Chemical Characterization of Isoprene- and Monoterpene-DerivedSecondary Organic Aerosol Tracers in Remote Marine Aerosols overa Quarter CenturyTianqu Cui,† Hilary S. Green,†,∥ Paul W. Selleck,‡ Zhenfa Zhang,† Rachel E. O’Brien,§,⊥ Avram Gold,†
Melita Keywood,‡ Jesse H. Kroll,§ and Jason D. Surratt*,†
†Department of Environmental Sciences and Engineering, Gillings School of Global Public Health, University of North Carolina atChapel Hill, Chapel Hill, North Carolina 27599, United States‡CSIRO Oceans and Atmosphere, Aspendale, Victoria 3195, Australia§Department of Civil and Environmental Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139,United States
*S Supporting Information
ABSTRACT: Isoprene and monoterpenes are ubiquitousprecursors of biogenic secondary organic aerosol (SOA) overcontinental regions; however, their contributions to organicaerosol (OA) mass and chemical processes in remote marineatmospheres remain uncertain. Determining exact sources oforganics in marine aerosol is needed to more accurately assessaerosol climate effects in remote locations through coupledatmospheric chemistry−climate simulations. Over 200archived ocean-originated particulate samples collected from1991 to 2015 at Cape Grim, Australia, were analyzed using asuite of chromatographic and mass spectrometric techniques.To our knowledge, this is one of the longest running continualarchives of marine aerosol samples collected under remotebackground conditions. Up to 6.75 and 1.11 ng m−3 of isoprene- and monoterpene-derived SOA, respectively, were quantifiedusing authentic standards. Although there was no clear temporal trend over the decades, consistent seasonal variations wereobserved, with higher biogenic SOA in summer, which was moderately to strongly correlated (r = 0.61−0.85) with marinebioactivity indicators, such as methanesulfonic acid and chlorophyll a. These correlations indicate that marine biota likely emitbiogenic SOA precursors that are able to contribute to remote background OA levels. On the basis of historical observations ofOA mass estimated in the marine baseline samples, these biogenic SOA tracers contributed up to 0.71% (0.26 ± 0.24% onaverage) of the total OA mass fraction. Our data suggest that larger sources of OA exist in remote marine atmospheres, such asprimary OA produced from bubble-bursting processes or SOA precursors derived from photosensitized reactions of dissolvedorganic matter present in the sea surface microlayer.
Atmospheric aerosol particles derived from marine sources canplay a role in the global climate system through direct andindirect effects.1 In recent decades, sources and chemicalconstituents of marine aerosol particles have been exam-ined.2−4 While primary aerosol is emitted directly from wind-driven bubble bursting on the ocean surface,5 marinesecondary aerosol particles originate largely from theatmospheric oxidation of biogenic volatile organic compounds(BVOCs),6,7 including phytoplankton-emitted dimethylsulfide(DMS)2 and well-established secondary organic aerosol (SOA)precursors, such as isoprene and monoterpenes.8−10 Whilecurrent debate focuses on the magnitude and spatial
distribution of global fluxes of isoprene and monoterpenes aswell as their contribution to sub-micrometer marine aerosolmass,11,12 the origins, chemical composition, and formationmechanisms of marine SOA derived from these precursorsremain poorly characterized.Since marine biota were unveiled as a source of isoprene and
monoterpene emissions,8,10 their production rates have been
Special Issue: New Advances in Organic Aerosol Chemistry
Received: March 18, 2019Revised: April 26, 2019Accepted: April 29, 2019Published: April 29, 2019
Article
http://pubs.acs.org/journal/aesccqCite This: ACS Earth Space Chem. 2019, 3, 935−946
determined from a variety of marine phytoplankton,heterotrophic bacteria, and seaweeds,13,14 with productionrates varying with plankton species, chlorophyll concentration,and environmental parameters (e.g., temperature, solarradiation, and nutrient abundance).11,15 Oceanic studies reportatmospheric mixing ratios of isoprene and monoterpenes up toseveral hundred parts per trillion by volume (pptv), likelyresulting in SOA derived from marine biological emis-sions.10,16,17 Specifically, significant levels of isoprene-derivedSOA (iSOA) and monoterpene-derived SOA (mSOA) tracers,up to 36 and 20 ng m−3, respectively, have been measuredfrom marine samples collected from the Arctic to Antarcticregions.18 Modeling estimates based on marine chlorophyll aextrapolate the global oceanic fluxes of isoprene to be <1−12Tg of C year−1 and α-pinene to be <0.2−30 Tg of C year−1
(from the “bottom-up” to “top-down” approaches).19,20 Inaddition to biogenic sources, recent studies revealed potentialaerosol sources involving dissolved organic matter frombubble-bursting processes and photosensitized reactions ofnon-volatile plankton-derived organics present in the seasurface microlayer, possibly resulting in subsequent SOAformation.21,22
Spatial distributions of oceanic emissions of isoprene andmonoterpenes as well as their SOA tracers have been examinedin a large number of field studies around the globe.18,23
However, few field studies have been able to assess temporaltrends of SOA quantity and composition.24,25 Moreover, mostfield measurements have been performed at coastal locations,and higher concentrations of SOA were observed than incentral or remote oceans. The difference is likely due to higherlevels of phytoplankton in coastal waters or continentaloutflows and coastal vessel emissions.18,26 Therefore, con-tinental and anthropogenic sources should be eliminated todetermine the precise contribution of the remote marine-derived BVOCs to the formation of SOA.In this study, we analyzed over 200 marine-derived aerosol
samples collected at Cape Grim, Australia, from australsummer and winter seasons during 1991−2015. To ourknowledge, this is one of the longest running filter samplearchives of marine-derived aerosol monitored under cleanbackground conditions, where continental outflows wereintentionally excluded. Known iSOA and mSOA tracers weremeasured at the molecular level in filter extracts by ultra-performance liquid chromatography interfaced to a high-resolution quadrupole time-of-flight mass spectrometerequipped with an electrospray ionization source (UPLC/ESI−HR-QTOFMS) and gas chromatography interfaced to aquadrupole mass spectrometer equipped with electronionization source (GC/EI−MS).27−32 Our study aimed toobtain the quantities and temporal trends of iSOA and mSOAfrom remote marine aerosol samples to gain insights into theirsources and contributions over the oceans. In addition,correlations of marine bioactivity indicators, includingmethanesulfonic acid (MSA), oxalic acid, and chlorophyll a,with iSOA and mSOA tracers were assessed to further establishmarine sources.2,33,34
2. MATERIALS AND METHODS2.1. Field Aerosol Collection under Baseline Con-
ditions at Cape Grim, Australia. The Cape Grim BaselineAir Pollution Station is located at the top of a 94 m cliff on thewestern side of the northwest tip of Tasmania, Australia (40°40′ 56″ S, 144° 41′ 18″ E), as shown in Figure S1 of the
Supporting Information. This station is surrounded by a rockyreef and nearby grassland. The station is a global monitoringstation in World Meteorological Organization (WMO)’sGlobal Atmospheric Watch Program and has been operatingsince 1976.35
Week-long aerosol filter samples were collected from 1991to 2015 using high-volume filter samplers (customizedinstrument in 1991−2002 and high-volume sampler, Ecotech3000, in 2003−2015) equipped with size-selective inlets,resulting in a particle size cut of coarse particulate matter(PM10, aerosol with aerodynamic diameters of ≤10 μm)during 2003−2015 and PM11−12 (aerosol with aerodynamicdiameters of ≤11−12 μm) during 1990−2003. The samplingflow rate was 54 m3 h−1, regulated by pressure transducers overthe 1990−2003 sampling period, and 67.8 m3 h−1, regulated bymass flow controllers over the 2003−2015 sampling period.The flow rates were audited and calibrated with a calibrationorifice plate every 3 months, and the size-selective inlet wascleaned and regreased monthly. Samplers were equipped withPallflex Emfab filters (Pall Life Sciences), 25 × 20 cm,constructed of pure borosilicate glass microfibers reinforcedwith woven glass cloth and bonded with polytetrafluoro-ethylene (PTFE). Monthly field blanks were taken by runningthe high-volume filter sampler for a few minutes.To ensure exclusive collection of ocean-originated aerosols,
the high-volume filter samplers were operated only duringperiods with on-shore air flow that originated from the openocean. From 1990 to 2003, the “baseline event switch 2” wasemployed for particulate sample collection: wind directionbetween 190° and 280° (Figure S1 of the SupportingInformation) and the condensation nucleus (CN) concen-tration of <600 particles cm−3. Monthly operation with“baseline event switch 3” commenced in 2003: wind directionbetween 190° and 280° and the CN concentration thresholddetermined by the 90th percentile of CN hourly medians over5 previous years at the 50 m level, interpolated using cubicsplines to give daily values. Post-analysis of air mass origin andback trajectory, standard deviation of ozone, and angular radon(an ideal indicator for continental emissions) distributionssuggests that the wind and CN conditions are appropriatetriggers for the switch to ensure baseline conditions. See theSupporting Information for details regarding sample collection,baseline criteria, and collocated data in annual or biennial CapeGrim baseline reports since 1976.Exposed filters were stored at 3 °C at Cape Grim until they
were transported to Commonwealth Scientific and IndustrialResearch Organization (CSIRO) laboratories at Aspendale,Victoria, Australia, where they underwent gravimetric massdetermination and ion chromatography (IC) analysis forsoluble ion concentrations, usually within 2 months of samplecollection. Then, the remaining non-extracted filters werestored at 3 °C until shipped to the University of NorthCarolina at Chapel Hill (UNC) in 2016 to be stored at −20 °Cuntil extracted for the subsequent GC/EI−MS and UPLC/ESI−HR-QTOFMS analyses.
2.2. Filter Extractions. 2.2.1. Strategy of Filter Extrac-tions. A total of 211 field samples and 26 field blank filterswere selected from 14 summer seasons (mid-December−mid-February) and 15 winter seasons (mid-June−mid-August)during the 1991−2015 sampling period for chemical analysisof known iSOA and mSOA tracers. Each season wasrepresented by compositing 5−9 week long filters collectedduring the season (summarized in Table S1 of the Supporting
ACS Earth and Space Chemistry Article
DOI: 10.1021/acsearthspacechem.9b00061ACS Earth Space Chem. 2019, 3, 935−946
Information), resulting in average seasonal concentrationsbeing determined for each SOA tracer. An aliquot of each filter(12.5 × 20 cm) was individually extracted and combined byseason to ensure sufficient mass for GC/EI−MS and UPLC/ESI−HR-QTOFMS analyses. This protocol provided sufficienttime resolution of each biogenic SOA tracer over the entiresampling period in a cost-effective manner.2.2.2. Filter Extraction Procedures. Filters were extracted
with 20 mL of high-purity methanol (LC−MS CHROMA-SOLV-grade, Sigma-Aldrich), followed by sonication for 45min. The methanol extracts were then blown dry under agentle N2 stream at room temperature (21−22 °C). Driedseasonal extracts were reconstituted in 3 mL of methanol, splitinto three equal aliquots, blown dry, and then reconstitutedusing solvents that were appropriate for the specific analyticaltechnique applied to the chemical characterization, asdescribed in subsequent sections. Immediately prior toanalysis, reconstituted seasonal extracts were filtered througha PTFE syringe filter (0.2 μm pore size, Agilent) to removeundissolved sea salt particles, insoluble particles, or filter fibers.2.3. Aerosol-Phase Chemical Characterization.
