Collateral sensitivity networks reveal evolutionary instability and novel treatment strategies in ALK mutated non-small cell lung cancer. Andrew Dhawan* 1,2 , Daniel Nichol* 3,4 , Fumi Kinose 5 , Mohamed E. Abazeed 1 , Andriy Marusyk 6 , Eric B. Haura 5 , and Jacob G. Scott† 1 1 Department of Translational Hematology and Oncology Research, Cleveland Clinic 2 Department of Oncology, University of Oxford 3 Department of Computer Science, University of Oxford 4 Department of Integrated Mathematical Oncology, H. Lee Moffitt Cancer Center and Research Institute 5 Department of Thoracic Oncology, Experimental Therapeutics Program, H. Lee Moffitt Cancer Center and Research Institute 6 Department of Cancer Imaging and Metabolism, H. Lee Moffitt Cancer Center and Research Institute September 18, 2016 * indicates equal contribution † to whom correspondence should be addressed, [email protected]1 . CC-BY-NC-ND 4.0 International license not peer-reviewed) is the author/funder. It is made available under a The copyright holder for this preprint (which was . http://dx.doi.org/10.1101/075846 doi: bioRxiv preprint first posted online Sep. 19, 2016;
.CC-BY-NC-ND 4.0 International licensenot peer-reviewed) is the author/funder. It is made available under aThe copyright holder for this preprint (which was. http://dx.doi.org/10.1101/075846doi: bioRxiv preprint first posted online Sep. 19, 2016;
Drug resistance remains an elusive problem in cancer therapy, particularly with novel
targeted therapy approaches. Much work is currently focused upon the development of
an increasing arsenal of targeted therapies, towards oncogenic driver genes such as ALK-
EML4, to overcome the inevitable resistance that develops as therapies are continued over
time. The current clinical paradigm after failure of first line ALK TKI is to administer
another drug in the same class. As to which drug however, the answer is uncertain, as
clinical evidence is lacking. To address this shortcoming, we evolved resistance in an ALK
rearranged non-small cell lung cancer line (H3122) to a panel of 4 ALK tyrosine kinase in-
hibitors used in clinic, and performed a collateral sensitivity analysis to each of the other
drugs. We found that all of the ALK inhibitor resistant cell lines displayed a significant
cross-resistance to all other ALK inhibitors. To test for the stability of the resistance pheno-
types, we evaluated the ALK-inhibitor sensitivities after drug holidays of varying length (1,
3, 7, 14, and 21 days). We found the resistance patterns to be stochastic and dynamic, with
few conserved patterns. This unpredictability led us to an expanded search for treatment
options for resistant cells. In this expansion, we tested a panel of 6 more anti-cancer agents
for collateral sensitivity among the resistant cells, uncovering a multitude of possibilities
for further treatment, including cross-sensitivity to several standard cytotoxic therapies as
well as the HSP-90 inhibitors. Taken together, these results imply that resistance to targeted
therapy in non-small cell lung cancer is truly a moving target; but also one where there are
many opportunities to re-establish sensitivities where there was once resistance.
2
.CC-BY-NC-ND 4.0 International licensenot peer-reviewed) is the author/funder. It is made available under aThe copyright holder for this preprint (which was. http://dx.doi.org/10.1101/075846doi: bioRxiv preprint first posted online Sep. 19, 2016;
Drug resistance in cancer is fundamentally an evolutionary problem [1]. Tumour cells, within
the varying microenvironment of a patient, are subjected to the selective pressures of the drugs
to which they are exposed, and respond in a manner governed by Darwinian dynamics [2]. As
a consequence of these stochastic evolutionary dynamics, the resultant population of drug-
resistant cancer cells may display features of cross-resistance, or conversely, collateral sensi-
tivity, to other chemotherapeutic agents; a knowledge of which, may be used to guide further
therapy. Collateral sensitivity is sensitivity towards a second drug which occurs after the evo-
lution of resistance to a first drug, when the resistant state causes a vulnerability to another
drug that was not previously present [3]. Clinically, a case of collateral sensitivity by resistance
mutations, or sensitization to a second drug in a state of resistance to a first-line therapy, has
been shown in a patient with ALK-positive NSCLC [4]. Further, the utility of a broader knowl-
edge of collateral sensitivity to panels of drugs has been shown in E. coli and lymphoma alike
[3, 5].
In addition to the concept of collateral sensitivity, drug holidays, or metronomic therapy,
have also been proposed to limit the development of resistance (or analagously, extend ef-
ficacy) in cancer treatment [6, 7, 8]. Upon removal of the selection pressure (therapy), it is
no longer necessarily advantageous to possess the changes associated with resistance. If the
changes associate with resistance come at a significant cost then, these traits may be selected
out of the population in the absence of drug. The molecular basis of the efficacy of drug hol-
idays, in a general sense, is thought to be due to reversible non-mutational mechanisms [6].
