Page 1
PERICYCLIC AND PSEUDOPERICYCLIC: [3, 3] AND [3, 5] REARRANGEMENTS
by
Deepali Butani, M.Sc.
A Dissertation
In
CHEMISTRY
Submitted to the Graduate Faculty
of Texas Tech University in
Partial Fulfillment of
the Requirements for
the Degree of
DOCTOR OF PHILOSOPHY
Approved
David M. Birney, Chairman
Jorge A. Morales
William L. Hase
Peggy Gordon Miller
Dean of the Graduate School
August, 2011
Page 2
Copyright 2011, Deepali Butani
Page 3
Texas Tech University, Deepali Butani, August 2011
ii
ACKNOWLEDGMENTS
I wish to thank my advisor, Professor David M. Birney, for his excellent
guidance, continued support, freedom and encouragement to pursue interesting and
challenging research problems. His enthusiasm and skill for suggesting efficient
solutions has been a big factor behind most of the work done by me in the last six
years. Even though I was a physical chemist he encouraged me a great deal about
synthetic chemistry and has broadened much of my synthetic point of view.
I would also like to express my sincere thanks to Professor Jorge A. Morales
and Professor William L. Hase for serving in my committee and providing constant
support and advice throughout my doctoral degree.
I am thankful to all the faculty and staff members of the Department of
Chemistry and Biochemistry, Texas Tech University for their support and help during
the period of six years. Their ever-helping nature made my student experience a
memorable one. My sincere thanks to Mr. David W. Purkiss for his help and
assistance with NMR spectroscopy related issues.
I would like to thank Texas Tech University and Robert A Welch Foundation
for the financial support for the research project.
I would like to thank all past and current group members of Dr, Birney’s
group: George Tamas, Shikha Sharma, Trideep Rajale, Jo Ramos, Ali Al-Khafaji,
Krishnaja Duvvuri, Rudhran Mehra, Fabrice Duvernay, Indra Reddy Gudipati, Tina
Page 4
Texas Tech University, Deepali Butani, August 2011
iii
Thomas, Hua Ji and Sherina Shahbazian who shared their willingness to listen to my
problems, both research and otherwise, and giving useful suggestions and advice.
I have had the fortune of making some great friends during my stay in
Lubbock. Last six years would have been impossible without constant help and
guidance from Arindam Mazumdar and Rahul Kanungoe. They have helped me
completely transforming into a better person. I would also like to thank Cole Seifert
for his unconditional support and helping me learn a lot about life. Also would like to
thank Chandrani Banerjee, Anuja Malvankar, Manav Gupta and Bipasha Deb for their
company during my early years; and Pillhun Son, Eric Clevenger, Sunil Paladugu,
Suresh Pindi and Sekhar Kunapareddy.
At the end, I would like to express my sincere gratitude to all my family
members and relatives for their unconditional support. My cousins, Sajjan Chuggani
and Chandramita Chuggani, Mahesh uncle and Asha aunty have been a constant
source of encouragement for me. I wish to express special gratitude to my father
Pritam Gobindram Butani and my sister, Jaya Butani without whose encouragement
and guidance, I would not have made it here. My mother Late Neena Butani has been
an inspiration to finish my doctoral program. Finally, I would like to dedicate this
thesis to my grandparents, Prem and Pushpa Chawla, who made sure I did something
meaningful in life, and for their love and affection, when it mattered the most.
Page 5
Texas Tech University, Deepali Butani, August 2011
iv
TABLE OF CONTENTS
ACKNOWLEDGMENTS ............................................................................................. ii
ABSTRACT ............................................................................................................. vi
LIST OF TABLES .................................................................................................. viii
LIST OF FIGURES ................................................................................................. xiv
I. INTRODUCTION .................................................................................................... 1
1.1 Pericyclic Reactions ..................................................................................... 1
1.2 Pseudopericyclic Reactions .......................................................................... 7
1.3 Sigmatropic Rearrangements ...................................................................... 15
1.4 References ................................................................................................... 23
II. COMPUTATIONAL STUDIES OF SIGMATROPIC REARRANGEMENT OF ALLYLIC
AND VINYLOGOUS AZIDES ................................................................................... 30
2.1 Azide Chemistry ......................................................................................... 30
2.2 Sigmatropic Rearrangement of Allylic Azide ............................................ 35
2.3 Computational Method ............................................................................... 40
2.4 Results and Discussion ............................................................................... 41
2.4.1 Study of [3, 3] Sigmatropic Rearrangement of Allylic Azide ............ 41
2.4.2 Study of [3, 5] Sigmatropic Rearrangement of Vinylogous
(pentadienyl) Azide ........................................................................... 58
2.5 Conclusion .................................................................................................. 73
2.6 References .................................................................................................. 74
III. SYNTHESIS OF PENTADIENYL ALCOHOL DERIVATIVES AND STUDY OF THEIR
POSSIBLE [3, 3] AND [3, 5] SIGMATROPIC REARRANGEMENTS ........................... 82
3.1 Rearrangement of Esters ............................................................................. 82
3.1.1 Background .......................................................................................... 82
Page 6
Texas Tech University, Deepali Butani, August 2011
v
3.1.2 Computational Study on Rearrangement of Esters .............................. 84
3.1.3 Proposed alcohol molecules ................................................................. 88
3.1.4 Proposed synthesis of phenyl(2-vinylcyclopent-1-enyl)methyl
acetate .................................................................................................. 91
3.1.5 Proposed synthesis of phenyl(2-vinylcyclohex-1-enyl)methyl
acetate .................................................................................................. 95
3.1.6 Flash Vacuum Pyrolysis ....................................................................... 99
3.1.7 Result and Discussion ........................................................................ 103
3.2 Rearrangement of Trichloroacetimidates ................................................... 109
3.2.1 Background ........................................................................................ 109
3.2.2 Proposed synthesis of phenyl(2-vinylcyclopent-1-enyl)methyl 2,2,2-
trichloroacetimidate.............................................................................. 112
3.2.3 Proposed synthesis of phenyl(2-vinylcyclohex-1-enyl)methyl 2,2,2-
trichloroacetimidate ............................................................................ 115
3.2.4. Results and Discussion ....................................................................... 118
3.3 Rearrangement of Xanthates ....................................................................... 119
3.3.1 Background .......................................................................................... 119
3.3.2 Proposed synthesis of S-methyl O-phenyl(2-vinylcyclopent-1-enyl)methyl
carbonodithioate ................................................................................... 121
3.3.3 Proposed synthesis of S-methyl O-phenyl(2-vinylcyclohex-1-enyl)methyl
carbonodithioate ................................................................................... 123
3.3.4. Results and Discussion ......................................................................... 126
3.4 Conclusion .................................................................................................. 127
3.5 Experimental Section ................................................................................... 128
3.6 References .................................................................................................... 155
APPENDICES ........................................................................................................ 161
A. OPTIMIZED CARTESIAN COORDINATES FOR THE
THEORETICAL CALCULATIONS ............................................................. 161
B. 1H-NMR,
13C-NMR, HMQC AND COSY SPECTRA ............................. 196
Page 7
Texas Tech University, Deepali Butani, August 2011
vi
ABSTRACT
Sigmatropic rearrangements have been known over 100 years. For almost 60
years the [3, 3] sigmatropic rearrangement was known only for the Claisen (one
oxygen present) and the Cope (all carbons) rearrangements, which constituted an
exceptionally versatile class of bond organization processes with many obvious
applications in organic synthesis. More recently, other types of [3, 3] sigmatropic
rearrangements have been studied. Even though [3, 3] sigmatropic rearrangements
have been known for quite a long period of time, there has been very little study done
on [3, 5] sigmatropic rearrangements. In this study, the main aim is to study various
types of reactions undergoing [3, 3] and [3, 5] sigmatropic rearrangements and classify
them as pericyclic or pseudopericyclic.
In the first part of this dissertation, computational studies were carried on allyl
azide and the vinylogous azide to study possible sigmatropic rearrangements of them.
There has been numerous syntheses using [3, 3] rearrangements of allylic azides but
there were not any previous computational studies. Computational studies using
Gaussian 03 at the RB3LYP/6-31G(d,p) level of theory for [3,3] sigmatropic
rearrangements of allylic azides as well as different possible conformers of allyl azide
were calculated. At this level, the activation energy barrier was predicted to be 23.1
kcal/mol and a rate constant of 1.09E-05 s-1
was calculated. Also, as the rearrangement
proceeded through six-centered transition state, the geometry of which suggests that
the reaction is pericyclic in nature. Similar studies were done for the sigmatropic
Page 8
Texas Tech University, Deepali Butani, August 2011
vii
rearrangement of the vinylogous azide. The vinylogous azide was proposed to have to
two possible thermal pathways, [3, 3] and [3, 5] sigmatropic rearrangements. The
studies clearly showed the formation of eight-centered transition state, with an orbital
disconnection on the azide, indicating it is pseudopericyclic. However, the activation
energy for the [3, 5] rearrangement was calculated to be 42.4 kcal/mol with rate
constant of 7.30E-20 s-1
, which is much higher than the competing [3, 3]
rearrangement, 15.2 kcal/mol.
In the second part of this dissertation, we designed suitable alcohol molecules,
to prepare acetate, trichloroacetimidate and methyl carbonodithioate derivatives, in
such a manner that they could have six- and eight- member transition structures to
possibly see [3, 3] and [3, 5] sigmatropic rearrangements. The reason to synthesize
such molecule was to test the earlier prediction of [3, 3] and [3, 5] rearrangement of
esters. Earlier computational studies done by Birney’s group showed that for a [3, 5]
rearrangement to be allowed the distance between reactive centers need to be close
enough for rearrangement to occur. Preliminary results suggest [3, 3] or [3, 5]
sigmatropic rearrangements can be observed in different molecules.
Page 9
Texas Tech University, Deepali Butani, August 2011
viii
LIST OF TABLES
2.1 The calculated absolute energies (Hartree), zero-point energies (ZPE, kcal/mol),
relative energies (kcal/mol with respect to ground state), relative energies with
zero-point energy correction (kcal/mol) of different models at different
constrained C-N bond length (Å) at the RB3LYP/6-31G(d,p) level of theory ..... 42
2.2 Dihedral angles (C-C-C-N and C-C-N-N) of ground state, transition state and
different conformers using RB3LYP/6-31G(d,p) level of theory ......................... 44
2.3 The calculated absolute energies (AE, Hartree), dipole moment (DM, Debye), low
or imaginary frequencies (LF, cm-1
), zero-point energies (ZPE, kcal/mol),
absolute energies with zero-point energy correction (AE+ZPE, kcal/mol) and
relative energies with zero-point energy correction (RE, kcal/mol) for the
stationary points of the [3, 3] sigmatropic rearrangement reaction of allyl azide at
the RB3LYP/6-31G(d,p) level of theory ............................................................... 45
2.4 The calculated absolute energies (AE, Hartree), dipole moment (DM, Debye), low
or imaginary frequencies (LF, cm-1
), zero-point energies (ZPE, kcal/mol),
absolute energies with zero-point energy correction (AE+ZPE, kcal/mol) and
relative energies with zero-point energy correction (RE, kcal/mol) for the
stationary points of the [3, 3] sigmatropic rearrangement reaction of allyl azide at
the RHF/6-31G(d,p) level of theory ..................................................................... 46
2.5 The calculated absolute energies (AE, Hartree), dipole moment (DM, Debye), low
or imaginary frequencies (LF, cm-1
), zero-point energies (ZPE, kcal/mol),
Page 10
Texas Tech University, Deepali Butani, August 2011
ix
absolute energies with zero-point energy correction (AE+ZPE, kcal/mol) and
relative energies with zero-point energy correction (RE, kcal/mol) for the
stationary points of the [3, 3] sigmatropic rearrangement reaction of allyl azide at
the RMP2/6-31G(d,p) level of theory................................................................... 47
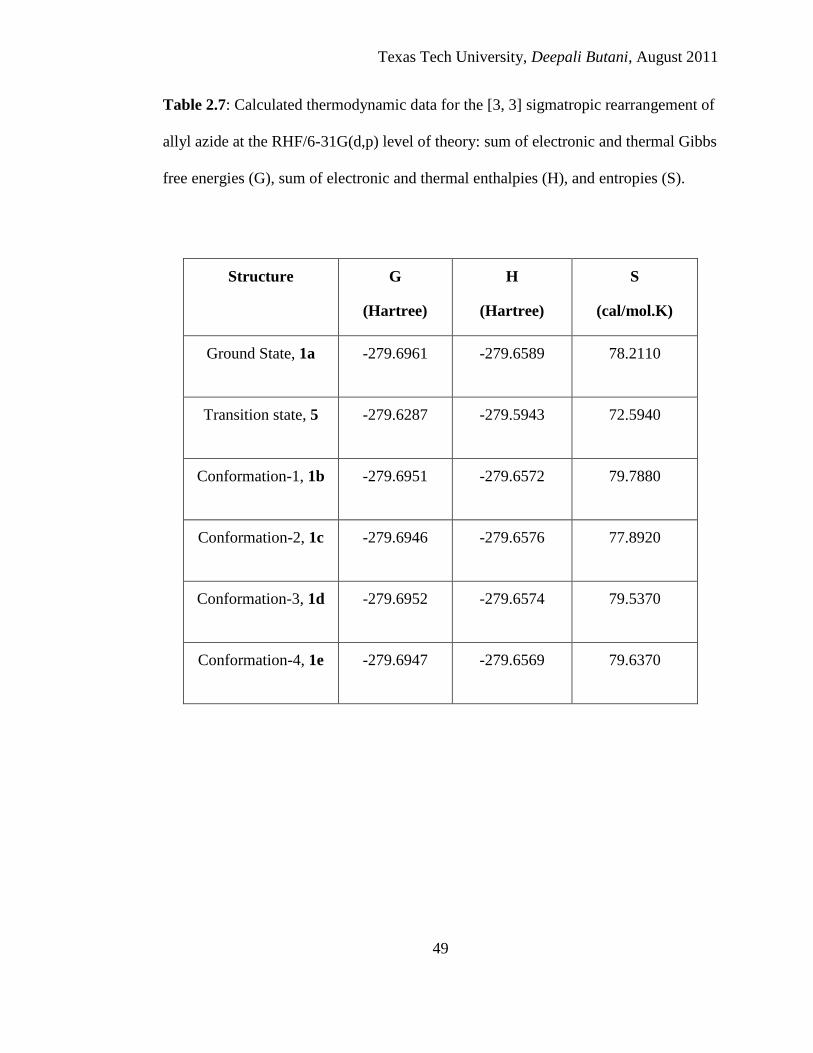
2.6 Calculated thermodynamic data for the [3, 3] sigmatropic rearrangement of allyl
azide at the RB3LYP/6-31G(d,p) level of theory: sum of electronic and thermal
Gibbs free energies (G), sum of electronic and thermal enthalpies (H), and
entropies (S) ........................................................................................................... 48
2.7 Calculated thermodynamic data for the [3, 3] sigmatropic rearrangement of allyl
azide at the RHF/6-31G(d,p) level of theory: sum of electronic and thermal Gibbs
free energies (G), sum of electronic and thermal enthalpies (H), and entropies
(S) .......................................................................................................................... 49
2.8 Calculated thermodynamic data for the [3, 3] sigmatropic rearrangement of allyl
azide at the RMP2/6-31G(d,p) level of theory: sum of electronic and thermal
Gibbs free energies (G), sum of electronic and thermal enthalpies (H), and
entropies (S) ............................................................................................................ 50
2.9 Activation parameters for the [3, 3] sigmatropic rearrangement of allyl azide at
three different level of theory using 6-31G(d.p) basis set using ground state
conformation (1a) as reference. (Gibbs activation free energy, (ΔG≠, kcal/mol),
Enthalpies of activation, (ΔH≠, kcal/mol), Entropy of activation, (ΔS
≠, cal/mol.K),
Activation energy, (Ea, kcal/mol) and Rate constant, (k, s-1
)) ................................ 51
Page 11
Texas Tech University, Deepali Butani, August 2011
x
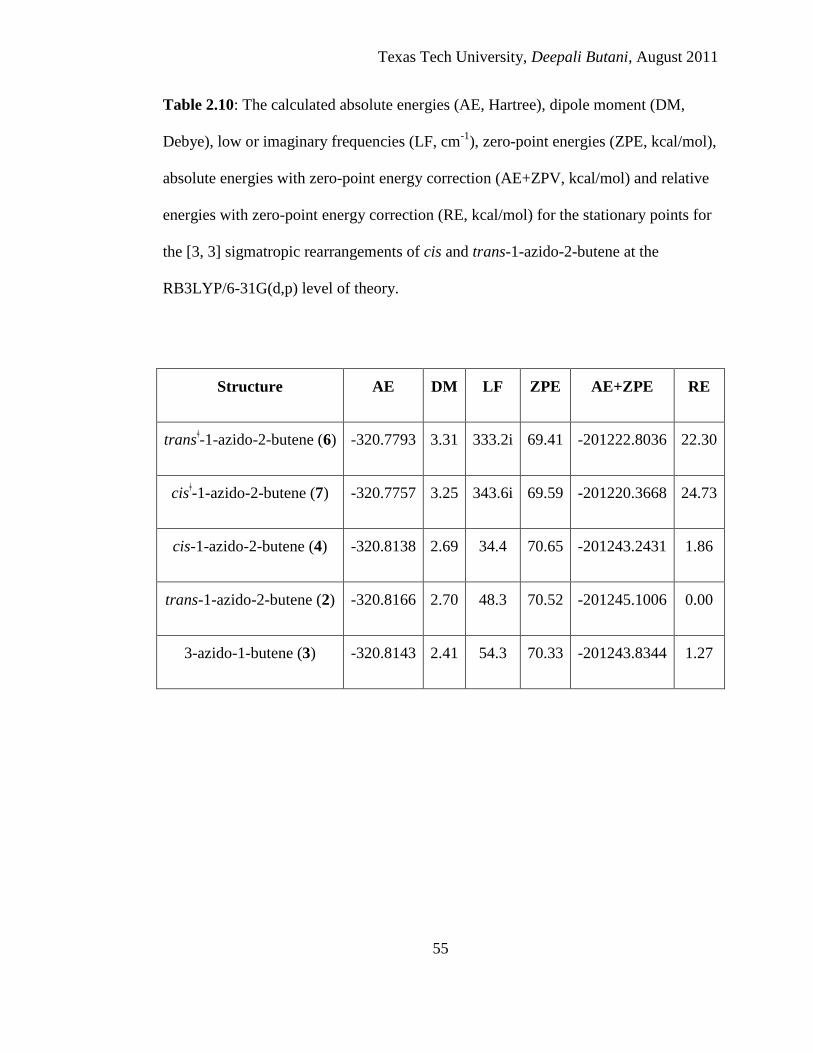
2.10 The calculated absolute energies (AE, Hartree), dipole moment (DM, Debye),
low or imaginary frequencies (LF, cm-1
), zero-point energies (ZPE, kcal/mol),
