1 Chemical Structure Representation and Search Systems Lecture 3. Nov 4, 2003 John Barnard Barnard Chemical Information Ltd Chemical Informatics Software & Consultancy Services Sheffield, UK
Transcript
1Chemical Structure Representation
and Search Systems
Lecture 3. Nov 4, 2003
John Barnard
Barnard Chemical Information LtdChemical Informatics Software & Consultancy Services
Sheffield, UK
2 Lecture 3: Topics to be Covered
More Graph Theory Structure Analysis and Processing
• canonicalisation and symmetry perception• ring perception• functional group identification• structure fingerprints and fragments• structure depiction• principles of structure searching
3 Graph Terminology
degree of a nodenumber of edges
meeting at it
leaf nodea node of degree 1
pathconnected sequence
of edges between two nodes
1
3
2
3
33
2
1
1
1
2
2
2
4 Graph Terminology
cyclepath which returns
to its starting node
treegraph with no cycles
subgraphgraph containing a
subset of the nodes and edges of another graph
5 Graph Terminology
spanning treea tree subgraph that
contains all the nodes(but not necessarilyall the edges) of a graph
6 Graph Terminology
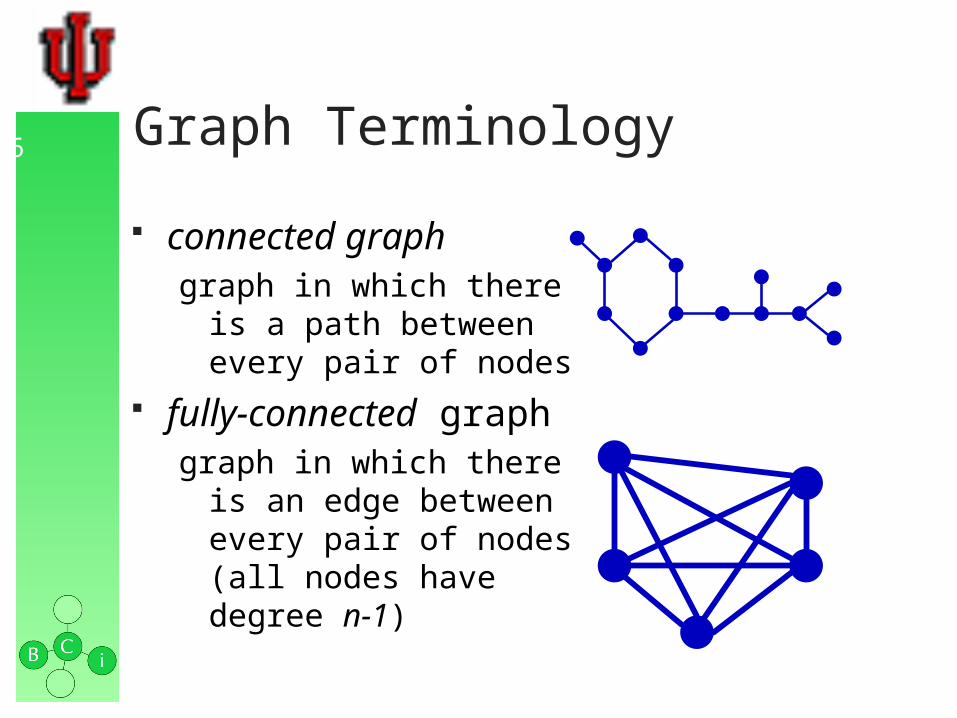
connected graphgraph in which there
is a path between every pair of nodes
fully-connected graphgraph in which there
is an edge between every pair of nodes(all nodes have degree n-1)
7 Graph Terminology
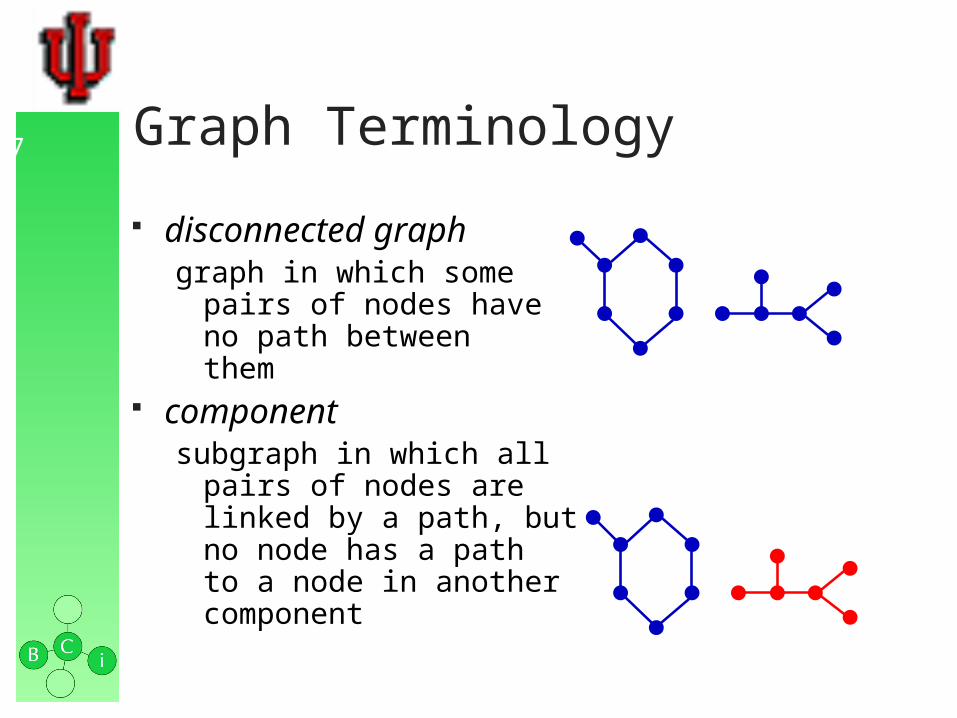
disconnected graphgraph in which some
pairs of nodes have no path betweenthem
