metabolism, glucose homeostasis, and inflammatory response. Mol. Pharm. 5:35–41
97. Wada T, Gao J, Xie W. 2009. PXR and CAR in energy metabolism. Trends Endocrinol. Metab. 20:273–7998. Kretschmer XC, Baldwin WS. 2005. CAR and PXR: xenosensors of endocrine disrupters? Chem. Biol.
Interact. 155:111–2899. Ren H, Aleksunes LM, Wood C, Vallanat B, George MH, et al. 2010. Characterization of peroxisome
proliferator-activated receptor α–independent effects of PPARα activators in the rodent liver: Di-(2-ethylhexyl) phthalate also activates the constitutive-activated receptor. Toxicol. Sci. 113:45–59
100. Eveillard A, Mselli-Lakhal L, Mogha A, Lasserre F, Polizzi A, et al. 2009. Di-(2-ethylhexyl)-phthalate(DEHP) activates the constitutive androstane receptor (CAR): a novel signaling pathway sensitive tophthalates. Biochem. Pharmacol. 77:1735–46
101. Eveillard A, Lasserre F, de Tayrac M, Polizzi A, Claus S, et al. 2009. Identification of potential mech-anisms of toxicity after di-(2-ethylhexyl)-phthalate (DEHP) adult exposure in the liver using a systemsbiology approach. Toxicol. Appl. Pharmacol. 236:282–92
102. Hernandez JP, Huang W, Chapman LM, Chua S, Moore DD, Baldwin WS. 2007. The environmentalestrogen, nonylphenol, activates the constitutive androstane receptor. Toxicol. Sci. 98:416–26
103. Morere P, Nouvet G, Stain JP, Paillot B, Metayer J, Hemet J. 1975. Information obtained by liver biopsyin 100 tuberculous patients. Sem. Hop. 51:2095–102 (In French)
104. Calandre EP, Rodriquez-Lopez C, Blazquez A, Cano D. 1991. Serum lipids, lipoproteins and apolipopro-teins A and B in epileptic patients treated with valproic acid, carbamazepine or phenobarbital. Acta Neurol.Scand. 83:250–53
105. Lahtela JT, Arranto AJ, Sotaniemi EA. 1985. Enzyme inducers improve insulin sensitivity in noninsulin-dependent diabetic subjects. Diabetes 34:911–16
www.annualreviews.org • Pollutants and Metabolic Disruption 159
Ann
u. R
ev. P
hysi
ol. 2
011.
73:1
35-1
62. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by K
arol
insk
a In
stitu
tet o
n 07
/26/
11. F
or p
erso
nal u
se o
nly.

PH73CH07-Desvergne ARI 3 January 2011 16:15
106. Piriou A, Warnet JM, Jacqueson A, Claude JR, Truhaut R. 1979. Fatty liver induced by high doses ofrifampicin in the rat: possible relation with an inhibition of RNA polymerases in eukariotic cells. Arch.Toxicol. Suppl. 1979:333–37
107. Nakamura K, Moore R, Negishi M, Sueyoshi T. 2007. Nuclear pregnane X receptor cross-talk withFoxA2 to mediate drug-induced regulation of lipid metabolism in fasting mouse liver. J. Biol. Chem.282:9768–76
108. Zhou J, Zhai Y, Mu Y, Gong H, Uppal H, et al. 2006. A novel pregnane X receptor-mediated and sterolregulatory element-binding protein-independent lipogenic pathway. J. Biol. Chem. 281:15013–20
109. Argaud D, Halimi S, Catelloni F, Leverve XM. 1991. Inhibition of gluconeogenesis in isolated rathepatocytes after chronic treatment with phenobarbital. Biochem. J. 280(Pt. 3):663–69
110. Kodama S, Koike C, Negishi M, Yamamoto Y. 2004. Nuclear receptors CAR and PXR cross talk withFOXO1 to regulate genes that encode drug-metabolizing and gluconeogenic enzymes. Mol. Cell. Biol.24:7931–40