Little is known, however, about the length of drug holiday necessary for the outgrowth of the
original, sensitive populations, and this is likely highly variable from cancer to cancer, and
from patient to patient, though indeed, small-scale clinical studies have shown benefit with
this technique [6, 9]. Recent work in this area has also shown that adaptive therapy, or the
careful titration of therapy to maintain a stable tumour burden, but not the eradication, to
prevent resistance and ultimate therapeutic failure, has significant clinical promise [10, 2].
In this work, we combine the ideas of drug holidays and collateral sensitivity to develop
strategies of overcoming TKI resistance in ALK-positive NSCLC. First described in 2007, re-
arrangements in the anaplastic lymphoma kinase (ALK) and the echinoderm microtubule-
3
.CC-BY-NC-ND 4.0 International licensenot peer-reviewed) is the author/funder. It is made available under aThe copyright holder for this preprint (which was. http://dx.doi.org/10.1101/075846doi: bioRxiv preprint first posted online Sep. 19, 2016;
associated protein-like 4 (EML4) genes have been found to drive approximately 5-10% of all
non-small cell lung cancer (NSCLC) cases, disproportionately affecting younger, generally
non- or light smoking, patients [11, 12, 13]. Clinically, targeted therapies inhibiting the ki-
nase activity of ALK have proven to be efficacious, significantly extending progression-free
survival compared to standard therapies. These trials have led to the ALK inhibitor Crizotinib
to be the first-line standard of care for metastatic tumours driven by this oncogene [14, 15]. Af-
ter widespread use began however, reports of resistance to ALK inhibition quickly emerged,
and it has since become apparent that within one year of starting such therapy, resistance
almost inevitably emerges [16, 17, 18]. The clinical question that arises thereafter is how to
proceed with therapy, and current National Comprehensive Cancer Network (NCCN) guide-
lines suggest that for a symptomatic patient, a second-line agent of the ALK-TKI class should
be used, such as Ceritinib or Alectinib [14]. Reportedly, these next generation ALK TKIs can
overcome resistance to Crizotinib [19], but there is little guidance as to how to choose the most
efficacious second line therapy. There are, however, alternative strategies not represented in
these guidelines that may provide avenues for further treatment. One such strategy to combat
therapeutic resistance, with reports of possible efficacy in lymphoma [5], involves exploiting
collateral sensitivity.
In this work we derive what is, to our knowledge, the first collateral sensitivity network
using targeted therapies in any solid cancer, and extend the analysis to the period after the
drug is removed. We posit that drug holidays are an important, yet overlooked aspect of
clinical treatment. Typically, when anti-cancer therapies are switched, after progression say,
there is a latency period in which the patient is not receiving any form of anti-cancer therapy,
on the order of weeks to months. This latency period, or holiday, has not been considered
in previous experimental or theoretical models. This led us to investigate the evolutionary
dynamics of collateral sensitivities in drug resistant cell lines after having undergone various
lengths of drug holidays. In particular, we examine the structure of the collateral sensitivity
network for the available ALK TKI in an ALKmut NSCLC line. We further investigate the effect
of drug holidays on this structure, and the potential for novel cycling regimens using a panel
of clinically relevant therapeutic agents. We show that evolution of resistance to a particular
ALK-TKI leads to a substantial cross-resistance to other ALK-TKIs, but this varies significantly
4
.CC-BY-NC-ND 4.0 International licensenot peer-reviewed) is the author/funder. It is made available under aThe copyright holder for this preprint (which was. http://dx.doi.org/10.1101/075846doi: bioRxiv preprint first posted online Sep. 19, 2016;
Table 1: Panel of drugs used in study, their abbreviations, and classification.