componentsubgraph in which all
pairs of nodes are linked by a path, but no node has a path to a node in another component
8 Graph Terminology
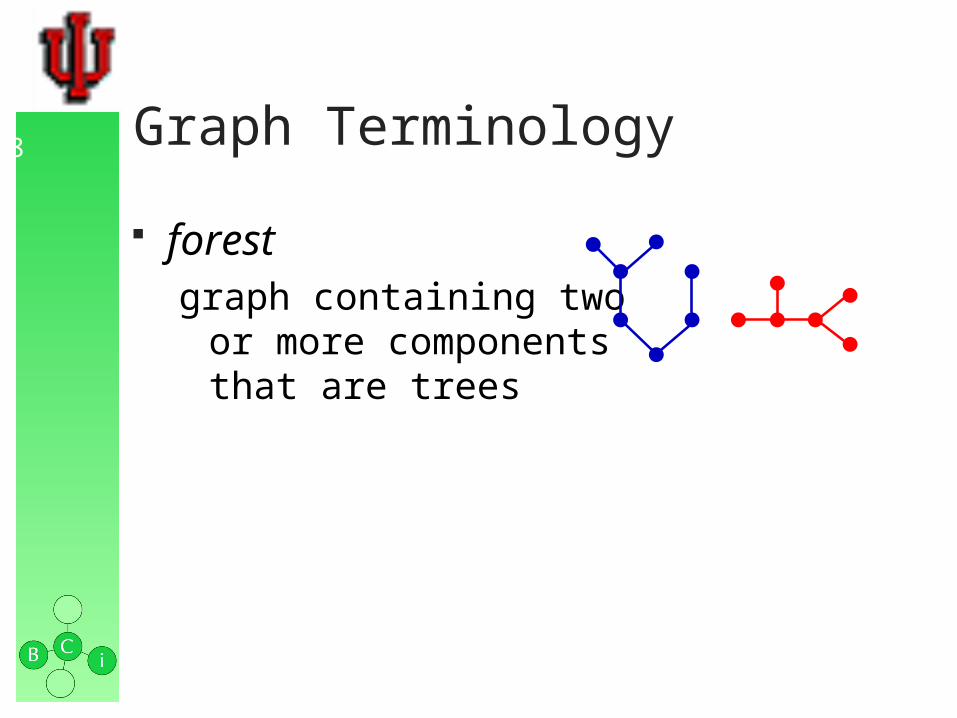
forestgraph containing two
or more components that are trees
9 Canonicalisation
a given chemical structure (or graph) can have many valid and unambiguous representations• different order of rows in connection table• different order of atoms in SMILES
for comparison purposes it would be useful to have a single unique or “canonical” representation
process of converting input representation to canonical form is called “canonicalisation” or “canonisation”• process of applying “rules” (i.e. an algorithm)
10 Canonicalisation
an obvious approach:• generate all possible valid SMILES• choose the one that comes first alphabetically
this would be very slow, but effective, and there is a danger of missing one• principle was used for canonicalising
Wiswesser Line Notation
11 Canonicalisation
most methods in use today involve renumbering the atoms in some unique and reproducible way• can be used to number rows in connection table• can determine order of atoms in SMILES
normally involve a node labelling technique called “relaxation”• example is Morgan’s algorithm (1965)
12 Morgan’s algorithm
1. Label each node with its degree
2. Count number ofdifferent values
1
3
2
3
33
2
1
1
1
2
2
2
3 d iffe re nt v a lu e s{ 1 , 2 , 3 }
13 Morgan’s algorithm
3. Recalculate labelsby summing labelvalues at neighbournodes
