

SUPPORTING INFORMATION
A Copper-mediated reverse aromatic Finkelstein reaction in ionic liquid
Anh T. H. Nguyen, Dat P. Nguyen, Ngan T. K. Phan, Dung T. T. Lam, Nam T. S. Phan,
Thanh Truong*
Department of Chemical Engineering, Ho Chi Minh University of Technology, VNU-HCM,
268 Ly Thuong Kiet, District 10, Ho Chi Minh City, Vietnam
*Email: [email protected]
*To whom correspondence should be addressed: [email protected]
S1

Table of Contents
Section S1 Materials and Instrumentation S3
Section S2 Synthesis and Characterization of Ionic Liquids S4
Section S3 Calibration curve preparation S7
Section S4 Catalysis S9
Section S5 References S14
S2

Section S1. Materials and Instrumentation:
Reactions were performed in 2-dram vials using screw caps with 17 mm hole and white
silicone septum with white Teflon face (from SUPELCO). Column chromatography was
performed on 60Å silica gel (Sigma Aldrich). Gas chromatographic (GC) analyses were
performed using a Shimadzu GC 2010-Plus equipped with a flame ionization detector (FID)
and an SPB-5 column (length = 30 m, inner diameter = 0.25 mm, and film thickness = 0.25
µm). The temperature program for GC analysis heated samples from 100 °C for 1.0 minutes;
heated from 100 to 280 °C at 40 °C/min; held at 280 °C for 3 min. Inlet and detector
temperature were set constant at 280 °C. Diphenyl ether was used as internal standard to
calculate the GC yield of reaction. GC-MS analyses were performed on a Shimadzu GCMS-
QP2000 Plus chromatograph equipped with a Restek column (Rtx-XLB, 30 m × 0.25 mm
I.D.). The 1H and 13C NMR were recorded on a Brucker AV 500 MHz using residual solvent
peak as a reference. All procedures were performed under air atmosphere unless otherwise
noted.
All reagents and starting materials were obtained commercially from Merck, Sigma-Aldrich,
and Acros Organics and were used as received without any further purification unless
otherwise noted.
Table S1. ReagentsChemicals Origin Specification
1-methylimidazole Merck 99%
1-Bromobutane Merck 98%
1-Bromohexane Merck 98%
1-Bromooctane Merck 98%
Ethyl acetate Baker 99%
S3

Diethyl ether Baker 99%
Hexafluorophosphoric acid Acros Organics 60%
Tetrafluorophosphoric acid Acros Organics 48%
1-methylimidazole Merck 99%
p-xylene Merck 99%
4-bromoanisole Merck 98%
Methanol China 99.5%
Dichloromethane Merck 99.8%
Toluene Merck 99.9%
1-bromo-4-nitrobezene Sigma 99%
1-bromo-3-nitrobenzene Sigma 99%
1-bromo-2-nitrobenzene Sigma 99%
4-bromoanisole Acros Organics 98%
Dimethylformamide (DMF) Merck 99,8%
DMSO Merck 99.5%
DMA Merck 99.5%
NMP Merck 99.5%
Diglymer Merck 99.5%
4-iodoacetonphenone Sigma 99%
3-iodoacetonphenone Sigma 98%
2-iodoacetonphenone Sigma 98%
CuBr Sigma 99.9%
Iodo benzene Sigma 99%
1-iodo-4-nitrobenzene Sigma 98%
2-iodotoluene Sigma 98%
4-iodotoluene Sigma 99%
Diphenyl ether Sigma 98%
Na2SO4 Acros Organics 99%
S4

1,4-diiodobenzene Acros Organics 99%
1-iodonaphthalene Sigma 98%
1-bromo-4-fluorobenzene Sigma 99%
4-iodopyridine Sigma 99%
3-iodoindole Acros Organics 99%
All reactions or entries were run at least two times to minimize errors and the reported
numbers were the average values.
S5

