Research paper CCT128930 induces cell cycle arrest, DNA damage, and autophagy independent of Akt inhibition Feng-Ze Wang a, b , Zheng-Yao Chang a , Hong-Rong Fei c , Ming-Feng Yang b , Xiao-Yi Yang b , Bao-Liang Sun b, d, * a School of Biological Science, Taishan Medical University, Taian 271016, PR China b Key Lab of Cerebral Microcirculation in Universities of Shandong, Taishan Medical University, Taian 271000, PR China c School of Pharmacology, Taishan Medical University, Taian 271016, PR China d Department of Neurology, Affiliated Hospital of Taishan Medical University, Taian, Shandong 271000, China article info Article history: Received 5 March 2014 Accepted 22 April 2014 Available online 1 May 2014 Keywords: CCT128930 Akt Cell cycle DNA damage Autophagy abstract PI3K/Akt/mTOR pathway plays an important role in tumor progression and anti-cancer drug resistance. The aim of the present study is to determine the antitumor effect of CCT128930, a novel small molecule inhibitor of Akt, in the HepG2 hepatoma cancer cells. Our results showed that at low concentrations, CCT128930 increased, but not inhibited, the phosphorylation of Akt in HepG2 and A549 cells. CCT128930 inhibited cell proliferation by inducing cell cycle arrest in G1 phase through downregulation of cyclinD1 and Cdc25A, and upregulation of p21, p27 and p53. A higher dose (20 mM) of CCT128930 triggered cell apoptosis with activation of caspase-3, caspase-9, and PARP. Treatment with CCT128930 increased phosphorylation of ERK and JNK in HepG2 cells. CCT128930 activated DNA damage response of HepG2 cell characterized by phosphorylation of H2AX, ATM (ataxia-telangiectasia mutated), Chk1 and Chk2. Upon exposure to CCT128930 at a higher concentration, HepG2 cells exhibited autophagy was accom- panied by an increase the levels of LC3-II and Beclin-1. Blocking autophagy using chloroquine magnified CCT128930-induced apoptotic cell death and the phosphorylation of H2AX. The results in this study have advanced our current understandings of the anti-cancer mechanisms of CCT128930 in cancer cells. Ó 2014 Elsevier Masson SAS. All rights reserved. 1. Introduction It is widely accepted that PI3K/Akt/mTOR pathway plays a key role in controlling cell growth, apoptosis and cell differentiation. Mutations or genetic alterations of many components of the PI3K/ Akt pathway frequently occur in human cancers [1]. Signaling through PI3K drives a number of cell functions by the activation of the Akt protein (also known as protein kinase B, PKB). The down- stream effector of PI3K, Akt, increases the cell cycle progression by blocking FOXO-mediated transcription of cell cycle inhibitors that lead to the accumulation and activation of the mTOR-raptor kinase complex, which in turn mediates protein kinase p70S6K1 activation to regulate protein synthesis and cell proliferation [2]. mTOR functions as a central element in a signaling pathway involved in the control of cell proliferation, apoptosis and autophagy, an ubiquitous and evolutionarily conserved process that degrades cytosolic components via the lysosomes and allows cells to survive various forms of stress [3]. Many studies have shown that DNA damage or replication errors results in the inhibition of Cdks activity via activation of the cell cycle checkpoint, which is essential for preventing entry into mitosis in the presence of genomic defects [4,5]. The key regulators of the checkpoint pathways in eukaryotic cell DNA damage response are ataxia-telangiectasia mutated (ATM) and ATM-related (ATR) protein kinases, which are activated in response to DNA damaging agents such as ionizing radiation, ultraviolet light and DNA replication inhibitors [6]. The downstream target kinases of ATR/ATM in response to DNA damage are Chk1 and Chk2. ATR/ATM activate Chk1 and Chk2 protein kinases responsible for the inhibi- tion via phosphorylation of the Cdc25 phosphatase. Although Chk1 or Chk2 are known to trigger G2/M phase cell cycle arrest, these kinases are also known to mediate ATM-dependent G1 checkpoint due to activating p53 tumor suppressor protein by phosphorylation [7,8]. Autophagy is a conservative process of protein degradation through autophagosome and lysosome system, which generally * Corresponding author. Department of Neurology, Affiliated Hospital of Taishan Medical University, Taian, Shandong 271000, PR China. E-mail address: [email protected](B.-L. Sun). Contents lists available at ScienceDirect Biochimie journal homepage: www.elsevier.com/locate/biochi http://dx.doi.org/10.1016/j.biochi.2014.04.008 0300-9084/Ó 2014 Elsevier Masson SAS. All rights reserved. Biochimie 103 (2014) 118e125
CCT128930 induces cell cycle arrest, DNA damage, and autophagyindependent of Akt inhibition
Feng-Ze Wang a,b, Zheng-Yao Chang a, Hong-Rong Fei c, Ming-Feng Yang b, Xiao-Yi Yang b,Bao-Liang Sun b,d,*
a School of Biological Science, Taishan Medical University, Taian 271016, PR ChinabKey Lab of Cerebral Microcirculation in Universities of Shandong, Taishan Medical University, Taian 271000, PR Chinac School of Pharmacology, Taishan Medical University, Taian 271016, PR ChinadDepartment of Neurology, Affiliated Hospital of Taishan Medical University, Taian, Shandong 271000, China
a r t i c l e i n f o
Article history:Received 5 March 2014Accepted 22 April 2014Available online 1 May 2014
http://dx.doi.org/10.1016/j.biochi.2014.04.0080300-9084/� 2014 Elsevier Masson SAS. All rights re
a b s t r a c t
PI3K/Akt/mTOR pathway plays an important role in tumor progression and anti-cancer drug resistance.The aim of the present study is to determine the antitumor effect of CCT128930, a novel small moleculeinhibitor of Akt, in the HepG2 hepatoma cancer cells. Our results showed that at low concentrations,CCT128930 increased, but not inhibited, the phosphorylation of Akt in HepG2 and A549 cells. CCT128930inhibited cell proliferation by inducing cell cycle arrest in G1 phase through downregulation of cyclinD1and Cdc25A, and upregulation of p21, p27 and p53. A higher dose (20 mM) of CCT128930 triggered cellapoptosis with activation of caspase-3, caspase-9, and PARP. Treatment with CCT128930 increasedphosphorylation of ERK and JNK in HepG2 cells. CCT128930 activated DNA damage response of HepG2cell characterized by phosphorylation of H2AX, ATM (ataxia-telangiectasia mutated), Chk1 and Chk2.Upon exposure to CCT128930 at a higher concentration, HepG2 cells exhibited autophagy was accom-panied by an increase the levels of LC3-II and Beclin-1. Blocking autophagy using chloroquine magnifiedCCT128930-induced apoptotic cell death and the phosphorylation of H2AX. The results in this study haveadvanced our current understandings of the anti-cancer mechanisms of CCT128930 in cancer cells.
