177 Chapter 7 HDAC2 in COPD 7.1 Introduction As already described in detail in Chapter 2, COPD is characterized by progressive inflammation of the airways (and destruction of the lung parenchyma) mediated by increased expression of inflammatory genes, in response to noxious particles, gases and especially tobacco smoke (Barnes, 2003). In COPD a specific pattern of airway inflammation is mainly characterized by increased numbers of neutrophils, macrophages and T-lymphocytes, predominantly cytotoxic (CD8+) cells (Barnes et al., 2005; Saetta et al., 1993; Saetta et al., 1998; Saetta et al., 1997; Saetta, Turato, Maestrelli, Mapp, & Fabbri, 2001). The increased expression of these inflammatory genes is regulated by acetylation of core histones around which DNA is wound, allowing access of pro-inflammatroy transcription factors to transcription-regulatory sites. On the other hand these activated genes are switched off by deacetylation of these histones (Chapter 2) (Barnes et al., 2005). Examples of the transcription factors involved in this regulatory system in airway diseases are nuclear factor-Kappa B (NF-κB), activator protein-1 (AP-1) and also the activated glucocorticoid receptors (Barnes, 2006d). These are all at least partly controlled by this histone acetylation/deacetylation mechanism. COPD responds relatively poorly to therapeutic corticosteroids, even though steroids have been shown to have effects both in the short term and long term use. As discussed in Chapter 2 there is emerging evidence that corticosteroids resistance in COPD is due to decrease in histone deacetylase -2 (or HDAC-2) (Barnes, 2006c; Ito et al., 2001). Though exact mechanisms are still not clearly understood, it is suggested that it may involve modulation of HDACs by nitrosylation on distinct tyrosine residues in response to tobacco smoke (Barnes et al., 2005). Since 1960, it has been known that acetylation of DNA-associated histone proteins and remodelling of the tightly packed chromatin structure is associated with
Transcript
177
Chapter 7
HDAC2 in COPD
7.1 Introduction
As already described in detail in Chapter 2, COPD is characterized by progressive
inflammation of the airways (and destruction of the lung parenchyma) mediated by
increased expression of inflammatory genes, in response to noxious particles, gases
and especially tobacco smoke (Barnes, 2003). In COPD a specific pattern of airway
inflammation is mainly characterized by increased numbers of neutrophils,
macrophages and T-lymphocytes, predominantly cytotoxic (CD8+) cells (Barnes et
al., 2005; Saetta et al., 1993; Saetta et al., 1998; Saetta et al., 1997; Saetta, Turato,
Maestrelli, Mapp, & Fabbri, 2001). The increased expression of these inflammatory
genes is regulated by acetylation of core histones around which DNA is wound,
allowing access of pro-inflammatroy transcription factors to transcription-regulatory
sites. On the other hand these activated genes are switched off by deacetylation of
these histones (Chapter 2) (Barnes et al., 2005). Examples of the transcription factors
involved in this regulatory system in airway diseases are nuclear factor-Kappa B
(NF-κB), activator protein-1 (AP-1) and also the activated glucocorticoid receptors
(Barnes, 2006d). These are all at least partly controlled by this histone
acetylation/deacetylation mechanism.
COPD responds relatively poorly to therapeutic corticosteroids, even though steroids
have been shown to have effects both in the short term and long term use. As
discussed in Chapter 2 there is emerging evidence that corticosteroids resistance in
COPD is due to decrease in histone deacetylase -2 (or HDAC-2) (Barnes, 2006c; Ito
et al., 2001). Though exact mechanisms are still not clearly understood, it is
suggested that it may involve modulation of HDACs by nitrosylation on distinct
tyrosine residues in response to tobacco smoke (Barnes et al., 2005).
Since 1960, it has been known that acetylation of DNA-associated histone proteins
and remodelling of the tightly packed chromatin structure is associated with
178
induction of genes (Littau et al., 1965). Histone and chromatin remodelling is central
to gene expression and regulation through the process of acetylation, deacetylation
and also methylation, though this is even less understood (Barnes et al., 2005; Rice &
Allis, 2001). Research regarding the exact role of histone acetylation and
deacetylation in chronic inflammatory disease is only in its infancy, and even more
so in COPD, where the picture has probably been made rather over-simplistic, as
indeed the degree of absolute ICS insensitivity has been exaggerated. However, there
is significant body of literature (as explained in Chapter 2) suggesting that expression
and activity of anti-inflammatory HDAC2 are reduced in COPD lungs, airways and
alveolar macrophages and becomes worse with severity of the disease (Barnes,
2006c; Ito et al., 2001).