2 Results
2.1 Significant cross-resistance among ALK-TKI resistant cells
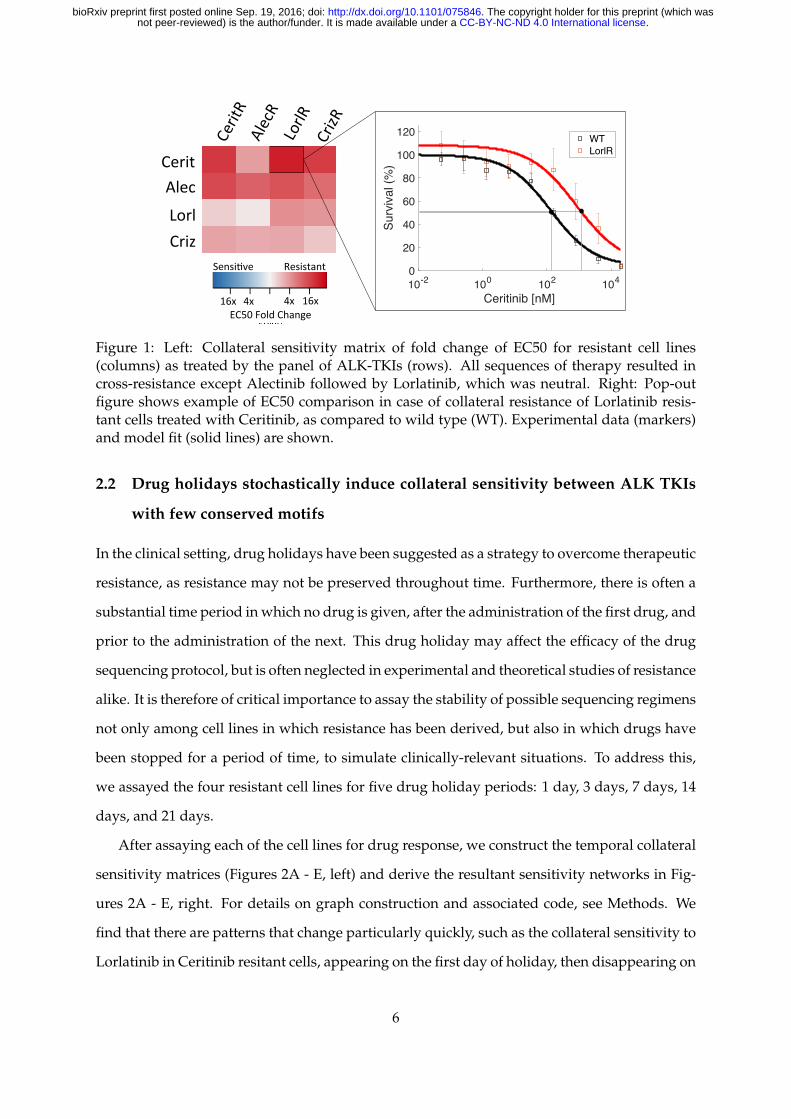
Evolution of resistance to ALK inhibitors (Crizotinib, Alectinib, Lorlatinib, and Ceritinib), was
induced over a 16 week period beginning with wild type H3122 ALK-positive NSCLC cell
line, and cell lines were subjected to a collateral sensitivity analysis, as described in Methods.
From the obtained EC50 values in the dose-response experiments for each of the resistant cell
lines, we compare the collateral resistances and sensitivities between the cell lines. The data
is presented as a heat map of fold changes of the EC50 values compared to the values for the
treatment naive parental cells (Figure 1). Contrary to the accepted clinical guidelines, none of
the 4 resistant cell lines obtained displayed collateral sensitivity to any other ALK TKI at the
time of maximum drug exposure. Further, with the exception of Lorlatinib treatment following
Alectinib resistance, which was neutral, cross-resistance was observed among all four resistant
cell lines.
5
.CC-BY-NC-ND 4.0 International licensenot peer-reviewed) is the author/funder. It is made available under aThe copyright holder for this preprint (which was. http://dx.doi.org/10.1101/075846doi: bioRxiv preprint first posted online Sep. 19, 2016;
Figure 1: Left: Collateral sensitivity matrix of fold change of EC50 for resistant cell lines(columns) as treated by the panel of ALK-TKIs (rows). All sequences of therapy resulted incross-resistance except Alectinib followed by Lorlatinib, which was neutral. Right: Pop-outfigure shows example of EC50 comparison in case of collateral resistance of Lorlatinib resis-tant cells treated with Ceritinib, as compared to wild type (WT). Experimental data (markers)and model fit (solid lines) are shown.