2.3.1. Instrumentation. Chemical characterization of theiSOA and mSOA constituents was performed on a GC/EI−MS (Hewlett-Packard 5890 Series II gas chromatographinterfaced to a HP 5971A Series mass selective detector) andan UPLC/ESI−HR-QTOFMS (Agilent 6520 Series) operatedin the negative ion mode. Operating details of both GC/EI−MS and UPLC/ESI−HR-QTOFMS have been describedelsewhere and are briefly described below.36
2.3.2. GC/EI−MS for Determination of iSOA. For analysisby GC/EI−MS, the GC column (Econo-Cap EC-5, 30 m ×0.25 mm × 0.25 μm) was heated up to 310 °C and theionization source operated at 70 eV; the full temperatureelution program for the GC/EI−MS has been describedpreviously by Surratt et al.37 One aliquot of the dried seasonalextract was derivatized by the addition of 200 μL of N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA) + trimethyl-chlorosilane (99:1, v/v, Supleco) and 100 μL of pyridine(98% anhydrous, Sigma-Aldrich), followed by heating for 1 hat 70 °C. Analysis was performed within 24 h followingtrimethylsilylation. iSOA standards were synthesized in-houseby published procedures: 2-methylglyceric acid (2-MG), 2-methyltetrols (2-methylthreitol and 2-methylerythritol), andcis- and trans-3-methyltetrahydrofuran-3,4-diols (3-MeTHF-3,4-diols).38,39 In addition, high-purity commercially availablechemicals (>99%, all from Sigma-Aldrich), including levoglu-cosan and ketopinic acid, were used as authentic or surrogateexternal standards for identification and quantification.2.3.3. UPLC/ESI−HR-QTOFMS for Determination of
mSOA. For UPLC/ESI−HR-QTOFMS analysis, an aliquotof the dried seasonal extract was reconstituted with 300 μL of50:50 (v/v) methanol (LC−MS CHROMASOLV-grade,Sigma-Aldrich) and high-purity deionized water (18.2 MΩ,Milli-Q). Aliquots (5 μL) were injected onto a reverse-phaseUPLC column (Waters ACQUITY UPLC HSS T3 column,2.1 × 100 mm, 1.8 μm particle size) eluted at a flow rate of 0.3mL min−1 with a mobile phase of methanol containing 0.1%acetic acid (LC−MS CHROMASOLV-grade, Sigma-Aldrich)and water containing 0.1% acetic acid (LC−MS CHROMA-SOLV-grade, Sigma-Aldrich). The gradient elution programused to separate these mSOA components has been previouslydescribed by Kristensen et al.40
A number of carboxylic acids and organosulfates (OSs)derived from monoterpenes and/or isoprene were examinedand quantified by the UPLC/ESI−HR-QTOFMS operated innegative ion mode using authentic or surrogate standards.BVOC-derived OSs that were closely examined includedglycolic acid sulfate (C2H3O6S
−), lactic acid sulfate(C3H6O6S
−), 2-MG OS (C4H7O7S−), 2-methyl-3-buten-2-ol
(MBO)-derived OS (C5H11O6S−), 2-methyltetrol sulfates
(C5H1 1O7S−) , and a monote rpene -de r i v ed OS
(C10H17O5S−);32,41−43 however, no OSs were detected in
any of the composited seasonal samples. On the basis of thesimilarity of structures, terebic acid (98%, Tokyo ChemicalIndustry) was used to quantify terebic and terpenylic acids,both of which are known mSOA constituents.28,31 In addition,authentic pinic acid (99%, Sigma-Aldrich) was used to quantifypinic acid, whereas authentic 3-methyl-1,2,3-butanetricarbox-ylic acid (MBTCA, 98%, Toronto Research Chemicals) wasused to quantify MBTCA but used as a surrogate standard toquantify diaterpenylic acid acetate (DTAA). Pinic acid,MBTCA, and DTAA are also known as mSOA tracers.28−30,44
2.3.4. IC for Water-Soluble Particle-Phase Constituents. A6.25 cm2 section of each high-volume filter sample was cut andextracted to measure major water-soluble constituents, such asMSA and oxalic acid, by IC (Dionex ICS-3000), performed atthe CSIRO laboratories within 2 months after collection (seethe Supporting Information for details).
2.3.5. Quality Control and Corrections. Six-point calibra-tion curves were constructed for each standard over theconcentration range of 0.25−50 μg mL−1 for GC/EI−MS and0.01−50 μg mL−1 for UPLC/ESI−HR-QTOFMS. SOA tracersquantified from the field samples were not detected from anylaboratory or field blanks. The extraction efficiency for allcompounds was >94%, confirmed by a second extraction andanalysis of selected filters. The recovery rates of spikedstandards were 85 ± 15% (average ± one standard deviation)for GC/EI−MS and 65 ± 20% for UPLC/ESI−HR-QTOFMS. The results reported in this study have not beencorrected for recovery. Carryover of SOA constituentsquantified by GC/EI−MS and UPLC/ESI−HR-QTOFMSwas found to be negligible by analyzing solvent blanks betweeninjections of seasonal samples from Cape Grim. The analyticalprecision determined by quintuplicate injections of selectsamples and standards was 1.8 ± 0.2% for GC/EI−MS and 3.6± 1.2% for UPLC/ESI−HR-QTOFMS.Possible changes in the chemical composition of archived
filter samples were also assessed during this study. A total of 24selected filter samples covering 1990−2011 were reanalyzedusing IC in 2016 to identify changes in inorganic or organicconstituents. The following average increases from time ofcollection to 2016 were observed: Na+, 4 ± 20%; Mg2+, 6 ±24%; SO4
2−, 11 ± 18%; oxalate, 18 ± 40%; and MSA, 1 ±29%. Different fractions of a set of filters (n = 6) were analyzedby UPLC/ESI−HR-QTOFMS in 2011 and again in 2014. Nosignificant change was observed over the 3 years of storageunder dark conditions at −20 °C; however, this cannot fullyrule out that some degradation of the iSOA and mSOA tracersdid not occur during the years of storage at Cape Grim,CSIRO, and UNC. It is worth noting here that the presence ofiSOA tracers, such as 2-MG and the 2-methyltetrols, wasrecently reported in ice core samples, representing severalhundred years of atmospheric aerosol deposition.24,25 Resultsof the analysis were interpreted under the assumption that theiSOA tracers were stable over time. Recent work has shown
ACS Earth and Space Chemistry Article
DOI: 10.1021/acsearthspacechem.9b00061ACS Earth Space Chem. 2019, 3, 935−946
that iSOA is low-volatility in nature.45 Further, as will bediscussed in subsequent sections, the quantities of the iSOAand mSOA tracers are in good agreement with recent marineaerosol composition studies.
3. RESULTS AND DISCUSSION
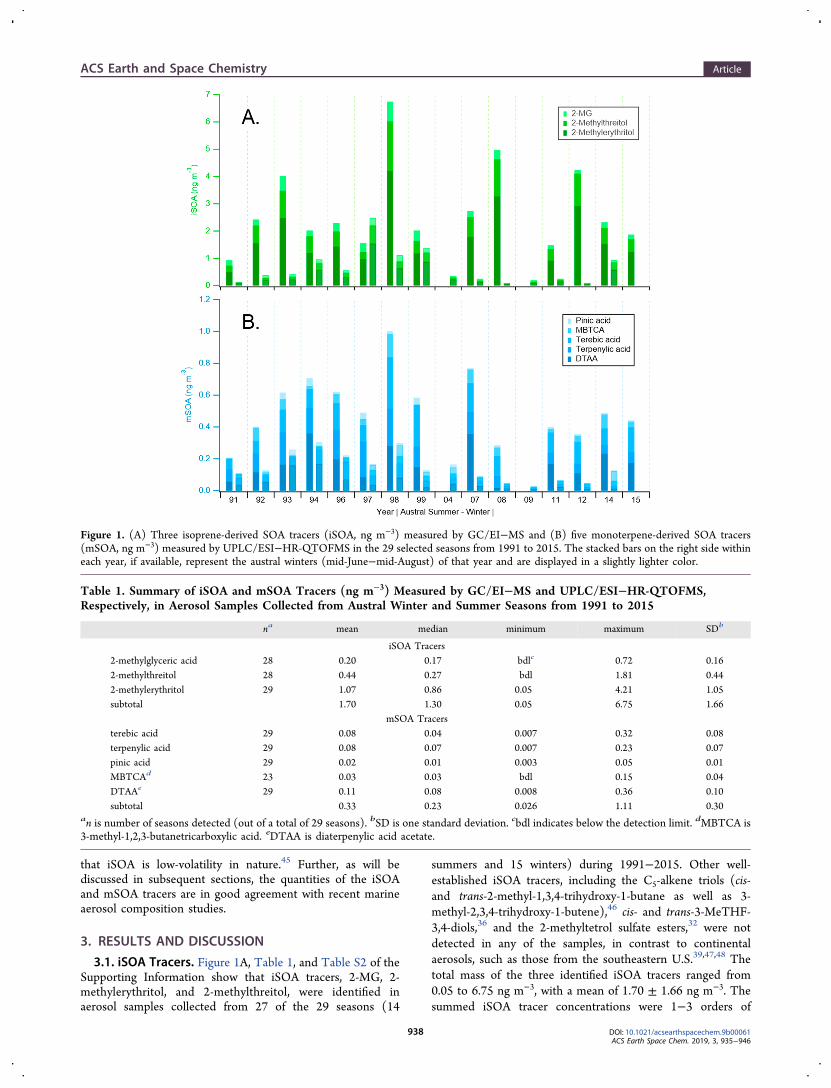
3.1. iSOA Tracers. Figure 1A, Table 1, and Table S2 of theSupporting Information show that iSOA tracers, 2-MG, 2-methylerythritol, and 2-methylthreitol, were identified inaerosol samples collected from 27 of the 29 seasons (14
summers and 15 winters) during 1991−2015. Other well-established iSOA tracers, including the C5-alkene triols (cis-and trans-2-methyl-1,3,4-trihydroxy-1-butane as well as 3-methyl-2,3,4-trihydroxy-1-butene),46 cis- and trans-3-MeTHF-3,4-diols,36 and the 2-methyltetrol sulfate esters,32 were notdetected in any of the samples, in contrast to continentalaerosols, such as those from the southeastern U.S.39,47,48 Thetotal mass of the three identified iSOA tracers ranged from0.05 to 6.75 ng m−3, with a mean of 1.70 ± 1.66 ng m−3. Thesummed iSOA tracer concentrations were 1−3 orders of
Figure 1. (A) Three isoprene-derived SOA tracers (iSOA, ng m−3) measured by GC/EI−MS and (B) five monoterpene-derived SOA tracers(mSOA, ng m−3) measured by UPLC/ESI−HR-QTOFMS in the 29 selected seasons from 1991 to 2015. The stacked bars on the right side withineach year, if available, represent the austral winters (mid-June−mid-August) of that year and are displayed in a slightly lighter color.