Section S2. Synthesis of Ionic Liquids
1–Butyl–3–methylimidazolium bromide ([BMIM]Br
1H NMR (500 MHz, DMSO): δ = 0.887 (t, 3H; CH3), 1.249 – 1.257 (m, 2H; CH2CH3), 1.765
– 1.779 (m, 2H; CH2CH2CH3), 3.882 (s, 3H; N-CH3), 4.198 – 4.228 (m, 2H; N-CH2), 7.778
(t, 1H; N-CH=C), 7.856 (t, 1H; N-CH=C), 9.340 (s, 1H, N-CH=N).
1–Hexyl–3–methylimidazolium bromide ([HMIM]Br: 1-methylimidazole (20.5 g, 0.25 mol)
was mixed with 1-bromohexane (45.9 g, 0.28 mol) in a 250 mL round bottom flask equipped
with a flux condenser. The mixture was then irradiated in a microwave oven (Sanyo – EM
S2086W – 800W) at 80W, and stirred vigorously during the reaction time by the magnetic
stirrer. The irradiation was paused every 10 seconds to prevent overheating. The irradiation
was repeated for a total time of 6 minutes. After completion, the resulting mixture was
cooled down to room temperature, washed with ethyl acetate (100 mL × 3), and with diethyl
ether (100 mL × 3) to remove starting materials and undesired products. The residue of
volatile solvents was removed by a vacuum rotary evaporation at 50 oC to deliver 56.8 g of
product (93 % yield).
1H NMR (500 MHz, DMSO): δ = 0.848 (t, 3H, CH3); 1.248 – 1.280 (m, 6H, CH2CH2CH2);
1.769 – 1.790 (m, 2H, CH2); 3.863 (s, 3H, N–CH3); 4.171 (t, 2H,N–CH2); 7.730 – 7.740 (m,
1H, N–CH=C); 7.801 – 7.810 (m, 1H, N–CH=C); 9.240 (s, 1H, N–CH=N)
1–Octyl–3–methylimidazolium bromide ([OMIM]Br: 1-methylimidazole (20.5 g, 0.25 mol)
was mixed with 1-bromooctane (53.8 g, 0.28 mol) in a 250 mL round bottom flask equipped
with a flux condenser. The mixture was then irradiated in a microwave oven (Sanyo – EM
S2086W – 800W) at 80W, and stirred vigorously during the reaction time by the magnetic
stirrer. The irradiation was paused every 10 s to prevent overheating. The irradiation was
S6

repeated for a total time of 9 min. After completion, the resulting mixture was cooled down
to room temperature, washed with ethyl acetate (100 mL × 3), and with diethyl ether (100
mL × 3) to remove starting materials and undesired products. The residue of volatile solvents
was removed by a vacuum rotary evaporation at 50 oC to deliver 62.8 g of product (83 %
yield).
1H NMR (500 MHz, DMSO): δ = 0.840 (t, 3H, CH3); 1.211 – 1.250 (m, 10H,
CH2CH2CH2CH2CH2); 1.699 – 1.778 (m, 2H, CH2); 3.870 (s, 3H, N–CH3); 4.178 (t, 2H,N–
CH2); 7.740 – 4.780 (m, 1H, N–CH=C); 7.801 – 7.840 (m, 1H, N–CH=C); 9.295 (s, 1H, N–
CH=N)
In a typical procedure for preparation of 1–butyl–3–methylimidazolium
hexafluorophosphate: hexafluorophosphoric acid (40 mL, 0.288 mol) of was added to a
plastic conical flask containing 50 mL of cold distilled water. This mixture was stirred and
immersed in an ice bath (mixture I). The mixture of 1–butyl–3–methylimidazolium bromide
(50 g, 0.228 mol) with 50 mL of cold distilled water was also stirred and immersed in
another ice bath (mixture II). Next, the mixture I was added dropwise to mixture II in 10
minutes. The resulting mixture was continuously stirred in ice bath for 24 h. After
completion, the upper acidic aqueous layer was almost separated by decanting and the
bottom layer was washed by cold water until the almost excess acid was removed. The
acidity was tested by pH paper. The excess water was removed by a vacuum rotary
evaporation at 70 oC to delivery 53.3 g of product (83 % yield).
1H NMR (500 MHz, DMSO): δ = 0.905 (t, 3H, CH3); 1.262 (m, 2H, CH2CH3); 1.771 (m, 2H,
CH2CH2CH3); 3.846 (s, 3H, N–CH3); 4.157 (t, 2H, N-CH2); 7.668 (m, 1H, N–CH=C); 7.733
(m, 1H, N-CH=C); 9.071 (s, 1H, N-CH=N).
S7

