

Steeps needed to move forward in Spain
3rd European Symposium in rare anaemias
Dra. Ana VillegasPresidente grupo español estudio
Talasemias y hemoglobinopatiasEritropatología
19 Noviembre 2010

Steeps needed to move forward in Spain
• Estudios epidemiológicos• Diagnóstico de talasemias• Consejo genético• Diagnóstico prenatal• Manejo terapéutico de las talasemias

Talasemias • Disminución o ausencia de síntesis de
una o mas cadenas de globina
Hemoglobinopatias Estructurales• Síntesis de una cadena
estructuralmente anormal. Variantes de Hb normal
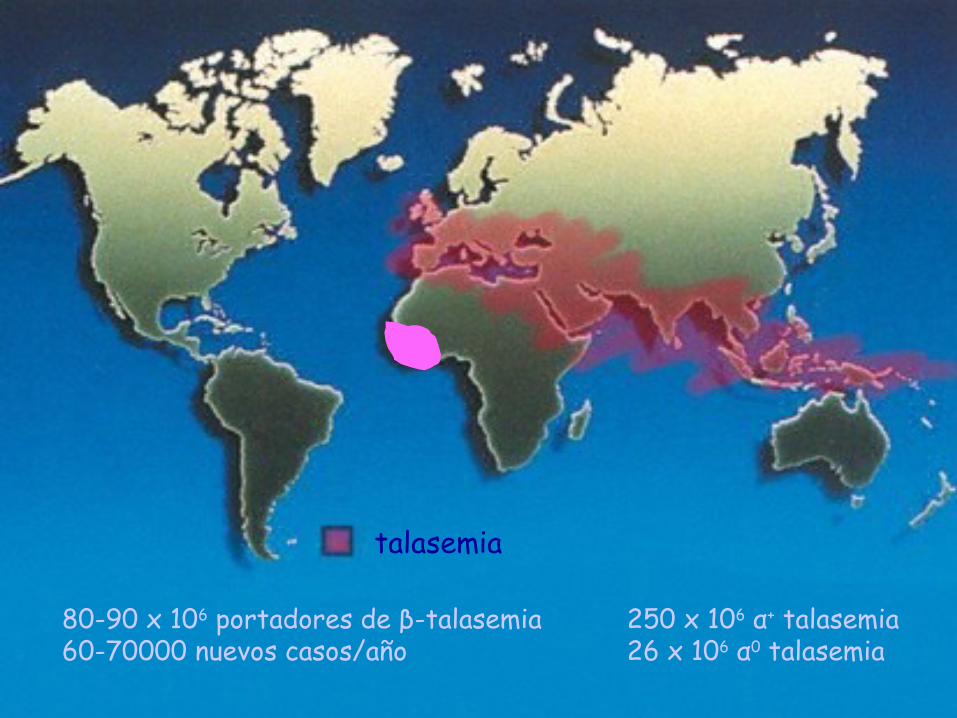
talasemia
80-90 x 106 portadores de β-talasemia60-70000 nuevos casos/año
250 x 106 α+ talasemia26 x 106 α0 talasemia

Datos epidemiológicos en EspañaTalasemias
• Incidencia β-Talasemia 0,2—2%• Incidencia α-Talasemia 4,79% *
α+ talasemia 4,49%α0 talasemia 0,29%ααα genes 0,72%
*determinado mediante biología molecular
Baiget M. Biol Clin Hematol 1981;251:8-10*Sanchez J Tesis Doctoral Madrid 1997
Martin Nuñez G Sangre 1995;40:459-64 Oliva E Med Clin 1998;110:361-4
*Villegas A Hematologica 1998;83:99-103

Datos epidemiológicos en EspañaHemoglobinopatias estructurales
• Incidencia 0,77% HPLC variant * 1,08% HPLC fase reversa (cadenas de globina)* 1,47% HPLC variant**
Población española no Hb S, C, D * Gonzalez AF Haematologica 2002;87:368-372 ** Mañu M Med Clin 2006;126(8)181-5
* Joyanes B Med Clin 2006;126(8)290-2

