42
Dr. Gangadhar Chatterjee MBBS;MD Assistant Professor RCSM Govt. Medical college, Kolhapur, MH, India
| Date post: | 21-Jan-2018 |
| Category: |
Health & Medicine |
| Upload: | gangadhar-chatterjee |
| View: | 392 times |
| Download: | 5 times |

Dr. Gangadhar ChatterjeeMBBS;MD
Assistant ProfessorRCSM Govt. Medical college, Kolhapur, MH, India


• The ionized forms of uric acid,
predominante in plasma,
extracellular fluid and synovial
fluid.
• Approximately 98% existing as
monosodium urate at pH 7.4
• Plasma is saturated with
monosodium urate at a
concentration of 6.8 mg/dl.
• At higer concentrations, plasma
is therfore supersaturated,
creating the potential for urate
crystal precipitation.

• Urate production varies with the purine content of the diet and the rates of purine biosyntesis, degradation and salvage.
• 2/3 to ¾ of urate is excreted by kidneys, and most of theremainder is elimínated through the intestines.
Renal handling• Glomerular filtration
• Tubular reabsorption
• Secretion
• Postsecretory reabsorption

• Serum urate levels vary with age and sex.
• Children: 3 to 4 mg/dl
• Adult men: 6 to 6.8 mg/dl

• Defined as a plasma urate concentration > 7.0 mg/dl
• Can result from:
• Increased production of uric acid
• Decreased excretion of uric acid
• Combination of the two processes.
Increased Urate Production
Diet provides an exogenous source of purines and, accordingly, contributes
to the serum urate in proportion to its purine content.
• Foods high in nucleic acid: liver, thymus and pancreas, kidney
and anchovy.
• Restriction intake: reduces: 1 mg/dl

• Endogenous sources:
• De novo purine biosynthesis: 11 step
• Increased PRPP synthetase activity and HPRT deficiency are
associated with overproduction of purine, hyperuricemia and
hyperuricaciduria.
Decreased Uric Acid Excretion
Alterated uric acid excretion could result from decreased
glomerular filtration, decreased tubular secretion or
enhanced tubular reabsorption.
• Decreased tubular secretion of urate causes the secondary
hyperuricemia of acidosis.
• Diabetic ketoacidosis, starvation, ethanol intoxication, lactic acidosis,
and salicylate intoxication are accompanied by accumulations of
organic acids (B-hydroxybutyrate, acetoacetate, lactate or salicylates)
that compete with urate for tubular secretion.

Combined Mechanisms
• Alcohol intake promotes hyperuricemia:
• Fast hepatic breakdown of ATP and increases urateproduction.
• Can induce hyperlacticacidemia, and inhibition of uricacid secretion.
• The higher purine content in some alcoholic beverages suchas beer may also be a factor.

Primary
• No recognized cause
• Hypoxanthine
phosphoribosyltransferas
e deficiency
• Increased phosphoribosyl
pyrophosphatase activity.
Secondary
• Hereditary fructose
intolerance
• Myeloproliferative
disease
• Lymphoproliferative
disease
• Hemolyticc anemia
• Drugs: Low-doses salicylate,
diuretis, pyrazinamide,
ethambutol, nicotinamide, etanol

• Hyperuricemia does not represent a disease.
• Is not an specific indication for therapy.
• The finding of hyperuricemia is an indication to determine
its cause.
• The hyperuricemia of individuals who excrete uric acid
above this level while on a purine-free diet is due to purine
overproduction, whereas it is due to decreased excretion in
those who excrete lower amounts on the purine-free diet.