Similar procedure was applied for the synthesis of 1–butyl–3–methylimidazolium
tetrafluoroborate ([BMIM]BF4) with 61 % yield.
Section S3. Calibration curve preparation
4’-Chloroacetophenone (16.3 mg) and diphenyl ether (14.8 mg) were weighted into two
distinct 50 mL volumetric flasks. Next, ethyl acetate was added into the flasks until the
volume reached 50 mL to dilute these substances. Portions of the two solutions were
withdrawn and added into five 8 mL vials as showed in Table S2.
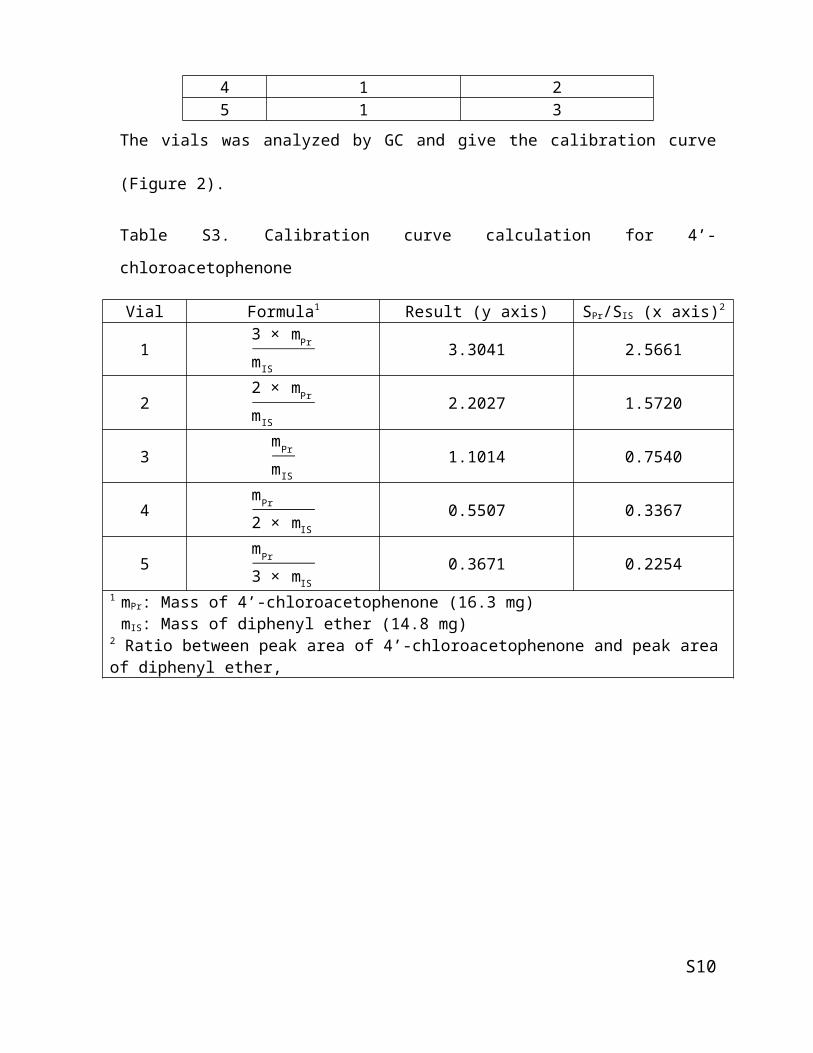
Table S2. Calibration curve preparation for 4’-chloroacetophenone
Vial Volume (mL)4’-Chloroacetophenone Diphenyl ether
1 3 12 2 13 1 14 1 25 1 3
The vials was analyzed by GC and give the calibration curve (Figure 2).
Table S3. Calibration curve calculation for 4’-chloroacetophenone
Vial Formula1 Result (y axis) SPr/SIS (x axis)2
13 × mPr
m IS3.3041 2.5661
22 × mPr
m IS2.2027 1.5720
3m Pr
m IS1.1014 0.7540
4m Pr
2 × mIS0.5507 0.3367
5m Pr
3 × mIS0.3671 0.2254
1 mPr: Mass of 4’-chloroacetophenone (16.3 mg)mIS: Mass of diphenyl ether (14.8 mg)
2 Ratio between peak area of 4’-chloroacetophenone and peak area of diphenyl ether,
S8

