

Dr Mohit Goel
JR I
28/3/13

Grey and white matter of CNS differ in morphology, water content & macromolecular content mainly membrane lipids.
Grey matter primarily contain neurons & their process.
White matter predominantly composed of myelinated axons.
Oligodendroglial cell membrane is the source of the myelin sheath.

Cholesterol, galactocerebrosidase, spingomyelin & phospholipids are found in fully formed white matter and account for stability & strength of the myelin membrane.
Normal myelination… Hyperintense on T1 & Hypointense on T2.
T1W parallel increase in lipids while T2W correlates to the period of maturation of myelin sheath.

General rules of myelination
Caudad to cephalad.
Central to peripheral.
Posterior to anterior.

INFRATENTORIAL
Dorsal medulla /
midbrain
Inferior / superior
cerebellar peduncles
Middle cerebellar
peduncle
Cerebellar white
matter
T1WI T2WI
First appear at
Birth Birth
Birth Birth
1 mth 3 mth
1 – 3 mth 8 – 18 mth

SUPRATENTORIAL
Internal capsule
Posterior limb
Anterior limb
Thalamus
Pre / postcentral gyri
Corpus callosum
Splenium
Genu
Centrum semiovale
Optic radiations
Subcortical U fibers
T1WI T2WI
Birth Birth
3 mth 3 – 6 mth
Birth Birth
1 mth 8 – 12 mth
3 – 4 mth 6 mth
6 mth 8 mth
Birth – 1 mth 3 mth
3 mth 3 mth
3 – 8 mth 8 – 18 mth
Birth
Dorsal medulla / mid brain.
Inferior / superior cerebellar peduncles.
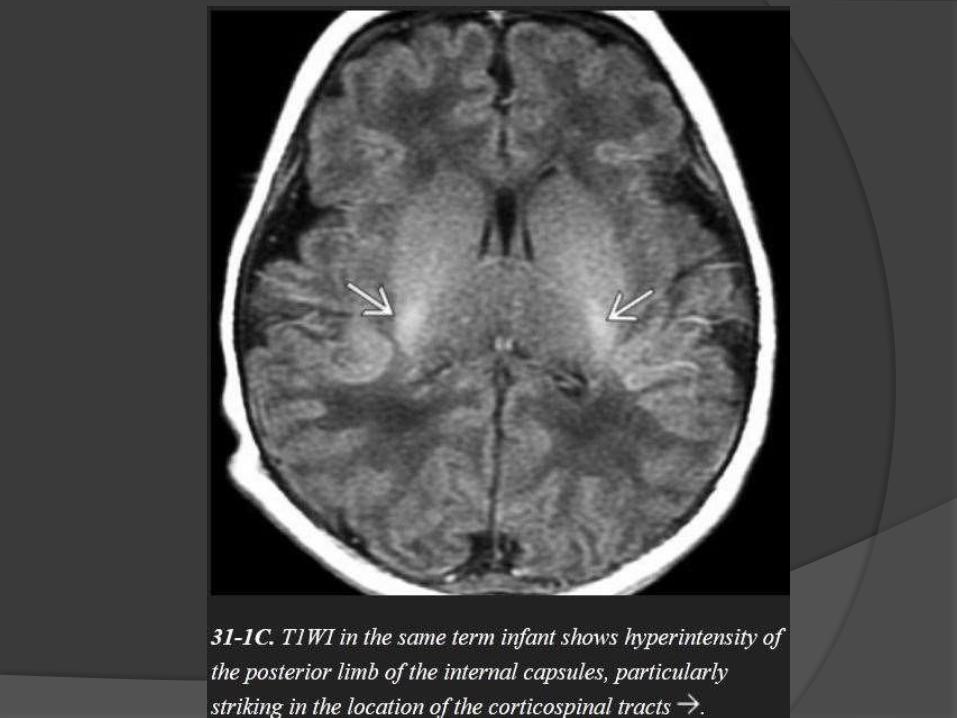
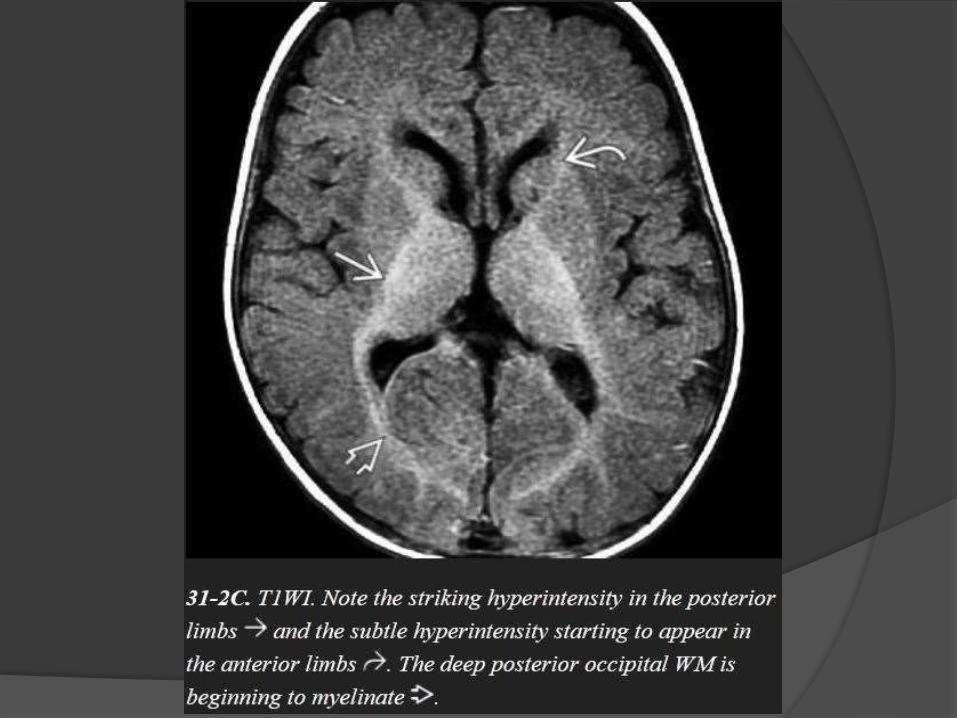
Posterior limb of internal capsule.
Ventrolateral thalamus.
One month
Deep cerebellar white matter.
Corticospinal tract.
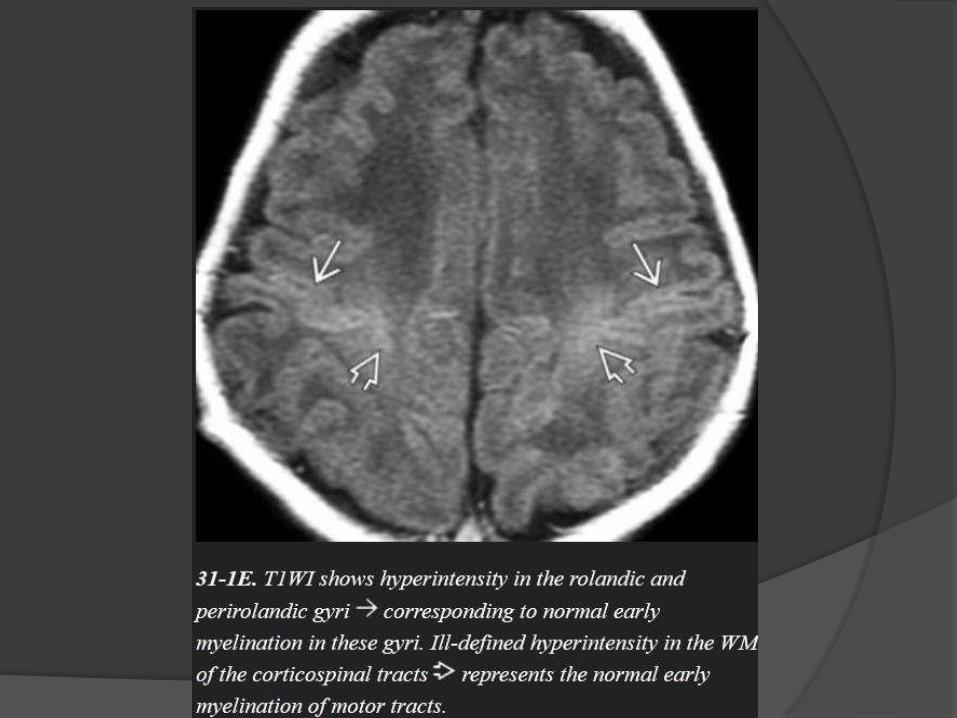
Pre / post central gyri.
Optic nerve / tracts.

Three month
Cerebellar folia.
Ventral brainstem.
Optic radiation.
Anterior limb of internal capsule.
Occipital subcortical U fibers.
Corpus callosum splenium.
Six month
Corpus callosum genu.
Paracentral subcortical U fibers.
Centrum semioval ( Partial ).

Eight month
Centrum semiovale ( complete except
frontoteporal area ).
Subcortical U fibers ( complete except most
rostral frontal area ).
Eighteen month
Essentially like adult.



