33
D esign A nd A nalysis O f C linicalTrials M artin L.Lesser,Ph.D . Biostatistics U nit Feinstein Institute forM edical Research N orth Shore – Long Island Jew ish Health System
| Date post: | 26-Dec-2015 |
| Category: |
Documents |
| Upload: | godwin-shepherd |
| View: | 219 times |
| Download: | 1 times |

Design And Analysis Of Clinical Trials
Martin L. Lesser, Ph.D.
Biostatistics Unit
Feinstein Institute for Medical Research North Shore – Long Island Jewish
Health System

CME Disclosure Statement• The North Shore LIJ Health System adheres to the ACCME's new
Standards for Commercial Support. Any individuals in a position to control the content of a CME activity, including faculty, planners and managers, are required to disclose all financial relationships with commercial interests. All identified potential conflicts of interest are thoroughly vetted by the North Shore-LIJ for fair balance and scientific objectivity and to ensure appropriateness of patient care recommendations.
• Course Director, Kevin Tracey, has disclosed a commercial interest in Setpoint, Inc. as the cofounder, for stock and consulting support. He has resolved his conflicts by identifying a faculty member to conduct content review of this program who has no conflicts.
• The speaker, Martin L. Lesser, PhD, has no conflicts.
2

Types of Clinical Trials
Phase I - exploratory; assessment of toxicity; determination of safe dosage;
pharmacokinetics Phase II - evaluation of efficacy in a select group of
patients; estimation of treatment effect Phase III - comparative trial; hypothesis testing Phase IV - establish new indication;
post-marketing surveillance

Phase I Designs
• “3+3” dose escalation design for determining maximum tolerated dose (MTD)
• Fixed multiple dose design (e.g., randomize 5 subjects to each of 5 doses)
• Goal: design should protect subjects from harm, especially in a trial for which safe dosing, pharmacokinetics, and potential toxicities are unknown or poorly understood
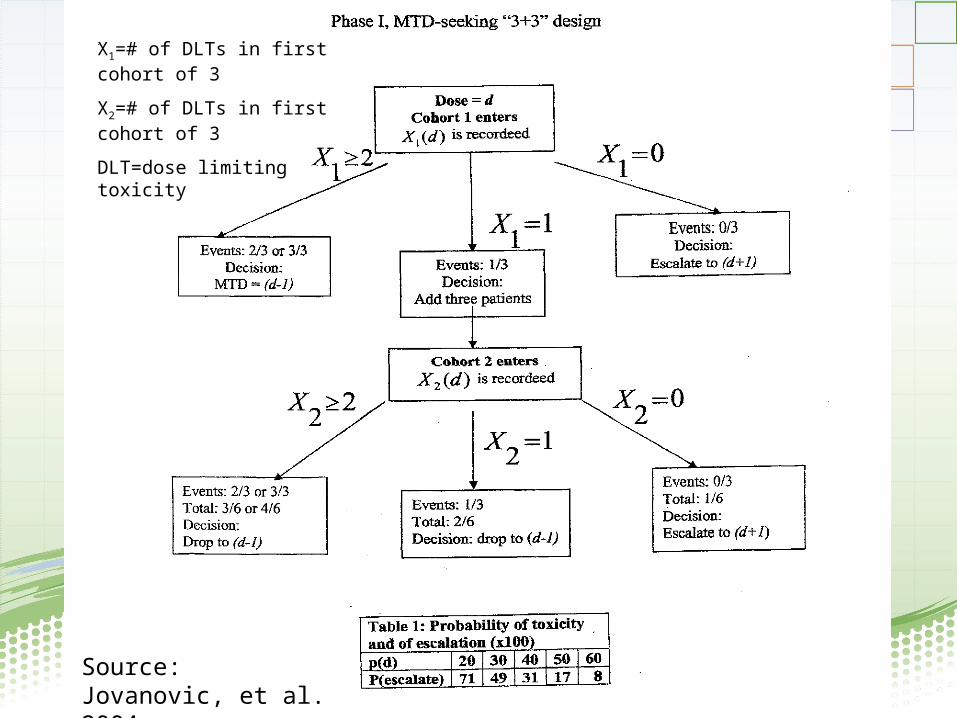
Source: Jovanovic, et al. 2004
X1=# of DLTs in first cohort of 3
X2=# of DLTs in first cohort of 3
DLT=dose limiting toxicity