111. Bhalla S, Ozalp C, Fang S, Xiang L, Kemper JK. 2004. Ligand-activated pregnane X receptor interfereswith HNF-4 signaling by targeting a common coactivator PGC-1α. Functional implications in hepaticcholesterol and glucose metabolism. J. Biol. Chem. 279:45139–47
112. Gao J, He J, Zhai Y, Wada T, Xie W. 2009. The constitutive androstane receptor is an antiobesitynuclear receptor that improves insulin sensitivity. J. Biol. Chem. 284:25984–92
113. Dong B, Saha PK, Huang W, Chen W, Abu-Elheiga LA, et al. 2009. Activation of nuclear receptor CARameliorates diabetes and fatty liver disease. Proc. Natl. Acad. Sci. USA 106:18831–36
114. Pascussi JM, Gerbal-Chaloin S, Duret C, Daujat-Chavanieu M, Vilarem MJ, Maurel P. 2008. The tangleof nuclear receptors that controls xenobiotic metabolism and transport: crosstalk and consequences.Annu. Rev. Pharmacol. Toxicol. 48:1–32
115. Deleted in proof116. Bock KW, Kohle C. 2006. Ah receptor: dioxin-mediated toxic responses as hints to deregulated physi-
ologic functions. Biochem. Pharmacol. 72:393–404117. Kohle C, Bock KW. 2009. Coordinate regulation of human drug-metabolizing enzymes, and conjugate
transporters by the Ah receptor, pregnane X receptor and constitutive androstane receptor. Biochem.Pharmacol. 77:689–99
118. Barnes-Ellerbe S, Knudsen KE, Puga A. 2004. 2,3,7,8-Tetrachlorodibenzo-p-dioxin blocks androgen-dependent cell proliferation of LNCaP cells through modulation of pRB phosphorylation. Mol. Phar-macol. 66:502–11
119. Sato S, Shirakawa H, Tomita S, Ohsaki Y, Haketa K, et al. 2008. Low-dose dioxins alter gene expressionrelated to cholesterol biosynthesis, lipogenesis, and glucose metabolism through the aryl hydrocarbonreceptor-mediated pathway in mouse liver. Toxicol. Appl. Pharmacol. 229:10–19
120. Arsenescu V, Arsenescu RI, King V, Swanson H, Cassis LA. 2008. Polychlorinated biphenyl-77 inducesadipocyte differentiation and proinflammatory adipokines and promotes obesity and atherosclerosis.Environ. Health Perspect. 116:761–68
121. Matthews J, Gustafsson JA. 2006. Estrogen receptor and aryl hydrocarbon receptor signaling pathways.Nucl. Recept. Signal. 4:e016
122. Swedenborg E, Ruegg J, Makela S, Pongratz I. 2009. Endocrine disruptive chemicals: mechanisms ofaction and involvement in metabolic disorders. J. Mol. Endocrinol. 43:1–10
123. Cimafranca MA, Hanlon PR, Jefcoate CR. 2004. TCDD administration after the proadipogenic dif-ferentiation stimulus inhibits PPARγ through a MEK-dependent process but less effectively suppressesadipogenesis. Toxicol. Appl. Pharmacol. 196:156–68
124. Feige JN, Gelman L, Michalik L, Desvergne B, Wahli W. 2006. From molecular action to physiologicaloutputs: Peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellularfunctions. Prog. Lipid. Res. 45:120–59
125. Michalik L, Wahli W. 2007. Peroxisome proliferator-activated receptors (PPARs) in skin health, repairand disease. Biochim. Biophys. Acta 1771:991–98
126. Barish GD, Narkar VA, Evans RM. 2006. PPARδ: a dagger in the heart of the metabolic syndrome.J. Clin. Investig. 116:590–97
160 Casals-Casas · Desvergne
Ann
u. R
ev. P
hysi
ol. 2
011.
73:1
35-1
62. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by K
arol
insk
a In
stitu
tet o
n 07
/26/
11. F
or p
erso
nal u
se o
nly.