• The most recognized complication of hyperuricemia is gouty
arthritis
• Nephrolithiasis
• Urate Nephropathy
• Uric Acid Nephropathy

• The prevalence of nephrolithiasis correlates with the serum and
urinary uric acid levels.
• Serum urate levels 13 mg/dl
• Urinary uric acid excretion > 1100 mg/d

• Deposits of monosodium urate crystals surrounded by a giant
cell inflammatory reaction in the medullary intrerstitium and
pyramids.
• Clinically: silent or cause proteinuria, hypertension and renal
insufficiency

• Precipitation in renal tubules and collecting ducts
cause obstruction to urine flow.
• Following sudden urate overproduction and marked
hyperuricaciduria:• Dehydration and acidosis
• Lymphoma
• Cytolytic therapy

Crystal-induced arthritides
• MSU (monosodium urate)
• CPPD (calcium pyrophosphate dihydrate)
• HA (calcium hydroxyapatite)
• Calcium oxalate (CaOx)

• Affecting middle-aged to elderly men.
• Women represent only 5 to 17% of all patients.
• Associated with an increased uric acid, hyperuricemia, episodic acuteand chronic arthritis, and deposition of MSU crystals in connectivetissue tophi and kidneys.
Acute and chronic arthritis
• Acute arthritis is the most frequent early clinical manifestation of MSU gout.
• Usually only one joint is affected initially
• Polyarticular acute gout is also seen in male hypertensive patientswith ethanol abuse as well as in postmenopausal women.

• “Disease of Kings”• Alexander the Great
• King Henry VIII
• Benjamin Franklin
• Alexander Hamilton
• Voltaire
• The metatarso phalangealjoint of the first toe is ofteninvolved.
• Ankles, and knees are alsocommonly affected.
• In elderly patients, fingerjoints may be inflamed.
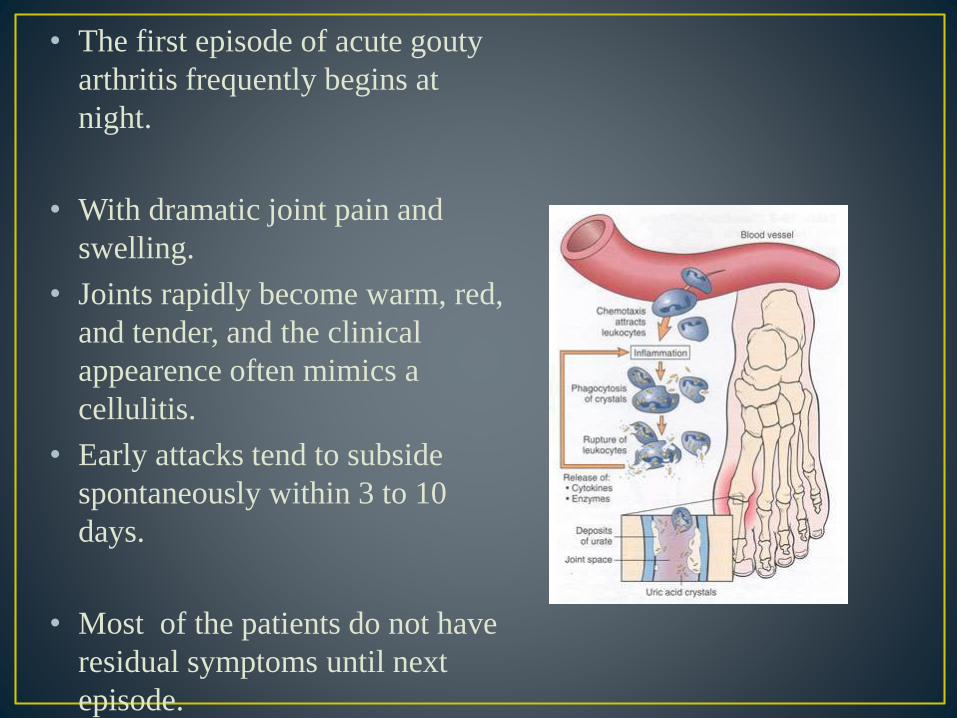
• The first episode of acute gouty
arthritis frequently begins at
night.
• With dramatic joint pain and
swelling.
• Joints rapidly become warm, red,
and tender, and the clinical
appearence often mimics a
cellulitis.
• Early attacks tend to subside
spontaneously within 3 to 10
days.
• Most of the patients do not have
residual symptoms until next
episode.
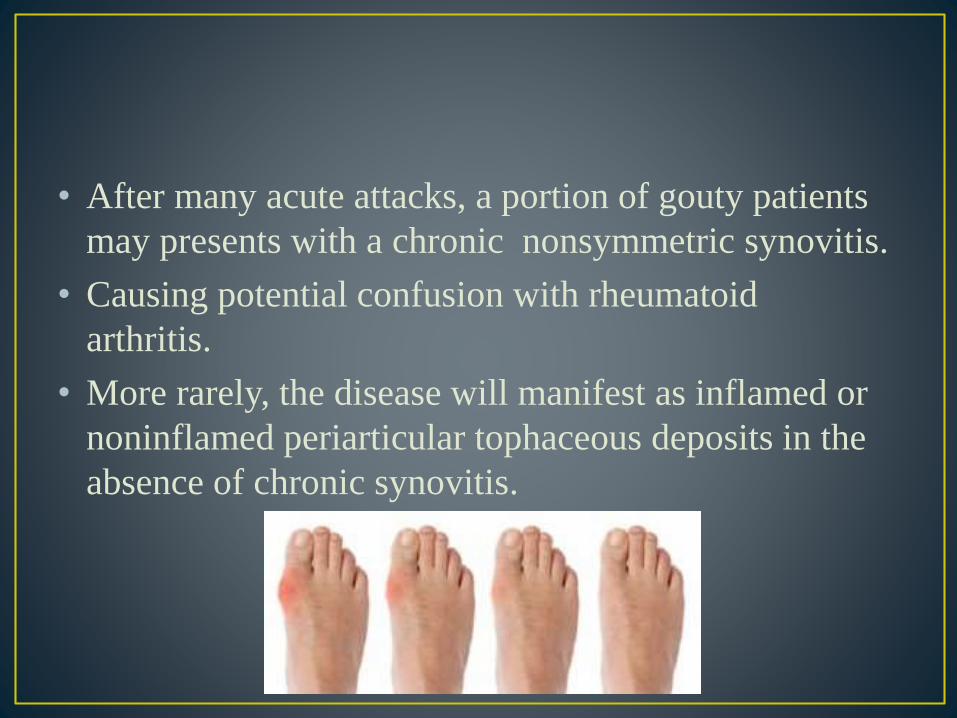
• After many acute attacks, a portion of gouty patients
may presents with a chronic nonsymmetric synovitis.
• Causing potential confusion with rheumatoid
arthritis.
• More rarely, the disease will manifest as inflamed or
noninflamed periarticular tophaceous deposits in the
absence of chronic synovitis.