Table 1. Summary of iSOA and mSOA Tracers (ng m−3) Measured by GC/EI−MS and UPLC/ESI−HR-QTOFMS,Respectively, in Aerosol Samples Collected from Austral Winter and Summer Seasons from 1991 to 2015
an is number of seasons detected (out of a total of 29 seasons). bSD is one standard deviation. cbdl indicates below the detection limit. dMBTCA is3-methyl-1,2,3-butanetricarboxylic acid. eDTAA is diaterpenylic acid acetate.
ACS Earth and Space Chemistry Article
DOI: 10.1021/acsearthspacechem.9b00061ACS Earth Space Chem. 2019, 3, 935−946
magnitude lower than those typically observed in continentallocations influenced by high isoprene emissions fromdeciduous trees and large amounts of acidic sulfate aerosolthat promote iSOA formation.27,36,39,49−54 The summed iSOAconcentrations are comparable to results from previous studiesof concentrations over the southern Pacific and Indian Oceansfrom September to March18 and over oceans at low latitudes inthe Northern Hemisphere from October to March23 andapproximately 1 order of magnitude higher than in theCanadian High Arctic from February to June.55
Among the three measured iSOA tracers, the 2-methyltetrolswere the most abundant iSOA constituents, with 2-methylerythritol being more abundant than 2-methylthreitol(Table 1 and Figure 1A). The ratio of 2-methylerythritol to 2-methylthreitol was approximately 5:2, similar to previousoceanic observations.18 2-MG contributed only a small massfraction (12% on average) to the total mass of the iSOA tracersmeasured during this study. Assuming that the organic aerosol(OA) mass measured in the 2002−2003 samples isrepresentative of other years during 1991−2015, Table S3 ofthe Supporting Information shows a ratio of total identifiediSOA tracers to the total PM10 OA mass averaged to 0.40% (upto 0.65%) in summer and 0.07% in winter (see the SupportingInformation for OA estimation). The mass fractions are muchlower than at continental sites, especially in the summertimesoutheastern U.S., where isoprene emissions combine withanthropogenic-derived acidic sulfate aerosol to yield largequantities of these SOA constituents (e.g., ∼9% of total OAmass at Look Rock, TN, U.S.A.).39
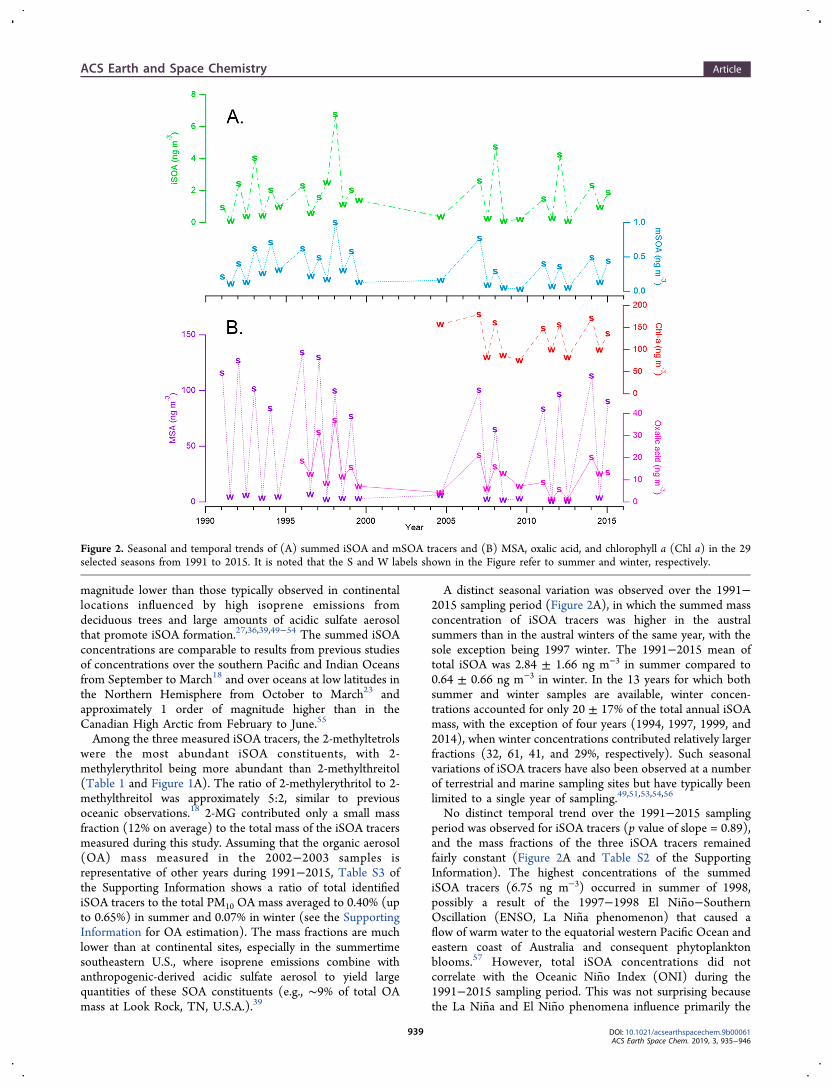
A distinct seasonal variation was observed over the 1991−2015 sampling period (Figure 2A), in which the summed massconcentration of iSOA tracers was higher in the australsummers than in the austral winters of the same year, with thesole exception being 1997 winter. The 1991−2015 mean oftotal iSOA was 2.84 ± 1.66 ng m−3 in summer compared to0.64 ± 0.66 ng m−3 in winter. In the 13 years for which bothsummer and winter samples are available, winter concen-trations accounted for only 20 ± 17% of the total annual iSOAmass, with the exception of four years (1994, 1997, 1999, and2014), when winter concentrations contributed relatively largerfractions (32, 61, 41, and 29%, respectively). Such seasonalvariations of iSOA tracers have also been observed at a numberof terrestrial and marine sampling sites but have typically beenlimited to a single year of sampling.49,51,53,54,56
No distinct temporal trend over the 1991−2015 samplingperiod was observed for iSOA tracers (p value of slope = 0.89),and the mass fractions of the three iSOA tracers remainedfairly constant (Figure 2A and Table S2 of the SupportingInformation). The highest concentrations of the summediSOA tracers (6.75 ng m−3) occurred in summer of 1998,possibly a result of the 1997−1998 El Nino−SouthernOscillation (ENSO, La Nina phenomenon) that caused aflow of warm water to the equatorial western Pacific Ocean andeastern coast of Australia and consequent phytoplanktonblooms.57 However, total iSOA concentrations did notcorrelate with the Oceanic Nino Index (ONI) during the1991−2015 sampling period. This was not surprising becausethe La Nina and El Nino phenomena influence primarily the
Figure 2. Seasonal and temporal trends of (A) summed iSOA and mSOA tracers and (B) MSA, oxalic acid, and chlorophyll a (Chl a) in the 29selected seasons from 1991 to 2015. It is noted that the S and W labels shown in the Figure refer to summer and winter, respectively.
ACS Earth and Space Chemistry Article
DOI: 10.1021/acsearthspacechem.9b00061ACS Earth Space Chem. 2019, 3, 935−946
equatorial Pacific region, while the Cape Grim site is locatedbelow the −40° latitude. We are unable to conclude from ourdata set how strongly (and by what mechanism) the 1997−1998 ENSO event enhanced iSOA levels.iSOA tracers typically observed over the continents were not
identified in our marine PM10 samples. For instance, the threeisomeric C5-alkene triols were not detected concurrently withthe 2-methyltetrols observed by GC/EI−MS at Cape Grim.This finding is consistent with previous oceanic measurementsof average levels of C5-alkene triols summed over a worldwideocean cruise, reporting the C5-alkene triols to be less abundantthan the 2-methyltetrols and 2-MG.18 The apparent absence ofC5-alkene triols in the GC/EI−MS analysis of Cape Grimsample extracts suggests that their concentrations were at orbelow the detection limits of our GC/EI−MS technique. Inthis regard, it is important to note recent work that hasdemonstrated 2-methyltetrol sulfates (if present) decomposeduring GC/EI−MS analysis to yield the C5-alkene triols,58
indicating that C5-alkene triols reported by the GC/EI−MSanalytical protocol may be artifacts of the analytical procedure.Given this suggestion, the absence of C5-alkene triols in theCape Grim samples analyzed by GC/EI−MS are in accordancewith the fact that the isomeric 2-methyltetrol sulfate esterswere not detected by UPLC/ESI−HR-QTOFMS, a sensitiveand definitive protocol for detection of OSs. Consistent withour findings, we note that, during an austral summer, Claeys etal. did not detect the C5-alkene triol or 2-methyltetrol markersat Amsterdam Island, a pristine marine environment in thesouthern Indian Ocean.59 Our inability to detect C5-alkenetriols, 2-methyltetrol sulfate esters, and 2-MG sulfate at CapeGrim over a lengthy period of sampling might be explained by(1) insufficient ambient concentrations as a result of the lowisoprene emission/flux as well as low yield relative to 2-MGand 2-methyltetrols, (2) oceanic formation pathways of 2-MGand 2-methyltetrol sulfates differing from those overcontinental locations,39,60 and (3) degradation (hydrolysis ofisoprene-derived OSs61) over long-term storage. Becauseformation of C5-alkene triols has been associated with acidicsulfate aerosol,36,37 our lack of detection may also suggest thatthe remote marine aerosol collected at Cape Grim are not asacidic as continental aerosol.39,62
Laboratory studies have demonstrated that 2-MG and 2-methyltetrols are formed through different chemical pathwaysfrom the hydroxyl radical (OH)-initiated oxidation and/or
ozonolysis of isoprene. Specifically, 2-methyltetrols are themajor products of isoprene photochemical oxidation by OHunder conditions of low nitrogen oxide concentrations (low-NOx conditions) via the isoprene epoxydiol (IEPOX)pathway.63 Over continental areas, acid-catalyzed multiphasereaction of IEPOX on acidic sulfate aerosol yields these iSOAtracers. Over oceanic regions, 2-methyltetrols may formthrough IEPOX hydrolysis in non-acidic aqueous inorganicaerosol or through cloud processing.64,65 Chamber studies byKleindienst et al. and Riva et al. have shown that isopreneozonolysis can also yield these iSOA tracers;66,67 however,average seasonal O3 concentrations at Cape Grim ranged from∼35 ppb in winter to ∼15 ppb in summer (Figure S2 of theSupporting Information) and did not correlate with theseasonal trend of iSOA, likely ruling out ozonolysis as a sourceof these iSOA marker compounds. 2-MG is formedpredominantly by OH-initiated oxidation of isoprene underhigh-NOx conditions via a pathway through methacrolein(MACR) and methacryloylperoxynitrate (MPAN).37,68 Inremote oceans, the mixing ratios of NOx are typically below10 pptv within the marine boundary layer,69 and 2-MG, ifpresent, must occur through alternative chemical pathways.For example, Kleindienst et al. demonstrated that both 2-MGand 2-methyletrols could form in the absence of both NOx andacidic sulfate aerosol when isoprene was photochemicallyoxidized during smog chamber studies; however, the exactmechanisms of their formations under these conditions remainunclear.70 All in all, under low-NOx conditions, OH-initiatedoxidation of isoprene could lead to the 2-methyltetrols and 2-MG, the iSOA markers that we detected, and might explain theabsence of their corresponding OSs (i.e., 2-methytetrol and 2-MG sulfates) in remote marine aerosol.