4. Count number ofdifferent values
1
3
2
3
33
2
1
1
1
2
2
2
3 d iffe re nt v a lu e s{ 1 , 2 , 3 }
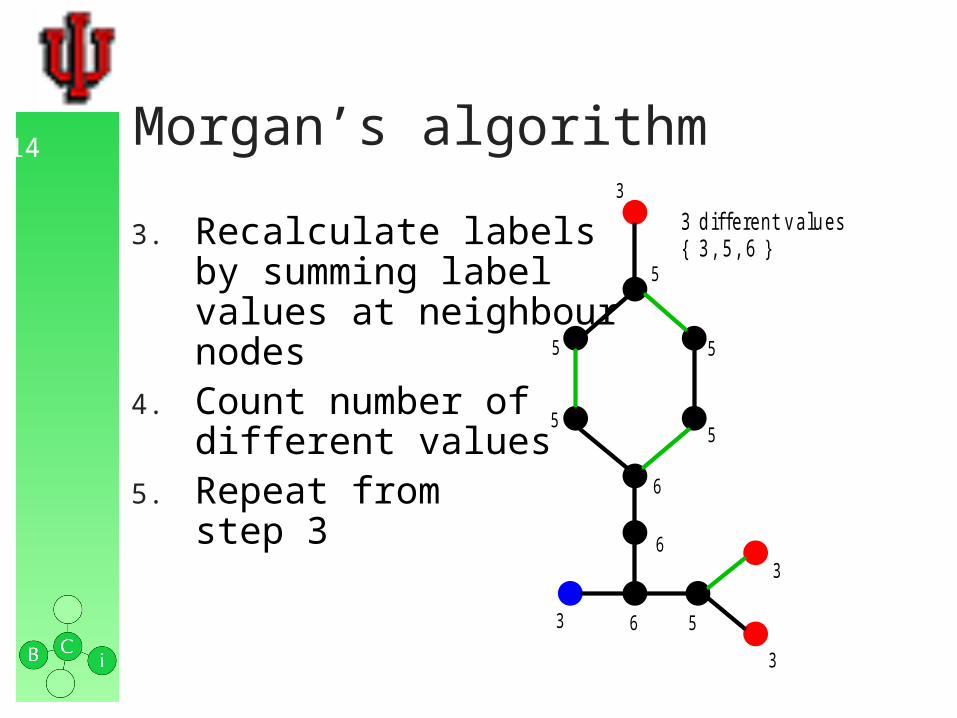
14 Morgan’s algorithm
3. Recalculate labelsby summing labelvalues at neighbournodes
4. Count number ofdifferent values
5. Repeat fromstep 3
3
5
5
6
56
5
3
3
3
5
5
6
3 d iffe re nt v a lu e s{ 3 , 5 , 6 }
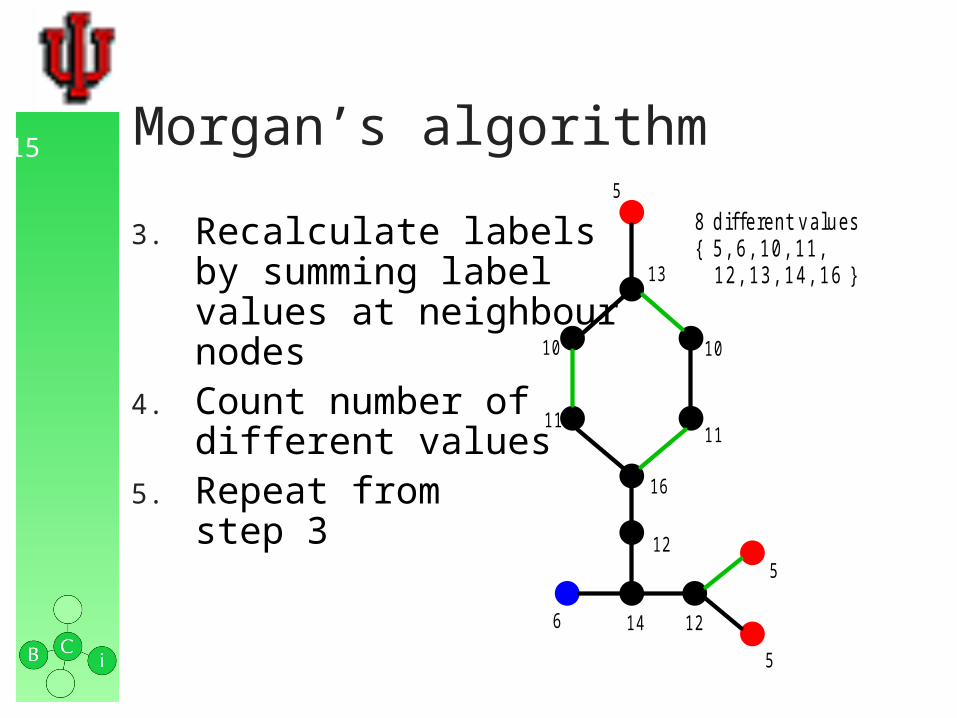
15 Morgan’s algorithm
3. Recalculate labelsby summing labelvalues at neighbournodes
4. Count number ofdifferent values
5. Repeat fromstep 3
5
13
10
16
1214
11
5
5
6
10
11
12
8 d iffe re nt v a lu e s{ 5 , 6 , 1 0 , 11 , 1 2 , 1 3 , 1 4 , 1 6 }
16 Morgan’s algorithm
3. Recalculate labelsby summing labelvalues at neighbournodes
4. Count number ofdifferent values
5. Repeat fromstep 3
13
25
24
34
2418
26
12
12
14
24
26
30
9 d iffe re nt v a lu e s{ 1 2 , 1 3 , 1 4 , 1 8 , 2 4 , 2 5 , 2 6 , 3 0 , 3 4 }
17 Morgan’s algorithm
3. Recalculate labelsby summing labelvalues at neighbournodes
4. Count number ofdifferent values
5. Repeat fromstep 3
25
61
51
82
4268
48
24
24
18
51
48
42
9 d iffe re nt v a lu e s{ 1 8 , 2 4 , 2 5 , 4 2 , 4 8 5 1 , 6 1 , 6 8 , 8 2 }
18 Morgan’s algorithm
3. Recalculate labelsby summing labelvalues at neighbournodes
4. Count number ofdifferent values
5. Repeat fromstep 3 until thereis no increase in thenumber of differentvalues
61
127
109
138
116102
133
42
42
68
109
133
150
1 0 d iffe re nt v a lu e s{ 4 2 , 6 1 , 6 8 , 1 0 2 , 1 0 9 , 11 6 , 1 2 7 , 1 3 3 , 1 3 8 , 1 5 0 }
19 Morgan’s algorithm