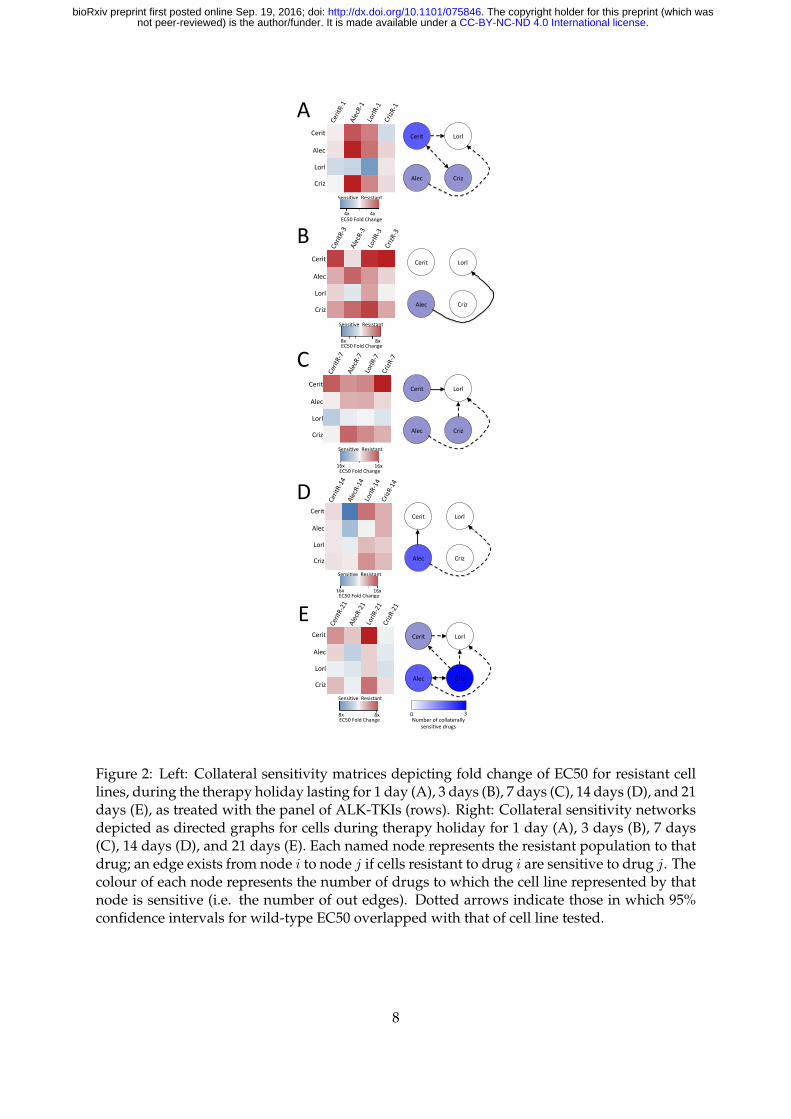
2.2 Drug holidays stochastically induce collateral sensitivity between ALK TKIs
with few conserved motifs
In the clinical setting, drug holidays have been suggested as a strategy to overcome therapeutic
resistance, as resistance may not be preserved throughout time. Furthermore, there is often a
substantial time period in which no drug is given, after the administration of the first drug, and
prior to the administration of the next. This drug holiday may affect the efficacy of the drug
sequencing protocol, but is often neglected in experimental and theoretical studies of resistance
alike. It is therefore of critical importance to assay the stability of possible sequencing regimens
not only among cell lines in which resistance has been derived, but also in which drugs have
been stopped for a period of time, to simulate clinically-relevant situations. To address this,
we assayed the four resistant cell lines for five drug holiday periods: 1 day, 3 days, 7 days, 14
days, and 21 days.
After assaying each of the cell lines for drug response, we construct the temporal collateral
sensitivity matrices (Figures 2A - E, left) and derive the resultant sensitivity networks in Fig-
ures 2A - E, right. For details on graph construction and associated code, see Methods. We
find that there are patterns that change particularly quickly, such as the collateral sensitivity to
Lorlatinib in Ceritinib resitant cells, appearing on the first day of holiday, then disappearing on
6
.CC-BY-NC-ND 4.0 International licensenot peer-reviewed) is the author/funder. It is made available under aThe copyright holder for this preprint (which was. http://dx.doi.org/10.1101/075846doi: bioRxiv preprint first posted online Sep. 19, 2016;
day 3, re-appearing on day 7, disappearing on day 14, and again re-appearing on day 21. Like-
wise, there are also stable patterns that are highly conserved, though few in number, such as
the cross-resistance of Lorlatinib resistant cells towards Ceritinib. Further implication of these
experimental results indicates that Lorlatinib might be a poor choice for first-line therapy, as
the cross-resistance it generates is significant and stable through drug holidays. Lorlatinib, as
indicated by these results, for this cell line, may be a better choice in the second-line, as many
resistant lines are sensitized towards it after a drug holiday.
Another implication of these experimental results lies in the design of metronomic chemother-
apy schedules. That is, there appears to be a dynamic sensitization of Alectinib resistant cells
to Alectinib, appearing only after 14 days of drug holiday, and maintained after 21 days of
holiday as well. This suggests the design of a schedule in which one treats in regular intervals,
but with a 14 or 21 day holiday in between cycles, as in metronomic chemotherapy.
