

Mag. Martina Gantschacher Director of the Department for
Health Law & Science of the
Sigmund Freud Private University Vienna, Paris
Inspection and Audit Findings –
Quality in Studies

Inspection and Audit Findings – Quality in Studies
Introduction
Inspection and Audit Findings – Quality in Studies
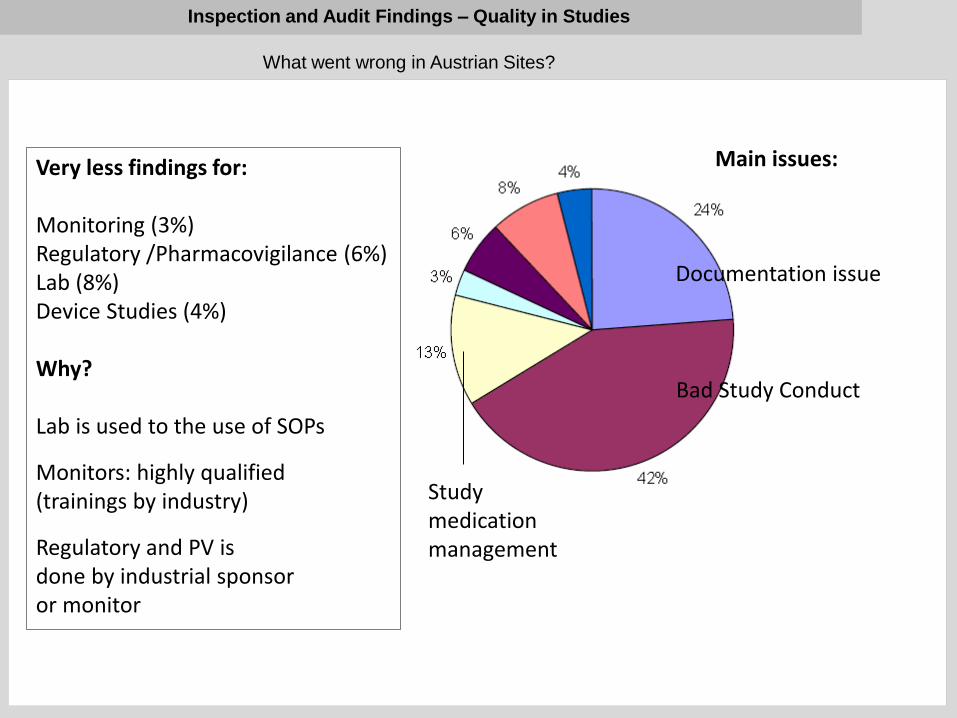
What went wrong in Austrian Sites?
Documentation issue
Bad Study Conduct
Study medication management
Very less findings for: Monitoring (3%) Regulatory /Pharmacovigilance (6%) Lab (8%) Device Studies (4%) Why? Lab is used to the use of SOPs
Monitors: highly qualified (trainings by industry)
Regulatory and PV is done by industrial sponsor or monitor
Main issues:

Inspection and Audit Findings – Quality in Studies
Qualification of findings
10% of the centres had more than 5 critical findings 50% of the centres more than 4 major findings 10% more than 10

Inspection and Audit Findings – Quality in Studies
Qualification of findings
50% of the centres more than 4 minor findings 10% more than 8

Inspection and Audit Findings – Quality in Studies
Main problems at the Conduct of the study and Documentation
Data protection!
Organisational failure! (no SOPs or regulatives at site)
Data not pseudonymized handed over to sponsor Informed Consent (dated by physician or not signed at all) Protocol violation In general or inclusion or exclusion criteria failed Incomplete initiation (missing training) Delegation of responsibilities ( to unknown staff or not qualified or delegation didn´t work) Patient notes do not show Source data that were filled into the CRF
Proof of Authenticy!

Inspection and Audit Findings – Quality in Studies
Main problems at the Management of IMP
Main problems with electronical CRFs
Quality defect? Used for trial Participants only? Compliance? Error by Missleading Labelling?
Storage of IMP (no temperature check , missing documentation)
Drug accountability (received-to patient- from patient –back to sponsor)
GMP Issues Re-labelling at centres, bad labelling Different versions Print outs missing Read only modus

Inspection and Audit Findings – Quality in Studies

Inspection and Audit Findings – Quality in Studies
2 LEGAL AND ADMINISTRATIVE ASPECTS The aim is to determine if all legal and administrative aspects of the clinical trial have been accomplished. The inspector should examine the legal and administrative aspects related to the implementation, progress and termination of the clinical trial. This includes the following points: 2.1 Communication with the IEC (Independent Ethics Committee)
Verification of accreditation / authorisation of the IEC by national authorities adequate composition of the IEC whether IEC approval/favourable opinion (signed and dated) was obtained before starting the trial and implementing any amendments at the centre and clearly identifies the trial, the investigator, the documents reviewed and their versions. (also amendments of the protocol, if applicable) Annual reports have been submitted to the IEC.
Conduct of GCP Inspection – Guidance document