• Presentation:
• Tophus: urate deposits
• Fingers>olecranon bursae>forearm>achillestendon>knees>wrists>hand
• Late complication of gout, uncommon in the general population
• Complications:
• Joint deformities and destruction
• Pain
• Damage to surrounding tissue
• Nerve compression

• Even the clinical appearance strongly suggests gout. The
diagnosis should be confirmed by needle aspiration of acute or
chronically inflamed joints or tophaceous deposits.
• Acute septic arthritis several of the other crystalline –
associated arthropathies, and psoriatic arthritis may present
with similar clinical features.
• Effusion appear cloudy due to leukocytes and a large amounts
crystals ocassionally produce a thick pasty or chalky joint fluid.

• Acute attack:
• Anti-inflammatory drug:
• Colchicine
• Nonsteroidal anti-inflamtory drugs
• Glucocorticoids
• Depending on the age of the patient and comorbid conditions.
• Uricosuric agents
• Probenecid
• Allopurinol


•Also known as Nyhan's syndrome, Kelley-Seegmiller syndrome and Juvenile gout
•It is a hereditary disorder of purine metabolism, characterized by menntalretardation, self-mutilation of the fingers and lips by biting, impaired renalfunction, and abnormal physical development.
• It is a recessive disease that is linked to the X chromosome
• It is caused by a deficiency of the enzyme hypoxanthine-guaninephosphoribosyltransferase (HPRT)
The hyperuricemia in Lesch-Nyhan patients is ex-plained, at least in part, on the basis of intracellular accu-mulation of PRPP leading to increased purine nucleotidebiosynthesis de novo and increased production of uric acid.