Talasemias
• α talasemia• β talasemia• δβ talasemia• γδβtalasemia• hemoglobinopatías estructurales con
fenotipo talasémico
• εγδβ talasemia• δ talasemia• γ talasemia

Talasemias
• Talasemia mayor • Talasemia minor • Talasemia intermedia• Talasemia silente

TIPOS DE α-TALASEMIAS Y BASE MOLECULAR
Tipo de lesión Genotipo Fenotipo
Deleción
Talasemia α+ heterocigota Talasemia α0 heterocigota Talasemia α+ homocigota Enfermedad de la HbH Hidropesia fetal por Hb Bart (α0-talasemia homocigota)
-α/αα --/αα -α/- α-α/----/--
Portador silenteRasgo talasémicoRasgo talasémicoEnfermedad de la HbHHidropesía fetal por Hb Bart
No deleción

Diagnóstico de la talasemia¿Cómo?
• 1º nivel de sospecha (clínica, hematimetria, frotis SP)
• 2º nivel presunción (EEF, Hb A2, Hb F, Hb H, Hb Bart, cuerpos de inclusión)
• 3º nivel de confirmación molecular

trasmisión de infecciones
expansión ósea
hipopituitarismo
hiperpigmentación
hipotiroidismohipoparatiroidismo
hipertensión pulmonary embolismo
miocardiopatía
trombosis venosa
hemosiderosisy cirrosis hepática
hemopoyesis extramedular
esplenomegalia
diabetes mellitus
artropatía
Retraso puberal
hipogonadismo
osteoporosis
baja estatura
Endocrinopatas tto hormonal sustitutivo
Transfusión leucoeplección tests virales
Osteoporosis bifosfonatos vitamina D
Sobrecarga férrica deferoxamina deferiprona deferasirox
Tratamiento de soporte Complicaciones
Tratamiento curativo
Tratamiento futuro
Traspalnte de progenitores médula ósea cordon umbilical no relacionado no mieloablativo
Tratamiento experimental eritropoyetina moduladores de Hb F (hidroxiurea, butiratos) antioxidantes
Terapia génica