� 2014 Elsevier Masson SAS. All rights reserved.
1. Introduction
It is widely accepted that PI3K/Akt/mTOR pathway plays a keyrole in controlling cell growth, apoptosis and cell differentiation.Mutations or genetic alterations of many components of the PI3K/Akt pathway frequently occur in human cancers [1]. Signalingthrough PI3K drives a number of cell functions by the activation ofthe Akt protein (also known as protein kinase B, PKB). The down-stream effector of PI3K, Akt, increases the cell cycle progression byblocking FOXO-mediated transcription of cell cycle inhibitors thatlead to the accumulation and activation of the mTOR-raptor kinasecomplex, which in turnmediates protein kinase p70S6K1 activationto regulate protein synthesis and cell proliferation [2]. mTORfunctions as a central element in a signaling pathway involved inthe control of cell proliferation, apoptosis and autophagy, anubiquitous and evolutionarily conserved process that degrades
Affiliated Hospital of Taishana.un).
served.
cytosolic components via the lysosomes and allows cells to survivevarious forms of stress [3].
Many studies have shown that DNA damage or replication errorsresults in the inhibition of Cdks activity via activation of the cellcycle checkpoint, which is essential for preventing entry intomitosis in the presence of genomic defects [4,5]. The key regulatorsof the checkpoint pathways in eukaryotic cell DNA damageresponse are ataxia-telangiectasia mutated (ATM) and ATM-related(ATR) protein kinases, which are activated in response to DNAdamaging agents such as ionizing radiation, ultraviolet light andDNA replication inhibitors [6]. The downstream target kinases ofATR/ATM in response to DNA damage are Chk1 and Chk2. ATR/ATMactivate Chk1 and Chk2 protein kinases responsible for the inhibi-tion via phosphorylation of the Cdc25 phosphatase. Although Chk1or Chk2 are known to trigger G2/M phase cell cycle arrest, thesekinases are also known to mediate ATM-dependent G1 checkpointdue to activating p53 tumor suppressor protein by phosphorylation[7,8].
Autophagy is a conservative process of protein degradationthrough autophagosome and lysosome system, which generally
F.-Z. Wang et al. / Biochimie 103 (2014) 118e125 119
functions as a homeostasis mechanism to maintain a balance be-tween the synthesis and degradation of cellular products. The roleof autophagy in cancer and cell death is apparently quite complexand poorly understood. Evidence indicates that the predominantfunction of autophagy in cancer cells is to confer stress tolerance,which contributes to tumor cell survival during cancer progression.On the other hand, induction of autophagic cell death has beenproposed as a mechanism of cell death, given that characteristics ofautophagy have been discovered in dying cells [9]. Similar to itspotential to either promote cell survival or induce cell death, a largebody of research shows a paradoxical role of autophagy duringchemotherapy [10]. There are increasing evidences indicates thatPI3K/Akt/mTOR inhibitors initiate autophagy as a survival programthat may interfere with their antitumor activity [11]. Therefore,autophagy may be a potential resistance mechanism and its abro-gation may be increase the cytotoxicity of mTOR inhibition.
The prevalence of PI3K/Akt/mTOR signaling abnormalities incancer cells has suggested the potential use of this pathway as anovel therapeutic target. Inhibitors of this signaling pathway arebeing actively developed and tested for tumor therapy [12e14]. Inthis study, we investigate a potential antitumor activity ofCCT128930 in vitro models. We demonstrate that CCT128930 in-hibits the cell proliferation by inducing G1 cell cycle arrest. At highconcentrations, CCT128930 causes apoptosis. Activation of g-H2AX,ATM, Chk1 and Chk2 may be responsible for the DNA damage inCCT128930-treated HepG2 cells. CCT128930 also triggers the auto-phagy, as evidenced by increased levels of the autophagy marker,LC3-II. Inhibitionof autophagybychloroquine (CQ) further increasesthe cytotoxicity of CCT128930. Our results indicate that CCT128930might represent a promising drug for the treatment of cancer.