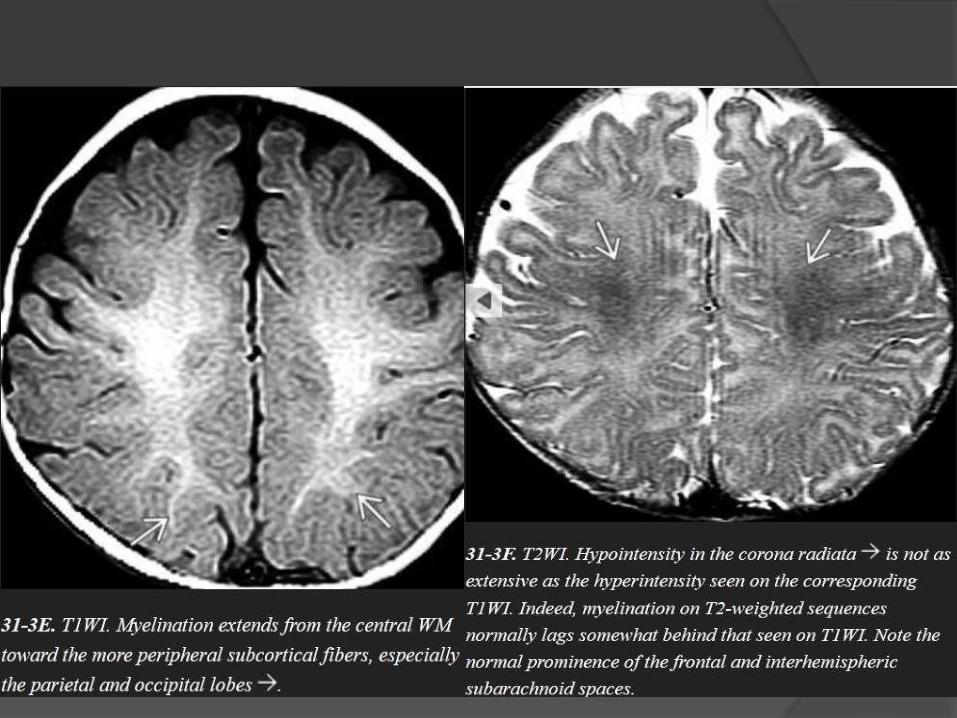
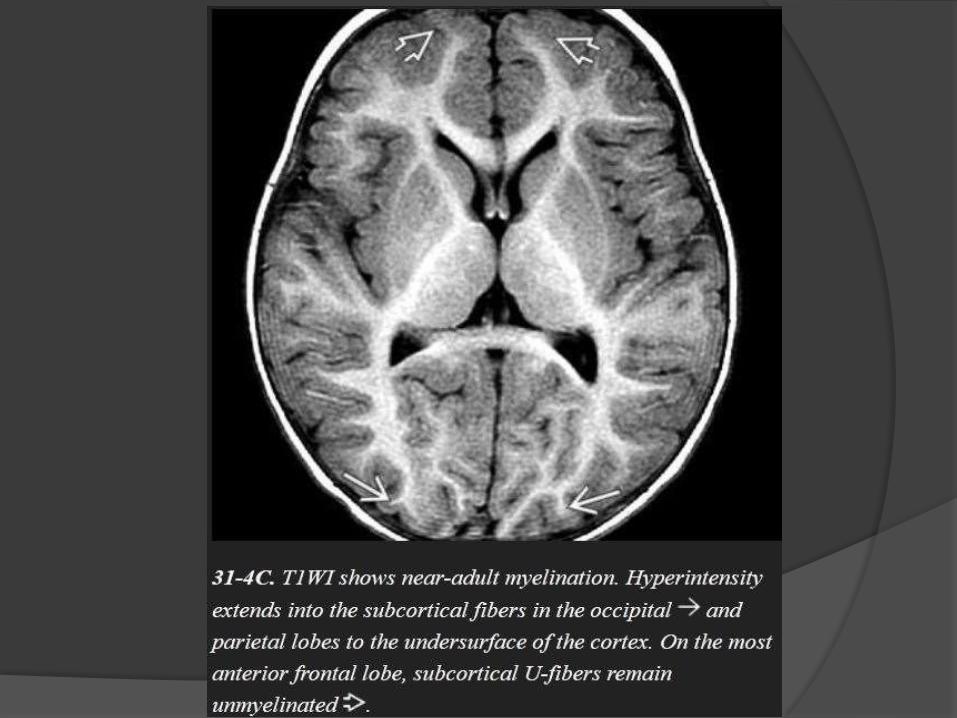

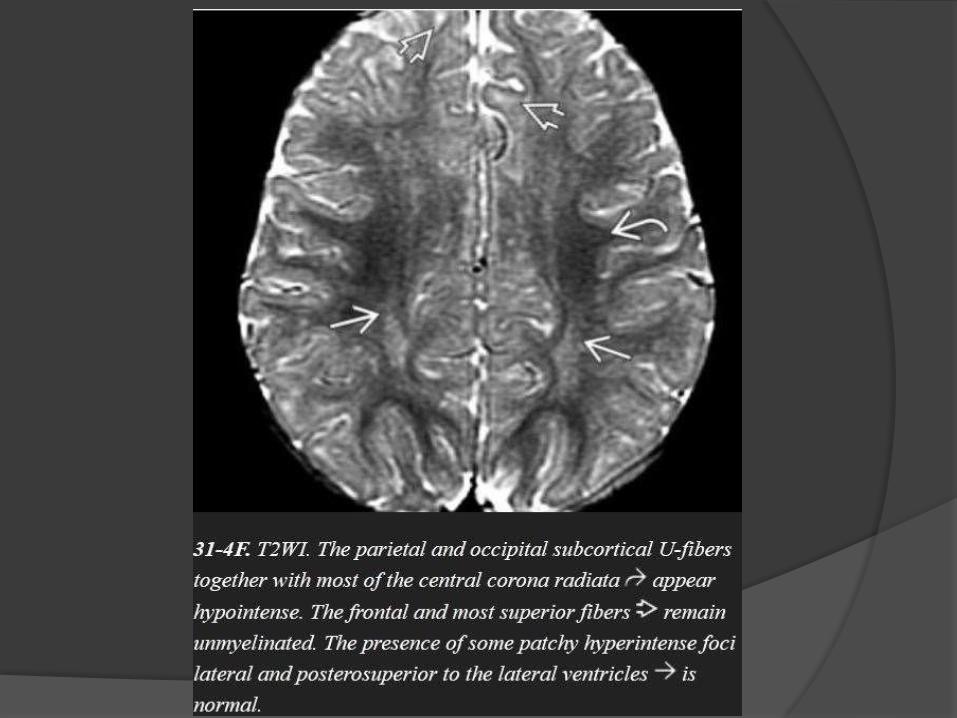

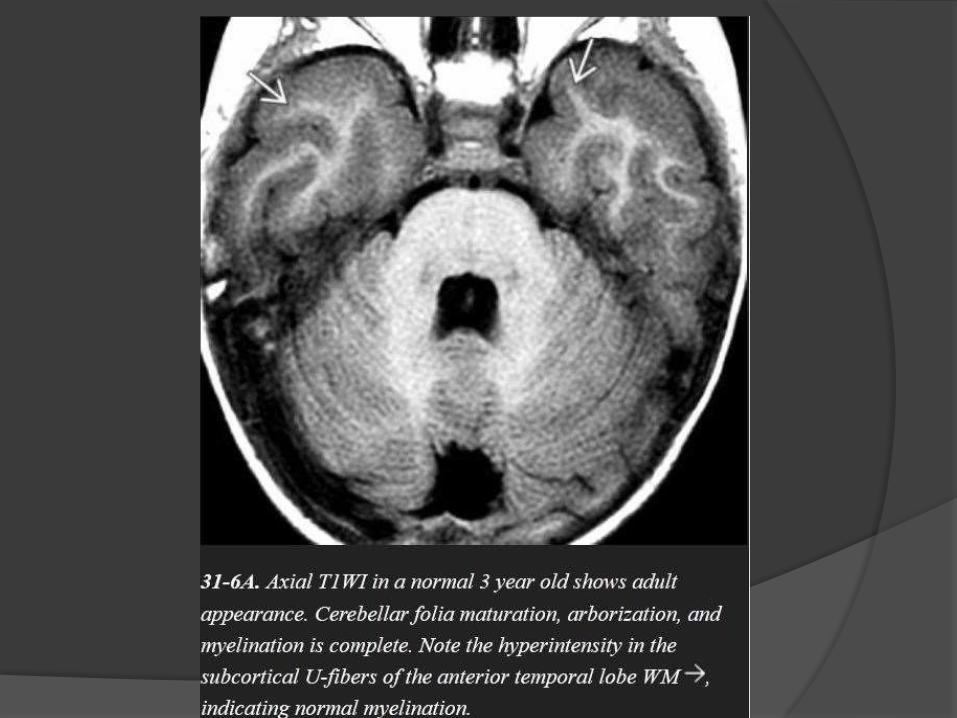
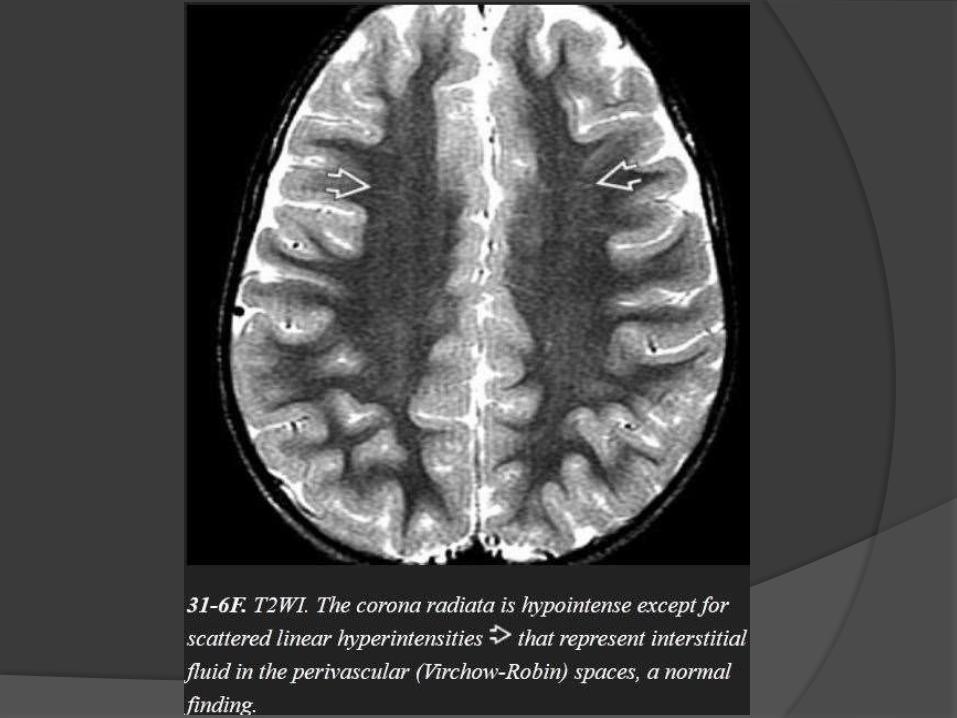

Myelination milestones after
birth
T1WI
3 m cerebellar white
matter
4 m corpus callosum
splenium
6 m corpus callosum
genu
T2WI
6 m corpus callosum
splenium
8 m corpus callosum
genu
11 m anterior limb
internal capsule
14 m frontal white
matter

White matter diseases are traditionally divided into two
categories:
• Dysmyelinating diseases
• Demyelinating diseases
• Hypomyelinating diseases
Dysmyelinating diseases - also known as leukodystrophies,
result from an inherited enzyme deficiency that causes
abnormal formation, destruction, or turnover of myelin.
Demyelinating diseases - involve destruction of intrinsically
normal myelin.
Hypomyelinating diseases - the WM may partially myelinate
but never myelinates completely).