PH73CH07-Desvergne ARI 3 January 2011 16:15
127. Auwerx J. 1999. PPARγ, the ultimate thrifty gene. Diabetologia 42:1033–49128. Takeuchi S, Matsuda T, Kobayashi S, Takahashi T, Kojima H. 2006. In vitro screening of 200 pesticides
for agonistic activity via mouse peroxisome proliferator-activated receptor (PPAR)α and PPARγ andquantitative analysis of in vivo induction pathway. Toxicol. Appl. Pharmacol. 217:235–44
129. Palut D, Ludwicki JK, Kostka G, Kopec-Szlezak J, Wiadrowska B, Lembowicz K. 2001. Studies of earlyhepatocellular proliferation and peroxisomal proliferation in Wistar rats treated with herbicide diclofop.Toxicology 158:119–26
130. Ward JM, Peters JM, Perella CM, Gonzalez FJ. 1998. Receptor and nonreceptor-mediated organ-specifictoxicity of di(2-ethylhexyl)phthalate (DEHP) in peroxisome proliferator-activated receptor α-null mice.Toxicol. Pathol. 26:240–46
131. Bility MT, Thompson JT, McKee RH, David RM, Butala JH, et al. 2004. Activation of mouse and humanperoxisome proliferator-activated receptors (PPARs) by phthalate monoesters. Toxicol. Sci. 82:170–82
132. Hurst CH, Waxman DJ. 2003. Activation of PPARα and PPARγ by environmental phthalate mo-noesters. Toxicol. Sci. 74:297–308
133. Feige JN, Gelman L, Rossi D, Zoete V, Metivier R, et al. 2007. The endocrine disruptor monoethyl-hexyl-phthalate is a selective peroxisome proliferator-activated receptor γ modulator that promotesadipogenesis. J. Biol. Chem. 282:19152–66
134. Lapinskas PJ, Brown S, Leesnitzer LM, Blanchard S, Swanson C, et al. 2005. Role of PPARαin mediatingthe effects of phthalates and metabolites in the liver. Toxicology 207:149–63
135. Zoete V, Grosdidier A, Michielin O. 2007. Peroxisome proliferator-activated receptor structures: ligandspecificity, molecular switch and interactions with regulators. Biochim. Biophys. Acta 1771:915–25
136. Feige JN, Gerber A, Casals-Casas C, Yang Q, Winkler C, et al. 2010. The pollutant diethylhexylphthalate regulates hepatic energy metabolism via species-specific PPARα-dependent mechanisms. En-viron. Health Perspect. 118:234–41
137. Inagaki T, Dutchak P, Zhao G, Ding X, Gautron L, et al. 2007. Endocrine regulation of the fastingresponse by PPARα-mediated induction of fibroblast growth factor 21. Cell Metab. 5:415–25
138. Boberg J, Metzdorff S, Wortziger R, Axelstad M, Brokken L, et al. 2008. Impact of diisobutyl phthalateand other PPAR agonists on steroidogenesis and plasma insulin and leptin levels in fetal rats. Toxicology250:75–81
139. Takacs ML, Abbott BD. 2007. Activation of mouse and human peroxisome proliferator-activated recep-tors (α, β/δ, γ) by perfluorooctanoic acid and perfluorooctane sulfonate. Toxicol. Sci. 95:108–17
140. Xie Y, Yang Q, Nelson BD, DePierre JW. 2003. The relationship between liver peroxisome proliferationand adipose tissue atrophy induced by peroxisome proliferator exposure and withdrawal in mice. Biochem.Pharmacol. 66:749–56
141. Asakawa A, Toyoshima M, Harada KH, Fujimiya M, Inoue K, Koizumi A. 2008. The ubiquitous environ-mental pollutant perfluorooctanoic acid inhibits feeding behavior via peroxisome proliferator-activatedreceptor-α. Int. J. Mol. Med. 21:439–45
142. Hines EP, White SS, Stanko JP, Gibbs-Flournoy EA, Lau C, Fenton SE. 2009. Phenotypic dichotomyfollowing developmental exposure to perfluorooctanoic acid (PFOA) in female CD-1 mice: Low dosesinduce elevated serum leptin and insulin, and overweight in mid-life. Mol. Cell. Endocrinol. 304:97–105
143. Nakamura T, Ito Y, Yanagiba Y, Ramdhan DH, Kono Y, et al. 2009. Microgram-order ammoniumperfluorooctanoate may activate mouse peroxisome proliferator-activated receptor α, but not humanPPARα. Toxicology 265:27–33
144. le Maire A, Grimaldi M, Roecklin D, Dagnino S, Vivat-Hannah V, et al. 2009. Activation of RXR-PPARheterodimers by organotin environmental endocrine disruptors. EMBO Rep. 10:367–73
145. Peters AK, Nijmeijer S, Gradin K, Backlund M, Bergman A, et al. 2006. Interactions of polybrominateddiphenyl ethers with the aryl hydrocarbon receptor pathway. Toxicol. Sci. 92:133–42
146. Sanders JM, Burka LT, Smith CS, Black W, James R, Cunningham ML. 2005. Differential expressionof CYP1A, 2B, and 3A genes in the F344 rat following exposure to a polybrominated diphenyl ethermixture or individual components. Toxicol. Sci. 88:127–33
147. Pacyniak EK, Cheng X, Cunningham ML, Crofton K, Klaassen CD, Guo GL. 2007. The flame retar-dants, polybrominated diphenyl ethers, are pregnane X receptor activators. Toxicol. Sci. 97:94–102
www.annualreviews.org • Pollutants and Metabolic Disruption 161
Ann
u. R
ev. P
hysi
ol. 2
011.
