27
80 Chapter 5 “SORTING OUT OF INTERFERENCE IN DETECTION OF ENDOTOXINS IN BIOTHERAPEUTIC DRUGS, LACTOBIONIC ACID & MEDICAL DEVICES”

80
Chapter 5
“SORTING OUT OF INTERFERENCE IN
DETECTION OF ENDOTOXINS IN
BIOTHERAPEUTIC DRUGS,
LACTOBIONIC ACID & MEDICAL DEVICES”

81
5.1 Biotherapeutic drugs
Abstract:
The effect of varying interference factors responsible for false positive
results was investigated. The interfering factors inhibiting the Endotoxins in
activated lymphocytes sample was sorted by heat denaturation. These
activated lymphocytes are Biotherapeutic drugs which are used for
autologous immune enhancement therapy. The technique used for
denaturation can be applied to protein samples as well, to get rid of inhibition
while quantifying Endotoxins.
Introduction:
Water is the principal source of Endotoxin in parenteral products. In
general, as per the United State Pharmacopeias (USP) the threshold
pyrogenic dose is 5 EU/kg/hr for parenteral drugs and 0.2 EU/kg/hr for
intrathecal drugs. When Endotoxin enters into human blood these toxins
induces white blood cells (WBC) to release cytokines, such as Tissue Necrosis
Factor (TNF), Interleukin-1 and Interleukin-8, which mediate a complex
biological response including pyrogensity, shock, coagulation and
inflammation (Bang FB, 1956). So it is mandatory to check the presence of
Endotoxin level in Biothereaputic drugs before passing the product into
market.
Bacterial Endotoxin is one of the most potent activator of mammalian
immune system. Gram negative bacterial outer membrane
Lipopolysaccharide (LPS) induces a cascade of defense mechanism that is
known as fever and inflammation (C.M.Good and H.E. Lane, 1977). In
incomparable fashion, for parenteral drugs administered into cerebrospinal
fluid (CSF) known as intrathecal administration, K is reduced to 0.2 EU/kg. The
most common toxic route of entry of Endotoxin into human system via
intrathecal administration. Endotoxins are negatively charged
macromolecules as small as 20-30 kDa, varying in size due to bacterial origin,
the presence of divalent cations or biological detergents. Bacterial Endotoxin
is the significant pyrogen that has been identified as a contaminant in

5.1 Biotherapeutic drugs
82
parenteral products. The LAL reaction with Endotoxin requires pH neutrally and
optimum levels of divalent cations82. A uniform temperature of 37°C optimizes
the rate of reaction. Most Biotherapeutic drug products require dilution with
LAL Reagent Water (LRW) (J.F. Cooper, 1990) before testing to avoid
interference. Testing of serum, plasma, protein sample is subjected to
inhibition from serine protease inhibitors and this interference creates
problems in both Biotech and research fields58.
Materials and Methods:
Materials:
Limulus Amoebocyte Lysate(LAL), Control Standard Endotoxin(CSE), LAL
Reagent Water (LRW) were purchased from Endosafe U.S, 76,77Depyrogenated
10 X 75 mm assay tubes, 16X100mm dilution tubes and pyrogen free
Micropipette tips.
Principle of BET (Bacterial Endotoxins test):
The principle of Bacterial Endotoxin test is to detect presence of
Endotoxin in the given sample to which the test is applied using Lysate derived
from the animal Limulus Polyphemus. Limulus polyphemus is also called as
Horseshoe crab. It is found in the eastern cost of America. This animal is living
fossil as since last 150 million years there has been no physical or physiological
change taken place in the animal. The blood of the animal is blue in colour
due to the presence of Hemocyanin instead of Hemoglobin. The unique
property of the blood is to react with Lipopolysaccharide (LPS) present in the
membrane of Gram-negative bacteria.
Methods:
Procedure of LAL test:
Equal volume of test sample and LAL reagent is added in a
depyrogenated test tube of 10 X 75 mm and incubate this mixture at 37± 1°C
for 60±2 min. Then invert the tube by 180° and look for gel formation. If a gel
inside the test tube is able to maintain its integrity after inverting the tube to
180° then it is a positive reaction which indicates presence of Endotoxin in the
sample. Other than this any condition is considered as negative which

5.1 Biotherapeutic drugs
83
Indicates absence of Endotoxin in the sample (lesser than the lysate
sensitivity).
Endotoxin Limit:
Since Endotoxin is ubiquitous in nature there has to be some safety limit
to pass the product. The Endotoxin limit should be such that it will not cause
any harmful effects to patient. Appendix E of USFDA (United States Food and
Drug Administration) gives Endotoxin limits for various products. It also gives a
formula through which Endotoxin limit for new product can be calculated.
The formula is as follows.
Endotoxin Limit = K/ M (USP 32, 2009)
Where, ‘K’ is threshold pyrogenic dose in humans and animals. 5 EU/kg body
weight for parenteral drug and 0.2EU/kg body weight for intrathecal drug.
‘M’ Maximum dose administered to a patient per Kg body weight per hour
(Not heroic dose).
The limit formula for radiopharmaceuticals is:
175/V except for intrathecally-administered products.
14/V for intrathecal drugs.
‘V’ equals the maximum recommended dose, in ml, at the expiration date or
time. For drugs administered on a per Square Meter of Body Surface:
5 EU/ [(dose * 1.8 sq. m.)/ 70 Kg]
Product Testing:
For testing products equal volume of drug (sample) and LAL reagent is
taken and following tubes are prepared
Negative Product Control (NPC) - Sample + LAL
Positive Product Control (PPC) - Sample + CSE (2λ) + LAL
Negative Water Control (NWC) - LRW + LAL
Positive Water Control (PWC) - LRW + CSE (2λ) + LAL
Majority of times it has been a common observation that if a product is tested
directly it inhibits the LAL test and thus shows interference (J. van Noordwijk et
al., 1997).

