

of November 27, 2018.This information is current as Syndrome
R-Independent NephroticγComplexes and FcFragment Induces Subepithelial Immune
3(IV) CollagenαImmunization with Murine Membranous Nephropathy:
Dorin-Bogdan BorzaSado, Laurence Heidet, Sandra Kleinau, Agnes B. Fogo andFlorina Olaru, Xu-Ping Wang, Maimouna Bah, Yoshikazu Jun-Jun Zhang, Mahdi Malekpour, Wentian Luo, Linna Ge,
ol.1103368http://www.jimmunol.org/content/early/2012/02/26/jimmun
published online 27 February 2012J Immunol
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2012 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on Novem
ber 27, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on N
ovember 27, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
Murine Membranous Nephropathy: Immunization witha3(IV) Collagen Fragment Induces Subepithelial ImmuneComplexes and FcgR-Independent Nephrotic Syndrome
Jun-Jun Zhang,*,1 Mahdi Malekpour,*,2 Wentian Luo,* Linna Ge,* Florina Olaru,*
Xu-Ping Wang,* Maimouna Bah,* Yoshikazu Sado,† Laurence Heidet,‡ Sandra Kleinau,x
Agnes B. Fogo,{ and Dorin-Bogdan Borza*,{
Membranous nephropathy (MN) is a leading cause of nephrotic syndrome in adults and a significant cause of end-stage renal dis-
ease, yet current therapies are nonspecific, toxic, and often ineffective. The development of novel targeted therapies requires a de-
tailed understanding of the pathogenic mechanisms, but progress is hampered by the lack of a robust mouse model of disease. We
report that DBA/1 mice as well as congenic FcgRIII2/2 and FcRg2/2 mice immunized with a fragment of a3(IV) collagen
developed massive albuminuria and nephrotic syndrome, because of subepithelial deposits of mouse IgG and C3 with correspond-
ing basement membrane reaction and podocyte foot process effacement. The clinical presentation and histopathologic findings
were characteristic of MN. Although immunized mice produced genuine anti-a3NC1 autoantibodies that bound to kidney and
lung basement membranes, neither crescentic glomerulonephritis nor alveolitis ensued, likely because of the predominance of
mouse IgG1 over IgG2a and IgG2b autoantibodies. The ablation of activating IgG Fc receptors did not ameliorate injury,
implicating subepithelial deposition of immune complexes and consequent complement activation as a major effector pathway.
We have thus established an active model of murine MN. This model, leveraged by the availability of genetically engineered mice
and mouse-specific reagents, will be instrumental in studying the pathogenesis of MN and evaluating the efficacy of novel
experimental therapies. The Journal of Immunology, 2012, 188: 000–000.
Various forms of glomerulonephritis (GN) collectively area major cause of end-stage renal disease. A prominentpathogenic mechanism is the glomerular deposition or in
situ formation of immune complexes (ICs), which induce tissue
injury by activating complement or engaging Fc receptors oneffector cells. The pattern of injury, effector mechanisms, histo-pathologic findings, and clinical course are largely determined bythe glomerular location of ICs (1).Membranous nephropathy (MN) is a leading cause of nephrotic
syndrome in adults. The hallmark feature is the subepithelial de-position of electron-dense ICs containing Ag, IgG, and comple-ment with glomerular basement membrane (GBM) thickening andpodocyte foot process effacement, but little glomerular inflam-mation. Subepithelial ICs form in situ when circulating Abs bind toAgs expressed on podocytes or to Ags planted at this site, althoughdeposition of small circulating ICs is also possible (1, 2). Podocyteautoantigens include phospholipase A2 receptor, targeted by auto-antibodies in ∼70% of patients with idiopathic MN (3), neutralendopeptidase, implicated in antenatal MN because of maternalalloimmunization (4), as well as aldose reductase, manganese sup-eroxide dismutase, and probably other autoantigens (5, 6). Cat-ionic BSA, likely of dietary origin, has been identified as the Agin some forms of early childhood MN (7). Pathogenesis of MN isdriven by subepithelial ICs activating complement and sublyticpodocyte injury by C5b-9, leading to proteinuria (8).Autoantibodies to a3a4a5(IV) collagen, the major component
of the GBM, mediate rapidly progressive crescentic GN (9, 10). Inanti-GBM Ab-mediated GN, including Goodpasture (GP) disease,autoantibodies bind to conserved epitopes in the noncollagenousdomain 1 (NC1) of a3(IV) collagen (11–14). In Alport posttrans-plant nephritis, anti-GBM alloantibodies bind to distinct epitopesin the NC1 domains of a3(IV) or a5(IV) collagen, present only inthe kidney allograft (14–18). A characteristic finding of anti-GBMdisease is the smooth linear deposition of IgG and often com-plement along the GBM, with a crescentic and necrotizing injury.Tissue-bound anti-GBM autoantibodies elicit a type II inflamma-
*Division of Nephrology, Department of Medicine, Vanderbilt University School ofMedicine, Nashville, TN 37232; †Shigei Medical Research Institute, 701 Okayama,Japan; ‡Hopital Necker-Enfants Malades, 75743 Paris, France; xDepartment of Celland Molecular Biology, Uppsala University, SE-75124 Uppsala, Sweden; {Depart-ment of Pathology, Microbiology, and Immunology, Vanderbilt University School ofMedicine, Nashville, TN 37232
1Current address: Department of Nephrology, First Affiliated Hospital of ZhengzhouUniversity, Zhengzhou, China.
2Current address: Department of Medicine, Tehran University of Medical Sciences,Tehran, Iran.
Received for publication November 23, 2011. Accepted for publication January 23,2012.
This work was supported by National Institutes of Health Grant R01 DK080799 (toD.B.B.). F.O. is supported by a postdoctoral fellowship from the American HeartAssociation South-East Affiliate (11POST7300008). M.B. has been a short-termtrainee in the Vanderbilt Medical Student Research Training Program in Nephrologyand Hypertension. Electron microscopy was performed by the Washington UniversityO’Brien Center for Kidney Disease Research, supported by National Institutes ofHealth Grant P30 DK079333. Silver staining was performed at the Histology andMorphometry Core of the Vanderbilt University O’Brien Kidney Disease Center,supported by National Institutes of Health Grant P30 DK079341.
Address correspondence and reprint requests to Dr. Dorin-Bogdan Borza, S-3223Medical Center North, Division of Nephrology, Department of Medicine, VanderbiltUniversity Medical Center, 1161 21st Avenue South, Nashville, TN 37232. E-mailaddress: [email protected]
Abbreviations used in this article: ACR, albumin-to-creatinine ratio; BM, basementmembrane; BUN, blood urea nitrogen; EM, electron microscopy; GBM, glomerularbasement membrane; GN, glomerulonephritis; GP, Goodpasture; IC, immune com-plex; IF, immunofluorescence; mIgG, mouse IgG; MN, membranous nephropathy;NC1, noncollagenous domain 1.
Copyright� 2012 by The American Association of Immunologists, Inc. 0022-1767/12/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1103368
Published February 27, 2012, doi:10.4049/jimmunol.1103368 by guest on N
ovember 27, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

tory response, inducing complement-mediated and Fcg receptor-mediated recruitment and activation of inflammatory cells. Thecontribution of various effector pathways has not been fully elu-cidated.Activating Fcg receptors have been identified as essential
mediators in some mouse models of Ab-mediated GN. Mice havethree activating IgG Fc receptors: FcgRI, FcgRIII, and FcgRIV(19). Each receptor comprises an a-chain associated with a com-mon FcRg subunit, which initiates signal transduction upon re-ceptor cross-linking by ICs. Ablation of activating IgG Fc re-ceptors in FcgR2/2 mice prevents nephrotoxic nephritis inducedby heterologous anti-GBM Abs (20, 21) and lupus nephritis inNZB/NZW mice (22), but does not protect against lupus nephritisin MRL/lpr mice (23) or cryoglobulinemia-associated mem-branoproliferative GN (24). The role of individual FcgRs has beenstudied in murine nephrotoxic nephritis, and variable require-ments for FcgRI, FcgRIII, and FcgRIV were reported by differentgroups (25–28). However, the importance of activating FcgRs inactive models of Ab-mediated nephritis elicited by immunizationwith glomerular Ags has not been determined, as C57BL/6 micecommonly used for these studies are relatively resistant to activelyinduced GN (29).Our goal was to determine the contribution of activating IgG Fc
receptors to the pathogenesis of murine autoimmune GN inducedby immunization with a well-defined GBM autoantigen. For thispurpose, we chose to use DBA/1 mice because of their greatersusceptibility to GN relative to C57BL/6 mice (30, 31) and theavailability of congenic FcgRIII2/2 and FcRg2/2 mice (32). Wildtype, FcgRIII2/2 and FcRg2/2 mice immunized with the NC1domain of a3(IV) collagen, regardless of the expression of acti-vating Fcg receptors, developed massive albuminuria and nephro-tic syndrome, associated with subepithelial IgG and C3 depositsby immunofluorescence (IF) and electron microscopy (EM) andcharacteristic GBM spike reaction without crescentic injury. Wehave thus established a novel active model of murine MN, whichwill be valuable for new studies of the disease mechanisms andpreclinical studies of experimental therapies.
