Arterial stiffening is one of the aging-related disorders, which can be accelerated by metabolic syndromes, diabe-
tes mellitus, and arteriosclerosis.1,2 Arterial stiffening leads to isolated systolic hypertension.1 Pulse wave velocity (PWV) is a noninvasive measure of arterial stiffening.3 Abundant epide-miological studies have demonstrated that arterial stiffening is an independent predictor of cardiovascular outcomes such as myocardial infarction, cognitive decline in aging, stroke, and chronic kidney diseases.4–7 Two longitudinal studies indicated that arterial stiffness predicts an increase in systolic blood pres-sure and incident hypertension.8,9
Klotho (KL) gene was originally identified as a puta-tive antiaging gene in mice.10 The Klotho gene was named after the purported Greek goddess Klotho who spins the thread of life.11 KL homozygous deficient mice carry a dis-ruption in the promoter region of the KL gene, leading to extensive premature aging phenotypes including severe
hyperphosphatemia, ectopic soft tissue calcification, and early death (<10 weeks).10,11 Overexpression of KL, how-ever, extended life span in mice.12,13 The mouse full-length KL gene encodes a single-pass transmembrane domain of 1014 amino acids (130 kDa).11 The short-form KL (≈65 kDa) can be generated by alternative RNA splicing or pro-teolytic cleavage.11 KL protein is predominantly expressed in the kidney and slightly expressed in parathyroid glands and brain choroid plexus.11 Both human and mouse KL gene are alternatively spliced after exon 3,14 which encodes the KL1 repeat of Klotho, called secreted Klotho (sKL). Soluble KL might include the truncated extracellular domain (KL1 and KL2), the KL1 fragment, and the KL2 fragment.11,15 sKL and soluble KL circulates in the blood and regulates function in organs and cells (eg, vascular cells) that do not express KL.11 KL plays important roles in a variety of physiologi-cal and pathological processes including modulation of Wnt
Abstract—Klotho was originally discovered as an aging-suppressor gene. The objective of this study is to investigate whether klotho gene deficiency affects high-fat diet (HFD)–induced arterial stiffening. Heterozygous Klotho-deficient (KL+/−) mice and WT littermates were fed on HFD or normal diet. HFD increased pulse wave velocity within 5 weeks in KL+/− mice but not in wild-type mice, indicating that klotho deficiency accelerates and exacerbates HFD-induced arterial stiffening. A greater increase in blood pressure was found in KL+/− mice fed on HFD. Protein expressions of phosphorylated AMP-activated protein kinase-α (AMPKα), phosphorylated endothelial nitric oxide synthase (eNOS), and manganese-dependent superoxide dismutase (Mn-SOD) were decreased, whereas protein expressions of collagen I, transforming growth factor-β1, and Runx2 were increased in aortas of KL+/− mice fed on HFD. Interestingly, daily injections of an AMPKα activator, 5-aminoimidazole-4-carboxamide-3-ribonucleoside, abolished the increases in pulse wave velocity, blood pressure, and blood glucose in KL+/− mice fed on HFD. Treatment with 5-aminoimidazole-4-carboxamide-3-ribonucleoside for 2 weeks not only abolished the downregulation of phosphorylated AMPKα, phosphorylated eNOS, and Mn-SOD levels but also attenuated the increased levels of collagen I, transforming growth factor-β1, Runx2, superoxide, elastic lamellae breaks, and calcification in aortas of KL+/− mice fed on HFD. In cultured mouse aortic smooth muscle cells, cholesterol plus KL-deficient serum decreased phosphorylation levels of AMPKα and LKB1 (an important upstream regulator of AMPKα activity) but increased collagen I synthesis, which can be eliminated by activation of AMPKα by 5-aminoimidazole-4-carboxamide-3-ribonucleoside. In conclusions, Klotho deficiency promoted HFD-induced arterial stiffening and hypertension via downregulation of AMPKα activity. (Hypertension. 2016;67:564-573. DOI: 10.1161/HYPERTENSIONAHA.115.06825.) • Online Data Supplement
Received November 20, 2015; first decision November 30, 2015; revision accepted December 24, 2015.From the Department of Physiology, College of Medicine, University of Oklahoma Health Sciences Center.The online-only Data Supplement is available with this article at http://hyper.ahajournals.org/lookup/suppl/doi:10.1161/HYPERTENSIONAHA.
115.06825/-/DC1.Correspondence to Zhongjie Sun, Department of Physiology, College of Medicine, BMSB 662A, Box 26901, University of Oklahoma Health Sciences
Center (OUHSC), 940 Stanton L Young Blvd, Oklahoma City, OK 73126. E-mail [email protected]
Antiaging Gene Klotho Deficiency Promoted High-Fat Diet–Induced Arterial Stiffening via Inactivation of
AMP-Activated Protein KinaseYi Lin, Jianglei Chen, Zhongjie Sun
Lin et al Klotho Deficiency and Arterial Stiffening 565
signal transduction, antioxidation, and renal ion channels and transporters.11,15,16
Aging and aging-related medical complications (meta-bolic syndrome, hypertension, diabetes mellitus, and chronic kidney disease) are associated with a decreased ratio of elas-tin/collagen (arterial remodeling) and arterial calcification (elastocalcinosis), which contribute significantly to arterial stiffening.17–20 Transforming growth factor β1 (TGF-β1) via binding to its receptors induces a variety of gene expressions in vascular smooth muscle cells that may result in colla-gen deposition and calcification.21,22 The prevalence of arte-rial stiffening is increased with age, whereas antiaging gene klotho levels are decreased with age.1 However, whether KL deficiency plays a role in the development of arterial stiffening is not fully understood.
Adenosine monophosphate–activated protein kinase (AMPK) has been shown to be essential in regulating vascular homeostasis. AMPKα is a serine/threonine kinase that regulates cellular energy homeostasis through its enzymatic activity stim-ulated by phosphorylation of threonine-172 in the catalytic α subunit.23,24 The phosphorylation status at threonine-172 is often used as an indicator of the activation state of AMPKα.25 In addi-tion, the activation of AMPKα through 5-aminoimidazole-4-car-boxamide-3-ribonucleoside (AICAR), an adenosine mimetic, was shown to decrease mean arterial pressure and vascular tone in hypertensive rats.26 AMPKα inactivation has been found in a model of calcification in rat aortic smooth muscle cells, and metformin has been shown to inhibit calcification via the acti-vation of the AMPKα pathway.27 However, whether AMPKα is involved in the pathogenesis of arterial stiffening is not clear.
The purpose of the study is to investigate whether Klotho deficiency plays a role in the pathogenesis of arterial stiffen-ing in animals fed on high-fat diet (HFD). We found that KL deficiency plus HFD accelerated and exacerbated arterial stiff-ening, which was associated with AMPKα inactivation in the arteries. In addition, we further assessed the effect of activa-tion of AMPKα by AICAR on HFD-induced arterial stiffen-ing in KL-deficient mice.
Methods
Animal StudiesHeterozygous KL+/− mutant mice with >9 generations in 129/Sv back-ground were kindly provided Dr Kuro-o et al.10 Briefly, all mice were housed in cages at room temperatures (25±1°C) and were provided with laboratory chow (Cat 5053; PicoLab) and tap water ad libitum. This proj-ect was approved by the Institutional Animal Care and Use Committee at the University of Oklahoma Health Sciences Center (details are avail-able in the Methods in the online-only Data Supplement).
Statistical AnalysisData were analyzed using a 1-way ANOVA. The Newman–Keuls procedure was used to assess differences between means. Data were expressed as mean±SEM. A P<0.05 were considered significant.