Classification of Leukodystrophies on the Basis of Organelle Disorder


Metachromatic Leukodystrophy
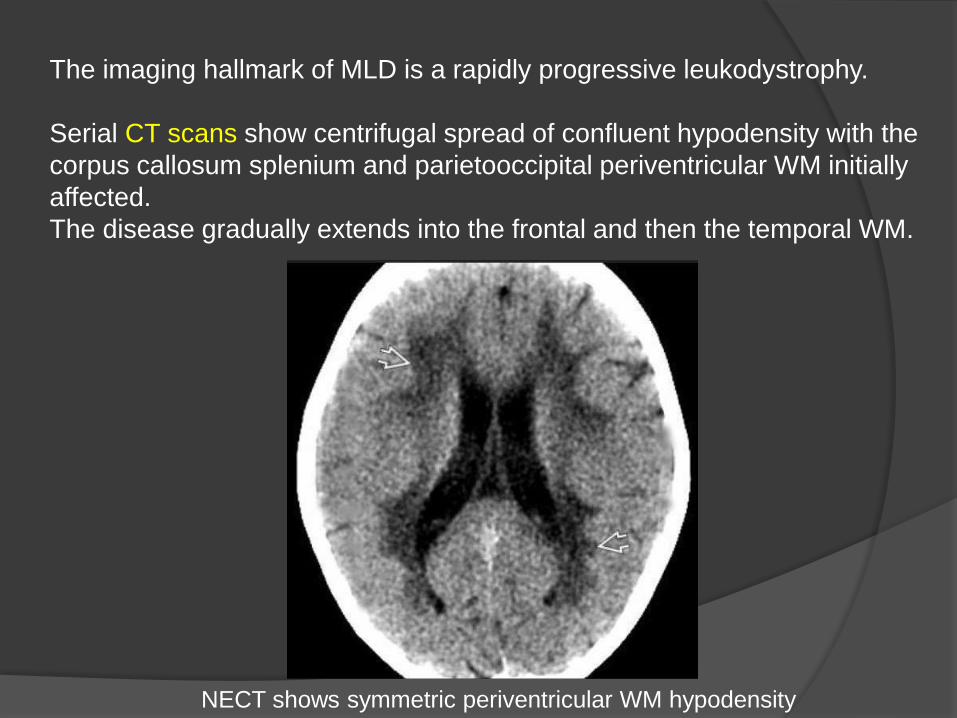
The imaging hallmark of MLD is a rapidly progressive leukodystrophy.
Serial CT scans show centrifugal spread of confluent hypodensity with the
corpus callosum splenium and parietooccipital periventricular WM initially
affected.
The disease gradually extends into the frontal and then the temporal WM.
NECT shows symmetric periventricular WM hypodensity

The typical MR appearance is confluent, symmetric, butterfly-shaped
T2/FLAIR hyperintensity in the periventricular WM.
The subcortical U-fibers and cerebellum are typically spared until late in the
disease.
Islands of normal myelin around medullary veins in the WM may produce a
striking "tiger" or "leopard" pattern with linear hypointensities in a sea of
confluent hyperintensity. No enhancement is seen on T1 C+.
A few cases of MLD have been reported with enlarged, enhancing cranial
nerves and/or cauda equina nerve roots.
Restricted diffusion is common.
MRS typically shows elevated choline with variable increase in myoinositol.

Metachromatic Leukodystrophy

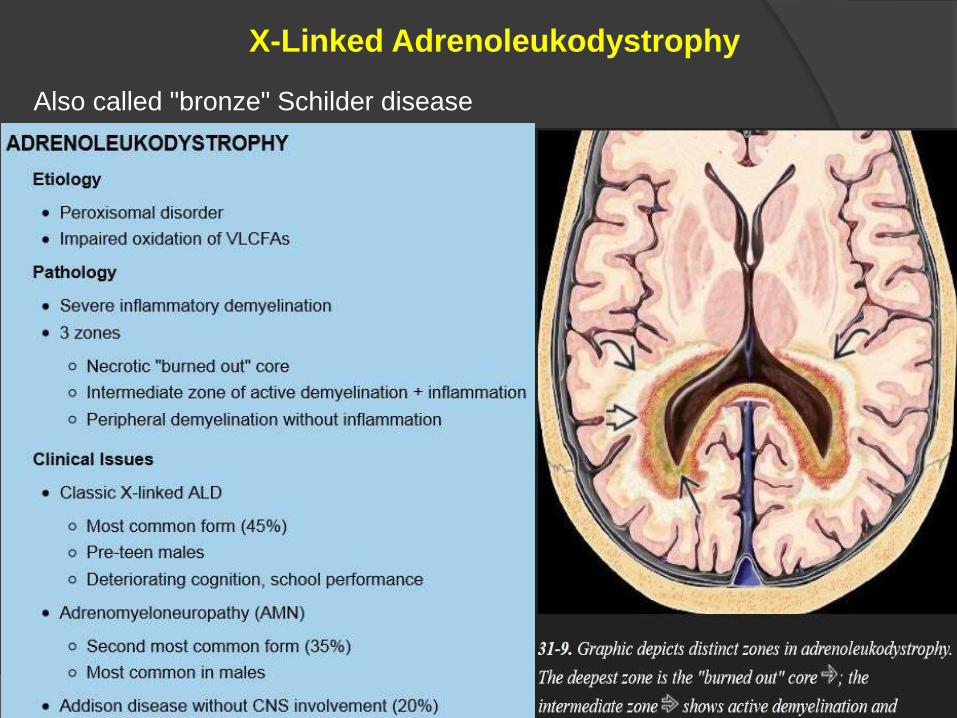
X-Linked Adrenoleukodystrophy
Also called "bronze" Schilder disease
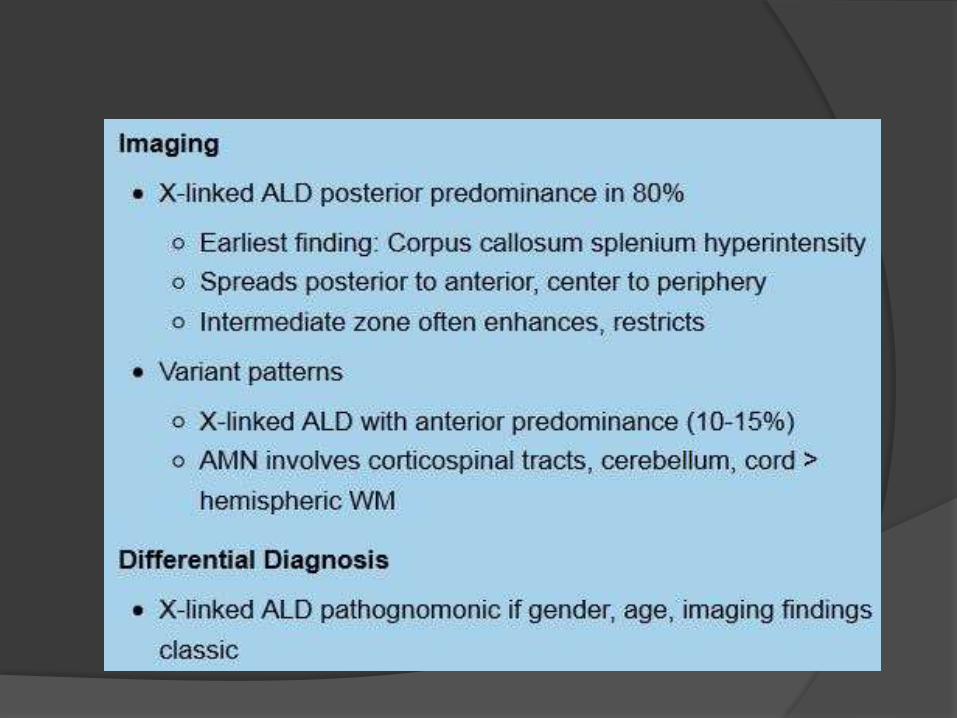
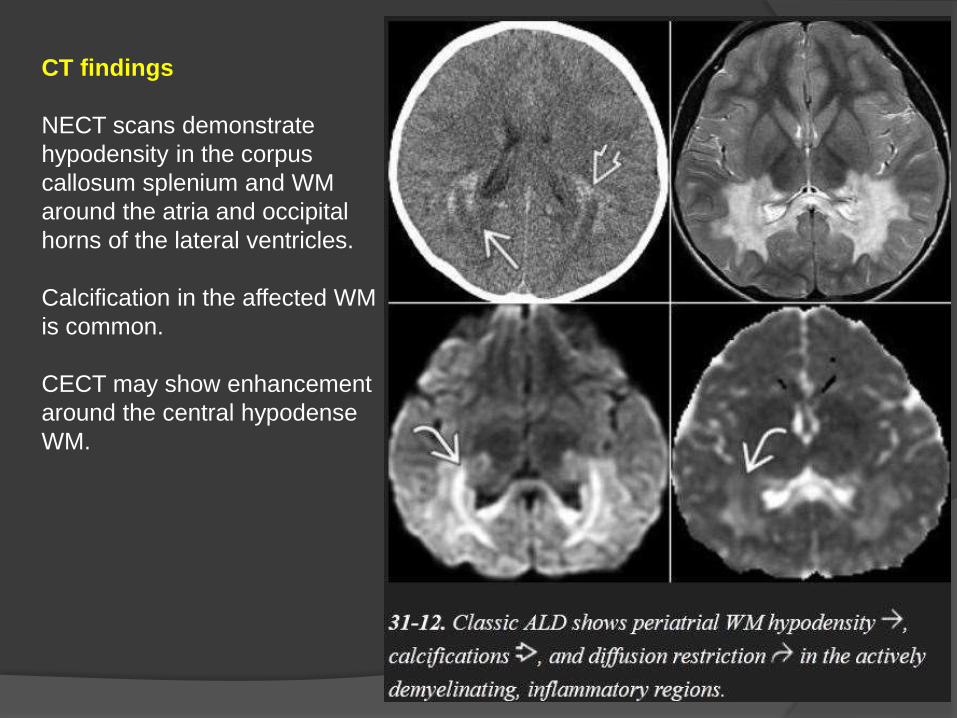
CT findings
NECT scans demonstrate
hypodensity in the corpus
callosum splenium and WM
around the atria and occipital
horns of the lateral ventricles.
Calcification in the affected WM
is common.
CECT may show enhancement
around the central hypodense
WM.