Materials and MethodsMaterials
The recombinant NC1 domain of human a3(IV) collagen (rh-a3NC1) wasexpressed in HEK293 cells and purified as described (33). Mouse kidney(Pel-Freez Biologicals, Rogers, AR) cortex basement membranes (BMs),including the GBM, were digested with bacterial collagenase (Wor-thington, Lakewood, NJ). The solubilized NC1 hexamers were purified bypassage through a DE-52 ion-exchange column and gel-filtration chro-matography (34). The a3a4a5NC1 and a1a2NC1 hexamers from mouseGBM were affinity-purified using immobilized mAb b14 (34). NC1 do-mains from mouse GBM were separated by reverse-phase HPLC on aC18 column (Vydac 201TP C18 10u) using a gradient of acetonitrile in0.1% trifluoroacetic acid, as described previously (35). Rat IgG mAbs H11(to a1NC1), H31 (to a3NC1), RH34 (to human a3NC1), RH42 (toa4NC1), b14 (to a5NC1), as well as murine mAbs TF51 (mIgG1), TF90(IgG2a), and TF86 (IgG2b) binding to the NC1 domains of mouse a3a4a5(IV) collagen were provided by Dr. Yoshikazu Sado.
Animal experiments
DBA/1 mice were purchased from The Jackson Laboratory. Breeding pairsof FcgRIII2/2 and FcRg2/2 mice backcrossed onto the DBA/1 back-ground for more than 12 generations were obtained from Dr. SandraKleinau. Col4a32/2 mice expressing a human COL4A3/COL4A4 YACtransgene (36) were obtained from Dr. Laurence Heidet. B6.Col4a32/2
mice (37) were provided by Dr. Jeffrey Miner (Washington University, St.Louis, MO). Mice were housed in a specific pathogen-free facility withfree access to food and water. All procedures were approved by the In-stitutional Animal Care and Use Committee and conducted in accordancewith the Guidelines for Animal Care and Use Program of VanderbiltUniversity.
Groups of four to six wild type, FcgRIII2/2 and FcRg2/2 DBA/1 mice,both males and females, between 6 and 10 wk old, were immunized s.c. attwo sites on the back with a3NC1 (30 mg in 50 ml PBS) emulsified inequal volume of CFA (Sigma, St. Louis, MO), then boosted 3 wk later withthe same amount of Ag in incomplete Freund’s adjuvant (Sigma). Incontrol mice, the Ag was replaced with vehicle (PBS). Blood was collectedevery 2 wk from the saphenous vein. Spot urine was collected every 2 wkusing a urine collection station (38). Mice were regularly checked for signsof disease and were euthanized if they developed edema, ascites, abnor-mally high blood urea nitrogen (BUN) levels, or excessive loss of bodyweight (.10% in a week) or became lethargic. The remaining mice werekilled at ∼18 wk after immunization, except for several mice from eachgroup sacrificed at 10 wk for comparison purposes. Blood, kidneys, andlungs were collected for further analysis. Three separate experiments wereperformed for each strain.
Evaluation of kidney function and renal histopathology
Urinary albumin excretion was measured in spot urine samples by cap-ture ELISA using a mouse albumin quantitation kit (Bethyl, Montgomery,TX). Urine creatinine and BUNwere measured using Infinity creatinine andurea liquid stable reagents (Thermo Fisher Scientific, Middletown, VA),according to the manufacturer’s protocols. Albuminuria was expressed asurinary albumin-to-creatinine ratio (ACR). The proteins in serum and urinesamples were separated by SDS-PAGE electrophoresis under nonreducingconditions and stained with Coomassie Brilliant Blue. Total plasma cho-lesterol and triglycerides were measured with standard enzymatic assays atthe Vanderbilt Mouse Metabolic Phenotyping Center. Portions of mousekidneys or lungs were fixed in 10% buffered formalin, dehydrated throughgraded ethanol, embedded in paraffin, and kidney sections (2 mm thick)were stained with periodic acid-Schiff (PAS) or Jones silver stain. Lungsections were stained with H&E. At least 50 glomeruli from each mousewere observed to assess lesions. Mesangial proliferation, global or seg-mental sclerosis, spike formation, necrosis, or crescents were assessed. Fortransmission EM, kidney cortex was fixed in 4% paraformaldehyde in 0.1M cacodylate buffer (pH 7.4), postfixed in aqueous 1.25% osmium te-troxide, dehydrated through an ethanol series, embedded in plastic, sec-tioned with a diamond knife, and stained with 4% uranyl acetate and leadcitrate. All assessments were made without knowledge of the experimentalgroup.
Direct and indirect IF
Portions of snap-frozen mouse kidneys or lung embedded in OCT werecryosectioned (5 mm), fixed in acetone for 10 min at 220˚C, and blockedwith 1% BSA. For direct IF, frozen sections were stained with FITC-conjugated goat anti-mouse IgG, IgG1, IgG2a, IgG2b (BD BiosciencePharmingen, San Jose, CA), or FITC-conjugated goat anti-mouse C3c(Nordic Immunology, Tilburg, the Netherlands). For indirect IF staining ofcollagen IV chains, rat IgG mAbs were used as described (36). Stainingwith mAbs H11, H31, and RH34 was performed both with and withoutprior treatment of the sections with acid urea (6 M urea in 0.1 M glycine,pH 3.0). Rabbit anti-C5b9 (Abcam, Cambridge, MA) was used at 5 mg/ml.Secondary Abs were Alexa Fluor 488-conjugated goat anti-rabbit and anti-rat IgG (Invitrogen, Carlsbad, CA) or FITC-goat anti-rat IgG (BD Bio-science Pharmingen, San Jose, CA). Sections were mounted with anti-fadereagent (Invitrogen, Carlsbad, CA) and examined under a Nikon EclipseE800 epifluorescence microscope. Photomicrographs were recorded witha charge-coupled device digital camera, using the same exposure settingsfor each primary Ab.
Analysis of circulating and kidney-bound mouse IgGautoantibodies
Kidney-bound Abs were eluted from homogenized mouse kidney cortexwith 0.1 M glycine, pH 2.8. Serum and kidney-eluted mouse IgG (mIgG)Abs were analyzed by ELISA. Maxisorp plastic plates were coated over-night with rh-a3NC1 (100 ng/well) or mouse GBM NC1 hexamers (300ng/well) and blocked with 1% BSA. Mouse sera were diluted as indicated.Secondary Abs were alkaline phosphatase-conjugated goat anti-mouse IgG(Rockland Immunochemicals, Gilbertsville, PA) and HRP-conjugated goatanti-mouse IgG1, IgG2a, and IgG2b (Bethyl, Montgomery, TX). The dilu-tions of secondary Abs were chosen so that standard mAbs (TF51 formIgG1, TF90 for mIgG2a, TF86 for mIgG2b at 1 mg/ml each) yieldedcomparable ELISA readings when assayed for binding to total mouseGBM NC1 hexamers. After the addition of substrate for phosphatase(Sigma, St. Louis, MO) or peroxidase (Biorad, Hercules, CA), color de-velopment was measured with a SpectraMax Plus 384 ELISA plate reader(Molecular Devices, Sunnyvale, CA).
2 NOVEL MOUSE MODEL OF MEMBRANOUS NEPHROPATHY
by guest on Novem
ber 27, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
Statistical analyses
Data are shown as mean 6 SEM. Statistical analyses were performedusing GraphPad Prism version 5.01. The significance of differences amonggroups was evaluated with one-way ANOVA test, followed by post hoctests for pairwise comparisons. Survival curves were compared with log-rank test. A p value of less than 0.05 was considered statistically significant.