3.2. mSOA Tracers. Five mSOA tracers (terebic acid,terpenylic acid, DTAA, MBTCA, and pinic acid) aresummarized in Table 1, Table S2 of the SupportingInformation, and Figure 1B. The mSOA tracers were identifiedin almost all of the 29 seasons. The sum of the five mSOAtracers ranged from 0.03 to 1.11 ng m−3, with an average of0.33 ± 0.30 ng m−3. This is approximately one-fifth of thequantified iSOA tracers, probably as a result of less significantmarine sources of monoterpenes compared to isoprene in thisregion.11
The mSOA tracer concentrations were up to 3 orders ofmagnitude lower than those measured at many continental
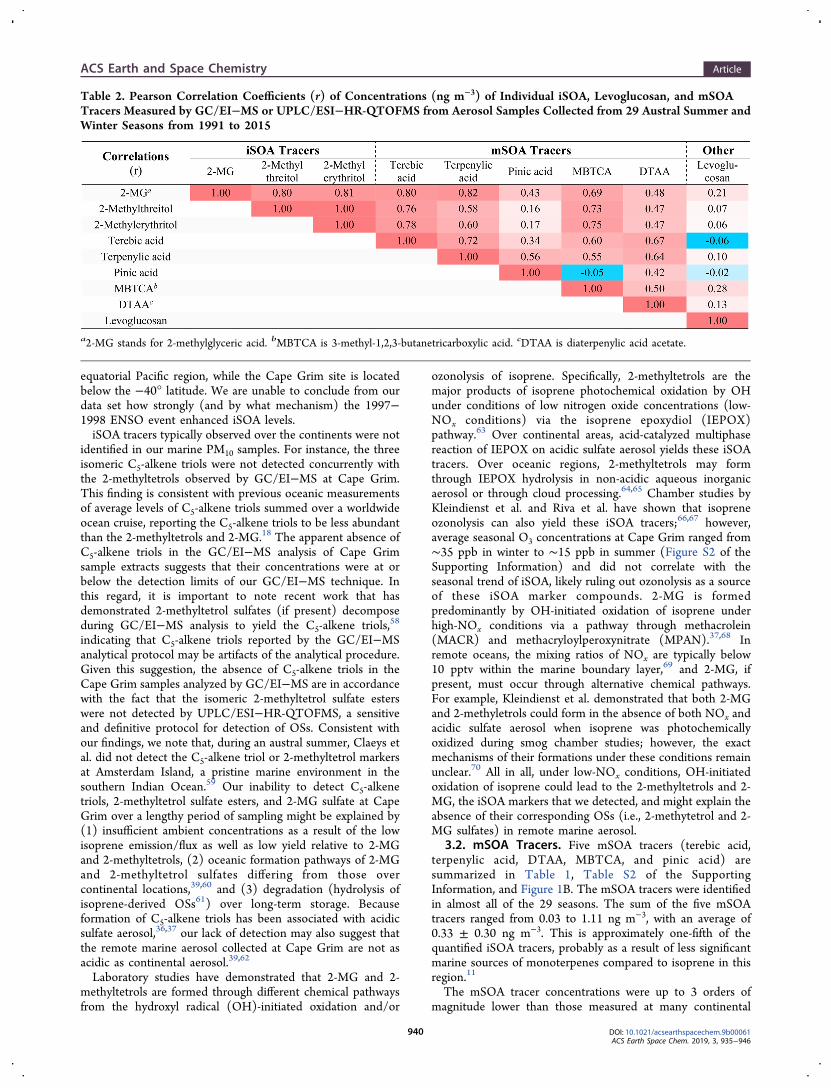
Table 2. Pearson Correlation Coefficients (r) of Concentrations (ng m−3) of Individual iSOA, Levoglucosan, and mSOATracers Measured by GC/EI−MS or UPLC/ESI−HR-QTOFMS from Aerosol Samples Collected from 29 Austral Summer andWinter Seasons from 1991 to 2015
a2-MG stands for 2-methylglyceric acid. bMBTCA is 3-methyl-1,2,3-butanetricarboxylic acid. cDTAA is diaterpenylic acid acetate.
ACS Earth and Space Chemistry Article
DOI: 10.1021/acsearthspacechem.9b00061ACS Earth Space Chem. 2019, 3, 935−946
sites49−51,71 but comparable to the oceanic concentrations overthe Southern Ocean and seas adjacent to the Australiancontinent ranging from latitudes of 30° to 60° S18 and overoceans at low latitudes in the Northern Hemisphere.23 UnlikeiSOA tracers, the measured concentrations of mSOA tracerswere much lower than those in the Canadian High Arctic,55
which is probably influenced by the continental emissions ofmonoterpenes from boreal forests.In addition to the five reported mSOA tracers, correspond-
ing ions of additional mSOA tracers, such as 3-hydroxyglutaric,3-hydroxy-4,4-dimethylglutaric, and 2-hydroxy-4-isopropyla-dipic acids,71 were detected but not quantified because ofthe lack of authentic standards (and lack of appropriatesurrogates). Other known mSOA tracers, such as hydrox-yterpenylic acid,72 α-pinene-derived organosufates,32 anddimer esters, such as pinyl-diaterebyl ester [molecular weight(MW) of 344], pinyl-diaterpenyl ester (MW of 358), andpinonyl-pinyl ester (MW of 368),31,40,73−75 were also targetedfor analysis but were below the detection limit of the UPLC/ESI−HR-QTOFMS.Similar to the iSOA tracers, an obvious seasonal variation
was observed throughout the quarter century period of filtercollection (Figure 2B), with higher concentrations of mSOAtracers in austral summers than in winters. The seasonal meanconcentration of the total mSOA tracers was 0.53 ± 0.21 ngm−3 in summer and 0.15 ± 0.09 ng m−3 in winter. In the 13years with both summer and winter samples available, winterconcentrations accounted for only one-fifth (22 ± 7%) of thesummed mSOA mass. Moreover, the individual and summediSOA concentrations measured by the GC/EI−MS and mSOAmeasured by the UPLC/ESI−HR-QTOFMS agreed well witheach other over the decades (Tables 2 and 3) but neither
correlated with levoglucosan (Table 2). Although levogluco-san, a well-known biomass burning tracer, was not correlatedwith either iSOA or mSOA, long-range transport ofcontinental-derived levoglucosan could contribute from timeto time to the remote marine OA collected at Cape Grim (seebaseline report information in the Supporting Information).No distinct general trend for the measured mSOA tracers
was observed during the entire sampling period (p value =0.29). As in the case of iSOA tracers, the highest concentrationof total mSOA tracers (1.00 ng m−3) occurred in summer of1998, which may indicate the same source or event leading tophytoplankton blooms and the enhanced formation of mSOAand iSOA tracers.In contrast to the iSOA tracers, the relative fractions of the
five mSOA tracers did not remain constant over time. Of the
five reported mSOA tracers, DTAA was the most abundantspecies, followed by terpenylic acid, terebic acid, MBTCA, andpinic acid. These five mSOA tracers have also been reported byUPLC/ESI−HR-QTOFMS as the most abundant mSOAtracers in laboratory-generated α-pinene SOA.40,73 Consistencybetween the field observations and the laboratory studiesabove, including the relative abundance of the tracers aboveand absence of dimer esters, suggests that these mSOA tracerswere primarily produced from OH-initiated oxidation overozonolysis of α-pinene.
3.3. Correlation of iSOA and mSOA with Particle-Phase Bioactivity Indicators. Phytoplankton chemically fixcarbon and release metabolites into seawater and theatmosphere, including DMS, isoprene, and monoter-penes.2,8,10,76 DMS is oxidized to MSA, and isoprene isoxidized to oxalic acid (through glyoxal, methylglyoxal, andpyruvic acid).2,77−79 Studies suggest that aqueous-phaseoxidation of marine biota-derived unsaturated fatty acids andphenolic compounds also contribute to the presence of oxalicacid in marine aerosol.59,80,81 Hence, MSA and oxalic acid areconsidered to be bioactivity tracers for phytoplankton.2,4,33,77
Because isoprene and monoterpenes are co-emitted with DMSby many phytoplankton species,14 the iSOA and mSOA tracersmight be anticipated to correlate with MSA and oxalic acid inthe particulate phase.82 Table 3 and Figure S3 of theSupporting Information show the correlation of summediSOA tracers (∑iSOA) with summed mSOA tracers(∑mSOA) and MSA over 29 seasons from 1991 to 2015and with oxalic acid over 21 season from 1996 to 2015.∑iSOA is moderately correlated with ∑mSOA (r = 0.74),MSA (r = 0.61), and oxalic acid (r = 0.62), while ∑mSOA issomewhat more strongly correlated with MSA (r = 0.75) andoxalic acid (r = 0.81).The seasonal variations of ∑iSOA and ∑mSOA tracers
were described in Figure 2A and MSA, oxalic acid, andchlorophyll a in Figure 2B. Overall, Figure 2 shows a consistentpattern of seasonal variations (with the exception of ∑iSOAduring 1997−1998). The most pronounced pattern isdisplayed by MSA with all winter concentrations of <10 ngm−3 and all summer concentrations of >60 ng m−3. Theseasonal pattern of oxalic acid was less pronounced than MSA,possibly as a result of more sources of oxalic acid as justdescribed. However, the correlations of oxalic acid and MSAwith ∑iSOA were almost the same as those with ∑mSOA.Such moderate correlations highlight the likely role ofphytoplankton as a marine source of the BVOCs (isopreneand monoterpenes), indicating that iSOA and mSOA might beformed through the atmospheric processes similar to those ofother marine biogenic aerosol components (i.e., MSA andoxalic acid) originating from phytoplankton. It is noted herethat the sodium ion (Na+) concentration measured from theCape Grim aerosol samples had no significant correlation witheither the iSOA or mSOA tracers (r = −0.30 and −0.29,respectively, and with p values of >0.05). The lack ofsignificant correlation with the Na+ aerosol mass concentrationfurther suggests that the measured iSOA and mSOA tracers aresecondary rather than primary in nature.