most nodes nowhave differentlabels
choose node withhighest label asnode 1
number its neighbours in orderof label values
61
127
109
138
116102
133
42
42
68
109
133
150
1 0 d iffe re nt v a lu e s{ 4 2 , 6 1 , 6 8 , 1 0 2 , 1 0 9 , 11 6 , 1 2 7 , 1 3 3 , 1 3 8 , 1 5 0 }
20 Morgan’s algorithm
most nodes nowhave differentlabels
choose node withhighest label asnode 1
number its neighbours in orderof label values
61
127
109
138
116102
133
42
42
68
109
133
150
1 0 d iffe re nt v a lu e s{ 4 2 , 6 1 , 6 8 , 1 0 2 , 1 0 9 , 11 6 , 1 2 7 , 1 3 3 , 1 3 8 , 1 5 0 }
1
2
3
21 Morgan’s algorithm
move to node 2 number its remaining
neighbours in orderof label values
• because label valuesare tied, choose one with higher bond order (green) first
move to node 3
61
127
109
138
116102
133
42
42
68
109
133
1501
2
3
45
22 Morgan’s algorithm
continue till all nodesare numbered
we now have a numbering for the rowsof the connection table
“breadth-first” trace• nodes are dealt with
in a “queue” (first in,first out)
61
127
109
138
116102
133
42
42
68
109
133
1501
2
3
45
67
89
10
11
12
13
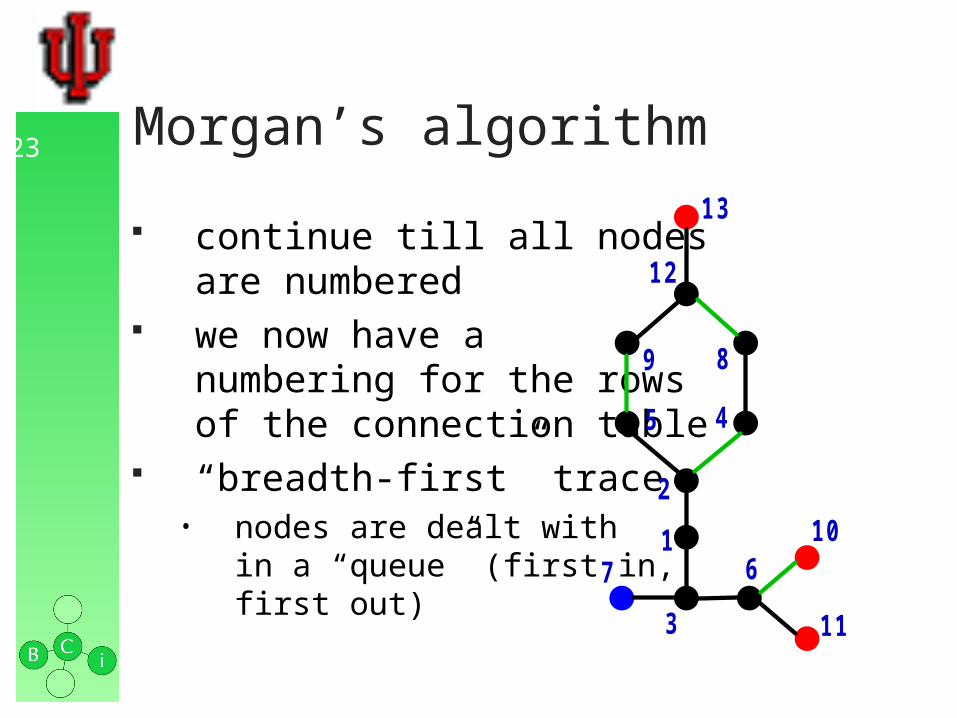
23 Morgan’s algorithm
continue till all nodesare numbered
we now have a numbering for the rowsof the connection table
“breadth-first” trace• nodes are dealt with
in a “queue” (first in,first out)
1
2
45
67
89
10
11
12
13
3
24 Morgan’s algorithm
“depth-first” trace isalso possible
• nodes are dealt with ina “stack” (last in, first out)