Phase II Designs
• Applied to a specific disease entity• Fixed dose is used• Simple primary outcome: response,
measurement of some parameter• Single arm, open label (traditional)• Single arm, blinded evaluator (uncommon)• Simon 2-stage design• Randomized Phase II trial (for selection of best
therapy)

Simon 2-Stage Optimal Design
• H0: p ≤ p0 vs. HA: p ≥ p1
• Where response rate ≤ p0 is uninteresting and response rate ≥ p1 is the desired target
• Simon’s “Optimal Design”: Observe n1 subjects in stage 1. If response rate r1≤ a1/n1, then stop the trial and reject the drug.
• If r1> a1/n1, then study an additional n2 subjects in stage 2, for a total of n=n1+n2. If the “total” response rate r ≤ a/n, then reject the drug. If r > a/n, then consider the drug for further testing and Phase III trials.
Simon: Controlled Clin Trials, 10:1-10, 1989.

Simon 2-Stage Optimal Design (cont’d)
• For given α, β, p0, and p1, this design minimizes EN(p0), the expected number of subjects studied under H0 .
• Example: Let α=0.05 β=0.20 p0= 0.30 p1= 0.45Stage 1: Enter 27 subjects; stop trial and reject drug if r1≤ 9/27.
If r1 > 9/27, then go on to Stage 2.Stage 2: Enter 54 additional subjects (total=81). If r ≤ 30/81, then reject the drug. If r > 30/81, then trial is favorable toward drug.
Note: E(N(p0)) = 41.7. Prob(early termination)=0.73

Simon 2-Stage Minimax Design
• Similar to the 2-stage optimal design• Minimizes the maximum total sample size (n) among all
optimal designs• Minimax design is attractive when subject accrual is low• Previous example worked with minimax:
r1≤ 16/46, r ≤ 25/65, EN(p0)=49.6, PET(p0)=0.81
(Optimal design had n=81.)
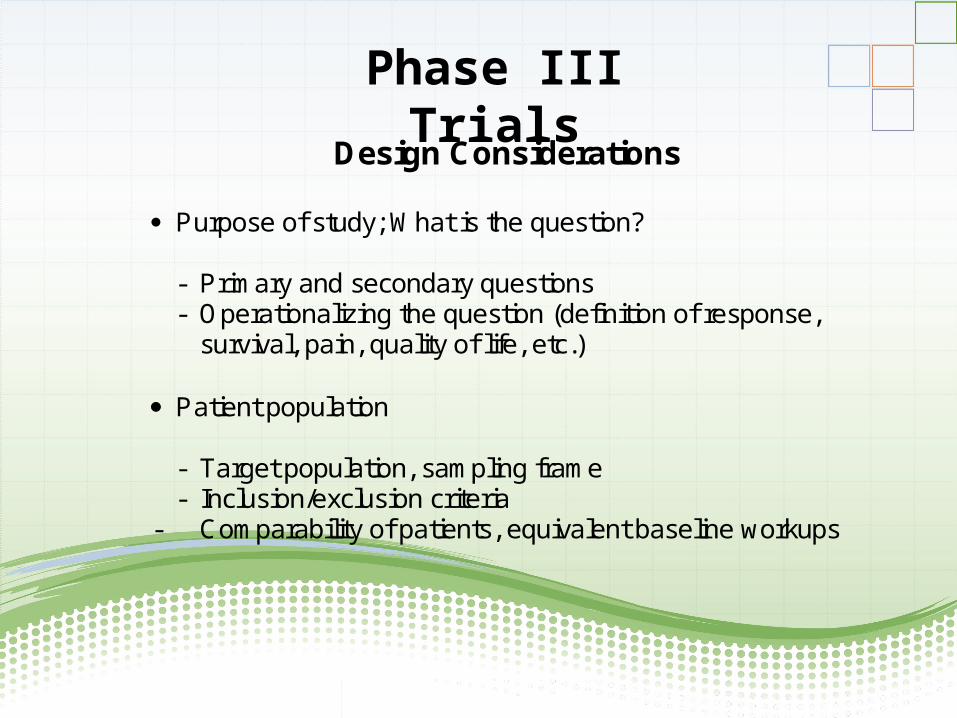
Design Considerations
Purpose of study; What is the question?
- Primary and secondary questions - Operationalizing the question (definition of response,
survival, pain, quality of life, etc.) Patient population
- Target population, sampling frame - Inclusion/exclusion criteria
- Comparability of patients, equivalent baseline workups
Phase III Trials