5.1 Biotherapeutic drugs
84
Interference:
Interference is defined as a significant difference between the end
points of positive water control and positive product control using standard
Endotoxin.
This interference could be either inhibition wherein the recovery of
Endotoxin is below than the expected or enhancement wherein the recovery
of Endotoxin is higher than expected.
Interference may occur due to following reasons:
Suboptimal pH:
The optimal pH for LAL reaction is in between about 6.8 to 7.4. If the
sample is too acidic or too basic it will inhibit enzymes involved in LAL reaction.
Hence it is very necessary to bring pH of reaction to neutral range. This can be
done using 1N Hcl or 1N NaOH for basic and acidic product respectively82.
Endotoxin modification:
Purified Endotoxin has tendency to form micelle formation which is due
to hydrophilic and hydrophobic interactions between LPS and water. This
aggregated Endotoxin escapes LAL test. Hence to avoid such problem vortex
mixing for samples is performed.
Container effects:
75Adsorption of Endotoxin on tube wall causes poor recovery of
Endotoxin in LAL test. So it is suggested to use high quality borosilicate glass
tubes because of its inert nature hence adsorption of Endotoxin is least.
Unbalanced cation levels:
Divalent cations play important role in LAL reactivity and dispersion of
Endotoxins. LAL test requires optimum concentration of Ca++and Mg++ ions. If
divalent cations are insufficient then Ca++and Mg++ ions are added externally.
Protein or enzyme modification:
Enzyme inactivation due to oxidants, proteolytic agents or specific
inactivators will cause inhibition.
Non-specific LAL activation:
Some molecules other than Endotoxin are known to react with LAL
reagent and give gel formation. This is enhancement reaction and is very
rare.

5.1 Biotherapeutic drugs
85
Subjects:
Activated lymphocyte sample which is used for autologous immune
enhancement therapy for myeloid leukemia has been tested for sorting out of
interference problem.
90% of interference problems are solved by just diluting the sample. But how
much sample can be diluted so that it can still detect Endotoxin limit is given
by formula for Maximum Valid Dilution (MVD).
MVD = concentration of sample X Endotoxin Limit / Lysate sensitivity
Example :
Drug : Activated Lymphocytes
Endotoxin Limit : NMT 0.25 EU/mL
Lysate sensitivity : 0.03125 EU/mL
MVD = potency x E.L
λ
= 1mL/ mL X 0.25 EU/ml
0.03125EU/mL
MVD = 8
Product Validation:
Product needs to be validated before start for routine testing. Validation is
a test condition where an Endotoxin standard is detected with the same
efficiency in a test sample as it is in LRW. This validation study consists of two
different phases wherein in Phase I (Preliminary screening) involve interference
testing and Phase II consists of validation of product.
Significance of product validation is that it gives information on
whether there are any interfering factors in the drug product to the LAL test
and also it gives an idea of the approximate levels of Endotoxin content in the
drug product. It also covers manufacturing of product and formulation of the
product.
It is always advisable to carry out revalidation if product formulation is
changed and which is likely to affect the interference pattern of the product
for LAL test. Also revalidation is to be conducted for any product if there is any
change in manufacturing procedures or in vendor.

5.1 Biotherapeutic drugs
86
Phase I: Preliminary Screening / interference Study
In this two identical series of product dilutions (two-fold dilutions), one spiked
with 2λ, and one left unspiked. The result of Phase I will tell you the non-
interfering dilution (NID) of the product, which is used for the actual validation
(Phase II). The non-interfering dilution (NID) is the first set of PPC that shows a
gel.
Example :
Drug : Activated Lymphocytes
Endotoxin Limit : NMT 0.25 EU/mg
Lysate sensitivity: 0.03125 EU/mL
MVD = potency x E.L
λ
= 1mL/mL X0.25 EU/ml
0.03125EU/mL
MVD = 8
Results and discussions: Sample Dilution 1:1 1:2 1:4 1:8
Unspiked -- -- -- --
Spiked -- -- -- --
Table: 1
This assay shows that there is inhibition up to 1:8 (MVD). Due to Inhibition LAL is
unable to detect the Endotoxins even in spiked sample After analyzing the
sample using different procedures, finally In order to sort out this inhibition
problem then the activated sample is heated at 55°C for 15min to coagulate
the proteins in the sample and this heat denaturation technique is applicable
to all protein samples. The denaturation won’t affect the Endotoxin because
Endotoxins can be denatured at 250°C for 30 minutes as per USP.
Assay results after Heat denaturation of the sample.
Sample Dilution 1:1 1:2 1:4 1:8
Unspiked -- ++ -- --
Spiked -- + + ++ ++
Table: 2

5.1 Biotherapeutic drugs
87
This assay shows that there is inhibition upto 1:1 dilution and the spike recovery
at 1:2 dilutions onwards. Therefore the NID is 1: 2. It is advisable to validate the
product at not less than 1:4 dilution to take care of any batch to batch
variation during regular production. So 1: 4 dilution is chosen for product
validation.
Phase II: Validation of Product
For validation, test and compare two identical series of Endotoxin dilutions
bracketingλ; One prepared in LRW and another prepared in product diluted
to the proposed test dilution. Here dilution selected for validation is 1:4. (Hot
spike method).
Example of results:
Endotoxin/product
Replicates 0.0625EU/mL 0.03125EU/mL 0.015 EU/mL 0.007 EU/mL
1 + + - -
2 + + - -
3 + + - -
4 + + - -
Table: 3
Negative product control: --Geometric Mean = 0.03 EU/ml
Endotoxin/LRW
Replicates 0.0625EU/mL 0.03125EU/mL 0.015 EU/mL 0.007 EU/mL
1 + + - -
2 + + - -
3 + + - -
4 + + - -
Table: 4
Blank: -- Geometric Mean = 0.03 EU/ml
Successful validation requires that both series confirm label claim (Geometric
mean) within +/- one two-fold dilution. Validation is conducted at this dilution
on three batches of product.