Results
KL Deficiency Accelerated and Exacerbated HFD-Induced Arterial Stiffening and HypertensionTo explore whether KL deficiency affects the development of arterial stiffening in animals with metabolic syndrome, KL
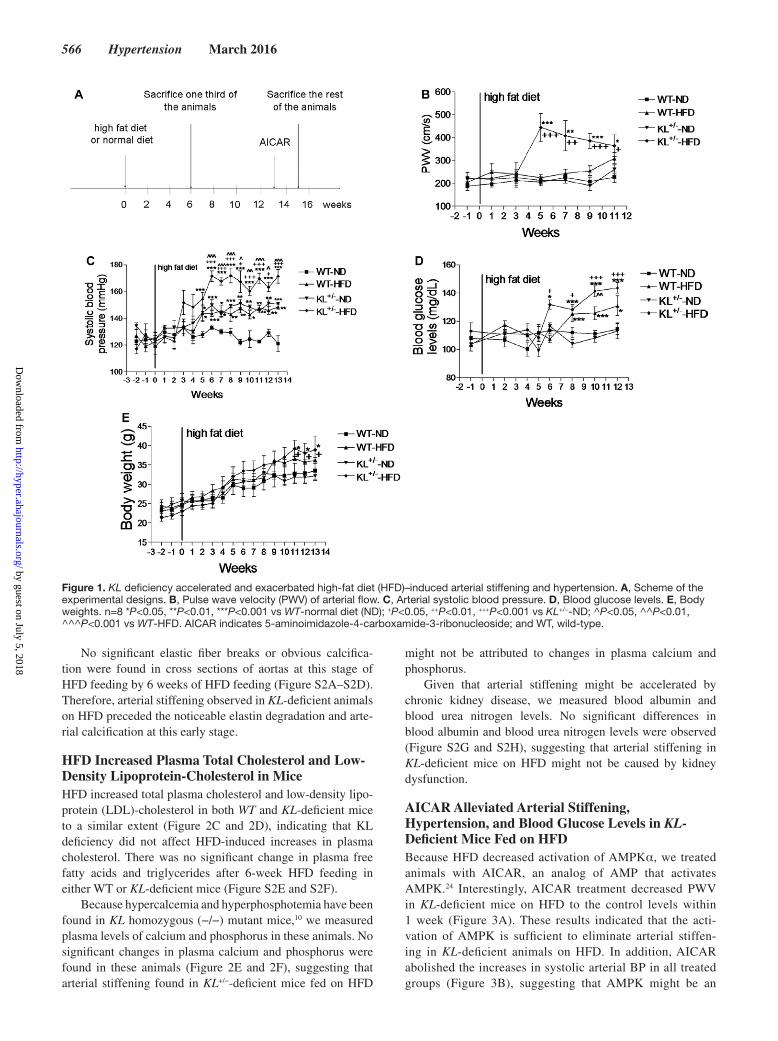
heterozygous mutant mice and their wild-type (WT) litter-mates were fed on HFD (Figure 1A). HFD increased PWV as early 5 weeks in KL+/−-deficient mice but not in WT mice (Figure 1B). These results suggested, for the first time, that klotho deficiency promotes HFD-induced arterial stiffening.
Interestingly, HFD induced a greater increase in systolic blood pressure (BP) by 5 weeks in KL-deficient mice than in WT mice fed on HFD (Figure 1C), indicating that KL defi-ciency exacerbates HFD-induced systolic hypertension. Systolic BP was elevated to a lesser extent in KL-deficient mice on normal diet (ND) and WT mice on HFD (Figure 1C). Similar changes were found in mean arterial BP (Figure S1A in the online-only Data Supplement). It is noticed, however, that diastolic BP was not elevated in KL+/− mice until 6 weeks after HFD (Figure S1B), suggesting that HFD-induced eleva-tion of systolic BP and diastolic BP in KL+/− mice is isolated.
Hyperglycemia is an important feature of metabolic syn-drome. HFD slightly increased blood glucose levels in both KL-deficient and WT mice (Figure 1D). Interestingly, KL-deficient mice with HFD developed an earlier increase in blood glucose levels than WT mice with HFD (Figure 1D). No significant changes in insulin sensitivity and glucose tolerance were found in these animals (Figure S1C and S1D). These data indicated that animals on HFD might be still in the early stage in the development of metabolic syndrome.
Obesity is an another hallmark of metabolic syndrome. HFD tended to increase body weight in both genotypes, but no significance was found until the 11th week. KL-deficient mice fed on HFD started to demonstrate a significantly greater increase in body weight than in the WT-ND and the KL+/−-ND (Figure 1E).
KL Deficiency Exacerbated HFD-Induced Expression of Aortic Collagen I and TGFβ1 but Decreased Levels of Aortic Phosphorylated AMPKα, Phosphorylated eNOS, and Mn-SOD Levels in Mice Fed on HFDDuring the sixth week of HFD, one third of the animals were euthanized for assessing the molecular changes associated with arterial stiffening. HFD increased collagen I and TGFβ1 expression in WT mice (Figure 2A). KL-deficient mice on ND had greater levels of TGFβ1 and collage I than WT mice on ND (Figure 2A). Interestingly, KL deficiency further increased HFD-induced TGFβ1 and collagen I expression (Figure 2A). An increase in TGFβ1 and collagen I expression could contribute to arterial stiffening. Unexpectedly, there was no significant difference in tropoelastin levels among differ-ent animal groups (Figure 2A). Taken together, these results suggested that KL deficiency further promotes HFD-induced collagen synthesis and arterial stiffening.
Interestingly, KL-deficient mice on HFD displayed decreased phosphorylation levels of AMPKα and eNOS (Figure 2B), suggesting that KL deficiency plus HFD down-regulates AMPKα and eNOS activities. KL-deficient mice on HFD also demonstrated lower levels of manganese-dependent superoxide dismutase (Mn-SOD) (Figure 2B). We confirmed that KL+/−-deficient mice had about half of short-form KL pro-tein in plasma (Figure S1E and S1F).
by guest on July 5, 2018http://hyper.ahajournals.org/
No significant elastic fiber breaks or obvious calcifica-tion were found in cross sections of aortas at this stage of HFD feeding by 6 weeks of HFD feeding (Figure S2A–S2D). Therefore, arterial stiffening observed in KL-deficient animals on HFD preceded the noticeable elastin degradation and arte-rial calcification at this early stage.
HFD Increased Plasma Total Cholesterol and Low-Density Lipoprotein-Cholesterol in MiceHFD increased total plasma cholesterol and low-density lipo-protein (LDL)-cholesterol in both WT and KL-deficient mice to a similar extent (Figure 2C and 2D), indicating that KL deficiency did not affect HFD-induced increases in plasma cholesterol. There was no significant change in plasma free fatty acids and triglycerides after 6-week HFD feeding in either WT or KL-deficient mice (Figure S2E and S2F).
Because hypercalcemia and hyperphosphotemia have been found in KL homozygous (−/−) mutant mice,10 we measured plasma levels of calcium and phosphorus in these animals. No significant changes in plasma calcium and phosphorus were found in these animals (Figure 2E and 2F), suggesting that arterial stiffening found in KL+/−-deficient mice fed on HFD
might not be attributed to changes in plasma calcium and phosphorus.
Given that arterial stiffening might be accelerated by chronic kidney disease, we measured blood albumin and blood urea nitrogen levels. No significant differences in blood albumin and blood urea nitrogen levels were observed (Figure S2G and S2H), suggesting that arterial stiffening in KL-deficient mice on HFD might not be caused by kidney dysfunction.
AICAR Alleviated Arterial Stiffening, Hypertension, and Blood Glucose Levels in KL-Deficient Mice Fed on HFDBecause HFD decreased activation of AMPKα, we treated animals with AICAR, an analog of AMP that activates AMPK.24 Interestingly, AICAR treatment decreased PWV in KL-deficient mice on HFD to the control levels within 1 week (Figure 3A). These results indicated that the acti-vation of AMPK is sufficient to eliminate arterial stiffen-ing in KL-deficient animals on HFD. In addition, AICAR abolished the increases in systolic arterial BP in all treated groups (Figure 3B), suggesting that AMPK might be an
Figure 1. KL deficiency accelerated and exacerbated high-fat diet (HFD)–induced arterial stiffening and hypertension. A, Scheme of the experimental designs. B, Pulse wave velocity (PWV) of arterial flow. C, Arterial systolic blood pressure. D, Blood glucose levels. E, Body weights. n=8 *P<0.05, **P<0.01, ***P<0.001 vs WT-normal diet (ND); +P<0.05, ++P<0.01, +++P<0.001 vs KL+/−-ND; ^P<0.05, ^^P<0.01, ^^^P<0.001 vs WT-HFD. AICAR indicates 5-aminoimidazole-4-carboxamide-3-ribonucleoside; and WT, wild-type.