Design Considerations(continued)
• Treatment Plan
• Blinding
• Use of Placebo Control
• Criteria for evaluation of treatment effect
(comparability of patient follow-up)

Design Considerations (continued)
General study design structure for comparative
studies
- randomized controls - concurrent non-randomized controls - historical controls (Phase II and III) - patient as own control (cross-over design)

Randomized Controls Advantages Reduces or eliminates bias because chance, alone, determines assignment Assures that most statistical methods will be valid Disadvantages Can be expensive, labor intensive Patients may refuse randomization, resulting in bias Potential ethical problems May upset the patient-physician relationship Not feasible if contamination is likely

Concurrent Non-Randomized Controls
Advantages Useful when randomization is not feasible Useful in group or community interventions Usually less cost/effort than randomized trials Disadvantages Assignment to treatment may be biased May require matching or post-hoc adjustments

Historical Controls Advantages Data already exist Relatively inexpensive Ethical problems of randomization are avoided Often requires fewer patients on new treatment Disadvantages HCs and current group subjects may differ on: Method/criteria for selection Diagnostic and/or follow-up criteria Disease epidemiology, etiology, or natural history may have changed Difficult to protect against unknown biases Some data elements may not be available in the HC era

Patient As Own Control
AdvantagesReduces variance, often resulting in
smaller required sample sizes
DisadvantagesOnly useful in certain disease settingsMay introduce "order" effectsNature of intervention may be influenced
by results of first study period

Design Considerations(continued)
• Blinding
• Placebo control
• Stratification
• The process of randomization
• Handling dropouts and non-compliance
• Statistical methods for data analysis
• Sample size and power
• Interim analysis and early stopping

Blinding
• Any attempt to make study participants unaware of which treatment is offered
• Is indicated when the occurrence and reporting of outcomes can be easily influenced by knowledge of treatment (subjective responses, behavior change)
• May be either single blind or double blind
• Blinding is not always feasible
• Blinding may be unsuccessful (ability to break the blind)

Placebo Control
• Appropriate when no effective standard treatment exists for the control group
• Makes subject’s attitudes to the trial as similar as possible in the treatment and control groups
• Major uses:
− Controls for psychological factors
− Maintains double blind design
− Controls for spontaneous disease variability
• Ethical issues:
- May be unethical to withhold treatment in order to administer placebo

Stratification
Randomization does not guarantee thatprognostic factors will be evenly distributedbetween treatment groups
Imbalance can be partly addressed bystratification prior to randomization
Imbalance can also be addressed by covariateadjustment at the time of analysis

S t r a t i f i c a t i o n : A n E x a m p l e
Randomize RT
Chemo
Observed difference is confounded by the prognostic factor
NO STRATIFICATION
Low Risk High Risk
27 (30%)
56 (80%)
83
62 (70%)
38 (20%)
100
89
94
183
Response Rate
Chemo 25%
RT 64%
Randomize RT
40 (45%)
43 (46%) Response
Chemo 25%
RT 64%
Chemo
Rate
Observed difference is not confounded by the prognostic factor
withinLow Risk
n=83
Randomize RT
49 (55%)
51 (54%)
Chemo
withinHigh Risk
n=100
RANDOMIZE WITHIN STRATA

The Process of Randomization
simple randomization
permuted block randomization
unbalanced randomization
randomized consent form
Examples of permuted block randomization
- B=1 AAAABABAAAAAABBB (11 A, 5 B)
- B=4 ABBA AABB BABA BABA ( 8 A, 8 B)
- B=6 AABABB ABABBA AAAB ( 9 A, 7 B)

Dropouts and Non-Compliance
Intention to Treat Principle
- analyze as randomized- evaluates the effect of a treatment "policy"
Analyze as Treated Principle
- exclude dropouts- adjust for compliance or dose received- evaluates the effect of the "active ingredient"
(but in a possibly biased subset of patients)