MR findings
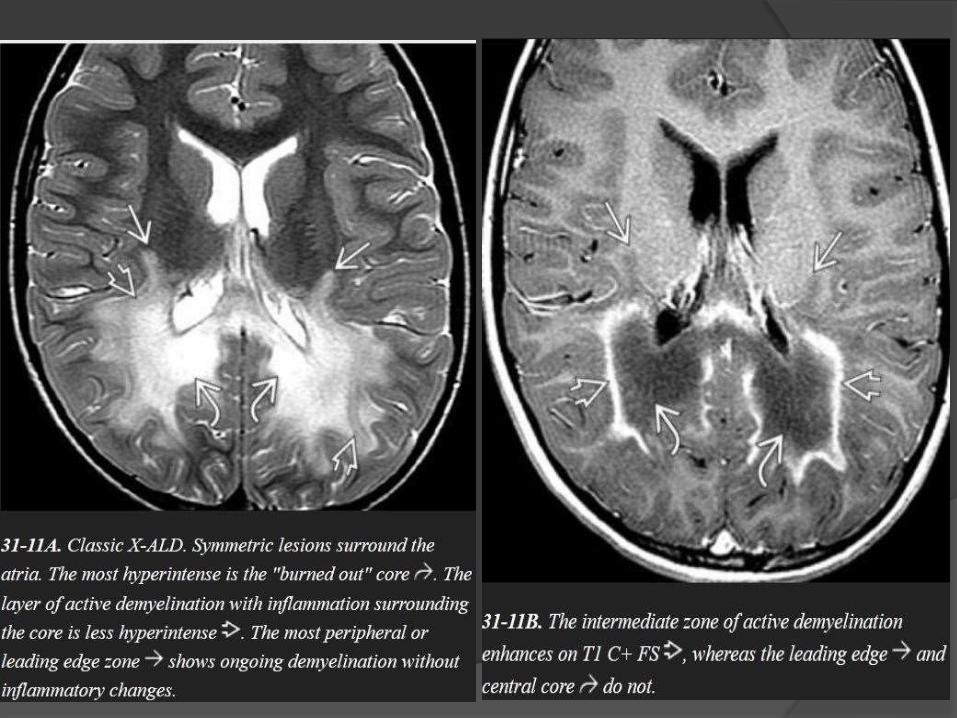
A posterior-predominant pattern is seen in 80% of patients with X-
ALD.
The earliest finding is T2/FLAIR hyperintensity in the middle of
the corpus callosum splenium.
As the disease progresses, hyperintensity spreads from posterior
to anterior and from the center to the periphery.
The peritrigonal WM, corticospinal tracts, fornix, commissural
fibers, plus the visual and auditory pathways can all eventually
become involved.

The leading edge of demyelination appears hyperintense on
T1WI but does not enhance.
The intermediate zone of active inflammatory demyelination
often enhances on T1 C+.
Diffusion restriction in the intermediate zone of inflammatory
demyelination may be present on DWI.
MRS shows decreased NAA even in normal-appearing WM.
Elevated choline, myoinositol, and lactate are common.
Globoid Cell Leukodystrophy (Krabbe Disease)
GLD is characterized by the presence of unique "globoid" cells in the demyelinating
lesions.
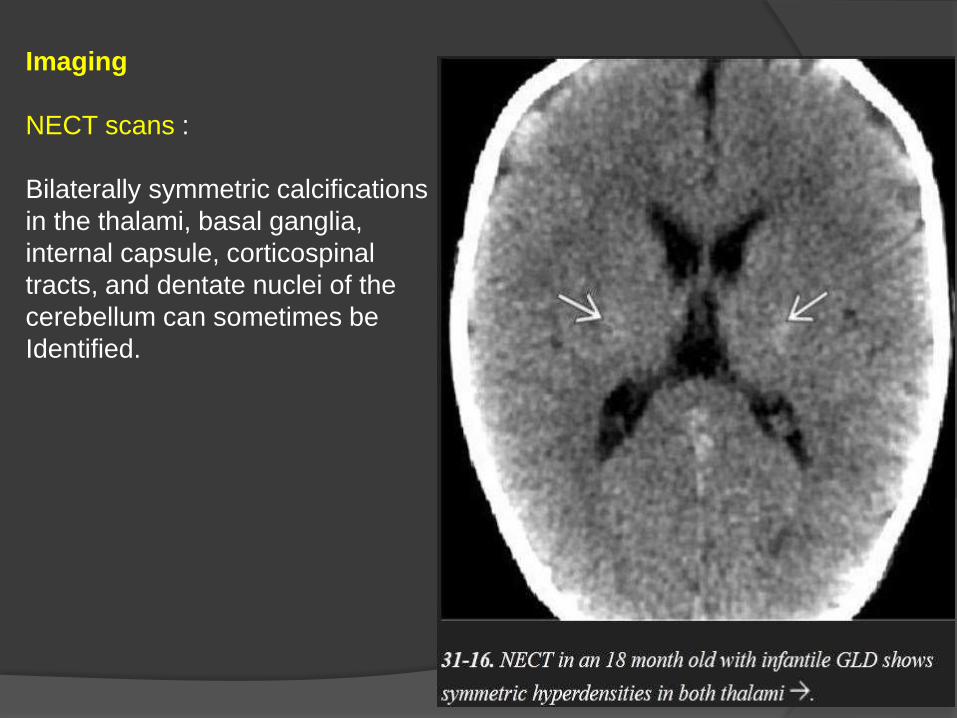
Imaging
NECT scans :
Bilaterally symmetric calcifications
in the thalami, basal ganglia,
internal capsule, corticospinal
tracts, and dentate nuclei of the
cerebellum can sometimes be
Identified.

Classic MR findings in GLD are corticospinal tract hyperintensity on T2/FLAIR
with confluent symmetric demyelination in the deep periventricular WM.
The subcortical U-fibers are typically spared. Bithalamic hypointensity on
T2WI is common.
Diffusion tensor imaging (DTI) may demonstrate reduced fractional anisotropy
in the corticospinal tracts before other abnormalities appear.
MRS findings of elevated choline and decreased NAA in the hemispheric WM
are characteristic but nonspecific.

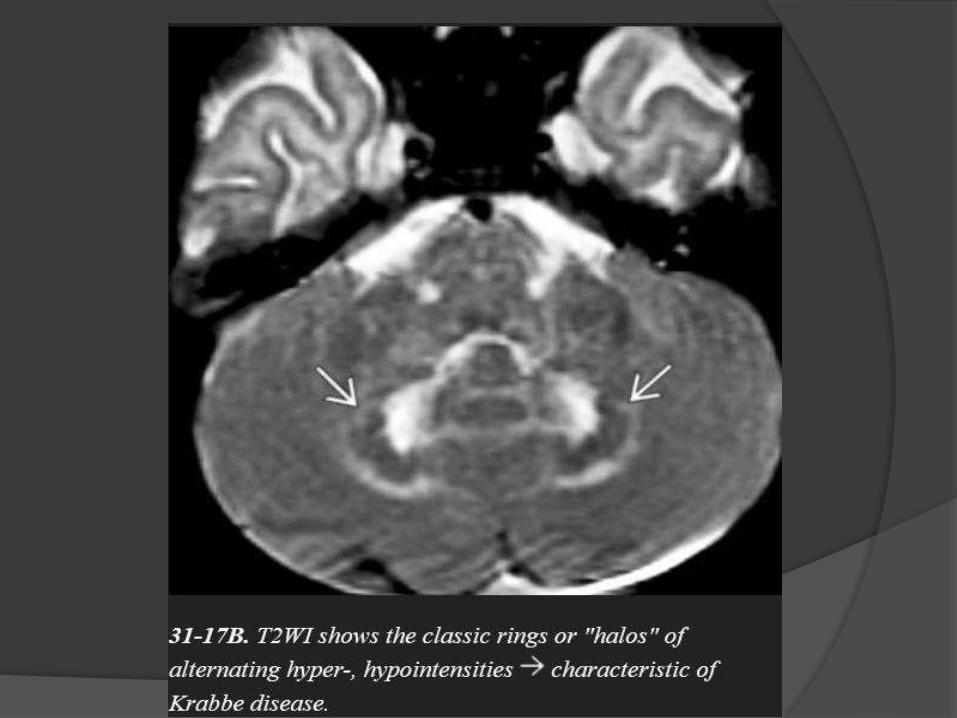
Cerebellar findings appear early in the disease course.
Alternating "halo" or ring-like hypointensities on T1WI and hyperintensities
on T2WI can be identified in the cerebellar WM surrounding the dentate
nuclei.
Another distinctive feature of GLD is enlargement of the intracranial optic
nerves and chiasm. Diffusely enlarged, enhancing cranial nerves and cauda
equina nerve roots have also been reported in GLD.
Differential Diagnosis
The WM changes in metachromatic leukodystrophy and vanishing white
matter disease may initially appear quite similar, but these disorders lack
the basal ganglia/thalamic calcifications typical of GLD.
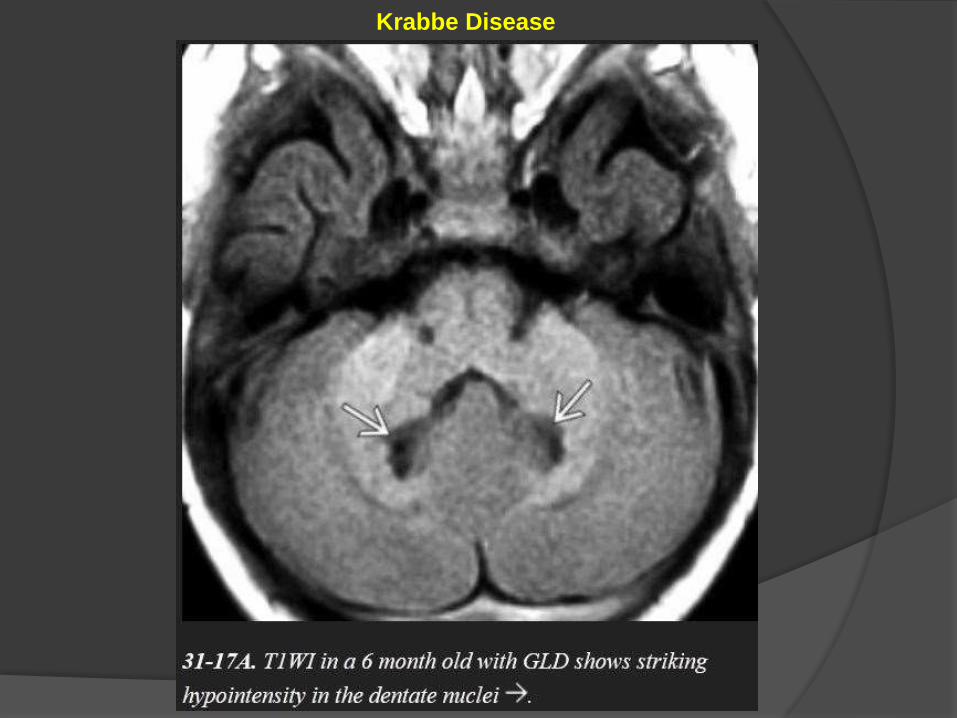
Krabbe Disease

Vanishing White Matter Disease
Characterized by diffusely abnormal white matter that literally "vanishes"
over time.
The deep frontoparietal WM is most severely affected with relatively lesser
involvement of the temporal lobes.
Classic VWM presents in children two to five years of age.
IMAGING
Extensive confluent WM T1 hypointensity with T2/FLAIR hyperintensity is
typical.