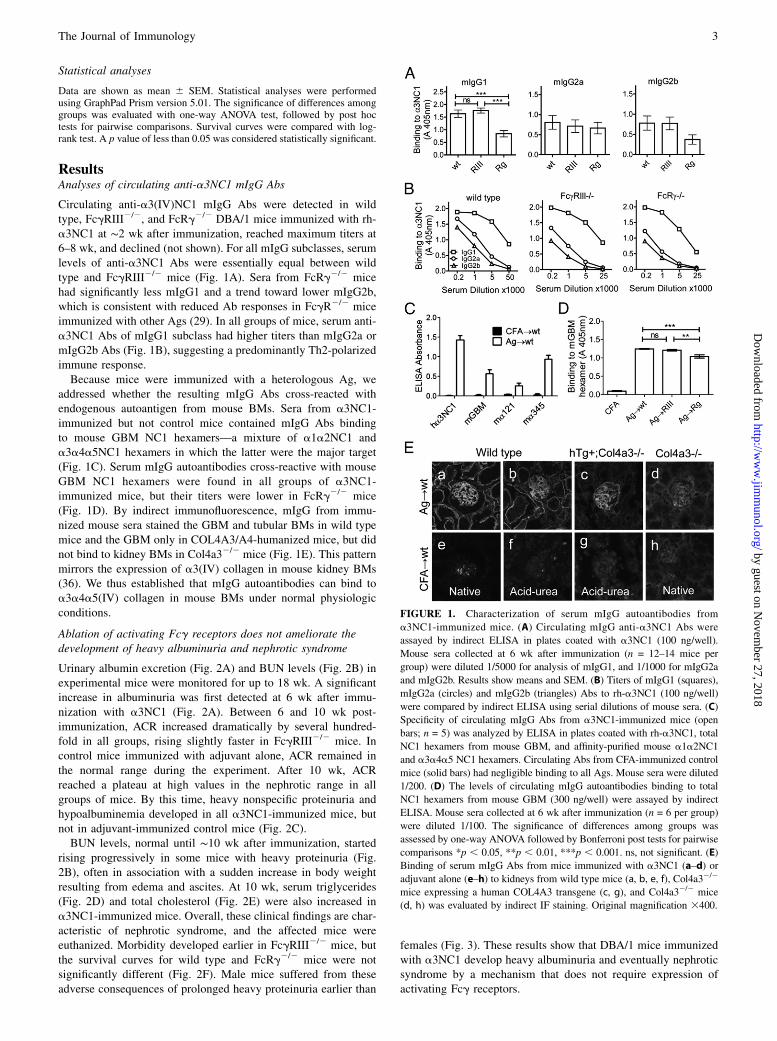
ResultsAnalyses of circulating anti-a3NC1 mIgG Abs
Circulating anti-a3(IV)NC1 mIgG Abs were detected in wildtype, FcgRIII2/2, and FcRg2/2 DBA/1 mice immunized with rh-a3NC1 at ∼2 wk after immunization, reached maximum titers at6–8 wk, and declined (not shown). For all mIgG subclasses, serumlevels of anti-a3NC1 Abs were essentially equal between wildtype and FcgRIII2/2 mice (Fig. 1A). Sera from FcRg2/2 micehad significantly less mIgG1 and a trend toward lower mIgG2b,which is consistent with reduced Ab responses in FcgR2/2 miceimmunized with other Ags (29). In all groups of mice, serum anti-a3NC1 Abs of mIgG1 subclass had higher titers than mIgG2a ormIgG2b Abs (Fig. 1B), suggesting a predominantly Th2-polarizedimmune response.Because mice were immunized with a heterologous Ag, we
addressed whether the resulting mIgG Abs cross-reacted withendogenous autoantigen from mouse BMs. Sera from a3NC1-immunized but not control mice contained mIgG Abs bindingto mouse GBM NC1 hexamers—a mixture of a1a2NC1 anda3a4a5NC1 hexamers in which the latter were the major target(Fig. 1C). Serum mIgG autoantibodies cross-reactive with mouseGBM NC1 hexamers were found in all groups of a3NC1-immunized mice, but their titers were lower in FcRg2/2 mice(Fig. 1D). By indirect immunofluorescence, mIgG from immu-nized mouse sera stained the GBM and tubular BMs in wild typemice and the GBM only in COL4A3/A4-humanized mice, but didnot bind to kidney BMs in Col4a32/2 mice (Fig. 1E). This patternmirrors the expression of a3(IV) collagen in mouse kidney BMs(36). We thus established that mIgG autoantibodies can bind toa3a4a5(IV) collagen in mouse BMs under normal physiologicconditions.
Ablation of activating Fcg receptors does not ameliorate thedevelopment of heavy albuminuria and nephrotic syndrome
Urinary albumin excretion (Fig. 2A) and BUN levels (Fig. 2B) inexperimental mice were monitored for up to 18 wk. A significantincrease in albuminuria was first detected at 6 wk after immu-nization with a3NC1 (Fig. 2A). Between 6 and 10 wk post-immunization, ACR increased dramatically by several hundred-fold in all groups, rising slightly faster in FcgRIII2/2 mice. Incontrol mice immunized with adjuvant alone, ACR remained inthe normal range during the experiment. After 10 wk, ACRreached a plateau at high values in the nephrotic range in allgroups of mice. By this time, heavy nonspecific proteinuria andhypoalbuminemia developed in all a3NC1-immunized mice, butnot in adjuvant-immunized control mice (Fig. 2C).BUN levels, normal until ∼10 wk after immunization, started
rising progressively in some mice with heavy proteinuria (Fig.2B), often in association with a sudden increase in body weightresulting from edema and ascites. At 10 wk, serum triglycerides(Fig. 2D) and total cholesterol (Fig. 2E) were also increased ina3NC1-immunized mice. Overall, these clinical findings are char-acteristic of nephrotic syndrome, and the affected mice wereeuthanized. Morbidity developed earlier in FcgRIII2/2 mice, butthe survival curves for wild type and FcRg2/2 mice were notsignificantly different (Fig. 2F). Male mice suffered from theseadverse consequences of prolonged heavy proteinuria earlier than
females (Fig. 3). These results show that DBA/1 mice immunizedwith a3NC1 develop heavy albuminuria and eventually nephroticsyndrome by a mechanism that does not require expression ofactivating Fcg receptors.
FIGURE 1. Characterization of serum mIgG autoantibodies from
a3NC1-immunized mice. (A) Circulating mIgG anti-a3NC1 Abs were
assayed by indirect ELISA in plates coated with a3NC1 (100 ng/well).
Mouse sera collected at 6 wk after immunization (n = 12–14 mice per
group) were diluted 1/5000 for analysis of mIgG1, and 1/1000 for mIgG2a
and mIgG2b. Results show means and SEM. (B) Titers of mIgG1 (squares),
mIgG2a (circles) and mIgG2b (triangles) Abs to rh-a3NC1 (100 ng/well)
were compared by indirect ELISA using serial dilutions of mouse sera. (C)
Specificity of circulating mIgG Abs from a3NC1-immunized mice (open
bars; n = 5) was analyzed by ELISA in plates coated with rh-a3NC1, total
NC1 hexamers from mouse GBM, and affinity-purified mouse a1a2NC1
and a3a4a5 NC1 hexamers. Circulating Abs from CFA-immunized control
mice (solid bars) had negligible binding to all Ags. Mouse sera were diluted
1/200. (D) The levels of circulating mIgG autoantibodies binding to total
NC1 hexamers from mouse GBM (300 ng/well) were assayed by indirect
ELISA. Mouse sera collected at 6 wk after immunization (n = 6 per group)
were diluted 1/100. The significance of differences among groups was
assessed by one-way ANOVA followed by Bonferroni post tests for pairwise
comparisons *p , 0.05, **p , 0.01, ***p , 0.001. ns, not significant. (E)
Binding of serum mIgG Abs from mice immunized with a3NC1 (a–d) or
adjuvant alone (e–h) to kidneys from wild type mice (a, b, e, f), Col4a32/2
mice expressing a human COL4A3 transgene (c, g), and Col4a32/2 mice
(d, h) was evaluated by indirect IF staining. Original magnification 3400.
The Journal of Immunology 3
by guest on Novem
ber 27, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

Kidney histopathology is typical of membranous nephropathy
PAS-stained kidney sections (Fig. 4Aa–h) from a3NC1-immunizedmice showed GBM thickening, but minimal glomerular inflam-mation and few crescents (56 3% in wild type mice and,26 1%in FcgRIII2/2 and FcRg2/2 mice; the difference was not signifi-cant by one-way ANOVA). Jones silver stain (Fig. 4Ai–l) showedprominent GBMs with capillary wall spikes (inset) characteristicof MN. Transmission EM (Fig. 4B) revealed severe irregularthickening of the GBM because of subepithelial electron-densedeposits surrounded by the expanded matrix, and diffuse efface-ment of podocyte foot processes in a3NC1-immunized mice.Glomerular deposition of mIgG and mC3 was analyzed by direct
IF staining. In all groups of a3NC1-immunized mice, glomerularcapillary loops showed intense staining (3+) for mIgG, in a gran-ular pattern characteristic of MN, with an underlying linear GBMpattern typical for anti-GBM autoantibodies (Fig. 5b–d). Weaklinear staining for mIgG was also observed in tubular BMs. C3was deposited in a fine granular pattern along the glomerularcapillary wall and focally in tubular BMs (Fig. 5f–h). Analysis ofkidney-bound mIgG subclasses showed intense staining formIgG1 (Fig. 5j–l) and weaker staining for mIgG2a (Fig. 5n–p) andmIgG2b (Fig. 5r–t).Indirect IF staining revealed C5b-9 deposition in the capillary
loops of a3NC1-immunized, but not CFA-immunized mice (Fig.
6A). Urinary excretion of C5b-9 was evaluated by Western blotanalysis. Rabbit Abs to mC5b-9 specifically stained an ∼250-kDaband (arrow) in urine samples collected at 8–10 wk (but not at 0–6wk) from a3NC1-immunized mice, but not CFA-immunized con-trol mice (Fig. 6B).