2. Materials and methods
2.1. Reagent
CCT128930 was purchased from Sata Cruz Biotechnology(Shanghai, China), and dissolved with DMSO. Crystal violet and 3-[4,5-dimethylthiazol-2-y-l]-2, 5-diphenyltetrazolium bromide(MTT) and chloroquine were obtained from SigmaeAldrich (St.Louis, USA). PD98059 was purchased from Promega (Madison,USA). SP600129 was obtained from Calbiochem (CA, USA). AnnexinV-FITC apoptosis detection kit was obtained from KeyGEN Biotech(Nanjing, China). Western Lightning Plus ECL was purchased fromperkinelmer (MA, USA).
Antibodies specific to b-actin, Beclin-1 and LC3-II were pur-chased from SigmaeAldrich. Rabbit anti-Akt, p-Akt, p70SK6,caspase-9, caspase-3, PARP, ATM, p-ATM, ATR, p-ATR, p-Chk2, Chk-1, p-Chk1, g-H2AX, p-p53, p-p38, JNK and Cdc2 polyclonal anti-bodies were obtained from Cell Signaling Technology (Shanghai,China). Mouse anti-Chk2, p21, p27 and p-JNK were purchased fromBD Biosciences. Anti-cyclinD1, Cdc25A, Cdc25B, Cdc25C, cyclinB1,p53, ERK and p-ERKwere purchased fromSanta cruz Biotechnology.
2.2. Cells and cell culture
HepG2 cell and A549 cell were purchased from the Type CultureCollection of the Chinese Academy of Sciences (Shanghai, China),and was cultured in RMPI 1640 medium containing 10% fetal calfserum (Gibco, USA), 100 U/mL penicillin and 100 mg/mL strepto-mycin in a humidified atmosphere (37 �C; 5% CO2).
2.3. Cell viability assay and cell colony formation assay
MTT assay was used to detect cell viability, as reported else-where [15]. Briefly, cells were plated in 96-well plates for 24 h. The
medium was then removed, and cells were treated with variousconcentrations of CCT128930. An amount of 20 mL MTT (5 mg/mL)was added for 4 h. After removing supernatant, 150 mL DMSO wasadded to resolve formazan crystals, and the absorbance wasdetected at 490 nm. For cell colony formation assay, HepG2 cellswere seeded at 1500 cells per well (6-well cell culture plate) andtreated with different concentrations of CCT128930 for 2 weeks.Cells were fixed in 1% glutaraldehyde and stained with 0.5% crystalviolet. Colonies with >30 cells were counted under an invertedmicroscope.
2.4. Cell cycle analysis
Cells were treated with CCT128930 for 24 h. At the end of thetreatment, cells were harvested and washed with ice-cold phos-phate-buffered saline (PBS), and fixed overnight in cold 70%ethanol at 4 �C. After washing with PBS, the cells was digested withRNase A and stained with PI. Samples were analyzed for their DNAcontent by using a FACSCalibur flow cytometry with CellQuestsoftware (Becton Dickinson, USA).
2.5. Annexin V-FITC/PI staining
Annexin V-FITC/PI apoptosis detection kit was used to detectcell apoptosis. To assess the extent of apoptosis induction, cellswere harvested (adherent and suspended cells) and stained ac-cording to the manufacturer’s instructions. Samples were analyzedwith the FACScan flow cytometry and CellQuest analysis software(Becton Dickinson).
2.6. Western blot analysis
After cells were lysed with ice-cold lysis buffer (150 mM NaCl,20 mM TriseHCl, 1% NP-40, 0.5% Na-DOC, 0.1% SDS, 0.2 mmol/LPMSF, and protease inhibitor cocktails) for 20 min on ice. Cell su-pernatants were obtained by centrifugation and the protein con-centration was determined. 30 mg protein was used for westernblotting analysis. Detection of the target proteins on the mem-branes was performed using the ECL Western Blotting DetectionReagents.
2.7. Immunoflourescence assay
Cultured cells were fixed with cold 100% methanol at 4 �C for10 min and permeabilized with PBS-0.1%Triton X-100 for 10 min.After blocked with 3% BSA, the cells were incubated with anti-g-H2AX antibody. After washing, cells were incubated with FITC-conjugated secondary antibodies (Santa cruz, 1:200 dilution),and DAPI was used to stain the nuclei. Fluorescence images wereobtained using a fluorescence microscope with appropriate filtersets.
2.8. Statistical analysis
All data were expressed as mean � SD. Statistical analysis wasperformed by the student’s t-test. P < 0.05 was indicated to bestatistical significant.
3. Results
3.1. CCT128930 inhibits cell proliferation in a concentration-dependent manner
CCT128930 was reported to be a PI3K/mTOR inhibitor [16], andthen we detect the effects of CCT128930 on the phosphorylation of
Fig. 1. The effect of CCT128930 on the phosphorylation state of Akt and p70S6K in HepG2 and A549 cells. Cells were incubated with CCT128930 at the indicated concentrations for24 h and then processed for western blotting analysis. b-actin was used as the loading control.
F.-Z. Wang et al. / Biochimie 103 (2014) 118e125120
Akt, an activator of mTOR, and its downstream effector p70 ribo-somal protein S6 kinase (p70S6K) in HepG2 and A549 cells. Asshown in Fig. 1, CCT128930 treatment did not inhibit the phos-phorylation of Akt (Ser473) and p70S6K (Thr389) at indicatedconcentrations. Much to our surprise, when the cells were treatedwith lower concentration of CCT128930, an increase in phosphor-ylation of Akt was observed.