88
5.2 Lactobionic Acid
Abstract:
Lactobionic acid used in organ preservative during organ transplant. The
effect of varying the pH and ionic strength in Lactobionic acid was
investigated. The interfering factors inhibiting the Endotoxins in Lactobionic
acid while quantifying with Limulus Amoebocyte Lysate was sorted out by
neutralizing the ionic concentration in sample using alkali and validated the
test sample. Lactobionic acid is weak acid and having pH in the range 1 to 2.
Since it is a weak acid its ionic dissociation is very less. As we add strong alkali
like NaOH in to it, the pH rises immediately and after some time it comes down
to acidic range. After adjusting the pH the sample to be analyzed within an
hour to avoid interference.
Introduction:
Bacterial Endotoxin is one of the most potent activator of mammalian
immune system. In general, as per the United State Pharmacopeias (USP) the
threshold pyrogenic dose is 5 EU/kg/hr for parenteral drugs and 0.2 EU/kg/hr
for intrathecal drugs5. When Endotoxin enters into human blood these toxins
induces white blood cells (WBC) to release cytokines, such as tissue necrosis
factor (TNF), interleukin-1 and interleukin-8, which mediate a complex
biological response including pyrogensity, shock, coagulation and
inflammation5,4&7. Gram negative bacterial outer membrane
Lipopolysacchride (LPS) induces a cascade of defense mechanism that is
known as fever and inflammation2. So it is mandatory to check the presence
of Endotoxin level in Organ preservative before using it for preservation of
organs during transplant.
The LAL reaction with Endotoxin requires pH neutrality and optimum levels of
Na+ and divalent cations. A uniform temperature of 37°C optimizes the rate of
reaction. Most therapeutic drug products require dilution with LAL Reagent
Water (LRW) before testing to avoid interference, where inhibition is failure to
recover the positive control, and enhancement is excess recovery. There are
3 principle causes of invalid or inhibitory results in gel clot testing are 1. Loss of
purified Endotoxin used for product positive controls (PPC). 2. Adverse

5.2 Lactobionic Acid
89
chemical conditions such as non-neutral pH or sub optimal levels of sodium
ions and divalent cations (Mg++ and Ca++). 3. Inadequate controlled test
parameters including testing accessories, reagents and analyst proficiency.
The aim of the study is to sort out the interfering factors which lead to the
diverse results in Lactobionic acid. The false positive results may cause severe
complication in the patients as discussed in literature.
Materials and Methods:
Materials:
Lyophilized Limulus Amoebocyte Lysate of 0.0312 sensitivity (LAL), Control
Standard Endotoxin 5 Eu/ng (CSE), LAL Reagent Water (LRW) of Endosafe US,
Depyrogenated (250°C for 30 min) 10 X 75 mm assay tubes, 16X100 mm
dilution tubes, pyrogen free Micropipette tips, vortex mixture, 1N NaoH and
Lactobionic acid were used for determination of Endotoxin content by the gel
clot technique.
The sensitivity of the Lysate (labeled 0.0312 Eu/mL) was determined by using
known amount of E.coli Control Standard Endotoxin.
In the gel-clot techniques, the reaction end point is determined from dilutions
of the material under test in direct comparison with parallel dilutions or a
reference Endotoxin, and quantities of Endotoxins are expressed in Endotoxin
units.
1. Preparation of Standard stock solution and standard solutions: The CSE
having a defined potency of 50 EU/Vial was reconstituted with 5ml of LRW
and mixed intermittently for 30 minutes using a vortex mixture and this
concentrate was used to prepare 2λ, λ, λ/2 & λ/4, where λ is the labeled
claim sensitivity of Lysate.
2. Preparation of sample solution: Test samples were diluted to the required
concentrations based on the formulae MVD. MVD is the maximum valid
dilution, which is allowable dilution of the specimen at which the Endotoxin
limit can be determined. The general equation to determine MVD is
MVD = (Endotoxin limit X Concentration of sample solution)/ (λ). Where E.L is
the Endotoxin limit of the test sample, which is specified in the individual
monograph/ based on the E.L formula if not mentioned in the monograph, in
terms of volume or units of active drug (in EU/mg).

5.2 Lactobionic Acid
90
3. Lactobionic acid sample preparation: Batch No: LBA-0109,
Potency=100mg/mL, E.L=0.005 Eu/mg, Lysate sensitivity is 0.0312 Eu/mL and
MVD = 16. The following test dilutions are prepared by 1:16 (6.25 mg/mL), 1:8
(12.5 mg/mL), 1:4 (25 mg/mL) & 1:2 (50 mg/mL). Lactobionic acid is weak acid
and having pH in the range 1 to 2.
Method:
Equal volume of test sample and LAL reagent is added in a depyrogenated
test tube of 10 X 75 mm and incubate this mixture at 37± 1°C for 60±2 min.
Then invert the tube by 180° and look for gel formation. If a gel inside the test
tube is able to maintain its integrity after inverting the tube to 180° then it is a
positive reaction which indicates presence of Endotoxin in the sample greater
than the limit. Other than this any condition is considered as negative which
indicates absence of Endotoxin in the sample (lesser than the lysate
sensitivity).
Product Testing:
For testing products equal volume of drug (sample) and LAL reagent is taken
and following tubes are prepared6.
Negative Product Control (NPC) - Sample + LAL
Positive Product Control (PPC) - Sample + CSE (2λ) + LAL
Negative Water Control (NWC) - LRW + LAL
Positive Water Control (PWC) - LRW + CSE (2λ) + LAL
Majority of times it has been a common observation that if a product is tested
directly it inhibits the LAL test and thus shows interference1&3.
Interference: Interference is defined as a significant difference between the
end points of positive water control and positive product control using
standard Endotoxin.
This interference could be either inhibition wherein the recovery of Endotoxin is
below than the expected or enhancement wherein the recovery of Endotoxin
is higher than expected.