Analyses of kidney-eluted mIgG autoantibodies
We next characterized the specificity of mIgG Abs eluted frommouse kidneys. Compared with serum Abs, kidney-eluted Abs hadgreater reactivity toward total mouse GBM NC1 hexamers (Fig.7A, right) and less toward rh-a3NC1 (Fig. 7A, left). The Agsin solution inhibited the binding of eluted Abs to rh-a3NC1 by50–75% and to mouse GBM hexamers by ∼80%, confirming thecross-reactivity of kidney-bound mIgG autoantibodies. In contrast,mouse GBM hexamers in solution inhibited the binding of serumAbs to immobilized mouse GBM hexamers but not to rh-a3NC1,indicating that the bulk of circulating Abs specifically target rh-a3NC1. Using murine NC1 domains separated by reverse-phaseHPLC, we further established that kidney-eluted mIgG Abs re-acted exclusively with mouse a3NC1 (Fig. 7B), thus ruling out anepitope spreading to other NC1 domains. By indirect IF, kidney-eluted Abs bound in a linear pattern to kidney BM from wildtype mice, but did not stain Col4a32/2 mouse kidneys (Fig. 7C).The latter suggests that intermolecular epitope spreading to nor-mal podocyte Ags, other than a3(IV) collagen, is unlikely.However, we cannot rule out the production of autoantibodies topodocyte Ags induced or upregulated during disease, or to neo-epitopes generated by oxidative modification of podocyte or GBMproteins. By ELISA, kidney-eluted IgG autoantibodies bindingto mouse GBM NC1 hexamers were predominantly mIgG1 (Fig.7D), as found for serum Abs (Fig. 1) and consistent with theanalysis of kidney-bound mIgG subclasses by direct IF staining(Fig. 5). Overall, these results show that kidney-bound mIgGautoantibodies are a subset of serum Abs that specifically targeta3NC1 epitopes shared by the human and mouse Ags, and whichare accessible in native NC1 hexamers of a3a4a5(IV) collagen.
Expression of collagen IV chains and glomerular deposition ofextrinsic Ag
Increased production of collagen IV or other BM components byinjured podocytes alters the GBM composition in experimental MN(39, 40). We analyzed the expression of collagen IV chains byindirect IF. In a3NC1-immunized mice, the GBM was stainedmore intensely by mAbs to a3, a4, and a5(IV) collagen chains,whereas the staining of tubular BMs was largely unchangedcompared with control mice(Fig. 8d–f, j–l, p–r, v–x). Glomerularexpression of a1(IV) collagen, mostly in a mesangial pattern, wasnot altered (Fig. 8b, h, n, t). These results reveal that a3-immunizedmice have increased production or reduced turnover, or both, ofa3a4a5(IV) collagen in the GBM, whereas a1a2(IV) collagendoes not appear to contribute to GBM expansion in this model.Unexpectedly, we found that mAb H31 specifically stained the
GBM (but not tubular BMs) in native kidney sections from a3NC1-immunized mice (Fig. 8i, u). As the staining pattern in kidneysfrom control mice shows, mAb H31 targets an a3NC1 epitope thatis normally cryptic in native NC1 hexamers in tissues (Fig. 8c, n),but is unmasked by acid-urea treatment (Fig. 8d, p). This findingimplies that endogenous a3(IV) collagen in the GBM is alteredduring the disease or that rh-a3NC1 monomers deposit in theglomeruli—a possibility addressed next.Indirect immunofluorescence staining using mAb RH34, which
specifically binds to human but not mouse a3NC1 (36), showedgranular deposition of rh-a3NC1 in the glomerular capillaryloops, but no staining of tubular BMs (Fig. 9b–d). Staining with
FIGURE 2. Mice immunized with a3NC1 develop heavy proteinuria and
nephrotic syndrome independent of Fcg receptors. Renal function was
monitored in wild type mice (squares), FcgRIII2/2 mice (triangles), and
FcRg2/2 mice (circles) immunized with rh-a3NC1 (n = 15–16 mice in each
group, from three separate experiments). Mice in the control group (3),
were immunized with adjuvant alone (n = 14; including 4–5 mice for each
strain). (A) Urinary albumin creatinine ratio (means and SEM). (B) Blood
urea nitrogen levels (means and SEM). (C) SDS-PAGE analysis of urine
(au–hu) and serum (as–hs) samples (2 ml/lane) from a3NC1-immunized
wild type mice (a, b), FcgRIII2/2 mice (c, d), FcRg2/2 mice (e, f) and
CFA-immunized control mice (g, h), collected before the onset of albu-
minuria and when mice were euthanized. Serum triglycerides (D) and total
cholesterol (E) were assayed at 10 wk after immunization with rh-a3NC1 or
adjuvant. The graphs show means and SEM (n = 7–9 mice in each group).
Differences among groups were significant by one-way ANOVA. (F) Sur-
vival curves show the proportion of mice not euthanized due to nephropa-
thy-associated morbidity. The difference between FcgRIII2/2 mice and
either wild type or FcRg2/2 mice was significant by log-rank test.
4 NOVEL MOUSE MODEL OF MEMBRANOUS NEPHROPATHY
by guest on Novem
ber 27, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

GP autoantibodies, which do not bind to a3(IV) collagen in nativemouse BMs, produced the same results (Fig. 9f–h). Overall, thecolocalization of rh-a3NC1 with IgG and C3 in the capillary loopssuggests that the extrinsic Ag used for immunization may par-ticipate in the formation of subepithelial IC deposits.
Absence of lung inflammation despite mIgG binding to alveolarBMs
Because a3(IV) collagen is a normal component of alveolar BMsand is targeted by autoantibodies mediating pulmonary hemor-rhage in GP patients, we assessed the lung phenotype in experi-mental mice. In a3NC1-immunized and control mice, lungsappeared grossly normal without overt hemorrhage. By light mi-croscopy, H&E-stained lung sections showed normal alveoli withno inflammation (Fig. 10d, g, j). Direct immunofluorescencestaining showed strong linear mIgG deposition along the alveolarBMs in a3NC1-immunized mice, but not in control mice (Fig.10e, h, k). Negative staining by mAb RH34 indicated the absenceof rh-a3NC1 in the lungs (Fig. 10f, i, l). These results show thatmIgG autoantibodies bound to alveolar BMs in vivo, but did notinduce inflammation.
DiscussionMembranous nephropathy is a common cause of nephrotic syn-drome in adults. Up to 40% of patients develop end-stage renaldisease (41). Current treatments are nonspecific, toxic, and oftenineffective (42, 43). Development of novel targeted therapiesrequires a thorough understanding of the pathogenic mechanisms.Rat Heymann nephritis has emerged as the best model forstudying the disease mechanisms, because it faithfully recapit-ulates human MN (44). However, further progress is hampered bythe lack of a mouse model that can take full advantage of theavailability of genetically engineered strains and mouse-specificreagents.In this study, we describe an active mouse model of membranous
nephropathy induced by immunization with a fragment of a3(IV)collagen, a normal GBM component, and target of GP autoanti-bodies. Wild type, FcgRIII2/2, and FcRg2/2 DBA/1 mice im-munized with rh-a3NC1 developed massive albuminuria followedby hypoalbuminemia, hyperlipidemia, and often edema—typicalfeatures of nephrotic syndrome. Evaluation of kidney histopa-thology revealed GBM thickening with subepithelial spikes,granular deposits of mIgG and mC3 along the glomerular capillary
FIGURE 3. For each strain, the time course for al-
buminuria (A–C), BUN (D–F), changes in body weight
(G–I), and the incidence of morbidity requiring eu-
thanasia (J–L) were compared among male (triangles)
and female (circles) mice immunized with rh-a3NC1
and adjuvant-immunized control mice of either sex
(squares). The kinetics of albuminuria was similar be-
tween males and females in all mouse strains (A–C).
The increase in BUN levels (D–F) was associated with
a sudden increase in body weight (G–I) because of
edema and ascites in nephrotic mice. Males (continu-
ous line) developed edema and ascites or their BUN
levels doubled earlier than female (dashes) mice (J–L),
or both. The significance of differences in the survival
curves was assessed by log-rank test.
The Journal of Immunology 5
by guest on Novem
ber 27, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

loops by IF, with subepithelial electron-dense immune complexesdeposits, and diffuse effacement of podocyte foot processes byEM. These clinical and histopathologic findings recapitulate thehallmark features of membranous nephropathy.The susceptibility of mouse strains to experimental GN induced
by immunization with rh-a3NC1 has been compared by Hopferet al. (30). C57BL/6 develop milder glomerular injury than doDBA/1 mice, whereas AKR and NOD mice are relatively resis-tant, despite mIgG deposition in the kidneys. Consistent with ourfindings, DBA/1 mice exhibit linear-granular GBM staining formIgG and mC3 and electron-dense subepithelial deposits at 11 wkpostimmunization. A higher proportion of crescents (∼23 versus∼5% in our model) may be the result of a more aggressive im-munization protocol (mice were immunized in the footpad andboosted three times). Whether the kidney phenotype in mousestrains studied by Hopfer et al. (30) recapitulates human MNcannot be ascertained, because albuminuria measurements orother features of nephrotic syndrome have not been reported.