absolute energies with zero-point energy correction (AE+ZPV, kcal/mol) and
relative energies with zero-point energy correction (RE, kcal/mol) for the
stationary points for the [3, 3] sigmatropic rearrangements of cis and trans-1-
azido-2-butene at the RB3LYP/6-31G(d,p) level of theory .................................... 55
2.11 Calculated thermodynamic data for the [3, 3] sigmatropic rearrangements of cis
and trans-1-azido-2-butene at the RB3LYP/6-31G(d,p) level of theory: sum of
electronic and thermal Gibbs free energies (G), sum of electronic and thermal
enthalpies (H), and entropies (S) ............................................................................. 56
2.12 Activation parameters for the [3, 3] sigmatropic rearrangements of trans-1-azido-
2-butene (2) and cis-1-azido-2-butene (4) from 3-azido-1-butene (3) at RB3LYP/6-
31G(d.p) level of theory using thermodynamic parameters from Table 2.11. (Gibbs
activation free energy, (ΔG≠, kcal/mol), Enthalpies of activation, (ΔH
≠, kcal/mol),
Entropy of activation, (ΔS≠, cal/mol.K), Activation energy, (Ea, kcal/mol) and Rate
constant, (k, s-1
)) ...................................................................................................... 56
2.13 The calculated absolute energies (AE, Hartree), dipole moment (DM, Debye),
low or imaginary frequencies (LF, cm-1
), zero-point energies (ZPE, kcal/mol),
absolute energies with zero-point energy correction (AE+ZPE, kcal/mol) and
relative energies with zero-point energy correction (RE, kcal/mol) for the
Page 12
Texas Tech University, Deepali Butani, August 2011
xi
stationary points of the [3, 5] and [3, 3] sigmatropic rearrangements of the
vinylogous azide at the RB3LYP/6-31G(d,p) level of theory ................................ 62
2.14 The calculated absolute energies (AE, Hartree), dipole moment (DM, Debye),
low or imaginary frequencies (LF, cm-1
), zero-point energies (ZPE, kcal/mol),
absolute energies with zero-point energy correction (AE+ZPE, kcal/mol) and
relative energies with zero-point energy correction (RE, kcal/mol) for the
stationary points of the [3, 5] and [3, 3] sigmatropic rearrangements of the
vinylogous azide at the RHF/6- 31G(d,p) level of theory ....................................... 63
2.15 The calculated absolute energies (AE, Hartree), dipole moment (DM, Debye),
low or imaginary frequencies (LF, cm-1
), zero-point energies (ZPE, kcal/mol),
absolute energies with zero-point energy correction (AE+ZPE, kcal/mol) and
relative energies with zero-point energy correction (RE, kcal/mol) for the
stationary points of the [3, 5] and [3, 3] sigmatropic rearrangements of the
vinylogous azide at the RMP2/6-31G(d,p) level of theory ..................................... 64
2.16 Calculated thermodynamic data for the [3, 5] and [3, 3] sigmatropic
rearrangements of the vinylogous azide at the RB3LYP/6-31G(d,p) level of theory:
sum of electronic and thermal Gibbs free energies (G), sum of electronic and
thermal enthalpies (H), and entropies (S) ............................................................... 65
2.17 Calculated thermodynamic data for the [3, 5] and [3, 3] sigmatropic
rearrangements of the vinylogous azide at the RHF/6-31G(d,p) level of theory:
Page 13
Texas Tech University, Deepali Butani, August 2011
xii
sum of electronic and thermal Gibbs free energies (G), sum of electronic and
thermal enthalpies (H), and entropies (S) ............................................................... 66
2.18 Calculated thermodynamic data for the [3, 5] and [3, 3] sigmatropic
rearrangements of the vinylogous azide at the RMP2/6-31G(d,p) level of theory:
sum of electronic and thermal Gibbs free energies (G), sum of electronic and
thermal enthalpies (H), and entropies (S) ............................................................... 67
2.19 Activation parameters for the [3, 5] and [3, 3] sigmatropic rearrangements of
vinylogous azide 16 and 19 from 15 at three different levels of theory using 6-
31G(d,p) basis set. (Gibbs activation free energy, (ΔG≠, kcal/mol), Enthalpies of
activation, (ΔH≠, kcal/mol), Entropy of activation, (ΔS
≠, cal/mol.K), Activation
energy, (Ea, kcal/mol) and Rate constant, (k, s-1
)) .................................................. 69
2.20 Dihedral angles of various structures of vinylogous azide rearrangements using
RB3LYP/6-31G(d,p) level of theory....................................................................... 70
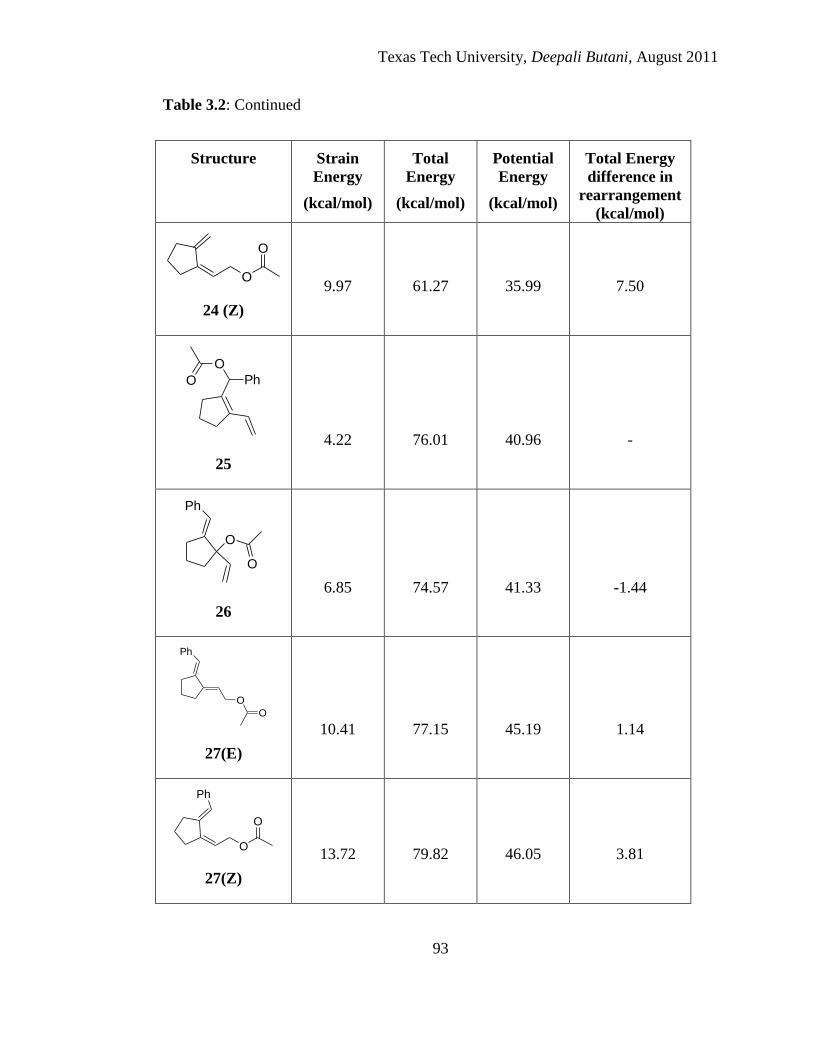
3.1 Calculated strain energy, total energy and potential energy of minimized structure
of a series of pentadienyl alcohols using MM2 ............................................................ 89
3.2 Calculated strain energy, total energy and potential energy of minimized structures
of various possible acetates and their possible [3, 3] and [3, 5] rearranged products
using MM2 .................................................................................................................... 91
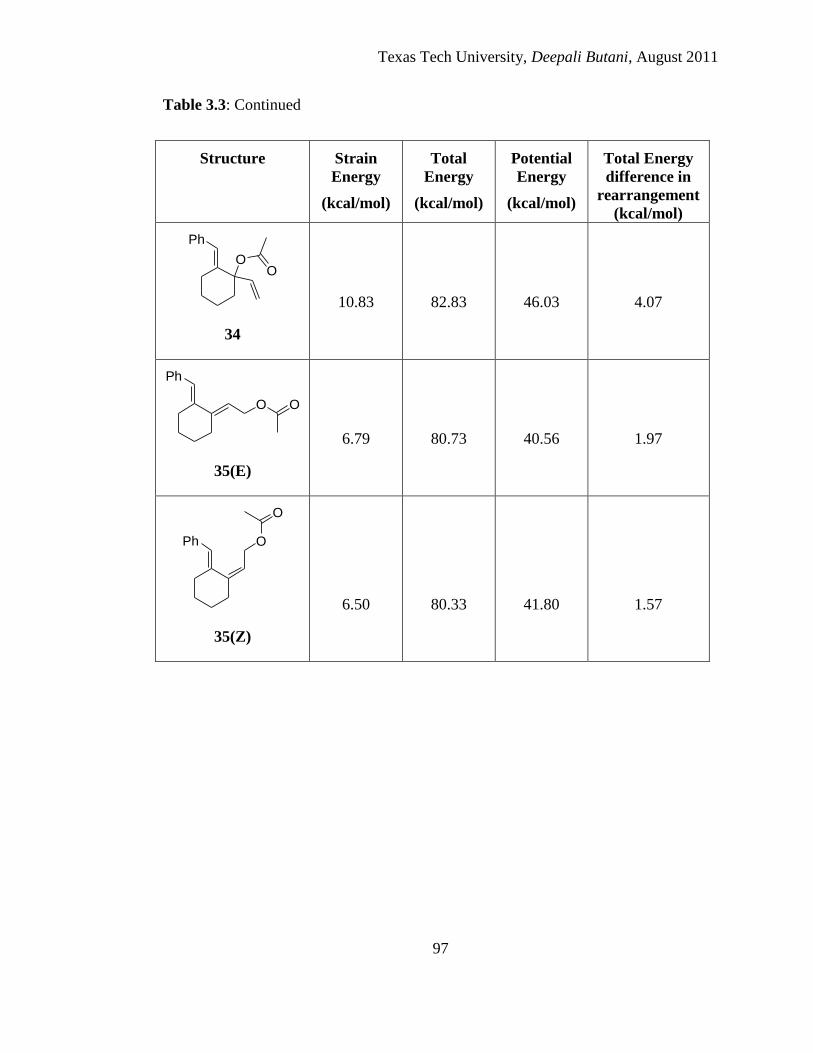
3.3 Calculated strain energy, total energy and potential energy of minimized structures
of various possible acetates and their possible [3, 3] and [3, 5] rearranged products
using MM2 .................................................................................................................... 96
Page 14
Texas Tech University, Deepali Butani, August 2011
xiii
3.4 Flash Vacuum Pyrolysis (FVP) experimental setup ............................................. 102
3.5 Products formed on pyrolysis of phenyl(2-vinylcyclopent-1-enyl)methyl acetate
(25) at two different temperatures ............................................................................... 104
3.6 Products formed on pyrolysis of phenyl (2-vinylcyclohex-1-enyl) methyl acetate
(33) at three different temperatures ............................................................................. 107
3.7 Calculated strain energy, total energy and potential energy of minimized structures
of trichloroacetimidate, 41 and their possible [3, 3], (42) and [3, 5], (43) rearranged
products using MM2 ................................................................................................... 114
3.8 Calculated strain energy, total energy and potential energy of minimized structures
of trichloroacetimidate, 44 and their possible [3, 3], (45) and [3, 5], (46)
rearrangement products using MM2 ........................................................................... 117
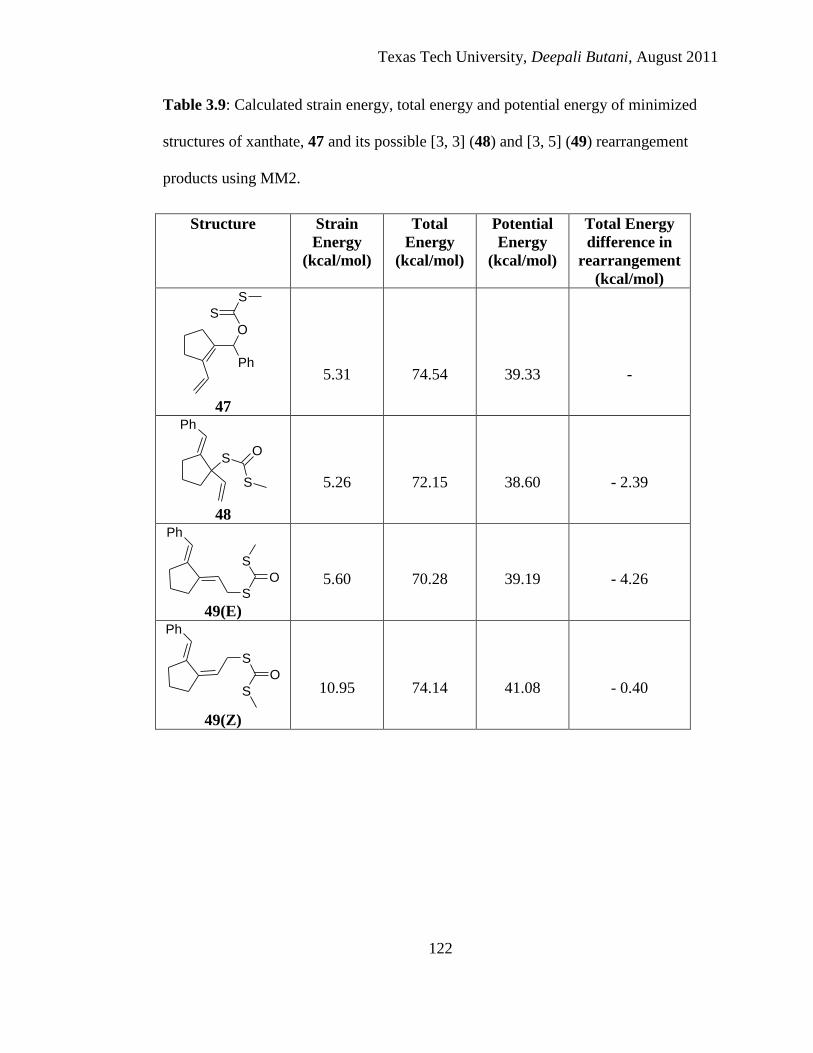
3.9 Calculated strain energy, total energy and potential energy of minimized structures
of xanthate, 47 and its possible [3, 3] (48) and [3, 5] (49) rearrangement products
using MM2 .................................................................................................................. 122
3.10 Calculated strain energy, total energy and potential energy of minimized
structures of xanthate, 50 and its possible [3, 3], (51) and [3, 5], (52) rearrangement
products using MM2 ................................................................................................... 125
Page 15
Texas Tech University, Deepali Butani, August 2011
xiv
LIST OF FIGURES
1.1 Complete orbital symmetry correlation diagram for the formation of cyclobutane
from two molecules of ethylene ................................................................................ 2
1.2 Orbital symmetry correlation diagram for the allowed conrotatory ring opening of
3,4-dimethylcyclobutene. .......................................................................................... 3
1.3 Orbital correlation diagram for the allowed disrotatory ring opening of 5,6-
dimethylcyclohexa-1,3-diene .................................................................................... 4
1.4 [3, 3] Sigmatropic rearrangement of octa-2,6-diene ................................................. 5
1.5 Example of Group Transfer reaction between an alkene and enophile .................... 5
1.6 (a) Addition of sulfur dioxide to butadiene; (b) Thermal cheletropic
decarbonylation of 3-cyclopentenone ....................................................................... 6
1.7 Dyotropic reactions (a) Type 1 reaction (b) Type 2 reaction .................................... 7
1.8 Degenerate rearrangement of PFDTSO .................................................................... 8
1.9 Proposed pseudopericyclic orbital interaction in the rearrangement of PFDTSO .... 8
1.10 Prototropy in internally hydrogen bonded enols of β-dicarbonyl compounds ........ 9
1.11 Orbitals and their interactions in the pseudopericyclic reaction of the addition of
the water to formylketene........................................................................................ 11
1.12 Orbital interactions in the pseudopericyclic decarbonylation of transition state of
furandione ............................................................................................................... 12
1.13 Examples of low or no barrier for pseudopericyclic reactions. ............................ 14
1.14 [1, 3] shift in the bicyclo[3.2.0] hept-ene system .................................................. 16
Page 16
Texas Tech University, Deepali Butani, August 2011
xv
1.15 Possible pathways for concerted [1, 3] migration ................................................. 17
1.16 Antara-antara [1, 5] methylene sigmatropic shift in propenylidene
cyclopropane ........................................................................................................... 18
1.17 1, 7] sigmatropic rearrangement of in o-butadienylphenols where it has one
orbital disconnection ............................................................................................... 19
1.18 General mechanism of [2, 3]-sigmatropic rearrangement..................................... 19
1.19 [2, 3]-sigmatropic rearrangement of benzyl allyl ether ......................................... 20
1.20 (a) Claisen and Cope rearrangements, (b) Transition state of the [3, 3] Claisen
rearrangement showing effects of stereochemistry, (c) Transition state of the [3, 3]
Cope rearrangement showing effects of stereochemistry ....................................... 21
1.21 [5, 5] shift of phenyl pentadienyl ether ................................................................. 22
2.1 Representative resonance structures of azides ........................................................ 31
2.2 Products from unimolecular decomposition of azides ............................................ 32
2.3 Nitrene products from azides .................................................................................. 32
2.4 Rearrangement products from azides ...................................................................... 32
2.5 Zwittazido cleavage of azides (a) general mechanisms, (b) a specific example ..... 33
2.6 Mechanism of acid-catalyzed decomposition ......................................................... 34
2.7 Mechanism of Staudinger Reaction ........................................................................ 34
2.8 Mechanism of Curtius Rearrangement.................................................................... 34
Page 17
Texas Tech University, Deepali Butani, August 2011
xvi
2.9 Reaction of Schmidt Rearrangement ...................................................................... 34
2.10 Reaction showing reduction .................................................................................. 35
2.11 Reaction showing Cycloaddition .......................................................................... 35
2.12 Mechanism showing nucleophilic attack at the azide terminus ............................ 35
2.13 [3, 3] Sigmatropic rearrangement of allyl azide (1) .............................................. 36
2.14 Rearrangement of an allylic azide ......................................................................... 37
2.15 Concerted rearrangement vs. SN2ʹ attack. Top: the expected SN2 pathway of
nucleophilic opening of epoxide, 1; middle: allylic azide rearrangement of 2
leading to 4-azido-2-buten-1-ol, 3; bottom: alternative SN2ʹ pathway leading to 4-
azido-2-buten-1-ol, 2 ............................................................................................... 38
2.16 Equilibrium between α- and γ-methylallyl azide .................................................. 38
2.17 Possible mechanistic alternatives for the allylic rearrangement ........................... 39
2.18 (a) [3, 3] Sigmatropic Rearrangement of allylic azide (1); (b) Molecular orbital
diagram of azide ...................................................................................................... 41
2.19 Energy profile showing relative energy and constrained C-N bond distance. This
helps in determining which structure should be taken to optimize at ground state
and transition state................................................................................................... 43
2.20 IRC calculation of transition state of the allylic azide using the RB3LYP/6-
31G(d,p) level of theory .......................................................................................... 44
Page 18
Texas Tech University, Deepali Butani, August 2011
xvii
2.21 Energy profile (RE from Table 2.3) of the [3,3] sigmatropic rearrangement of the
allyl azide and different conformers of the allyl azide at B3LYP/6-31G(d,p) level
of theory, where GS stands for ground state (1a), TS for transition state (5), C1 for
conformation1 (1b), C2 for conformation-2 (1c), C3 for conformation-3 (1d) and
C4 for conformation-4 (1e). Their relative energies in kcal/mol with respect to
ground state are provided in parentheses ................................................................ 52
2.22 Three different views of the transition state of the [3, 3] sigmatropic
rearrangement of allyl azide. Bond Lengths: C(6)-N(1) = 2.07 Å, C(4)-N(3) = 2.07
Å; Bond Angles: N(1)-N(2)-N(3) = 163.13°, C(4)-C(5)-C(6) = 120.39° ............... 53
2.23 Energy profile (RE from Table 2.11) for the [3, 3] sigmatropic rearrangements of
cis and trans-1-azido-2-butene at the RB3LYP/6-31G(d,p) level of theory, where
trans-1-azido-2-butene (2), 3-azido-1-butene (3), cis-1-azido-2-butene (4), trans-
transition state (6) and cis-transition state (7). Their relative energies in kcal/mol
with respect to 2 are provided in parentheses ......................................................... 57
2.24 Structure of trans-transition state (6). Bond Lengths: C(6)-N(1) = 2.11 Å, C(4)-
N(3) = 2.13 Å; Bond Angles: N(1)-N(2)-N(3) = 164.6°, C(4)-C(5)-C(6) = 121.3°,
C(5)-C(6)-C(7) = 123.1°, N(1)-C(6)-C(7) = 153.6° ............................................... 57
2.25 Structure of cis-transition state (7). Bond Lengths: C(6)-N(1) = 2.13 Å, C(4)-N(3)
= 2.11 Å; Bond Angles: N(1)-N(2)-N(3) = 164.5°, C(4)-C(5)-C(6) = 122.9°, C(5)-
C(6)-C(7) = 125.2°, N(1)-C(6)-C(7) = 97.0° .......................................................... 58
2.26 The [3,3] and [3,5] sigmatropic rearrangement of vinylogous azide (8) .............. 59
Page 19
Texas Tech University, Deepali Butani, August 2011
xviii
2.27 IRC run for transition state-1 (11): (a) IRC run in forward direction towards
possible product and (b) IRC run in reverse direction towards possible reactant
using RB3LYP/6-31G(d,p) level of theory. ............................................................ 60
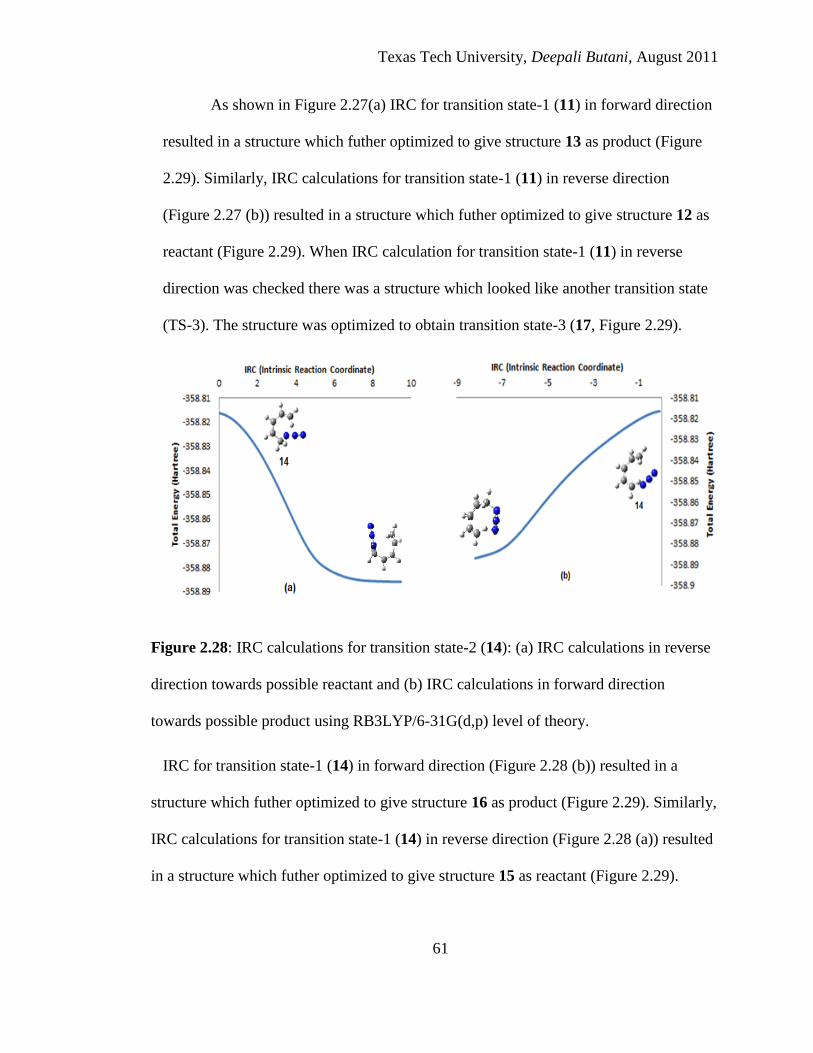
2.28 IRC calculations for transition state-2 (14): (a) IRC calculations in reverse
direction towards possible reactant and (b) IRC calculations in forward direction
towards possible product using RB3LYP/6-31G(d,p) level of theory .................... 61
2.29 Energy profile (RE from Table 2.13) for the [3, 5] and [3, 3] sigmatropic
rearrangements of vinylogous azide at the RB3LYP/6-31G(d,p) level of theory.