5.2 Lactobionic Acid
91
Product Validation:
Product needs to be validated before start for routine testing. Validation is a
test condition where an Endotoxin standard is detected with the same
efficiency in a test sample as it is in LRW. This validation study consists of two
different phases wherein in Phase I (Preliminary screening) involve interference
testing and Phase II consists of validation of product.
Significance of product validation is that it gives information on
whether there are any interfering factors in the drug product to the LAL test
and also it gives an idea of the approximate levels of Endotoxin content in the
drug product. It also covers manufacturing of product and formulation of the
product.
It is always advisable to carry out revalidation if product formulation is
changed and which is likely to affect the interference pattern of the product
for LAL test. Also revalidation is to be conducted for any product if there is any
change in manufacturing procedures or in vendor.
Phase I: Preliminary Screening / interference Study9
In this two identical series of product dilutions (two-fold dilutions), one
spiked with 2λ, and one left unspiked. The result of Phase I will tell you the non-
interfering dilution (NID) of the product, which is used for the actual validation
(Phase II). The non-interfering dilution (NID) is the first set of PPC that shows a
gel.
Results and discussions:
Lactobionic acid:
Sample Dilution 1:2 1:4 1:8 1:16
Unspiked -- -- -- --
Spiked -- -- -- --
Table: 1

5.2 Lactobionic Acid
92
This assay shows that there is inhibition up to 1:16 (MVD) in Lactobionic acid.
Due to Inhibition LAL is unable to detect the Endotoxins even in spiked sample
After analyzing the sample using different procedures, finally In order to sort
out this inhibition problem the acidic pH of the Lactobionic acid (1-2) is
adjusted to 7-8 with 1N NaoH.
Lactobionic acid: (Results after adjusting the Acidic pH to the range of 7-8
with 1 N NaoH).
Sample Dilution 1:2 1:4 1:8 1:16
Unspiked ++ -- -- --
Spiked ++ ++ ++ ++
Table: 2
This assay shows no inhibition upto 1:2 dilution in Lactobionic acid and the
spike recovery at 1:2 dilutions onwards. Therefore the NID is 1:2 (Lactobionic
acid). It is advisable to validate the product at not less than MVD/4 to take
care of any batch to batch variation. So MVD/4 dilution is chosen for product
validation.
Phase II:
Validation of Product
For validation, test and compare two identical series of Endotoxin dilutions
bracketing λ; One prepared in LRW and another prepared in product diluted
to the proposed test dilution. Here dilution selected for validation is 1:4. (Hot
spike method).
Example of results:
Endotoxin/product
Replicates 0.0625 Eu/mL 0.0312 Eu/mL 0.0156 Eu/mL 0.0078 Eu/mL
1 + + - -
2 + + - -
3 + + - -
4 + + - -
Table: 3
Negative product control: --Geometric Mean = 0.0312 EU/ml
5.2 Lactobionic Acid
93
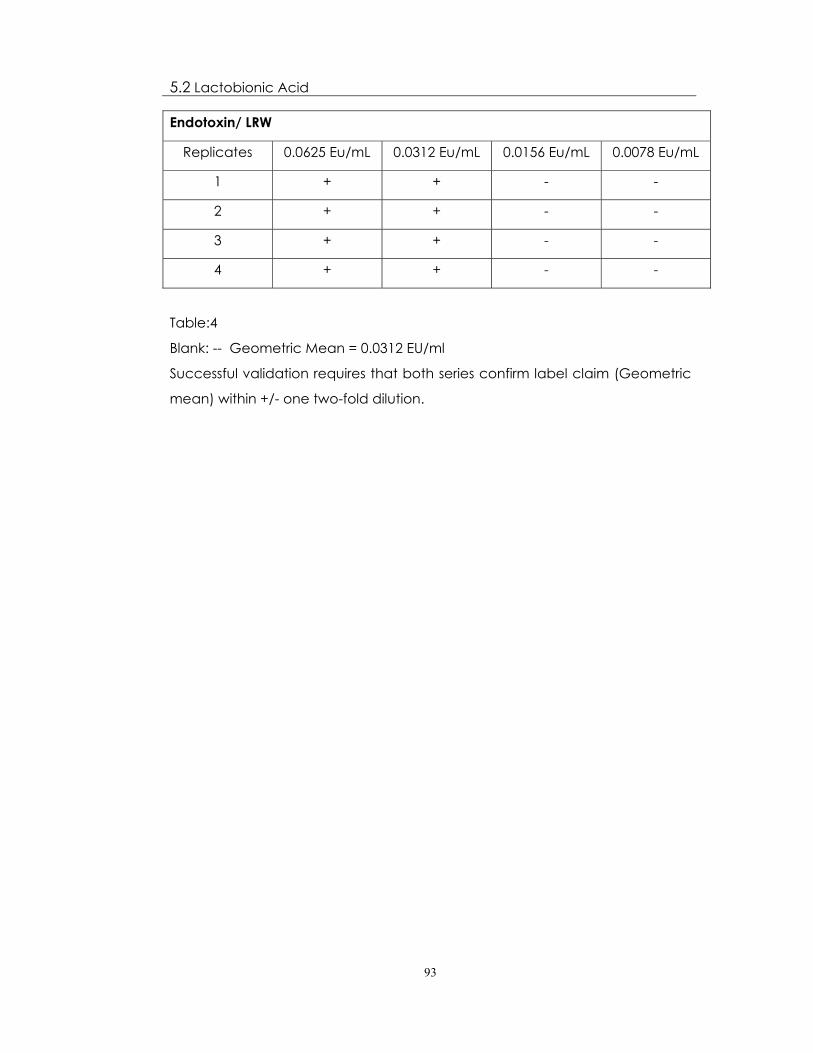
Endotoxin/ LRW
Replicates 0.0625 Eu/mL 0.0312 Eu/mL 0.0156 Eu/mL 0.0078 Eu/mL
1 + + - -
2 + + - -
3 + + - -
4 + + - -
Table:4
Blank: -- Geometric Mean = 0.0312 EU/ml
Successful validation requires that both series confirm label claim (Geometric
mean) within +/- one two-fold dilution.