Finally, we note that variation in these graphs highlights significant temporal instability
in collateral sensitivities over time. These dynamics have largely not been taken into account
in previous studies of collateral sensitivity and resistance, in both the fields of microbiology
and oncology [3, 22, 23, 24, 25]. Based on these experiments, we pose that this is a critical
phenomenon that must be accounted for when designing treatment schedules, and may have
significant implications for previously obtained results of collateral sensitivity.
7
.CC-BY-NC-ND 4.0 International licensenot peer-reviewed) is the author/funder. It is made available under aThe copyright holder for this preprint (which was. http://dx.doi.org/10.1101/075846doi: bioRxiv preprint first posted online Sep. 19, 2016;
Figure 2: Left: Collateral sensitivity matrices depicting fold change of EC50 for resistant celllines, during the therapy holiday lasting for 1 day (A), 3 days (B), 7 days (C), 14 days (D), and 21days (E), as treated with the panel of ALK-TKIs (rows). Right: Collateral sensitivity networksdepicted as directed graphs for cells during therapy holiday for 1 day (A), 3 days (B), 7 days(C), 14 days (D), and 21 days (E). Each named node represents the resistant population to thatdrug; an edge exists from node i to node j if cells resistant to drug i are sensitive to drug j. Thecolour of each node represents the number of drugs to which the cell line represented by thatnode is sensitive (i.e. the number of out edges). Dotted arrows indicate those in which 95%confidence intervals for wild-type EC50 overlapped with that of cell line tested.
8
.CC-BY-NC-ND 4.0 International licensenot peer-reviewed) is the author/funder. It is made available under aThe copyright holder for this preprint (which was. http://dx.doi.org/10.1101/075846doi: bioRxiv preprint first posted online Sep. 19, 2016;
2.3 Expanded drug panel provides opportunities for significant collateral sensitiv-
ity to ALK TKIs
After finding significant cross resistance between all ALK TKIs, we expanded our search by
testing the sensitivities to clinically relevant chemotherapeutic agents and heat shock protein
inhibitors. While clinical trials have shown the superiority of targeted therapy in the first-line
setting, there is currently little to no data about the relative efficacy of non-targeted chemother-
apy in the setting of therapeutic resistance. This avenue presents significant opportunity for
intervention in the case of resistance to the ALK TKIs, as we have found that there are a num-
ber of drugs, not in the class of TKIs, to which a number of the resistant cell lines are collat-
erally sensitive, as depicted in Figures 3A and B. We find that the cell lines resistant to the
first-line therapy of TKIs can often by sensitized towards non-TKIs in the resistant state. In or-
der to evaluate the various drugs in a general sense, we may consider the number of resistant
cell lines that display collateral sensitivity or cross-resistance towards a given drug, which we
plot in Figure 3B. While not an exhaustive, or mechanistic explanation, this may be used to
consider the potential for the use of a given therapy as a second-line agent in a probabilistic
sense, though we recognize that in a clinical setting this probability is adjusted (conditioned)
by knowledge of the first-line agent. Ranking drugs by the probability that a given resistant
cell line is collaterally sensitized towards them, as is done in Figures 3C and D, we observe
that Etoposide and Pemetrexed are the optimal choices for second line therapy, as the great-
est number of resistant cell lines are sensitized towards them. Further, ranking by number
of drugs to which cross-resistance exists in Figure 3D, we observe that the HSP-90 inhibitors,
Ganetespib and IPI504, are the poorest choices for second-line therapy after the emergence of
resistance, as they carry the greatest burden of cross-resistance.
Beyond assaying efficacy of these agents in the second line setting, or as drugs to prime a
cancer to be more sensitive to a targeted agent, this expanded panel of collateral sensitivities
and resistances can be taken advantage of to design drug cycling protocols. Because of the
patterns of collateral sensitivity observed in Figure 3A, a connected graph of 10 nodes, each
node representing a resistant cell line, can be generated (Figure 4A). From this, determining
drug cycling protocols consists of identifying graph cycles, which represent sequences of drugs
wherein sensitization towards the drugs repeats, and resistance to the final drug results in
9
.CC-BY-NC-ND 4.0 International licensenot peer-reviewed) is the author/funder. It is made available under aThe copyright holder for this preprint (which was. http://dx.doi.org/10.1101/075846doi: bioRxiv preprint first posted online Sep. 19, 2016;
Figure 3: (A): Collateral sensitivity matrix of fold change of EC50 for resistant cell lines(columns) as treated by the panel of anti-cancer therapies (rows). Grey boxes are indetermi-nate due to significant resistance to drug. (B): Bar graphs depicting the number of collaterallysensitive or resistant cell lines to each drug. (C): Ranked probabilities of collateral sensitivity ofeach drug to the resistant cell lines, when the drug is used as second-line therapy. (D): Rankedprobabilities of cross-resistance of each drug to resistant cell lines, when the drug is used assecond-line therapy.