3.4. Correlation of iSOA and mSOA with Chlorophylla. As another proxy for phytoplankton activity, integratedchlorophyll a content was obtained over 13 seasons from 2004to 2015, corresponding to the same period of filter collection,averaged for the region within 30−60° S and 90−150° E(Figure 2B). This region was selected on the basis of back-
Table 3. Pearson Correlation Coefficients (r) ofConcentrations of Summed iSOA Tracers (∑iSOA),Summed mSOA Tracers (∑mSOA), Oxalic Acid, MSA, andChlorophyll a Determined by GC/EI−MS, UPLC/ESI−HR-QTOFMS, IC, or Satellite, Respectively, from AustralSummer and Winter Seasons from 1991 to 2015
ACS Earth and Space Chemistry Article
DOI: 10.1021/acsearthspacechem.9b00061ACS Earth Space Chem. 2019, 3, 935−946
trajectory studies (Supporting Information) for suitable timescales (∼3.8 days) of SOA formation and transport. Thechlorophyll a content, measured daily over the entire planet bythe moderate resolution imaging spectroradiometer (MODIS)instrument aboard National Aeronautics and Space Admin-istration (NASA)’s Terra and Aqua satellites, was retrievedfrom a NASA Earth Observations’ online resource with highresolution (0.1° latitude × 0.1° longitude, monthly or 8 days).The trend of satellite-measured chlorophyll a concentrations(seasonal average = 0.126 ± 0.011 mg m−3) closely matchedlocal measurements from 2003 to 2006 in seawater samplescollected offshore at Couta Rocks, approximately 50 km southof Cape Grim (Figure S4 of the Supporting Information).Chlorophyll a concentrations exhibited seasonal variation,
ranging between 0.075 and 0.180 mg m−3 (Figure 2B), withhigher levels in summer than winter, consistent with the∑iSOA and ∑mSOA during the same period and inaccordance with expectation based on activity of phytoplank-ton as a source. This source assignment is supported by amoderate to strong correlation with ∑iSOA (r = 0.74) and∑mSOA (r = 0.85) (Table 3 and Figure S3 of the SupportingInformation) and is consistent with the role of phytoplanktonemissions as a source of marine isoprene and monoterpenes.As suggested for MSA, low concentrations of iSOA and mSOAover remote marine locations may contribute to cloudcondensation nuclei (CCN).83 More work is warranted toinvestigate to what extent marine iSOA and mSOA can serveas CCN; however, laboratory studies by Engelhart et al.demonstrated that pure SOA derived from the OH-initiatedoxidation of isoprene in the absence of sulfate aerosol hadsimilar CCN activation kinetics to pure ammonium sulfateaerosol.84 Furthermore, a recent study over the midwesternU.S. reported isoprene-derived OSs, which are also knowniSOA tracers, in cloudwater samples.85 Taken together, theseprior laboratory and field measurements of iSOA indicate thatiSOA may contribute (in part) to CCN activity of aerosol.
4. CONCLUSIONBecause the marine aerosol samples chemically characterizedin this study originated over the open ocean, where terrestrialemissions were largely absent, our results provide importantquantitative background levels of marine biogenic SOA over a25 year period (i.e., 1991−2015). Spatial distributions ofmarine iSOA and mSOA tracers were previously observed athigher concentrations over the coastal/tropical regions thanthe open oceans.23 In the southern mid-latitudes (30−60° S),where Cape Grim is located, iSOA and mSOA were 1−2orders of magnitude lower than those in the other oceanicregions.18 Our observations suggest that there are marinesources of isoprene and monoterpenes derived fromphytoplankton activity leading to biogenic SOA formationover the open oceans. Although isoprene, monoterpenes, andchlorophyll a were not as abundant at Cape Grim as othercoastal and oceanic sites,11 oxidation of isoprene andmonoterpenes yields measurable seasonal and temporal trendsof iSOA and mSOA. iSOA and mSOA do not contributesubstantial mass to the total OA mass (0.26 ± 0.24%, up to0.71%) measured at this site, possibly because PM10 sampleswere collected instead of sub-micrometer particles to whichiSOA and mSOA mainly contribute.86 Notably, recent Arcticmeasurements have also shown that SOA in that remotemarine atmosphere does not have chemical characteristics thatare typical of continental SOA (i.e., the OA is secondary but
arising from a different set of precursors than isoprene andmonoterpenes);87 specifically, it was shown that SOA isrequired to form to fit the aerosol size distributions measuredduring the Arctic summer, even though the mean isoprene andmonoterpenes mixing ratios were 1.5 and 5 pptv, respec-tively.86,88 The latter supports our implication that secondaryOA sources other than isoprene and monoterpenes exist in theremote marine atmosphere.It is noted that the summed mass concentrations of oxalic
acid and MSA were ∼28 times higher than the summedconcentrations of iSOA and mSOA. Additionally, the sum ofiSOA, mSOA, oxalic acid, and MSA mass concentrationscontributed up to 19.0% of the total OA mass (Table S3 of theSupporting Information), which is consistent with Claeys et al.at Amsterdam Island during summertime.59 Consequently,more work is needed to chemically characterize other OAconstituents (e.g., saccharides and fatty acids) that additionallycontribute to marine aerosol to reveal their sources. Inparticular, more information is required to fully understand theOA constituents associated with phytoplankton blooms tomore clearly identify the processes that generate OA, such asbubble-bursting processes and SOA precursors likely derivedfrom the sea surface microlayer.88 Preliminary findingsobtained from the UPLC/ESI−HR-QTOFMS method oper-ated in both negative and positive ion modes revealed thepresence of nitrogen (N)-containing organic species (Table S4of the Supporting Information). The tentatively identified N-containing species (as well as CHO and CHOS compounds)may also contribute to the uncharacterized OA mass thatremains in this study; however, we did not include thesepreliminary findings of the N-containing, CHO, and CHOScompounds because we did not have appropriate standards tofully identify their structures as well as accurately quantify theirmass concentrations. As a result, characterization of thesecompounds is beyond the scope of the present study. Ourresults provide observations and motivation for futurelaboratory, field, and modeling studies to assess the potentialof the marine sources leading to biogenic SOA and OAformation over the open oceans.
■ ASSOCIATED CONTENT
*S Supporting InformationThe Supporting Information is available free of charge on theACS Publications website at DOI: 10.1021/acsearthspace-chem.9b00061.
List of analyzed samples and field blank filters (TableS1), location of Cape Grim site and “baseline” sector ofwind direction (Figure S1), “baseline” conditions andsupporting data in baseline reports, concentrations ofindividual iSOA and mSOA tracers, levoglucosan, oxalicacid, MSA, and Chl a (Table S2), mass of collectedPM10, IC analysis, organic carbon, elemental carbon, andtotal carbon estimation, summed iSOA, mSOA, oxalicacid, and MSA as a fraction of OA mass (Table S3),surface ozone at Cape Grim, monthly average surfaceozone concentrations (Figure S2), correlation of iSOAand mSOA with MSA/oxalic acid/Chl a (Figure S3),Chl a data usage and description, satellite and in situmeasurements of Chl a (Figure S4), and list of identifiedN-containing species (Table S4) (PDF)
ACS Earth and Space Chemistry Article
DOI: 10.1021/acsearthspacechem.9b00061ACS Earth Space Chem. 2019, 3, 935−946
Rachel E. O’Brien: 0000-0001-8829-5517Jason D. Surratt: 0000-0002-6833-1450Present Addresses∥Hilary S. Green: Agricultural and Environmental ChemistryGraduate Group, Department of Food Science and Technol-ogy, University of California, Davis, Davis, California 95616,United States.⊥Rachel E. O’Brien: Department of Chemistry, College ofWilliam and Mary, Williamsburg, Virginia 23185, UnitedStates.
NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTS
This work was funded by the National Oceanic andAtmospheric Administration (NOAA) Climate ProgramOffice’s AC4 Program, Award NA13OAR4310064. Continuedsupport for the Cape Grim Program from the AustralianBureau of Meteorology and Commonwealth Scientific andIndustrial Research Organisation (CSIRO) and the personaleffort of numerous Cape Grim support staff maintainingequipment and collecting samples over the program lifetime isgratefully acknowledged. Ian Galbally and Suzie Molloy fromCSIRO are thanked for providing the monthly ozone data fromCape Grim, and Nada Derek from CSIRO is thanked forassistance with graphics. Steven Sai Hang Ho and Judith Chowfrom Desert Research Institute and Bim Graham are thankedfor the collection and analysis of total OA mass measured inthe Cape Grim 2002−2003 samples.
■ REFERENCES(1) Meskhidze, N.; McClain, C. R.; Petters, M. D.; Vignati, E.;Stetzer, O.; Osburn, C.; Kieber, D. J. Marine Aerosol−Cloud−Climate Interaction. Adv. Meteorol. 2010, 2010, 1−2.(2) Charlson, R. J.; Lovelock, J. E.; Andreae, M. O.; Warren, S. G.Oceanic Phytoplankton, Atmospheric Sulphur, Cloud Albedo andClimate. Nature 1987, 326 (16), 655−661.(3) O’Dowd, C. D.; Facchini, M. C.; Cavalli, F.; Ceburnis, D.;Mircea, M.; Decesari, S.; Fuzzi, S.; Yoon, Y. J.; Putaud, J.-P.Biogenically Driven Organic Contribution to Marine Aerosol. Nature2004, 431 (7009), 676−680.(4) Rinaldi, M.; Decesari, S.; Finessi, E.; Giulianelli, L.; Carbone, C.;Fuzzi, S.; O’Dowd, C. D.; Ceburnis, D.; Facchini, M. C. Primary andSecondary Organic Marine Aerosol and Oceanic Biological Activity:Recent Results and New Perspectives for Future Studies. Adv.Meteorol. 2010, 2010, 310682.(5) Quinn, P. K.; Collins, D. B.; Grassian, V. H.; Prather, K. A.;Bates, T. S. Chemistry and Related Properties of Freshly Emitted SeaSpray Aerosol. Chem. Rev. 2015, 115 (10), 4383−4399.(6) Hallquist, M.; Wenger, J. C.; Baltensperger, U.; Rudich, Y.;Simpson, D.; Claeys, M.; Dommen, J.; Donahue, N. M.; George, C.;Goldstein, A. H.; Hamilton, J. F.; Herrmann, H.; Hoffmann, T.;Iinuma, Y.; Jang, M.; Jenkin, M. E.; Jimenez, J. L.; Kiendler-Scharr, A.;Maenhaut, W.; McFiggans, G.; Mentel, Th. F.; Monod, A.; Prevot, A.S. H.; Seinfeld, J. H.; Surratt, J. D.; Szmigielski, R.; Wildt, J. TheFormation, Properties and Impact of Secondary Organic Aerosol:Current and Emerging Issues. Atmos. Chem. Phys. 2009, 9 (14),5155−5236.