94
5.3 Quantification of Bacterial Endotoxins in Medical devices:
Abstract: Like drug products, each lot of medical devices need to be
checked for bacterial Endotoxins for compliance before releasing into the
market. Endotoxins are found to be variable absorptive to the device surfaces
and leads to false negative results, to overcome this issue the desired test
device need to be filled with WFI/LRW and incubate it at 37°C for 30 min, so
that the Endotoxins stick to the walls of the medical device will be extracted
into the WFI/LRW. Then the WFI/LRW rinse will be tested and report the value
per ml of rinse. Then the reported value will be multiplies by total volume and
divide the same with the number of devices taken will gives us the Endotoxin
content in each medical device.
Materials and Methods:
Materials:
Lyophilized Limulus Amoebocyte Lysate of 0.125 sensitivity (LAL), Control
Standard Endotoxin 5 Eu/ng (CSE), LAL Reagent Water (LRW) of Endosafe US,
Depyrogenated (250°C for 30 min) 10 X 75 mm assay tubes, 16X100 mm
dilution tubes, pyrogen free Micropipette tips, vortex mixture, Blood sets, I.V
Set, 1mL Syringe, 5mL Syringe and Sutures were used for determination of
Endotoxin content by the gel clot technique.
The sensitivity of the Lysate (labeled 0.125 Eu/mL) was determined by using
known amount of E.coli Control Standard Endotoxin.
In the gel-clot techniques, the reaction end point is determined from dilutions
of the material under test in direct comparison with parallel dilutions or a
reference Endotoxin, and quantities of Endotoxins are expressed in Endotoxin
units.
1. Preparation of Standard stock solution and standard solutions: The CSE
having a defined potency of 50 EU/Vial was reconstituted with 5ml of LRW
and mixed intermittently for 30 minutes using a vortex mixture and this

5.3 Quantification of Bacterial endotoxins in Medical devices
95
concentrate was used to prepare 2λ, λ, λ/2 & λ/4, where λ is the labeled
claim sensitivity of Lysate.
2. Preparation of sample solution: Test samples were diluted to the required
concentrations based on the formulae MVD. MVD is the maximum valid
dilution, which is allowable dilution of the specimen at which the Endotoxin
limit can be determined. The general equation to determine MVD is
MVD = (Endotoxin limit X Concentration of sample solution)/ (λ). Where E.L is
the Endotoxin limit of the test sample, specified for Medical devices.
3. Blood set rinse sample preparation: Potency 1 mL/mL , E.L= 0.73 Eu/mL ,
Lysate sensitivity is 0.125 Eu/mL and MVD = 96.The following test dilutions are
prepared by 1:1, 1:2, 1:3, 1:4 & 1:5.
4. I.V set rinse sample preparation: Potency 1 mL/mL, E.L= 1.25 Eu/mL , Lysate
sensitivity is 0.125 Eu/mL and MVD = 10. The following test dilutions are
prepared by 1:1, 1:2, 1:4, 1:8 & 1:10.
5. 1mL Syringe rinse sample preparation: Potency 1 mL/mL, E.L= 18.75 Eu/mL,
Lysate sensitivity is 0.125 Eu/mL and MVD = 150. The following test dilutions are
prepared by 1:9, 1:18, 1:37, 1:75 & 1:150.
6. 5mL Syringe rinse sample preparation: Potency 1 mL/mL, E.L= 3.75 Eu/mL,
Lysate sensitivity is 0.125 Eu/mL and MVD = 30. The following test dilutions are
prepared by 1:2, 1:4, 1:8, 1:16 & 1:30.
7. Sutures rinse sample preparation: Potency 1 mL/mL, E.L= 12 Eu/mL, Lysate
sensitivity is 0.125 Eu/mL and MVD = 30. The following test dilutions are
prepared by 1:6, 1:12, 1:24, 1:48 & 1:96.
Methods
Equal volume of test sample and LAL reagent is added in a
depyrogenated test tube of 10 X 75 mm and incubate this mixture at 37± 1°C
for 60±2 min. Then invert the tube by 180° and look for gel formation. If a gel

5.3 Quantification of Bacterial endotoxins in Medical devices
96
inside the test tube is able to maintain its integrity after inverting the tube to
180° then it is a positive reaction which indicates presence of Endotoxin in the
sample greater than the limit. Other than this any condition is considered as
negative which indicates absence of Endotoxin in the sample (lesser than the
lysate sensitivity).
Product Testing: For testing products equal volume of drug (sample) and LAL
reagent is taken and following tubes are prepared)
Negative Product Control (NPC) - Sample + LAL
Positive Product Control (PPC) - Sample + CSE (2λ) + LAL
Negative Water Control (NWC) - LRW + LAL
Positive Water Control (PWC) - LRW + CSE (2λ) + LAL
Product Validation: Product needs to be validated before start for routine
testing. Validation is a test condition where an Endotoxin standard is
detected with the same efficiency in a test sample as it is in LRW. This
validation study consists of two different phases wherein in Phase I (Preliminary
screening) involve interference testing and Phase II consists of validation of
product.
Significance of product validation is that it gives information on
whether there are any interfering factors in the drug product to the LAL test
and also it gives an idea of the approximate levels of Endotoxin content in
the drug product. It also covers manufacturing of product and formulation of
the product.
It is always advisable to carry out revalidation if product formulation is
changed and which is likely to affect the interference pattern of the product
for LAL test. Also revalidation is to be conducted for any product if there is any
change in manufacturing procedures or in vendor.