10
.CC-BY-NC-ND 4.0 International licensenot peer-reviewed) is the author/funder. It is made available under aThe copyright holder for this preprint (which was. http://dx.doi.org/10.1101/075846doi: bioRxiv preprint first posted online Sep. 19, 2016;
Figure 4: (A): Collateral sensitivity network depicted as a directed graph, with edges pointingfrom nodes at which resistance to a particular drug has developed to a node for which sensi-tivity of the drug has increased. Arrows in red indicate an example of a cycling regimen of 4drugs. (B): Graph of number of drug cycles of given length in the graph presented in (A).
3 Discussion
In this work, four cell lines individually resistant to a panel of first-line therapies for ALK-
positive NSCLC were studied to determine patterns of resistance and sensitivity, from which to
infer second-line treatments. From the data collected, we have constructed a collateral sensitiv-
ity ‘map’ for cell lines resistant to the ALK-TKIs. From this we immediately observe that there
is significant cross-resistance that evolves under treatment from the first ALK-TKI to the oth-
ers, suggesting that using any of these drugs as a second-line agent would not provide optimal
therapeutic benefit. This differs from previous results, which offer observations of collateral
sensitivity among drugs of the same class, as in the case of BCR-ABL driven leukaemia treated
with tyrosine kinase inhibitors [5]. Following this, we assay the stability of this cross-resistance
after a drug holiday of 1, 3, 7, 14, or 21 days and find significant variation in collateral sensi-
tivity network structure as time progresses, with collateral sensitivities arising transiently and
unpredictably with few conserved motifs. In contrast, related work had not considered this
possibility of temporal variation after removal of therapy, and instead found particular genetic
11
.CC-BY-NC-ND 4.0 International licensenot peer-reviewed) is the author/funder. It is made available under aThe copyright holder for this preprint (which was. http://dx.doi.org/10.1101/075846doi: bioRxiv preprint first posted online Sep. 19, 2016;
changes conserved among collaterally sensitive clones, which are assumed to be temporally
stable [5]. A critical implication of these results is that even after relatively short drug holi-
days, conserved motifs are rare among the collateral sensitivity networks constructed. This
suggests that strategies involving drug holidays and collateral sensitivities offer promise as
collateral sensitivity does arise, but we must carefully consider the dynamics if we are to use
these strategies in the clinical setting.
Given the significant cross-resistance which arose in our evolved cell lines, a larger panel
of drugs was tested for collateral sensitivity, with encouraging results. Though it has been
shown that clinically these drugs are inferior to ALK-TKIs as first-line therapy in patients with
NSCLC harboring the ALK rearrangement, we find that there may be utility in considering
these drugs after initial therapeutic resistance to ALK-TKIs occurs, with many opportunities
for collateral sensitivity (Figure 4). Further, in cases with greater degrees of collateral sensitiv-
ity, such as with this expanded panel, as has been reported in this work and many others, the
potential for steering or drug cycling regimens exists [3, 5, 26, 27, 28], which would allow us
to sequence targeted and non-targeted agents together to regain sensitivity where there was
once resistance. In this manner, resistance towards a drug sensitizes for the next drug, and so
on, until the drugs of the cycle repeat, and we have shown in Figure 4D that there are a large
number of possibilities for these cycling regimens, even for the relatively small panel of drugs
tested.
Overall, the number of possible drug cycling regimens in Figure 4D provides hope that
expanding the arsenal of drugs will give novel avenues for therapy, and opportunities to rein-
state sensitivity, not accounted for in current clinical practices. However, we note that as we
have shown in Figure 2, collateral sensitivity is highly dynamic, and truly represents a ‘mov-
ing target.’ In this sense, we note that any chosen drug cycling regimen should show stability
over time, through drug holidays of varying lengths, as may occur when the drugs of the cy-
cles are being switched. In recent work with E. coli, drug cycling protocols based on collateral
sensitivity are highlighted, but this distinction based on temporal instability was not raised [3]
in regards to timing of the second (and subsequent) therapies. Further, the observation that
the collateral sensitivity networks change over time in the absence of drug raises the question
as to the stability and repeatability of the generation of the collateral sensitivity matrix itself.