more suitable for assigningatom numbers in SMILES where we want consecutivenumbers to form a path
OC(=O)C(N)CC1C=CC(O)=CC=1
61
127
109
138
116102
133
42
42
68
109
133
1506
7
4
138
25
129
3
1
10
11
25 Symmetry perception
if ties between label values cannot beresolved on basis of atom/bond types, the atoms are symmetrically equivalent, andit doesn’t matter which is chosen next
Morgan’s algorithm is thus also useful for identifying symmetry in molecules
26 Morgan’s algorithm
Provides canonical numbering for the nodes in a graph that doesn’t depend on any original numbering
Works by taking more of the graph into account at each iteration
• essence of “relaxation” technique is iteratively updating a value by looking at its immediate neighbours
It is not infallible• some graphs are known where the algorithm cannot distinguish
nodes that are not symmetrically equivalent There are many variations on it
• and several theoretical papers analysing it mathematically• O. Ivanciuc, “Canonical numbering and constitutional symmetry”,
in J. Gasteiger (Ed.) Handbook of Chemoinformatics, Vol 1, pp. 139-160. Wiley, 2003
27 Canonicalisation
Algorithms are applied to graphs not chemical structures
Issues such as aromaticity, tautomerism and stereochemistry need to be addressed before canonical numbering of the graph• Daylight’s canonicalisation algorithm for SMILES
perceives aromatic rings (using its own definition of aromaticity) as first step
28 Ring perception
How many rings are there in these structures and which ones are they?