Inspection and Audit Findings – Quality in Studies
2.2 Communication with the regulatory authorities to check whether notification/ authorisation of the trial, changes to the protocol, information about adverse events, transmission of reports and any exchanges of information have been carried out according to the GCP principles and local regulations. 2.3 Other communications It may be necessary to check any other required authorisation to perform the trial at the site and whether adequate information about the trial was given to other involved parties at the trial site (director of the institution, pharmacy, etc.). The documentation of insurance and indemnification should be checked.
Conduct of GCP Inspection – Guidance document

Inspection and Audit Findings – Quality in Studies
3 ORGANISATIONAL ASPECTS
3.1 Implementation of the trial at the site Organisation and Personnel · Organisation charts (facility management and scientific organisation charts). · Documentation of delegation of responsibilities by the principal investigator · SOP system where available. · Disaster plans, e.g. handling of defective equipment and consequences. · Staff – qualification, responsibilities, experience, availability, training programmes,training records, CV. Check the conditions of implementation of the study at the site: · Contracts between the sponsor and the investigator. · Qualifications and experience of the investigator's team in the considered clinical area. · Organisation of the site for the study: organisation chart, GCP training, trial specific training, specific equipment, specific procedures, Compliance with the planned time schedule for the study. · Correct implementation of the correct versions of the protocol and its amendments. . dates of the first inclusion/selection of a patient at the site
Conduct of GCP Inspection – Guidance document

Inspection and Audit Findings – Quality in Studies
3.2 Facilities and equipment . proper use, adequacy and validation status of procedures · Equipment used · Facilities. 3.3 Management of biological samples · Collection: person in charge of this task, dates and handling procedures, including labelling. · Storage of the samples before analysis or shipping. · Shipping conditions. 3.4 Organisation of the documentation (Volume 10) content of the trial master file and archiving trial subjects’ documents are available, completed and archived at the trial site: · Source documents (patient’s charts, X-ray, etc.). · Informed consent documents. · Case Report Form (CRF). A sample of data should be verified from the study report and or CRF to the source documents
Conduct of GCP Inspection – Guidance document

Inspection and Audit Findings – Quality in Studies
4 INFORMED CONSENT OF TRIAL SUBJECTS obtained in accordance with EU requirements as or the subjects' legally acceptable representative, prior to their entry into the study. This needs to include patients whose medical records are reviewed. It will be necessary to check: · The signed and self-dated (by the subject and by the person who conducted the informed consent discussion) consent form actually used and approved by the IEC at the time of inclusion of the subjects. · The centre practice for giving a copy of the informed consent to the patient · Consent for access to medical records by the authorities.
Conduct of GCP Inspection – Guidance document

Inspection and Audit Findings – Quality in Studies
5 REVIEW OF THE TRIAL SUBJECT DATA source data verification whether corrections of the data recorded in the CRF was done according to the EU requirements and dated by the authorised person and providing justification, if necessary. For a number of subjects that will be determined within the inspection plan, (the sample might include the first and last patient enrolled, etc 5.1 Characteristics of the subjects included in the clinical trial The aim is to determine whether the inclusion of the subjects in the trial was performed in accordance with the approved protocol and/or that protocol violations are documented, and also described in the study report. It should be checked whether: · Subjects included in the clinical trial existed and participated in the clinical trial. · Subjects’ participation was recorded in their medical records. · Subjects included fulfilled the inclusion criteria and none of the exclusion criteria stated in the protocol were present. Appropriate medical records must support these criteria.
Conduct of GCP Inspection – Guidance document

Inspection and Audit Findings – Quality in Studies
5.2 Subjects’ visits calendar subjects’ visits calendar established in the protocol was followed, done on the correct dates. 5.3 Efficacy and safety assessment data efficacy and safety data recorded in the CRF are in agreement with the source data Data related to endpoints should be compared with source documents, if appropriate, according to the scope of the inspection. adverse events recorded in the site records are also recorded in the CRF and were reported to the sponsor, IEC and authorities in accordance with the discontinuation of treatment and drops outs. 5.4 Concomitant therapy and intercurrent illness It should be verified whether concomitant therapy and intercurrent illnesses were managed in compliance with the protocol and recorded in the CRF and source documents.