Their relative energies in kcal/mol with respect to 16 are provided in
parentheses .............................................................................................................. 68
2.30 Three different views of 11 using RB3LYP/6-31G(d,p) level of theory. Bond
Lengths: N(1)-C(8) = 1.69 Å, N(3)-C(4) = 1.48 Å; Bond Angles: N(1)-N(2)-N(3)
= 140.8°, N(2)-N(3)-C(4) = 121.7°, N(3)-C(4)-C(5) = 109.7°, C(6)-C(7)-C(8) =
129.3°; Dihedral angle: C(8)-N(1)-N(3)-C(4) = 25.7° ............................................ 71
2.31 Three different views of 14 using RB3LYP/6-31G(d,p) level of theory. Bond
Lengths: N(1)-C(8) = 2.47 Å, N(3)-C(4) = 2.67 Å; Bond Angles: N(1)-N(2)-N(3)
= 174.2°, C(4)-C(5)-C(6) = 128.4°, C(6)-C(7)-C(8) = 126.2°; Dihedral angle:
C(8)-N(1)-N(3)-C(4) = -59.4° ................................................................................. 71
2.32 Three different views of 17 using RB3LYP/6-31G(d,p) level of theory. Bond
Lengths: N(1)-C(8) = 1.79 Å, N(3)-C(4) = 1.44 Å; Bond Angles: N(1)-N(2)-N(3)
Page 20
Texas Tech University, Deepali Butani, August 2011
xix
= 123.1°, N(2)-N(3)-C(4) = 101.9° C(4)-C(5)-C(6) = 61.4°, C(6)-C(7)-C(8) =
126.1°; Dihedral angle: C(8)-N(1)-N(3)-C(4) = -53.1° .......................................... 72
2.33 Three different views of 18 using RB3LYP/6-31G(d,p) level of theory. Bond
Lengths: N(1)-C(6) = 2.13 Å, N(3)-C(4) = 2.28 Å; Bond Angles: N(1)-N(2)-N(3)
= 167.9°, N(2)-N(3)-C(4) = 93.9° C(4)-C(5)-C(6) = 122.1°, C(6)-C(7)-C(8) =
123.4°. Dihedral angle: C(6)-N(1)-N(3)-C(4) = -6.82° .......................................... 72
3.1 Energy profile showing the [3, 3] rearrangement of 2,4-cyclohexadienyl formate,
10 to 11 (right side) and the degenerate [3, 5] rearrangement of 2,4-
cyclohexadienyl formate, 10 (left side), with their transition states (10ǂ and 11
ǂ) in
between. The geometries were calculated at the MP2/6-31G** level of theory and
the relative energies were calculated at the MP4/6-31G** + ZPV (kcal/mol) level
of theory. ................................................................................................................. 86
3.2 Energy profile showing 12 forming a boat transition state leading to the [3, 3]
rearrangement of 13. The relative energies in kcal/mol were calculated at the
MP4/6-31G** level of theory ................................................................................. 87
3.3 MM2 minimized structure of phenyl(2-vinylcyclohex-1-enyl)methanol (16, side
viewed from two different directions) where blue are H-atoms, grey are carbon
atoms, red are oxygen atoms and pink are lone pair orbitals .................................. 90
3.4 MM2 minimized structure of phenyl(2-vinylcyclopent-1-enyl)methanol (18, side
viewed from two different directions) where blue are H-atoms, grey are carbon
atoms, red are oxygen atoms and pink are lone pair orbitals .................................. 90
Page 21
Texas Tech University, Deepali Butani, August 2011
xx
3.5 MM2 minimized structure of phenyl(2-vinylcyclopent-1-enyl)methyl acetate (25)
where blue are H-atoms, grey are carbon atoms, red are oxygen atoms and pink are
lone pair orbitals ...................................................................................................... 94
3.6 MM2 minimized structure of [3, 3] (26) and [3, 5] (27) rearrangement products
from phenyl(2-vinylcyclopent-1-enyl)methyl acetate (25); where blue are H-
atoms, grey are carbon atoms, red are oxygen atoms and pink are lone pair
orbitals ...................................................................................................................... 94
3.7 MM2 minimized structure of phenyl(2-vinylcyclohex-1-enyl)methyl acetate (33)
where blue are H-atoms, grey are carbon atoms, red are oxygen atoms and pink are
lone pair orbitals ...................................................................................................... 98
3.8 MM2 minimized structure of [3, 3], 34 and [3, 5], 35 rearrangement products from
phenyl(2-vinylcyclohex-1-enyl)methyl acetate (33); where blue are H-atoms, grey
are carbon atoms, red are oxygen atoms and pink are lone pair orbitals ................ 98
3.9 Flash Vacuum Pyrolysis (FVP) setup ................................................................... 102
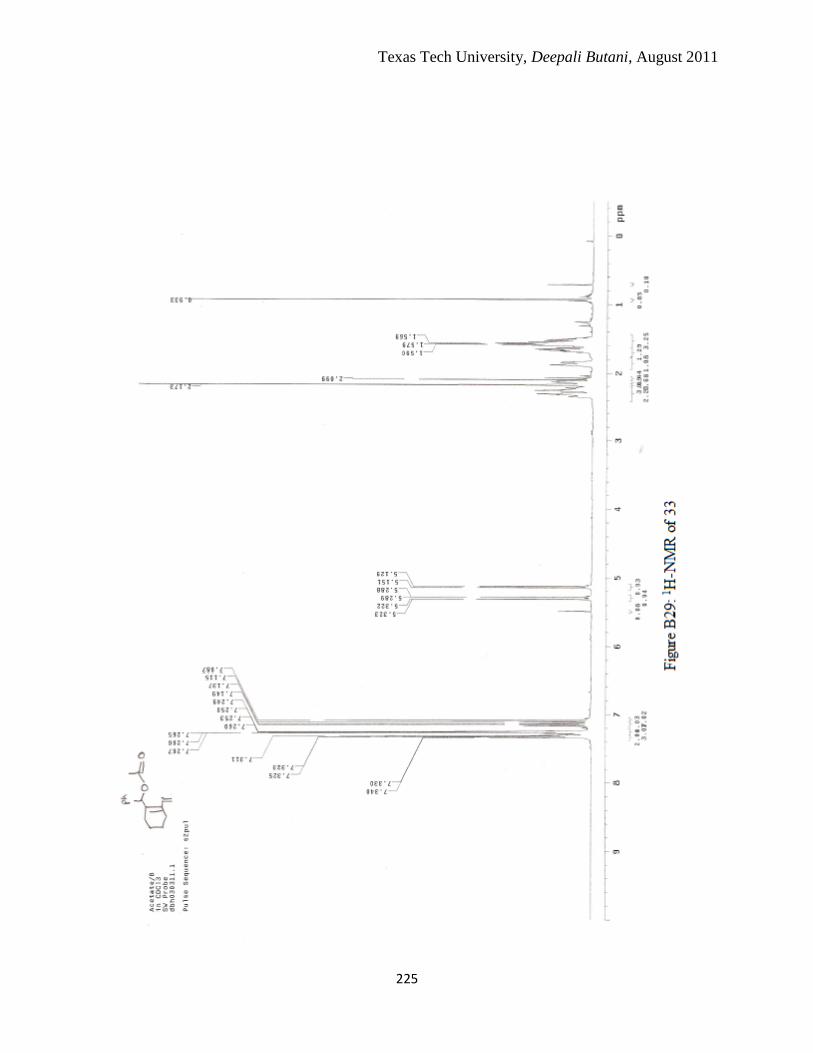
3.10 1H-NMR from FVP of 25, obtained column chromatography where boxed signals
are possibly from [3, 5] rearrangement ................................................................. 105
3.11 1H-NMR from FVP of 33, obtained column chromatography where boxed
signals are possibly from [3, 5] rearrangement ................................................... 108
3.12 Cyclic six-centered transition state of the [3, 3] rearrangement of allylic imidates
where R, R1, R2, R3, R4 are various alkyl groups ................................................ 111
Page 22
Texas Tech University, Deepali Butani, August 2011
xxi
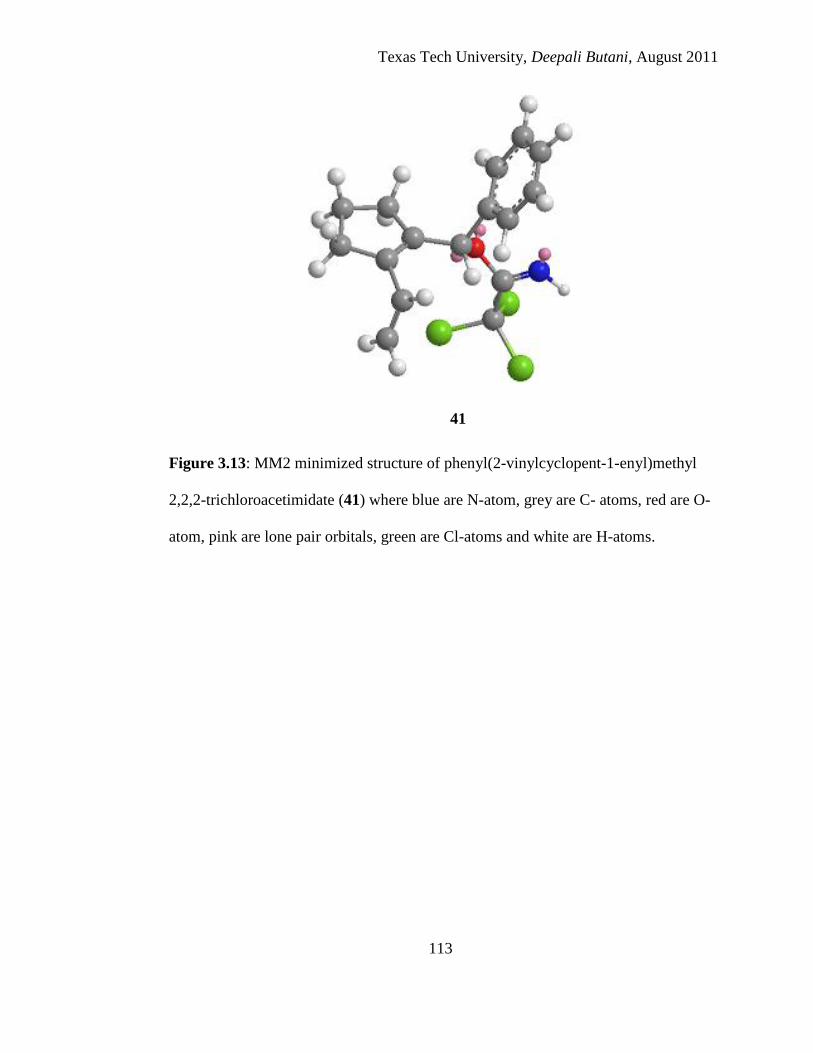
3.13 MM2 minimized structure of phenyl(2-vinylcyclopent-1-enyl)methyl 2,2,2-
trichloroacetimidate (41) where blue are N-atom, grey are C- atoms, red are O-
atom, pink are lone pair orbitals, green are Cl-atoms and white are H-atoms .... 113
3.14 MM2 minimized structure of phenyl(2-vinylcyclohex-1-enyl)methyl 2,2,2-
trichloroacetimidate (44) where blue are N-atom, grey are C- atoms, red are O-
atom, pink are lone pair orbitals, green are Cl-atoms and white are H-atoms .... 116
3.15 Energy profile showing the [3, 3] sigmatropic rearrangement of allylic xanthates
calculated at the MINDO/3 level of theory ......................................................... 120
3.16 Minimized structure of S-methyl O-phenyl (2-vinylcyclopent-1-enyl) methyl
carbonodithioate (47) where yellow are S-atom, grey are C- atoms, red are O-
atom, pink are lone pair orbitals, and white are H-atoms using MM2 ................ 121
3.17 Minimized structure of S-methyl O-phenyl(2-vinylcyclohex-1-enyl)methyl
carbonodithioate (50) where yellow are S-atom, grey are C- atoms, red are O-
atom, pink are lone pair orbitals, and white are H-atoms using MM2 level of
theory ................................................................................................................... 124
Page 23
Texas Tech University, Deepali Butani, August 2011
1
CHAPTER I
INTRODUCTION
1.1 PERICYCLIC REACTIONS
Pericyclic reactions represent an important class of concerted (single step)
processes involving σ- and π- systems. The fact that the reactions are concerted often
gives good stereochemical control of the product. By definition, pericyclic reactions
have a cyclic transition state. In the transition state, a concerted rearrangement of the
electrons takes place which causes σ- and π-bonds to simultaneously break and form.
Pericyclic reactivity can be understood in terms of frontier molecular orbital (FMO)
theory1 which can be explained by the favorable overlap of the Highest Occupied
Molecular Orbital (HOMO) and the Lowest Unoccupied Molecular Orbital (LUMO)
in the transition states. Alternatively, the outcome of reactions can be predicted by
considering the conservation of orbital symmetry. The conclusion of these analysis on
a variety of reactions are summarized in the Woodward-Hoffmann rules.2 These
reactions are popular with synthetic chemists because the reagents and conditions are
mild and the reactions are very “clean” unlike many organic chemical reactions that
results in the formation of large quantities of brown-black, smelly by-product of
unknown composition. Woodward and Hoffmann defined pericyclic reactions as ones
in which all the first order changes in bonding relationships take place in a concerted
closed curve.2 The following are six common types of pericyclic reactions. These
Page 24
Texas Tech University, Deepali Butani, August 2011
2
reactions can either be induced to occur under thermal conditions with simple heating
or under photochemical conditions.3,4,5
1. Cycloaddition: A cycloaddition is a pericyclic chemical reaction, in
which "two or more unsaturated molecules (or parts of the same molecule)
combine with the formation of a cyclic adduct in which there is a net
reduction of the bond multiplicity."6,7
Cycloaddition reactions can be
suprafacial / suprafacial (SS) or suprafacial/ antarafacial (AS). The orbital
correlation diagram for cycloaddition of two ethylene molecules to form
cyclobutane is shown in Figure 1.1 as a four-electron system, it is
thermally forbidden but photochemically allowed.
Figure 1.1: Complete orbital symmetry correlation diagram for the formation of
cyclobutane from two molecules of ethylene.2
Page 25
Texas Tech University, Deepali Butani, August 2011
3
2. Electrocyclic Reaction: An electrocyclic reaction is a type of pericyclic
rearrangement reaction where the net result is one π-bond being converted
into one σ- bond or vice-versa.7,8
The stereospecificity of the
rearrangement is determined by conrotatory or disrotatory mode of
transition state formation as predicted by the Woodward-Hoffmann rules.
The correlation diagram in Figure 1.2 shows how only a conrotatory ring
opening of 3,4-dimethylcyclobutene is symmetry allowed whereas the
correlation diagram in Figure 1.3 shows how only a disrotatory ring
opening of 5,6-dimethylcyclohexa-1,3-diene is symmetry allowed. This is
because only in these cases would maximum orbital overlap occur in the
transition state. Also, the product would be formed in the ground state
rather than an excited state.
Figure 1.2: Orbital symmetry correlation diagram for the allowed
conrotatory ring opening of 3,4-dimethylcyclobutene.9
Page 26
Texas Tech University, Deepali Butani, August 2011
4
Figure 1.3: Orbital correlation diagram for the allowed disrotatory ring
opening of 5,6-dimethylcyclohexa-1,3-diene.9
3. Sigmatropic Rearrangement: The sigmatropic rearrangement is a
pericyclic reaction wherein the net result is one σ-bond is changed to
another σ-bond in an uncatalyzed intramolecular process.10
In this type of
rearrangement reaction, a substituent moves from one part of a π-bonded
system to another part in an intramolecular reaction with simultaneous
rearrangement of the π system. In sigmatropic rearrangements the
Page 27
Texas Tech University, Deepali Butani, August 2011
5
transition state can be visualized as an association of two fragments
connected at their termini by two partial σ-bonds, one being broken and the
other being formed. Considering only atoms within the (real or
hypothetical) cyclic array undergoing reorganization, if the numbers of
these in the two fragments are designated i and j, then the rearrangement is
said to be a sigmatropic change of order [i, j] (conventionally [ i ] ≤ [ j ]).7
Figure 1.4 is an example showing the sigmatropic rearrangement of two
allyl fragments.11
Figure 1.4: [3, 3] Sigmatropic rearrangement of octa-2,6-diene.11
4. Group Transfer Reactions: Group transfer reactions are type of pericyclic
reactions where one π-bond is converted into one σ-bond, with migration
of σ- bond.12
Figure 1.5: Example of Group Transfer reaction between an alkene and
enophile. 12
Page 28
Texas Tech University, Deepali Butani, August 2011
6
5. Cheletropic reactions: Cheletropic reactions are pericyclic reactions which
involve formation of a transition state with a cyclic array of atoms and an
associated cyclic array of interacting orbitals. In other words, it is a
reorganization of σ and π bonds in a cyclic array.3
(a)
(b)
Figure 1.6: (a) Addition of sulfur dioxide to butadiene.3
(b) Thermal cheletropic decarbonylation of 3-cyclopentenone.88
6. Dyotropic reactions: A dyotropic reaction is a type of organic reaction and
more specifically a pericyclic valence isomerization in which two sigma
bonds simultaneously migrate intramolecularly.13
They can be either Type I
in which two migrating groups interchange their positions or Type II which
involves migration of new bonding sites without change of position (Figure
1.7)
Page 29
Texas Tech University, Deepali Butani, August 2011
7
Figure 1.7: Dyotropic reactions (a) Type 1 reaction (b) Type 2 reaction.13
1.2 PSEUDOPERICYCLIC REACTIONS
In 1976, Lemal and coworkers
14 were first to propose the name
“pseudopericyclic”. They proposed this name because they saw an extraordinarily
facile sigmatropic rearrangement for automerization while studying
perfluorotetramethyl Dewar thiophene exo-S-oxide (PFDTSO) by NMR at low
temperatures as shown in Figure 1.8. They found one signal at –100 °C (which could
be interpreted as structure c in Figure 1.8). Below –100 °C, the signal split into two,
corresponding to structure a (Figure 1.8). This led them to suggest that a rapid
exchange between the sulfoxide moiety and the rest of the molecule was taking place
in a degenerate rearrangement .
Page 30
Texas Tech University, Deepali Butani, August 2011
8
Figure 1.8: Degenerate rearrangement of PFDTSO.14
Since 1, 3-sigmatropic rearrangements of hydrocarbons are symmetry
forbidden, Lemal and coworkers
14 proposed what they termed a pseudopericyclic
mechanism. They suggested that the low activation energy (of exchange at -124 °C
was 6.8 ± 0.3 kcal/mol) of the reaction was due to a sulfur lone pair (i.e. nonbonding
orbital) forming a new bond to carbon, while the electrons from the cleavage of C-S
bond (i.e. orthogonal orbital) becoming a new lone pair as shown in Figure 1.9.14
This
avoided the cyclic orbital overlap of a pericyclic rearrangement.