Figure S1. Calibration curve for 4’-chloroacetophenone
From the calibration curve, GC yield of 4’-chloroacetophenone can be calculated by formula
(1):
GC yield (%) = mPr × 100%mPr '
= (SPr
SIS × 1.25815 + 0.13275) × mIS × 100%
mPr '
Where: mPr (mg): Mass of 4’-chloroacetophenone obtained
mPr’ (mg): Calculated mass of 4’-chloroacetophenone when yield = 100%
SPr: Peak area of 4’-chloroacetophenone in sample
SIS: Peak area of diphenyl ether in sample
(1)
S9
mIS (mg): Mass of diphenyl ether in sample
Section S4 Catalysis
Reaction kinetic
0 2 4 6 8 10 12 14 16 18 20 22 240.0
10.020.030.040.050.060.070.080.090.0
100.0
Reaction time (h)
GC
YIE
L (%
)
Fig. S2. Reaction kinetic
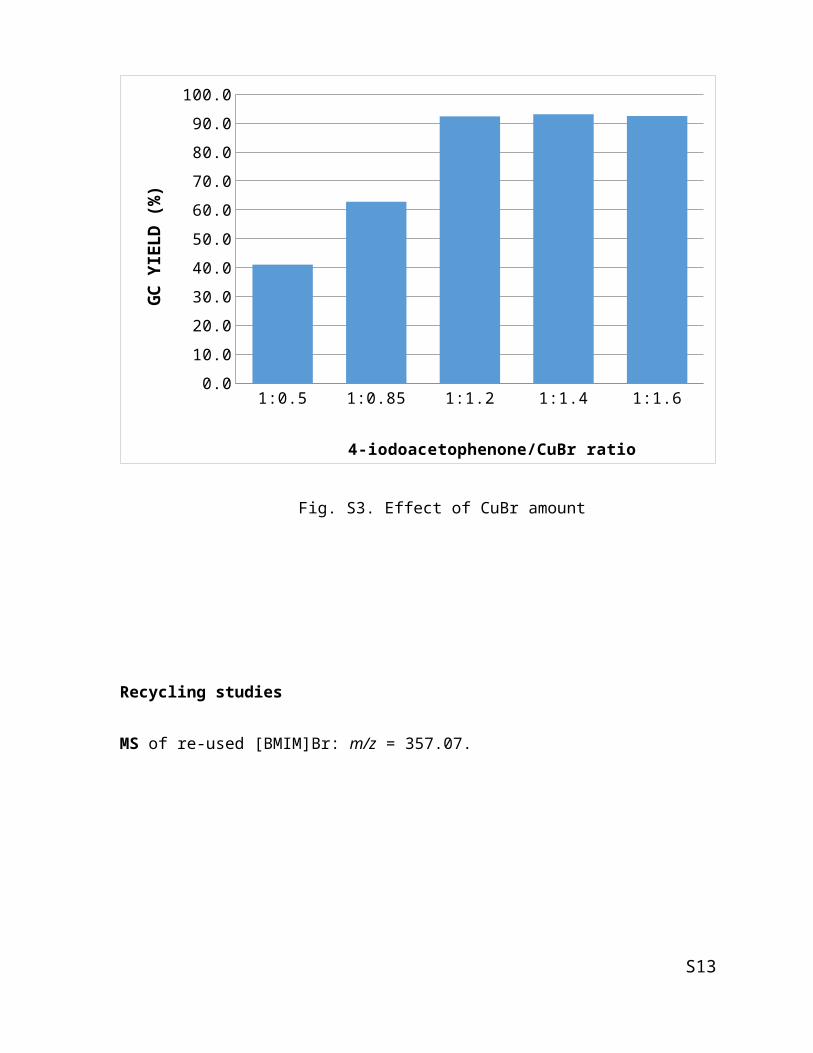
Effect of CuBr amount
1:0.5 1:0.85 1:1.2 1:1.4 1:1.60.0
10.0
20.0
30.0
40.0
50.0
60.0
70.0
80.0
90.0
100.0
4-iodoacetophenone/CuBr ratio
GC Y
IELD
(%)
Fig. S3. Effect of CuBr amount
S10