Inspection and Audit Findings – Quality in Studies
6 MANAGEMENT OF THE INVESTIGATIONAL MEDICINAL PRODUCT(S) · Instructions for handling of Investigational Medicinal Product(s) and trial related materials (if not included in protocol or investigator’s brochure). · Shipping records for Investigational Medicinal Product(s) and trial related material. Receipt date(s)of product delivery and quantity. expiration dates and codes assigned to the product and the trial subject. · allocation of treatment, randomisation and code breaking. · Investigational Medicinal Product(s) accountability at site (pharmacy or investigator): Date and quantity dispensed or returned, identification of recipients (patients’ code or authorised person’s). batch numbers, expiration dates and codes documentation about re-labelling, if applicable, date and quantity returned to the sponsor, documentation of destruction Treatment compliance suitability of storage conditions and their records (fridge, freezer and controlled substances, etc.).

Inspection and Audit Findings – Quality in Studies
Inspection Report (1)
Tentative format for an INSPECTION REPORT (IR) - Investigator site A. Administrative Information.
B. Background and general information 1. Reasons/cause for the inspection. 2. Reference texts List of the main legal references applicable within the context of this inspection 3. List of persons involved in the trial and contacted during the inspection: C. Administrative aspects of the trial. 1. Application/notification to competent authority. • Protocol, amendments and patient information and consent. • Contacts during the trial, i.e. adverse event reporting, change of expiry date(s), reports. 2. Contacts with the independent ethics committee • Approval of protocol, amendment(s) and patient information and consent. • List of IEC members and members present at the meeting. • Contacts during the trial, 3. Contacts with other committees, any other validation or authorisation.

Inspection and Audit Findings – Quality in Studies
• Authorisation by local ethics committee of the hospital • Authorisation by local authorities for particular studies or subjects included.
D. Trial documents • Protocol, version, date of signatures. • Amendments, dates, signatures. • Patient information and consent • Secrecy statement/agreement • Randomisation list, code envelopes • Investigator’s Brochure • Laboratories, technical departments, reference values • Investigators file • Quality management at the site • Archiving of trial documents, including archiving of hospital files • Other essential documents of the trial.
E. Conduct of trial. • Interview with principal investigator and key members of the trial team • Trial co-ordination • Trial subjects: examination, inclusion and follow up • Assessment and follow up of safety parameters
Inspection Report (2)

Inspection and Audit Findings – Quality in Studies
F. Documentation and reporting of data • Procedures, data and files examined • Informed consent • Inclusion, exclusion criteria and efficacy parameters • Recording and reporting of adverse events/reactions • Treatment discontinuation • Compliance, protocol and treatment. G. Accountability of medicinal products 1. At the pharmacy / investigators site • Documentation • Receipt and storage • (Randomisation, decoding) • Dispensing • Returns from clinic / trial subjects • Destruction/recovery by the sponsor 2. Administration to trial subjects • Documentation • Compliance • Returns
Inspection Report (3)

Inspection and Audit Findings – Quality in Studies
H. Laboratories, technical departments. • Certification or accreditation • Established quality control (external/internal) or other validation • Methods used • Reference data • Labelling and storage of samples • Transportation and samples examined • Documentation and archiving I. Monitoring and auditing 1. Monitoring • Monitoring visits and procedures used • Actions taken by the monitor • Monitor visit log 2. Auditing • Audit and audit certificate • Actions taken subsequent to the audit(s)
Inspection Report (4)

Inspection and Audit Findings – Quality in Studies
J. Summary, discussion and conclusions. • Closing meeting • List of observations made during the inspection with a reference to the GCP requirement not met and grading. • Summary and evaluation of observations • GCP compliance K. Dates and signature(s) of Lead inspector and other inspector(s) L. Response from the sponsor and investigator An evaluation by the inspectors of the response Other appendices as required
Inspection Report (5)