by guest on July 5, 2018http://hyper.ahajournals.org/
Lin et al Klotho Deficiency and Arterial Stiffening 567
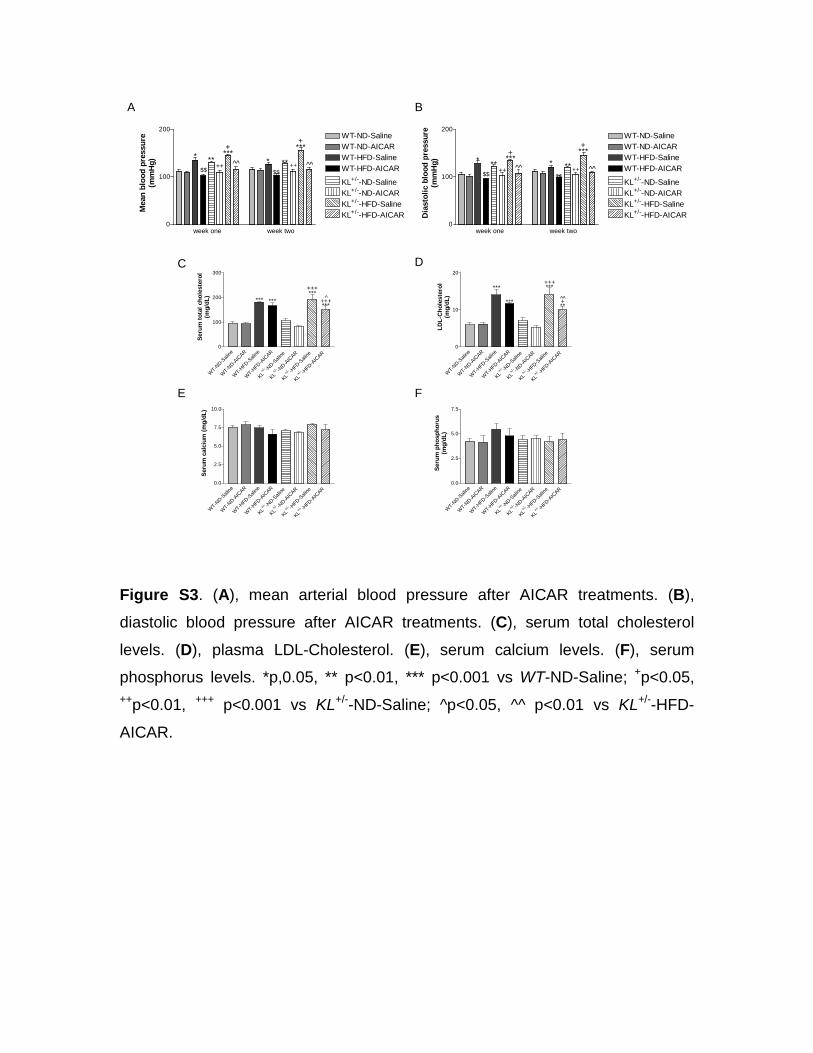
effective therapeutic target for hypertension. Similar results were observed for mean and diastolic arterial BP (Figure S3A and S3B). Furthermore, AICAR abolished the mild increase in blood glucose levels induce by HFD in both WT and KL-deficient mice (Figure 3C), suggesting that the activation of AMPK by AICAR might regulate glucose homeostasis in animals fed on HFD. Finally, AICAR also decreased body weight in KL-deficient mice fed on HFD (Figure 3D). Therefore, the activation of AMPKα improved not only arterial stiffening but also metabolic syndrome in KL-deficient mice.
AICAR Slightly Decreased Blood Total Cholesterol and LDL-Cholesterol in KL-Deficient Mice Fed on HFDAICAR treatments slightly but significantly decreased blood total cholesterol and LDL-cholesterol in KL-deficient mice but not in WT mice fed on HFD (Figure S3C and S3D). These data suggested that KL-deficient mice might be more sensi-tive to AICAR in terms of downregulation of HFD-induced hypercholesterolemia, which may partially contributes to a decrease in arterial stiffening by AICAR in KL-deficient mice. Although KL homozygous mice (−/−) have hypercalcemia and hyperphosphotemia,11 serum levels of calcium and phos-phorus were in normal range in KL heterozygous (+/−) mice
(Figure S3E and S3F). No significant changes in serum cal-cium and phosphorus were observed after AICAR treatments (Figure S3E and S3F).
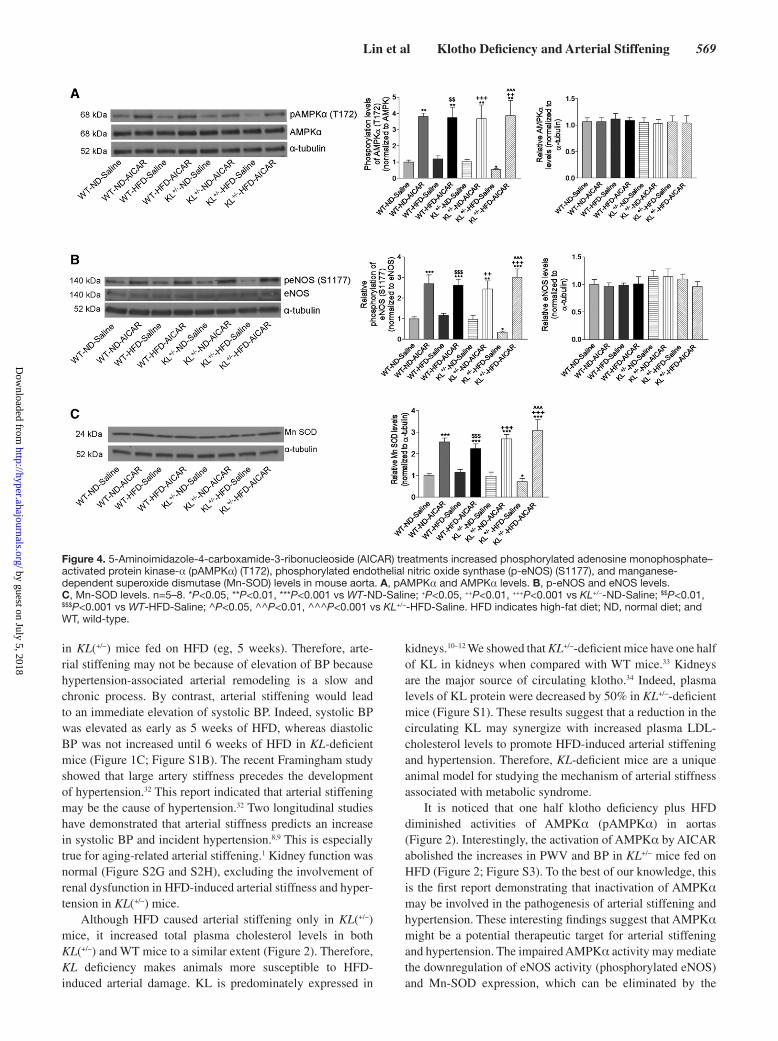
AICAR Increased Phosphorylated AMPKα, Phosphorylated eNOS, and Mn-SOD Levels and Decreased Superoxide Production, Collagen I Levels, and TGFβ1 Expression in AortasAICAR increased phosphorylation levels of AMPKα and eNOS in all treated groups although it did not alter the protein expression of AMPKα and eNOS (Figure 4A and 4B). These results suggested that AICAR treatments might decrease arterial stiffening and hypertension by activating AMPKα and eNOS. In addition, AICAR treatments also increased Mn-SOD in all 3 groups (Figure 4C). Interestingly, the urinary level of nitrite/nitrate (index of nitric oxide) was increased during weeks 1 and 2 of treatment with AICAR (Figure S4), suggesting that the activation of AMPKα enhances bioavail-ability of nitric oxide (NO).
Interestingly, KL deficiency plus HFD for 15 weeks sig-nificantly increased superoxide levels in aortas, which can be abolished by AICAR (Figure 5A and 5B). AICAR completely abolished klotho deficiency- and HFD-induced upregulation of collagen I and TGFβ1 in aortas (Figure 6A–6C). These data suggested that the beneficial effects of AICAR on arterial
Figure 2. KL deficiency exacerbated high-fat diet (HFD)–induced expression of aortic collagen I and transforming growth factor (TGF)-β1 and led to downregulation of aortic phosphorylated adenosine monophosphate–activated protein kinase-α (pAMPKα), phosphorylated endothelial nitric oxide synthase (p-eNOS), and manganese-dependent superoxide dismutase (Mn-SOD) levels in mice fed on HFD for 6 wk. A, Collagen I, tropoelastin, Runx2 protein levels in aorta. B, pAMPKα(T172), AMPKα, p-eNOS(S1177), eNOS, and Mn-SOD protein levels in aorta. C, Plasma total cholesterol levels. D, Plasma low-density lipoprotein (LDL)-cholesterol. E, Plasma calcium levels. F, Plasma phosphorus levels. n=5. *P<0.05, **P<0.01, ***P<0.001 vs WT-ND; +P<0.05, ++P<0.01, +++P<0.001 vs KL+/−-ND; ^P<0.05, ^^P<0.01, ^^^P<0.001 vs WT-HFD. ND indicates normal diet; and WT, wild-type.
by guest on July 5, 2018http://hyper.ahajournals.org/
stiffening and hypertension may be mediated via the AMPKα-eNOS-Mn-SOD-superoxide-TGFβ1-collagen I pathway.
AICAR Decreased Aortic Runx2 Levels and Abolished Arterial Calcification and Elastic Fiber Breaks in KL-Deficient Mice on HFDArterial calcification and elastic fiber breaks have been con-sidered as important factors contributing to the pathogenesis of arterial stiffening.28,29 AICAR significantly decreased pro-tein expression of Runx2 (a marker of osteoblasts) but not of tropoelastin in aortas in KL-deficient mice fed on HFD (Figure 6A, 6D, and 6E). Interestingly, KL-deficient mice fed on HFD for 15 weeks developed arterial calcification and more arterial elastic fiber breaks, which can be abolished by AICAR (Figure S5A–S5D). Therefore, these results demon-strated that HFD-induced calcification and elastic fiber frag-mentation in arteries of KL-deficient mice may be mediated by downrgulation of AMPKα. Furthermore, the activation of AMPKα by AICAR might alleviate arterial calcification via decreasing arterial Runx2, a key transcription factor in the regulation of bone formation.