Dropouts and Non-Compliance
Examples
- Patients with head and neck cancerrandomized to nasogastric feeding tube orgood oral nutrition;- Outcome=weight;- Some patients "cross-over" from NG tube
to oral nutrition arm
- Patients with familial polyposis randomizedto high fiber or low fiber diets;- Outcome=number and size of new polyps;- Some patients do not eat the required
amount of high fiber cereal; dose of fibervaries from patient to patient
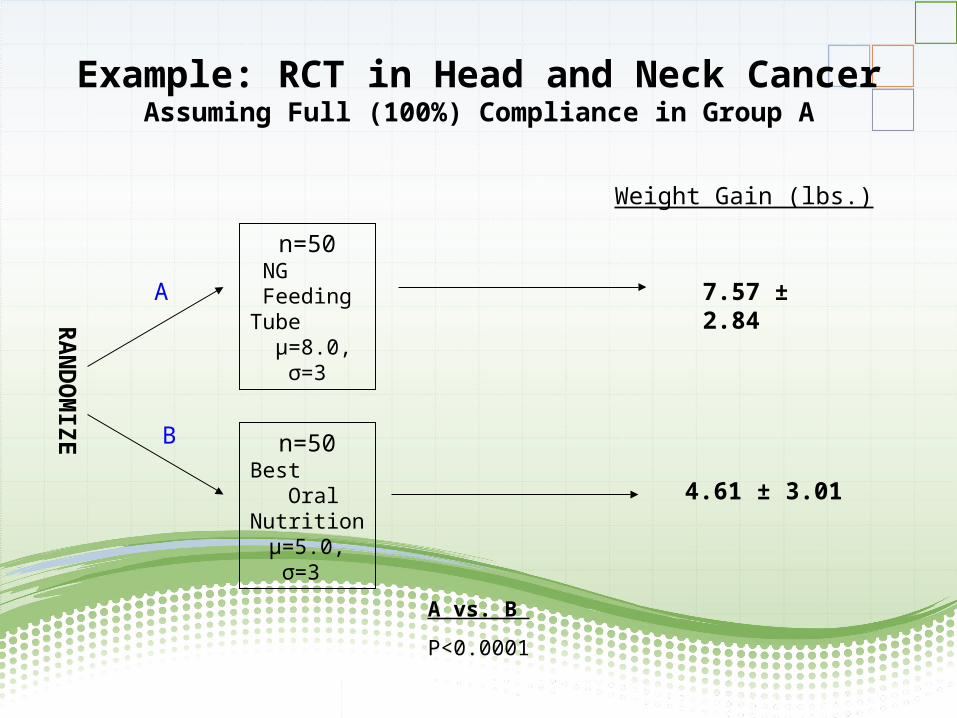
Example: RCT in Head and Neck CancerAssuming Full (100%) Compliance in Group A
RA
ND
OM
IZE
n=50NG
Feeding Tube
µ=8.0, σ=3
n=50Best
Oral Nutrition
µ=5.0, σ=3
A
B
7.57 ± 2.84
4.61 ± 3.01
Weight Gain (lbs.)
A vs. B
P<0.0001
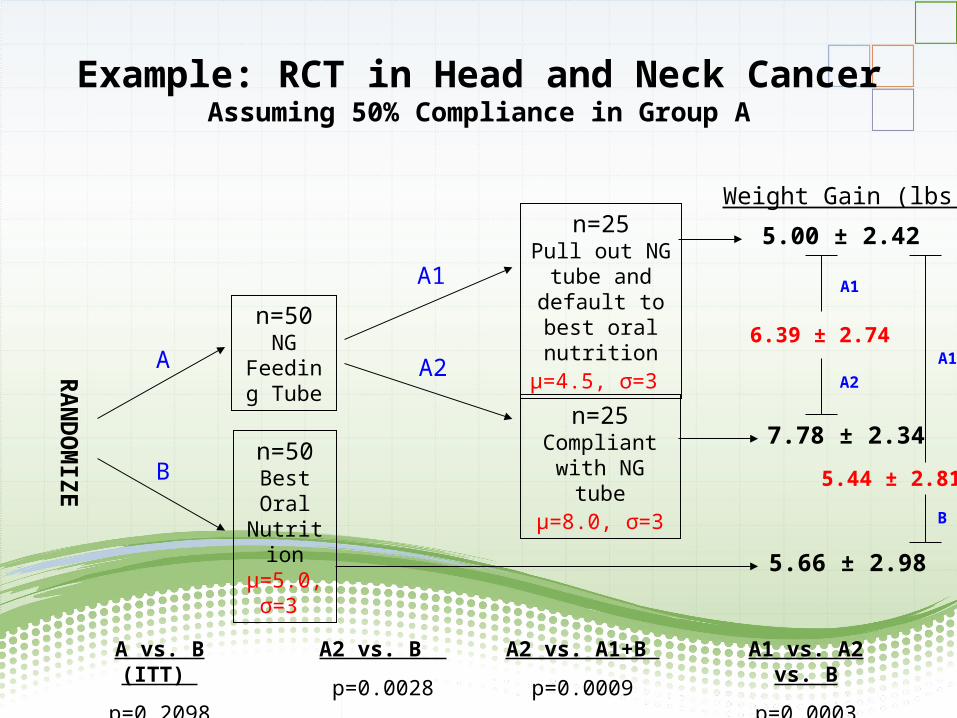
Example: RCT in Head and Neck CancerAssuming 50% Compliance in Group A
RA
ND
OM
IZE
n=50NG
Feeding Tube
n=50Best Oral
Nutritionµ=5.0, σ=3
n=25Pull out NG
tube and default to best oral nutritionµ=4.5, σ=3
n=25Compliant with
NG tubeµ=8.0, σ=3
A
B
A1
A2
5.00 ± 2.42
7.78 ± 2.34
5.66 ± 2.98
Weight Gain (lbs.)
6.39 ± 2.74
A1
A2
5.44 ± 2.81
A1
B
A vs. B (ITT)
p=0.2098
A2 vs. B
p=0.0028
A2 vs. A1+B
p=0.0009
A1 vs. A2 vs. B
p=0.0003