Figure 1.9: Proposed pseudopericyclic orbital interaction in the rearrangement of
PFDTSO.14
Hence, Lemal14
defined the term “pseudopericyclic reactions” as a concerted
transformation whose primary changes in bonding compassed a cyclic array of atoms,
Page 31
Texas Tech University, Deepali Butani, August 2011
9
at one (or more) of which nonbonding and bonding atomic orbitals interchange roles.
In the crucial sense the role interchange meant that there was a “disconnection” in the
cyclic array of overlapping orbitals because the atomic orbitals switching functions
were mutually orthogonal. This means that pseudopericyclic reactions cannot be
orbital symmetry forbidden.
Prototropy in internally hydrogen bonded enols of β-dicarbonyl compounds
(Figure 1.10) is an example of pseudopericyclic reaction.15
As the proton tunnels
between minima, lone pair and bonding orbitals formally interchange functions at
both oxygens in the planar chelate ring. Basically, a bonding p orbital and a
nonbonding lone pair orbital switches roles at left-hand oxygen, while a
complementary interchange occurs at the oxygen on the right. Although the
bonding/nonbonding distinction was not absolute, the separation of this concerted
process into persistently orthogonal σ and π components proved the fact that it was
not pericyclic.15
Figure 1.10: Prototropy in internally hydrogen bonded enols of β-dicarbonyl
compounds.15
Nearly 20 years later Birney and coworkers16
began a systematic study of a
series of pseudopericyclic reactions based on quantitative theory, transition state
calculations and experiments on a variety of thermal pseudopericyclic reactions,
Page 32
Texas Tech University, Deepali Butani, August 2011
10
including cycloadditions,17,20,21,38-42
sigmatropic rearrangements,18,40,43,44
electrocyclizations,17-19
cheletropic fragmentations,16,45
and group
transfers/eliminations.46
Based on these studies, a series of generalizations have been
developed. These are summarized as the following16-24
:
1. A pseudopericyclic reaction may be orbital symmetry allowed via a
pathway that maintains the orbital disconnections, regardless of the
number of electrons involved.
In pericyclic reactions aromatic orbitals overlap to form a
cyclic system and this is implicit in the pattern of alternating
allowed and forbidden predictions by Woodward-Hoffmann rules
and Frontier Molecular Orbital theory. These theories are useful but
are not directly applicable to pseudopericyclic reactions. In
pseudopericyclic reactions there are two kinds of orbital
interactions: in-plane orbital overlaps and out-of-plane ones. Since
these orbital interactions lead to disconnections between orbitals of
reactants, counting electrons to predict whether the reaction is
allowed becomes irrelevant. Examples illustrating the disconnection
of orbitals are:
Page 33
Texas Tech University, Deepali Butani, August 2011
11
Orbital topology is the [4 + 2] cycloaddition of water to
formylketene as shown in Figure 1.11.
Figure 1.11: Orbitals and their interactions in the pseudopericyclic
reaction of the addition of the water to formylketene.17
For the decarbonylation of furandione (Figure 1.12) a
pseudopericyclic orbital topology is possible, with two orbital
disconnections, i.e. two atoms where orthogonal sets of orbitals meet,
but do not overlap. Because no electrons are exchanged between the in-
plane and out-of-plane orbitals, the transition state for decarbonylation
of furandione is orbital symmetry allowed when the CO departs in the
plane of the molecule.
Page 34
Texas Tech University, Deepali Butani, August 2011
12
Figure 1.12: Orbital interactions in the pseudopericyclic
decarbonylation of transition state of furandione.16
2. Barriers to pseudopericyclic reactions can be very low or even
nonexistent
(a) If there is a good match between nucleophilic and
electrophilic sites in reactants
(b) If the geometrical constraints of the system allow for
appropriate angles in transition state, in close analogy to
Baldwin’s rules.35
(c) If the reaction is exothermic.
Page 35
Texas Tech University, Deepali Butani, August 2011
13
Pericyclic reactions are often easy to perform due to maximum
bonding orbital overlap arising in the concerted allowed pathway.
But the barrier for activation energy is roughly between 20-30
kcal/mol,47,48
which is substantially high. It is because pericyclic
reactions involve the unavoidable electron-electron repulsion
between interacting orbitals from the aromatic of transition states.49
Whereas, in case of pseudopericyclic reactions, there can be very
low or sometimes no barrier. This is because there is a lack of
cyclic orbital overlap which avoids electron-electron repulsion. In
addition, the planar transition state allows for a better orbital
overlap. For instance, in the addition of water to formylketene the
low barrier of 6.3 kcal/mol, because the lone pair of electrons on
oxygen in water attacks the carbon of ketene in plane and hence
there is no overlap of the out-of-plane p orbitals. The reaction also
has a match between the nucleophilic site of lone pairs on oxygen
of water and the electrophilic site of carbonyl carbon on ketene and
also the nucleophilic oxygen on ketene and electrophilic hydrogen
on water. As the lone pair on oxygen attacks the carbonyl carbon
the negative charge is dispersed to oxygen on ketene. This results in
an increased nucleophilicity of the oxygen, which ultimately helps
in abstracting the hydrogen from water easily. As there is no
accumulation of charge and also the electron-electron repulsion is
Page 36
Texas Tech University, Deepali Butani, August 2011
14
minimal, these have lower energy barriers. More examples of low
barrier pseudopericyclic reactions are shown in Figure 1.13.48
Figure 1.13: Examples of low or no barrier for pseudopericyclic
reactions.48
3. Pseudopericyclic reactions have a planar transition states if possible.
However, crowding at transition states can lead to small distortions
from planarity. This is in contrast to typically all hydrocarbon
pseudopericyclic reactions for which the need to maintain orbital
overlap leads to non-planar transition states.36,37
In pericyclic reactions it is essential that orbitals are non-planar so as to
maximize their overlap in the transition state. In contrast, for pseudopericyclic
reactions there is no or little overlap between out of plane (π) and in-plane (σ and π)
orbitals.
Birney16-24,50
and several other authors25-34
have been working in field of
pseudopericyclic for several years to show that a number of organic syntheses involve
Page 37
Texas Tech University, Deepali Butani, August 2011
15
this type of process. However, until now there is no universally accepted clear-cut,
absolute criterion for distinguishing a pseudopericyclic reaction from a normal
pericyclic reaction.
Pericyclic and pseudopericyclic reactions can be distinguished from each other using
several methods:
1. Density Functional Theory (DFT) and many other computational methods
are used to determine the barrier of reaction and also the pathway of
reaction. Often the geometry of the transition state requires an orbital
disconnection.
2. The study of magnetic properties and their relation with aromaticity. As
pericyclic reactions proceed via an aromatic transition state whereas in
pseudopericyclic arrangement aromaticity is avoided at transition state.
Two commonly used methods are Nucleus Independent Chemical Shift
(NCIS)51,52
and Anisotrop of Chemical-Induced Density (ACID).53,54
3. Pseudopericyclic reactions can occur regardless of the number of atoms
involved. Therefore observing both a six-centered and an eight-centered
reaction would suggest both are pseudopericyclic.
1.3 SIGMATROPIC REARRANGEMENTS
Sigmatropic reactions55,56
of neutral molecules are of special interest, as they are
controlled by the conservation of orbital symmetry.3 These reactions involve
intramolecular migration of a group from one carbon to another, and an obvious
Page 38
Texas Tech University, Deepali Butani, August 2011
16
experiment is to compare the migratory aptitudes of various groups in these reactions
with those found in other rearrangements. The purposes of such a comparison are to
provide predictive power over the facility of sigmatropic reactions and to gain a more
detailed knowledge of the transition-state structure for group migrations. Thermal and
photochemical [i, j] sigmatropic rearrangement can occur either via suprafacial or
antarafacial pathways, and the resultant product has stereochemical consequences.57-60
Classes of Sigmatropic Rearrangement are:
1. [1, 3] shift: A [1, 3] shift involves the shift of one atom (or substituent, -H
or -R) down three atoms of a π system. The Woodward-Hoffmann rules
dictate that a thermal [1, 3] shift would proceed via an antarafacial shift.
Although such a shift is symmetry allowed, the Mobius topology required
in the transition state prohibits such a shift because it is geometrically
impossible. Berson and Nelson have described an example of [1,3] shift in
the bicyclo[3.2.0] hept-ene system (Figure 1.14) demonstrating that
inversion does occur for the suprafacial path but with a high barrier,
presumably because the reaction is forbidden.61,62
Figure 1.14: [1, 3] shift in the bicyclo[3.2.0] hept-ene system.61,62
Page 39
Texas Tech University, Deepali Butani, August 2011
17
If the migrating atom is capable of undergoing stereochemical
inversion, then the selection rules allow two distinct suprafacial or
antarafacial routes with attendant retention or inversion at the migrating
center (Figure 1.15).62
Figure 1.15: Possible pathways for concerted [1, 3] migration.62
2. [1, 5] shift: A [1, 5] shift involves the shift of 1 substituent (-H, -R or -Ar)
down 5 atoms of a π system.63
The methylene sigmatropic shift in
propenylidene cyclopropane (Figure 1.16) occurs by Antara-Antara [1,5]
methylene shift and is pericyclic in nature but if heteroatoms like N and O
Page 40
Texas Tech University, Deepali Butani, August 2011
18
are introduced at both reacting termini of parent molecule then they can
become pseudopericyclic in nature.64
Figure 1.16: Antara-antara [1, 5] methylene sigmatropic shift in
propenylidene cyclopropane.64
3. [1, 7] shift: Thermal [1, 7] sigmatropic shifts are predicted by the
Woodward-Hoffmann rules to proceed in an antarafacial fashion, via a
Möbius topology transition state. The 4n-π-electron series explores the
competition between the antarafacial mode being manifested via inversion
at a single carbon, or antarafacial along a chain of atoms. Thus a [1, 7]
hydrogen shift can only occur antarafacially.65,66
Migration of hydrogen from oxygen to carbon in o-butadienylphenols
(Figure 17) 67,68
involves a [1, 7] shift. The [1, 7] shift is also responsible
for cis-trans isomerism 2, and competes favorably with electrocyclic ring
closure to 3.
Page 41
Texas Tech University, Deepali Butani, August 2011
19
Figure 1.17: [1, 7] sigmatropic rearrangement of in o-butadienylphenols where
it has one orbital disconnection.67,68
4. [2, 3] shift: The general scheme of [2, 3] sigmatropic rearrangement is
shown in Figure 1.18.
Figure 1.18: General mechanism of [2, 3]-sigmatropic rearrangement.75
Atom Y in Figure 1.18 can be oxygen, sulfur, selenium, or nitrogen. If Y is
nitrogen, the reaction is referred to as a 2, 3-Stevens rearrangement; if Y is
oxygen, then it is called a 2, 3-Wittig rearrangement. Because the reaction
is concerted, it exhibits a high degree of stereocontrol, and can be
employed early in a synthetic route to establish stereochemistry. The Wittig
rearrangement requires strongly basic conditions, however, as a carbanion
intermediate is essential.69-74
[2, 3] sigmatropic rearrangement is defined as
a thermal isomerization that proceeds via a six-electron, five-membered
cyclic transition state. [2, 3]-sigmatropic rearrangement of benzyl allyl
ether (as shown in Figure 1.19)75
where the new bond formed has a 2,3-
relationship to the old and the transition state is a five-membered ring. The
Page 42
Texas Tech University, Deepali Butani, August 2011
20
transition state can be quite chair-like so that the new π bond will be trans
if it has a choice.
Figure 1.19: [2, 3]-sigmatropic rearrangement of benzyl allyl ether.75
5. [3, 3] shift: [3, 3] sigmatropic rearrangements76-79
are the one which
follow the Woodward-Hoffmann rules predicting that the reaction proceeds
suprafacially with six electrons and it forms a Hückel topology transition
state. The [3, 3] sigmatropic processes are characterized by the formation
of highly ordered transition states where repulsive interactions are
minimized. The Claisen80-83
and the Cope84-86
rearrangement are known as
reliable protocols to generate defined configured tertiary and quaternary
carbon centers as well as complicated C atom-heteroatom bonds. The [3,
3] rearrangement defines the product olefin geometry. Also, the
stereospecific bond reorganization of the reactant helps in the prediction of
stereogenic properties of the product as shown in Figure 1.20 (a, b and c).
Page 43
Texas Tech University, Deepali Butani, August 2011
21
Figure 1.20: (a) Claisen and Cope rearrangements. (b) Transition state of the [3,
3] Claisen rearrangement showing effects of stereochemistry. (c) Transition state
of the [3, 3] Cope rearrangement showing effects of stereochemistry.
Page 44
Texas Tech University, Deepali Butani, August 2011
22
6. [5, 5] shift: [5, 5] Sigmatropic shifts also proceed suprafacially by Hückel
topology transition state. An example of such a rearrangement is the [5, 5]
shift is seen in rearrangement of phenyl pentadienyl ether (Figure 1.21).89
Figure 1.21: [5, 5] shift of phenyl pentadienyl ether.89
There are different classes of sigmatropic rearrangements classified and most
of them are pericyclic in nature. Our aim throughout this dissertation will be study
potential [3, 5] sigmatropic rearrangements which we expect will pseudopericyclic in
nature.
Page 45
Texas Tech University, Deepali Butani, August 2011
23
1.4 REFERENCES
1. Fukui, K.; Yonezawa, T.; Shingu, H. J. Chem. Phys. 1952, 20, 722-
725.
2. Woodward, R. B.; Hoffmann, R. Angew. Chem. 1969, 81, 797.
3. Woodward, R. B.; Hoffmann, R. Angew. Chem. Int. Ed. Engl. 1967, 8,
781.
4. Hoffmann, R.; Woodward, R. B. Science. 1970, 167, 825.
5. Hoffmann, R.; Woodward, R. B. Acc. Chem. Res. 1968, 1, 17.
6. Hoffmann, R.; Woodward, R. B. J. Am. Chem. Soc. 1965, 87, 2046.
7. IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold
Book”) 1997.
8. Woodward, R. B.; Hoffmann, R. J. Am. Chem. Soc. 1965, 87, 395.
9. http://en.wikipedia.org/wiki/Electrocyclic_reaction, 2011.
10. Carey, F. A.; R. J. Sundberg. Advanced Organic Chemistry Part A.
11. http://en.wikipedia.org/wiki/Sigmatropic_reaction, 2011.
12. http://en.wikipedia.org/wiki/Group_transfer_reaction, 2011.
13. ern ndez, . oss o, . P. ierra, . . Chem. Rev. 2009, 109,
6687.
Page 46
Texas Tech University, Deepali Butani, August 2011
24
14. (a) Ross, J. A.; Seiders, R. P.; Lemal, D. M. J. Am. Chem. Soc. 1976,
98, 4325. (b) Bushweller, C. H.; Ross, J. A.; Lemal, D. M. J. Am.
Chem. Soc. 1977, 99, 629.
15. Alexander, E. C., Uliana, J. J. Am. Chem. Soc. 1976, 14, 4325.
16. Birney, D. M.; Ham, S.; Unruh, G. R. J. Am. Chem. Soc. 1997, 119,
4509.
17. Birney, D. M.; Wagenseller, P. E. J. Am. Chem. Soc. 1994, 116, 6262.
18. Birney, D. M. J. Org. Chem. 1996, 61, 243.
19. Birney, D. M. J. Am. Chem. Soc. 2000, 122, 10917.
20. Shumway, W.; Ham, S.; Moer, J.; Whittlesey, B. R.; Birney, D. M.
J.Org. Chem. 2000, 65, 7731.
21. Shumway, W.; Dalley, N. K.; Birney, D. M. J. Org. Chem. 2001, 66,
5832.
22. Zhou, C.; Birney, D. M. J. Am. Chem. Soc. 2002, 124, 5231.
23. Birney, D. M. Org. Lett. 2004, 6, 851.
24. Zhou, C.; Birney, D. M. J. Org. Chem. 2004, 69, 86.
25. Luo, L.; Bartberger, M. D.; Dolbier, W. R. J. J. Am. Chem. Soc. 1997,
119, 12366.
26. Fabian, W. M. F.; Bakulev, V. A.; Kappe, C. O. J. Org. Chem. 1998,
63, 5801.
27. Fabian, W. M. F.; Kappe, C. O.; Bakulev, V. A. J. Org. Chem. 2000,
65, 47.
Page 47
Texas Tech University, Deepali Butani, August 2011
25
28. Alajarin, M.; Vidal, A.; Sanchez-Andrada, P.; Tovar, F.; Ochoa, G.
Org. Lett. 2000, 2, 965.
29. Rauhut, G. J. Org. Chem. 2001, 66, 5444.
30. Chamorro, E. J. Chem. Phys. 2003, 118, 8687.
31. Finnerty, J. J.; Wentrup, C. J. Org. Chem. 2004, 69, 1909.
32. Zora, M. J. Org. Chem. 2004, 69, 1940.
33. Kalcher, J.; Fabian, W. M. F. Theor. Chem. Acc. 2003, 109, 195.
34. Chamorro, E.; Notario, R. J. Phys. Chem. A 2004, 108, 4099.
35. Baldwin, J. E. ; Thomas, R.C. ; Kruse, L. I. ; Silberman, L. ; J. Org.
Chem. 1977, 42, 3846.
36. Woodward, R. B.; Hoffmann, R. The Conservation of Orbital
Symmetry; Verlag Chemie, GmbH: Weinheim, 1970.
37. Houk, K. N.; Li, Y.; Evanseck, J. D. Angew. Chem., Int. Ed. Engl.
1992, 31, 682.
38. Ham, S.; Birney, D.M. Tetrahedron Lett. 1994, 35, 8113.
39. Wagenseller, P. E.; Birney, D. M.; Roy, D. J. Org. Chem. 1995, 60,
2853.
40. Ham, S.; Birney, D. M. J. Org. Chem. 1996, 61, 3962.
41. Bartsch, R. A.; Chae, Y. M.; Ham, S.; Birney, D. M. J. Am. Chem.
Soc. 2001, 123, 7479.
42. Matsui, H.; Zuckerman, E. J.; Katagiri, N.; Sugihara, T.; Kaneko, C.;
Ham, S.; Birney, D. M. J. Phys. Chem. A 1997, 101, 3936.
Page 48
Texas Tech University, Deepali Butani, August 2011
26
43. Birney, D. M.; Xu, X.; Ham, S. Angew. Chem., Int. Ed. 1999, 38, 189.
44. Quideau, S.; Looney, M. A.; Pouységu, L.; Ham, S.; Birney, D.M.
Tetrahedron Lett. 1999, 40, 615.
45. Chamorro, E. J. Chem. Phys. 2003, 118, 8687.
46. Ji, H.; Li, L.; Xu, X.; Ham, S.; Hammad, L.A.; Birney, D.M. J. Am.
Chem. Soc., 2009, 131, 528.
47. Martin, J. G.; Hill, R. K. Chem. Rev. 1961, 61, 537.
48. (a) Kwart, H.; King, K. Chem.Rev. 1968, 69, 415. (b) Zhou, C. PhD.
Dissertation, Texas Tech University 2004.
49. Houk, K. N.; Gandour, R. W.; Strozier, R. W.; Rondan, N. G.;
Paquette, L. A. J. Am. Chem. Soc. 1979, 101, 6797.
50. Birney, D. M. Current Org. Chem. 2010, 14, 1658.
51. Schleyer, P. V.; Maerker, C.; Dransfeld, A.; Jiao, H.J.; Hommes, N. J.
Am. Chem. Soc. 1996, 118, 6317.
52. Chen, Z. F.; Wannere, C.S.; Corminboeuf, C.; Puchta, R.; Schleyer, P.
V. Chem. Rev. 2005, 105, 3842.
53. Herges, R. Chem. Rev. 2006, 106, 4820.
54. Geuenich, D.; Hess, K.; Kohler, F.; Herges, R. Chem. Rev. 2005, 105,
3758.
55. Gill, G. B. Quarr. Rev. (London) 1968, 22, 338.
56. Miller, L. L.; Greisinger, R.; Boyer, R. F. J. Am. Chem. Soc. 1969, 91,
1578.
Page 49
Texas Tech University, Deepali Butani, August 2011
27
57. Berson, J. A.; Willcott, M. R. Rec. Chem. Prog. 1966, 27, 139.
58. Berson, J. A. Acc. Chem. Res. 1968, 1, 152.
59. Berson, J. A. Ibid. 1972, 5, 406.
60. Doering, W. von E.; Roth, W. R. Angew. Chem., Int. Ed. Engl. 1963,
2, 115.
61. (a) Berson, J. A.; Nelson, G. L. J. Am. Chem. Soc. 1967, 89, 5503. (b)
Berson, J. A.; Nelson, G. L. J. Am. Chem. Soc. 1970, 92, 1096.