5.3 Quantification of Bacterial endotoxins in Medical devices
97
(i) Blood Set:
Preliminary Screening Test
Product : Blood set
Concentration : 1 ml / mL
Endotoxin Limit : K X N V
K= 20 EU/Device
N = No of Devices
V = Volume of solution rinsed/ taken
E.L = 20 EU/Device x 3 Devices
82mL
= 0.73 EU/ml
MVD = Potency x E.L
Sensitivity of Lysate
= 1ml/ml x 0.73 EU/ml
0.125 EU/ml
= 5
Test Dilution : 1:1, 1:2, 1:3, 1:4 and 1: 5
Test sample Rinse: Three blood bags from each lot are taken and
completely filled with WFI and incubated it at 37°C for 30 min, Then the
Endotoxins stick to the walls of the bag are extracted into the WFI. Then the
WFI rinse was tested and reported the value per ml of rinse. Then the
reported value was multiplied by total volume and divided the same with
the number of devices taken, it gives us the Endotoxin content in each
Blood bag.
Test Dilutions NPC PPC
1:1 − − + +
1:2 − − + +
1:3 − − + +
1:4 − − + +
1:5 − − + +
++: Gel Formation − − : No Gel Formation

5.3 Quantification of Bacterial endotoxins in Medical devices
98
Conclusion: Carried out screening and observed Non Interfering Dilution (NID)
at MVD/8 (1:1). Non Interfering dilution is 1ml/ mL. Endotoxins
content is < 4 EU/ device.
End-Product Endotoxins Test79:
Product : Blood set
Preparation : Product Concentration : 1mL / mL
Endotoxin Limit : 20 EU/Device
MVD : 5
Test Dilution : 1:2
Results : Test Endotoxin
Concentration EU/mL.
NEG
Control
NEG
Product
Control
Test
End
point.
Log of
End
point.
Geometric Mean
=A log (log of End
point.)
Material 2λ λ ½λ ¼λ EU/mL
+ + - − − 0.125 -0.9030
+ + - − − 0.125 -0.9030
+ + - − 0.125 -0.9030
CSE
Water
Control
+ + - − 0.125 -0.9030
+ + - − − 0.0625 -1.2041
+ + - − − 0.0625 -1.2041
+ + - − − 0.125 -0.9030
Positive
Product
Control
+ + - − − 0.125 -0.9030
Antilog
(-3.612/4)
=Antilog –0.903
=0.125EU/mL
Antilog
(-4.2142/4)
=Antilog–1.05355
=0.088EU/mL
Interpretation :
Test results are : Valid
Comments : Product validation for Blood set was carried out at MVD/8 (1:2).
The GM of the end points in Endotoxin / LRW was 0.125EU/mL and
Endotoxin/product was 0.088 EU/mL which is in between 2λ and ½ λ, which
indicates no inhibition and enhancement at test dilution 1:2.

5.3 Quantification of Bacterial endotoxins in Medical devices
99
(II) I.V SET
Preliminary Screening Test
Product : I.V Set
Concentration : 1 ml / mL
Endotoxin Limit : K X N
V
K= 20 EU/Device
N = No of Devices
V = Volume of solution rinsed/ taken
E.L = 20 EU/Device x 3 Devices
48mL
= 1.25 EU/ml
MVD = Potency x E.L
Sensitivity of Lysate
= 1ml/ml x 1.25 EU/ml
0.125 EU/ml
= 10
Test Dilution : 1:1, 1:2, 1:4, 1:8 and 1: 10
Test sample Rinse: Three I.V Sets from each lot are taken and completely
filled with WFI and incubated it at 37°C for 30 min, Then the Endotoxins stick
to the walls of the bag was extracted into the WFI. Then the WFI rinse was
tested and reported the value per ml of rinse. Then the reported value was
multiplied by total volume and divide the same with the number of devices
taken, it gives us the Endotoxin content in each I.V bag.
Test Dilutions NPC PPC
1:1 − − − −
1:2 − − + +
1:4 − − + +
1:8 − − + +
1:10 − − + +
++: Gel Formation − − : No Gel Formation