Statistical Methods Commonly Used in the Analysis of Clinical Trials Data
Binary response data
- chi square, Fisher exact test - multiple logistic regression
Survival, duration of response, and time until event data
- Kaplan-Meier product limit method - logrank test, Gehan-Wilcoxon test - Cox proportional hazards regression
Continuous-type data
- analysis of variance - ordinary multiple regression

Sample Size Considerations
Concept of power
Type of endpoint/outcome variable
Specification of clinically significant difference of interest
Estimation and confidence intervals
Multiple endpoints, Bonferroni correction
Tables of sample size and power

Patient Flow in Clinical Trials
Available
Considered
Eligible
Consented
Enrolled
Compliant
Adequately Followed

Sample Size/ Power Sample
Suppose the response rate using standard therapy (A) is assumed to be30%. The investigator would like to see an increase in the response rate toat least 50% (with treatment B) in order for it to be considered clinicallyuseful. A trial of A vs. B would require 125 patients in each group in order tohave a 90% chance (power) of detecting a difference of this magnitude orlarger (two-tailed test, 5% significance level).
Other calculations:n=93/group to achieve 80% powern=56/group to achieve 90% power to detect response rates of 30% vs. 60%n=42/group to achieve 80% power to detect response rates of 30% vs. 60%n=184/group to achieve 90% power to detect response rates of 30% vs. 35%

Interim Analysis and Early Stopping
Dangers of naive interim analysis
- increases Type I error rate (significance level) - increases bias with respect to "expected" results - data lags may influence interim results
Statistically sound stopping rules (i.e., rules that maintain the Type I error rate and desired power)
- group sequential analysis (O'Brien-Fleming, Pocock,
Lan-Demets, etc.) - curtailed sampling "individual" sequential testing - conditional power
Early stopping depends on formal statistics as well as on other factors

Example:The BHAT Trial
(Beta-blocker Heart Attack Trial)
• Randomized, double-blind, placebo-controlled trial to test the effect of propanolol (beta-blocker) on total mortality
• n = 3837 patients randomized to propanolol or placebo
• Trial was stopped 1 year early (on the 6th interim analysis) using the O-F group sequential approach when logrank X2 =2.82 > 2.23

O’Brien-Fleming Boundaries Applied to the BHAT Trial

