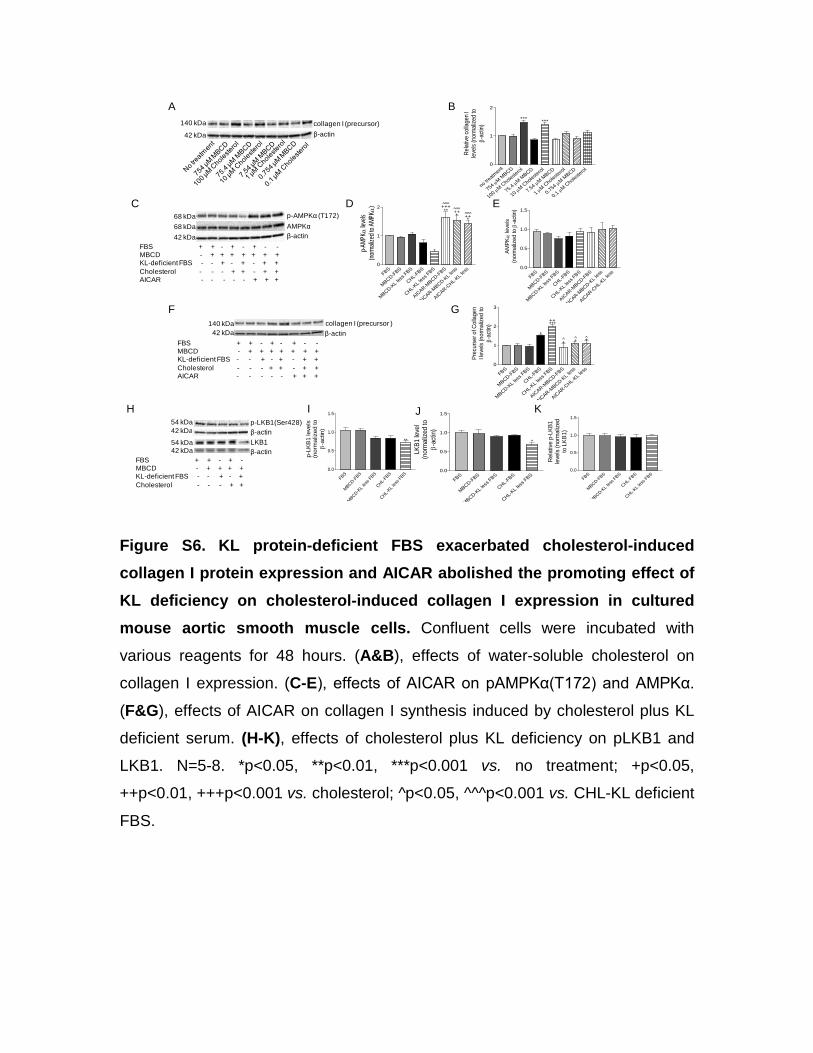
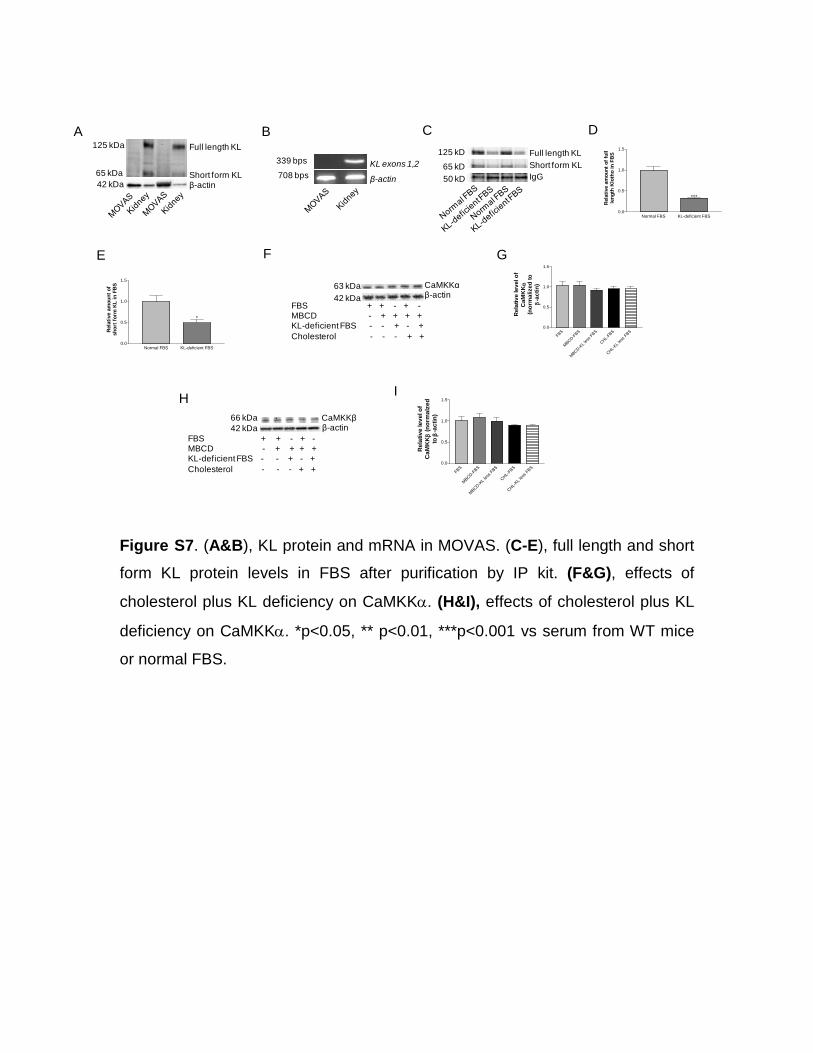
KL Protein Deficiency Plus Cholesterol Inactivate AMPKα via Downregulation of LKB1 ActivityTo further study the molecular mechanisms of direct regula-tion of collagen I protein expression by KL and to avoid in vivo complications, we used a cell line of mouse vascular smooth muscle cells (MOVAS). Interestingly, we found that water-soluble cholesterol dose dependently increased precursor of collagen I in MOVAS (Figure S6A and S6B). Neither full-length KL nor short-form KL was detected in MOVAS (Figure S7A). KL gene was not detected in MOVAS (Figure S7B). Because fetal bovine serum used in cell culture contained both
full-length and short-form KL, we removed ≈50% of KL in both forms from fetal bovine serum using an IP purification kit (Figure S7C–S7E). Interestingly, cholesterol plus KL defi-ciency indeed decreased phosphorylation of AMPKα (Figure S6C–S6E), whereas AICAR treatments increased phos-phorylated AMPKα (pAMPKα) levels (Figure S6C–S6E). As shown in Figure S6F and S6G, KL-deficient fetal bovine serum potentiated the cholesterol-induced increase in colla-gen I. AICAR treatments abolished the increase in collagen I induced by cholesterol alone or KL-deficient fetal bovine serum plus cholesterol in MOVAS (Figure S6F and S6G). Interestingly, cholesterol plus KL deficiency also attenuated phosphorylation level of LKB1 and protein level of LKB1 in MOVAS (Figure S6H–S6K). Cholesterol plus KL deficiency did not affect calcium/calmodulin-dependent protein kinase α (CaMKKα) and CaMKKβ levels in MOVAS (Figure S7F–S7I). LKB1 and CaMKKβ are considered key activators of AMPKα in various mammalian cells.30 Taken together, these results indicated that cholesterol plus KL deficiency might inactivate AMPKα likely via decreasing LKB1 activity, result-ing in collagen I accumulation in aortic smooth muscle cells.
DiscussionAging is associated with a decline in klotho levels and an increase in prevalence of arterial stiffening and hypertension.1 At the age of 70 years, the serum level of klotho is about one half of what it was at 40 years.11,31 This study demonstrated, for the first time, that one half deficiency of klotho, an aging-suppressor gene, accelerated and exacerbated HFD-induced arterial stiffening and hypertension (Figure 1B and 1C). This finding advances the current understanding of aging-related arterial stiffness and hypertension.1 An increase in PWV and elevation of systolic BP occur at approximately the same time
Figure 3. 5-Aminoimidazole-4-carboxamide-3-ribonucleoside (AICAR) alleviated arterial stiffening, hypertension, and high blood glucose levels in KL-deficient mice fed with high-fat diet (HFD). A, Pulse wave velocity (PWV) of arterial flow. B, Arterial systolic blood pressure. C, Blood glucose levels. D, Body weights. n=5–8. *P<0.05, **P<0.01 vs WT-ND-Saline; +P<0.05, ++P<0.01 vs KL+/−-ND-Saline; $$P<0.01 vs WT-HFD-Saline; ^P<0.05, ^^P<0.01 vs. KL+/−-HFD-Saline. ND indicates normal diet; and WT, wild-type.
by guest on July 5, 2018http://hyper.ahajournals.org/
Lin et al Klotho Deficiency and Arterial Stiffening 569
in KL(+/−) mice fed on HFD (eg, 5 weeks). Therefore, arte-rial stiffening may not be because of elevation of BP because hypertension-associated arterial remodeling is a slow and chronic process. By contrast, arterial stiffening would lead to an immediate elevation of systolic BP. Indeed, systolic BP was elevated as early as 5 weeks of HFD, whereas diastolic BP was not increased until 6 weeks of HFD in KL-deficient mice (Figure 1C; Figure S1B). The recent Framingham study showed that large artery stiffness precedes the development of hypertension.32 This report indicated that arterial stiffening may be the cause of hypertension.32 Two longitudinal studies have demonstrated that arterial stiffness predicts an increase in systolic BP and incident hypertension.8,9 This is especially true for aging-related arterial stiffening.1 Kidney function was normal (Figure S2G and S2H), excluding the involvement of renal dysfunction in HFD-induced arterial stiffness and hyper-tension in KL(+/−) mice.