Recycling studies
MS of re-used [BMIM]Br: m/z = 357.07.
S11

Fig. S4. MS of reused [BMIM]Br
4-Bromoacetophenone
This compound is known.[1] Column chromatography using hexane: ethyl acetate = 9:1.
Product was achieved with 176.1 mg (89 % of yield).
1H NMR (500 MHz, CDCl3) δ, 2.585 (s, 3H), 7.6 (d, 2H, J = 9.0 Hz), 7.8 (d, 2H, J = 9.0 Hz).
MS: m/z = 200 [p-Br-C6H4-COCH3]+
1-Bromo-4-nitrobenzene
This compound is known [1]. Column chromatography using hexane: ethyl acetate = 4:1.
Product was achieved with 152.7 mg (71 % of yield).
1H NMR (500 MHz, CDCl3) δ 7.68 (2H, d, J=8.6 Hz), 8.08 (2H, d, J=8.6 Hz).
MS: m/z = 203.
Methyl-4-bromobenzoate
This compound is known [1]. Column chromatography using hexane: ethyl acetate = 7:1.
Product was achieved with 173.2 mg (81 % of yield).
1H NMR (500 MHz, CDCl3) δ 3.84 (s, 3H), 7.51 (d, J = 8.7 Hz, 2H), 7.83(d, J = 8.7 Hz, 2H)
MS: m/z = 216.
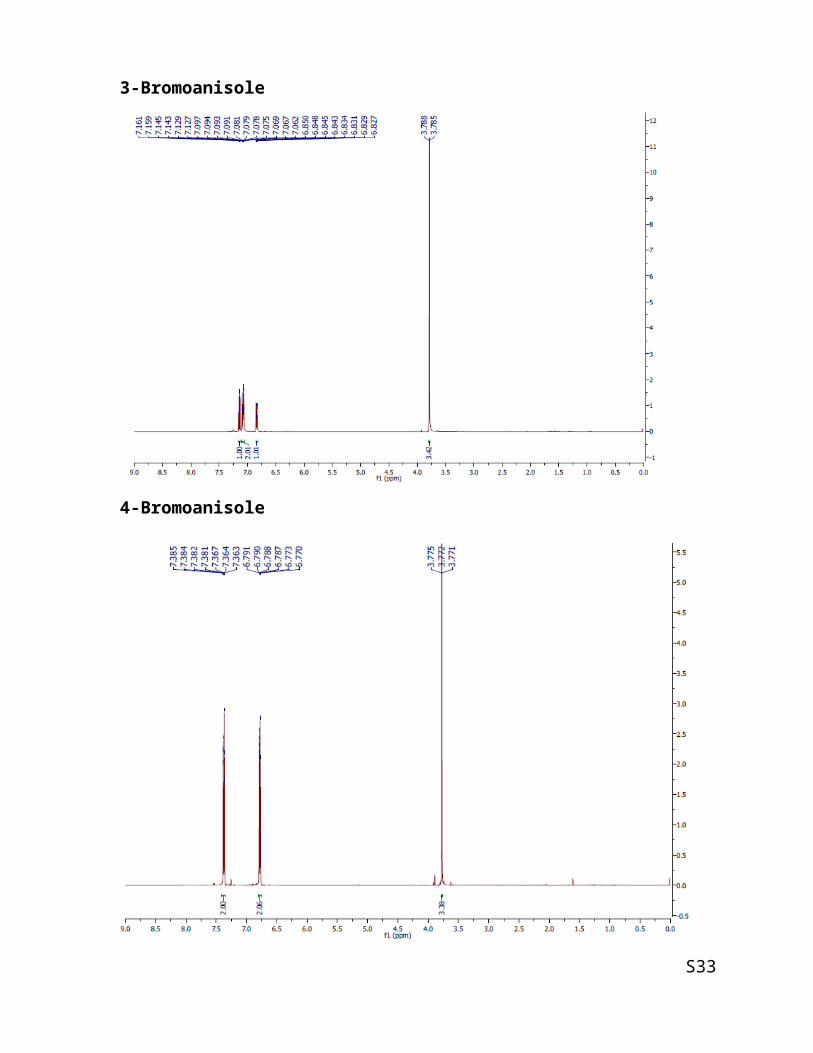
4-Bromoanisole
This compound is known [2]. Column chromatography using hexane: diethyl ether = 15:1.
Product was achieved with 137.4 mg (74 % of yield).