rings are important features of chemical structures• nomenclature generation• aromaticity perception• synthetic significance• fragment descriptor generation
29 Rings and ring systems
A ring system is a subgraph in which every edge is part of a cycle
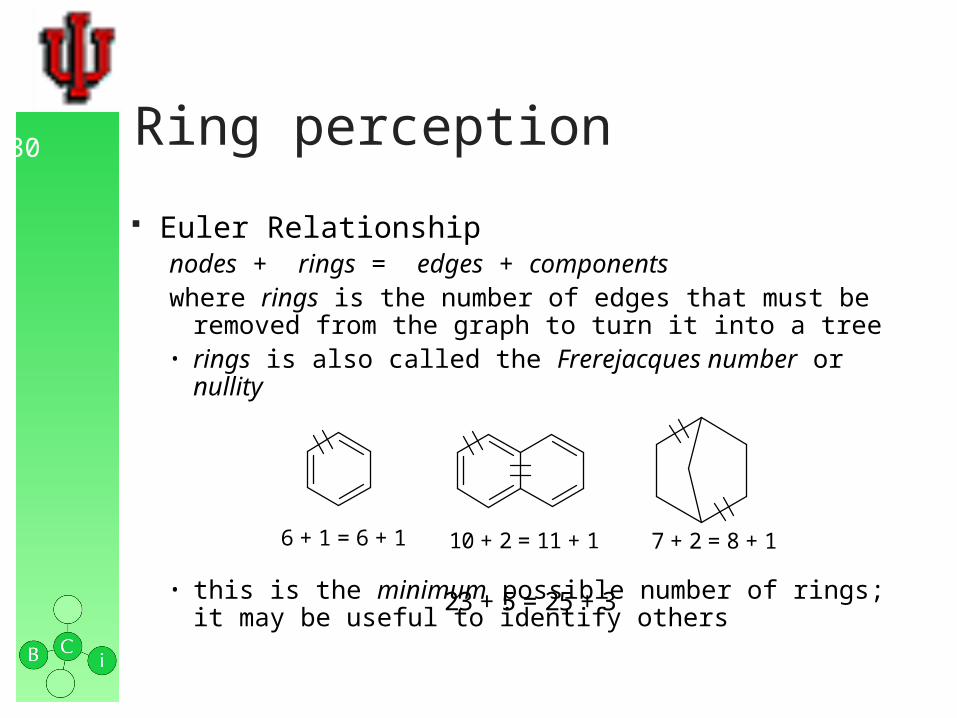
30 Ring perception
Euler Relationship nodes + rings = edges + componentswhere rings is the number of edges that must be removed from
the graph to turn it into a tree• rings is also called the Frerejacques number or nullity
• this is the minimum possible number of rings; it may be useful to identify others