12
.CC-BY-NC-ND 4.0 International licensenot peer-reviewed) is the author/funder. It is made available under aThe copyright holder for this preprint (which was. http://dx.doi.org/10.1101/075846doi: bioRxiv preprint first posted online Sep. 19, 2016;
cell line for each of the 10 drugs. H3122 cells were then seeded at 70% confluence and drugs
were added at a starting concentration of 20% of the determined IC50 value. Media/drug was
changed every 3 days and cells were passaged once they reached 90% confluency. After two
13
.CC-BY-NC-ND 4.0 International licensenot peer-reviewed) is the author/funder. It is made available under aThe copyright holder for this preprint (which was. http://dx.doi.org/10.1101/075846doi: bioRxiv preprint first posted online Sep. 19, 2016;
Figure 5: Experimental design diagram. Panel (A) depicts the evolution of resistance by expo-sure for 16 weeks to drug to create a drug-resistant population (DrugR) and in (B) the popula-tion is removed from drug (‘relaxed’).
4.2 Mathematical analysis
Experimentally, dose-response curves for each cell line to each drug were obtained at varying
concentration levels. To extract meaningful comparison from these sets of curves, these were
fit using non-linear optimization in R to the following mathematical model. Key model pa-
rameters are EC50, which for the purposes of this experiment is defined as the concentration
of the drug at which the half-maximal effect of cell kill was observed , and the hill coefficient,
n. We then model the survival S(d) after the administration of a particular drug dose d as:
14
.CC-BY-NC-ND 4.0 International licensenot peer-reviewed) is the author/funder. It is made available under aThe copyright holder for this preprint (which was. http://dx.doi.org/10.1101/075846doi: bioRxiv preprint first posted online Sep. 19, 2016;
where n is the Hill coefficient, H is the maximum survival proportion observed, and L is
the minimal survival proportion observed. In certain cases of data fitting using this procedure,
results obtained for the EC50 are reported as indeterminate. These curves represented cases
in which cellular resistance was so significant that growth continued in the presence of drug,
resulting in a survival function that was not of Hill-type, and could therefore not be meaning-
fully fit to obtain an EC50.
We note that while there is no perfect measure of drug resistance or sensitivity, the EC50
was chosen in this experiment for the primary purpose of comparability with existing litera-
ture, and because of the strong degree of agreement of this model to experimental data, under-
scoring its utility in comparing dose-response curves. Importantly, we note that there are cases
in which the EC50 may mislead as to whether a cell line is resistant or sensitive. For instance,
there is the possibility that the reported EC50 for a cell line is lower than the wild type case, but
the value of L is much greater, meaning that a much smaller proportion of cells were actually
eliminated, even at maximal drug concentrations.
4.3 Detection of cycles
Once the graph defined by the collateral sensitivity network was reconstructed, cycles were
counted from this graph. This was done computationally in Python, with code as provided in
the supplementary files. The algorithm relies upon a depth-first search to traverse the graph,
iterating through the graph, not revisiting any nodes it has already visited in that iteration of
the search. The ‘depth’ of the search is restricted to be the cycle length. The search is then
iterated over the graph, and if the starting node is reached at exactly the depth of the desired
cycle length, that path is saved as a cycle. We note that this method, by its definition, counts
every cycle of length n, n times, so we divide by this when recording the number of unique
cycles.
15
.CC-BY-NC-ND 4.0 International licensenot peer-reviewed) is the author/funder. It is made available under aThe copyright holder for this preprint (which was. http://dx.doi.org/10.1101/075846doi: bioRxiv preprint first posted online Sep. 19, 2016;
The authors would like to thank Dr. Nima Sharifi and Mr. Artem Kaznatcheev for helpful
discussions and commentary on the manuscript during preparation.
16
.CC-BY-NC-ND 4.0 International licensenot peer-reviewed) is the author/funder. It is made available under aThe copyright holder for this preprint (which was. http://dx.doi.org/10.1101/075846doi: bioRxiv preprint first posted online Sep. 19, 2016;
[1] Lauren MF Merlo, John W Pepper, Brian J Reid, and Carlo C Maley. Cancer as an evolu-
tionary and ecological process. Nature Reviews Cancer, 6(12):924–935, 2006.
[2] Robert J Gillies, Daniel Verduzco, and Robert A Gatenby. Evolutionary dynamics of car-
cinogenesis and why targeted therapy does not work. Nature Reviews Cancer, 12(7):487–
493, 2012.
[3] Lejla Imamovic and Morten OA Sommer. Use of collateral sensitivity networks to design
drug cycling protocols that avoid resistance development. Science Translational Medicine,
5(204):204ra132–204ra132, 2013.