1H NMR (500 MHz, CDCl3) δ 3.77 (s, 3H), 7.33 (d, J = 8.1 Hz, 2H), 6.79 (d, J = 8.1 Hz, 2H);
MS: m/z = 187.
S12
4-Bromotoluene
This compound is known. Column chromatography using hexane. Product was achieved
with 107.9 mg (64 % of yield). GC and GC-MS are identical with authentic compound.
3-Bromoacetophenone
This compound is known [3]. Column chromatography using hexane: ethyl acetate = 9:1.
Product was achieved with 154.1 mg (78 % of yield).
1H NMR (500 MHz, CDCl3) δ: 2.62 (s, 3H), 7.36 (t, J = 7.8 Hz, 1 H), 7.66 – 7.70 (m, 1H),
7.84 – 7.89 (m, 1H), 8.09 (s, 1H).
MS: /z = 200 [m-Br-C6H4-COCH3]+
2-Bromobenzonitrile
This compound is known [3]. Column chromatography using hexane: ethyl acetate = 9:1.
Product was achieved with 110.2 mg (61 % of yield).
1H NMR (500 MHz, CDCl3) δ: 7.50-7.41 (m, 2H), 7.69 (m, 2H).
MS: m/z = 183.
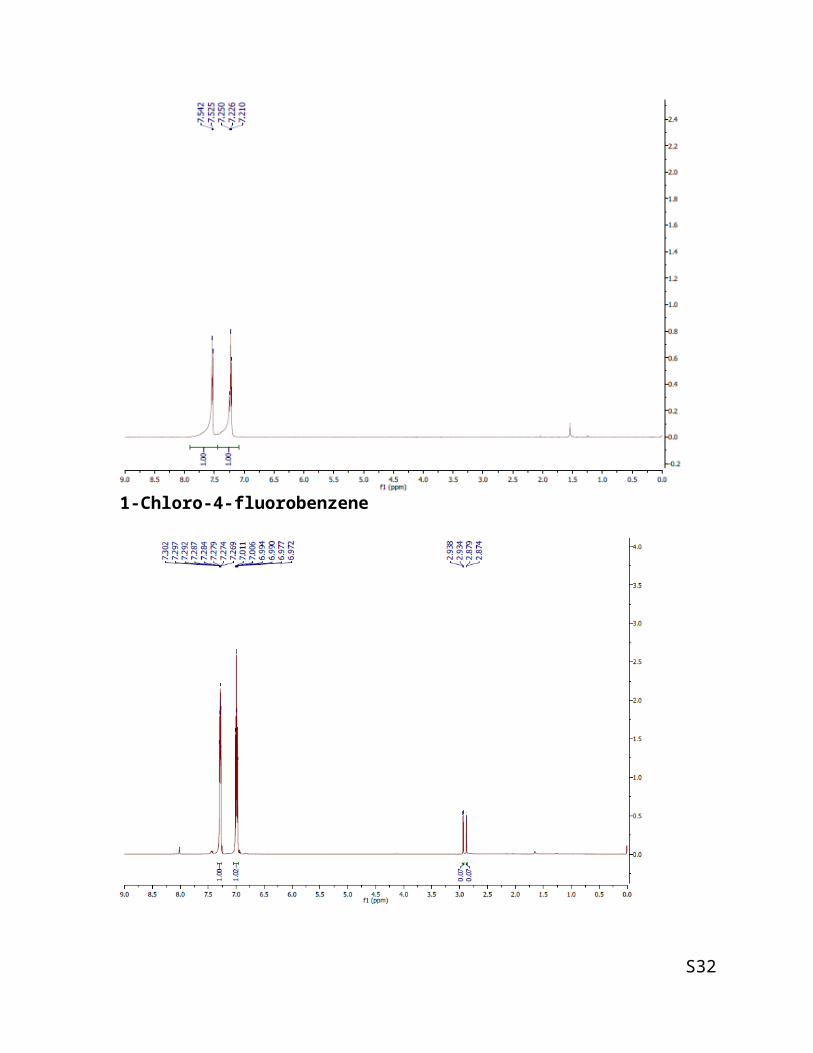
3-Bromoanisole
This compound is known [2]. Column chromatography using hexane: ethyl acetate = 15:1.
Product was achieved with 124.7 mg (67 % of yield).
1H NMR (500 MHz, CDCl3) δ 3.80 (s, 3H), 6.80 - 6.85 (m, 1H), 7.05- 7.08 (m, 1H), 7.10 -
7.21 (m, 2H).
MS: m/z = 188.
S13