ANEMIA Microcítica (VCM<80fl) (HCM<27pg)Historia familiar, raza,edad,sexo gestación, sangrado.
Ferritina,Sideremia, TBIC, IST.
Ferritina N o ↑,Sideremia N o ↑, TBIC N.
Ferritina ↓ ,Sideremia ↓, TBIC ↑.
Ferritina N o ↑,Sideremia N o ↓ , TBIC ↓ .
Ferritina ↑,Sideremia N o ↑, TBIC N.
Talasemia
Estudio de hemoglobinas
Consejo genético
Ferropenia
Estudio de causadigestivo, ginecológico
Tratamiento
Enf. crónica
Signos de E:C:VSG, rouleaux,
neutrofilia, linfopenia,↓ albumina
Diagnósticoenf. de base
A sideroblástica
MO: ↑ hemosiderinasideriblastos en
anillo
Investigar causa
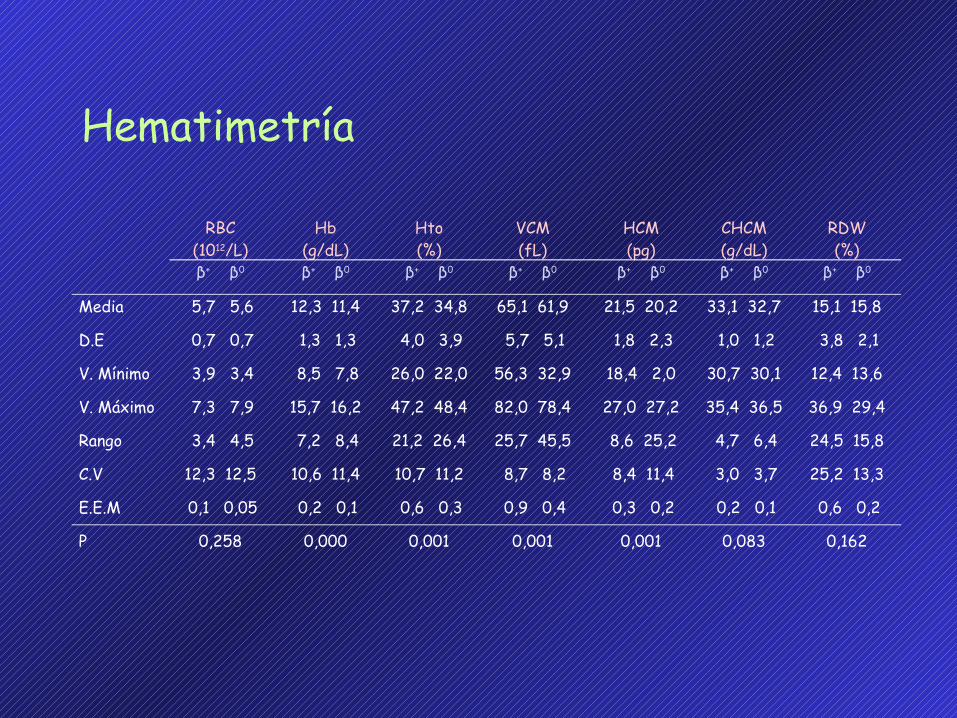
RBC(1012/L)
Hb(g/dL)
Hto(%)
VCM(fL)
HCM(pg)
CHCM(g/dL)
RDW(%)
β+ β0 β+ β0 β+ β0 β+ β0 β+ β0 β+ β0 β+ β0
Media
D.E
V. Mínimo
V. Máximo
Rango
C.V
E.E.M
P
5,7 5,6
0,7 0,7
3,9 3,4
7,3 7,9
3,4 4,5
12,3 12,5
0,1 0,05
0,258
12,3 11,4
1,3 1,3
8,5 7,8
15,7 16,2
7,2 8,4
10,6 11,4
0,2 0,1
0,000
37,2 34,8
4,0 3,9
26,0 22,0
47,2 48,4
21,2 26,4
10,7 11,2
0,6 0,3
0,001
65,1 61,9
5,7 5,1
56,3 32,9
82,0 78,4
25,7 45,5
8,7 8,2
0,9 0,4
0,001
21,5 20,2
1,8 2,3
18,4 2,0
27,0 27,2
8,6 25,2
8,4 11,4
0,3 0,2
0,001
33,1 32,7
1,0 1,2
30,7 30,1
35,4 36,5
4,7 6,4
3,0 3,7
0,2 0,1
0,083
15,1 15,8
3,8 2,1
12,4 13,6
36,9 29,4
24,5 15,8
25,2 13,3
0,6 0,2
0,162
Hematimetría
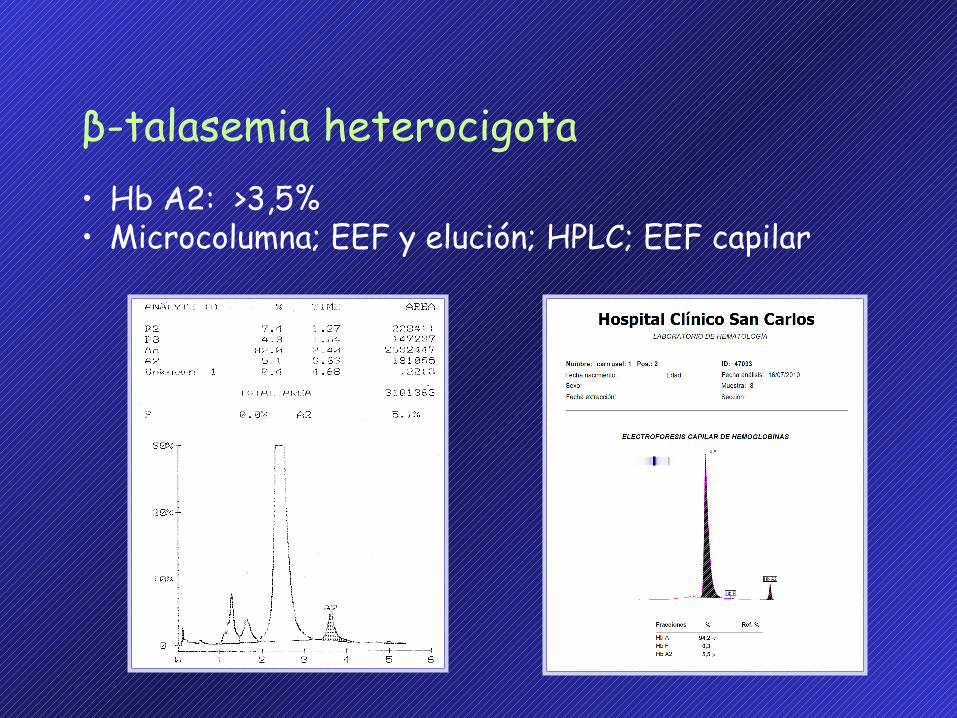
β-talasemia heterocigota• Hb A2: >3,5%• Microcolumna; EEF y elución; HPLC; EEF capilar