Inspection and Audit Findings – Quality in Studies
Solution to some problems might be:
QM-System at the investigator´s site that involves hospital procedures,
Advanced training
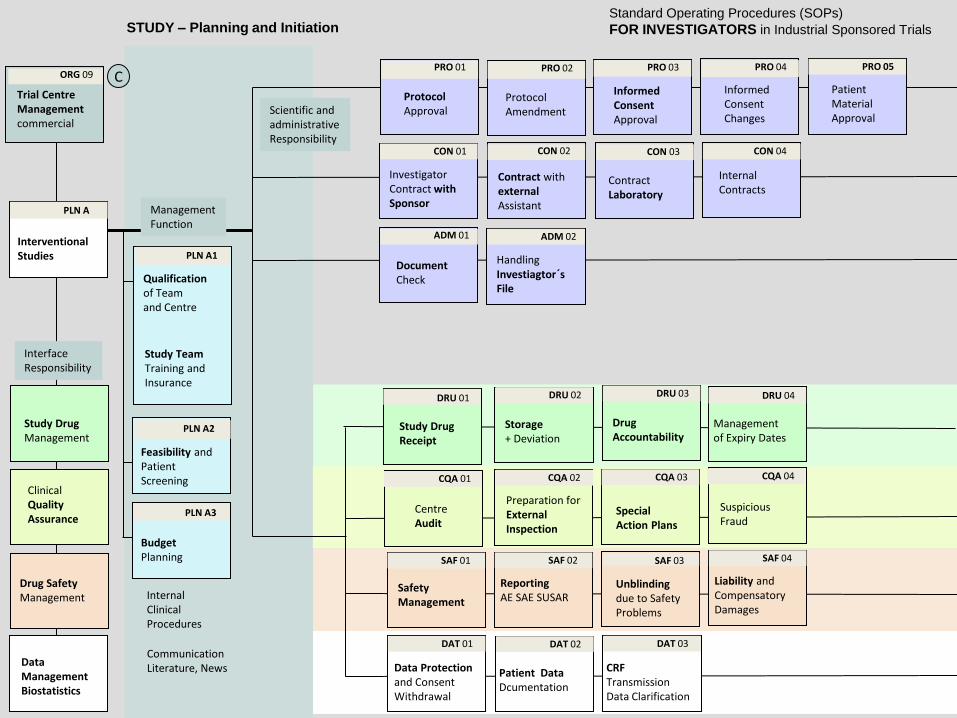
STUDY – Planning and Initiation
ADM 01
PRO 01
Protocol Approval
PRO 02 PRO 03
Protocol Amendment
Clinical Quality Assurance
Study Drug Management
Drug Safety Management
Data Management Biostatistics
Document Check
Qualification of Team and Centre
Budget Planning
Study Team Training and Insurance
CON 01
Investigator Contract with Sponsor
CON 02 CON 03
Contract with external Assistant
Interventional Studies
PLN A
Informed Consent Approval
Contract Laboratory
PRO 04
Informed Consent Changes
ADM 02
Handling Investiagtor´s File
CON 04
Internal Contracts
CQA 02
Preparation for External Inspection
CQA 03
Special Action Plans
DRU 01
Study Drug Receipt
DRU 02
Storage + Deviation
DRU 03
Drug Accountability
CQA 04
Suspicious Fraud
SAF 02
Reporting AE SAE SUSAR
DAT 01
Data Protection and Consent Withdrawal
SAF 01
Safety Management
SAF 04
Liability and Compensatory Damages
SAF 03
Unblinding due to Safety Problems
DAT 02
Patient Data Dcumentation
Standard Operating Procedures (SOPs)
FOR INVESTIGATORS in Industrial Sponsored Trials
PLN A3
PLN A2
Feasibility and Patient Screening
PLN A1
PRO 05
Patient Material Approval
ORG 09 C Trial Centre Management commercial
Management Function
Interface Responsibility
Scientific and administrative Responsibility
CQA 01
Centre Audit
DAT 03
CRF Transmission Data Clarification
DRU 04
Management of Expiry Dates
Internal Clinical Procedures
Communication Literature, News

SAF 05
Safety Follow up
Final Control Project Management
Payments
ADM 04 ADM 05
Premature Study Termination
Regular Study Termination
Study Report Writing and Approval
Publication Guidelines
PRO 08 PRO 09
ADM 07
Archiving and Destruction
Termination
ADM 06
Site Closure
Study Duration > One Year EC Report
DRU 04
Final Drug Accountability
DRU 05
Drug Destruction (optional)
ADM 03
PRO 06 PRO 07
CON 05
Amendment and dissolution of a Contract
Unplanned Follow-up
Randomization + Deviation
Study – Conduct
Prestudy Visit
Monitoring Visit Support
INV C
Prestudy Visit
Study Initiation Preparation
Close Out Visit Preparation
INV D
INV B
Prestudy Visit
Prestudy Visit Planning
INV A
Patient Recruitment Plan
Patient Information
Prestudy Visit
Forms and Logistics
Source Data Management
Patient Time Management
Patient Compliance Check
Drug Accountability, Temperature
INV 01
INV 03
INV 02
NUR 01
NUR 02
NUR 03
NUR 04
INV 03
Patient Follow up
INV 04
Study Subject Management
Study Assistance
Rescue Management Plan
Patient Notes
Clinical Assessments
INV 05
INV 06
NUR 07
Handling Emergency envelopes
NUR 08
Medical Management
Study Management PLN A4
PLN A5

Mag. Martina Gantschacher Director of the Department for
Health Law & Science of the
Sigmund Freud Private University Vienna, Paris
0043 664 2819 578 Martina.gantschacher@esqh .at