6 + 1 = 6 + 1 10 + 2 = 11 + 1 7 + 2 = 8 + 1
23 + 5 = 25 + 3
31 Which rings to perceive?
Usually the smallest set of smallest rings• two 6-membered rather than
one 6- and one 10-membered• two 5-membered rather than
one 5- and one 6-membered
But there may be more than one SSSR• C-S-C-C-C-C• C-C-C-C-O-C• C-S-C-C-O-C
three different 6-membered rings
S
O
S
OO
S
32 Which rings to perceive?
Sometimes a large envelopering may be aromatic, whensmaller rings are not
Ring perception is a complex area where there are no right answers• there is a lot of literature on the subject
33 Ring perception by spanning tree
start at an arbitrary node “grow a spanning tree”
• add neighbours of current node to a queue
o provided they are not already in it
• move to the next node in the queue• repeat until queue is empty
those edges from original graph not in the spanning tree are ring closures
1
2
3
45
67
89
10
11
12
13
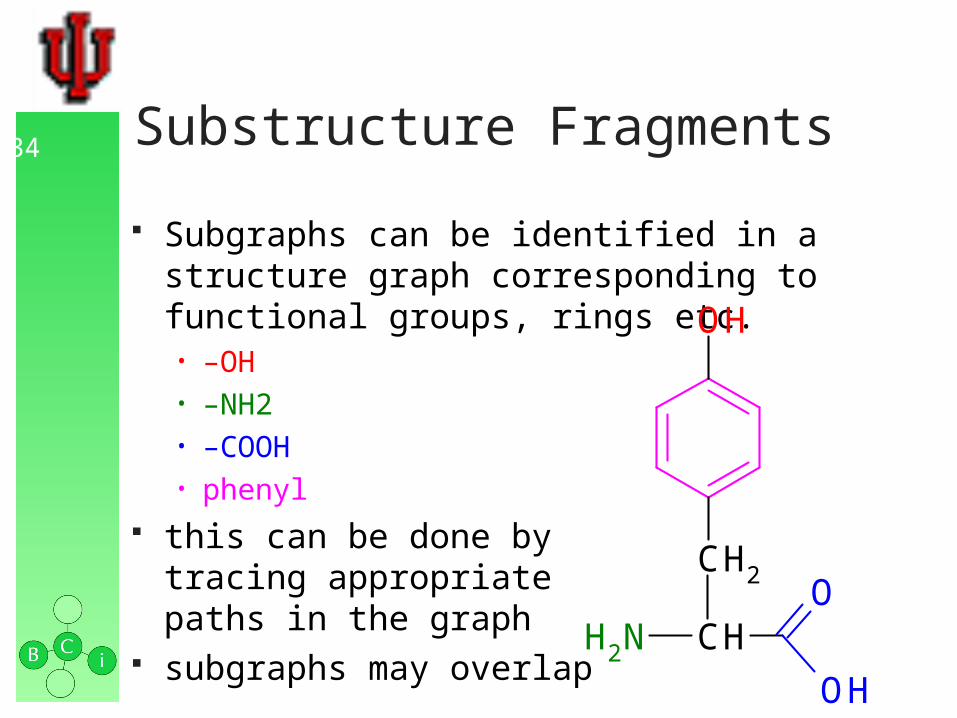
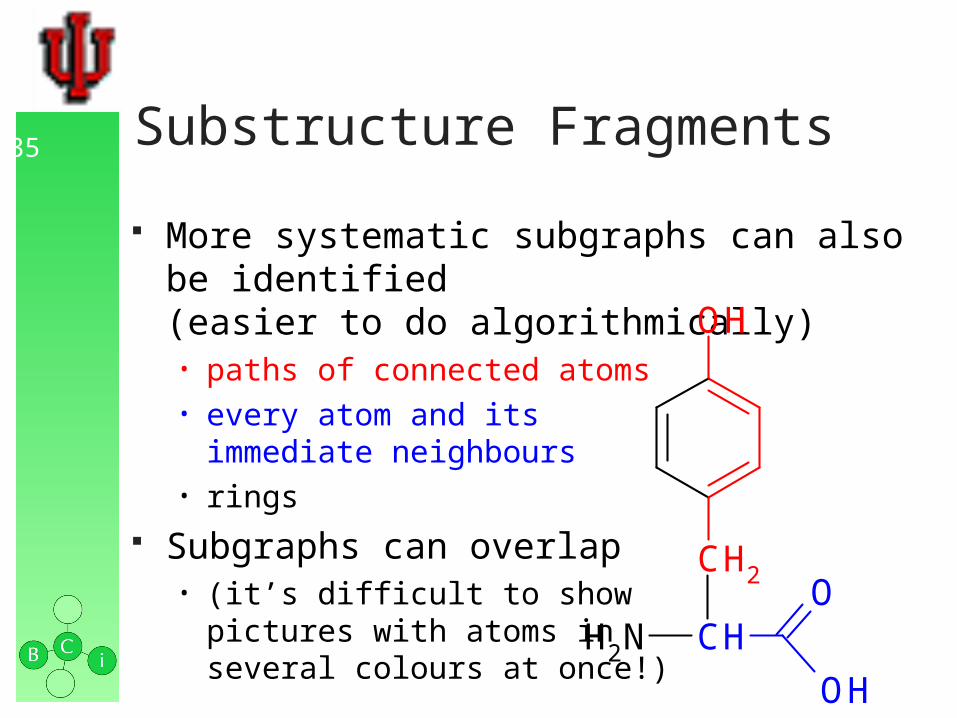
34 Substructure Fragments
Subgraphs can be identified in a structure graph corresponding to functional groups, rings etc. • –OH• –NH2• –COOH• phenyl
this can be done bytracing appropriatepaths in the graph
subgraphs may overlap
OH
CH2
CHNH2
OH
O
35 Substructure Fragments
More systematic subgraphs can also be identified(easier to do algorithmically)• paths of connected atoms• every atom and its
immediate neighbours• rings
Subgraphs can overlap• (it’s difficult to show
pictures with atoms inseveral colours at once!)