To investigate the effects of CCT128930 on the growth of cancercells, MTT assay was used to evaluate the HepG2 and A549 cellviability. The results showed that CCT128930 decreased theviability of cancer cells in a dose-dependent manner(IC50 ¼ 30.03 mM in HepG2 cell and IC50 ¼ 32.94 mM in A549 cell)(Fig. 2A). Colony formation assay revealed that treatment with10 mM CCT128930 totally blocked the formation of colonies,
Fig. 2. CCT128930 inhibited the growth of cancer cells. (A) CCT128930 reduced the viabilitypolystyrene culture plates at 37 �C with 5% (v/v) CO2 for one day. After 24 h of incubation,CCT128930 inhibits the colony formation ability of HepG2 cell. The histogram represents thecompared with the control. (C) Morphological changes in CCT128930-treated HepG2 cells. Mconcentrations of CCT128930.
confirming the anti-cancer activity of CCT128930 against cancercells (Fig. 2B). Light microscopy observation of CCT128930-treatedHepG2 cells revealed morphological changes accompanyingshrinkage of the cell size and constriction of cell shape, which maybe an early indicator of responses to cell growth inhibition orapoptotic stimuli (Fig. 2C).
3.2. CCT128930 induces G1 phase arrest in HepG2 and A549 cells
For finding whether the growth inhibitory function ofCCT128930 was associated with its effect on cell cycle progression,we investigated changes of cell cycle distribution in CCT128930-treated cancer cells by flow cytometric analysis. As shown inFig. 3A, CCT128930 treatment significantly increased G1 phase cells
of HepG2 and A549 cells. Cells were seeded at a density of 1 � 106 cells/mL in 96-wellcells were incubated with CCT128930 for 24 h and then processed for MTT assay. (B)mean colony number of 3 independent experiments (mean � SD). *P < 0.05, **P < 0.01orphological changes observed in the HepG2 cells after 24 h of treatment with various
Fig. 3. CCT128930 induced G1 phase arrest in HepG2 and A549 cells. (A) CCT128930 was observed to induce G1 phase arrest. Upon exposure to CCT128930 at indicated con-centrations for 24 h, both HepG2 cells and A549 cells all exhibited G1 phase arrest. (B) Expression of the cell cycle-associated proteins. HepG2 cells were treated with CCT128930 forthe indicated concentrations. Cell lysates were fractionated on SDS-polyacrylamide gels and analyzed by western blotting with antibodies against cell cycle associated proteins. b-actin was shown as loading control.
F.-Z. Wang et al. / Biochimie 103 (2014) 118e125 121
in HepG2 and A549 cells. Investigation of the pivotal proteinsinvolved in cell cycle transition by CCT128930 showed thatexpression of p21, p27 and p53 was increased by CCT128930treatment (Fig. 3B).
3.3. High doses of CCT128930-induced HepG2 cell apoptosis
Next, we performed experiments to determine whether thisinhibitory effect of CCT128930 on cell viability resulted fromapoptotic cell death. Apoptotic or necrotic cells were detected afterincubation with increasing concentrations of CCT128930 for 24 h.
Annexin V/PI flow cytometry results revealed that apoptotic rate in20 mM CCT128930 treatment HepG2 cells increased from 5.33% to12.62% (Fig. 4A), these results indicated that high dose ofCCT128930 induced the cell death in HepG2 cells.
To elucidate whether CCT128930-treated hepG2 cells expresstypical apoptotic markers, we used western blotting analysis todetermine the cleavage of apical pro-caspase-3, pro-caspase-9, andPARP in HepG2 cells. As shown in Fig. 4B, HepG2 cells exhibitedsignificantly increased the cleavage of caspase-3, caspase-9 andPARP after exposure to 20 mM CCT128930, suggesting enhancedapoptotic activity in HepG2 cells.
Fig. 4. High dose of CCT128930 induced cell apoptosis in HepG2 cells. (A) Annexin V/FITC-PI FACS assay of HepG2 cells exposed to various doses of CCT128930 (0e20 mM) for 24 h.The percentage of early apoptotic cells with exposed CCT128930 is located in the lower right quadrant and the percentage of necrotic/apoptosis in terminal stages is located in theupper right quadrant. Data are representative of one of three repeats. (B) Western blotting analysis of apoptosis-related proteins. HepG2 cells were exposed to CCT128930 for 24 h,and western blot analysis was carried out to detect the cleavage of caspases-9, caspase-3 and PARP.
F.-Z. Wang et al. / Biochimie 103 (2014) 118e125122
3.4. ERK and JNK are required for CCT128930-induced cytotoxicityin HepG2 cells
Mitogen-activated protein kinase (MAPK) signaling pathwayactivation contributes to decrease cell proliferation and inducingapoptosis in response to a variety of extra cellular stresses [17], wethen determined whether the activity of ERK, JNK and p38 MAPKwere upregulated in HepG2 cells after treatment with CCT128930.Using western blotting analysis, we found that phosphorylation ofERK and JNK proteinwas increased in HepG2 cells after exposure toCCT128930 for 24 h (Fig. 5A). However, no significant changes ofphosphorylated p38 MAPK were observed from the western blot-ting results.
To assess whether CCT128930-induced cell viability inhibitionwas dependent on ERK and JNK activation, HepG2 cells werecultured with CCT128930 (20 mM) either alone or in combinationwith inhibitors of ERK (PD98059) and JNK1/2 (SP600129) for 24 h,and MTT assays were carried out. As shown in Fig. 5B, inhibition ofcell viability by CCT128930 was partially alleviated in the presenceof PD98059 or SP600125, suggesting a dependence on ERK andJNK signaling for the CCT128930 mediates the cell growth andsurvival.