Properties of anti-a3NC1 mIgG Abs: effect on the renalphenotype
Because IgG autoantibodies are central mediators of both anti-GBM/GP disease and MN, we have characterized in detail theproperties of circulating and tissue-bound mIgG Abs elicited byimmunization with rh-a3NC1. We showed that bona fide mIgGanti-a3NC1 autoantibodies are produced, which cross-react with
native mouse GBM NC1 hexamers in vitro and bind to mouseGBM and alveolar BM in vivo. Hence, these mIgG autoantibod-ies must bind to a3NC1 epitopes accessible in the murinea3a4a5NC1 hexamers, similar to mIgG Abs produced in Co-l4a32/2 mice immunized with rh-a3NC1 and to human IgG
FIGURE 4. GBM thickening with the formation of spikes, and sub-
epithelial electron-dense deposits are characteristic of membranous ne-
phropathy. (A) Kidney sections from adjuvant-immunized control mice
and wild type, FcgRIII2/2, and FcRg2/2 mice immunized with rh-a3NC1,
stained with PAS (a–h) or Jones silver (i–l) show GBM thickening but
little glomerular inflammations or crescents, with occasional proteinaceous
casts and mild interstitial fibrosis and inflammation. Jones silver stain
shows GBM spikes (inset, arrows). Original magnification 3200 (a–d),
3400 (e–h), 3630 (i–l). (B) Transmission EM shows subepithelial elec-
tron-dense deposits (red arrowheads), thickened GBM, and podocyte foot
process effacement in a3NC1-immunized wild type (a), FcgRIII2/2 (b)
and FcRg2/2 (c) mice. Original magnification 32850.
FIGURE 5. Granular deposition of mIgG and mC3 in the glomerular
capillary loops. Direct IF microscopy shows kidneys from adjuvant-im-
munized control mice (a, e, i, m, q) and wild type (b, f, j, n, r), FcgRIII2/2
(c, g, k, o, s) and FcRg2/2 (d, h, l, p, t) mice euthanized at 10 wk after
immunization with rh-a3NC1. Sections were stained with fluorophore-
conjugated Abs to mIgG (a–d), mC3c (e–h), mIgG1 (i–l), mIgG2a (m–p),
mIgG2b (q–t). Original magnification3400 . The granular staining pattern
is apparent at high magnification shown in the inset (d, h). Indirect im-
munofluorescence staining of kidney sections from the CFA control mice
with mIgG1 mAb TF51 (i, inset), mIgG2a mAb TF90 (m, inset) and
mIgG2b mAb TF86 (q, inset), all specific for mouse a3a4a5 NC1
domains, provided a reference for assessing the intensity of staining for
each mIgG subclass deposited in the kidneys.
FIGURE 6. Kidney deposition and urinary excretion of mC5b-9. (A)
Indirect IF staining shows granular GBM deposition of mC5b-9 in a3NC1-
immunized wild type (a) and FcRg2/2 (b) mice, but not in adjuvant-
immunized mice (c). Staining was negative when the primary Ab was
omitted (d). Original magnification 3400. (B). Western blot analysis of
mouse urine samples (5 ml/lane) separated by SDS-PAGE under nonre-
ducing conditions using Abs to mC5b-9 showed specific staining of an
∼250-kDa band (arrow) in urine collected at 8 wk (f), but not at 0, 2, 4, or
6 wk after immunization with a3NC1 (b–e, respectively), nor in urine
collected at 8 wk after immunization with CFA (a). Urine from a 7-mo-old
B6.Col4a32/2 mouse with heavy albuminuria (ACR = 103) was used as an
additional control. The band at ∼150 kDa was also stained when the pri-
mary Ab was omitted, suggesting it is due to secondary Ab cross-reacting
with excreted mIgG in mouse urine.
6 NOVEL MOUSE MODEL OF MEMBRANOUS NEPHROPATHY
by guest on Novem
ber 27, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

alloantibodies mediating posttransplant anti-GBM nephritis inautosomal recessive Alport syndrome (16). In contrast, GP auto-antibodies cross-react only with isolated mouse a3NC1 mono-mers or dimers, but do not bind to native mouse a3a4a5NC1hexamers, which are extensively cross-linked and thus protectedagainst autoantibody-induced dissociation (45–47). Although wecannot rule out that mice also produced GP-like autoantibodies,such autoantibodies would not bind to mouse tissues in vivo.Production of mIgG autoantibodies that bind in vivo to murineBMs containing a3a4a5(IV) collagen indicates that immune self-tolerance toward endogenous a3(IV) collagen is breached in miceimmunized with a3NC1 monomers.Although anti-a3NC1 mIgG autoantibodies bound to GBM and
alveolar BMs, they did not mediate crescentic glomerulonephritisor alveolitis, suggesting a limited ability to elicit inflammation.The simplest explanation is the prevalence of mIgG1 over mIgG2aand mIgG2b among anti-a3NC1 autoantibodies. Only mIgG2aand mIgG2b but not mIgG1 mediate neutrophil influx and glo-merular injury in a mouse model of acute neutrophil-mediated
glomerular inflammation (25). In particular, anti-a3NC1 mAbsof the mIgG1 subclass that bind to mouse GBM in vivo are notnephritogenic (48). The lattice structure of collagen IV limits themaximum amount and density of autoantibodies that can bind tothe NC1 domains. Therefore, competition by mIgG1 could reducethe density of GBM-bound IgG2a and IgG2b autoantibodies be-
FIGURE 7. Specificity of kidney-bound mIgG autoantibodies. (A)
ELISA shows the binding of serum (open bars) and kidney-eluted (closed
bars) mIgG Abs to immobilized rh-a3NC1 (hAg, left) and total NC1
hexamers from mouse GBM (mAg, right), and the inhibition of this
binding by hAg (5 mg/ml) or mAg (50 mg/ml) in solution. (B) NC1
domains from mouse GBM were separated by reverse-phase HPLC on
a C18 column (top), and the composition of the major peaks (a–d) was
analyzed by Western blotting with chain-specific mAbs (bottom). Kidney-
eluted mIgG autoantibodies reacted only with the fraction containing
murine a3NC1. (C) Indirect IF staining shows the binding of mIgG eluted
from the kidneys of a3NC1-immunized mice (a, b) or control mice (c, d)
to kidney cryostat sections from wild type mice (a, c) and Col4a32/2
Alport mice (b, d). (D) ELISA analysis of subclasses of mIgG eluted from
kidneys at ∼10 wk after immunization with rh-a3NC1 (diagonal lines) or
CFA (solid bars) showed the prevalence of mIgG1 autoantibodies binding
to immobilized mouse GBM NC1 hexamers. Anti-NC1 mAbs TF51, TF90,
and TF86 (1 mg/ml each) were used as standards for mIgG1, mIgG2a, and
mIgG2b, respectively (open bars). A similar distribution of mIgG sub-
classes was found for kidney-eluted autoantibodies binding to rh-a3NC1
(not shown). D, dimers; M, monomers.
FIGURE 8. Increased collagen IV deposition in the GBM. Kidneys from
wild type (a–l) and FcRg2/2 (m–x) mice at 10 wk after immunization with
adjuvant (a–f, m–r) or rh-a3NC1 (g–l, s–x) were analyzed by indirect IF
microscopy. Acetone-fixed cryostat sections were stained with mAbs H11
specific for a1NC1 (a, g, m, s), H31 specific for a3NC1 (c, i, n, u), RH42
specific for a4NC1 (e, k, q, w), and b14 specific for a5NC1 (f, l, r, x).
Some sections were treated with acid urea (AU) before staining with mAbs
H11 (b, h, n, t) and H31 (d, j, p, v). Original magnification 3400.
FIGURE 9. Deposition of rh-a3NC1 in the glomerular capillary loops.
Indirect IF staining shows the binding of mAb RH34 (a–d) and affinity-
purified human GP autoantibodies (e–h) to kidney sections from adjuvant-
immunized control mice (a, e) and from wild type mice (b, f), FcgRIII2/2
mice (c, g), and FcRg2/2 mice (d, h) at 10 wk after immunization with rh-
a3NC1. A detailed view (insets) illustrates the granular staining pattern.
Control sections stained with secondary Ab but omitting the primary Ab
were negative (not shown). Binding of mAb RH34 was slightly reduced
but not abolished by acid urea treatment (not shown). Original magnifi-
cation 3400.