OH
CH2
CHNH2
OH
O
36 Substructure fragments
• fragments provide “index terms” for a chemical structureo analogous to keywords in a text document
• they can be used in searching for structureso retrieved structures must contain the same fragments as the
query
• “ambiguous” representationso many different structures can have the same fragments,
connected together in different ways
• fragments to be used may be a closed listo controlled “vocabulary” (dictionary) of structural features
• or an open-ended list (like free text searching)o e.g. all unbranched paths of up to 6 atoms
37 Fragment codes
• many early chemical information systems were based on identifying fragments of this sort
o originally the fragments were identified manuallyo and represented on punched cards
• special fragment codes (dictionaries of fragments) were devised for different systems
o some of these are still in use, though with automated encoding of structures
o particularly important are the systems for “Markush” structures in patents (e.g. Derwent WPI code)
38 Fingerprints
the fragments present in a structure can be represented as a sequence of 0s and 1s
00010100010101000101010011110100• 0 means fragment is not present in structure• 1 means fragment is present in structure (perhaps
multiple times)
each 0 or 1 can be represented as a single bit in the computer (a “bitstring”)
for chemical structures often called structure “fingerprints”
39 Fingerprints
fingerprints are typically 150-2500 bits long where a fixed dictionary of fragments is used there
can be a 1:1 relationship between fragment and bit position in fingerprint• sometimes several related fragments will “set” the same
bit
disadvantage is that if structure contains no fragments from the dictionary, no bits are set• can be avoided if “generalised” fragments are used
(involving e.g. “any atom”, “any ring bond” types)
40 Fingerprints
if fragment set is open-ended, the fragment description (e.g. C-C-N-C-C-O) can be “hashed” to a number in fixed range (e.g. 1 to 1024) and this is the bit number to be set
disadvantages:• different and unrelated fragments may “collide” at the
same bit position• difficult to work back from bit position to fragment• this usually causes only slight degradation in search
performance (false hits), but can be more of a problem in other applications of fingerprints
41 Fingerprints
Hashed fingerprints• typically used in software from Daylight
Chemical Information Systems Inc. Dictionary fingerprints
• Chemical Abstracts Service• MDL Information Systems Inc
o ISIS or MACCS keys (166 and 960 bits)
• Barnard Chemical Information Ltdo customised dictionaries
42 2D structure depiction
if structures are stored without 2D display coordinates, we need to generate them• SMILES
“depiction” algorithms are used for this identify and lay out ring systems first
• complications over orientation of some systems• Chemical Abstracts stores “standard depictions” of all
ring systems it has encountered
then add side chains, avoiding collisions• many features can be added to improve appearance
43 3D structure depiction
much more complicated than 2D need to store standard bond lengths and angles need to distinguish atoms in different hybridisation states
(sp2 vs sp3 carbon) need rotate single bonds to avoid “bumps” sophisticated “conformation generation” programs identify
low-energy conformers• very useful for identifying molecules with the correct shape to fit
into biological receptor sites
J. Sadowski, “3D structure generation”, in J. Gasteiger (Ed.) Handbook of Chemoinformatics, Vol 1, pp. 231-261. Wiley, 2003
44 Nomenclature generation
most systematic nomenclature is based on ring systems• need to identify/prioritise ring systems first• identify standard numbering for system
o frequently need to store this
• add side chains and substituents with appropriate locants
J. L. Wisniewski, “Chemical nomenclature and structure representation: algorithmic generation and conversion”, in J. Gasteiger (Ed.) Handbook of Chemoinformatics, Vol 1, pp. 139-160. Wiley, 2003
45 Conclusions from Lecture 3
there are several important jargon terms used in graph theory, which crop up in chemical informatics
canonicalisation provides a unique numbering for the atoms in a molecule
• Morgan algorithm can be used to achieve it it’s not always obvious how many rings there are, or which
ones they are fingerprints represent the presence or absence of
substructure fragments in a molecule• they are ambiguous representations of structure
46 Topic for Lecture 4: Structure searching
two main varieties of search • full structure search
o query is is complete moleculeo is this molecule in the database?
• or tautomers, stereoisomers etc. of it,
• substructure searcho query is a pattern of atoms and bondso does this pattern occur as a substructure (subgraph)