The disease is initially periventricular but later spreads to involve the
subcortical arcuate fibers. Over time, the affected WM undergoes
rarefaction.
Cavitary foci of CSF-like signal intensity may develop.
Diffuse volume loss with enlarged ventricles and sulci is seen on serial
studies.
VWM does not enhance.


Maple Syrup Urine Disease
Symptoms usually develop within a few days after birth and
include poor feeding, lethargy, vomiting, and seizures.
In severe cases, the urine smells like maple syrup or burnt
sugar.
NECT scans
show profound hypodensity in the myelinated WM with
vasogenic edema in the (Areas of early myelination) dorsal
brainstem, cerebellum, cerebral peduncles, and posterior limb
of the internal capsule.
MR scans show striking T2/FLAIR hyperintensity with relatively
crisp margins.
DWI shows restricted diffusivity
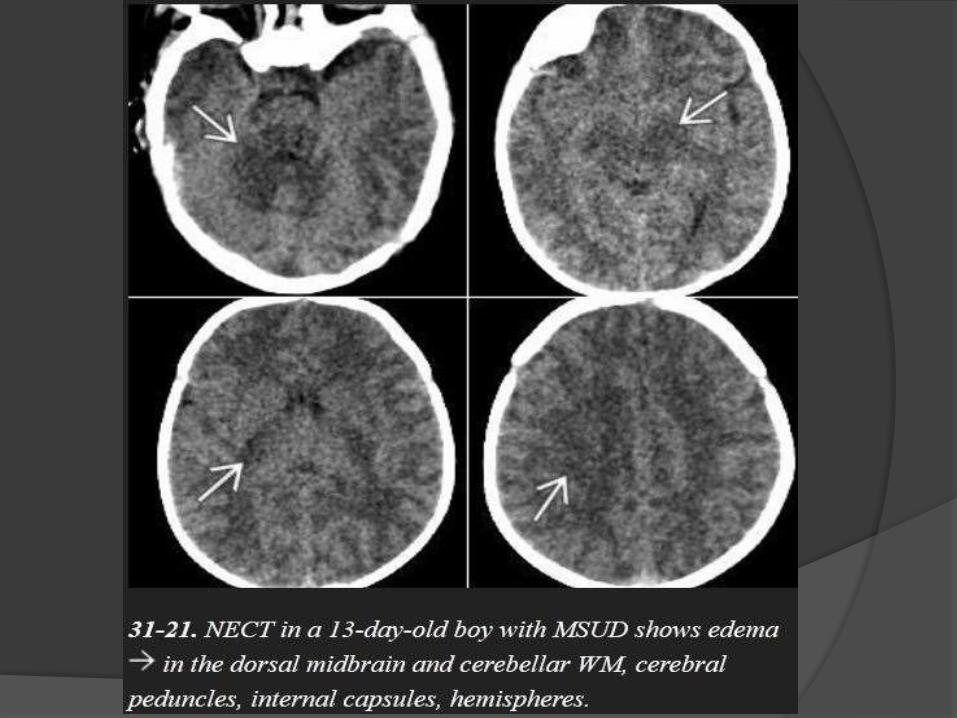
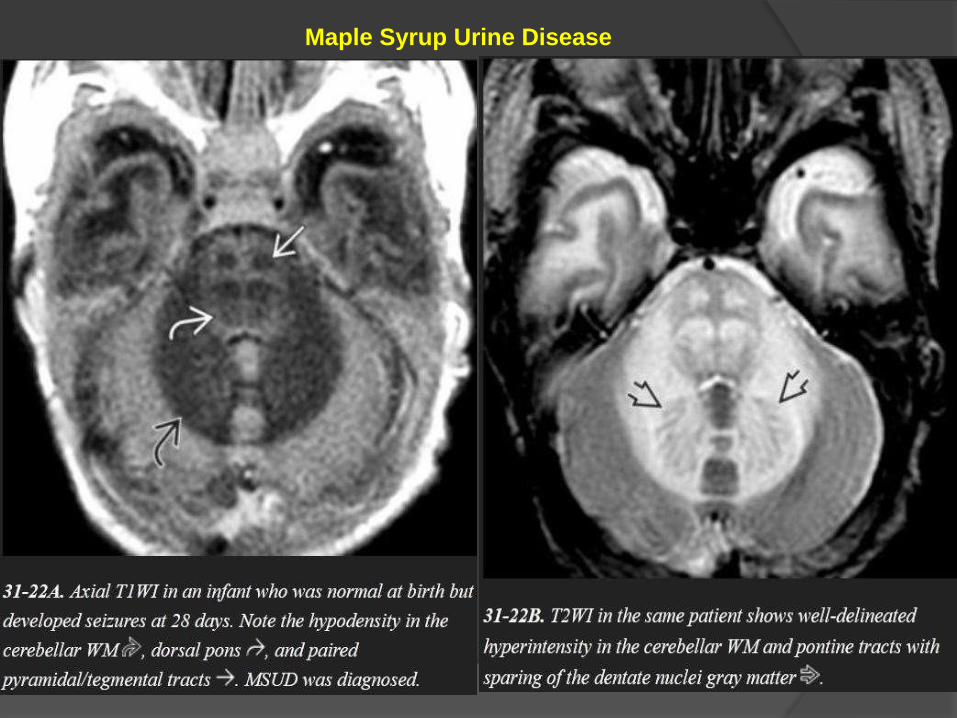
Maple Syrup Urine Disease

Maple Syrup Urine Disease
Subcortical White Matter Predominance
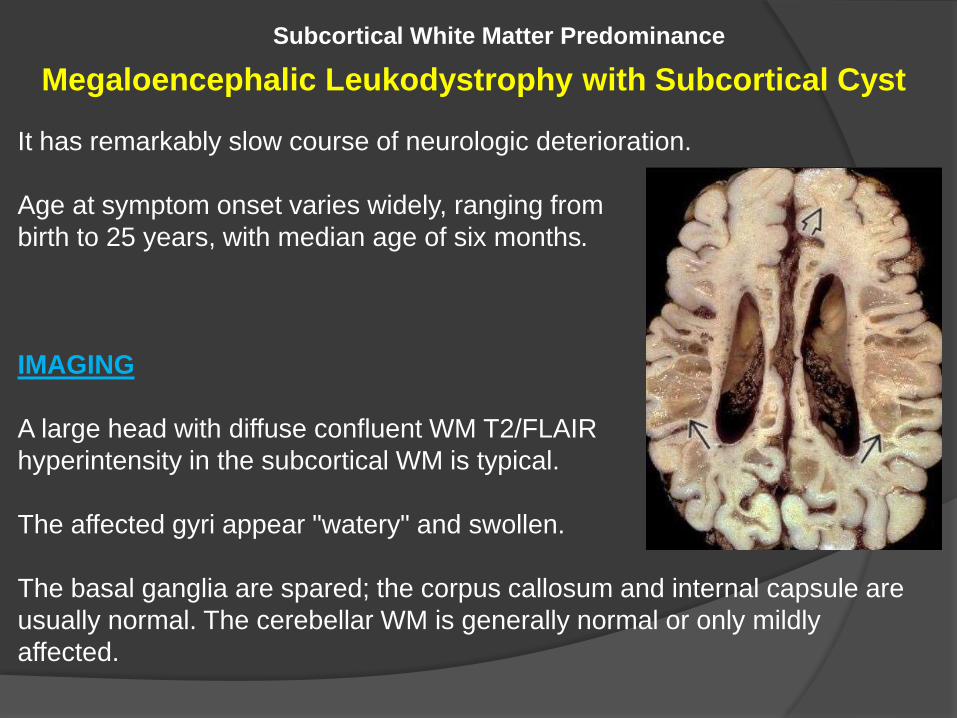
Megaloencephalic Leukodystrophy with Subcortical Cyst
It has remarkably slow course of neurologic deterioration.
Age at symptom onset varies widely, ranging from
birth to 25 years, with median age of six months.
IMAGING
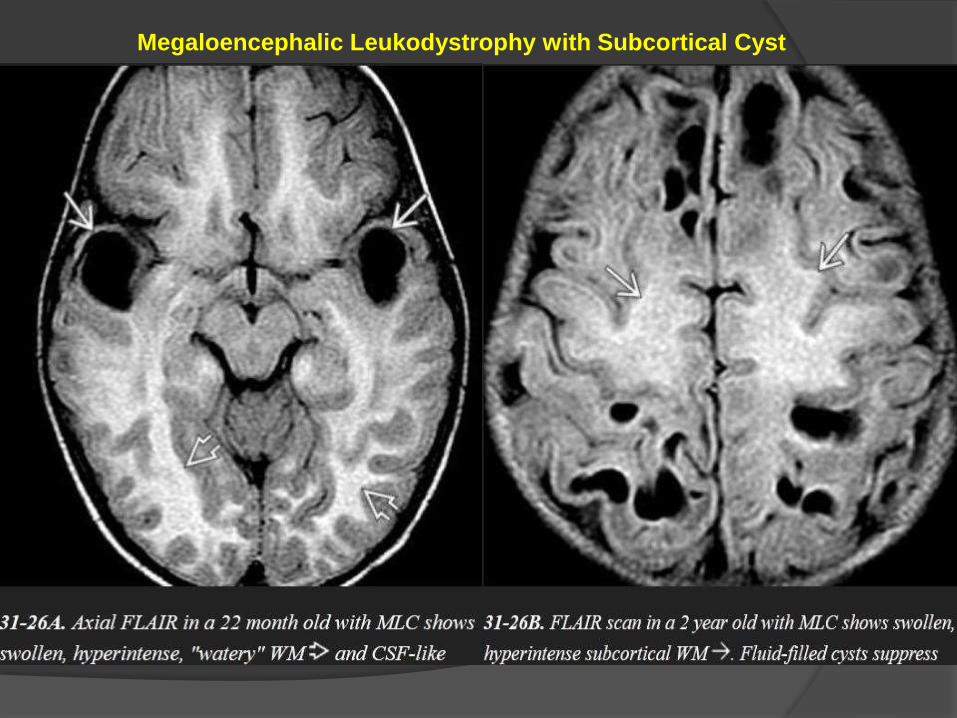
A large head with diffuse confluent WM T2/FLAIR
hyperintensity in the subcortical WM is typical.
The affected gyri appear "watery" and swollen.
The basal ganglia are spared; the corpus callosum and internal capsule are
usually normal. The cerebellar WM is generally normal or only mildly
affected.