62. Spangler, C.W. Chem. Rev. 1976, 76, 187.
63. Mironov, V. A.; Fedorovich, A. D.; Akhem, A. A. Russ. Chem. Rev.
1981, 50, 666.
64. Lopez, C. S.; Faza, O. N.; Souto, J. A.; Alvarez, R.; Lera, A. R. J.
Comp. Chem. 2007, 28, 1411.
65. Elnagar, H. Y.; Okamura, W. H. J. Org. Chem. 1988, 53, 3060.
66. Hoeger, C. A.; Okamura, W. H. J. Am. Chem. Soc. 1985, 107, 268.
67. Hug, R.; Hansen, H. J.; Schmid, H. Helv. Chim. Acta. 1972, 55, 1828.
68. Schweizer, E. E.; Crouse, D. M.; Dalrymple, D. L. J. Chem. Soc. D
1969, 354.
69. Nakai, I. Chem. Rev. 1986, 86, 885.
70. Mikami, K.; Nakai, T. Synthesis 1991, 594.
71. Nakai, T.; Mikami, K. Org. React. 1995, 46, 105.
72. Nakai, T.; Tomooka, K. Pure Appl. Chem. 1997, 69, 595.
73. Vogel, C. Synthesis 1997, 497.
Page 50
Texas Tech University, Deepali Butani, August 2011
28
74. (a) Hoffmann, R. W. Angew. Chem., Int. Ed. Engl. 1979, 18, 563. (b)
Hiersemann, M.; Abraham, L.; Pollex, A. Org. Biomol. Chem. 2003,
1, 1088.
75. McNally, A.; Evans, B.; Gaunt, M. J. Angew. Chem. Int. Ed. 2006, 45,
2116-2119.
76. Hansen, H. J.; Schmid, H. Tetrahedron 1974, 30, 1959.
77. Andreev, V. G.; Kolomiets, A. F. Russ. Chem. Rev. 1993, 62, 553.
78. Enders, D.; Knopp, M.; Schiffers, R. Tetrahedron: Asymmetry 1996,
7, 1847.
79. Nubbemeyer, U. Synlett. 2003, 961.
80. Castro, A. M. M. Chem. Rev. 2004, 104, 2939.
81. Claisen, L.; Ber. 1912, 45, 3157.
82. Claisen, L.; Tietze, E.; Ber. 1925, 58, 275.
83. Claisen, L.; Tietze, E.; Ber. 1926, 59, 2344.
84. Cope, A. C.; Hardy, E. M. J. Am. Chem. Soc. 1940, 62, 441.
85. Hoffmann, R.; Stohrer, W. D. J. Am. Chem. Soc. 1971, 93, 6941.
86. Dupuis, M.; Murray, C.; Davidson, E. R. J. Am. Chem. Soc. 1991, 113,
9756.
87. Klarner, F.G. Topics in Stereochem. 1984, 15, 1.
88. Unruh, G. R.; Birney, D. M., J. Am. Chem. Soc. 2003, 125, 8529 –
8533.
Page 51
Texas Tech University, Deepali Butani, August 2011
29
89. Miller, B. Advanced Organic Chemistry. 2nd Ed. Upper Saddle River:
Pearson Prentice Hall. 2004.
Page 52
Texas Tech University, Deepali Butani, August 2011
30
CHAPTER II
COMPUTATIONAL STUDIES OF SIGMATROPIC REARRANGEMENT OF
ALLYLIC AND VINYLOGOUS AZIDES
2.1 AZIDE CHEMISTRY
Azide chemistry was first introduced in the 1864 with discovery of phenyl
azide by Peter Griess. 1,2
The chemistry of these electron rich and flexible
intermediates broadened after important contributions by Curtius who developed
hydrogen azide and discovered the rearrangement of acyl azide to corresponding
isocyanates (Curtius rearrangement).3,4
However, the organic azides started receiving
considerable attention in the 1950s and 1960s5,6
with new applications in the
chemistry of the acyl, aryl and alkyl azides. Synthesis of hetrocycles such as triazoles
and tetrazoles as well as with their use as blowing agents and as functional groups in
pharmaceuticals led to the extensive use of organic azide compounds for industrial
purposes.7-11
AZT (an azidonucleoside) is an organic azide compound that is used for
treatment of AIDS.12
Azides are considered as the first and foremost energy-rich molecules which
often exhibit explosive properties. The azido group is considered to be the highly
energetic functional group. The N3 π-bond easily polarizes which consequently
results in strong exothermic dissociation reactions leading to release of molecular
nitrogen and reactive nitrene groups. It has been reported that the introduction of
azido group into an organic compound increases its energy content by approximately
Page 53
Texas Tech University, Deepali Butani, August 2011
31
290-355 kJ/mol13,14
which makes organic azides useful for the preparation of
energetic materials such as energetic polymers or high-energy-density materials in
explosives or propellant formation (NaN3 is the propellant used in the air bags of
automobiles).15,16
However, the poor thermal and mechanical stability of many
organic azides sometimes make them impractical to use.
Organic azides have a variety of chemical diversity because of the
physicochemical properties of azides. Some of these properties of organic azides are
explained by the resonance structures of azides, which have different dipolar
characteristics as shown in Figure 2.1.
Figure 2.1: Representative resonance structures of azides.
The structures 1c and 1d which were proposed by Pauling17,18
explain the
facile decomposition into corresponding nitrene and dinitrogen as well as the
reactivity as a 1,3-dipole. The regioselectivity of the reaction with an electrophile or
nucleophile can be explained by the mesomeric structure 1d. Azide ions are known as
pseudohalides as the electronegativitiy value of N3 (7.7eV) which is very close to that
of Cl (8.3eV) and Br (7.5eV).19,20
The most common types of reactions encountered by azides are: 21
Page 54
Texas Tech University, Deepali Butani, August 2011
32
1. Unimolecular Decomposition by Light or Heat: It generates singlet or triplet
nitrenes. The possible reaction pathways are summarized in Figure 2.2 and
discussed in more detail below.
Figure 2.2: Products from unimolecular decomposition of azides.
a. Nitrene-Derived Products: 21
The more electron-attracting is R, the
more electrophilic will be the singlet nitrene, so promoting its
reactions relative to those of the triplet nitrene.
Figure 2.3: Nitrene products from azides.
b. Rearrangement followed by Nucleophilic Attack22
Figure 2.4: Rearrangement products from azides.
Page 55
Texas Tech University, Deepali Butani, August 2011
33
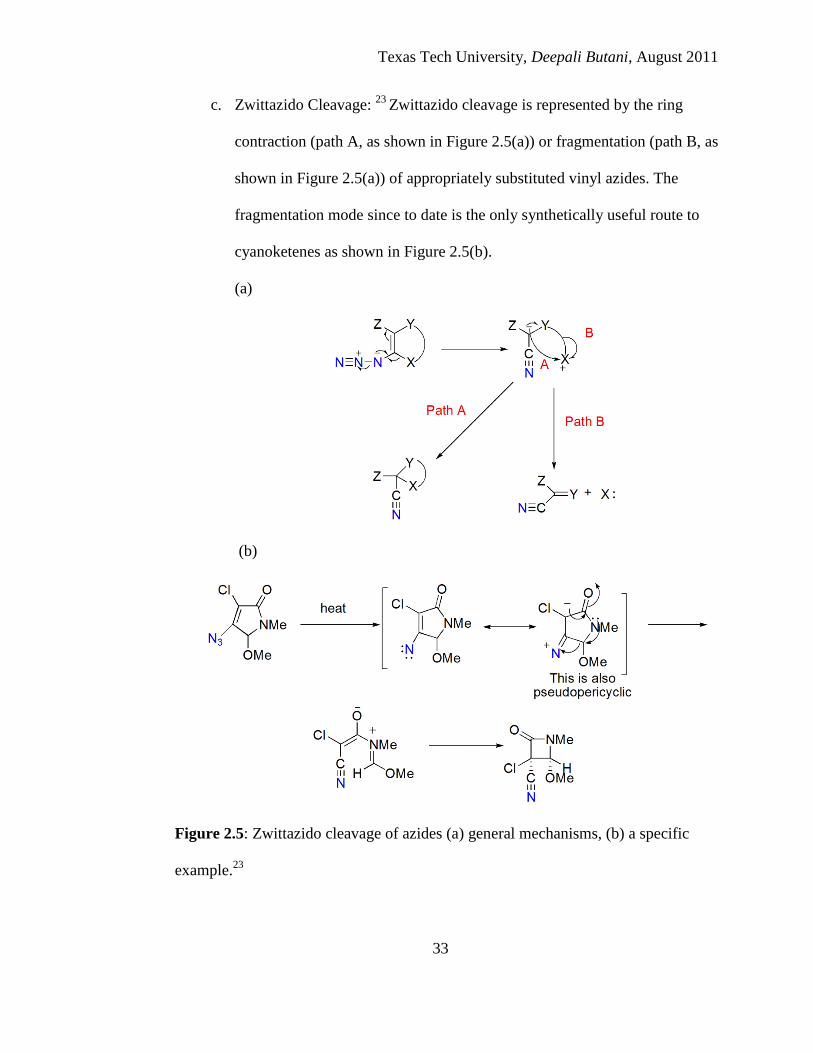
c. Zwittazido Cleavage: 23
Zwittazido cleavage is represented by the ring
contraction (path A, as shown in Figure 2.5(a)) or fragmentation (path B, as
shown in Figure 2.5(a)) of appropriately substituted vinyl azides. The
fragmentation mode since to date is the only synthetically useful route to
cyanoketenes as shown in Figure 2.5(b).
(a)
(b)
Figure 2.5: Zwittazido cleavage of azides (a) general mechanisms, (b) a specific
example.23
Page 56
Texas Tech University, Deepali Butani, August 2011
34
2. Acid- Catalyzed Decomposition24
Figure 2.6: Mechanism of acid-catalyzed decomposition.
3. Staudinger Reaction25
Figure 2.7: Mechanism of Staudinger Reaction.
4. Curtius Rearrangement26
Figure 2.8: Mechanism of Curtius Rearrangement.
5. Schmidt Rearrangement27
Figure 2.9: Reaction of Schmidt Rearrangement.
Page 57
Texas Tech University, Deepali Butani, August 2011
35
6. Reduction28
Figure 2.10: Reaction showing reduction.
7. Cycloadditions29
Figure 2.11: Reaction showing cycloaddition.
8. Nucleophilic Attack at the Azide Terminus30
Figure 2.12: Mechanism showing nucleophilic attack at the azide terminus.
2.2 SIGMATROPIC REARRANGEMENT OF ALLYLIC AZIDES
As mentioned in Section 2.1 organic azides represent an important class of
compounds for organic synthesis and material sciences. There have been extensive
studies on the reactivity of organic azides. However, among this family, allylic azides
have not been explored as much. The synthesis of allylic azides is complicated as they
undergo [3, 3] sigmatropic rearrangement as shown in Figure 2.13.31
Page 58
Texas Tech University, Deepali Butani, August 2011
36
Figure 2.13: [3, 3] Sigmatropic rearrangement of allyl azide (1).
[3, 3] Sigmatropic rearrangements of acyclic allylic azides have been reported
at or below room temperature.32
But cyclic allylic azides seem to require higher
temperatures to rearrange. Indeed, it has been seen that most allylic azides exist as
mixtures of regioisomers that interconvert rapidly at ambient temperature. This is a
major drawback which hampers the use of allylic azides in synthesis. It has also been
reported that, in general, tertiary and secondary allylic azides rearrange much more
rapidly than primary allylic azides.33
In addition, the regioisomer with the more
substituted alkene are usually thermodynamically more favored. Hence, a high degree
of regioselectivity can be obtained in cases where the double bond is conjugated with
an unsaturation34
while some degree of regiochemical control is achieved using
competitive reactivity of either the azide or alkene moiety.35
Olefins are often considered stable in most acid/base environments, therefore
one expects that the special case of allylic azides might possess the familiar reactivity
profile, and it does, even though the azide and the olefins groups are engaged in
dynamic [3,3] sigmatropic equilibrium process as shown in Figure 2.14.36
Page 59
Texas Tech University, Deepali Butani, August 2011
37
Figure 2.14: Rearrangement of an allylic azide.36
[3, 3] Sigmatropic rearrangements like the Cope and the Claisen reactions have
been known for over 60 years. But it was not until 1954, when Vander Werf and
coworkers37
while studying the reaction of sodium azide with epoxides, observed that
the allylic case gave a mixture of regioisomers. Although the possibility of as SN2ʹ
attack could not be ruled out (Figure 2.15), they were the first to propose the
hypothesis that the resulting mixture could be result of sigmatropic rearrangement.
Page 60
Texas Tech University, Deepali Butani, August 2011
38
Figure 2.15: Concerted rearrangement vs. SN2ʹ attack. Top: the expected SN2 pathway
of nucleophilic opening of epoxide, 1; middle: allylic azide rearrangement of 2 leading
to 4-azido-2-buten-1-ol, 3; bottom: alternative SN2ʹ pathway leading to 4-azido-2-
buten-1-ol, 2.37
In 1960, Gagneux, Winstein and Young reported that allylic azides exist as
equilibrium mixture of regioisomers. They showed that α- and γ-methylallyl azide
rapidly form an equilibrium mixture of the two isomers (Figure 2.16).31a
Figure 2.16: Equilibrium between α- and γ-methylallyl azide.
The rates of the allylic azide rearrangements were found to be remarkably
insensitive to methyl substitution in the substrate azide or to solvent change. It was
also seen that the change from ground state to transition state in azide isomerization
involved a very little increase in polar character. And finially, no detectable solvolysis
competes with azide isomerization, even in 70% aqueous acetone.
Page 61
Texas Tech University, Deepali Butani, August 2011
39
The two mechanisms that were postulated to rationalize this equilibrium are
shown in Figure 2.17:38(a)
1. A concerted [3, 3] -sigmatropic rearrangement (Path A) in
which the sterochemical integrity of molecule is preserved.
2. A dissociative process involving ion-pair formation (Path B)
whereby the initial stereochemistry can be lost.
Figure 2.17: Possible mechanistic alternatives for the allylic rearrangement.
The remarkable insensitivity of the rearrangement to changes in solvent as well
as to alkyl substitution are indicative of very little charge separation in the transition
state, and it is generally assumed that the equilibration occurs via a cyclic transition
state (i.e., Path A). No definitive proof has been presented to date, however, which
unequivocally distinguishes between the above two mechanistic possibilities. Lacking
well-defined regio- and stereochemistry, the rearrangement has been underutilized and
no general approach to affecting a controllable stereoselective isomerization has been
reported.
Page 62
Texas Tech University, Deepali Butani, August 2011
40
Since then various groups38(b-i)
are using the concept of sigmatropic
rearrangement of allylic azide to provide mechanistic route and use them for the
purpose of syntheses of different useful compounds. Our main aim in this chapter is to
study [3, 3] sigmatropic rearrangement of the allylic azide through computational
studies and also our aim here is to study [3, 5] sigmatropic rearrangement of the
vinologous azide and distinguish them as pericylic or pseudopericyclic.
2.3 COMPUTATIONAL METHOD
All the computational calculations were carried out using Gaussian 03
program.39
The 6-31G(d,p) basis set40
was used throughout. Geometries were
completely optimized at using RHF/6-31G(d,p) , RMP2/6-31G(d,p) (ab initio second-
order Møller-Plesset perturbation)41-45
and Density Functional Theory using
RB3LYP/6-31G(d,p) (the hybrid three parameter function developed by Becke with
Lee-Yang-Parr correlation function).46-55
A systematic search with constrained
distance was performed at the RB3LYP/6-31G(d,p) level. Frequency calculations
verified the identity of each stationary point as a minimum or transition state. Zero-
point vibrational energies have been computed and have not been scaled. All energies
discussed here are the results of calculations at the RB3LYP/6-31G(d,p) + ZPE level.
Page 63
Texas Tech University, Deepali Butani, August 2011
41
2.4 RESULTS AND DISCUSSIONS
2.4.1 Study of the [3, 3] Sigmatropic Rearrangement of Allylic Azide
The geometry of allyl azide as shown in Figure 2.18 (a) was calculated in
different conformations. A transition state for degenerate rearrangement was also
located as described below. Molecular orbitals in Figure 2.18 (b) shows that three N-
atoms in azide are in same plane.
Initially, the C-N distance was constrained at various lengths to obtain an
energy profile for the reaction (Figure 2.19). This indicated a minimum energy
conformation near 1.50 Å and a transition state near 2.00 Å. The values of all relative
energies with constrained bond lengths are given in Table 2.1.
Figure 2.18: (a) [3, 3] Sigmatropic Rearrangement of allylic azide (1).
(b) Molecular orbital diagram of azide.
Page 64
Texas Tech University, Deepali Butani, August 2011
42
Table 2.1: The calculated absolute energies (Hartree), zero-point energies (ZPE,
kcal/mol), relative energies (kcal/mol with respect to ground state), relative energies
with zero-point energy correction (kcal/mol) of different models at different
constrained C-N bond length (Å) at the RB3LYP/6-31G(d,p) level of theory.
Constrained C-N
Bond Length
(Å)
Absolute
Energy
(Hartree)
Absolute
Energy
(kcal/mol)
ZPE
(kcal/mol)
Relative
Energy + ZPE
(kcal/mol)
1.50 -281.4936 -176640.04 52.80 40.49
1.75 -281.4785 -176630.59 51.83 49.46
2.00 -281.4601 -176619.01 51.36 62.47
2.25 -281.4659 -176622.65 52.79 59.05
2.50 -281.4755 -176628.69 53.01 0.00
Page 65
Texas Tech University, Deepali Butani, August 2011
43
Figure 2.19: Energy profile showing relative energy and constrained C-N bond
distance. This helps in determining which structure should be taken to optimize at
ground state and transition state.
The optimization was done for ground state using the structure at 1.50 Å,
removing the constraint on the bond length. The transition state was similarly found
beginning with the optimized geometry of the structure with a C-N bond length near
2.00 Å. Later an IRC (Intrinsic Reaction Coordinate) was run from transition state to
confirm the connection to the product (i.e. degeneracy of rearrangement). The other
conformations (1b-e) were found by rotating around the C-C and the C-N single
bonds. Four other conformations were located (Table 2.2).
Page 66
Texas Tech University, Deepali Butani, August 2011
44
Figure 2.20: IRC calculation of transition state of the allylic azide using the
RB3LYP/6-31G(d,p) level of theory.
Table 2.2: Dihedral angles (C-C-C-N and C-C-N-N) of ground state, transition state
and different conformers using RB3LYP/6-31G(d,p) level of theory.
Structure Dihedral angle
C-C-C-N
Dihedral angle
C-C-N-N
Ground State, 1a -120.7 64.8
Transition state, 5 -67.7 28.4
Conformation-1, 1b 0.02 -179.9
Conformation-2, 1c -1.8 -84.7
Conformation-3, 1d -127.1 -169.4
Conformation-4, 1e -118.8 -78.6
Page 67
Texas Tech University, Deepali Butani, August 2011
45
The results of the optimization of the ground state, the transition state, and four
other conformations at three different levels of theory are provided in Tables 2.3, 2.4
and 2.5. Also all the calculated thermodynamic quantities are given in Tables 2.6, 2.7
and 2.8.
Table 2.3: The calculated absolute energies (AE, Hartree), dipole moment (DM,
Debye), low or imaginary frequencies (LF, cm-1
), zero-point energies (ZPE, kcal/mol),
absolute energies with zero-point energy correction (AE+ZPE, kcal/mol) and relative
energies with zero-point energy correction (RE, kcal/mol) for the stationary points of
the [3, 3] sigmatropic rearrangement reaction of allyl azide at the RB3LYP/6-31G(d,p)
level of theory.
Structure AE DM LF ZPE AE+ZPE RE
Ground State, 1a -281.4936 2.40 63.2 52.82 -176587.2352 0.00
Transition state, 5 -281.4555 2.96 372.3i 51.91 -176564.2434 22.99
Conformation-1, 1b -281.4925 2.20 27.4 52.64 -176586.6936 0.54
Conformation-2, 1c -281.4926 2.31 46.6 52.79 -176586.6001 0.64
Conformation-3, 1d -281.4920 2.41 32.7 52.59 -176586.4489 0.79
Conformation-4, 1e -281.4922 2.41 38.8 52.75 -176586.4392 0.80
Page 68
Texas Tech University, Deepali Butani, August 2011
46
Table 2.4: The calculated absolute energies (AE, Hartree), dipole moment (DM,
Debye), low or imaginary frequencies (LF, cm-1
), zero-point energies (ZPE, kcal/mol),
absolute energies with zero-point energy correction (AE+ZPE, kcal/mol) and relative
energies with zero-point energy correction (RE, kcal/mol) for the stationary points of
the [3, 3] sigmatropic rearrangement reaction of allyl azide at the RHF/6-31G(d,p)
level of theory.