Fig. 5. Pretreatment ERK and JNK inhibitor partially reduced CCT128930-inhbited cell viaexpression was determined by western blotting analysis. (B) To investigate the effect of thtreated as before with 20 mM PD98059 (PD) and 20 mM SP600125 (SP), respectively. Aftassay. *Indicates that P < 0.05 versus control cells.
3.5. CCT128930 activates DNA damage sensor kinases andassociates cellular checkpoints in HepG2 cells
To further elucidate the possible mechanism of CCT128930-induced cell cycle arrest and apoptosis, we detected the DNAdamage after CCT128930 exposure by examined the expression ofphosphorylation of H2AX (g-H2AX), an indicator of the presence ofDNA damage. We observed an increased phosphorylation of H2AXat Ser-139 in CCT128930-treated HepG2 cells (Fig. 6A). Immuno-fluorescence staining also showed an increased formation of g-H2AX foci in CCT128930-treated HepG2 cells (Fig. 6B).
Both ATM and ATR play a central role in coordinating the DNAdamage response, including cell cycle checkpoint control andapoptosis [18].We thereforemeasured theeffectofCCT128930onthekey signaling proteins of this pathway. TreatmentofHepG2 cellswithindicated concentrations of CCT128930 increased the phosphoryla-tionofATMat Ser-1981without anychange in the total levels of ATM.However, we did not observe any changes in the phosphorylation ofATR (Ser428) in CCT128930-treated cells (Fig. 6C). Since phosphory-lation of Chk1 and Chk2 also serves as an indicator of checkpointactivation in response to DNA damage, we determined whether thephosphorylation of Chk1 and Chk2 were affected in CCT128930-
bility. (A) Cells were treated with different doses of CCT128930 for 24 h and proteine ERK and JNK inhibitor on CCT128930-inhibited cell viability, HepG2 cells were pre-er 24 h incubation with CCT128930 (20 mM), cell viability was analyzed using MTT
Fig. 6. Effect of CCT128930 on DNA damage responses. (A) DNA double-strand breaks, as measured by g-H2AX, were induced by CCT128930 treatment. Whole cell lysates wereprepared and subjected to western blotting using antibodies against indicated proteins. b-actin antibody was used as an internal control to show equal protein loading. (B) Detectionof g-H2AX by immunofluorescence. Cells were treated with the indicated concentrations of CCT128930 for 24 h, stained with rabbit anti-g-H2AX (Ser139) antibody and goat anti-rabbit FITC-conjugated secondary antibody (green), and then counterstained with DAPI (blue). (C) CCT128930 regulates the phosphorylation of ATM, Chk1, and Chk2 in HepG2 cells.Cells were treated with different doses of CCT128930 for 24 h, and then western blotting analysis was performed with antibodies against the indicated proteins. b-actin antibodywas used as a loading control.
F.-Z. Wang et al. / Biochimie 103 (2014) 118e125 123
treatedcells.As showninFig. 6C, exposure toCCT128930significantlyincreased the phosphorylation of Chk1 and Chk2 in HepG2 cells.
3.6. Inhibition of autophagy exacerbates the cytotoxic activity ofCCT128930
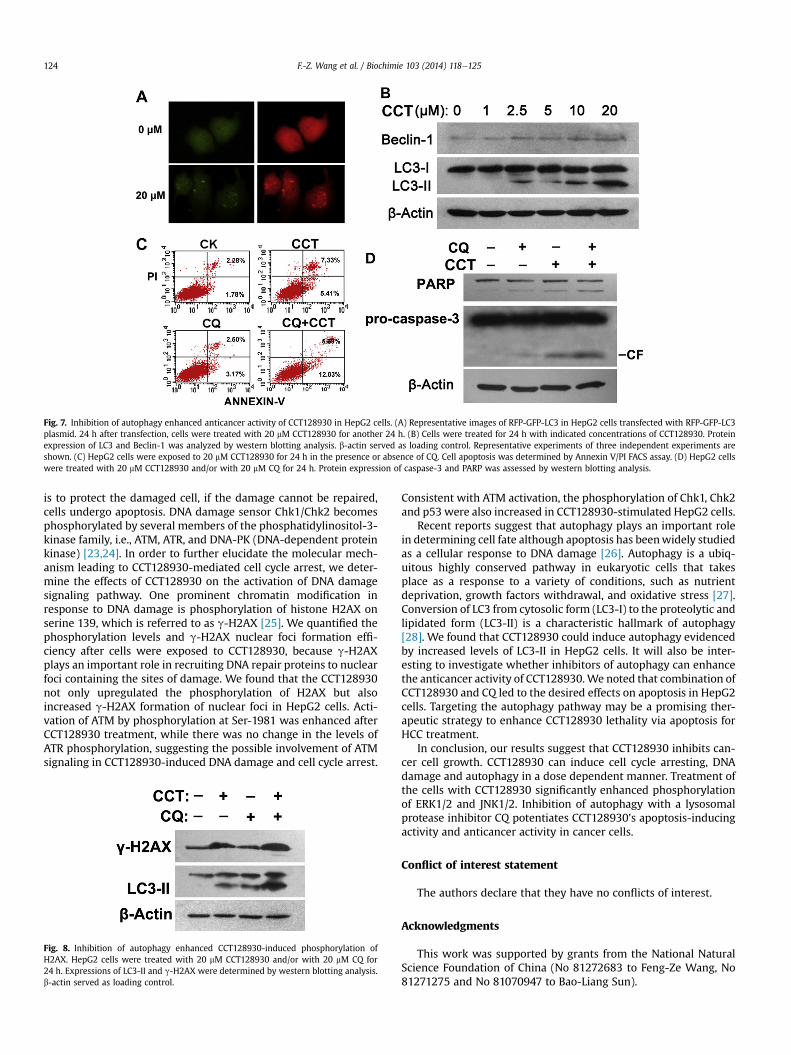
Autophagy is the basic catabolic mechanism by mediating theturnover of intracellular organelles and protein complexes; we thendetermined whether CCT128930 induced autophagy in HepG2cells. We detected the levels of LC3-II for monitoring autophagybecause it has been widely used for estimating the abundance ofthe autophagosome or autophagy [19]. HepG2 cells were trans-fected with RFP-GFP-LC3 for 24 h; the cells were then treated withCCT128930 for 24 h. As presented in Fig. 7A, CCT128930 inducedthe redistribution of GFP-LC3 from a diffuse pattern to punctatestructures. Moreover, an increase in LC3-II expressionwas observedafter treatment of CCT128930 in HepG2 cells. Beclin1 (Atg6), awell-known key regulator of autophagy, was also upregulated inCCT128930-treated HepG2 cells (Fig. 7B).