5.3 Quantification of Bacterial endotoxins in Medical devices
100
Conclusion: Carried out screening and observed Non Interfering Dilution (NID)
at 1:2 dilution, at 1:1 the rinse of the I.V set is showing inhibition.
Non Interfering dilution is 1:2 dilution. Endotoxins content is < 2.5
EU/ device.
End-Product Endotoxins Test:
Product : I.V set
Preparation : Product Concentration : 1mL / mL
Endotoxin Limit : 20 EU/Device
MVD : 10
Test Dilution : 1:2
Results : Test Endotoxin
Concentration EU/mL.
NEG
Control
NEG
Product
Control
Test
End
point.
Log of
End
point.
Geometric Mean
=A log (log of End
point.)
Material 2λ λ ½λ ¼λ EU/mL
+ + - − − 0.125 -0.9030
+ + - − − 0.125 -0.9030
+ + - − 0.125 -0.9030
CSE
Water
Control
+ + - − 0.125 -0.9030
+ + - − − 0.0625 -1.2041
+ + - − − 0.0625 -1.2041
+ + - − − 0.125 -0.9030
Positive
Product
Control
+ + - − − 0.125 -0.9030
Antilog
(-3.612/4)
=Antilog –0.903
=0.125EU/mL
Antilog
(-4.2142/4)
=Antilog–1.05355
=0.088EU/mL
Interpretation :
Test results are : Valid
Comments : Product validation for I.V set was carried out at 1:4 dilution. The
GM of the end points in Endotoxin / LRW was 0.125EU/mL and
Endotoxin/product was 0.088 EU/mL which is in between 2λ and ½ λ, which
indicates no inhibition and enhancement at test dilution 1:4 dilution.
5.3 Quantification of Bacterial endotoxins in Medical devices
101
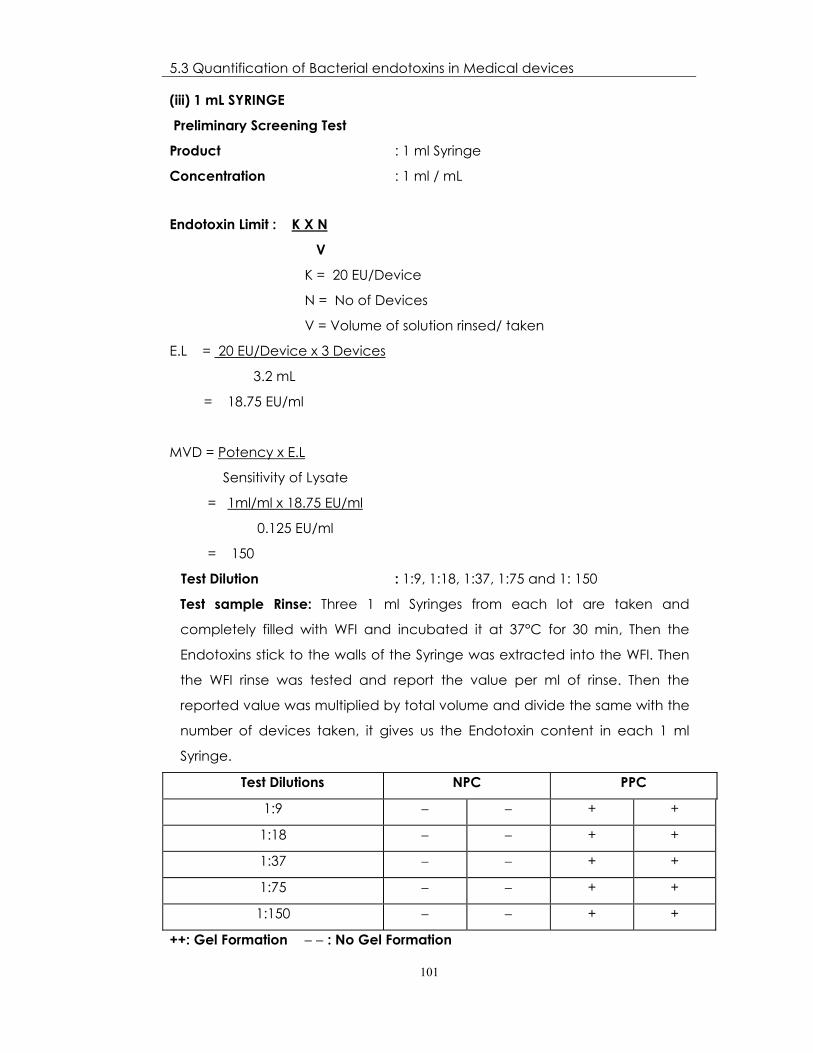
(iii) 1 mL SYRINGE
Preliminary Screening Test
Product : 1 ml Syringe
Concentration : 1 ml / mL
Endotoxin Limit : K X N
V
K = 20 EU/Device
N = No of Devices
V = Volume of solution rinsed/ taken
E.L = 20 EU/Device x 3 Devices
3.2 mL
= 18.75 EU/ml
MVD = Potency x E.L
Sensitivity of Lysate
= 1ml/ml x 18.75 EU/ml
0.125 EU/ml
= 150
Test Dilution : 1:9, 1:18, 1:37, 1:75 and 1: 150
Test sample Rinse: Three 1 ml Syringes from each lot are taken and
completely filled with WFI and incubated it at 37°C for 30 min, Then the
Endotoxins stick to the walls of the Syringe was extracted into the WFI. Then
the WFI rinse was tested and report the value per ml of rinse. Then the
reported value was multiplied by total volume and divide the same with the
number of devices taken, it gives us the Endotoxin content in each 1 ml
Syringe.
Test Dilutions NPC PPC
1:9 − − + +
1:18 − − + +
1:37 − − + +
1:75 − − + +
1:150 − − + +
++: Gel Formation − − : No Gel Formation

5.3 Quantification of Bacterial endotoxins in Medical devices
102
Conclusion: Carried out screening and observed Non Interfering Dilution
(NID) at 1:9 dilutions. Non Interfering dilution is 1:9 dilution.
Endotoxins content is < 1.5 EU/ device.
End-Product Endotoxins Test:
Product : 1 ml Syringe
Preparation : Product Concentration : 1mL / mL
Endotoxin Limit : 20 EU/Device
MVD : 150
Test Dilution : 1:18
Results :
Test Endotoxin
Concentration EU/mL.
NEG
Control
NEG
Product
Control
Test
End
point.
Log of
End
point.
Geometric Mean
=A log (log of End
point.)
Material 2λ λ ½λ ¼λ EU/mL
+ + - − − 0.125 -0.9030
+ + - − − 0.125 -0.9030
+ + - − 0.125 -0.9030
CSE
Water
Control
+ + - − 0.125 -0.9030
+ + - − − 0.0625 -1.2041
+ + - − − 0.0625 -1.2041
+ + - − − 0.125 -0.9030
Positive
Product
Control
+ + - − − 0.125 -0.9030
Antilog
(-3.612/4)
=Antilog –0.903
=0.125EU/mL
Antilog
(-4.2142/4)
=Antilog–1.05355
=0.088EU/mL
Interpretation :
Test results are : Valid
Comments : Product validation for 1 ml Syringe was carried out at 1:8
dilution. The GM of the end points in Endotoxin / LRW was 0.125EU/mL and
Endotoxin/product was 0.088 EU/mL which is in between 2λ and ½ λ, which
indicates no inhibition and enhancement at test dilution 1:8 dilution.