[4] Alice T Shaw, Luc Friboulet, Ignaty Leshchiner, Justin F Gainor, Simon Bergqvist, Alexei
Brooun, Benjamin J Burke, Ya-Li Deng, Wei Liu, Leila Dardaei, et al. Resensitization to
crizotinib by the lorlatinib alk resistance mutation l1198f. New England Journal of Medicine,
374(1):54–61, 2016.
[5] Boyang Zhao, Joseph C Sedlak, Raja Srinivas, Pau Creixell, Justin R Pritchard, Bruce Tidor,
Douglas A Lauffenburger, and Michael T Hemann. Exploiting temporal collateral sensi-
tivity in tumor clonal evolution. Cell, 165(1):234–246, 2016.
[6] A Becker, L Crombag, DAM Heideman, FB Thunnissen, AW van Wijk, PE Postmus, and
EF Smit. Retreatment with erlotinib: regain of tki sensitivity following a drug holiday
for patients with nsclc who initially responded to egfr-tki treatment. European Journal of
Cancer, 47(17):2603–2606, 2011.
[7] Jasmine Foo and Franziska Michor. Evolution of resistance to targeted anti-cancer thera-
pies during continuous and pulsed administration strategies. PLoS Computational Biology,
5(11):e1000557, 2009.
[8] Irina Kareva, David J Waxman, and Giannoula Lakka Klement. Metronomic chemother-
apy: an attractive alternative to maximum tolerated dose therapy that can activate anti-
tumor immunity and minimize therapeutic resistance. Cancer Letters, 358(2):100–106,
2015.
17
.CC-BY-NC-ND 4.0 International licensenot peer-reviewed) is the author/funder. It is made available under aThe copyright holder for this preprint (which was. http://dx.doi.org/10.1101/075846doi: bioRxiv preprint first posted online Sep. 19, 2016;
et al. Eml4-alk mutations in lung cancer that confer resistance to alk inhibitors. New
England Journal of Medicine, 363(18):1734–1739, 2010.
[17] Robert C Doebele, Amanda B Pilling, Dara L Aisner, Tatiana G Kutateladze, Anh T Le,
Andrew J Weickhardt, Kimi L Kondo, Derek J Linderman, Lynn E Heasley, Wilbur A
18
.CC-BY-NC-ND 4.0 International licensenot peer-reviewed) is the author/funder. It is made available under aThe copyright holder for this preprint (which was. http://dx.doi.org/10.1101/075846doi: bioRxiv preprint first posted online Sep. 19, 2016;
Hidetoshi Hayashi, Hiroyasu Kaneda, Ken Takezawa, Kiyoko Kuwata, Haruka Yam-
aguchi, et al. Activation of her family signaling as a mechanism of acquired resistance
to alk inhibitors in eml4-alk–positive non–small cell lung cancer. Clinical Cancer Research,
18(22):6219–6226, 2012.
[21] Jim Sang, Jaime Acquaviva, Julie C Friedland, Donald L Smith, Manuel Sequeira, Chao-
hua Zhang, Qin Jiang, Liquan Xue, Christine M Lovly, John-Paul Jimenez, et al. Targeted
inhibition of the molecular chaperone hsp90 overcomes alk inhibitor resistance in non–
small cell lung cancer. Cancer Discovery, 3(4):430–443, 2013.
[22] Matthew D Hall, Misty D Handley, and Michael M Gottesman. Is resistance useless? mul-
tidrug resistance and collateral sensitivity. Trends in Pharmacological Sciences, 30(10):546–
556, 2009.
[23] Kristen M Pluchino, Matthew D Hall, Andrew S Goldsborough, Richard Callaghan, and
Michael M Gottesman. Collateral sensitivity as a strategy against cancer multidrug resis-
tance. Drug Resistance Updates, 15(1):98–105, 2012.
[24] Csaba Pal, Balazs Papp, and Viktoria Lazar. Collateral sensitivity of antibiotic-resistant
microbes. Trends in Microbiology, 23(7):401–407, 2015.
19
.CC-BY-NC-ND 4.0 International licensenot peer-reviewed) is the author/funder. It is made available under aThe copyright holder for this preprint (which was. http://dx.doi.org/10.1101/075846doi: bioRxiv preprint first posted online Sep. 19, 2016;
.CC-BY-NC-ND 4.0 International licensenot peer-reviewed) is the author/funder. It is made available under aThe copyright holder for this preprint (which was. http://dx.doi.org/10.1101/075846doi: bioRxiv preprint first posted online Sep. 19, 2016;
.CC-BY-NC-ND 4.0 International licensenot peer-reviewed) is the author/funder. It is made available under aThe copyright holder for this preprint (which was. http://dx.doi.org/10.1101/075846doi: bioRxiv preprint first posted online Sep. 19, 2016;