

Homology Modeling
• comparative modeling vs. ab initio folding• alignment (check gaps)• threading• loop building• re-packing side-chains in core, DEE, SCWRL• fold evaluation/scoring • statistical potentials (pot. of mean force) - DFIRE • minimization• servers: Swiss-model

• ab initio folding – Rosetta (Baker)– MONSSTER (Skolnick)– I-TASSER (Zhang)

Sequence Alignment
• critical step• gaps should be in loops (check in final model)• dynamic programming (Smith-Waterman)
– LALIGN: http://www.ch.embnet.org/software/LALIGN_form.html
– adjust gap parameters: gap-open penalty>x?gap-extension penalty<x? x=average match score
– could also adjust substitution matrix (PAM250, BLOSUM62)
• use PSI-Blast to include info from homologs– iterative: retrieves homologs, refines search...
• use HMM to align to family

Threading• use info about 3D structure to
improve alignment• local secondary structure,
solvent-accessibility• 3D profiles (Eisenberg)• 3D-PSSM/Phyre (Sternberg,
Lawrence Kelley)• THREADER• RAPTOR

MODELLER (Sali)
• references– A. Šali and T. L. Blundell. Comparative
protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779-815, 1993.
– A. Fiser, R. K. G. Do and A. Š ali. Modeling of loops in protein structures. Protein Science 9, 1753-1773, 2000.
– Fiser A, Sali A. (2003). Modeller: generation and refinement of homology-based protein structure models. Methods Enz. 374:461-91.
• loop-modeling via dynamics • evaluation:
– >30% identity?– stereochemistry: Procheck – contacts/exposure: ProSA (Sippl,
1993) – distance-based pair potentials

Side-chain re-packing• mutations cause steric conflicts (and voids)
– changing rotamers can relieve conflicts – adjacent side-chains are coupled– multiple changes might be required– combinatorial search: exhaustive versus Monte Carlo (Holm &
Sander, 1992)
• DEE (Dead-End Elimination) – pruning method – pre-processing, singles, pairs– Desmet, Mayo– reduction in branching factor?
• interesting application: use DEE todetermine rotamer populations for tryptophans; use to predict fluorescence quenching times (Hellings 2003, BiophysJ)
• rigid backbone assumption– how important is backbone flexibility?
– also sample alternative backbone conformations at each site
– (Georgiev and Donald, 2007)

SCWRL 3.0• Canutescu et al. (2003)
– Dunbrack BBdep rotamer library– de-couple interaction graph into
bi-connected componentsrepresenting local dependencies
• TreePack (Xu and Berger, JACM 2006)– geometric neighborhood graph decomposition; up to 90x faster
side-chain interactions:
energy of configuration:

Loop Modeling• two approaches:
1. MD/conformational sampling
2. templates from loop library
• accuracy depends on length: 2-4 (turns), 4-8, >8 (ab initio)
• importance in immunoglobulins (hyper-variable loops in antigen-binding region)
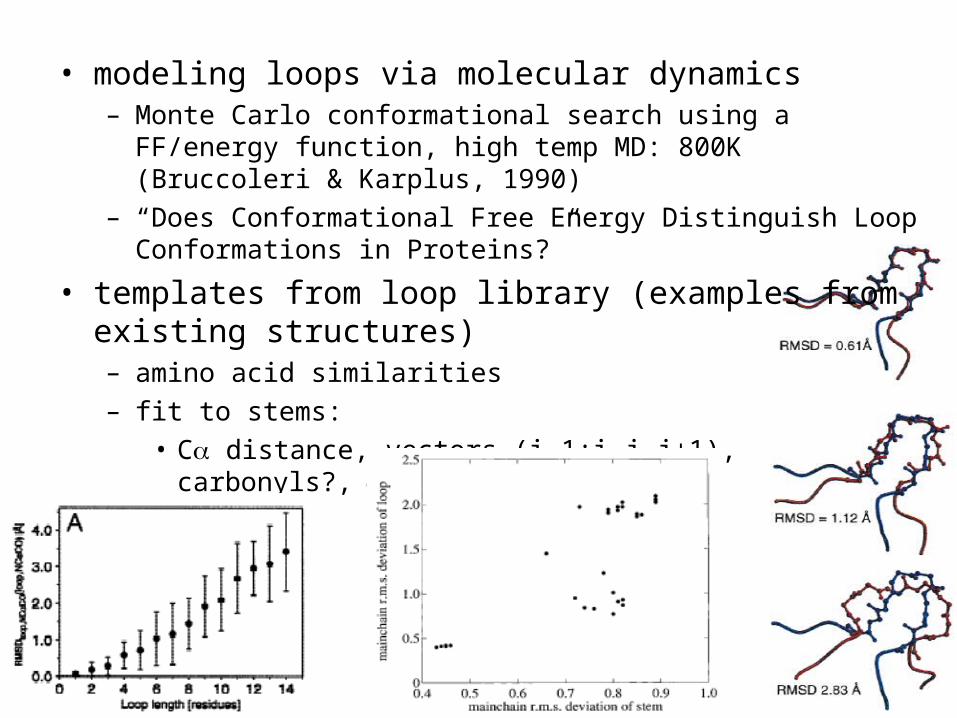
• modeling loops via molecular dynamics – Monte Carlo conformational search using a FF/energy function,
high temp MD: 800K (Bruccoleri & Karplus, 1990)– “Does Conformational Free Energy Distinguish Loop
Conformations in Proteins?”
• templates from loop library (examples from existing structures)– amino acid similarities– fit to stems:
• C distance, vectors (i-1:i,j,j+1), carbonyls?, angles
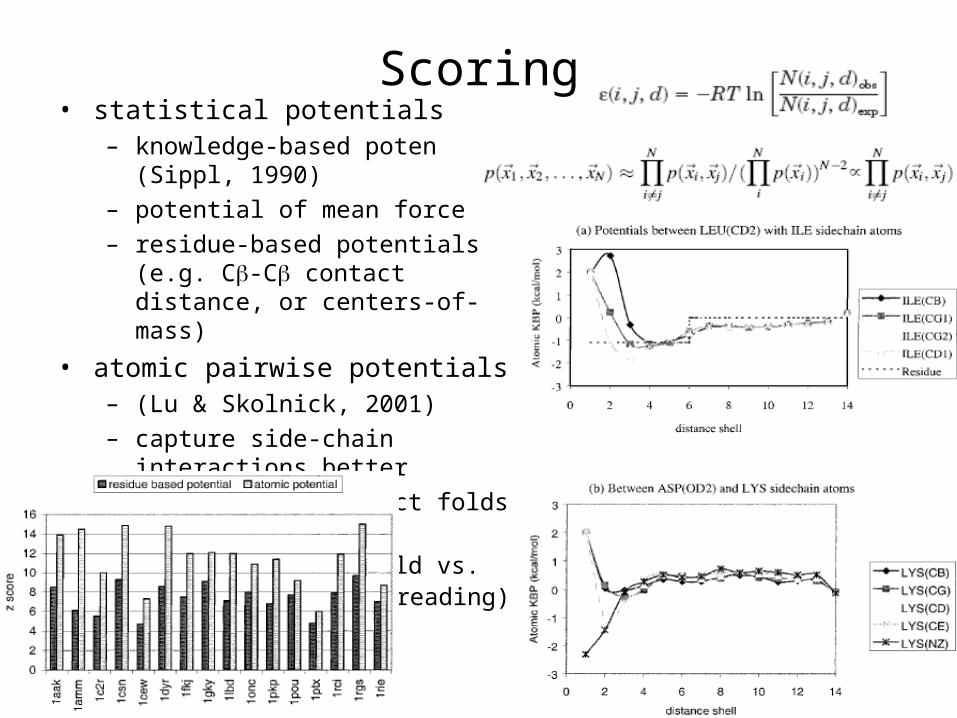
Scoring• statistical potentials
– knowledge-based poten (Sippl, 1990)
– potential of mean force
– residue-based potentials (e.g. C-C contact distance, or centers-of-mass)
• atomic pairwise potentials – (Lu & Skolnick, 2001)
– capture side-chain interactions better
– discriminate correct folds better
– z-score of true fold vs. decoys (gapless threading)

DFIRE (Yaoqi Zhou)
• Distance-scaled Finite Ideal-gas REference state – Zhou and Zhou (Prot. Sci, 2002)– all-atom potential– Nexp(i,j,r) will not increase in r2 as in an infinite system– =1.57 gives best correlation with density in radial shells– improves ability to recognize correct fold versus decoys
– see also: • RAPDF (Samudrala and Moult, 1998)• DOPE (Shen and Sali, 2006)
fair???
instead, assume:

Minimization
• a logical step, however...• one of the conclusions from CASP4 (Baker):
– minimization generally made models worse (took predicted structures farther from native)
– threshold: • minimization works if rmsd<2Å, • but ab initio models are often 4-6Å rmsd
– backbone adjustments required?