Although HFD caused arterial stiffening only in KL(+/−) mice, it increased total plasma cholesterol levels in both KL(+/−) and WT mice to a similar extent (Figure 2). Therefore, KL deficiency makes animals more susceptible to HFD-induced arterial damage. KL is predominately expressed in
kidneys.10–12 We showed that KL+/−-deficient mice have one half of KL in kidneys when compared with WT mice.33 Kidneys are the major source of circulating klotho.34 Indeed, plasma levels of KL protein were decreased by 50% in KL+/−-deficient mice (Figure S1). These results suggest that a reduction in the circulating KL may synergize with increased plasma LDL-cholesterol levels to promote HFD-induced arterial stiffening and hypertension. Therefore, KL-deficient mice are a unique animal model for studying the mechanism of arterial stiffness associated with metabolic syndrome.
It is noticed that one half klotho deficiency plus HFD diminished activities of AMPKα (pAMPKα) in aortas (Figure 2). Interestingly, the activation of AMPKα by AICAR abolished the increases in PWV and BP in KL+/− mice fed on HFD (Figure 2; Figure S3). To the best of our knowledge, this is the first report demonstrating that inactivation of AMPKα may be involved in the pathogenesis of arterial stiffening and hypertension. These interesting findings suggest that AMPKα might be a potential therapeutic target for arterial stiffening and hypertension. The impaired AMPKα activity may mediate the downregulation of eNOS activity (phosphorylated eNOS) and Mn-SOD expression, which can be eliminated by the
Figure 4. 5-Aminoimidazole-4-carboxamide-3-ribonucleoside (AICAR) treatments increased phosphorylated adenosine monophosphate–activated protein kinase-α (pAMPKα) (T172), phosphorylated endothelial nitric oxide synthase (p-eNOS) (S1177), and manganese-dependent superoxide dismutase (Mn-SOD) levels in mouse aorta. A, pAMPKα and AMPKα levels. B, p-eNOS and eNOS levels. C, Mn-SOD levels. n=5–8. *P<0.05, **P<0.01, ***P<0.001 vs WT-ND-Saline; +P<0.05, ++P<0.01, +++P<0.001 vs KL+/−-ND-Saline; $$P<0.01, $$$P<0.001 vs WT-HFD-Saline; ^P<0.05, ^^P<0.01, ^^^P<0.001 vs KL+/−-HFD-Saline. HFD indicates high-fat diet; ND, normal diet; and WT, wild-type.
by guest on July 5, 2018http://hyper.ahajournals.org/
activation of AMPKα by AICAR (Figure 4). Normalization of eNOS activity and Mn-SOD expression was associated with abolishment of the increased superoxide levels in aortas of KL+/− mice fed on HFD (Figure 5). The activation of AMPKα also attenuated the increased levels of TGFβ1 and collagen I in aortas from KL+/−-deficient mice fed on HFD. These results strongly suggest that the inactivation of AMPKα is an impor-tant upstream factor in the regulation of arterial stiffening in KL-deficient mice, and that the activation of AMPKα is suf-ficient to restore arterial compliance which is associated with normalization of TGFβ1 and collagen I levels. Arterial col-lagen deposition has been believed to be an important factor contributing to the pathogenesis of arterial stiffening.2 Thus, one of ultimate goals in the treatment of arterial stiffening is to block collagen synthesis.
It is expected that eNOS activity was increased by 1-week treatment with AICAR because the activation of AMPKα by AICAR would functionally interact with eNOS and upregu-late its activity (phosphorylation) via LKB1,30 which occurs quickly. Indeed, the nitric oxide (NO) level was increased within 1 week of treatment with AICAR (Figure S4). Therefore, the early and quick drop in PWV and BP within 1 week of treatment may be partially attributed to relaxation of blood vessels caused by increased bioavailability of NO.
Thus, there may be a functional component of arterial stiffness caused by increased vascular tension in KL+/− mice treated with HFD. By contrast, the structural recovery of blood ves-sels is a relatively slow process although it contributes to the attenuation of arterial stiffening and hypertension by AICAR.
KL deficiency did not significantly affect metabolic parameters (cholesterol, body weight, and phosphorus) under normal or HFD diets (Figure 2). AICAR only slightly decreased the HFD-induced increase in plasma cholesterol levels (Figure S3). It has been well documented that AICAR increases fatty acid oxidation via the activation of the ACC1 and ACC2 pathways in skeletal muscles and liver.35–38 AICAR may also inhibit cholesterol synthesis in the liver.39 Metformin, another AMPK activator, also decreases the cho-lesterol levels.40 It is noticed, however, that the slight drop of plasma cholesterol levels cannot explain the significant attenuating effect of AICAR on HFD-induced arterial stiff-ening and hypertension (Figure 3). On the contrary, AICAR can directly activate AMPKα in vascular cells which, in turn, increases eNOS activity (Figure 4) leading to improvement in arterial stiffening and hypertension in KL+/− mice fed on HFD. Indeed, AICAR almost abolished the increase in PWV and BP, suggesting that the rescuing effect of AICAR may be primarily mediated by its vascular effect.
Figure 5. 5-Aminoimidazole-4-carboxamide-3-ribonucleoside (AICAR) alleviated arterial superoxide production in KL-deficient mice with high-fat diet (HFD). A, Representative images of aortic dihydroethidium (DHE) staining. B, Quantification of DHE staining for mouse aorta. n=5–8. ***P<0.001 vs WT-ND-Saline; +++P<0.001 vs KL+/−-ND-Saline; ^^^P<0.001 vs KL+/−-HFD-Saline. HFD indicates high-fat diet; ND, normal diet; and WT, wild-type.
by guest on July 5, 2018http://hyper.ahajournals.org/
Lin et al Klotho Deficiency and Arterial Stiffening 571
KL gene deficiency or mutation is associated with arterial stiffening and hypertension.1,11 A decrease in plasma Klotho is used as a biomarker of arterial stiffening in patients with chronic kidney disease.41 Aging-related hypertension is largely caused by arterial stiffening or remodeling.1 Unfortunately, the current antihypertensive drugs are mainly designed to reduce peripheral resistance and are not adequate to alter the patho-logical process of arterial stiffening or even selectively reduce systolic BP in isolated systolic hypertension.18 Thus, this study suggests that supplementation with Klotho protein or pharmacological activation of AMPKα may be a novel thera-peutic strategy to alleviate arterial stiffening and hypertension. The antihypertensive effect of AICAR may be mediated by the activation of eNOS and decreased arterial stiffening.
To assess the direct effect of cholesterol in SMCs, water-soluble cholesterol was added to the cultured SMCs. Cholesterol loading by water-soluble cholesterol has been shown to induce mouse aortic SMCs into a macrophage-like state.42 Water-soluble cholesterol delivered cholesterol rap-idly and directly to the plasma membrane.43 Thus, cholesterol loading to cultured aortic SMCs might recapture the effects of high LDL-cholesterol on aortic SMCs in animals fed on HFD. KL protein-deficient serum exacerbated cholesterol-induced collagen I protein expression, and AICAR abolished the promoting effects of KL deficiency on cholesterol-induced collagen I expression in SMCs (Figure S6). AMPK has been reported to inhibit TGFβ-induced fibrogenic responses (col-lagen I) in hepatic stellate cells by targeting transcriptional coactivator p300.44 Adenoviral transduction of constitutively active AMPKα was sufficient to prevent TGFβ-induced colla-gen I and fibronectin in cultured fibroblasts.45 Therefore, these
data strongly suggest that AICAR decreased arterial stiffen-ing largely via suppressing cholesterol-induced collagen I synthesis.
Interestingly, KL deficiency plus cholesterol loading also attenuated the phosphorylation level of LKB1 and protein expression of LKB1 in SMCs (Figure S6). LKB1 is one of the key activators of AMPKα in various mammalian cells.30 Therefore, these results suggest that KL deficiency plus cho-lesterol might inactivate AMPKα likely through decreasing LKB1 activity.