(7) Ziemann, P. J.; Atkinson, R. Kinetics, Products, and Mechanismsof Secondary Organic Aerosol Formation. Chem. Soc. Rev. 2012, 41(19), 6582.(8) Bonsang, B.; Polle, C.; Lambert, G. Evidence for MarineProduction of Isoprene. Geophys. Res. Lett. 1992, 19 (11), 1129−1132.(9) Guenther, A.; Hewitt, C. N.; Erickson, D.; Fall, R.; Geron, C.;Graedel, T.; Harley, P.; Klinger, L.; Lerdau, M.; Mckay, W. A.; Pierce,T.; Scholes, B.; Steinbrecher, R.; Tallamraju, R.; Taylor, J.;Zimmerman, P. A Global-Model of Natural Volatile Organic-Compound Emissions. J. Geophys. Res.: Atmos. 1995, 100 (D5),8873−8892.(10) Yassaa, N.; Peeken, I.; Zollner, E.; Bluhm, K.; Arnold, S.;Spracklen, D.; Williams, J. Evidence for Marine Production ofMonoterpenes. Environ. Chem. 2008, 5 (6), 391−401.(11) Shaw, S. L.; Gantt, B.; Meskhidze, N. Production and Emissionsof Marine Isoprene and Monoterpenes: A Review. Adv. Meteorol.2010, 2010 (1), 1−24.(12) Booge, D.; Marandino, C. A.; Schlundt, C.; Palmer, P. I.;Schlundt, M.; Atlas, E. L.; Bracher, A.; Saltzman, E. S.; Wallace, D. W.R. Can Simple Models Predict Large-Scale Surface Ocean IsopreneConcentrations? Atmos. Chem. Phys. 2016, 16 (18), 11807−11821.(13) Broadgate, W. J.; Malin, G.; Kupper, F. C.; Thompson, A.; Liss,P. S. Isoprene and Other Non-Methane Hydrocarbons fromSeaweeds: A Source of Reactive Hydrocarbons to the Atmosphere.Mar. Chem. 2004, 88 (1−2), 61−73.(14) Shaw, S. L.; Chisholm, S. W.; Prinn, R. G. Isoprene Productionby Prochlorococcus, a Marine Cyanobacterium, and OtherPhytoplankton. Mar. Chem. 2003, 80 (4), 227−245.(15) Foster, P. N.; Prentice, I. C.; Morfopoulos, C.; Siddall, M.; VanWeele, M. Isoprene Emissions Track the Seasonal Cycle of CanopyTemperature, Not Primary Production: Evidence from RemoteSensing. Biogeosciences 2014, 11 (13), 3437−3451.(16) Liakakou, E.; Vrekoussis, M.; Bonsang, B.; Donousis, C.;Kanakidou, M.; Mihalopoulos, N. Isoprene above the EasternMediterranean: Seasonal Variation and Contribution to the OxidationCapacity of the Atmosphere. Atmos. Environ. 2007, 41 (5), 1002−1010.(17) Yokouchi, Y.; Li, H.-J.; Machida, T.; Aoki, S.; Akimoto, H.Isoprene in the Marine Boundary Layer (Southeast Asian Sea, EasternIndian Ocean, and Southern Ocean): Comparison with DimethylSulfide and Bromoform. J. Geophys. Res.: Atmos. 1999, 104 (D7),8067−8076.(18) Hu, Q.-H.; Xie, Z.-Q.; Wang, X.-M.; Kang, H.; He, Q.-F.;Zhang, P. Secondary Organic Aerosols over Oceans via Oxidation ofIsoprene and Monoterpenes from Arctic to Antarctic. Sci. Rep. 2013, 3(1), 2280.(19) Luo, G.; Yu, F. A Numerical Evaluation of Global OceanicEmissions of α-Pinene and Isoprene. Atmos. Chem. Phys. 2010, 10 (4),2007−2015.(20) Hackenberg, S. C.; Andrews, S. J.; Airs, R. L.; Arnold, S. R.;Bouman, H. A.; Cummings, D.; Lewis, A. C.; Minaeian, J. K.; Reifel,K. M.; Small, A.; Tarran, G. A.; Tilstone, G. H.; Carpenter, L. J. Basin-Scale Observations of Monoterpenes in the Arctic and AtlanticOceans. Environ. Sci. Technol. 2017, 51 (18), 10449−10458.(21) Ciuraru, R.; Fine, L.; van Pinxteren, M.; D’Anna, B.; Herrmann,H.; George, C. Unravelling New Processes at Interfaces: Photo-chemical Isoprene Production at the Sea Surface. Environ. Sci. Technol.2015, 49 (22), 13199−13205.(22) Bernard, F.; Ciuraru, R.; Boreave, A.; George, C. Photo-sensitized Formation of Secondary Organic Aerosols above the Air/Water Interface. Environ. Sci. Technol. 2016, 50 (16), 8678−8686.(23) Fu, P.; Kawamura, K.; Miura, K. Molecular Characterization ofMarine Organic Aerosols Collected during a Round-the-WorldCruise. J. Geophys. Res.: Atmos. 2011, 116 (D13), 1−14.(24) Pokhrel, A.; Kawamura, K.; Ono, K.; Seki, O.; Fu, P.; Matoba,S.; Shiraiwa, T. Ice Core Records of Monoterpene- and Isoprene-SOATracers from Aurora Peak in Alaska since 1660s: Implication for
ACS Earth and Space Chemistry Article
DOI: 10.1021/acsearthspacechem.9b00061ACS Earth Space Chem. 2019, 3, 935−946
Climate Change Variability in the North Pacific Rim. Atmos. Environ.2016, 130, 105−112.(25) Fu, P.; Kawamura, K.; Seki, O.; Izawa, Y.; Shiraiwa, T.;Ashworth, K. Historical Trends of Biogenic SOA Tracers in an IceCore from Kamchatka Peninsula. Environ. Sci. Technol. Lett. 2016, 3(10), 351−358.(26) Capaldo, K.; Corbett, J. J.; Kasibhatla, P.; Fischbeck, P.; Pandis,S. N. Effects of Ship Emissions on Sulphur Cycling and RadiativeClimate Forcing over the Ocean. Nature 1999, 400 (6746), 743−746.(27) Claeys, M.; Graham, B.; Vas, G.; Wang, W.; Vermeylen, R.;Pashynska, V.; Cafmeyer, J.; Guyon, P.; Andreae, M. O.; Artaxo, P.;Maenhaut, W. Formation of Secondary Organic Aerosols ThroughPhotooxidation of Isoprene. Science (Washington, DC, U. S.) 2004,303 (5661), 1173−1176.(28) Claeys, M.; Iinuma, Y.; Szmigielski, R.; Surratt, J. D.; Blockhuys,F.; Van Alsenoy, C.; Boge, O.; Sierau, B.; Gomez-Gonzalez, Y.;Vermeylen, R.; Van der Veken, P.; Shahgholi, M.; Chan, A. W. H.;Herrmann, H.; Seinfeld, J. H.; Maenhaut, W. Terpenylic Acid andRelated Compounds from the Oxidation of α-Pinene: Implications forNew Particle Formation and Growth above Forests. Environ. Sci.Technol. 2009, 43 (18), 6976−6982.(29) Szmigielski, R.; Surratt, J. D.; Gomez-Gonzalez, Y.; Van derVeken, P.; Kourtchev, I.; Vermeylen, R.; Blockhuys, F.; Jaoui, M.;Kleindienst, T. E.; Lewandowski, M.; Offenberg, J. H.; Edney, E. O.;Seinfeld, J. H.; Maenhaut, W.; Claeys, M. 3-Methyl-1,2,3-Butane-tricarboxylic Acid: An Atmospheric Tracer for Terpene SecondaryOrganic Aerosol. Geophys. Res. Lett. 2007, 34 (24), 2−7.(30) Glasius, M.; Lahaniati, M.; Calogirou, A.; Di Bella, D.; Jensen,N. R.; Hjorth, J.; Kotzias, D.; Larsen, B. R. Carboxylic Acids inSecondary Aerosols from Oxidation of Cyclic Monoterpenes byOzone. Environ. Sci. Technol. 2000, 34 (6), 1001−1010.(31) Yasmeen, F.; Vermeylen, R.; Szmigielski, R.; Iinuma, Y.; Boge,O.; Herrmann, H.; Maenhaut, W.; Claeys, M. Terpenylic Acid andRelated Compounds: Precursors for Dimers in Secondary OrganicAerosol from the Ozonolysis of α-and β-Pinene. Atmos. Chem. Phys.2010, 10 (19), 9383−9392.(32) Surratt, J. D.; Gomez-Gonzalez, Y.; Chan, A. W. H.; Vermeylen,R.; Shahgholi, M.; Kleindienst, T. E.; Edney, E. O.; Offenberg, J. H.;Lewandowski, M.; Jaoui, M.; Maenhaut, W.; Claeys, M.; Flagan, R. C.;Seinfeld, J. H. Organosulfate Formation in Biogenic SecondaryOrganic Aerosol. J. Phys. Chem. A 2008, 112 (36), 8345−8378.(33) Rinaldi, M.; Decesari, S.; Carbone, C.; Finessi, E.; Fuzzi, S.;Ceburnis, D.; O’Dowd, C. D.; Sciare, J.; Burrows, J. P.; Vrekoussis,M.; Ervens, B.; Tsigaridis, K.; Facchini, M. C. Evidence of a NaturalMarine Source of Oxalic Acid and a Possible Link to Glyoxal. J.Geophys. Res.: Atmos. 2011, 116 (D16), 1−12.(34) Meskhidze, N.; Nenes, A. Phytoplankton and Cloudiness in theSouthern Ocean. Science (Washington, DC, U. S.) 2006, 314 (5804),1419−1423.(35) Crawford, J.; Cohen, D. D.; Stelcer, E.; Atanacio, A. J. LongTerm Fine Aerosols at the Cape Grim Global Baseline Station: 1998to 2016. Atmos. Environ. 2017, 166, 34−46.(36) Lin, Y. H.; Zhang, Z.; Docherty, K. S.; Zhang, H.;Budisulistiorini, S. H.; Rubitschun, C. L.; Shaw, S. L.; Knipping, E.M.; Edgerton, E. S.; Kleindienst, T. E.; Gold, A.; Surratt, J. D.Isoprene Epoxydiols as Precursors to Secondary Organic AerosolFormation: Acid-Catalyzed Reactive Uptake Studies with AuthenticCompounds. Environ. Sci. Technol. 2012, 46 (1), 250−258.(37) Surratt, J. D.; Chan, A. W. H.; Eddingsaas, N. C.; Chan, M.;Loza, C. L.; Kwan, A. J.; Hersey, S. P.; Flagan, R. C.; Wennberg, P. O.;Seinfeld, J. H. Reactive Intermediates Revealed in Secondary OrganicAerosol Formation from Isoprene. Proc. Natl. Acad. Sci. U. S. A. 2010,107 (15), 6640−6645.(38) Zhang, Z.; Lin, Y. H.; Zhang, H.; Surratt, J. D.; Ball, L. M.;Gold, A. Technical Note: Synthesis of Isoprene AtmosphericOxidation Products: Isomeric Epoxydiols and the RearrangementProducts Cis-and Trans-3-Methyl-3,4- Dihydroxytetrahydrofuran.Atmos. Chem. Phys. 2012, 12 (18), 8529−8535.