The Journal of Immunology 7
by guest on Novem
ber 27, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

low the threshold required for efficient crosslinking of activatingFcgRs on effector cells, while augmenting signaling through theinhibitory IgG receptor, FcgRIIB.Though present in smaller amounts in mouse sera and kidneys,
mIgG2a and mIgG2b may be essential mediators of murine MN.Only mIgG2a and mIgG2b but not mIgG1 are complement fixing,and a3NC1-immunized mice had glomerular deposition of mC3and C5b-9. C5b-9 mediated podocyte injury is considered essen-tial for developing proteinuria in experimental and possibly hu-man MN (8, 49). We note that two mouse strains resistant toa3NC1-induced GN, AKR, and NOD (30) are naturally deficientin complement C5 (50). Because the ablation of activating FcgRsdid not prevent the development of heavy albuminuria and ne-phrotic syndrome in our model, the major effector pathway ofsubepithelial ICs is most likely complement activation, thus im-plying a role for mIgG2a and mIgG2b.As mIgG1 is the murine equivalent of human IgG4, our model
recapitulates the prevalence of human IgG4 autoantibodies in id-iopathic MN (51). Because mIgG1 is associated with Th2 andmIgG2a with Th1 polarization, the prevalence of mIgG1 ina3NC1-immunized DBA/1 mice implies a Th2 bias. Th subsetsaffect the pattern of injury and outcomes in GN, with Th1 pre-dominance implicated in proliferative and crescentic glomerulo-nephritis and Th2 predominance associated with membranousnephropathy (52). In idiopathic MN, Th2 cytokines increase andstimulate B cells to produce IgG4 (53, 54). A strong Th2 polari-zation has been reported in murine MN induced by cationic BSA
(55). Th2 deviation induced by disruption of theWsx1 gene, whichencodes a subunit of IL-27R, changed the renal pathophysiologyfrom proliferative to membranous lupus in MRL/lpr mice (56).Hence, the development of murine MN may depend on features ofthe Ag and host factors that favor a Th2 polarization.
Formation of subepithelial ICs and the role of GBM Ags in MN
Given the central role of subepithelial ICs in the pathogenesis ofMN, we addressed their origin in our model. Colocalization of theAg (rh-a3NC1) with mIgG and mC3 in the GBM (though not inkidney tubular or alveolar BMs) is consistent with a planted Agmechanism or deposition of small circulating ICs. Glomerulardeposition of extrinsic Ags from circulation occurs in other activemodels of MN, such as rats immunized with human brush borderAg and rabbits immunized with rat tubular Ag (57, 58). Proto-typical planted Ags are exogenous cationic proteins, which bind tonegative sites in the GBM and on podocytes. Cationic BSA is anAg in early childhood MN (7), and repeated injections with cat-ionized BSA induce experimental MN (59, 60). Human a3NC1has a high isoelectric point of ∼9, which may contribute to itsdeposition in glomeruli. Alternatively, ha3NC1 may bind directlyto its receptors on the podocyte cell surface, such as a3b1 or avb3integrins (61, 62).Because a3(IV) collagen deposited in the GBM is normally
expressed by podocytes (63), subepithelial ICs may also form whenanti-a3NC1 autoantibodies bind to nascent a3(IV) collagen beforeit is stably incorporated into the GBM. Indeed, the increased de-position of a3a4a5(IV) collagen in the GBM implies augmentedproduction of a3(IV) collagen by podocytes in murine MN. Sev-eral GBM Ags have been implicated in various models of MN.Administration of Abs to GBM heparan sulfate proteoglycans intopresensitized rats induces subepithelial IC deposits and MN (64).Anti-laminin Abs play a central role in HgCl2-induced nephropa-thy in Brown-Norway rats, including formation of subepithelialdeposits and heavy proteinuria (65). These studies raise the ques-tion of whether GBM Ags are likewise involved in human MN.A possible role of a3(IV) collagen in the pathogenesis of some
forms of MN is suggested by the association between anti-GBM/GP disease and MN. At least 25 cases have been reported (66).Anti-GBM/GP disease can either precede or is followed by MN,although simultaneous presentation is more common. In thesepatients, the linear GBM IgG staining found on the renal biopsy islikely due to classic GP autoantibodies (anti-a3NC1), typicallypositive by serology. Much less is known about how subepithe-lial ICs form; they may consist of GP autoantibodies bound topodocyte-associated a3(IV) collagen or to nascent a3(IV) colla-gen not yet incorporated in the GBM, or to a3(IV) collagenfragments released from the GBM. The same mechanisms mayaccount for the formation of subepithelial IC in our mouse model.
Comparison with other models of experimental MN
Our mouse model recapitulates hallmark features of human MN aswell as rat Heymann nephritis, the “gold standard” for experi-mental MN. Compared with the widely used passive model, activeHeymann nephritis more closely resembles human disease be-cause it involves the host’s own immune system. Its major dis-advantages are longer duration, much greater variability, andanimal discomfort because of footpad immunizations (67). Bycomparison, in our active mouse model, nephropathy is inducedmore rapidly and reliably. In addition, unlike rats, mouse modelscan take advantage of existing transgenic mice and mouse-specificreagents.The development of a robust murine model of MN has been
a work in progress for decades (67). Early models had various
FIGURE 10. In vivo binding of mIgG autoantibodies to alveolar BMs
does not induce lung inflammation. Lungs from adjuvant-immunized
control mice (a–c) and a3NC1-immunized wild type (d–f), FcgR2/2
(g–i), and FcgRIII2/2 (j–l) mice were collected at ∼10–14 wk after im-
munization. Paraffin sections were stained with H&E and observed by light
microscopy (a, d, g, j). In vivo binding of mIgG to alveolar BMs was
evaluated by direct IF (b, e, h, k). Indirect IF staining for rh-a3NC1 using
mAb RH34 was negative in all lung sections (c, f, i, l). Shown for com-
parison is the deposition of mIgG (m) and RH34 staining (n) in kidneys
from FcgRIII2/2 mice at 10 wk after immunization with rh-a3NC1.
Original magnification 3200.
8 NOVEL MOUSE MODEL OF MEMBRANOUS NEPHROPATHY
by guest on Novem
ber 27, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

limitations, such as minimal or transient proteinuria or comple-ment independence. Severe proteinuria, diffuse GBM thickening,subepithelial deposits, and GBM spikes are induced in susceptiblemouse strains by repeated injections with cationized BSA (59);however, this model is cumbersome, requiring numerous i.v.injections (every other day for 6 wk). Characteristic featuresof MN are also induced in mice after passive immunization withsheep Abs to murine podocytes (6). This model may depend onthe quality of the heterologous antisera or the immunogen prep-aration used, or both, because other groups have found that sheepAbs to mouse podocyte Ags cause little glomerular damage inwild type mice despite in vivo binding to glomerular capillarywall (68).We anticipate that the murine MN model described in this study,
leveraged by the availability of numerous strains of knockoutmice, will help to address new questions about the disease patho-mechanisms, such as the role of various pathways of complementactivation. A potential drawback is the possible need for back-crossing mice to the DBA/1 background. Knockout mice are mostoften available on the C57BL/6 background. In our hands, C57BL/6J mice immunized with a3NC1 according to the protocol used inDBA/1 mice developed moderate albuminuria by 12–18 wk (ACRranging from 1.64 to 2.39, compared with 0.01–0.03 in controlmice; n = 3 mice in each group), but no features of nephroticsyndrome (data not shown). The susceptibility of other mousestrains to experimental MN is being addressed in ongoing studies.Nonetheless, our model of active MN in DBA/1 mice is readilyusable for testing the effectiveness on novel targeted drugs and isideal for preclinical studies of experimental therapies.
AcknowledgmentsWe thank Dr. Billy Hudson for providing cells expressing rh-a3NC1, Dr.
Elena Tchekneva for access to the urine collection station, Ellen Donnert for
performing the Jones silver stain, and Anjana Hassan for technical support.
DisclosuresThe authors have no financial conflicts of interest.
References1. Nangaku, M., and W. G. Couser. 2005. Mechanisms of immune-deposit for-
mation and the mediation of immune renal injury. Clin. Exp. Nephrol. 9: 183–191.
2. Glassock, R. J. 2010. The pathogenesis of idiopathic membranous nephropathy:a 50-year odyssey. Am. J. Kidney Dis. 56: 157–167.
3. Beck, L. H., Jr., R. G. Bonegio, G. Lambeau, D. M. Beck, D. W. Powell,T. D. Cummins, J. B. Klein, and D. J. Salant. 2009. M-type phospholipase A2receptor as target antigen in idiopathic membranous nephropathy. N. Engl. J.Med. 361: 11–21.
4. Debiec, H., V. Guigonis, B. Mougenot, F. Decobert, J. P. Haymann, A. Bensman,G. Deschenes, and P. M. Ronco. 2002. Antenatal membranous glomerulone-phritis due to anti-neutral endopeptidase antibodies. N. Engl. J. Med. 346: 2053–2060.
5. Prunotto, M., M. L. Carnevali, G. Candiano, C. Murtas, M. Bruschi,E. Corradini, A. Trivelli, A. Magnasco, A. Petretto, L. Santucci, et al. 2010.Autoimmunity in membranous nephropathy targets aldose reductase and SOD2.J. Am. Soc. Nephrol. 21: 507–519.
6. Meyer-Schwesinger, C., S. Dehde, P. Klug, J. U. Becker, S. Mathey, K. Arefi,S. Balabanov, S. Venz, K. H. Endlich, M. Pekna, et al. 2011. Nephrotic syndromeand subepithelial deposits in a mouse model of immune-mediated anti-podocyteglomerulonephritis. J. Immunol. 187: 3218–3229.