In this study, BP was measured using a computerized vol-ume–pressure recording tail-cuff method, a noninvasive and high-throughput measurement technique. It facilitates long-term monitoring of BP in unanesthetized animals. This method has been confirmed to be in good agreement with the radio-telemetry measurement and recommended by the American Heart Association.46 The repeatable measurements of BP over a 15-week period are a strong guarantee for the reliability of the BP data. This method can reliably monitor BP and is a common method for monitoring BP in our laboratory.47–50
KL deficiency and HFD did not affect aortic elastin pro-tein expression levels (Figures 2 and 6) but increased elas-tic lamellae breaks (Figure S5). The increased elastin streak fragmentation, which seems to be mediated by downregula-tion of AMPKα, would also contribute to arterial stiffening. Another interesting finding is that KL-deficient mice fed on HFD developed arterial calcification (Figure S5). Arterial calcification could contribute to arterial stiffening. RUNX2 is a reliable marker of osteoblasts and has been used as an indicator of bone formation. Therefore, KL deficiency plus HFD leads to arterial calcification likely via inactivation
Figure 6. 5-Aminoimidazole-4-carboxamide-3-ribonucleoside (AICAR) downregulated collagen I, Runx2, and transforming growth factor β1 (TGF-β1) levels in mouse aortas. A, Representative Western blots fro collagen I, tropoelastin, Runx2, and TGFβ1. Quantification of collagen I (B), TGFβ1 (C), Runx2 (D), and tropoelastin (E). n=5–8. *P<0.05, **P<0.01, ***P<0.001 vs WT-ND-Saline; +P<0.05, ++P<0.01, +++P<0.001 vs KL+/−-ND-Saline; $$P<0.01, $$$P<0.001 vs WT-HFD-Saline; ^P<0.05, ^^P<0.01, ^^^P<0.001 vs KL+/−-HFD-Saline. HFD indicates high-fat diet; ND, normal diet; and WT, wild-type.
by guest on July 5, 2018http://hyper.ahajournals.org/
of AMPKα because AICAR abolished the upregulation of RUNX2 and arterial calcification in KL-deficient mice fed on HFD (Figure 6; Figure S5).
The serum levels of klotho decrease with age after 40 years.31 By contrast, the prevalence of arterial stiffening and hypertension increases with age.1 KL+/− mice were used which mimics a half KL reduction in the aged population.31 KL homozygous (−/−) mice develop extensive aging pheno-types and die before the age of 8 weeks (body weight = 8 g).10 KL homozygous mice also demonstrate severe hyperphos-phatemia, emphysema, and soft tissue calcification.11,12 As a result, klotho homozygous mice were not used for this study.
PerspectivesThis study revealed a previously unidentified role of KL defi-ciency in promoting the development of HFD-induced arterial stiffening and hypertension. Protein expression of pAMPKα, phosphorylated eNOS, and Mn-SOD was downregulated, whereas protein expression of collagen I, Runx2, and TGFβ1 was upregulated in aortas of KL+/− mice fed on HFD, which can be abolished by the activation of AMPKα by AICAR. Therefore, KL deficiency might promote HFD-induced arte-rial stiffening via downregulation of AMPKα activity, which leads to upregulation of collagen I levels in aortas. Therapeutic activation of AMPKα might be a novel strategy for alleviating arterial stiffening and hypertension. It should be mentioned that hypertension is a complicated and multifactorial disor-der. The antihypertensive effect of AMPK activation may not be applied to other forms of hypertension that do not involve reduction of AMPK activity.
Sources of FundingThis work was supported by National Institutes of Health (NIH) HL R01 118558, DK093403, HL105302, and HL102074. This publica-tion was made possible by NIH grant number 9P20GM104934-06 from the Center of Biomedical Research Excellence (COBRE) Program of the National Institute of General Medical Sciences.
DisclosuresNone.
References 1. Sun Z. Aging, arterial stiffness, and hypertension. Hypertension.
2015;65:252–256. doi: 10.1161/HYPERTENSIONAHA.114.03617. 2. Lee HY, Oh BH. Aging and arterial stiffness. Circ J. 2010;74:2257–2262. 3. Adji A, O’Rourke MF, Namasivayam M. Arterial stiffness, its assess-
ment, prognostic value, and implications for treatment. Am J Hypertens. 2011;24:5–17. doi: 10.1038/ajh.2010.192.
4. Boutouyrie P, Tropeano AI, Asmar R, Gautier I, Benetos A, Lacolley P, Laurent S. Aortic stiffness is an independent predictor of primary coro-nary events in hypertensive patients: a longitudinal study. Hypertension. 2002;39:10–15.
5. Matsuoka H, Nakamura M. Cognitive dysfunction and electroencephalo-gram in schizophrenia. Seishin Shinkeigaku Zasshi. 2005;107:307–322.
6. Waldstein SR, Rice SC, Thayer JF, Najjar SS, Scuteri A, Zonderman AB. Pulse pressure and pulse wave velocity are related to cognitive decline in the Baltimore Longitudinal Study of Aging. Hypertension. 2008;51:99–104. doi: 10.1161/HYPERTENSIONAHA.107.093674.
8. Liao D, Arnett DK, Tyroler HA, Riley WA, Chambless LE, Szklo M, Heiss G. Arterial stiffness and the development of hypertension. The ARIC study. Hypertension. 1999;34:201–206.
9. Najjar SS, Scuteri A, Shetty V, Wright JG, Muller DC, Fleg JL, Spurgeon HP, Ferrucci L, Lakatta EG. Pulse wave velocity is an independent predic-tor of the longitudinal increase in systolic blood pressure and of incident hypertension in the Baltimore Longitudinal Study of Aging. J Am Coll Cardiol. 2008;51:1377–1383. doi: 10.1016/j.jacc.2007.10.065.
10. Kuro-o M, Matsumura Y, Aizawa H, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. doi: 10.1038/36285.
11. Xu Y, Sun Z. Molecular basis of Klotho: from gene to function in aging. Endocr Rev. 2015;36:174–193. doi: 10.1210/er.2013-1079.
12. Wang Y, Sun Z. Current understanding of klotho. Ageing Res Rev. 2009;8:43–51. doi: 10.1016/j.arr.2008.10.002.
13. Kurosu H, Yamamoto M, Clark JD, et al. Suppression of aging in mice by the hormone Klotho. Science. 2005;309:1829–1833. doi: 10.1126/science.1112766.
14. Huang CL. Regulation of ion channels by secreted Klotho: mechanisms and implications. Kidney Int. 2010;77:855–860. doi: 10.1038/ki.2010.73.
15. Hu MC, Kuro-o M, Moe OW. Secreted klotho and chronic kidney disease. Adv Exp Med Biol. 2012;728:126–157. doi: 10.1007/978-1-4614-0887-1_9.
16. Liu H, Fergusson MM, Castilho RM, Liu J, Cao L, Chen J, Malide D, Rovira II, Schimel D, Kuo CJ, Gutkind JS, Hwang PM, Finkel T. Augmented Wnt signaling in a mammalian model of accelerated aging. Science. 2007;317:803–806. doi: 10.1126/science.1143578.
17. Chung AW, Yang HH, Sigrist MK, Brin G, Chum E, Gourlay WA, Levin A. Matrix metalloproteinase-2 and -9 exacerbate arterial stiffening and angiogenesis in diabetes and chronic kidney disease. Cardiovasc Res. 2009;84:494–504. doi: 10.1093/cvr/cvp242.
18. Dao HH, Essalihi R, Bouvet C, Moreau P. Evolution and modulation of age-related medial elastocalcinosis: impact on large artery stiffness and isolated systolic hypertension. Cardiovasc Res. 2005;66:307–317. doi: 10.1016/j.cardiores.2005.01.012.
19. Jeffcoate WJ, Rasmussen LM, Hofbauer LC, Game FL. Medial arterial calcification in diabetes and its relationship to neuropathy. Diabetologia. 2009;52:2478–2488. doi: 10.1007/s00125-009-1521-6.
20. Temmar M, Liabeuf S, Renard C, Czernichow S, Esper NE, Shahapuni I, Presne C, Makdassi R, Andrejak M, Tribouilloy C, Galan P, Safar ME, Choukroun G, Massy Z. Pulse wave velocity and vascular calcification at different stages of chronic kidney disease. J Hypertens. 2010;28:163–169. doi: 10.1097/HJH.0b013e328331b81e.
21. Bouvet C, Moreau S, Blanchette J, de Blois D, Moreau P. Sequential acti-vation of matrix metalloproteinase 9 and transforming growth factor beta in arterial elastocalcinosis. Arterioscler Thromb Vasc Biol. 2008;28:856–862. doi: 10.1161/ATVBAHA.107.153056.
23. Ruderman NB, Carling D, Prentki M, Cacicedo JM. AMPK, insulin resis-tance, and the metabolic syndrome. J Clin Invest. 2013;123:2764–2772. doi: 10.1172/JCI67227.
24. Srivastava RA, Pinkosky SL, Filippov S, Hanselman JC, Cramer CT, Newton RS. AMP-activated protein kinase: an emerging drug target to regulate imbalances in lipid and carbohydrate metabolism to treat cardio-metabolic diseases. J Lipid Res. 2012;53:2490–2514. doi: 10.1194/jlr.R025882.
25. Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, Hardie DG. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem. 1996;271:27879–27887.
26. Ford RJ, Teschke SR, Reid EB, Durham KK, Kroetsch JT, Rush JW. AMP-activated protein kinase activator AICAR acutely lowers blood pressure and relaxes isolated resistance arteries of hypertensive rats. J Hypertens. 2012;30:725–733. doi: 10.1097/HJH.0b013e32835050ca.