73:1
35-1
62. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by K
arol
insk
a In
stitu
tet o
n 07
/26/
11. F
or p
erso
nal u
se o
nly.

PH73CH07-Desvergne ARI 3 January 2011 16:15
148. Darnerud PO. 2008. Brominated flame retardants as possible endocrine disrupters. Int. J. Androl. 31:152–60
149. Costa LG, Giordano G. 2007. Developmental neurotoxicity of polybrominated diphenyl ether (PBDE)flame retardants. Neurotoxicology 28:1047–67
150. Soto AM, Rubin BS, Sonnenschein C. 2009. Interpreting endocrine disruption from an integrativebiology perspective. Mol. Cell. Endocrinol. 304:3–7
151. Vanderberg JP. 2009. Reflections on early malaria vaccine studies, the first successful human malariavaccination, and beyond. Vaccine 27:2–9
152. vom Saal FS, Akingbemi BT, Belcher SM, Birnbaum LS, Crain DA, et al. 2007. Chapel Hill bisphenol Aexpert panel consensus statement: integration of mechanisms, effects in animals and potential to impacthuman health at current levels of exposure. Reprod. Toxicol. 24:131–38
153. Crews D, McLachlan JA. 2006. Epigenetics, evolution, endocrine disruption, health, and disease.Endocrinology 147:S4–10
154. Vineis P, Khan AE, Vlaanderen J, Vermeulen R. 2009. The impact of new research technologies on ourunderstanding of environmental causes of disease: the concept of clinical vulnerability. Environ. Health8:54
155. Lin HK, Altuwaijri S, Lin WJ, Kan PY, Collins LL, Chang C. 2002. Proteasome activity is required forandrogen receptor transcriptional activity via regulation of androgen receptor nuclear translocation andinteraction with coregulators in prostate cancer cells. J. Biol. Chem. 277:36570–76
156. Hugo ER, Brandebourg TD, Woo JG, Loftus J, Alexander JW, Ben-Jonathan N. 2008. Bisphenol Aat environmentally relevant doses inhibits adiponectin release from human adipose tissue explants andadipocytes. Environ. Health Perspect. 116:1642–47
157. Moreno-Aliaga MJ, Matsumura F. 2002. Effects of 1,1,1-trichloro-2,2-bis( p-chlorophenyl)-ethane ( p,p′-DDT) on 3T3-L1 and 3T3-F442A adipocyte differentiation. Biochem. Pharmacol. 63:997–1007
158. World Health Organ. 2002. Evaluation of Certain Food Additives and Contaminants. Geneva: World HealthOrgan.
162 Casals-Casas · Desvergne
Ann
u. R
ev. P
hysi
ol. 2
011.
73:1
35-1
62. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by K
arol
insk
a In
stitu
tet o
n 07
/26/
11. F
or p
erso
nal u
se o
nly.

PH73-FrontMatter ARI 14 January 2011 10:0
Annual Review ofPhysiology
Volume 73, 2011Contents
PERSPECTIVES, David Julius, Editor
A Long Affair with Renal TubulesGerhard H. Giebisch � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 1
CARDIOVASCULAR PHYSIOLOGY, Jeffrey Robbins, Section Editor
Heart Valve Structure and Function in Development and DiseaseRobert B. Hinton and Katherine E. Yutzey � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � �29
Myocardial Remodeling: Cellular and Extracellular Events and TargetsJennifer A. Dixon and Francis G. Spinale � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � �47
ECOLOGICAL, EVOLUTIONARY, AND COMPARATIVE PHYSIOLOGY,Martin E. Feder, Section Editor
Ecological Physiology of Diet and Digestive SystemsWilliam H. Karasov, Carlos Martınez del Rio, and Enrique Caviedes-Vidal � � � � � � � � � � � � �69
Effects of Oxygen on Growth and Size: Synthesis of Molecular,Organismal, and Evolutionary Studies with Drosophila melanogasterJon F. Harrison and Gabriel G. Haddad � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � �95
LEA Proteins During Water Stress: Not Just for Plants AnymoreSteven C. Hand, Michael A. Menze, Mehmet Toner, Leaf Boswell,
and Daniel Moore � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 115
ENDOCRINOLOGY, Holly A. Ingraham, Section Editor
Endocrine Disruptors: From Endocrine to Metabolic DisruptionCristina Casals-Casas and Beatrice Desvergne � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 135
Endometriosis: The Role of NeuroangiogenesisAlbert Asante and Robert N. Taylor � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 163
Zebrafish in Endocrine Systems: Recent Advances and Implications forHuman DiseaseHeiko Lohr and Matthias Hammerschmidt � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 183
vii
Ann
u. R
ev. P
hysi
ol. 2
011.