(39) Budisulistiorini, S. H.; Li, X.; Bairai, S. T.; Renfro, J.; Liu, Y.;Liu, Y. J.; McKinney, K. A.; Martin, S. T.; McNeill, V. F.; Pye, H. O.T.; Nenes, A.; Neff, M. E.; Stone, E. A.; Mueller, S.; Knote, C.; Shaw,S. L.; Zhang, Z.; Gold, A.; Surratt, J. D. Examining the Effects ofAnthropogenic Emissions on Isoprene-Derived Secondary OrganicAerosol Formation during the 2013 Southern Oxidant and AerosolStudy (SOAS) at the Look Rock, Tennessee Ground Site. Atmos.Chem. Phys. 2015, 15 (15), 8871−8888.(40) Kristensen, K.; Cui, T.; Zhang, H.; Gold, A.; Glasius, M.;Surratt, J. D. Dimers in Alpha -Pinene Secondary Organic Aerosol:Effect of Hydroxyl Radical, Ozone, Relative Humidity and AerosolAcidity. Atmos. Chem. Phys. 2014, 14 (8), 4201−4218.(41) Olson, C. N.; Galloway, M. M.; Yu, G.; Hedman, C. J.; Lockett,M. R.; Yoon, T.; Stone, E. A.; Smith, L. M.; Keutsch, F. N.Hydroxycarboxylic Acid-Derived Organosulfates: Synthesis, Stability,and Quantification in Ambient Aerosol. Environ. Sci. Technol. 2011, 45(15), 6468−6474.(42) Zhang, H.; Worton, D. R.; Lewandowski, M.; Ortega, J.;Rubitschun, C. L.; Park, J. H.; Kristensen, K.; Campuzano-Jost, P.;Day, D. A.; Jimenez, J. L.; Jaoui, M.; Offenberg, J. H.; Kleindienst, T.E.; Gilman, J.; Kuster, W. C.; de Gouw, J.; Park, C.; Schade, G. W.;Frossard, A. A.; Russell, L.; Kaser, L.; Jud, W.; Hansel, A.; Cappellin,L.; Karl, T.; Glasius, M.; Guenther, A.; Goldstein, A. H.; Seinfeld, J.H.; Gold, A.; Kamens, R. M.; Surratt, J. D. Organosulfates as Tracersfor Secondary Organic Aerosol (SOA) Formation from 2-Methyl-3-Buten-2-Ol (MBO) in the Atmosphere. Environ. Sci. Technol. 2012, 46(17), 9437−9446.(43) Nguyen, T. B.; Bates, K. H.; Crounse, J. D.; Schwantes, R. H.;Zhang, X.; Kjaergaard, H. G.; Surratt, J. D.; Lin, P.; Laskin, A.;Seinfeld, J. H.; Wennberg, P. O. Mechanism of the Hydroxyl RadicalOxidation of Methacryloyl Peroxynitrate (MPAN) and Its Pathwaytoward Secondary Organic Aerosol Formation in the Atmosphere.Phys. Chem. Chem. Phys. 2015, 17 (27), 17914−17926.(44) Yasmeen, F.; Szmigielski, R.; Vermeylen, R.; Gomez-Gonzalez,Y.; Surratt, J. D.; Chan, A. W. H.; Seinfeld, J. H.; Maenhaut, W.;Claeys, M. Mass Spectrometric Characterization of IsomericTerpenoic Acids from the Oxidation of α-Pinene, β-Pinene, d-Limonene, and I“3-Carene in Fine Forest Aerosol. J. Mass Spectrom.2011, 46 (4), 425−442.(45) Hu, W.; Palm, B. B.; Day, D. A.; Campuzano-Jost, P.;Krechmer, J. E.; Peng, Z.; de Sa, S. S.; Martin, S. T.; Alexander, M. L.;Baumann, K.; Hacker, L.; Kiendler-Scharr, A.; Koss, A. R.; de Gouw,J. A.; Goldstein, A. H.; Seco, R.; Sjostedt, S. J.; Park, J.-H.; Guenther,A. B.; Kim, S.; Canonaco, F.; Prevot, A. S. H.; Brune, W. H.; Jimenez,J. L. Volatility and Lifetime against OH Heterogeneous Reaction ofAmbient Isoprene-Epoxydiols-Derived Secondary Organic Aerosol(IEPOX-SOA). Atmos. Chem. Phys. 2016, 16 (18), 11563−11580.(46) Wang, W.; Kourtchev, I.; Graham, B.; Cafmeyer, J.; Maenhaut,W.; Claeys, M. Characterization of Oxygenated Derivatives ofIsoprene Related to 2-Methyltetrols in Amazonian Aerosols UsingTrimethylsilylation and Gas Chromatography/Ion Trap MassSpectrometry. Rapid Commun. Mass Spectrom. 2005, 19 (10),1343−1351.(47) Lin, Y. H.; Knipping, E. M.; Edgerton, E. S.; Shaw, S. L.;Surratt, J. D. Investigating the Influences of SO2 and NH3 Levels onIsoprene-Derived Secondary Organic Aerosol Formation UsingConditional Sampling Approaches. Atmos. Chem. Phys. 2013, 13(16), 8457−8470.(48) Rattanavaraha, W.; Chu, K.; Budisulistiorini, S. H.; Riva, M.;Lin, Y. H.; Edgerton, E. S.; Baumann, K.; Shaw, S. L.; Guo, H.; King,L.; Weber, R. J.; Neff, M. E.; Stone, E. A.; Offenberg, J. H.; Zhang, Z.;Gold, A.; Surratt, J. D. Assessing the Impact of AnthropogenicPollution on Isoprene-Derived Secondary Organic Aerosol Formationin PM2.5 Collected from the Birmingham, Alabama, Ground Siteduring the 2013 Southern Oxidant and Aerosol Study. Atmos. Chem.Phys. 2016, 16 (8), 4897−4914.(49) Ding, X.; Zheng, M.; Yu, L.; Zhang, X.; Weber, R. J.; Yan, B.;Russell, A. G.; Edgerton, E. S.; Wang, X. Spatial and Seasonal Trendsin Biogenic Secondary Organic Aerosol Tracers and Water-Soluble
ACS Earth and Space Chemistry Article
DOI: 10.1021/acsearthspacechem.9b00061ACS Earth Space Chem. 2019, 3, 935−946
Organic Carbon in the Southeastern United States. Environ. Sci.Technol. 2008, 42 (14), 5171−5176.(50) Hu, D.; Bian, Q.; Li, T. W. Y.; Lau, A. K. H.; Yu, J. Z.Contributions of Isoprene, Monoterpenes, β-Caryophyllene, andToluene to Secondary Organic Aerosols in Hong Kong during theSummer of 2006. J. Geophys. Res.: Atmos. 2008, 113 (D22), 1−14.(51) Kleindienst, T. E.; Jaoui, M.; Lewandowski, M.; Offenberg, J.H.; Lewis, C. W.; Bhave, P. V.; Edney, E. O. Estimates of theContributions of Biogenic and Anthropogenic Hydrocarbons toSecondary Organic Aerosol at a Southeastern US Location. Atmos.Environ. 2007, 41 (37), 8288−8300.(52) Kourtchev, I.; Ruuskanen, T.; Maenhaut, W.; Kulmala, M.;Claeys, M. Observation of 2-Methyltetrols and Related Photo-Oxidation Products of Isoprene in Boreal Forest Aerosols fromHyytiala, Finland. Atmos. Chem. Phys. Discuss. 2005, 5 (3), 2947−2971.(53) Shen, R. Q.; Ding, X.; He, Q. F.; Cong, Z. Y.; Yu, Q. Q.; Wang,X. M. Seasonal Variation of Secondary Organic Aerosol Tracers inCentral Tibetan Plateau. Atmos. Chem. Phys. 2015, 15 (15), 8781−8793.(54) Xia, X.; Hopke, P. K. Seasonal Variation of 2-Methyltetrols inAmbient Air Samples. Environ. Sci. Technol. 2006, 40 (22), 6934−6937.(55) Fu, P.; Kawamura, K.; Chen, J.; Barrie, L. A. Isoprene,Monoterpene, and Sesquiterpene Oxidation Products in the HighArctic Aerosols during Late Winter to Early Summer. Environ. Sci.Technol. 2009, 43 (11), 4022−4028.(56) Fu, P.; Kawamura, K.; Chen, J.; Charriere, B.; Sempere, R.Organic Molecular Composition of Marine Aerosols over the ArcticOcean in Summer: Contributions of Primary Emission and SecondaryAerosol Formation. Biogeosciences 2013, 10 (2), 653−667.(57) Chavez, F. P.; Strutton, P. G.; Friederich, G. E.; Feely, R. A.;Feldman, G. C.; Foley, D. G.; McPhaden, M. J. Biological andChemical Response of the Equatorial Pacific Ocean to the 1997−98El Nino. Science (Washington, DC, U. S.) 1999, 286 (5447), 2126−2131.(58) Cui, T.; Zeng, Z.; Dos Santos, E. O.; Zhang, Z.; Chen, Y.;Zhang, Y.; Rose, C. A.; Budisulistiorini, S. H.; Collins, L. B.; Bodnar,W. M.; de Souza, R. A. F.; Martin, S. T.; Machado, C. M. D.; Turpin,B. J.; Gold, A.; Ault, A. P.; Surratt, J. D. Development of a HydrophilicInteraction Liquid Chromatography (HILIC) Method for theChemical Characterization of Water-Soluble Isoprene Epoxydiol(IEPOX)-Derived Secondary Organic Aerosol. Environ. Sci. Process.Impacts. 2018, 20 (11), 1524−1536.(59) Claeys, M.; Wang, W.; Vermeylen, R.; Kourtchev, I.; Chi, X.;Farhat, Y.; Surratt, J. D.; Gomez-Gonzalez, Y.; Sciare, J.; Maenhaut,W. Chemical Characterisation of Marine Aerosol at Amsterdam Islandduring the Austral Summer of 2006−2007. J. Aerosol Sci. 2010, 41 (1),13−22.(60) Xu, L.; Guo, H.; Boyd, C. M.; Klein, M.; Bougiatioti, A.;Cerully, K. M.; Hite, J. R.; Isaacman-Vanwertz, G.; Kreisberg, N. M.;Knote, C.; Olson, K.; Koss, A.; Goldstein, A. H.; Hering, S. V.; deGouw, J.; Baumann, K.; Lee, S.-H.; Nenes, A.; Weber, R. J.; Ng, N. L.Effects of Anthropogenic Emissions on Aerosol Formation fromIsoprene and Monoterpenes in the Southeastern United States. Proc.Natl. Acad. Sci. U. S. A. 2015, 112 (1), 37−42.(61) Darer, A. I.; Cole-Filipiak, N. C.; O’Connor, A. E.; Elrod, M. J.Formation and Stability of Atmospherically Relevant Isoprene-Derived Organosulfates and Organonitrates. Environ. Sci. Technol.2011, 45 (5), 1895−1902.(62) Budisulistiorini, S. H.; Baumann, K.; Edgerton, E. S.; Bairai, S.T.; Mueller, S.; Shaw, S. L.; Knipping, E. M.; Gold, A.; Surratt, J. D.Seasonal Characterization of Submicron Aerosol Chemical Compo-sition and Organic Aerosol Sources in the Southeastern United States:Atlanta, Georgia, and Look Rock, Tennessee. Atmos. Chem. Phys.2016, 16 (8), 5171−5189.(63) Paulot, F.; Crounse, J. D.; Kjaergaard, H. G.; Kurten, A.; St.Clair, J. M.; Seinfeld, J. H.; Wennberg, P. O. Unexpected Epoxide