To determine whether induction of autophagy by CCT128930 isa survival or death mechanism, we detected the CCT128930-induced apoptosis in the presence of CQ, a lysosomal protease in-hibitor. As shown in Fig. 7C, the presence of 20 mM CQ markedlyenhanced the HepG2 cells apoptosis, indicating that the inhibitionof autophagy enhanced CCT128930-induced apoptosis of HepG2cells. In agreement, the combination of CCT128930 with CQ wasalso more potent than CCT128930 alone in inducing apoptosisevidenced by increased cleavage of caspase-3 and PARP (Fig. 7D).These data indicate that inhibition of CCT128930-induced auto-phagy drives HepG2 cells to die of apoptosis.
3.7. Inhibition of autophagy enhances the CCT128930-inducedphosphorylation of H2AX
Last, we determined the changes of CCT128930-induced H2AXexpression in the presence of CQ. From thewestern blotting results,
we found that the inhibition of autophagy with CQ may bepotentiating CCT128930-evoked DNA damage response, becausethe inhibition of autophagy significantly enhanced CCT128930-induced phosphorylation of H2AX in HepG2 cells (Fig. 8).
4. Discussion
The poor results of chemotherapy in HCC combined with thesuccess of sorafenib have led to intensifying research efforts aimingon identification of novel molecular targets for HCC therapy [20].CCT128930, a novel ATP-competitive Akt inhibitor, is discovered byfragment-based in silico screening and structure-based design, andshows selectivity for Akt over PKA by targeting a single amino aciddifference [21]. Previous studies have showed the antitumor ac-tivity of CCT128930 in U87MG and BT474 human breast cancerxenografts. Here, we report the mechanism that CCT128930 in-hibits cell cycle procession and induces DNA damage and auto-phagy in a human hepatoma cell line HepG2.
Abnormal type of excessive proliferation is a key characteristicof tumorigenesis, and inhibiting the proliferative signals in tumorcells is an effective way in anti-cancer therapy. In this study, weshowed that exposure of HepG2 cells to CCT128930 resulted in theaccumulation of cells in the G1 phase. CCT128930 treatmentdecreased the expression of cyclinD1 and Cdc25A, and increasedthe levels of p21, p27 and p53. In addition to inhibiting the prolif-eration of HepG2 cells, high dose of CCT128930 showed an ability toinduce apoptosis. Caspase activation is generally considered to be akey mark of apoptosis, playing an important role in the inductionand regulation of apoptosis. Our study clearly showed that after24 h of treatment with CCT128930, the expression of cleavedcaspase-9, -3 and PARP was enhanced significantly.
One of the biochemical hallmarks of apoptotic cell death is theformation of DNA double-strand breaks (DSB), which producesoligonucleosomal DNA fragments. Under conditions unrelated toapoptosis, DSB induce the rapid activation of conserved DNAdamage response (DDR) pathways [22]. The biological goal of DDR
Fig. 7. Inhibition of autophagy enhanced anticancer activity of CCT128930 in HepG2 cells. (A) Representative images of RFP-GFP-LC3 in HepG2 cells transfected with RFP-GFP-LC3plasmid. 24 h after transfection, cells were treated with 20 mM CCT128930 for another 24 h. (B) Cells were treated for 24 h with indicated concentrations of CCT128930. Proteinexpression of LC3 and Beclin-1 was analyzed by western blotting analysis. b-actin served as loading control. Representative experiments of three independent experiments areshown. (C) HepG2 cells were exposed to 20 mM CCT128930 for 24 h in the presence or absence of CQ. Cell apoptosis was determined by Annexin V/PI FACS assay. (D) HepG2 cellswere treated with 20 mM CCT128930 and/or with 20 mM CQ for 24 h. Protein expression of caspase-3 and PARP was assessed by western blotting analysis.
F.-Z. Wang et al. / Biochimie 103 (2014) 118e125124
is to protect the damaged cell, if the damage cannot be repaired,cells undergo apoptosis. DNA damage sensor Chk1/Chk2 becomesphosphorylated by several members of the phosphatidylinositol-3-kinase family, i.e., ATM, ATR, and DNA-PK (DNA-dependent proteinkinase) [23,24]. In order to further elucidate the molecular mech-anism leading to CCT128930-mediated cell cycle arrest, we deter-mine the effects of CCT128930 on the activation of DNA damagesignaling pathway. One prominent chromatin modification inresponse to DNA damage is phosphorylation of histone H2AX onserine 139, which is referred to as g-H2AX [25]. We quantified thephosphorylation levels and g-H2AX nuclear foci formation effi-ciency after cells were exposed to CCT128930, because g-H2AXplays an important role in recruiting DNA repair proteins to nuclearfoci containing the sites of damage. We found that the CCT128930not only upregulated the phosphorylation of H2AX but alsoincreased g-H2AX formation of nuclear foci in HepG2 cells. Acti-vation of ATM by phosphorylation at Ser-1981 was enhanced afterCCT128930 treatment, while there was no change in the levels ofATR phosphorylation, suggesting the possible involvement of ATMsignaling in CCT128930-induced DNA damage and cell cycle arrest.