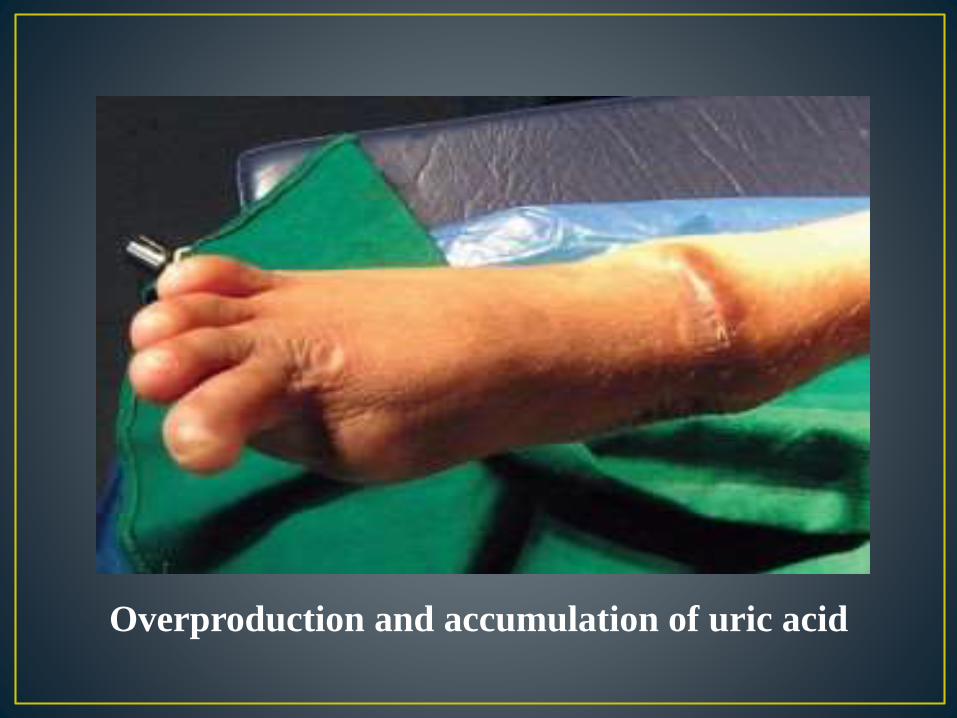
Overproduction of uric acid
• Urate crystal formations, which look like orange sand, are deposited in diapers of the babies
• Kidney stones
• Blood in the urine
• Dysphagia (difficulty swallowing)
• Swelling of the joints
• Vomiting
Behavioral Abnormalities
• Impaired cognitive
functon
• Self-mutation
• Aggression/Impulsion