4-Chloroacetophenone
This compound is known [4]. Column chromatography using hexane: ethyl acetate = 10:1.
Product was achieved with 126.1 mg (82 % of yield).
1H NMR (500 MHz, CDCl3) δ 2.586 (s, 3H), 7.4 (d, J = 9.0 Hz, 2H), 7.9 (d, J = 9.0 Hz,
2H).
MS: m/z = 154 [p-Cl-C6H4-COCH3]+
1-Chloronaphthalene
This compound is known [3]. Column chromatography using hexane: ethyl acetate = 30:1.
Product was achieved with 122.8 mg (76 % of yield).
1H NMR (500 MHz, CDCl3) δ 7.30 (t, J = 7.8 Hz, 1H), 7.44 – 7.47 (m, 1H), 7.50 -7.56 (m,
2H), 7.69 (d, J = 7.8 Hz, 1H), 7.80 (d, J = 8.0 Hz, 1H), 8.26 (d, J = 8.0 Hz, 1H).
MS: m/z = 162.
1,4-dibromobenzene
This compound is known [5]. Column chromatography using hexane: ethyl acetate = 25:1.
Product was achieved with 165.0 mg (70 % of yield).
1H NMR (500 MHz, CDCl3) δ 7.29 (s, 4H).
MS: m/z = 236.
1-bromo-4-iodobenzene
This compound is known [5]. Column chromatography using hexane: diethyl ether = 40:1.
Product was achieved with 180.2 mg (64 % of yield).
S14