alfa-talasemiaalfa-talasemiaVCM (fL)VCM (fL)
7675,9
74,2 70,2
69,2
67,7 68,2
65,963,9
61,361,1
61,7
01020304050607080
-a/aa -a/-a --/aa -a/--
hombres
mujeresniños
888 241 92 15 Nº casos:
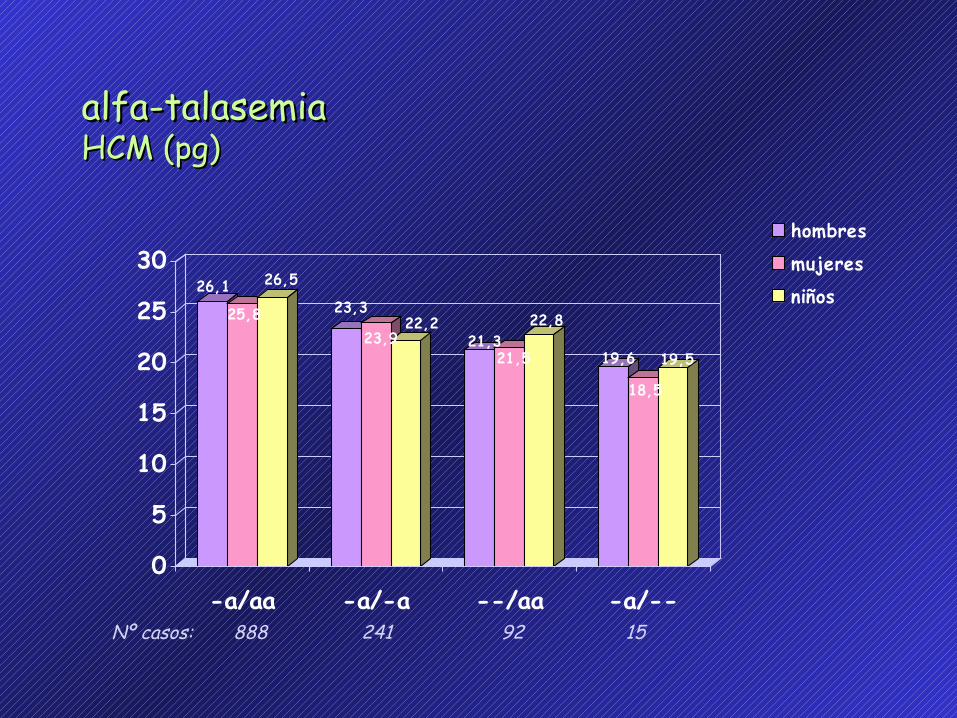
alfa-talasemiaalfa-talasemiaHCM (pg)HCM (pg)
26,1
25,8
26,5
23,3
23,922,2
21,321,5
22,8
19,6
18,5
19,5
0
5
10
15
20
25
30
-a/aa -a/-a --/aa -a/--
hombres
mujeresniños
888 241 92 15 Nº casos:

Aplicaciones de la Biología Molecular al Diagnóstico de las Talasemias
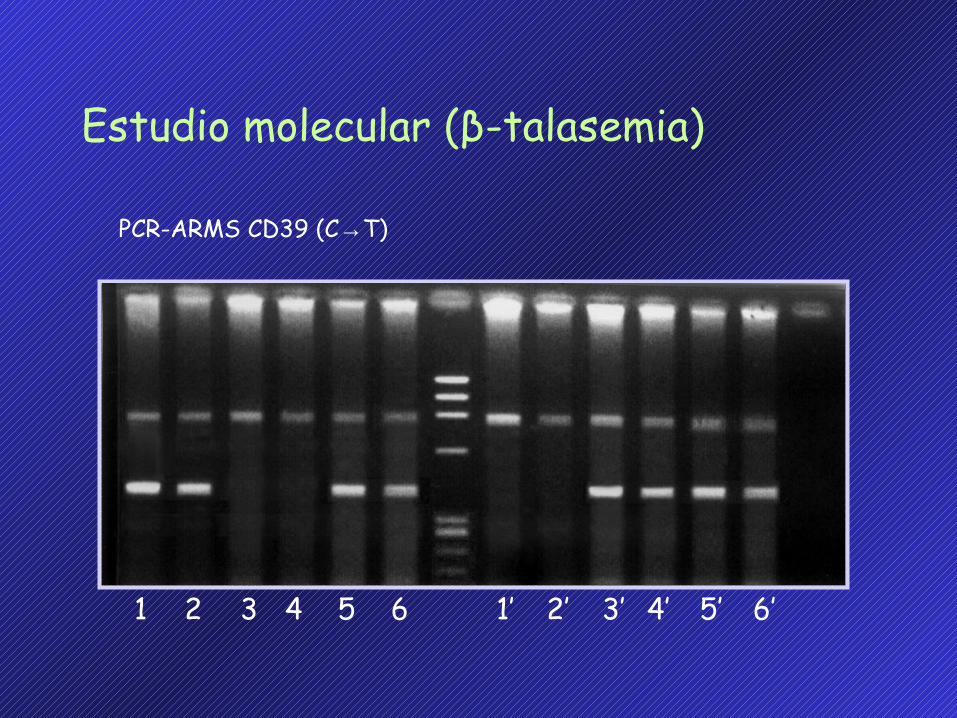
PCR-ARMS CD39 (C→T)
Estudio molecular (β-talasemia)
1 2 3 4 5 6 1’ 2’ 3’ 4’ 5’ 6’