7. Debiec, H., F. Lefeu, M. J. Kemper, P. Niaudet, G. Deschenes, G. Remuzzi,T. Ulinski, and P. Ronco. 2011. Early-childhood membranous nephropathy dueto cationic bovine serum albumin. N. Engl. J. Med. 364: 2101–2110.
8. Cunningham, P. N., and R. J. Quigg. 2005. Contrasting roles of complementactivation and its regulation in membranous nephropathy. J. Am. Soc. Nephrol.16: 1214–1222.
9. Borza, D. B. 2007. Autoepitopes and alloepitopes of type IV collagen: role in themolecular pathogenesis of anti-GBM antibody glomerulonephritis. Nephron.Exp. Nephrol. 106: e37–e43.
10. Hudson, B. G., K. Tryggvason, M. Sundaramoorthy, and E. G. Neilson. 2003.Alport’s syndrome, Goodpasture’s syndrome, and type IV collagen. N. Engl. J.Med. 348: 2543–2556.
11. Borza, D. B., K. O. Netzer, A. Leinonen, P. Todd, J. Cervera, J. Saus, andB. G. Hudson. 2000. The goodpasture autoantigen. Identification of multiplecryptic epitopes on the NC1 domain of the alpha3(IV) collagen chain. J. Biol.Chem. 275: 6030–6037.
12. Netzer, K. O., A. Leinonen, A. Boutaud, D. B. Borza, P. Todd, S. Gunwar,J. P. Langeveld, and B. G. Hudson. 1999. The Goodpasture autoantigen. Map-ping the major conformational epitope(s) of alpha3(IV) collagen to residues17-31 and 127-141 of the NC1 domain. J. Biol. Chem. 274: 11267–11274.
13. Hellmark, T., H. Burkhardt, and J. Wieslander. 1999. Goodpasture disease.Characterization of a single conformational epitope as the target of pathogenicautoantibodies. J. Biol. Chem. 274: 25862–25868.
14. Pedchenko, V., O. Bondar, A. B. Fogo, R. Vanacore, P. Voziyan, A. R. Kitching,J. Wieslander, C. Kashtan, D. B. Borza, E. G. Neilson, et al. 2010. Moleculararchitecture of the Goodpasture autoantigen in anti-GBM nephritis. N. Engl. J.Med. 363: 343–354.
15. Kang, J. S., C. E. Kashtan, A. N. Turner, L. Heidet, B. G. Hudson, andD. B. Borza. 2007. The alloantigenic sites of alpha3alpha4alpha5(IV) collagen:pathogenic X-linked alport alloantibodies target two accessible conformationalepitopes in the alpha5NC1 domain. J. Biol. Chem. 282: 10670–10677.
16. Wang, X. P., A. B. Fogo, S. Colon, G. Giannico, S. R. Abul-Ezz, J. H. Miner, andD. B. Borza. 2005. Distinct epitopes for anti-glomerular basement membranealport alloantibodies and goodpasture autoantibodies within the noncollagenousdomain of alpha3(IV) collagen: a janus-faced antigen. J. Am. Soc. Nephrol. 16:3563–3571.
17. Kalluri, R., A. Torre, C. F. Shield, III, E. D. Zamborsky, M. C. Werner,E. Suchin, G. Wolf, U. M. Helmchen, L. P. van den Heuvel, R. Grossman, et al.2000. Identification of alpha3, alpha4, and alpha5 chains of type IV collagen asalloantigens for Alport posttransplant anti-glomerular basement membraneantibodies. Transplantation 69: 679–683.
18. Brainwood, D., C. Kashtan, M. C. Gubler, and A. N. Turner. 1998. Targets ofalloantibodies in Alport anti-glomerular basement membrane disease after renaltransplantation. Kidney Int. 53: 762–766.
19. Nimmerjahn, F., and J. V. Ravetch. 2006. Fcgamma receptors: old friends andnew family members. Immunity 24: 19–28.
20. Wakayama, H., Y. Hasegawa, T. Kawabe, T. Hara, S. Matsuo, M. Mizuno,T. Takai, H. Kikutani, and K. Shimokata. 2000. Abolition of anti-glomerularbasement membrane antibody-mediated glomerulonephritis in FcRgamma-deficient mice. Eur. J. Immunol. 30: 1182–1190.
21. Park, S. Y., S. Ueda, H. Ohno, Y. Hamano, M. Tanaka, T. Shiratori, T. Yamazaki,H. Arase, N. Arase, A. Karasawa, et al. 1998. Resistance of Fc receptor- deficientmice to fatal glomerulonephritis. J. Clin. Invest. 102: 1229–1238.
22. Clynes, R., C. Dumitru, and J. V. Ravetch. 1998. Uncoupling of immune com-plex formation and kidney damage in autoimmune glomerulonephritis. Science279: 1052–1054.
23. Matsumoto, K., N. Watanabe, B. Akikusa, K. Kurasawa, R. Matsumura, Y. Saito,I. Iwamoto, and T. Saito. 2003. Fc receptor-independent development of auto-immune glomerulonephritis in lupus-prone MRL/lpr mice. Arthritis Rheum. 48:486–494.
24. Guo, S., A. S. Muhlfeld, T. A. Wietecha, C. J. Peutz-Kootstra, J. Kowalewska,K. Yi, M. Spencer, W. Pichaiwong, F. Nimmerjahn, K. L. Hudkins, andC. E. Alpers. 2009. Deletion of activating Fcgamma receptors does not conferprotection in murine cryoglobulinemia-associated membranoproliferative glo-merulonephritis. Am. J. Pathol. 175: 107–118.
25. Giorgini, A., H. J. Brown, H. R. Lock, F. Nimmerjahn, J. V. Ravetch,J. S. Verbeek, S. H. Sacks, and M. G. Robson. 2008. Fc gamma RIII and Fcgamma RIV are indispensable for acute glomerular inflammation induced byswitch variant monoclonal antibodies. J. Immunol. 181: 8745–8752.
26. Fujii, T., Y. Hamano, S. Ueda, B. Akikusa, S. Yamasaki, M. Ogawa, H. Saisho,J. S. Verbeek, S. Taki, and T. Saito. 2003. Predominant role of FcgammaRIII inthe induction of accelerated nephrotoxic glomerulonephritis. Kidney Int. 64:1406–1416.
27. Kaneko, Y., F. Nimmerjahn, M. P. Madaio, and J. V. Ravetch. 2006. Pathologyand protection in nephrotoxic nephritis is determined by selective engagement ofspecific Fc receptors. J. Exp. Med. 203: 789–797.
28. Tarzi, R. M., K. A. Davies, J. W. Claassens, J. S. Verbeek, M. J. Walport, andH. T. Cook. 2003. Both Fcgamma receptor I and Fcgamma receptor III mediatedisease in accelerated nephrotoxic nephritis. Am. J. Pathol. 162: 1677–1683.
29. Nakamura, A., T. Yuasa, A. Ujike, M. Ono, T. Nukiwa, J. V. Ravetch, andT. Takai. 2000. Fcgamma receptor IIB-deficient mice develop Goodpasture’ssyndrome upon immunization with type IV collagen: a novel murine model forautoimmune glomerular basement membrane disease. J. Exp. Med. 191: 899–906.
30. Hopfer, H., R. Maron, U. Butzmann, U. Helmchen, H. L. Weiner, and R. Kalluri.2003. The importance of cell-mediated immunity in the course and severity ofautoimmune anti-glomerular basement membrane disease in mice. FASEB J. 17:860–868.
31. Xie, C., R. Sharma, H. Wang, X. J. Zhou, and C. Mohan. 2004. Strain distri-bution pattern of susceptibility to immune-mediated nephritis. J. Immunol. 172:5047–5055.
32. Kleinau, S., P. Martinsson, and B. Heyman. 2000. Induction and suppression ofcollagen-induced arthritis is dependent on distinct fcgamma receptors. J. Exp.Med. 191: 1611–1616.
33. Boutaud, A., D. B. Borza, O. Bondar, S. Gunwar, K. O. Netzer, N. Singh,Y. Ninomiya, Y. Sado, M. E. Noelken, and B. G. Hudson. 2000. Type IV col-lagen of the glomerular basement membrane. Evidence that the chain specificityof network assembly is encoded by the noncollagenous NC1 domains. J. Biol.Chem. 275: 30716–30724.
The Journal of Immunology 9
by guest on Novem
ber 27, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

34. Kang, J. S., X. P. Wang, J. H. Miner, R. Morello, Y. Sado, D. R. Abrahamson,and D. B. Borza. 2006. Loss of alpha3/alpha4(IV) collagen from the glomer-ular basement membrane induces a strain-dependent isoform switch toalpha5alpha6(IV) collagen associated with longer renal survival in Col4a3-/-
Alport mice. J. Am. Soc. Nephrol. 17: 1962–1969.35. Saus, J., J. Wieslander, J. P. Langeveld, S. Quinones, and B. G. Hudson. 1988.
Identification of the Goodpasture antigen as the alpha 3(IV) chain of collagen IV.J. Biol. Chem. 263: 13374–13380.