1H NMR (500 MHz, CDCl3) δ 7.25 (d, J=8.2 Hz, 2H), 7.55 (d, J=8.2 Hz, 2H).
MS: m/z = 284.
1-Chloro-4-fluorobenzene
This compound is known. Column chromatography using pentane. Product was achieved
with 54.5 mg (42 % of yield). The identity of the product was confirmed by GC and GC-MS
analysis as compared to those of authentic sample.
1-Bromo-4-chlorobenzene
This compound is known.[4] Column chromatography using hexane. Product was achieved
with 112.0 mg (59 % of yield).
1H NMR (500 MHz, CDCl3) δ 7.08 (d, J=8.7 Hz, 2H), 7.25 (d, J=8.7 Hz, 2H).
MS: m/z = 192.
1-Bromo-2-phenylethylene
This compound is known [5]. Column chromatography using hexane: ethyl acetate = 30:1.
Product was achieved with 122.1 mg (67 % of yield).
1H NMR (500 MHz, CDCl3) δ 6.74 (d, J = 13.8 Hz, 1H), 7.15 (d, J = 13.8 Hz, 1H), 7.28 -
7.35 (m, 5H).
MS: m/z = 184.
4-Chloropyridine
This compound is known [6]. Column chromatography using hexane: diethyl ether = 15:1.
Product was achieved with 83.2 mg (74 % of yield).
1H NMR (500 MHz, DMSO-d6) δ 7.43 – 7.47 (m, 2H), 8.52 – 8.55 (m, 2H).
S15

MS: m/z = 113.
3-Bromoindole
This compound is known [6]. Column chromatography using hexane: diethyl ether = 5:1.
Product was achieved with 83.7 mg (43 % of yield).
1H NMR (500 MHz, CDCl3) δ 7.18 (s, 1H), 7.20 – 7.24 (m, 2H), 7.29 – 7.34 (m, 1H), 7.60 –
7.63 (m, 1H), 8.10 (s, 1H).
MS: m/z = 197.
Section S5. References
[1] Arvela RK, Leadbeater NE. Fast and easy halide exchange in aryl halides. Synlett 2003; 8:1145–1148.[2] Cant AA, Bhalla R, Pimlott SL, Sutherland A. Nickel-catalysed aromatic Finkelstein reaction of aryl and heteroaryl bromides. Chem Commun 2012; 48:3993–3995.[3] Klapars A, Buchwald SL. Copper-catalyzed halogen exchange in aryl halides: an aromatic finkelstein reaction. J Am Chem Soc 2002;124:14844–14845.[4] Jin X, Davies RP. Copper-catalysed aromatic-Finkelstein reactions with amine-based ligand systems. Catal Sci Technol 2017;7:2110–2117.[5] Li L, Liu W, Zeng H, Mu X, Cosa G, Mi Z, Li CJ, Photo-induced metal-catalyst-free aromatic finkelstein reaction. J Am Chem Soc, 2015;137:8328–8331.[6] Chen M, Ichikawa S, Buchwald SL. Rapid and efficient copper-catalyzed finkelstein reaction of (hetero)aromatics under continuous-flow conditions . Angew Chem Int Ed 2015;54:263 – 266.
S16

1H-NMR OF [BMIM]Br
S17

13C-NMR OF [BMIM]Br
S18

1H-NMR OF [HMIM]Br
S19

13C-NMR OF [HMIM]Br
S20

1H-NMR OF [OMIM]Br
S21

13C-NMR of [OMIM]Br
S22

1H-NMR OF [BMIM]PF6
S23

13C-NMR OF [BMIM]PF6
4-Bromoacetophenone
S24

1-bromo-4-iodobenzene
S25

1-Chloro-4-fluorobenzene
3-Bromoanisole
S26

4-Bromoanisole
4-Bromotoluene
S27

4-Chloroacetophenone
4-Chloroanisole
S28

1-Chloronapthalene
Methyl-4-bromobenzoate
S29

1-Bromo-4-nitrobenzene
Mass spectrometry
S30

4-Bromoanisole
4-Bromotoluene
S31
3-Bromoanisole
4-Chloroacetopheneone
S32
4-Chloroanisole
1-Chloro-4-fluorobenzene
S33

1-Bromo-4-iodobenzene
4-Bromoacetophenone
S34