Characteristic CSF-like subcortical cysts develop in the anterior temporal
lobes and then appear in the frontoparietal lobes.
Unlike the "watery" WM, the cysts suppress completely on FLAIR. The
number and size of the cysts may increase over time.
The abnormal WM and cysts do not enhance on T1 C+.
MRS shows mild to moderately decreased NAA and reduced NAA:Cr ratio.
Megaloencephalic Leukodystrophy with Subcortical Cyst

Hypomyelinating Disorders
Pelizaeus-Merzbacher Disease
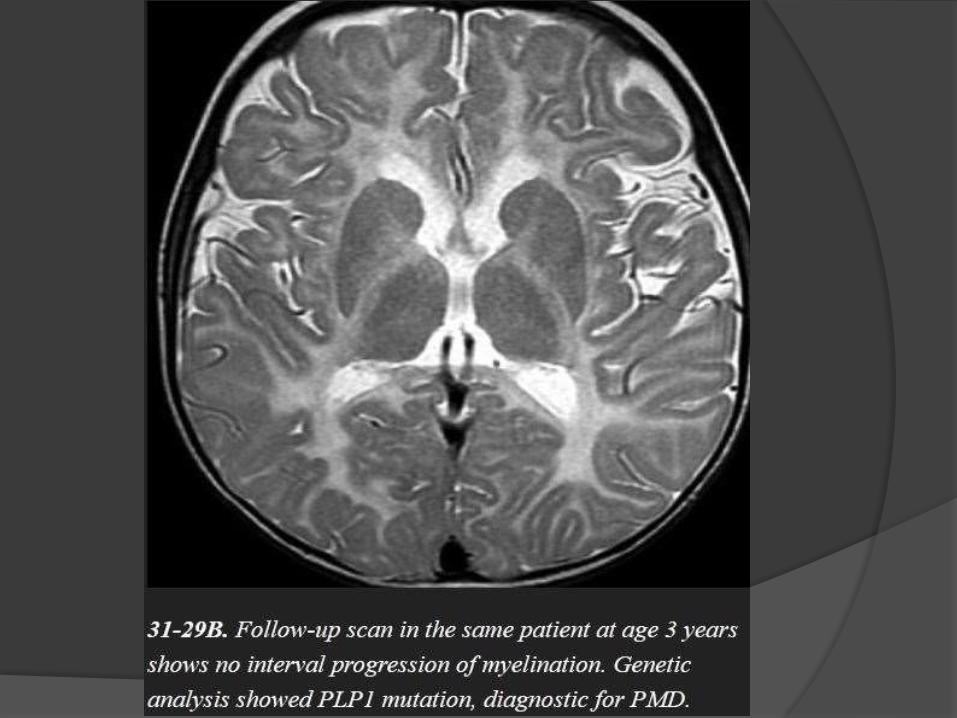
X-linked disorder that results in nearly complete lack of myelination.
One of the most common hypomyelinating disorders.
100% of classic PMD cases occur in males.
PMD is typically identified in infants under one year of age.
Imaging
The typical imaging appearance of PMD is nearly complete lack of
myelination.
The entire cerebral WM appears strikingly and homogeneously
hyperintense on T2WI.

Preserved myelin around perivascular spaces gives the WM a "tiger"
pattern.
Hyperintensity of the pyramidal tracts or entire pons is typically present.
Progressive WM and cerebellar volume loss are common.

Disorders Affecting Both Gray and White Matter
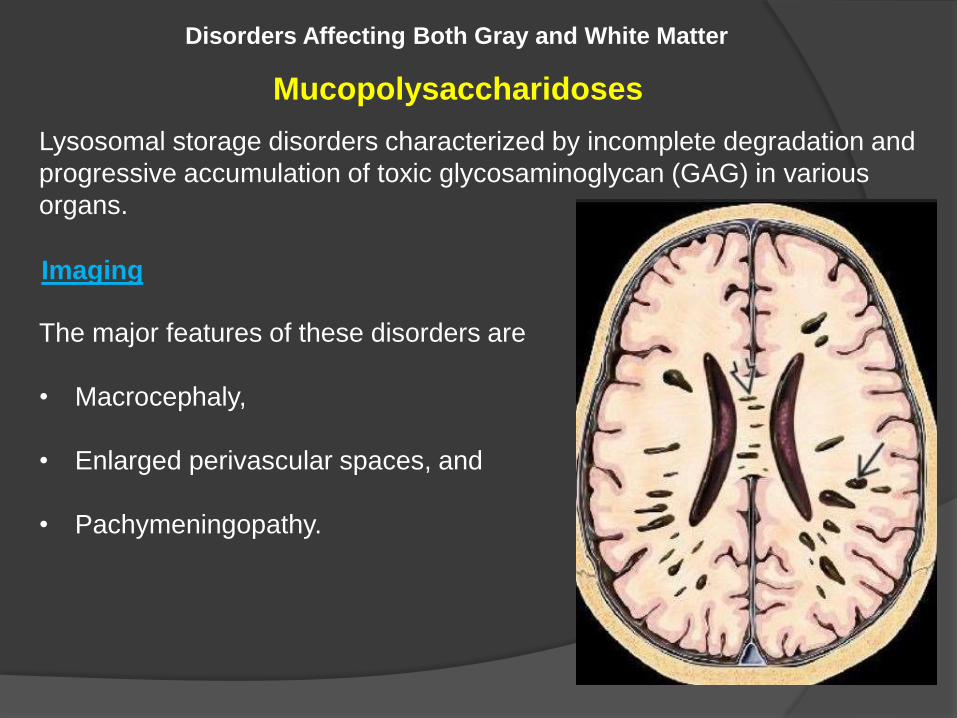
Mucopolysaccharidoses
Lysosomal storage disorders characterized by incomplete degradation and
progressive accumulation of toxic glycosaminoglycan (GAG) in various
organs.
The major features of these disorders are
• Macrocephaly,
• Enlarged perivascular spaces, and
• Pachymeningopathy.
Imaging

Macrocephaly
NECT scans show an enlarged head, often with metopic "beaking".
Progressive hydrocephalus and atrophy can be present.
Enlarged Perivascular Spaces
A striking sieve-like cribriform appearance in the posterior cerebral WM
and corpus callosum is characteristic and is caused by numerous dilated
PVSs (peri-vascular spaces)- also called as "Hurler holes," these
enlarged PVSs are typical of both Hurler and Hunter diseases. They are
much less common in the other MPSs.
NECT scans may show decreased density with multifocal CSF-like
hypodensities in the WM and basal ganglia.

T2 scans show CSF-like hyperintensity in the enlarged PVSs. The
surrounding WM may show patchy or confluent hyperintensity. The PVSs
themselves suppress completely on FLAIR.
The enlarged PVSs do not "bloom" on T2* and do not enhance following
contrast administration.
Pachymeningopathy
Thickened meninges can compress the medulla or upper cervical cord.
Odontoid dysplasia and a short C1 posterior arch—common in the MPSs—
can exacerbate the craniovertebral junction stenosis, causing progressive
myelopathy.
A lumbar gibbus with a "beaked" L1 vertebral body is common in Hurler
disease.
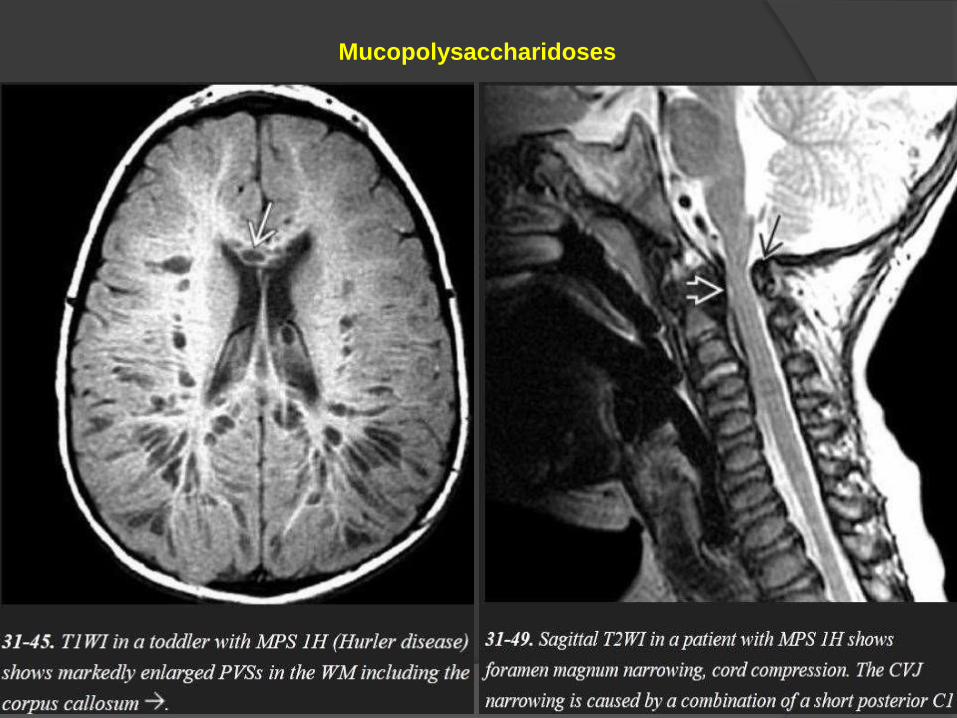
Mucopolysaccharidoses


MR shows virtually complete absence of myelination with confluent
T2/FLAIR hyperintensity throughout the WM and globi pallidi.
Early in the disease course, the subcortical arcuate fibers are initially
affected and the gyri may appear swollen.
As the disease progresses, diffuse volume loss with ventricular and sulcal
enlargement ensues.
The hemispheric and cerebellar WM, basal ganglia, and cortex are all
affected.
MRS - is the key to the definitive diagnosis of CD.
Markedly elevated NAA is seen in virtually all cases.
Cr is reduced. An elevated myoisitol peak is sometimes present.

Canavan Disease
Canavan disease is the only identified genetic disorder caused by a defect in
a metabolite— N-acetyl-L-aspartate (NAA) —that is produced exclusively in
the brain.
The most common form by far is infantile CD. Infantile CD presents between
three and six months and is characterized by hypotonia, macrocephaly, and
seizures.
Death between one or two years is typical.
Imaging
NECT shows a large head with diffuse WM hypodensity in the cerebral
hemispheres and cerebellum.
The globi pallidi also appear hypodense. CD does not enhance.