5.3 Quantification of Bacterial endotoxins in Medical devices
103
(iv) 5 mL SYRINGE
Preliminary Screening Test
Product : 5 ml Syringe
Concentration : 1 ml / mL
Endotoxin Limit : K X N
V
K = 20 EU/Device
N = No of Devices
V = Volume of solution rinsed/ taken
E.L = 20 EU/Device x 3 Devices
16 mL
= 3.75 EU/ml
MVD = Potency x E.L
Sensitivity of Lysate
= 1ml/ml x 3.75 EU/ml
0.125 EU/ml
= 30
Test Dilution : 1:1, 1:2, 1:4, 1:8 and 1: 16
Test sample Rinse: Three 5 ml Syringes from each lot are taken and
completely filled with WFI and incubated it at 37°C for 30 min, Then the
Endotoxins stick to the walls of the Syringe was extracted into the WFI. Then
the WFI rinse was tested and report the value per ml of rinse. Then the
reported value was multiplied by total volume and divide the same with the
number of devices taken, it gives us the Endotoxin content in each 5 ml
Syringe.
Test Dilutions NPC PPC
1:2 + + + +
1:4 − − + +
1:8 − − + +
1:16 − − + +
1:30 − − + +
++: Gel Formation − − : No Gel Formation

5.3 Quantification of Bacterial endotoxins in Medical devices
104
Conclusion: Carried out screening and observed Non Interfering Dilution
(NID) at MVD/8 (1:2) dilution. Non Interfering dilution is 1:2
dilution. Endotoxins content is < 1.6 EU/ device.
End-Product Endotoxins Test:
Product : 5 ml Syringe
Preparation : Product Concentration : 1mL / mL
Endotoxin Limit : 20 EU/Device
MVD : 30
Test Dilution : 1:4
Results :
Test Endotoxin
Concentration EU/mL.
NEG
Control
NEG
Product
Control
Test
End
point.
Log of
End
point.
Geometric Mean
=A log (log of End
point.)
Material 2λ λ ½λ ¼λ EU/mL
+ + - − − 0.125 -0.9030
+ + - − − 0.125 -0.9030
+ + - − 0.125 -0.9030
CSE
Water
Control
+ + - − 0.125 -0.9030
+ + - − − 0.0625 -1.2041
+ + - − − 0.0625 -1.2041
+ + - − − 0.125 -0.9030
Positive
Product
Control
+ + - − − 0.125 -0.9030
Antilog
(-3.612/4)
=Antilog –0.903
=0.125EU/mL
Antilog
(-4.2142/4)
=Antilog–1.05355
=0.088EU/mL
Interpretation :
Test results are : Valid
Comments : Product validation for 5ml Syringe was carried out at MVD/4
(1:4). The GM of the end points in Endotoxin / LRW was 0.125EU/mL and
Endotoxin/product was 0.088 EU/mL which is in between 2λ and ½ λ, which
indicates no inhibition and enhancement at test dilution 1:4 dilution.

5.3 Quantification of Bacterial endotoxins in Medical devices
105
(V) SUTURES
Preliminary Screening Test
Product : Suture
Concentration : 1 ml / mL
Endotoxin Limit : K X N
V
K= 20 EU/Device
N = No of Devices
V = Volume of solution rinsed/ taken
E.L = 20 EU/Device x 3 Devices
5mL
= 12 EU/ml
MVD = Potency x E.L
Sensitivity of Lysate
= 1ml/ml x 12 EU/ml
0.125 EU/ml
= 96
Test Dilution : 1:6, 1:12, 1:24, 1:48 and 1: 96
Test sample Rinse: Three Sutures sets from each lot are taken and soaked
them in 5 ml WFI and incubated it at 37°C for 30 min, and then the
Endotoxins stick to the suture roll was extracted into the WFI. Then the WFI
rinse was tested and report the value per ml of rinse. Then the reported
value was multiplied by total volume and divide the same with the number
of devices (Sutures), it gives us the Endotoxin content in each Suture roll.
Test Dilutions NPC PPC
1:6 − − + +
1:12 − − + +
1:24 − − + +
1:48 − − + +
1:96 − − + +
++: Gel Formation − − : No Gel Formation

5.3 Quantification of Bacterial endotoxins in Medical devices
106
Conclusion: Carried out screening and observed Non Interfering Dilution
(NID) at MVD/16 (1:1). Non Interfering dilution is 1:6 (I Part of
Rinse and 5 parts of LRW) Endotoxins content is < 1.25 EU/
device (Per suture roll).
End-Product Endotoxins Test:
Product : Suture
Preparation : Product Concentration : 1mL / mL
Endotoxin Limit : 20 EU/Device
MVD : 96
Test Dilution : 1:12
Results : Test Endotoxin
Concentration EU/mL.
NEG
Control
NEG
Product
Control
Test
End
point.
Log of
End
point.
Geometric Mean
=A log (log of End
point.)
Material 2λ λ ½λ ¼λ EU/mL
+ + - − − 0.125 -0.9030
+ + - − − 0.125 -0.9030
+ + - − 0.125 -0.9030
CSE
Water
Control
+ + - − 0.125 -0.9030
+ + - − − 0.0625 -1.2041
+ + - − − 0.0625 -1.2041
+ + - − − 0.125 -0.9030
Positive
Product
Control
+ + - − − 0.125 -0.9030
Antilog
(-3.612/4)
=Antilog –0.903
=0.125EU/mL
Antilog
(-4.2142/4)
=Antilog–1.05355
=0.088EU/mL
Interpretation :
Test results are : Valid
Comments : Product validation for Suture (roll) was carried out at MVD/8
(1:12). The GM of the end points in Endotoxin / LRW was 0.125EU/mL and
Endotoxin/product was 0.088 EU/mL which is in between 2λ and ½ λ, which
indicates no inhibition and enhancement at test dilution MVD / 8 (1:12).

