Fig. 8. Inhibition of autophagy enhanced CCT128930-induced phosphorylation ofH2AX. HepG2 cells were treated with 20 mM CCT128930 and/or with 20 mM CQ for24 h. Expressions of LC3-II and g-H2AX were determined by western blotting analysis.b-actin served as loading control.
Consistent with ATM activation, the phosphorylation of Chk1, Chk2and p53 were also increased in CCT128930-stimulated HepG2 cells.
Recent reports suggest that autophagy plays an important rolein determining cell fate although apoptosis has beenwidely studiedas a cellular response to DNA damage [26]. Autophagy is a ubiq-uitous highly conserved pathway in eukaryotic cells that takesplace as a response to a variety of conditions, such as nutrientdeprivation, growth factors withdrawal, and oxidative stress [27].Conversion of LC3 from cytosolic form (LC3-I) to the proteolytic andlipidated form (LC3-II) is a characteristic hallmark of autophagy[28]. We found that CCT128930 could induce autophagy evidencedby increased levels of LC3-II in HepG2 cells. It will also be inter-esting to investigate whether inhibitors of autophagy can enhancethe anticancer activity of CCT128930.We noted that combination ofCCT128930 and CQ led to the desired effects on apoptosis in HepG2cells. Targeting the autophagy pathway may be a promising ther-apeutic strategy to enhance CCT128930 lethality via apoptosis forHCC treatment.
In conclusion, our results suggest that CCT128930 inhibits can-cer cell growth. CCT128930 can induce cell cycle arresting, DNAdamage and autophagy in a dose dependent manner. Treatment ofthe cells with CCT128930 significantly enhanced phosphorylationof ERK1/2 and JNK1/2. Inhibition of autophagy with a lysosomalprotease inhibitor CQ potentiates CCT128930’s apoptosis-inducingactivity and anticancer activity in cancer cells.
Conflict of interest statement
The authors declare that they have no conflicts of interest.
Acknowledgments
This work was supported by grants from the National NaturalScience Foundation of China (No 81272683 to Feng-Ze Wang, No81271275 and No 81070947 to Bao-Liang Sun).
F.-Z. Wang et al. / Biochimie 103 (2014) 118e125 125
References
[1] E. Aksamitiene, A. Kiyatkin, B.N. Kholodenko, Cross-talk between mitogenicRas/MAPK and survival PI3K/Akt pathways: a fine balance, Biochem. Soc.Trans. 40 (2012) 139e146.
[2] J.A. Engelman, J. Luo, L.C. Cantley, The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism, Nat. Rev. Genet. 7 (2006)606e619.
[3] S. Saiki, Y. Sasazawa, Y. Imamichi, S. Kawajiri, T. Fujimaki, I. Tanida,H. Kobayashi, F. Sato, S. Sato, K. Ishikawa, M. Imoto, N. Hattori, Caffeine in-duces apoptosis by enhancement of autophagy via PI3K/Akt/mTOR/p70S6Kinhibition, Autophagy 7 (2011) 176e187.
[4] H.Y. Chang, M.H. Shih, H.C. Huang, S.R. Tsai, H.F. Juan, S.C. Lee, Middle infraredradiation induces G2/M cell cycle arrest in A549 lung cancer cells, PLoS One 8(2013) e54117.
[5] H.J. Cho, Y.J. Oh, S.H. Han, H.J. Chung, C.H. Kim, N.S. Lee, W.J. Kim, J.M. Choi,H. Kim, Cdk1 protein-mediated phosphorylation of receptor-associated pro-tein 80 (RAP80) serine 677 modulates DNA damage-induced G2/M checkpointand cell survival, J. Biol. Chem. 288 (2013) 3768e3776.
[6] J. Smith, L.M. Tho, N. Xu, D.A. Gillespie, The ATM-Chk2 and ATR-Chk1 pathwaysin DNA damage signaling and cancer, Adv. Cancer Res. 108 (2010) 73e112.
[7] A. Kulkarni, K.C. Das, Differential roles of ATR and ATM in p53, Chk1, andhistone H2AX phosphorylation in response to hyperoxia: ATR-dependentATM activation, Am. J. Physiol. Lung Cell. Mol. Physiol. 294 (2008) L998eL1006.
[8] A. Choudhury, A. Cuddihy, R.G. Bristow, Radiation and new molecular agentspart I: targeting ATM-ATR checkpoints, DNA repair, and the proteasome,Semin. Radiat. Oncol. 16 (2006) 51e58.
[9] Z.J. Yang, C.E. Chee, S. Huang, F.A. Sinicrope, The role of autophagy in cancer:therapeutic implications, Mol. Cancer Ther. 10 (2011) 1533e1541.
[10] X. Sui, R. Chen, Z. Wang, Z. Huang, N. Kong, M. Zhang, W. Han, F. Lou, J. Yang,Q. Zhang, X. Wang, C. He, H. Pan, Autophagy and chemotherapy resistance: apromising therapeutic target for cancer treatment, Cell Death Dis. 4 (2013)e838.