Canavan Disease

Canavan Disease

Alexander Disease
The infantile form, which is the most common,
patients younger than two years present with megalencephaly, progressive
psychomotor retardation, and seizures.
Imaging
NECT scans of infants with AxD show a large head with symmetric WM
hypodensity in the frontal lobes that extends posteriorly into the caudate
nuclei and internal/external capsules.
Intense bifrontal periventricular enhancement can be seen on CECT scans
early in the disease course.
MR shows T1 hypointensity and T2/FLAIR hyperintensity in the frontal WM,
caudate nuclei, and anterior putamina.
Subcortical U-fibers are involved early in the disease course.

A classic finding is a T1 hypointense, T2 hyperintense rim around the
frontal horns.
FLAIR scans may demonstrate cystic encephalomalacia in the frontal
WM in more severe, protracted cases.
Enlargement of the caudate heads and fornices, which appear swollen
and hyperintense.
The thalami, globi pallidi, brainstem, and cerebellum are less commonly
affected.
MRS shows decreased NAA, elevated myoinositol, and variably
increased choline and lactate.
DWI shows normal to increased diffusivity in the affected WM.

Alexander Disease


Zellweger syndrome spectrum (ZSS)
Imaging
ZSS is characterized by microgyria and pachygyria, often with bilaterally
symmetric parasylvian lesions.
Hypomyelinated WM is seen as confluent T2/FLAIR WM hyperintensity.
Subependymal (caudothalamic) germinolytic cysts are common findings.
Hyperbilirubinemia may cause increased T1 signal intensity in the globi
pallidi of older patients.
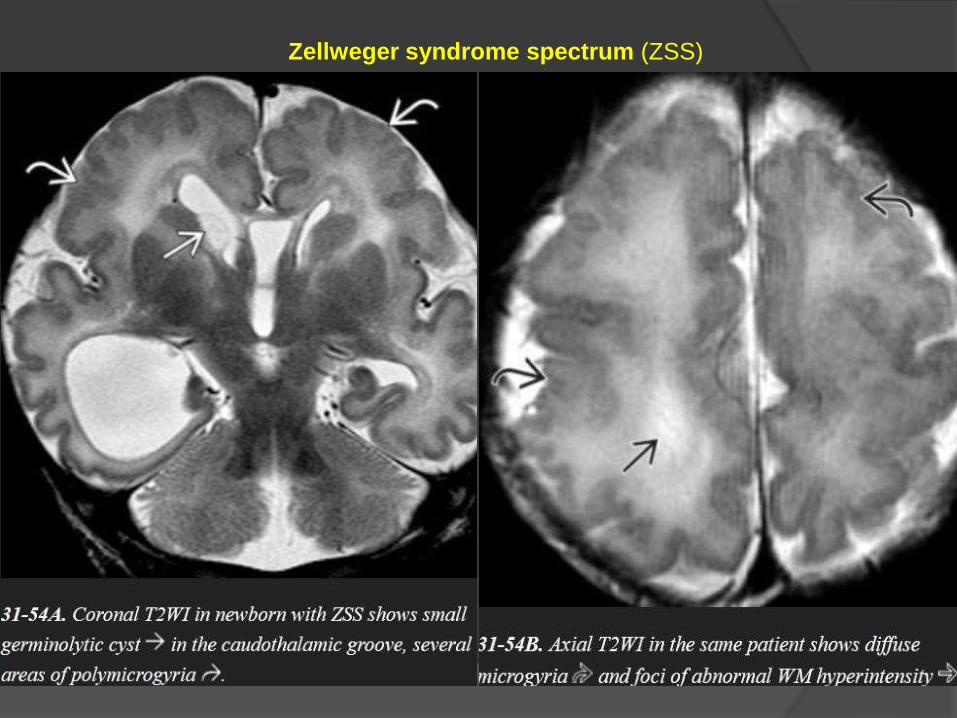
Zellweger syndrome spectrum (ZSS)

Leigh Disease
Serum and CSF lactate levels are elevated.
Imaging
Bilaterally symmetric areas of T2/FLAIR hyperintensity in the basal ganglia.
The putamina (especially the posterior segments) are consistently affected, as
are the caudate heads.
The dorsomedial thalami can also be involved whereas the globi pallidi are
less commonly affected.
Mid- and lower brainstem (pons/medulla) lesions are typical.
Leigh Disease

Symmetric lesions in the cerebral peduncles are common, and the
periaqueductal gray matter is frequently affected.
Acute lesions may restrict on DWI but do not enhance.
MRS of the brain parenchyma and CSF typically shows a prominent
lactate peak at 1.3 ppm.
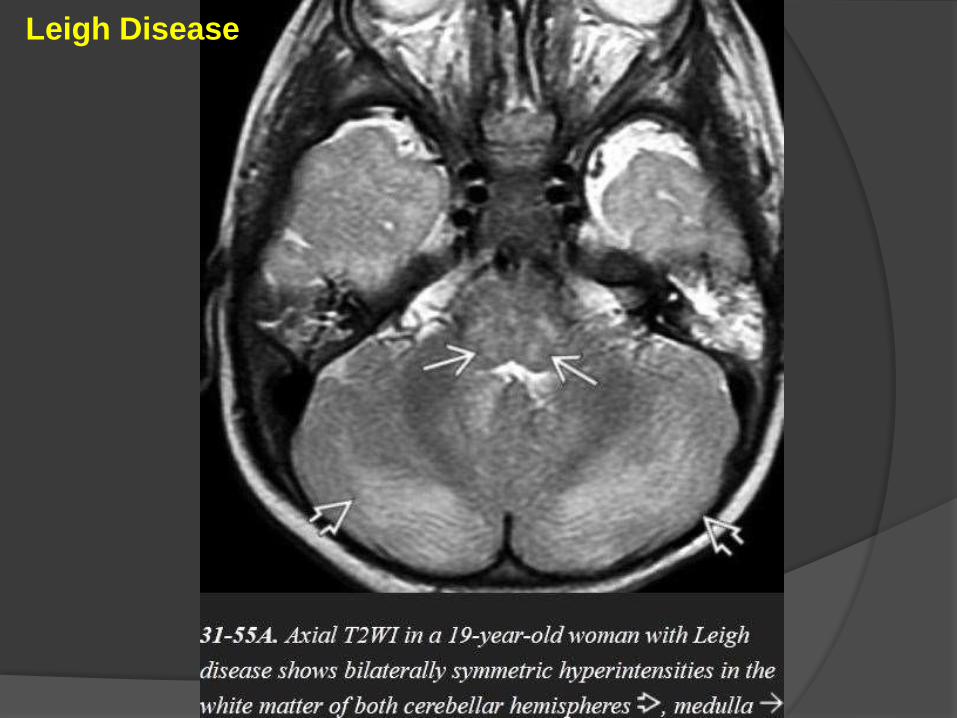
Leigh Disease

Leigh Disease

MELAS
Mitochondrial encephalomyopathy with lactic acidosis and stroke-like
episodes (MELAS)
The clinical triad of lactic acidosis, seizures, and stroke-like episodes is the
classic presentation.
Acute MELAS –
often shows swollen T2/FLAIR hyperintense gyri.
The underlying WM is normal
The cortical abnormalities cross vascular distribution territories,
distinguishing MELAS from acute cerebral infarction.
The parietal and occipital lobes are most commonly affected.
Gyral enhancement on T1 C+ is typical.
MRA shows no evidence of major vessel occlusion.

Chronic MELAS-
shows multifocal lacunar-type infarcts,
symmetric basal ganglia calcifications,
WM volume loss, and progressive
atrophy of the parietooccipital cortex.
MRS
Two-thirds of cases show a prominent lactate "doublet" at 1.3 ppm in
otherwise normal-appearing brain.
One-third of cases show no evidence for elevated lactate levels in the
brain parenchyma but may demonstrate a lactate peak in the ventricular
CSF.
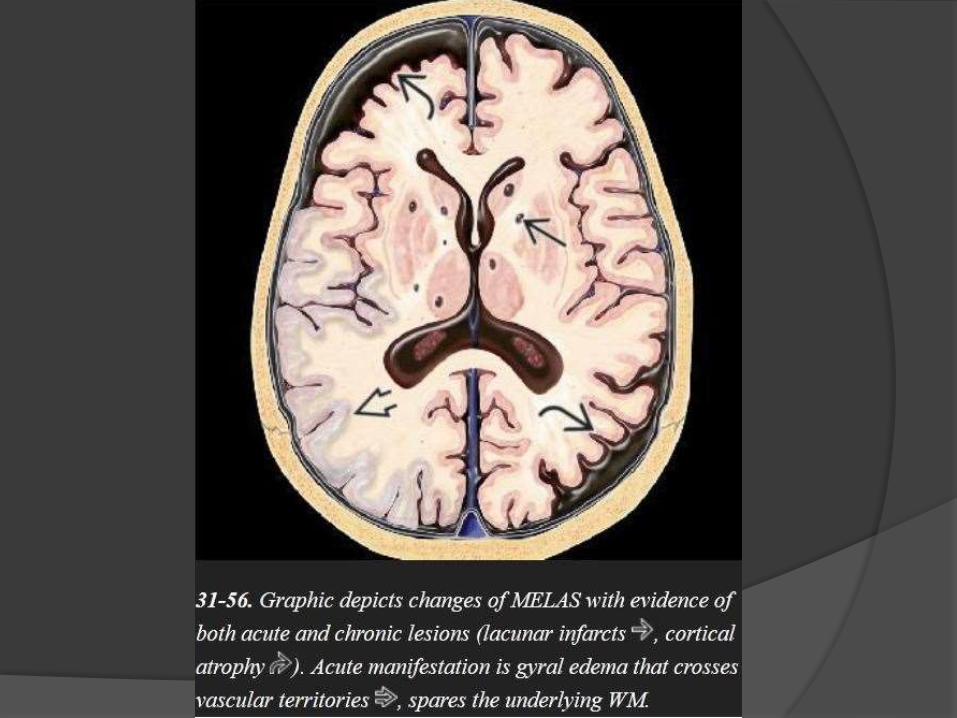