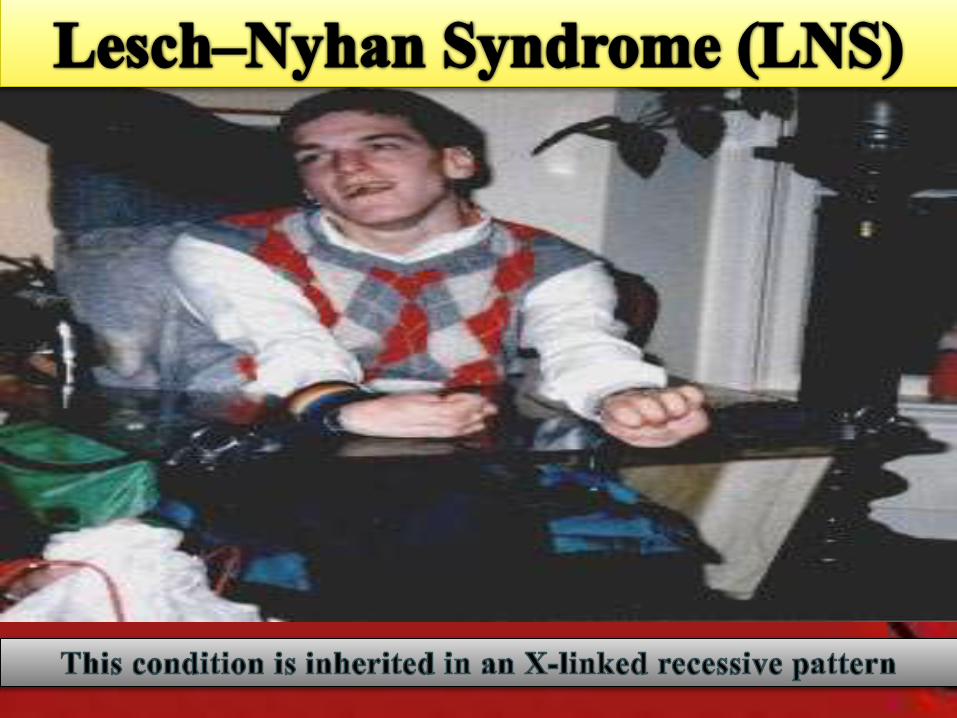


Behavioral Abnormalities
self-mutilation of the lips by biting

Behavioral Abnormalities
self-mutilation of the fingers by biting
Overproduction and accumulation of uric acid

• There may be a family history of this condition.
• The doctor will perform a physical exam. The exam may show:
• Overexaggerated reflexes
• Spacity
• Blood and urine tests may reveal high uric acid levels. A skin biopsy may show decreased levels of the HGP enzyme.
• Prenatal diagnosis is possible by DNA testing of fetaltissue drawn by amniocentesis or chorionic villussampling (CVS)

-LNS itself cannot be treated
-Only the symptoms of LNS can be treated.
-The drug allopurinol may be used to control
excessive amounts of uric acid.
-Kidney stones can be treated with lithotripsy
-To help reduce some of the problem behaviors and
neurological effects of LNS :
Diazepam (Diastat, Valium)
Haloperidol (Haldol)
Phenobarbital (Luminal)

-The prognosis for LNS is poor because there are no
treatments for the neurological effects of the syndrome.
-Persons with this syndrome usually require assistance
walking and sitting and generally need a wheelchair to get
around.
-The build-up of excessive uric acid in the body causes painful
episodes of self-mutilation and may result in severe retardation
and death.

• Adenosine deaminase (ADA) is an essential enzyme of purine metabolism and is highly conserved throughout phylogeny.
• investigations indicated that ADA deficiency accounts forapproximately 20% of cases of human SCID and that it is the most severe of the immunodeficiency diseases, affecting both cell-mediated and humoral immunity (Buckley et al., 1997; Hershfieldand Mitchell, 2001).
• Soon after their discovery that defects in ADA were associated with immunodeficiency, Giblett and colleagues examined other immunodeficient individuals for deficiencies in purine catabolic enzymes and found that defects in purine nucleoside phosphorylasealso result in immunodeficiency disease.

molecular weight of 41 kDa .
adenosine aminohydrolase, EC 3.5.4.4
monomeric, zinc-dependent enzyme.
encoded by 12 exons.
ADA gene is 32 kb.
The gene is on chromosome 20q13.11.
mostly an intracellular enzyme.
found throughout body.
part of the purine catabolism pathway.
most active in lymphocytes.
functions in eliminating adenosine and deoxyadenosine.

• Associated with the loss of ADA activity ,The thymus is absent or small and dysplastic in ADA-deficient individuals (Borzy et al., 1979).
• They have severely reduced numbers of peripheral T, B, and natural killer (NK) cells(Buckley et al., 1997).
• ADA-deficient SCID is the only immunodeficiency in which all three cell types are severely reduced in number.
• Autosomal recessive disease .
• In the absence of ADA lymphocytes are destroyed.