Formation in the Gas-Phase Photooxidation of Isoprene. Science(Washington, DC, U. S.) 2009, 325 (5941), 730−733.(64) Nguyen, T. B.; Coggon, M. M.; Bates, K. H.; Zhang, X.;Schwantes, R. H.; Schilling, K. A.; Loza, C. L.; Flagan, R. C.;Wennberg, P. O.; Seinfeld, J. H. Organic Aerosol Formation from theReactive Uptake of Isoprene Epoxydiols (IEPOX) onto Non-AcidifiedInorganic Seeds. Atmos. Chem. Phys. 2014, 14 (7), 3497−3510.(65) Lim, H.-J.; Carlton, A. G.; Turpin, B. J. Isoprene FormsSecondary Organic Aerosol through Cloud Processing: ModelSimulations. Environ. Sci. Technol. 2005, 39 (12), 4441−4446.(66) Kleindienst, T. E.; Lewandowski, M.; Offenberg, J. H.; Jaoui,M.; Edney, E. O. Ozone-Isoprene Reaction: Re-Examination of theFormation of Secondary Organic Aerosol. Geophys. Res. Lett. 2007, 34(1), L01805.(67) Riva, M.; Budisulistiorini, S. H.; Zhang, Z.; Gold, A.; Surratt, J.D. Chemical Characterization of Secondary Organic AerosolConstituents from Isoprene Ozonolysis in the Presence of AcidicAerosol. Atmos. Environ. 2016, 130, 5−13.(68) Surratt, J. D.; Murphy, S. M.; Kroll, J. H.; Ng, N. L.;Hildebrandt, L.; Sorooshian, A.; Szmigielski, R.; Vermeylen, R.;Maenhaut, W.; Claeys, M.; Flagan, R. C.; Seinfeld, J. H. ChemicalComposition of Secondary Organic Aerosol Formed from thePhotooxidation of Isoprene. J. Phys. Chem. A 2006, 110 (31),9665−9690.(69) Ridley, B. A.; Carroll, M. A.; Gregory, G. L. Measurements ofNitric Oxide in the Boundary Layer and Free Troposphere over thePacific Ocean. J. Geophys. Res.: Atmos. 1987, 92 (D2), 2025.(70) Kleindienst, T. E.; Lewandowski, M.; Offenberg, J. H.; Jaoui,M.; Edney, E. O. The Formation of Secondary Organic Aerosol fromthe Isoprene + OH Reaction in the Absence of NOx. Atmos. Chem.Phys. Discuss. 2009, 9 (2), 10015−10054.(71) Claeys, M.; Szmigielski, R.; Kourtchev, I.; Van Der Veken, P.;Vermeylen, R.; Maenhaut, W.; Jaoui, M.; Kleindienst, T. E.;Lewandowski, M.; Offenberg, J. H.; Edney, E. O. HydroxydicarboxylicAcids: Markers for Secondary Organic Aerosol from the Photo-oxidation of α-Pinene. Environ. Sci. Technol. 2007, 41 (5), 1628−1634.(72) Kahnt, A.; Iinuma, Y.; Blockhuys, F.; Mutzel, A.; Vermeylen, R.;Kleindienst, T. E.; Jaoui, M.; Offenberg, J. H.; Lewandowski, M.;Boge, O.; Herrmann, H.; Maenhaut, W.; Claeys, M. 2-Hydroxyterpe-nylic Acid: An Oxygenated Marker Compound for α-PineneSecondary Organic Aerosol in Ambient Fine Aerosol. Environ. Sci.Technol. 2014, 48 (9), 4901−4908.(73) Kristensen, K.; Watne, Å. K.; Hammes, J.; Lutz, A.; Petaja, T.;Hallquist, M.; Bilde, M.; Glasius, M. High-Molecular Weight DimerEsters Are Major Products in Aerosols from α-Pinene Ozonolysis andthe Boreal Forest. Environ. Sci. Technol. Lett. 2016, 3 (8), 280−285.(74) Zhang, X.; McVay, R. C.; Huang, D. D.; Dalleska, N. F.;Aumont, B.; Flagan, R. C.; Seinfeld, J. H. Formation and Evolution ofMolecular Products in α-Pinene Secondary Organic Aerosol. Proc.Natl. Acad. Sci. U. S. A. 2015, 112 (46), 14168−14173.(75) Gao, Y.; Hall, W. A.; Johnston, M. V. Molecular Compositionof Monoterpene Secondary Organic Aerosol at Low Mass Loading.Environ. Sci. Technol. 2010, 44 (20), 7897−7902.(76) Keller, M. D.; Bellows, W. K.; Guillard, R. R. L. DimethylSulfide Production in Marine Phytoplankton. Biogenic Sulfur in theEnvironment. 1989, 393, 167−182.(77) Carlton, A. G.; Turpin, B. J.; Altieri, K. E.; Seitzinger, S.; Reff,A.; Lim, H. J.; Ervens, B. Atmospheric Oxalic Acid and SOAProduction from Glyoxal: Results of Aqueous PhotooxidationExperiments. Atmos. Environ. 2007, 41 (35), 7588−7602.(78) Altieri, K. E.; Seitzinger, S. P.; Carlton, A. G.; Turpin, B. J.;Klein, G. C.; Marshall, A. G. Oligomers Formed through In-CloudMethylglyoxal Reactions: Chemical Composition, Properties, andMechanisms Investigated by Ultra-High Resolution FT-ICR MassSpectrometry. Atmos. Environ. 2008, 42 (7), 1476−1490.(79) Carlton, A. G.; Turpin, B. J.; Lim, H.-J.; Altieri, K. E.;Seitzinger, S. Link between Isoprene and Secondary Organic Aerosol
ACS Earth and Space Chemistry Article
DOI: 10.1021/acsearthspacechem.9b00061ACS Earth Space Chem. 2019, 3, 935−946
(SOA): Pyruvic Acid Oxidation Yields Low Volatility Organic Acidsin Clouds. Geophys. Res. Lett. 2006, 33 (6), L06822.(80) Kawamura, K.; Sakaguchi, F. Molecular Distributions of WaterSoluble Dicarboxylic Acids in Marine Aerosols over the Pacific OceanIncluding Tropics. J. Geophys. Res.: Atmos. 1999, 104, 3501−3509.(81) Wang, H.; Kawamura, K.; Yamazaki, K. Water-SolubleDicarboxylic Acids, Ketoacids and Dicarbonyls in the AtmosphericAerosols over the Southern Ocean and Western Pacific Ocean. J.Atmos. Chem. 2006, 53 (1), 43−61.(82) Bikkina, S.; Kawamura, K.; Miyazaki, Y.; Fu, P. HighAbundances of Oxalic, Azelaic, and Glyoxylic Acids and Methylglyoxalin the Open Ocean with High Biological Activity: Implication forSecondary OA Formation from Isoprene. Geophys. Res. Lett. 2014, 41(10), 3649−3657.(83) Berresheim, H.; Eisele, F. L.; Tanner, D. J.; McInnes, L. M.;Ramsey-Bell, D. C.; Covert, D. S. Atmospheric Sulfur Chemistry andCloud Condensation Nuclei (CCN) Concentrations over theNortheastern Pacific Coast. J. Geophys. Res.: Atmos. 1993, 98 (D7),12701.(84) Engelhart, G. J.; Moore, R. H.; Nenes, A.; Pandis, S. N. CloudCondensation Nuclei Activity of Isoprene Secondary Organic Aerosol.J. Geophys. Res.: Atmos. 2011, 116 (D2), D02207.(85) Pratt, K. A.; Fiddler, M. N.; Shepson, P. B.; Carlton, A. G.;Surratt, J. D. Organosulfates in Cloud Water above the Ozarks’Isoprene Source Region. Atmos. Environ. 2013, 77, 231−238.(86) Croft, B.; Martin, R. V.; Leaitch, W. R.; Burkart, J.; Chang, R.Y.-W.; Collins, D. B.; Hayes, P. L.; Hodshire, A. L.; Huang, L.;Kodros, J. K.; Moravek, A.; Mungall, E. L.; Murphy, J. G.; Sharma, S.;Tremblay, S.; Wentworth, G. R.; Willis, M. D.; Abbatt, J. P. D.; Pierce,J. R. Arctic Marine Secondary Organic Aerosol ContributesSignificantly to Summertime Particle Size Distributions in theCanadian Arctic Archipelago. Atmos. Chem. Phys. 2019, 19 (5),2787−2812.(87) Willis, M. D.; Kollner, F.; Burkart, J.; Bozem, H.; Thomas, J. L.;Schneider, J.; Aliabadi, A. A.; Hoor, P. M.; Schulz, H.; Herber, A. B.;Leaitch, W. R.; Abbatt, J. P. D. Evidence for Marine BiogenicInfluence on Summertime Arctic Aerosol. Geophys. Res. Lett. 2017, 44(12), 6460−6470.(88) Mungall, E. L.; Abbatt, J. P. D.; Wentzell, J. J. B.; Lee, A. K. Y.;Thomas, J. L.; Blais, M.; Gosselin, M.; Miller, L. A.; Papakyriakou, T.;Willis, M. D.; Liggio, J. Microlayer Source of Oxygenated VolatileOrganic Compounds in the Summertime Marine Arctic BoundaryLayer. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (24), 6203−6208.
ACS Earth and Space Chemistry Article
DOI: 10.1021/acsearthspacechem.9b00061ACS Earth Space Chem. 2019, 3, 935−946