Structure AE DM LF ZPE AE+ZPE RE
Ground State, 1a -279.7562 2.01 69.1 56.70 -175493.0823 0.00
Transition state, 5 -279.6890 3.41 563.3i 55.68 -175451.9910 41.09
Conformation-1, 1b -279.7543 1.85 24.7 56.51 -175492.1108 0.97
Conformation-2, 1c -279.7548 1.93 56.5 56.69 -175492.2329 0.85
Conformation-3, 1d -279.7544 1.96 35.6 56.47 -175492.2470 0.84
Conformation-4, 1e -279.7541 2.06 32.9 56.61 -175491.8871 1.20
Page 69
Texas Tech University, Deepali Butani, August 2011
47
Table 2.5: The calculated absolute energies (AE, Hartree), dipole moment (DM,
Debye), low or imaginary frequencies (LF, cm-1
), zero-point energies (ZPE, kcal/mol),
absolute energies with zero-point energy correction (AE+ZPE, kcal/mol) and relative
energies with zero-point energy correction (RE, kcal/mol) for the stationary points of
the [3, 3] sigmatropic rearrangement reaction of allyl azide at the RMP2/6-31G(d,p)
level of theory.
Structure AE DM LF ZPE AE+ZPE RE
Ground State, 1a -280.6730 2.27 58.0 53.94 -176071.1552 0.00
Transition state, 5 -280.6443 3.12 184.9i 53.28 -176053.8247 17.33
Conformation-1, 1b -280.6707 2.10 30.1 53.74 -176069.9310 1.22
Conformation-2, 1c -280.6717 2.22 71.1 53.94 -176070.3321 0.82
Conformation-3, 1d -280.6707 2.29 43.7 53.77 -176069.8737 1.28
Conformation-4, 1e -280.6713 2.38 57.2 53.94 -176070.1169 1.04
Page 70
Texas Tech University, Deepali Butani, August 2011
48
Table 2.6: Calculated thermodynamic data for the [3, 3] sigmatropic rearrangement of
allyl azide at the RB3LYP/6-31G(d,p) level of theory: sum of electronic and thermal
Gibbs free energies (G), sum of electronic and thermal enthalpies (H), and entropies
(S).
Structure G
(Hartree)
H
(Hartree)
S
(cal/mol.K)
Ground State, 1a -281.4401 -281.4022 79.7530
Transition state, 5 -281.4015 -281.3664 73.9800
Conformation-1, 1b -281.4396 -281.4013 80.6570
Conformation-2, 1c -281.4390 -281.4013 79.4370
Conformation-3, 1d -281.4392 -281.4008 80.9190
Conformation-4, 1e -281.4392 -281.4009 80.5790
Page 71
Texas Tech University, Deepali Butani, August 2011
49
Table 2.7: Calculated thermodynamic data for the [3, 3] sigmatropic rearrangement of
allyl azide at the RHF/6-31G(d,p) level of theory: sum of electronic and thermal Gibbs
free energies (G), sum of electronic and thermal enthalpies (H), and entropies (S).
Structure G
(Hartree)
H
(Hartree)
S
(cal/mol.K)
Ground State, 1a -279.6961 -279.6589 78.2110
Transition state, 5 -279.6287 -279.5943 72.5940
Conformation-1, 1b -279.6951 -279.6572 79.7880
Conformation-2, 1c -279.6946 -279.6576 77.8920
Conformation-3, 1d -279.6952 -279.6574 79.5370
Conformation-4, 1e -279.6947 -279.6569 79.6370
Page 72
Texas Tech University, Deepali Butani, August 2011
50
Table 2.8: Calculated thermodynamic data for the [3, 3] sigmatropic rearrangement of
allyl azide at the RMP2/6-31G(d,p) level of theory: sum of electronic and thermal
Gibbs free energies (G), sum of electronic and thermal enthalpies (H), and entropies
(S).
Structure G
(Hartree)
H
(Hartree)
S
(cal/mol.K)
Ground State, 1a -280.6176 -280.5798 79.6140
Transition state, 5 -280.5879 -280.5532 73.1060
Conformation-1, 1b -280.6161 -280.5777 80.7730
Conformation-2, 1c -280.6159 -280.5785 78.6710
Conformation-3, 1d -280.6158 -280.5776 80.3100
Conformation-4, 1e -280.6160 -280.5781 79.7030
Using the thermodynamic values in Table 2.6, 2.7 and 2.8, activation energy
Ea. and rate constant k were calculated using following formulas:
-
------ (eq.1)
Page 73
Texas Tech University, Deepali Butani, August 2011
51
( ) --------- (eq.2)
where Boltzmann constant, kb=1.38E-23 J K-1
, Plank’s constant, h=6.626E-34 J s,
T=298.15 K, Gas constant, R=1.98 cal K−1
mol−1
, concentration, c0=1, enthalpies of
activation (ΔH≠), Gibbs activation free energies (ΔG
≠).
Table 2.9: Activation parameters for the [3, 3] sigmatropic rearrangement of allyl
azide at three different level of theory using 6-31G(d.p) basis set using ground state
conformation (1a) as reference. (Gibbs activation free energy, (ΔG≠, kcal/mol),
Enthalpies of activation, (ΔH≠, kcal/mol), Entropy of activation, (ΔS
≠, cal/mol.K),
Activation energy, (Ea, kcal/mol) and Rate constant, (k, s-1
))
From Table 2.9 it is observed that the RHF method predicts a higher energy barrier,
RMP2 predicts a lower energy barrier while RB3LYP predicts an intermediate energy
barrier. These results are in agreement with previous studies56,57
which shows that RB3LYP
Level of theory ΔG≠ ΔH
≠ ΔS
≠ Ea k
RB3LYP 24.209 22.488 -5.773 23.08 1.09E-05
RHF 42.243 40.567 -5.617 41.16 6.43E-19
RMP2 18.667 16.727 -6.508 17.32 1.26E-01
Page 74
Texas Tech University, Deepali Butani, August 2011
52
level of theory are in closer agreement with experimental results as observed in many
pericyclic reactions.
After computing all the data an energy profile is made using calculation results
from RB3LYP/6-31G(d,p) as shown in Figure 2.21. The calculated energy barrier
(RE) for the [3, 3] sigmatropic rearrangement of the allyl azide with respect to ground
state (1a) is 23.08 kcal/mol (Table 2.9).
Figure 2.21: Energy profile (RE from Table 2.3) of the [3,3] sigmatropic
rearrangement of the allyl azide and different conformers of the allyl azide at
B3LYP/6-31G(d,p) level of theory, where GS stands for ground state (1a), TS for
transition state (5), C1 for conformation1 (1b), C2 for conformation-2 (1c), C3 for
conformation-3 (1d) and C4 for conformation-4 (1e). Their relative energies in
kcal/mol with respect to ground state are provided in parentheses.
Page 75
Texas Tech University, Deepali Butani, August 2011
53
The transition state geometry of the [3, 3] sigmatropic rearrangement of allyl
azide suggest that it is pericyclic in nature. Figure 2.22 shows different views of the
transition state.
Figure 2.22: Three different views of the transition state of the [3, 3] sigmatropic
rearrangement of allyl azide. Bond Lengths: C(6)-N(1) = 2.07 Å, C(4)-N(3) = 2.07 Å;
Bond Angles: N(1)-N(2)-N(3) = 163.13°, C(4)-C(5)-C(6) = 120.39°.
Page 76
Texas Tech University, Deepali Butani, August 2011
54
A reaction is pericyclic in name if the reaction proceeds in concerted manner
involving no charged intermediates with a single cyclic transition state.58
Furthermore,
textbook examples of pericyclic reactions have cyclic overlap. As seen in Figure 2.22
the [3, 3] sigmatropic rearrangement of allyl azide fits these criteria, including cyclic
orbital overlap between the allylic fragment and one of the two π-systems on the azide
fragment, hence it is pericyclic in nature.
Figure 2.14: Rearrangement of an allylic azide.36
As shown in Figure 2.14, 3-azido-1-butene (3) is an intermediate in the
interconversion of cis-1-azido-2-butene (4) and trans-1-azido-2-butene (2), via a
sequential [3, 3] sigmatropic rearrangements. RB3LYP/6-31G(d,p) calculations were
performed on this system; the results are summarized in Table 2.10 and Figure 2.23.
The transition state (6) for formation of trans-1-azido-2-butene from 3-azido-1-butene
(3) is 2.36 kcal/mol lower than that (7) for the cis-1-azido-2-butene (Table 2.12) but
both are low enough in energy to account for the rapid interconversion observed at
room temperature.32
Page 77
Texas Tech University, Deepali Butani, August 2011
55
Table 2.10: The calculated absolute energies (AE, Hartree), dipole moment (DM,
Debye), low or imaginary frequencies (LF, cm-1
), zero-point energies (ZPE, kcal/mol),
absolute energies with zero-point energy correction (AE+ZPV, kcal/mol) and relative
energies with zero-point energy correction (RE, kcal/mol) for the stationary points for
the [3, 3] sigmatropic rearrangements of cis and trans-1-azido-2-butene at the
RB3LYP/6-31G(d,p) level of theory.
Structure AE DM LF ZPE AE+ZPE RE
transǂ-1-azido-2-butene (6) -320.7793 3.31 333.2i 69.41 -201222.8036 22.30
cisǂ-1-azido-2-butene (7) -320.7757 3.25 343.6i 69.59 -201220.3668 24.73
cis-1-azido-2-butene (4) -320.8138 2.69 34.4 70.65 -201243.2431 1.86
trans-1-azido-2-butene (2) -320.8166 2.70 48.3 70.52 -201245.1006 0.00
3-azido-1-butene (3) -320.8143 2.41 54.3 70.33 -201243.8344 1.27
Page 78
Texas Tech University, Deepali Butani, August 2011
56
Table 2.11: Calculated thermodynamic data for the [3, 3] sigmatropic rearrangements
of cis and trans-1-azido-2-butene at the RB3LYP/6-31G(d,p) level of theory: sum of
electronic and thermal Gibbs free energies (G), sum of electronic and thermal
enthalpies (H), and entropies (S).
Structure G
(Hartree)
H
(Hartree)
S
(cal/mol.K)
transǂ-1-azido-2-butene (6) -320.6995 -320.6607 81.7940
cisǂ-1-azido-2-butene (7) -320.6956 -320.6569 81.3410
cis-1-azido-2-butene (4) -320.7346 -320.6926 88.4150
trans-1-azido-2-butene (2) -320.7371 -320.6955 87.6360
3-azido-1-butene (3) -320.7347 -320.6936 86.5010
Table 2.12: Activation parameters for the [3, 3] sigmatropic rearrangements of trans-
1-azido-2-butene (2) and cis-1-azido-2-butene (4) from 3-azido-1-butene (3) at
RB3LYP/6-31G(d.p) level of theory using thermodynamic parameters from Table
2.11. (Gibbs activation free energy, (ΔG≠, kcal/mol), Enthalpies of activation, (ΔH
≠,
kcal/mol), Entropy of activation, (ΔS≠, cal/mol.K), Activation energy, (Ea, kcal/mol)
and Rate constant, (k, s-1
))
Structure ΔG≠ ΔH
≠ ΔS
≠ Ea k
transǂ-1-azido-2-butene (6) 22.052 20.649 -4.707 21.24 4.15E-04
cisǂ-1-azido-2-butene (7) 24.541 23.003 -5.160 23.60 6.20E-06
Page 79
Texas Tech University, Deepali Butani, August 2011
57
Figure 2.23: Energy profile (RE from Table 2.11) for the [3, 3] sigmatropic
rearrangements of cis and trans-1-azido-2-butene at the RB3LYP/6-31G(d,p) level of
theory, where trans-1-azido-2-butene (2), 3-azido-1-butene (3), cis-1-azido-2-butene
(4), trans-transition state (6) and cis-transition state (7). Their relative energies in
kcal/mol with respect to 2 are provided in parentheses.
Page 80
Texas Tech University, Deepali Butani, August 2011
58
Figure 2.24: Structure of trans-transition state (6). Bond Lengths: C(6)-N(1) = 2.11 Å,
C(4)-N(3) = 2.13 Å; Bond Angles: N(1)-N(2)-N(3) = 164.6°, C(4)-C(5)-C(6) =
121.3°, C(5)-C(6)-C(7) = 123.1°, N(1)-C(6)-C(7) = 153.6°.
Figure 2.25: Structure of cis-transition state (7). Bond Lengths: C(6)-N(1) = 2.13 Å,
C(4)-N(3) = 2.11 Å; Bond Angles: N(1)-N(2)-N(3) = 164.5°, C(4)-C(5)-C(6) =
122.9°, C(5)-C(6)-C(7) = 125.2°, N(1)-C(6)-C(7) = 97.0°.
2.4.2 Study of the [3, 5] Sigmatropic Rearrangement of Vinylogous
(Pentadienyl) Azide
After examining the [3, 3] sigmatropic rearrangement of allyl azide, our next
aim was to add a vinyl group to allylic structure. With a vinyl group added, the
Page 81
Texas Tech University, Deepali Butani, August 2011
59
vinylogous (pentadienyl) azide and the orthogonal π-system of the azide present the
possibility of an eight-centered pseudopericyclic transition state for a [3, 5]
sigmatropic rearrangement. The [3, 3] rearrangement of vinylogous azides are still
possible, these reactions are shown in Figure 2.26.
Figure 2.26: The [3, 3] and [3, 5] sigmatropic rearrangement of vinylogous azide (8).
The initial calculations were done using the RB3LYP/6-31G(d,p) level of
theory by constraining the C-N distance to find structures close to the possible
transition states. The calculations were done at bond lengths of 1.50 Å, 1.80 Å and
2.00 Å. The optimization for the transiton state using the structure calculated at 1.80
Å, removing the constrain on bond length, resulted in the structure of transition state-1
(11, Figure 2.27). Similar optimization of structure obtained at 2.00 Å resulted in the
structure of transition state-2 (14, Figure 2.28). After obtaining the transition state-1
(11) and transition state-2 (14), IRC (Intrinsic Reaction Coordinate) calculations
(Figure 2.27 a,b and Figure 2.28 a,b) were run on transition states in both directions to
obtain possible reactants and products. The structures obtained through the IRC
calculations were optimized to reactants and products. Optimizations were done to
Page 82
Texas Tech University, Deepali Butani, August 2011
60
obtain structures of possible [3, 3] rearrangement, product-4 (19) and transition state-
4 (18), Figure 2.29.
Figure 2.27: IRC run for transition state-1 (11): (a) IRC run in forward direction
towards possible product and (b) IRC run in reverse direction towards possible
reactant using RB3LYP/6-31G(d,p) level of theory.
Page 83
Texas Tech University, Deepali Butani, August 2011
61
As shown in Figure 2.27(a) IRC for transition state-1 (11) in forward direction
resulted in a structure which futher optimized to give structure 13 as product (Figure
2.29). Similarly, IRC calculations for transition state-1 (11) in reverse direction
(Figure 2.27 (b)) resulted in a structure which futher optimized to give structure 12 as
reactant (Figure 2.29). When IRC calculation for transition state-1 (11) in reverse
direction was checked there was a structure which looked like another transition state
(TS-3). The structure was optimized to obtain transition state-3 (17, Figure 2.29).
Figure 2.28: IRC calculations for transition state-2 (14): (a) IRC calculations in reverse
direction towards possible reactant and (b) IRC calculations in forward direction
towards possible product using RB3LYP/6-31G(d,p) level of theory.
IRC for transition state-1 (14) in forward direction (Figure 2.28 (b)) resulted in a
structure which futher optimized to give structure 16 as product (Figure 2.29). Similarly,
IRC calculations for transition state-1 (14) in reverse direction (Figure 2.28 (a)) resulted
in a structure which futher optimized to give structure 15 as reactant (Figure 2.29).
Page 84
Texas Tech University, Deepali Butani, August 2011
62
The results of all optimizations done for different reactants, transition states
and products at three levels of theory are provided in Tables 2.13, 2.14 and 2.15. Also
all the calculated thermodynamic quantities are given in Tables 2.16, 2.17 and 2.18.
Table 2.13: The calculated absolute energies (AE, Hartree), dipole moment (DM,
Debye), low or imaginary frequencies (LF, cm-1
), zero-point energies (ZPE, kcal/mol),
absolute energies with zero-point energy correction (AE+ZPE, kcal/mol) and relative
energies with zero-point energy correction (RE, kcal/mol) for the stationary points of
the [3, 5] and [3, 3] sigmatropic rearrangements of the vinylogous azide at the
RB3LYP/6-31G(d,p) level of theory.
Structure AE DM LF ZPE AE+ZPE RE
transition state-1 (11) -358.8145 4.11 512.8i 73.57 -225086.1357 48.79
reactant-1 (12) -358.8627 3.60 164.5 75.57 -225114.3378 20.59
product-1 (13) -358.8926 2.62 44.3 74.05 -225134.6329 0.29
transition state-2 (14) -358.8163 4.74 450.5i 71.97 -225088.8464 46.08
reactant-2 (15) -358.8861 2.34 35.5 73.68 -225130.9458 3.98
product-2 (16) -358.8932 2.48 29.7 74.14 -225134.9240 0.00
transition state-3 (17) -358.8143 3.49 443.4i 72.44 -225087.1339 47.79
transition state-4 (18) -358.8603 3.56 277.1i 72.51 -225115.9120 19.01
product-4 (19) -358.8882 2.42 44.9 73.27 -225132.6676 2.26
Page 85
Texas Tech University, Deepali Butani, August 2011
63
Table 2.14: The calculated absolute energies (AE, Hartree), dipole moment (DM,
Debye), low or imaginary frequencies (LF, cm-1
), zero-point energies (ZPE, kcal/mol),
absolute energies with zero-point energy correction (AE+ZPE, kcal/mol) and relative
energies with zero-point energy correction (RE, kcal/mol) for the stationary points of
the [3, 5] and [3, 3] sigmatropic rearrangements of the vinylogous azide at the RHF/6-
31G(d,p) level of theory.
Structure AE DM LF ZPE AE+ZPE RE
transition state-1 (11) -356.5322 4.89 841.9 79.35 -223648.1933 66.96
reactant-1 (12) -356.6148 3.86 192.1 82.12 -223697.2306 17.93
product-1 (13) -356.6390 2.28 48.5 79.41 -223715.1560 0.00
transition state-2 (14) -356.5260 7.69 -500.9 77.52 -223646.0824 69.07
reactant-2 (15) -356.6148 3.86 41.9 79.39 -223699.9588 15.20
product-2 (16) -356.6386 2.05 28.2 79.51 -223714.7548 0.40
transition state-3 (17) -356.5231 5.82 -715.7 77.53 -223644.3062 70.85
transition state-4 (18) -356.5785 4.01 -471.4 77.88 -223678.6938 36.46
product-4 (19) -356.6371 1.96 53.4 78.72 -223714.6357 0.52
Page 86
Texas Tech University, Deepali Butani, August 2011
64
Table 2.15: The calculated absolute energies (AE, Hartree), dipole moment (DM,
Debye), low or imaginary frequencies (LF, cm-1
), zero-point energies (ZPE, kcal/mol),
absolute energies with zero-point energy correction (AE+ZPE, kcal/mol) and relative
energies with zero-point energy correction (RE, kcal/mol) for the stationary points of
the [3, 5] and [3, 3] sigmatropic rearrangements of the vinylogous azide at the
RMP2/6-31G(d,p) level of theory.
Structure AE DM LF ZPE AE+ZPE RE
transition state-1 (11) -357.7337 4.91 494.8i 75.14 -224406.3472 56.21
reactant-1 (12) -357.7875 3.93 181.4 77.05 -224438.1787 24.38
product-1 (13) -357.8218 2.56 53.2 75.49 -224461.2655 1.29
transition state-2 (14) -357.7404 5.23 730.7i 73.34 -224412.3471 50.21
reactant-2 (15) -357.8234 2.17 21.9 75.21 -224462.5557 0.00
product-2 (16) -357.8221 2.29 40.6 75.58 -224461.3380 1.22
transition state-3 (17) -357.7496 4.42 214.4i 74.43 -224416.9983 45.56
transition state-4 (18) -357.7978 3.47 239.8i 74.05 -224447.6564 14.90
product-4 (19) -357.8199 2.28 54.9 74.65 -224460.9034 1.65
Page 87
Texas Tech University, Deepali Butani, August 2011
65
Table 2.16: Calculated thermodynamic data for the [3, 5] and [3, 3] sigmatropic
rearrangements of the vinylogous azide at the RB3LYP/6-31G(d,p) level of theory:
sum of electronic and thermal Gibbs free energies (G), sum of electronic and thermal
enthalpies (H), and entropies (S).