• Deoxyadenosine and deoxyguanosine are toxic to human lymphoid cells in culture and have been implicated in the pathogenesis of the immunodeficiency states associated with adenosine deaminase and purine nucleoside phosphorylase deficiency, respectively.
• deoxyadenosine is not destroyed, is converted to dAMP and then into dATP.
• There marked increase in cellular concentrations of dATP due to the lack of conversion of excess deoxyadenosine to deoxyinosine and hypoxanthine .
• dATP is a potent feedback inhibitor ofdeoxynucleotide biosynthesis and DNA replication
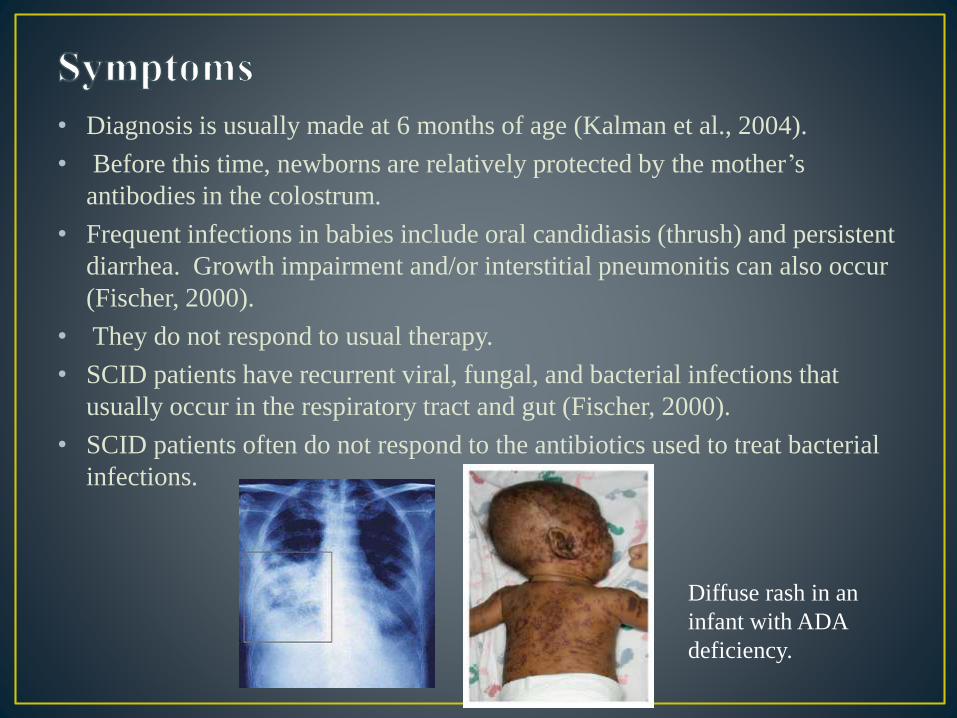
• Diagnosis is usually made at 6 months of age (Kalman et al., 2004).
• Before this time, newborns are relatively protected by the mother’s
antibodies in the colostrum.
• Frequent infections in babies include oral candidiasis (thrush) and persistent
diarrhea. Growth impairment and/or interstitial pneumonitis can also occur
(Fischer, 2000).
• They do not respond to usual therapy.
• SCID patients have recurrent viral, fungal, and bacterial infections that
usually occur in the respiratory tract and gut (Fischer, 2000).
• SCID patients often do not respond to the antibiotics used to treat bacterial
infections.
Diffuse rash in an
infant with ADA
deficiency.

Lymphopenia
Defect in T-cell activation
low serumimmunoglobulins
beware – antibody transferred from mother
Elevated IgE
Peripheral eosinophilia
Elevated plasma adenosine
Elevated plasma and urine 2-deoxyadenosine levels
Elevated dATP levels in erythrocytes.

• A successful biochemical approach for the treatment of ADA deficiency involves the use of enzyme replacement therapy wherein a polyethylene glycol–modified form of bovine ADA (PEG–ADA) is provided to patients by twice weekly intramuscular injection (Hershfield et al., 1993).
gene therapy
• the hope is that efficient transfer of a recombinant ADA gene into hematopoietic cells will result in the outgrowth of a genetically repaired immune system.
• For these and other reasons, ADA gene therapy studies were the first to use ex vivo approaches to stably introduce new genetic information into patients.











![Nucleotide Metabolism in Plants1[OPEN] · Nucleotide Metabolism in Plants1[OPEN] Claus-Peter Witte,2,3 and Marco Herde Leibniz Universität Hannover, Department of Molecular Nutrition](https://static.documents.pub/doc/80x56/5f07196d7e708231d41b4ced/nucleotide-metabolism-in-plants1open-nucleotide-metabolism-in-plants1open-claus-peter.jpg)