36. Heidet, L., D. B. Borza, M. Jouin, M. Sich, M. G. Mattei, Y. Sado, B. G. Hudson,N. Hastie, C. Antignac, and M. C. Gubler. 2003. A human-mouse chimera ofthe alpha3alpha4alpha5(IV) collagen protomer rescues the renal phenotype inCol4a3-/- Alport mice. Am. J. Pathol. 163: 1633–1644.
37. Andrews, K. L., J. L. Mudd, C. Li, and J. H. Miner. 2002. Quantitative trait lociinfluence renal disease progression in a mouse model of Alport syndrome. Am. J.Pathol. 160: 721–730.
38. Tchekneva, E., M. Dikov, V. Kadkina, and D. Polosukhina. 2011. Collectionstation for accelerated collection of the specimens from laboratory animals.United States patent application 20110239953. 2011 Oct 6.
39. Kim, Y., R. Butkowski, B. Burke, M. M. Kleppel, J. Crosson, A. Katz, andA. F. Michael. 1991. Differential expression of basement membrane collagen inmembranous nephropathy. Am. J. Pathol. 139: 1381–1388.
40. Minto, A. W., R. Kalluri, M. Togawa, E. C. Bergijk, P. D. Killen, andD. J. Salant. 1998. Augmented expression of glomerular basement membranespecific type IV collagen isoforms (alpha3-alpha5) in experimental membranousnephropathy. Proc. Assoc. Am. Physicians 110: 207–217.
41. Fervenza, F. C., S. Sethi, and U. Specks. 2008. Idiopathic membranous ne-phropathy: diagnosis and treatment. Clin. J. Am. Soc. Nephrol. 3: 905–919.
42. Glassock, R. J. 2004. The treatment of idiopathic membranous nephropathy:a dilemma or a conundrum? Am. J. Kidney Dis. 44: 562–566.
43. Waldman, M., and H. A. Austin, III. 2009. Controversies in the treatment ofidiopathic membranous nephropathy. Nat. Rev. Nephrol. 5: 469–479.
44. Cybulsky, A. V., R. J. Quigg, and D. J. Salant. 2005. Experimental membranousnephropathy redux. Am. J. Physiol. Renal Physiol. 289: F660–F671.
45. Luo, W., X. P. Wang, C. E. Kashtan, and D. B. Borza. 2010. Alport alloanti-bodies but not Goodpasture autoantibodies induce murine glomerulonephritis:protection by quinary crosslinks locking cryptic a3(IV) collagen autoepitopesin vivo. J. Immunol. 185: 3520–3528.
46. Borza, D. B., O. Bondar, S. Colon, P. Todd, Y. Sado, E. G. Neilson, andB. G. Hudson. 2005. Goodpasture autoantibodies unmask cryptic epitopes byselectively dissociating autoantigen complexes lacking structural reinforcement:novel mechanisms for immune privilege and autoimmune pathogenesis. J. Biol.Chem. 280: 27147–27154.
47. Vanacore, R. M., A. J. Ham, J. P. Cartailler, M. Sundaramoorthy, P. Todd,V. Pedchenko, Y. Sado, D. B. Borza, and B. G. Hudson. 2008. A role for collagenIV cross-links in conferring immune privilege to the Goodpasture autoantigen:structural basis for the crypticity of B cell epitopes. J. Biol. Chem. 283: 22737–22748.
48. Summers, S. A., O. M. Steinmetz, M. Li, J. Y. Kausman, T. Semple,K. L. Edgtton, D. B. Borza, H. Braley, S. R. Holdsworth, and A. R. Kitching.2009. Th1 and Th17 cells induce proliferative glomerulonephritis. J. Am. Soc.Nephrol. 20: 2518–2524.
49. Nangaku, M., S. J. Shankland, and W. G. Couser. 2005. Cellular response toinjury in membranous nephropathy. J. Am. Soc. Nephrol. 16: 1195–1204.
50. Nilsson, U. R., and H. J. Muller-Eberhard. 1967. Deficiency of the fifth com-ponent of complement in mice with an inherited complement defect. J. Exp.Med. 125: 1–16.
51. Oliveira, D. B. 1998. Membranous nephropathy: an IgG4-mediated disease.Lancet 351: 670–671.
52. Holdsworth, S. R., A. R. Kitching, and P. G. Tipping. 1999. Th1 and Th2 Thelper cell subsets affect patterns of injury and outcomes in glomerulonephritis.Kidney Int. 55: 1198–1216.
53. Kuroki, A., M. Iyoda, T. Shibata, and T. Sugisaki. 2005. Th2 cytokines increaseand stimulate B cells to produce IgG4 in idiopathic membranous nephropathy.Kidney Int. 68: 302–310.
54. Hirayama,K., I. Ebihara, S.Yamamoto,H.Kai, K.Muro,K.Yamagata,M.Kobayashi,and A. Koyama. 2002. Predominance of type-2 immune response in idiopathicmembranous nephropathy. Cytoplasmic cytokine analysis. Nephron 91: 255–261.
55. Wu, C. C., J. S. Chen, S. J. Chen, S. H. Lin, A. Chen, L. C. Chang, H. K. Sytwu,and Y. F. Lin. 2007. Kinetics of adaptive immunity to cationic bovine serumalbumin-induced membranous nephropathy. Kidney Int. 72: 831–840.
56. Shimizu, S., N. Sugiyama, K. Masutani, A. Sadanaga, Y. Miyazaki, Y. Inoue,M. Akahoshi, R. Katafuchi, H. Hirakata, M. Harada, et al. 2005. Membranousglomerulonephritis development with Th2-type immune deviations in MRL/lprmice deficient for IL-27 receptor (WSX-1). J. Immunol. 175: 7185–7192.
57. Edgington, T. S., R. J. Glassock, and F. J. Dixon. 1967. Autologous immune-complex pathogenesis of experimental allergic glomerulonephritis. Science 155:1432–1434.
58. Barabas, A. Z., and R. Lannigan. 1981. Immune-complex nephritis in the rabbitproduced by injections of rat renal tubular fraction 3 antigen. Br. J. Exp. Pathol.62: 94–102.
59. Chen, J. S., A. Chen, L. C. Chang, W. S. Chang, H. S. Lee, S. H. Lin, andY. F. Lin. 2004. Mouse model of membranous nephropathy induced by cationicbovine serum albumin: antigen dose-response relations and strain differences.Nephrol. Dial. Transplant. 19: 2721–2728.
60. Border, W. A., H. J. Ward, E. S. Kamil, and A. H. Cohen. 1982. Induction ofmembranous nephropathy in rabbits by administration of an exogenous cationicantigen. J. Clin. Invest. 69: 451–461.
61. Borza, C. M., D. B. Borza, V. Pedchenko, M. A. Saleem, P. W. Mathieson,Y. Sado, H. M. Hudson, A. Pozzi, J. Saus, D. R. Abrahamson, et al. 2008. Humanpodocytes adhere to the KRGDS motif of the alpha3alpha4alpha5 collagen IVnetwork. J. Am. Soc. Nephrol. 19: 677–684.
62. Borza, C. M., A. Pozzi, D. B. Borza, V. Pedchenko, T. Hellmark, B. G. Hudson,and R. Zent. 2006. Integrin alpha3beta1, a novel receptor for alpha3(IV) non-collagenous domain and a trans-dominant Inhibitor for integrin alphavbeta3. J.Biol. Chem. 281: 20932–20939.
63. Abrahamson, D. R., B. G. Hudson, L. Stroganova, D. B. Borza, and P. L. St John.2009. Cellular origins of type IV collagen networks in developing glomeruli. J.Am. Soc. Nephrol. 20: 1471–1479.
64. Makino, H., B. Lelongt, and Y. S. Kanwar. 1988. Nephritogenicity of proteo-glycans. II. A model of immune complex nephritis. Kidney Int. 34: 195–208.
65. Icard, P., L. Pelletier, M. C. Vial, C. Mandet, R. Pasquier, A. Michel, andP. Druet. 1993. Evidence for a role of antilaminin-producing B cell clones thatescape tolerance in the pathogenesis of HgCl2-induced membranous glomerul-opathy. Nephrol. Dial. Transplant. 8: 122–127.
66. Basford, A. W., J. Lewis, J. P. Dwyer, and A. B. Fogo. 2011. Membranous ne-phropathy with crescents. J. Am. Soc. Nephrol. 22: 1804–1808.
67. Jefferson, J. A., J. W. Pippin, and S. J. Shankland. 2010. Experimental Models ofMembranous Nephropathy. Drug Discov. Today Dis. Models 7: 27–33.
68. Bao, L., M. Haas, J. Pippin, Y. Wang, T. Miwa, A. Chang, A. W. Minto,M. Petkova, G. Qiao, W. C. Song, et al. 2009. Focal and segmental glomer-ulosclerosis induced in mice lacking decay-accelerating factor in T cells. J. Clin.Invest. 119: 1264–1274.
10 NOVEL MOUSE MODEL OF MEMBRANOUS NEPHROPATHY
by guest on Novem
ber 27, 2018http://w
ww
.jimm
unol.org/D
ownloaded from