The methodology that has been used was not comprehensive enough to identify the
changes in total cellularity in the lamina propria and largely dependent on molecular
RNA quantitation and protein analysis which could not take into account relative
differences in cellular profiles in airway tissues in different disease and control
groups, where biases could arise due to differences in total and differential
cellularity. There has been a serious lack of comprehensive biopsy studies to confirm
the extent of suppressed HDAC2 levels by immunostaining within the airways of
COPD; which can take into account cell numbers and type, its reversibility with ICS
or smoking cessation. Therefore, I designed a detailed cross-sectional study and used
material collected in a longitudinal study (D. W. Reid et al., 2008), for looking in
detail at the status of HDAC2 expression in COPD airways.
179
7.2 Hypothesis
I hypothesized that the current literature is correct in that HDAC2 is down-regulated
in COPD airways, and that these reduced HDAC2 levels are normalised by
aggressive ICS therapy and also smoking cessation in patients with COPD.
7.2.1 Aims
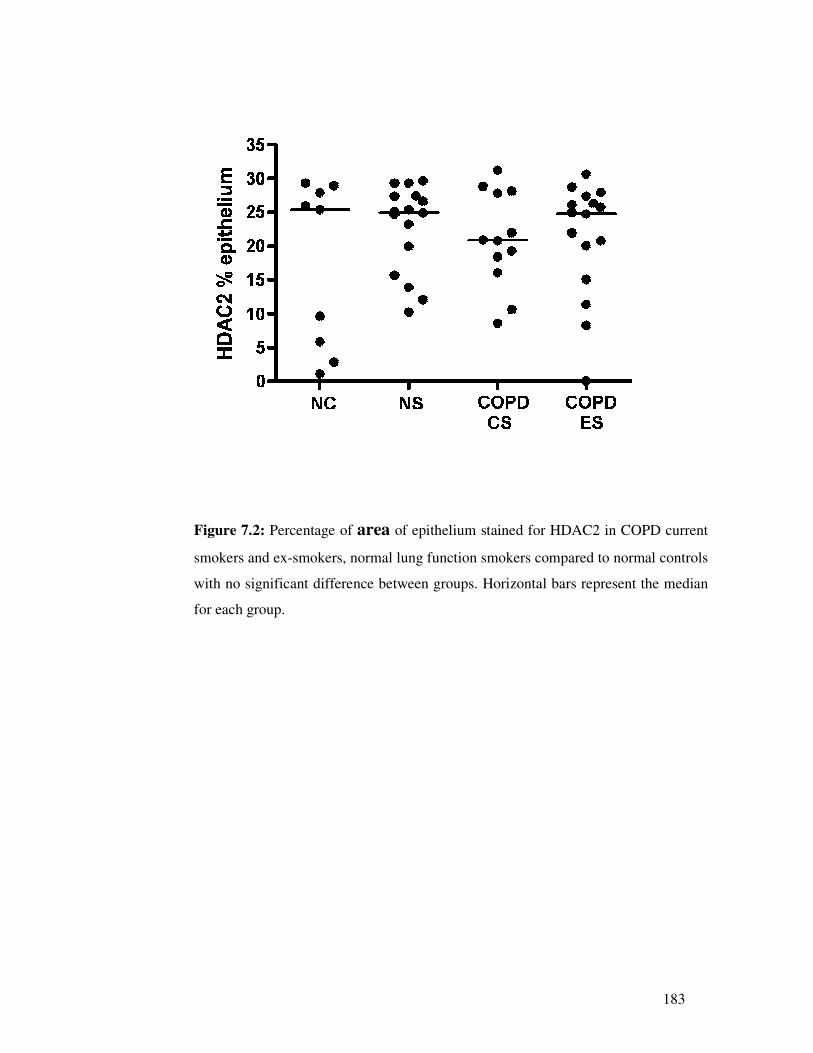
1. To confirm the extent of suppressed HDAC2 by immunostaining airway biopsies
from COPD current smokers and also from normal lung function smokers to
understand whether changes in HDAC2 are solely related to COPD or smoking
or both.
2. To confirm the potential of smoking cessation to raise HDAC2 expression by
comparing airway biopsies from COPD ex-smokers versus current smokers with
COPD, and the potential for HDAC2 expression to be normalised by aggressive
ICS therapy.
7.3 Methods and materials
7.3.1 Subjects and study design
The studies involved both detailed cross-sectional and longitudinal analyses. In the
cross-sectional study, 17 current smokers with established COPD (CS), 16 current
smokers with normal lung function (NS), 17 ex-smokers with COPD (ES) and 15
normal healthy, never-smoking controls (NC) were recruited for bronchoscopy and
airway biopsy. In longitudinal analysis the COPD subjects further entered a double