Estudio molecular (α-talasemia deleción)
2: ααα3.7/αα 1: -α3.7/αα
3: -α3.7/-α3.7
1 2 2 2 3 4 2 4
4: αα/αα
Southern blot, con las endonucleasa de restricción Bam HIy sonda α (1,5 Kb. Pst I).
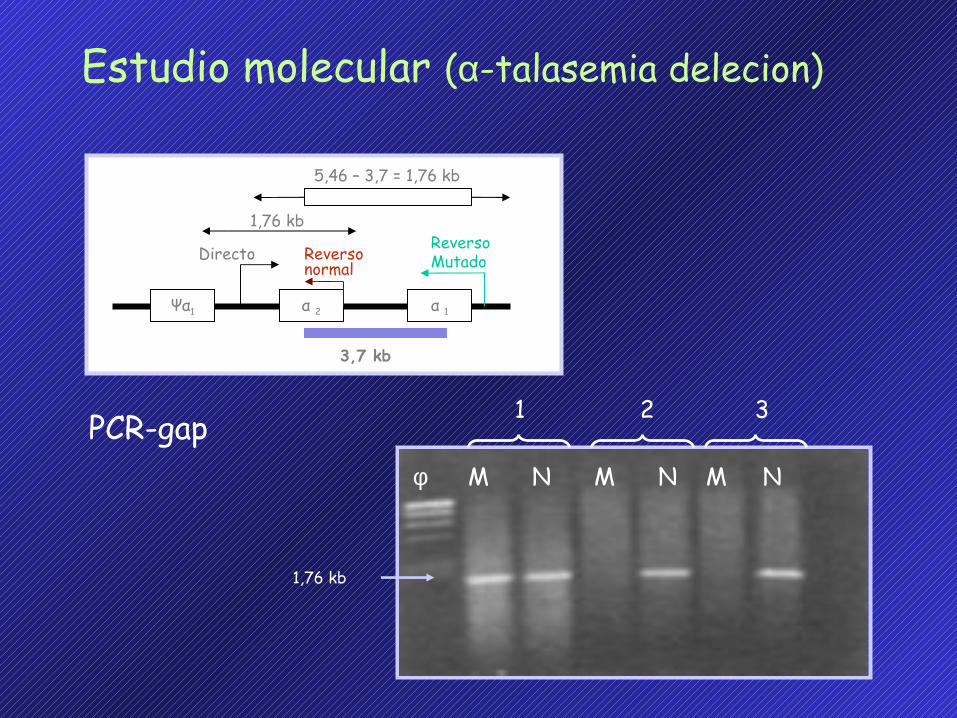
1,76 kb
5,46 – 3,7 = 1,76 kb
3,7 kb
ReversoMutadoDirecto Reverso
normal
Ψα1 α 1α 2
1,76 kb
φ M N M N M N
1 2 3
Estudio molecular (α-talasemia delecion)
PCR-gap

φ 1 32 4 65 7 8 119 10
PCR-RFLP (Hph y Nco I)C8: 5’-GAGCCTGGCCAAACCATCAC-3’ / C3: 5’-CCATTGTTGGCACATTCCGG-3’
Codon iniciación(αNcoα): ATG → ACG
IVS-1 (-5 nt) donador(αHphα): GGTGAGGCT → GGCT
1094 bp -849 bp -
1943 bp -
- 1078 bp- 1395 bp
αNcoα/αααNcoα/αNcoα
αHphα/αααHphα/αHphα
* * *
**
* ** *
**
Estudio molecular (α-talasemia no delecion)

Utilidad de la biología molecular en el estudio de las talasemias
• Esencial en el diagnóstico de la α –talasemia
• Diagnóstico con expresión severa de β-talasemia– Asociación β-talasemia y
hemoglobinopatias estructurales• Asociaciones de α y β talasemia• Permitir un consejo genético adecuado• Indispensable para realizar el
diagnóstico prenatal y preimplantación

Enero 1996-Abril 2008 Mayo 2008-Octubre 2010
Total Dx. prenatales 36 21
Procedencia
Españoles βT/βT 23 (63,9%) 9 (42,8%)
Marroquíes βT/βT 4 (11,1%)
Sub-saharianos βS/βS 9 (25%) 12 (57,0%)
¿Cuándo pidieron asesoramiento genético?
Antes del embarazo 35 21
Durante el embarazo 1
Resultados
βT/βT 4 2
βS/βS 2 3
Diagnostico Prenatal

Agradecimientos
Fernando Ataúlfo GonzálezPaloma RoperoLara VinuesaFélix de la FuenteJorge MartínezPersonal del Laboratorio
Grupo Español de Eritropatología de la SEHH