Structure G
(Hartree)
H
(Hartree)
S
(cal/mol.K)
transition state-1 (11) -358.7279 -358.6896 80.5850
reactant-1 (12) -358.7724 -358.7350 78.6660
product-1 (13) -358.8085 -358.7653 90.9770
transition state-2 (14) -358.7341 -358.6925 87.3980
reactant-2 (15) -358.8034 -358.7592 93.1260
product-2 (16) -358.8092 -358.7658 91.2090
transition state-3 (17) -358.7299 -358.6908 82.4240
transition state-4 (18) -358.7772 -358.7359 86.8320
product-4 (19) -358.8058 -358.7619 92.2700
Page 88
Texas Tech University, Deepali Butani, August 2011
66
Table 2.17: Calculated thermodynamic data for the [3, 5] and [3, 3] sigmatropic
rearrangements of the vinylogous azide at the RHF/6-31G(d,p) level of theory: sum of
electronic and thermal Gibbs free energies (G), sum of electronic and thermal
enthalpies (H), and entropies (S).
Structure G
(Hartree)
H
(Hartree)
S
(cal/mol.K)
transition state-1 (11) -356.4360 -356.3986 78.6130
reactant-1 (12) -356.5137 -356.4772 76.6230
product-1 (13) -356.5462 -356.5036 89.5970
transition state-2 (14) -356.4346 -356.3938 86.0230
reactant-2 (15) -356.5474 -356.5046 90.0320
product-2 (16) -356.5462 -356.5036 89.5970
transition state-3 (17) -356.4307 -356.3917 81.9690
transition state-4 (18) -356.5137 -356.4772 76.6230
product-4 (19) -356.5137 -356.4772 76.6230
Page 89
Texas Tech University, Deepali Butani, August 2011
67
Table 2.18: Calculated thermodynamic data for the [3, 5] and [3, 3] sigmatropic
rearrangements of the vinylogous azide at the RMP2/6-31G(d,p) level of theory: sum
of electronic and thermal Gibbs free energies (G), sum of electronic and thermal
enthalpies (H), and entropies (S).
Structure G
(Hartree)
H
(Hartree)
S
(cal/mol.K)
transition state-1 (11) -357.6442 -357.6066 79.1130
reactant-1 (12) -357.6947 -357.6576 78.1670
product-1 (13) -357.7351 -357.6923 89.9530
transition state-2 (14) -357.6551 -357.6149 84.6110
reactant-2 (15) -357.7382 -357.6942 92.7230
product-2 (16) -357.7352 -357.6925 89.9580
transition state-3 (17) -357.6616 -357.6231 80.9250
transition state-4 (18) -357.6947 -357.6576 78.1670
product-4 (19) -357.6947 -357.6576 78.1670
Page 90
Texas Tech University, Deepali Butani, August 2011
68
An energy profile summarizing the computational results at the RB3LYP/6-
31G(d,p) as shown in Figure 2.29.
Figure 2.29: Energy profile (RE from Table 2.13) for the [3, 5] and [3, 3] sigmatropic
rearrangements of vinylogous azide at the RB3LYP/6-31G(d,p) level of theory. Their
relative energies in kcal/mol with respect to 16 are provided in parentheses.
Optimization of structure 14 at transition state using UB3LYP/6-31G(d,p)
level of theory gave the same energy values hence showing that it does not have any
biradical character.
From Figure 2.29 it can be seen that the lowest energy transition state for [3, 5]
rearrangement is 14, hence 14 is used for calculating the activation energy and rate
constant for the [3, 5] rearrangement. Using thermodynamic quantities from Tables
2.16, 2.17 and 2.18 and eq. 1 and eq. 2, the rate constant (k) and activation energy (Ea)
for [3, 3] and [3, 5] sigmatropic rearrangements of vinylogous azide were calculated as
summarized in Table 2.19.
Page 91
Texas Tech University, Deepali Butani, August 2011
69
Table 2.19: Activation parameters for the [3, 5] and [3, 3] sigmatropic rearrangements
of vinylogous azide 16 and 19 from 15 at three different levels of theory using 6-
31G(d,p) basis set. (Gibbs activation free energy, (ΔG≠, kcal/mol), Enthalpies of
activation, (ΔH≠, kcal/mol), Entropy of activation, (ΔS
≠, cal/mol.K), Activation
energy, (Ea, kcal/mol) and Rate constant, (k, s-1
))
To study the rearrangement of vinylogous azide in detail, the dihedral angles
(C-C-C-N, C-C-N-N and C-C-C-C (Table 2.20)) of each structure was considered. As
our main interest in studying rearrangement depended on the transition states, Figures
2.30, 2.31, 2.32 and 2.34 were used to show different views of transition states.
Rearrangement Level of theory ΔG≠ ΔH
≠ ΔS
≠ Ea k
[3.5] RB3LYP 43.532 41.824 -5.728 42.42 7.30E-20
[3.3] RB3LYP 16.480 14.604 -6.294 15.20 5.07E+00
[3.5] RHF 70.766 69.570 -4.009 70.16 7.71E-40
[3.3] RHF 38.329 36.749 -5.298 37.34 4.78E-16
[3.5] RMP2 52.150 49.731 -8.112 50.32 3.48E-26
[3.3] RMP2 16.467 14.440 -6.797 15.03 5.18E+00
Page 92
Texas Tech University, Deepali Butani, August 2011
70
Table 2.20: Dihedral angles of various structures of vinylogous azide rearrangements
using RB3LYP/6-31G(d,p) level of theory.
Structure Dihedral Angle
(N3-C4-C5-C6)
Dihedral Angle
(N2-N3-C4-C5)
Dihedral Angle
(C5-C6-C7-C8)
transition state-1 (11) -79.29 96.85 4.31
reactant-1 (12) -65.92 65.55 -3.07
product-1 (13) 118.71 -65.09 -2.25
transition state-2 (14) -53.22 118.76 -12.58
reactant-2 (15) 90.14 66.92 -1.95
product-2 (16) -46.64 126.92 -3.73
transition state-3 (17) -96.54 115.04 7.92
transition state-4 (18) 70.21 -27.58 -160.91
product-4 (19) 126.45 -173.70 125.23
Page 93
Texas Tech University, Deepali Butani, August 2011
71
Figure 2.30: Three different views of 11 using RB3LYP/6-31G(d,p) level of theory.
Bond Lengths: N(1)-C(8) = 1.69 Å, N(3)-C(4) = 1.48 Å; Bond Angles: N(1)-N(2)-
N(3) = 140.8°, N(2)-N(3)-C(4) = 121.7°, N(3)-C(4)-C(5) = 109.7°, C(6)-C(7)-C(8) =
129.3°; Dihedral angle: C(8)-N(1)-N(3)-C(4) = 25.7°.
Figure 2.31: Three different views of 14 using RB3LYP/6-31G(d,p) level of theory.
Bond Lengths: N(1)-C(8) = 2.47 Å, N(3)-C(4) = 2.67 Å; Bond Angles: N(1)-N(2)-
N(3) = 174.2°, C(4)-C(5)-C(6) = 128.4°, C(6)-C(7)-C(8) = 126.2°; Dihedral angle:
C(8)-N(1)-N(3)-C(4) = -59.4°.
Page 94
Texas Tech University, Deepali Butani, August 2011
72
Figure 2.32: Three different views of 17 using RB3LYP/6-31G(d,p) level of theory.
Bond Lengths: N(1)-C(8) = 1.79 Å, N(3)-C(4) = 1.44 Å; Bond Angles: N(1)-N(2)-
N(3) = 123.1°, N(2)-N(3)-C(4) = 101.9° C(4)-C(5)-C(6) = 61.4°, C(6)-C(7)-C(8) =
126.1°; Dihedral angle: C(8)-N(1)-N(3)-C(4) = -53.1°.
Figure 2.33: Three different views of 18 using RB3LYP/6-31G(d,p) level of theory.
Bond Lengths: N(1)-C(6) = 2.13 Å, N(3)-C(4) = 2.28 Å; Bond Angles: N(1)-N(2)-
N(3) = 167.9°, N(2)-N(3)-C(4) = 93.9° C(4)-C(5)-C(6) = 122.1°, C(6)-C(7)-C(8) =
123.4°. Dihedral angle: C(6)-N(1)-N(3)-C(4) = -6.82°.
From Figure 2.31 it can be seen that the sigmatropic rearrangement of the
vinylogous azide is via a eight-centered transition state. Also, structure 14 is orbital
symmetry allowed due to two orbital disconnections involving azide fragement, where
Page 95
Texas Tech University, Deepali Butani, August 2011
73
lone pair from nitrogen at azide forms a new C-N bond becoming a new bonded pair
with simultaneous cleavage of existing C-N bond to form a new lone pair. Hence, it
shows that it is pseudopericyclic in nature.
2.5 CONCLUSION
The [3, 3] sigmatropic rearrangement of allyl azide was found to be pericyclic
with low barrier of 23.08 kcal/mol. The low barrier can be explained from the fact that
the geometry of transition state of allyl azide is appropriate for cyclic orbital overlap.
The high nucleophilicity of azide may also be a factor in the low barrier. Calculations
for the [3, 3] rearrangements of cis and trans-1-azido-2-butene also shows that the
barrier of their interconversion is very low. Hence, this offers an explanation of the
experimentally observed rapid interconversion of cis and trans at room temperature.
The vinylogous azide was calculated to undergo [3, 5] as well [3, 3]
sigmatropic rearrangement. However, the barrier for transition state (14) undergoing
[3, 5] rearrangement was found to be much higher than the barrier for transition state
(18) of [3, 3] rearrangement. The eight-centered transition state-2 (14) of vinylogous
azide was found to be pseudopericyclic, with two orbital disconnections on the azide
fragment. The high barrier for the [3, 5] rearrangement can be explained due to the
unfavorable geometry of transition state which is required to allow the orthogonal π-
orbitals of the azide to participate in the pseudopericyclic rearrangement.
Page 96
Texas Tech University, Deepali Butani, August 2011
74
2.6 REFERENCES
1. Griess, P. Philos. Trans. R. Soc. London 1864, 13, 377.
2. Griess, P. Justus Liebigs Ann. Chem. 1865, 135,131.
3. Curtius, T. Ber. Dtsch. Chem. Ges. 1890, 23, 3023.
4. Curtius, T. J. Prakt. Chem. 1894, 50, 275.
5. Smith, P. A. S. Org. React. 1946, 3, 337.
6. Boyer, J. H.; Canter, F. C. Chem. Rev. 1954, 54, 1.
7. Bräse, S.; Gil, C.; Knepper, K.; Zimmermann, V. Angew. Chem. Int. Ed.
2005, 44, 5188.
8. Scriven, E. F. V.; Turnbull, K. Chem. Rev. 1988, 88, 297.
9. Scriven, E. F. V. Azides and Nitrenes: Reactivity and Utility 1984,
Academic Press, Orlando, FL, USA.
10. Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Angew. Chem. Int. Ed. 2001, 40,
2004.
11. Binder, W. H.; Kluger, C. Curr. Org. Chem. 2006, 10, 1791.
12. Lin, T. S.; Prusoff, H. J. Med. Chem. 1978, 21, 109.
13. Dagley, I. J.; Spear, R. J. Organic Energetic Compounds 1996, Nova
Science Publishers Inc., New York, USA.
Page 97
Texas Tech University, Deepali Butani, August 2011
75
14. Haiges, R.; Boatz, A.; Vij, A.; Gerken, M.; Schneider, S.; Schroer, T.;
Christie, K.O. Angew. Chem. Int. Ed. 2003, 42, 5847.
15. Agrawal, J. P.; Hodgson, R.D. Organic Chemistry of Explosives 2007, John
Wiley & Sons, Inc., New York, USA .
16. Huynh , M. H. V. ; Hiskey, M. A.; Chavez, D. E.; Naud, D. L.; Gilardi, R.
D. J. Am. Chem. Soc.2005, 127, 12537.
17. Pauling, L.; Brockway, L. O. J. Am. Chem. Soc. 1937, 59, 13.
18. Brockway, L. O.; Pauling, L. Proc. Natl. Acad. Sci. USA 1933, 19, 860.
19. Tornieporth-Oetting, I. C.; Klapötke, T. M. Angew. Chem. 1995, 107, 559.
20. Tornieporth-Oetting, I. C.; Klapötke, T. M. Angew. Chem. Int. Ed. Engl.
1995, 34, 511.
21. Scriven, E. F. V.; Turnbull, K. Chem. Rev. 1988, 88, 297.
22. Cadogan, J. I. G. Acc. Chem. Res. 1972, 5, 303.
23. Moore, H. W. Acc. Chem. Res. 1979, 12, 125.
24. L'abbe G. Chem. Rev. 1969, 69 (3), 345–363.
25. Staudinger, H.; Meyer, J. Helv. Chim. Acta. 1919, 2, 635.
26. Reichen, W. Chem. Rev. 1978, 78, 569.
27. Ege, S. N.; Sherk, K. W. J. Am. Chem. Soc. 1953, 75, 354.
28. (a) Bose, A. K.; Kistner, J. F.; Farber, L. J. Org. Chem. 1962, 27, 2925. (b)
Boyer, J. H. J. Am. Chem. Soc. 1951, 73, 5865. (c) Brimacombe, J. S.;
Page 98
Texas Tech University, Deepali Butani, August 2011
76
Bryan, J. G. H.; Husain, A.; Stacey, M.; Telley, M. S. Carbohydr. Res.
1967, 3, 318. (d) Hedeyatullah, M.; Guy, A. Tetrahedron Lett. 1975, 2455.
(e) Smith, P. A. S.; Hall, J. H.; Kan, R. O. J. Am. Chem. Soc. 1962, 84, 485.
(f) Casini, G.; Goodman, L. Ibid. 1964, 86, 1427. (g) Nicolaides, E. D.;
Westland, R. D.; Wittle, E. L. J. Am. Chem. Soc. 1954, 76, 2887. (h)
Paulsen, H.; Kobernick, W.; Autschbach, E. Chem. Ber. 1972, 105, 1524. (i)
Secrist, J. A.; Logue, M. W. J. Org. Chem. 1972, 37, 335. (j) Mungall, W.
S.; Greene, G. L.; Heavner, G. A.; Letsinger, R. L. J. Org. Chem. 1975, 40,
1659. (k) Corey, E. J.; Nicolau, K. C.; Balanson, R. D.; Machida, Y.
Synthesis 1975, 590. (l) Staudinger, H.; Hauser, E. Helu. Chim. Acta 1921,
4, 861. (m) Horner, L.; Gross, A. Liebigs Ann. Chem. 1955, 591, 117. (n)
Vaultier, M.; Knouzi, N.; Carrie, R. Tetrahedron Lett. 1983, 24, 763. (o)
Sanevoshi. M.: Adachi, T.: Yamada, Y.; Inoue, I. Synthesis. 1977, 45. (p)
Bayley, H.; Standring, D. N.; Knowles, J. R. Tetrahedron Lett. 1978, 3633.
(q) Belinka, B. A., Jr.; Hassner, A. J. Org. Chem. 1979, 44, 4712. (r)
Hassner, A.; Levy, L. A. J. Am. Chem. Soc. 1965, 87, 4203. (s) Kirk, D. N.;
Wilson, M. A. J. Chem. Soc., Chem. Commun. 1970, 64. (t) Kirk, D. N.;
Wilson, M. A. J. Chem. Soc. 1971, 414. (u) Ho, T. L.; Henninger, M.; Olah,
G. A. Synthesis 1976, 815. (v) Stanovnik. B.: Tisler. M.: Polanc. S.:
Gracner. M. Svnthesis. 1978, 65. (w) Polanc, S.; Stanovnik, B.; Tisler, M.
Synthesis 1980, 830. (x) Frankel, M.; Wagner, D.; Gertner, D.; Zilkha, A. J.
Organomet.Chem. 1967, 7, 518. (y) Kloosterman, M.; Kuyl-Yeherskiely,
Page 99
Texas Tech University, Deepali Butani, August 2011
77
E.; van Boom, J. H. Recl. Trau. Chim. Pavs-Bas 1985, 104, 291. (z) Boyer,
J. H.; Ellzey, S. E., Jr. J. Org. Chem. 1958, 23, 127.
29. Lwowski, W. In 1, 3-Dipolar Cycloaddition Chemistry; Padwa, A., Ed.;
Wiley: New York, 1984, 1, 559.
30. (a) Regitz, M. In Methods of Preparative Organic Chemistry; Foerst, W.,
Ed.; Academic: New York, 1971, 4. (b) Reagents for Organic Synthesis 1,
1178; 2, 415; 3, 291; 4,510; 6, 597; 9, 472.
31. (a) Gageaux, A; Winstein, S; Young, W.G. J. Am. Chem. Soc. 1960, 82,
5956. (b) Vander Werf, C. A.; Heasley, V.L. J. Org. Chem. 1966, 31, 3534.
(c) Arimoto, M..; Yamaguchi, H.; Fujita, E.; Ochai, M.; Nagao, Y.
Tetrahedron Lett. 1987, 28, 6289. (d) Chida, N.; Tobe, T.; Murai, K.;
Yamazaki, K.; Ogawa, S. Heterocycles 1994, 38, 2383. (e) Trost, B.M.;
Pulley, S.R. Tetrahedron Lett. 1995, 36, 8737.
32. (a) Murahashi, S. I.; Tanigushi, Y.; Imada, Y.; Tanigawa, Y. J. Org. Chem.
1989, 54, 3292–3303. (b) Safi, M.; Fahrang, R.; Sinou, D. Tetrahedron Lett.
1990, 31, 527–530. (c) Arimoto, M.; Yamaguchi, H.; Fujita, E. Tetrahedron
Lett. 1987, 28, 6289–6292.
33. Lauzon, S.; Tremblay, F.; Gagnon, D.; Godbout, G.; Chabot, C.; Mercier-
Shanks, C.; Perreault, S.; DeSéve, H.; Spino, C. J. Org. Chem. 2008, 73,
6239–6250.
34. Panek, J. S.; Yang, M.; Muler, I. J. Org. Chem. 1992, 57, 4063–4064.
Page 100
Texas Tech University, Deepali Butani, August 2011
78
35. Feldman, A. K.; Colasson, B.; Sharpless, K. B.; Fokin, V. V. J. Am.Chem.
Soc. 2005, 127, 13444–13445.
36. Lauzon, S.; Tremblay, F.; Gagnon, D.; Godbout, C.; Chabot, C.; Mercier-
Shanks, C.; Perreault, S.; DeSéve, H.; Spino, C. J. Org. Chem. 2008, 73,
6239–6250.
37. Vander Werf, C. A.; Heisler, R. Y.; McEwen, W. E. J. Am. Chem. Soc.
1954, 76, 1231.
38. (a) Padwa, A.; Sá, M. M. Tetrahedron Letters 1997, 38 (29), 5083. (b) Lee,
E.E. Angew. Chem. Int. Ed. 2004, 43, 1865-1868. (c) Takasu, H.; Tsuji, Y.;
Sajiki, H.; Hirota, K. Tetrahedron 2005, 61, 11027-11031. (d) Chang, Y.
K.; Lo, H. J.; Yan, T. H. Org. Lett. 2009, 11, 4278-4281. (e) Gagnon, D.;
Lauzon, S.; Godbout, C.; Spino, C. Org. Lett. 2005, 7, 4769-4771. (f)
Cardillo, G.; Fabbroni, S.; Gentilucci, L.; Perciaccante, R.; Piccinelli, F.;
Tolomelli, A. Org. Lett. 2005, 7, 533-536. (g) Sá, M.M. J. Braz. Chem. Soc.
2003, 14, 1005-1010. (h) Klepper, F.; Jahn, E.M.; Hickmann, V.; Carell, T.
Angew. Chem. Int. Ed. 2007, 46, 2325-2327. (i) Fava, C.; Galeazzi, R.;
Mobbilli, G.; Orena, M. Tetrahedron: Asymmetry 2001, 12, 2731-2741.
39. The full reference for Gaussian 03, Revision C.02, Frisch, M. J.; Trucks, G.
W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.;
Montgomery, Jr., J. A.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J.
M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.;
Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara,
Page 101
Texas Tech University, Deepali Butani, August 2011
79
M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda,
Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.;
Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.;
Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.;
Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.;
Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain,
M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.;
Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.;
Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.;
Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng,
C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.;
Chen, W.; Wong, M. W.; Gonzalez, C.; and Pople, J. A.; Gaussian, Inc.,
Wallingford CT, 2004.
40. Hariharan, P. C.; Pople, J. A. Theor. Chim. Acta 1973, 28, 213.
41. Head-Gordon, M.; Pople, J. A.; Frisch, M. J. Chem. Phys. Lett. 1988, 153,
503.
42. Frisch, M. J.; Head-Gordon, M.; Pople, J. A. Chem. Phys. Lett. 1990, 166,
275.
43. Frisch, M. J.; Head-Gordon, M.; Pople, J. A. Chem. Phys. Lett. 1990, 166,
281.