27. Cao X, Li H, Tao H, Wu N, Yu L, Zhang D, Lu X, Zhu J, Lu Z, Zhu Q. Metformin inhibits vascular calcification in female rat aortic smooth muscle cells via the AMPK-eNOS-NO pathway. Endocrinology. 2013;154:3680–3689. doi: 10.1210/en.2013-1002.
28. Cecelja M, Chowienczyk P. Role of arterial stiffness in cardiovascular disease. JRSM Cardiovasc Dis. 2012;1:. doi: 10.1258/cvd.2012.012016.
29. Zhou RH, Vendrov AE, Tchivilev I, Niu XL, Molnar KC, Rojas M, Carter JD, Tong H, Stouffer GA, Madamanchi NR, Runge MS. Mitochondrial oxidative stress in aortic stiffening with age: the role of smooth muscle cell function. Arterioscler Thromb Vasc Biol. 2012;32:745–755. doi: 10.1161/ATVBAHA.111.243121.
by guest on July 5, 2018http://hyper.ahajournals.org/
Lin et al Klotho Deficiency and Arterial Stiffening 573
30. Carling D, Thornton C, Woods A, Sanders MJ. AMP-activated protein kinase: new regulation, new roles? Biochem J. 2012;445:11–27. doi: 10.1042/BJ20120546.
31. Xiao NM, Zhang YM, Zheng Q, Gu J. Klotho is a serum factor related to human aging. Chin Med J (Engl). 2004;117:742–747.
32. Kaess BM, Rong J, Larson MG, Hamburg NM, Vita JA, Levy D, Benjamin EJ, Vasan RS, Mitchell GF. Aortic stiffness, blood pressure progression, and incident hypertension. JAMA. 2012;308:875–881. doi: 10.1001/2012.jama.10503.
33. Zhou X, Chen K, Lei H, Sun Z. Klotho gene deficiency causes salt-sen-sitive hypertension via monocyte chemotactic protein-1/CC chemokine receptor 2-mediated inflammation. J Am Soc Nephrol. 2015;26:121–132. doi: 10.1681/ASN.2013101033.
34. Lindberg K, Amin R, Moe OW, Hu MC, Erben RG, Östman Wernerson A, Lanske B, Olauson H, Larsson TE. The kidney is the principal organ mediating klotho effects. J Am Soc Nephrol. 2014;25:2169–2175. doi: 10.1681/ASN.2013111209.
35. Velasco G, Geelen MJ, Guzmán M. Control of hepatic fatty acid oxidation by 5’-AMP-activated protein kinase involves a malonyl-CoA-dependent and a malonyl-CoA-independent mechanism. Arch Biochem Biophys. 1997;337:169–175. doi: 10.1006/abbi.1996.9784.
36. Smith AC, Bruce CR, Dyck DJ. AMP kinase activation with AICAR simulta-neously increases fatty acid and glucose oxidation in resting rat soleus mus-cle. J Physiol. 2005;565(pt 2):537–546. doi: 10.1113/jphysiol.2004.081679.
37. Viollet B, Guigas B, Leclerc J, Hébrard S, Lantier L, Mounier R, Andreelli F, Foretz M. AMP-activated protein kinase in the regulation of hepatic energy metabolism: from physiology to therapeutic perspectives. Acta Physiol (Oxf). 2009;196:81–98. doi: 10.1111/j.1748-1716.2009.01970.x.
38. O’Neill HM, Lally JS, Galic S, et al. AMPK phosphorylation of ACC2 is required for skeletal muscle fatty acid oxidation and insulin sen-sitivity in mice. Diabetologia. 2014;57:1693–1702. doi: 10.1007/s00125-014-3273-1.
39. Sriwijitkamol A, Musi N. Advances in the development of AMPK-activating compounds. Expert Opin Drug Discov. 2008;3:1167–1176. doi: 10.1517/17460441.3.10.1167.
40. Xu T, Brandmaier S, Messias AC, et al. Effects of metformin on metabo-lite profiles and LDL cholesterol in patients with type 2 diabetes. Diabetes Care. 2015;38:1858–1867. doi: 10.2337/dc15-0658.
41. Kitagawa M, Sugiyama H, Morinaga H, Inoue T, Takiue K, Ogawa A, Yamanari T, Kikumoto Y, Uchida HA, Kitamura S, Maeshima Y, Nakamura K, Ito H, Makino H. A decreased level of serum soluble Klotho is an indepen-dent biomarker associated with arterial stiffness in patients with chronic kid-ney disease. PLoS One. 2013;8:e56695. doi: 10.1371/journal.pone.0056695.
42. Rong JX, Shapiro M, Trogan E, Fisher EA. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc Natl Acad Sci U S A. 2003;100:13531–13536. doi: 10.1073/pnas.1735526100.
43. Christian AE, Haynes MP, Phillips MC, Rothblat GH. Use of cyclo-dextrins for manipulating cellular cholesterol content. J Lipid Res. 1997;38:2264–2272.
44. Lim JY, Oh MA, Kim WH, Sohn HY, Park SI. AMP-activated protein kinase inhibits TGF-β-induced fibrogenic responses of hepatic stel-late cells by targeting transcriptional coactivator p300. J Cell Physiol. 2012;227:1081–1089. doi: 10.1002/jcp.22824.
46. Feng M, Whitesall S, Zhang Y, Beibel M, D’Alecy L, DiPetrillo K. Validation of volume-pressure recording tail-cuff blood pressure measure-ments. Am J Hypertens. 2008;21:1288–1291. doi: 10.1038/ajh.2008.301.
47. Crosswhite P, Sun Z. Ribonucleic acid interference knockdown of interleukin 6 attenuates cold-induced hypertension. Hypertension. 2010;55:1484–1491. doi: 10.1161/HYPERTENSIONAHA.109.146902.
48. Sun Z, Cade R, Zhang Z, Alouidor J, Van H. Angiotensinogen gene knockout delays and attenuates cold-induced hypertension. Hypertension. 2003;41:322–327.
49. Zhou X, Chen K, Wang Y, Schuman M, Lei H, Sun Z. Antiaging gene Klotho regulates adrenal CYP11B2 expression and aldosterone synthesis [published online ahead of print October 15, 2015]. J Am Soc Nephrol. doi: 10.1681/ASN.2015010093. http://jasn.asnjournals.org/content/early/2015/10/14/ASN.2015010093.full. Accessed October 15, 2015.
50. Wang Y, Sun Z. Klotho gene delivery prevents the progression of sponta-neous hypertension and renal damage. Hypertension. 2009;54:810–817. doi: 10.1161/HYPERTENSIONAHA.109.134320.
What Is New?• It is new and interesting that haplodeficiency of klotho gene accelerates
the development of high-fat diet–induced arterial stiffening and hyper-tension.
•This study demonstrates, for the first time, that klotho gene deficiency plus high-fat diet downregulates vascular AMPKα expression and activity.
What Is Relevant?• It is significant that klotho deficiency promotes high-fat diet -induced
arterial stiffening and hypertension which are prevalent cardiovascular disorders associated with aging.
•This study reveals that activation of AMP-activated protein kinase-α may be a new therapeutic strategy for arterial stiffening, an independent risk factor for cardiovascular mortality and morbidity.
Summary
Klotho deficiency promoted high-fat diet–induced arterial stiffening and hypertension. The promoting effects of Klotho deficiency on arterial stiffening might be mediated by downregulation of AMP-activated protein kinase-α activity.
Novelty and Significance
by guest on July 5, 2018http://hyper.ahajournals.org/
is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Hypertension doi: 10.1161/HYPERTENSIONAHA.115.068252016;67:564-573; originally published online January 18, 2016;Hypertension.
http://hyper.ahajournals.org/content/67/3/564World Wide Web at:
The online version of this article, along with updated information and services, is located on the
is online at: Hypertension Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer this process is available in the
click Request Permissions in the middle column of the Web page under Services. Further information aboutOffice. Once the online version of the published article for which permission is being requested is located,
can be obtained via RightsLink, a service of the Copyright Clearance Center, not the EditorialHypertensionin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on July 5, 2018http://hyper.ahajournals.org/
Anti-aging Gene Klotho Deficiency Promoted High Fat Diet-induced Arterial Stiffening via Inactivation of AMP-activated Protein Kinase
Yi Lin, Jianglei Chen, Zhongjie Sun
Department of Physiology, College of Medicine, University of Oklahoma Health Sciences Center, Oklahoma City, OK73104, USA
Running Title: Klotho deficiency and arterial stiffening Total characters: 6,337; Total Words: 1,157 Address Correspondence to: Zhongjie Sun, MD, PhD, FAHA Professor and Vice Chair Chair, Research Committee Professor of Physiology Director, The Robert & Mary Cade Laboratory BMSB 662A, Box 26901 Department of Physiology, BMSB 662A College of Medicine University of Oklahoma Health Sciences Center (OUHSC) 940 Stanton L. Young Blvd. Oklahoma City, OK 73126-0901 USA [email protected] Tel. 405-271-2226 x56237 Fax. 405-271-3181
28456-R, Santa Cruz Biotechnology), LKB1 (sc-5638, Santa Cruz
Biotechnology), CaMKKα (sc-11370, Santa Cruz Biotechnology), and CaMKKβ
(sc-50341, Santa Cruz Biotechnology. The blot was then rinsed and reprobed
with antibodies against α-tubulin (2135, Cell Signaling), β-actin (Abcam), AMPKα
(2193, Cell Signaling), or eNOS (612664, BD Transduction) for the loading
control.