[11] C. Seitz, M. Hugle, S. Cristofanon, A. Tchoghandjian, S. Fulda, The dual PI3K/mTOR inhibitor NVP-BEZ235 and chloroquine synergize to trigger apoptosisvia mitochondrial-lysosomal cross-talk, Int. J. Cancer 132 (2013) 2682e2693.
[12] V. Papadimitrakopoulou, Development of PI3K/AKT/mTOR pathway inhibitorsand their application in personalized therapy for non-small-cell lung cancer,J. Thorac. Oncol. 7 (2012) 1315e1326.
[13] P. Wu, Y.Z. Hu, PI3K/Akt/mTOR pathway inhibitors in cancer: a perspective onclinical progress, Curr. Med. Chem. 17 (2010) 4326e4341.
[14] M. Mazzoletti, M. Broggini, PI3K/AKT/mTOR inhibitors in ovarian cancer, Curr.Med. Chem. 17 (2010) 4433e4447.
[15] H.R. Fei, L.Y. Cui, Z.R. Zhang, Y. Zhao, F.Z. Wang, Caudatin inhibits carcinomichuman alveolar basal epithelial cell growth and angiogenesis through
modulating GSK3b/b-catenin pathway, J. Cell. Biochem. 113 (2012) 3403e3410.
[16] T.A. Yap, M.I. Walton, L.J. Hunter, M. Valenti, A. de Haven Brandon, P.D. Eve,R. Ruddle, S.P. Heaton, A. Henley, L. Pickard, G. Vijayaraghavan, J.J. Caldwell,N.T. Thompson, W. Aherne, F.I. Raynaud, S.A. Eccles, P. Workman, I. Collins,M.D. Garrett, Preclinical pharmacology, antitumor activity, and developmentof pharmacodynamic markers for the novel, potent AKT inhibitor CCT128930,Mol. Cancer Ther. 10 (2011) 360e371.
[17] J. Wang, L. Yuan, H. Xiao, C. Xiao, Y. Wang, X. Liu, Momordin Ic induces HepG2cell apoptosis through MAPK and PI3K/Akt-mediated mitochondrial path-ways, Apoptosis 18 (2013) 751e765.
[18] Y. Shiloh, Y. Ziv, The ATM protein kinase: regulating the cellular response togenotoxic stress, and more, Nat. Rev. Mol. Cell. Biol. 4 (2013) 197e210.
[19] C.B. McLeland, J. Rodriguez, S.T. Stern, Autophagy monitoring assay: qualita-tive analysis of MAP LC3-I to II conversion by immunoblot, Methods Mol. Biol.697 (2011) 199e206.
[20] A. Psyrri, N. Arkadopoulos, M. Vassilakopoulou, V. Smyrniotis, G. Dimitriadis,Pathways and targets in hepatocellular carcinoma, Expert Rev. AnticancerTher. 12 (2012) 1347e1357.
[21] J.J. Caldwell, T.G. Davies, A. Donald, T. McHardy, M.G. Rowlands, G.W. Aherne,L.K. Hunter, K. Taylor, R. Ruddle, F.I. Raynaud, M. Verdonk, P. Workman,M.D. Garrett, I. Collins, Identification of 4-(4-aminopiperidin-1-yl)-7H-pyrrolo[2,3-d]pyrimidines as selective inhibitors of protein kinase B through frag-ment elaboration, J. Med. Chem. 51 (2008) 2147e2157.
[22] S. Solier, O. Sordet, K.W. Kohn, Y. Pommier, Death receptor-induced activationof the Chk2- and histone H2AX-associated DNA damage response pathways,Mol. Cell. Biol. 29 (2009) 68e82.
[23] P. Bouwman, J. Jonkers, The effects of deregulated DNA damage signalling oncancer chemotherapy response and resistance, Nat. Rev. Cancer 12 (2012)587e598.
[24] S. Solier, Y.W. Zhang, A. Ballestrero, Y. Pommier, G. Zoppoli, DNA damageresponse pathways and cell cycle checkpoints in colorectal cancer: currentconcepts and future perspectives for targeted treatment, Curr. Cancer Drug.Targets 12 (2012) 356e371.
[25] H. Cha, J.M. Lowe, H. Li, J.S. Lee, G.I. Belova, D.V. Bulavin, A.J. Fornace Jr., Wip1directly dephosphorylates gamma-H2AX and attenuates the DNA damageresponse, Cancer Res. 70 (2010) 4112e4122.
[26] N.S. Deen, S.J. Huang, L. Gong, T. Kwok, R.J. Devenish, The impact of autophagicprocesses on the intracellular fate of Helicobacter pylori: more tricks from anenigmatic pathogen? Autophagy 9 (2013) 639e652.
[27] B.J. Altman, S.R. Jacobs, E.F. Mason, R.D. Michalek, A.N. MacIntyre, J.L. Coloff,O. Ilkayeva, W. Jia, Y.W. He, J.C. Rathmell, Autophagy is essential to suppresscell stress and to allow BCR-Abl-mediated leukemogenesis, Oncogene 30(2011) 1855e1867.
[28] O. Florey, M. Overholtzer, Autophagy proteins in macroendocytic engulfment,Trends Cell. Biol. 22 (2012) 374e380.











![Esophageal squamous cell carcinoma invasion is inhibited by ......induces cell cycle arrest and inhibits growth [10, 18]. This “dual role” phenomenon has also been observed in](https://static.documents.pub/doc/80x56/60e90ff818aa1f1c7803f0f6/esophageal-squamous-cell-carcinoma-invasion-is-inhibited-by-induces-cell.jpg)