73:1
35-1
62. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by K
arol
insk
a In
stitu
tet o
n 07
/26/
11. F
or p
erso
nal u
se o
nly.

PH73-FrontMatter ARI 14 January 2011 10:0
GASTROINTESTINAL PHYSIOLOGY, James M. Anderson, Section Editor
Mesenchymal Cells of the Intestinal Lamina PropriaD.W. Powell, I.V. Pinchuk, J.I. Saada, Xin Chen, and R.C. Mifflin � � � � � � � � � � � � � � � � � � � � 213
Niemann-Pick C1-Like 1 (NPC1L1) Protein in Intestinal and HepaticCholesterol TransportLin Jia, Jenna L. Betters, and Liqing Yu � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 239
Regulation of Electroneutral NaCl Absorption by the Small IntestineAkira Kato and Michael F. Romero � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 261
Tight Junction Pore and Leak Pathways: A Dynamic DuoLe Shen, Christopher R. Weber, David R. Raleigh, Dan Yu, and Jerrold R. Turner � � � 283
NEUROPHYSIOLOGY, Roger Nicoll, Section Editor
How the Genetics of Deafness Illuminates Auditory PhysiologyGuy P. Richardson, Jacques Boutet de Monvel, and Christine Petit � � � � � � � � � � � � � � � � � � � � � � 311
RENAL AND ELECTROLYTE PHYSIOLOGY, Gerhard H. Giebisch, Section Editor
Mechanisms Underlying Rapid Aldosterone Effects in the KidneyWarren Thomas and Brian J. Harvey � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 335
Regulation of Renal NaCl Transport by Nitric Oxide, Endothelin,and ATP: Clinical ImplicationsJeffrey L. Garvin, Marcela Herrera, and Pablo A. Ortiz � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 359
Renin Release: Sites, Mechanisms, and ControlArmin Kurtz � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 377
Terminal Differentiation in Epithelia: The Role of Integrinsin Hensin PolymerizationQais Al-Awqati � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 401
RESPIRATORY PHYSIOLOGY, Richard C. Boucher, Jr., Section Editor
Epithelial-Mesenchymal Interactions in Pulmonary FibrosisHarold A. Chapman � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 413
Interaction of Cigarette Exposure and Airway EpithelialCell Gene ExpressionJerome S. Brody and Katrina Steiling � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 437
The Lung: The Natural Boundary Between Nature and NurtureMax A. Seibold and David A. Schwartz � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 457
viii Contents
Ann
u. R
ev. P
hysi
ol. 2
011.
73:1
35-1
62. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by K
arol
insk
a In
stitu
tet o
n 07
/26/
11. F
or p
erso
nal u
se o
nly.

PH73-FrontMatter ARI 14 January 2011 10:0
Role of Chitin and Chitinase/Chitinase-Like Proteins in Inflammation,Tissue Remodeling, and InjuryChun Geun Lee, Carla A. Da Silva, Charles S. Dela Cruz, Farida Ahangari,
Bing Ma, Min-Jong Kang, Chuan-Hua He, Seyedtaghi Takyar,and Jack A. Elias � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 479
SPECIAL TOPIC, THROMBOSIS, Charles T. Esmon, Special Topic Editor
The Link Between Vascular Features and ThrombosisCharles T. Esmon and Naomi L. Esmon � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 503
Role of Tissue Factor in Venous ThrombosisDavid A. Manly, Jeremiah Boles, and Nigel Mackman � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 515
Venous Valvular Stasis–Associated Hypoxia and Thrombosis: What Isthe Link?Edwin G. Bovill and Albert van der Vliet � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 527
Indexes
Cumulative Index of Contributing Authors, Volumes 69–73 � � � � � � � � � � � � � � � � � � � � � � � � � � � 547
Cumulative Index of Chapter Titles, Volumes 69–73 � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 550
Errata
An online log of corrections to Annual Review of Physiology articles may be found athttp://physiol.annualreviews.org/errata.shtml
Contents ix
Ann
u. R
ev. P
hysi
ol. 2
011.
73:1
35-1
62. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by K
arol
insk
a In
stitu
tet o
n 07
/26/
11. F
or p
erso
nal u
se o
nly.