RNA Isolation and RT-PCR. Total RNA was purified from mouse kidney and
MOVAS using TRIzol® Reagent, followed by Qiagen RNeasy® Mini Kit. RNA
(500 ng) was reverse-transcribed using SuperScriptTM III Reverse Transcriptase
with OligodT20 in the presence of 10ul dNTP for 1h at 50ºC. The resulting
cDNAs were used as templates for PCR with oligonucleotides primers to amplify
Klotho mRNA and β-actin mRNA. One specific pair of primers for mouse KL
mRNA was used, which target exons 1 and 2 of mouse Klotho cDNA (F: 5’-
CCTGGTCGACCATTTCAG-3’ and R: 5’-
AGCACAAAGTCGACAGACTTCTGGC-3’).9 The primers for β-actin gene were
used as the internal control. The PCR product for β-actin was 708 bps. PCR
reactions (50-µL volume) contained 3 µl of above cDNA, 0.2 µM of appropriate
oligonucleotide primer pair, and 1 x of New England Biolabs Taq 2X Mater Mix.
PCR amplification conditions were as follows: 5 min at 95°C followed by 30
cycles of 95°C for 1min, optimized annealing temperature for each primer pair for
1 min, and 68°C for 1 min. The PCR products were separated on 1.5% agarose
gels and stained with ethidium bromide. The bands were visualized using a
ChemiDoc System BioRad Imager.
References 1. Sriwijitkamol A, Musi N. Advances in the development of AMPK-activating compounds.
Expert Opin Drug Discov. 2008;3:1167-1176. 2. Reddy AK, Taffet GE, Li YH, Lim SW, Pham TT, Pocius JS, Entman ML, Michael LH, Hartley
CJ. Pulsed Doppler signal processing for use in mice: applications. IEEE Trans Bio-med Eng. 2005;52:1771-1783.
3. Crosswhite P, Sun Z. Ribonucleic acid interference knockdown of interleukin 6 attenuates cold-induced hypertension. Hypertension. 2010;55:1484-1491.
4. Whitesall SE, Hoff JB, Vollmer AP, D'Alecy LG. Comparison of simultaneous measurement of mouse systolic arterial blood pressure by radiotelemetry and tail-cuff methods. Am J Physiol Heart Circ Physiol. 2004;286:H2408-2415.
5. Feng M, Whitesall S, Zhang Y, Beibel M, D'Alecy L, DiPetrillo K. Validation of volume-pressure recording tail-cuff blood pressure measurements. Am J Hypertens. 2008;21:1288-1291.
6. Sun Z, Cade R, Zhang Z, Alouidor J, Van H. Angiotensinogen gene knockout delays and attenuates cold-induced hypertension. Hypertension. 2003;41(2):322-327.
7. Sun Z, Bello-Roufai M, Wang X. RNAi inhibition of mineralocorticoid receptors prevents the development of cold-induced hypertension. Am J Physiol Heart Circ Physiol. 2008;294:H1880-1887.
8. Wang Y, Sun Z. Klotho gene delivery prevents the progression of spontaneous hypertension and renal damage. Hypertension. 2009;54:810-817.
9. Lin Y, Sun Z. Antiaging gene Klotho enhances glucose-induced insulin secretion by up-regulating plasma membrane levels of TRPV2 in MIN6 beta-cells. Endocrinology. 2012;153:3029-3039.
10. Wang X, Skelley L, Wang B, Mejia A, Sapozhnikov V, Sun Z. AAV-based RNAi silencing of NADPH oxidase gp91(phox) attenuates cold-induced cardiovascular dysfunction. Human Gene Ther. 2012;23:1016-1026.
A B
C D
-3 -2 -1 0 1 2 3 4 5 6 7 8 9 10 11 12 13 1475
100
125
150
175WT-normal dietWT-high fat diet
KL+/--normal dietKL+/--high fat diet
high fat diet***+++^^^
***+++^^^
*
*
***+++^^
**
*
***
****
+̂***+++^^^ ***
+++^^^
***+++^^^ ***
+++^^^
**
**
**
**
****
weeks
Mea
n bl
ood
pres
sure
(mm
Hg)
-3 -2 -1 0 1 2 3 4 5 6 7 8 9 10 11 12 13 1475
100
125
150 WT-normal dietWT-high fat diet
KL+/--normal dietKL+/--high fat diet
high fat diet
****
***+++̂
***
*******
+̂***+++^^^
***+++^^^ ***
+++^^^***+++^^^
***+++^^^
***+++^^^
weeks
Dia
stol
ic b
lood
pre
ssur
e(m
mH
g)
** *
* *
****
0 30 60 90 120 1500
100
200
300WT-normal dietWT-high fat diet
KL+/--normal dietKL+/--high fat diet
Time (minutes)
Blo
od g
luco
se le
vels
(nor
mal
ized
to th
eba
sal,
%)
E
65 kD Short form KL50 kD IgG
WT KL+/-0.0
0.5
1.0
1.5
**
Rel
ativ
e le
vels
of
shor
t for
m K
L in
mou
se s
erum
F
0 30 60 90 120 1500
25
50
75
100
125
150WT-normal dietWT-high fat diet
KL+/--normal dietKL+/--high fat diet
Time (minutes)
Blo
od g
luco
se le
vels
(nor
mal
ized
to th
e ba
sal
,%)
Supplemental Data
Figure S1. (A), mean arterial blood pressure for the first 13 weeks. (B), diastolic
blood pressure for the first 13 weeks. (C), insulin sensitivity test in the 11th week
of HFD feeding. (D), glucose tolerance test in the 9th week of HFD feeding.
(E&F), plasma KL protein levels in KL deficient and WT mice. *p<0.05, **p<0.01,
***p<0.001 vs WT-normal diet (ND); +p<0.05, +++p<0.001 vs KL+/--ND; ^p<0.05,
^^p<0.01, ^^^p<0.001 vs WT-HFD.
WT-NDH
&E
Aliz
arin
-Red
WT-HFD KL+/--ND KL+/--HFDA
WT-norm
al die
t
WT-high
fat d
iet
KL+/- -no
rmal
diet
KL+/- -hi
gh fa
t diet
0.0
0.1
0.2
Are
a fr
actio
n of
Aliz
arin
-red
sta
inin
g (%
of a
ortic
med
ium
)
10x
40x
WT-ND WT-HFD KL+/--ND KL+/--HFDC
WT-norm
al die
t
WT-high
fat d
iet
KL+/- -no
rmal
diet
KL+/- -hi
gh fa
t diet
0.00
0.25
0.50
0.75
1.00
Rup
ture
s of
ela
stic
lam
ella
e in
aor
ta (p
er10
4µ m
2 )
B
D
E F
WT-ND WT-HFD KL+/--ND KL+/--HFD0
1
2
3
4
Blo
od a
lbum
in (g
/dL)
WT-ND WT-HFD KL+/--ND KL+/--HFD0
10
20
30
Blo
od u
rea
nitr
ogen
(mg/
dL)
HG
WT-ND WT-HFD KL+/--ND KL+/--HFD0.00
0.25
0.50
0.75
Blo
od fr
ee fa
tty
acid
es(m
mol
/L)
WT-ND WT-HFD KL+/--ND KL+/--HFD0
25
50
75
Blo
od tr
igly
ceri
des
(mg/
dL)
Figure S2. (A), H&E staining and Alizarin-red staining of mouse aorta in the 6th
week of HFD feeding. (B), quantification of Alizarin-red staining in arterial
medium. (C). Verhoeff’s Elastic Lamellae staining in mouse aorta in the 6th week
of HFD feeding. (D), the number of ruptures of aortic elastic lamellae (per 104
Figure S6. KL protein-deficient FBS exacerbated cholesterol-induced collagen I protein expression and AICAR abolished the promoting effect of KL deficiency on cholesterol-induced collagen I expression in cultured mouse aortic smooth muscle cells. Confluent cells were incubated with
various reagents for 48 hours. (A&B), effects of water-soluble cholesterol on
collagen I expression. (C-E), effects of AICAR on pAMPKα(T172) and AMPKα.
(F&G), effects of AICAR on collagen I synthesis induced by cholesterol plus KL
deficient serum. (H-K), effects of cholesterol plus KL deficiency on pLKB1 and
LKB1. N=5-8. *p<0.05, **p<0.01, ***p<0.001 vs. no treatment; +p<0.05,
++p<0.01, +++p<0.001 vs. cholesterol; ^p<0.05, ^^^p<0.001 vs. CHL-KL deficient