MELAS

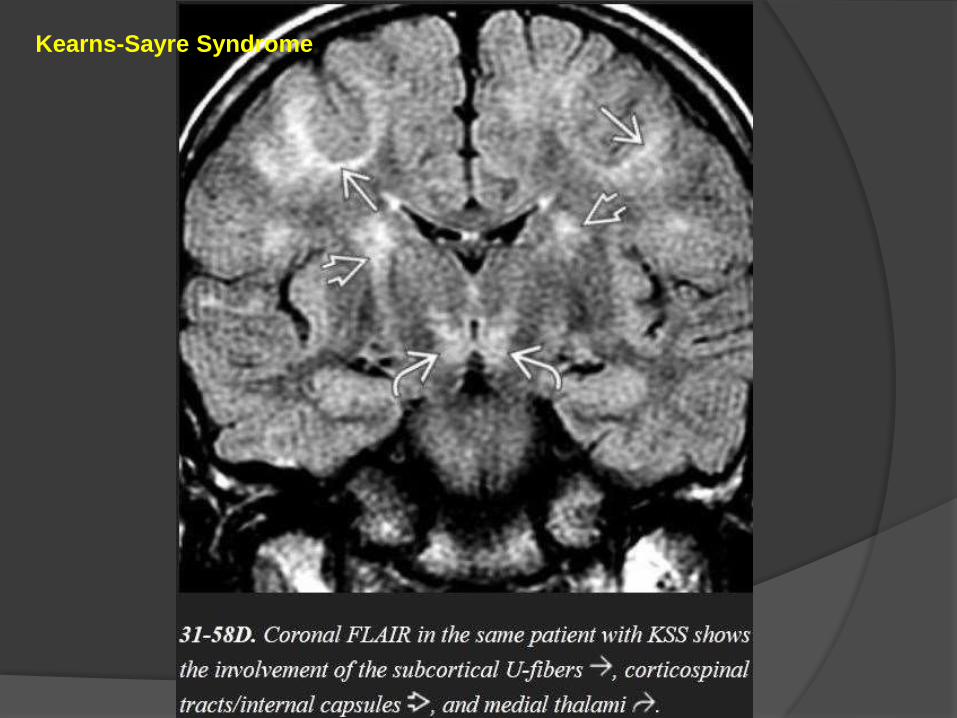
Kearns-Sayre Syndrome
KSS typically presents in older children or young adults and is characterized
by short stature, progressive external ophthalmoplegia, retinitis pigmentosa,
sensorineural hearing loss, and ataxia.
Imaging
CT scans show variable symmetric basal ganglia calcifications.
Mild cortical and cerebellar volume loss is common.
MR shows increased signal intensity in the basal ganglia, WM, and
cerebellum on T2/FLAIR.
The subcortical arcuate fibers, corticospinal tracts, cerebellum, and posterior
brainstem are involved early in the disease course while the periventricular
WM remains relatively spared.
DWI shows reduced diffusivity in the brainstem and subcortical WM.
MRS demonstrates elevated lactate.

Kearns-Sayre Syndrome

Kearns-Sayre Syndrome
Kearns-Sayre Syndrome

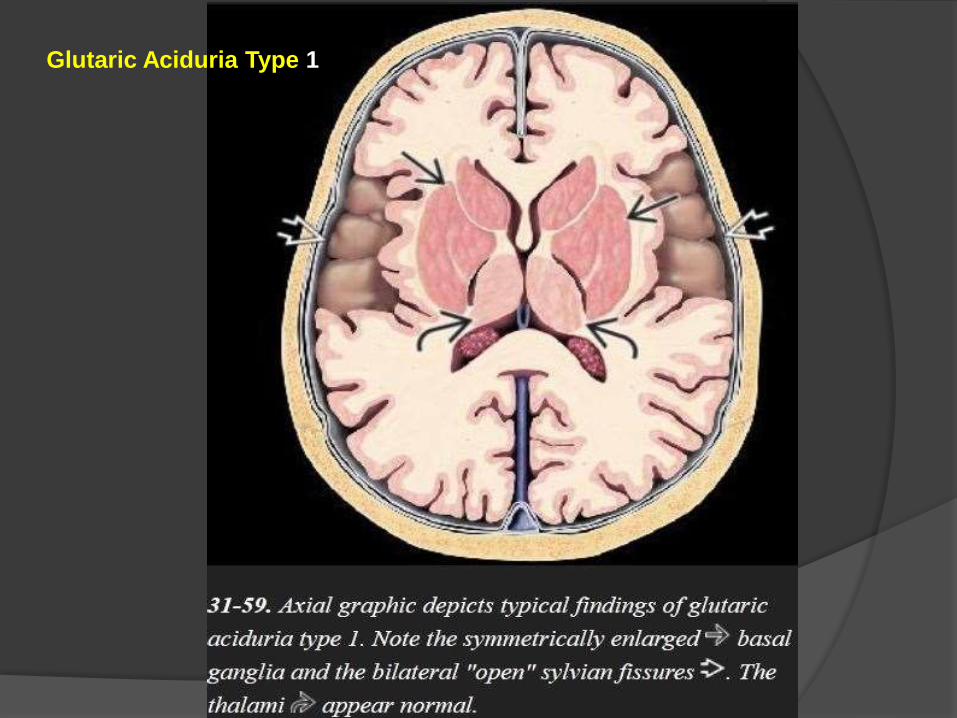
Glutaric Aciduria Type 1
Imaging
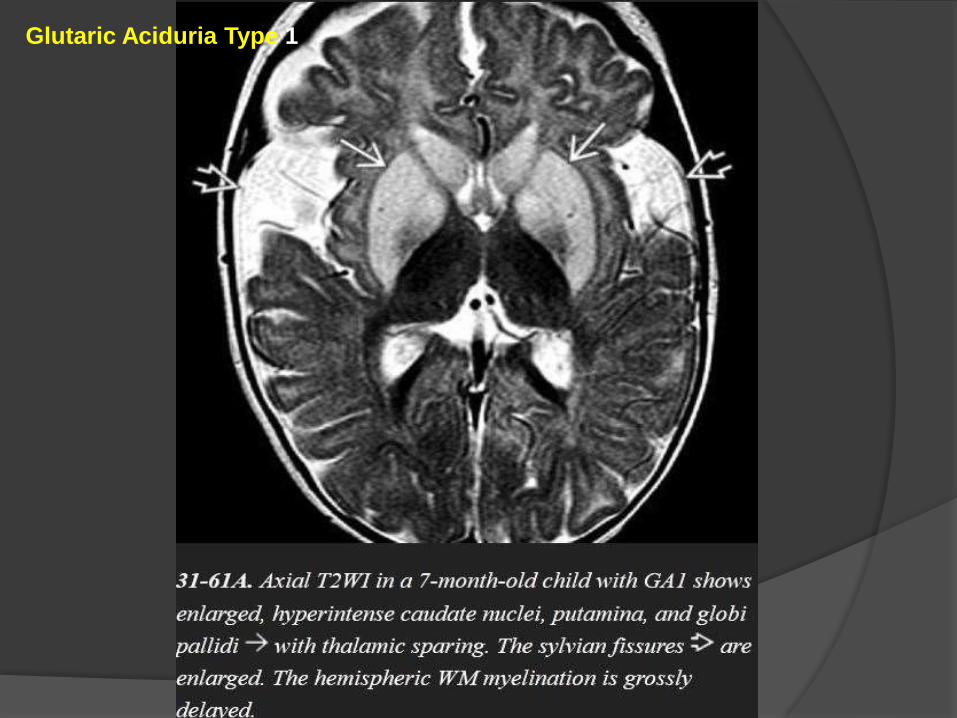
The three "signature" imaging findings of classic GA1 are
(1)macrocrania,
(2) bilateral widened ("open") sylvian fissures, and
(3) bilaterally symmetric basal ganglia lesions.
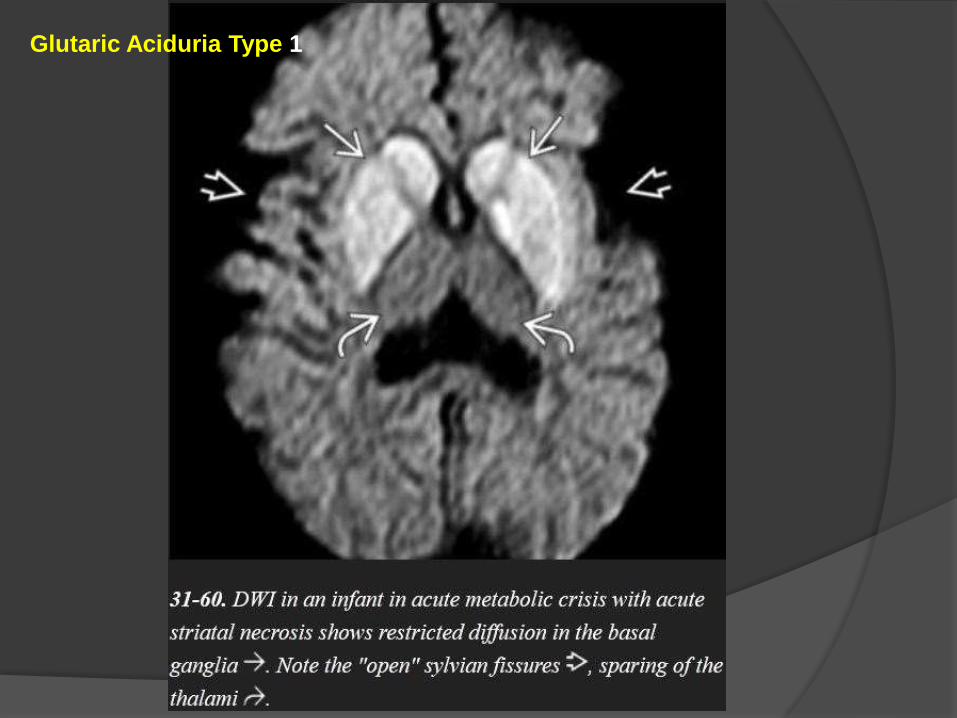
Severe GA1 may also cause diffuse hemispheric WM abnormalities.
GA1 infants in metabolic crisis often present with acute striatal necrosis.
Bilateral diffusely swollen basal ganglia that are T2/FLAIR hyperintense and
that restrict on DWI are typical.

Chronic GA1 causes enlarged CSF spaces and atrophy.
The volume loss may tear bridging veins that cross from the brain surface to
the dura, resulting in recurrent subdural hematomas.
GA1 does not enhance on T1 C+ scans.
MRS is nonspecific with decreased NAA, increased Cho:Cr ratio, and
(during crisis) elevated lactate level.
Glutaric aciduria type 2 (GA2)
Imaging studies show symmetric T2/FLAIR hyperintensity in the basal
ganglia and hemispheric WM, but the "open" sylvian fissures characteristic
of GA1 are absent.
Glutaric Aciduria Type 1
Glutaric Aciduria Type 1
Glutaric Aciduria Type 1

Glutaric Aciduria Type 1