44. Saebo, S.; Almlof, J. Chem. Phys. Lett. 1989, 154, 83.
45. Møller, C.; Plesset, M. S. Phys. Rev. 1934, 46, 618.
Page 102
Texas Tech University, Deepali Butani, August 2011
80
46. Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785.
47. Becke, A. D. J. Chem. Phys. 1993, 98, 5648.
48. Wodrich, M. D.; Corminboeuf, C.; Schreiner, P. R.; Fokin, A. A.; Schleyer,
P. V. R. Org. Lett. 2009, 9, 1851.
49. Tirado-Rives, J.; Jorgensen, W. L. J. Chem. Theory Comput. 2008, 4, 297.
50. Sousa, S. F.; Fernandes, P. A.; Ramos, M. J. J. Phys. Chem. A 2007, 111,
10439.
51. Wodrich, M. D.; Corminboeuf, C.; Schleyer, P. V. R. Org. Lett. 2006, 8,
3631.
52. Schreiner, P. R.; Fokin, A. A.; Robert A. Pascal, J.; Meijere, A. D. Org.
Lett. 2006, 8, 3635.
53. Wiest, O.; Montiel, D. C.; Houk, K. N. J. Phys. Chem. A 1997, 101, 8378.
54. Curtiss, L. A.; Raghavachari, K.; Redfern, P. C.; Pople, J. A. J. Chem.
Phys. 1997, 106, 1063-1079.
55. Houk, K. N.; Gonzalez, J.; Li, Y. Acc. Chem. Res. 1995, 28, 81.
56. Hermida-Ramón, J. M.; Rodríguez-Otero, J.; Cabaleiro-Lago, E. M. J. Phys.
Chem. A 2003, 107, 1651–1654.
57. Ji, H.; Li, L.; Xu, X.; Ham, S.; Hammad, L. A.; Birney, D. M. J. Am. Chem.
Soc. 2009, 131, 528–537.
58. (a) Woodward, R. B.; Hoffmann, R. Angew. Chem. 1969, 81, 797. (b)
Woodward, R. B.; Hoffmann, R. Angew. Chem. Int. Ed. Engl. 1967, 8, 781.
(c) Hoffmann, R.; Woodward, R. B. Science. 1970, 167, 825. (d)
Page 103
Texas Tech University, Deepali Butani, August 2011
81
Hoffmann, R.; Woodward, R. B. Acc. Chem. Res. 1968, 1, 17. (e)
Hoffmann, R.; Woodward, R. B. J. Am. Chem. Soc. 1965, 87, 2046.
Page 104
Texas Tech University, Deepali Butani, August 2011
82
CHAPTER III
SYNTHESIS OF PENTADIENYL ALCOHOL DERIVATIVES AND STUDY OF
THEIR POSSIBLE [3, 3] AND [3, 5] SIGMATROPIC REARRANGEMENTS
3.1 REARRANGEMENT OF ESTERS
3.1.1 Background
After initial discovery of [3, 3]-sigmatropic rearrangements their scope and
utility has been greatly expanded.1,2
A wide variety of methods have been reported for
promoting the Cope and the Claisen rearrangements, including the use of Brønsted
acids, Brønsted bases, Lewis acids, and transition metals. These developments led to
the use of milder reaction conditions and, therefore, the examination of more complex
and synthetically relevant substrates.3
Lewis and coworkers in late 1960’s were the first to observe the rearrangement
of allylic esters, also known as [1, 3]-dioxa-Cope rearrangement.4-6
They observed the
rearrangement in the gas phase and the reaction was proposed to be a sigmatropic
rearrangement, proceeding via a six-membered cyclic transition state as shown in
Structure 1 of Scheme 3.1. They also considered the possibility of a four-membered
cyclic transition state as shown in Structure 2 of Scheme 3.1.
Page 105
Texas Tech University, Deepali Butani, August 2011
83
Scheme 3.1
Unlike the closely related Claisen rearrangement, this reaction is an
equilibrium process, in which the product distribution is governed by the relative
thermodynamic stabilities of the two allylic isomers. Therefore, strategies that
recognize this are required for the [1, 3]-dioxa-Cope rearrangement to become
synthetically useful for high product selectivity. At the same time rearrangement of
allylic esters using a mild catalyst have shown some promising results.7
in related
work, Overman and co-workers found that both allylic acetates and allylic carbamates
rearranged in the presence of either mercury(II) 8, 9
or palladium(II) 10-12
catalysts gave
moderate to high levels of product selectivity as shown in Scheme 3.2. Use of
PdCl2(NCCH3)2 as catalyst gave a high level of chirality transfer with observed
enantiomeric selectivity of rearrangement of allylic acetates.10-12
This method has
been proven useful in the synthesis of several natural products, including steroids,13
amino acids,14
and prostaglandins.15
Overman classified the transition-metal-
catalyzed reactions as “cyclization-induced” rearrangements, proposing a unique
mechanism for rearrangement of esters. The key feature of this mechanism was that
the metal acted as an electrophilic catalyst, binding to the alkene and activating it
toward nucleophilic attack by the pendant carbonyl oxygen to generate a cyclic,
organometallic intermediate 3 as shown in Scheme 3.3.7
Page 106
Texas Tech University, Deepali Butani, August 2011
84
Scheme 3.2
Scheme 3.3: Proposed mechanism of “cyclization-induced” rearrangements.7
3.1.2 Computational Study on Rearrangement of Esters
Wessely and coworkers16
observed that acyloxycyclohexadione 4 undergoes
thermal rearrangements. The observed phenol products, 7 and 9 were proposed to arise
from tautomerization of the rearranged acyloxycyclohexadiones 5 and 8 (and the acyl
migration of 6 to 7, Scheme 3.4). These authors proposed competing [3, 3] and [3, 5]
sigmatropic rearrangements, to form 5 and 8, respectively. They could not rule out
sequential [3, 3] rearrangements of 4 to 8 to 5. This work was published prior to the
Woodward-Hoffmann rules.50
Subsequent publications assumed that, the [3, 5]
rearrangement was forbidden, in analogy to the hydrocarbon system, until Birney et al.
revisited the system.17
Page 107
Texas Tech University, Deepali Butani, August 2011
85
Scheme 3.4
Birney and coworkers17
who were interested in study of pseudopericyclic
reactions, choose 2,4-cyclohexadienyl formate (10) as a model system (Scheme 3.5)
for understanding the rearrangements of acyloxycyclohexadiones seen earlier. They
calculated that the barrier for the [3, 5] sigmatropic rearrangement of 10 to 10 was 3.0
kcal/mol lower than the calculated barrier for the related [3, 3] sigmatropic
rearrangement of 10 to 11. This is because the [3, 3] rearrangement has a boat-shaped
transition state whereas the [3, 5] rearrangement has a transition state,
pseudopericyclic in nature, with the breaking and forming bonds in the same plane of
the ester. Figure 3.1 shows the calculated geometries and energies of the structures
involved in the rearrangements.
Page 108
Texas Tech University, Deepali Butani, August 2011
86
Scheme 3.5
Figure 3.1: Energy profile showing the [3, 3] rearrangement of 2,4-cyclohexadienyl
formate, 10 to 11 (right side) and the degenerate [3, 5] rearrangement of 2,4-
cyclohexadienyl formate, 10 (left side), with their transition states (10ǂ and 11
ǂ) in
between. The geometries were calculated at the MP2/6-31G** level of theory and the
relative energies were calculated at the MP4/6-31G** + ZPV (kcal/mol) level of
theory.17
They also did studies on simplest formate ester ((Z)-penta-2,4-dienyl formate,
12, Scheme 3.6) to possibly see the [3, 5] rearrangement.17
But instead the
Page 109
Texas Tech University, Deepali Butani, August 2011
87
optimization of transition state constrained to Cs symmetry led not to a [3, 5]
rearrangement but to a boat transition state for a [3, 3] rearrangement, 13, Scheme 3.6.
Figure 3.2 shows the schematic representation of the rearrangement. One conclusion
from this study is that a [3, 5] rearrangement to be allowed the distance between
reactive centers needs to be close enough for rearrangement to occur.
Scheme 3.6
Figure 3.2: Energy profile showing 12 forming a boat transition state leading to the
[3, 3] rearrangement of 13. The relative energies in kcal/mol were calculated at the
MP4/6-31G** level of theory.17
Page 110
Texas Tech University, Deepali Butani, August 2011
88
3.1.3 Proposed alcohol molecules
After carefully reviewing the calculation on esters, we wanted to study
possible [3, 3] and [3, 5] sigmatropic rearrangements of esters experimentally. Our
main aim was to design such as ester which can undergo both [3, 3] and [3, 5]
rearrangements. Hence, the selection of alcohol became an important factor. The
alcohol molecule was designed in such a way that the ester formed from alcohol
should have the oxygen (which would be one of the reactive centers) closer to C=C
where the rearrangement would take place. So, we examined a series of pentadienyl
alcohols (14-18). Compound 15 has the methanol group attached to a six-membered
ring (cyclohexene ring) with a vinyl group at the other end of the double bond. Also,
we added a phenyl group to the carbon next to alcohol in 16. All this was done to
make sure that it can form an eight-centered transition state. Then we did the same for
the five-membered ring structures as well (17 and 18). Table 3.1 shows the strain
energy, total energy and potential energy of each molecule was calculated using
MM2.51
Figure 3.3 and Figure 3.4 show the minimized structure of the proposed
pentadienyl alcohols for further experiments.
Page 111
Texas Tech University, Deepali Butani, August 2011
89
Table 3.1: Calculated strain energy, total energy and potential energy of minimized
structure of a series of pentadienyl alcohols using MM2.
Structure Strain Energy
(kcal/mol)
Total Energy
(kcal/mol)
Potential Energy
(kcal/mol)
OH
14
-2.03 24.66 9.86
OH
15
-1.14 43.28 23.35
OHPh
16
0.92 64.26 32.81
OH
17
-1.17 39.38 18.81
OHPh
18
0.41 57.46 27.40
Page 112
Texas Tech University, Deepali Butani, August 2011
90
16
Figure 3.3: MM2 minimized structure of phenyl(2-vinylcyclohex-1-enyl)methanol
(16, side viewed from two different directions) where blue are H-atoms, grey are
carbon atoms, red are oxygen atoms and pink are lone pair orbitals.
18
Figure 3.4: MM2 minimized structure of phenyl(2-vinylcyclopent-1-enyl)methanol
(18, side viewed from two different directions) where blue are H-atoms, grey are
carbon atoms, red are oxygen atoms and pink are lone pair orbitals.
Page 113
Texas Tech University, Deepali Butani, August 2011
91
3.1.4 Proposed synthesis of phenyl(2-vinylcyclopent-1-enyl)methyl
acetate (25)
We proposed to synthesize phenyl(2-vinylcyclopent-1-enyl)methyl acetate, 25
(Figure 3.5) because one of the reactive centers (i.e. O from C=O) is closer to the
other (H attached to C=C). Also, it has enough strain to undergo not only [3, 3]-
sigmatropic rearrangement but also [3, 5]-sigmatropic rearrangement. The minimized
structures of proposed [3, 3] and [3, 5] rearranged products are shown in Figure 3.6.
Also, Table 3.2 shows the strain energy; total energy of molecule and potential energy
of different acetates and their rearranged products are calculated using MM2.
Table 3.2: Calculated strain energy, total energy and potential energy of minimized
structures of various possible acetates and their possible [3, 3] and [3, 5] rearranged
products using MM2.
Structure Strain
Energy
(kcal/mol)
Total
Energy
(kcal/mol)
Potential
Energy
(kcal/mol)
Total Energy
difference in
rearrangement
(kcal/mol)
O
O
19
1.40 35.30 17.76 -
O
O
20
2.52 39.57 18.94 4.26
Page 114
Texas Tech University, Deepali Butani, August 2011
92
Table 3.2: Continued
Structure Strain
Energy
(kcal/mol)
Total
Energy
(kcal/mol)
Potential
Energy
(kcal/mol)
Total Energy
difference in
rearrangement
(kcal/mol)
O
O
21-cis
1.93 44.28 23.36 8.98
O
O
21-trans
1.57 41.89 19.89 6.59
O
O
22
3.13 53.77 24.93 -
O
O
23
5.19 56.00 33.55 2.06
O
O
24 (E)
8.87 58.57 32.64 5.74
Page 115
Texas Tech University, Deepali Butani, August 2011
93
Table 3.2: Continued
Structure Strain
Energy
(kcal/mol)
Total
Energy
(kcal/mol)
Potential
Energy
(kcal/mol)
Total Energy
difference in
rearrangement
(kcal/mol)
O
O
24 (Z)
9.97 61.27 35.99 7.50
O
PhO
25
4.22 76.01 40.96 -
Ph
O
O
26
6.85 74.57 41.33 -1.44
Ph
O
O
27(E)
10.41 77.15 45.19 1.14
Ph
O
O
27(Z)
13.72 79.82 46.05 3.81
Page 116
Texas Tech University, Deepali Butani, August 2011
94
The results in Table 3.2 suggest that the [3, 5] sigmatropic rearrangement of acetate 25
is the least endothermic and might be anticipated to be observed.
25
Figure 3.5: MM2 minimized structure of phenyl(2-vinylcyclopent-1-enyl)methyl
acetate (25) where blue are H-atoms, grey are carbon atoms, red are oxygen atoms and
pink are lone pair orbitals.
Figure 3.6: MM2 minimized structure of [3, 3] (26) and [3, 5] (27) rearrangement
products from phenyl(2-vinylcyclopent-1-enyl)methyl acetate (25); where blue are H-
atoms, grey are carbon atoms, red are oxygen atoms and pink are lone pair orbitals.
Page 117
Texas Tech University, Deepali Butani, August 2011
95
The following scheme shows the methods used for the synthesis of phenyl(2-
vinylcyclopent-1-enyl)methyl acetate (25, Scheme 3.7).
Scheme 3.7
3.1.5 Proposed synthesis of phenyl(2-vinylcyclohex-1-enyl)methyl
acetate (33)
Along the same lines we decided to study the rearrangement on phenyl(2-
vinylcyclohex-1-enyl)methyl acetate (33, Figure 3.7). We examined the MM2
minimized structures of all different acetates with possible [3, 3] and [3, 5]
sigmatropic rearrangements as shown in Table 3.3.
Page 118
Texas Tech University, Deepali Butani, August 2011
96
Table 3.3: Calculated strain energy, total energy and potential energy of minimized
structures of various possible acetates and their possible [3, 3] and [3, 5] rearranged
products using MM2.
Structure Strain
Energy
(kcal/mol)
Total
Energy
(kcal/mol)
Potential
Energy
(kcal/mol)
Total Energy
difference in
rearrangement
(kcal/mol)
O
O
30
2.61 60.80 30.69 -
OO
31
7.65 63.31 35.84 2.51
O
O
32 (E)
5.95 61.78 35.54 0.98
O
O
32 (Z)
5.88 60.31 35.55 -0.49
PhO
O
33
8.01 78.76 44.68 -
Page 119
Texas Tech University, Deepali Butani, August 2011
97
Table 3.3: Continued
Structure Strain
Energy
(kcal/mol)
Total
Energy
(kcal/mol)
Potential
Energy
(kcal/mol)
Total Energy
difference in
rearrangement
(kcal/mol)
Ph
OO
34
10.83 82.83 46.03 4.07
Ph
O O
35(E)
6.79 80.73 40.56 1.97
Ph O
O
35(Z)
6.50 80.33 41.80 1.57
Page 120
Texas Tech University, Deepali Butani, August 2011
98
33
Figure 3.7: MM2 minimized structure of phenyl(2-vinylcyclohex-1-enyl)methyl
acetate (33) where blue are H-atoms, grey are carbon atoms, red are oxygen atoms and
pink are lone pair orbitals.
Figure 3.8: MM2 minimized structure of [3, 3], 34 and [3, 5], 35 rearrangement
products from phenyl(2-vinylcyclohex-1-enyl)methyl acetate (33); where blue are H-
atoms, grey are carbon atoms, red are oxygen atoms and pink are lone pair orbitals.
Page 121
Texas Tech University, Deepali Butani, August 2011
99
The synthesis of phenyl(2-vinylcyclohex-1-enyl)methyl acetate (33, Scheme 3.8) was
similar to that of 25 (above).
Scheme 3.8
3.1.6 Flash Vacuum Pyrolysis
Flash vacuum pyrolysis (FVP) is experimental technique involving the vacuum
sublimation or distillation of a substrate through a hot tube (generally at temperatures
between 300–1000 °C) to induce chemical change. After passing through the tube, the
products are quenched at low temperatures in a trap placed at the exit point of the
tube.18,19
The pyrolysis therefore takes place under dilute, short contact time
conditions in the gas phase such that individual molecules react intramolecularly in the
effective absence of other molecules of substrate, product or substantial amounts of
oxygen. The technique is therefore known to produce much cleaner results than other
forms of pyrolysis. Flash Vacuum Pyrolysis is a robust, highly reproducible method
and has found widespread use in applications ranging from matrix isolation and
Page 122
Texas Tech University, Deepali Butani, August 2011
100
mechanistic studies to preparative organic chemistry.20
As is the case with any method
which depends on gas flow, the residence time of substrate molecules in the hot
(reacting) zone is a key parameter,21
and this in turn depends on a number of variables
including the pressure and temperature, the dimensions of the tube, throughput rate
and the presence of any material.21
Whereas static pyrolysis which are usually carried out in a sealed tube or by
reflux in presence of some solvent, for a longer period of time, this sometimes does
not yield clean products. However, in a flow system for FVP, compounds are exposed
to heat for short time and the pyrolysates are cooled immediately to very low
temperatures. This prevents bimolecular reactions from happening. Therefore, flash
pyrolysis requires higher temperatures to compensate for the short contact times.22
The
FVP method is used to study highly reactive intermediates or very unstable organic
compounds. The compounds or fragments formed in the pyrolysis are immediately
cooled down to -198 °C or trapped by other compounds so they can be further studied.
The method can also be used preparatively for synthesis of larger amounts of
compounds. For example, FVP of indanetrione and of phthalic anhydride as shown in
Scheme 3.9.23
Page 123
Texas Tech University, Deepali Butani, August 2011
101
Scheme 3.9
In our experiment we were studying the rearrangement of phenyl(2-
vinylcyclopent-1-enyl)methyl acetate (25) and phenyl(2-vinylcyclohex-1-enyl)methyl
acetate (33) in which a C-O bond had to be broken. The enthalpy of C-O bond is 358
kJ/mol which meant that C-O bond breaking would require a large amount of energy.
Experimental Procedure used for flash vacuum pyrolysis
The Flash Vacuum Pyrolysis technique was used to study rearrangement of
phenyl(2-vinylcyclopent-1-enyl)methyl acetate (25) and phenyl(2-vinylcyclohex-1-
enyl)methyl acetate (33). The experiment was carried out between 270-370 °C on the
apparatus as shown in Figure 3.9.
Page 124
Texas Tech University, Deepali Butani, August 2011
102
Figure 3.9: Flash Vacuum Pyrolysis (FVP) setup.
Table 3.4: Flash Vacuum Pyrolysis (FVP) experimental setup.
Number Description
1 Quartz tube with one end closed, sample is placed directly in quartz tube- the
quartz tube used is 66 cm length and 2.54 cm in diameter.
2 Digital thermometer
3 Pyrolysis oven, Heavy duty Heating Equipment
4 U-shape product collector tube
5 Dewar flask- used for cooling the pyrolysates at -198 °C
Approximately 100-200 mg of sample (25 or 33) was dissolved in anhydrous
diethyl ether (because they are very viscous, they need to be diluted for transfer) was
placed in the quartz tube (1). A vacuum of 0.1 torr was drawn on the quartz tube to
remove residual solvents such as diethyl ether, hexane or ethyl acetate. Then the
Page 125
Texas Tech University, Deepali Butani, August 2011
103
equipment was set up as shown in Figure 3.9. The liquid nitrogen bath is kept under
the U-tube to cool the pyrolysates. The pyrolysis oven was adjusted to a proper
temperature and the digital thermometer was set in the middle of the oven to read the
temperature of the oven. After the oven had reached a required temperature, the closed
end of the quartz tube was slowly pushed inside the oven. This was continued until the
closed end of the tube had gone inside the oven and the sample had vaporized the
quartz tube. The products were collected either at the end of the end of quartz tube or
in the U-tube. After the pyrolysis tube cooled, the system was filled with nitrogen. The
U-tube was removed from the system and warmed up to room temperature. A 1H-
NMR spectrum was obtained for crude the pyrolysis products collected both at the end
of the pyrolysis tube and in the U-tube. The samples obtained were then
chromatographed on silica gel column using 2% ethyl acetate solution in hexane to
separate the different products. The verification of products formed was done by 1H-
NMR and 13
C-NMR.
3.1.7 Result and Discussion
Although Flash Vacuum Pyrolysis experiments are highly reproducible using
one set of apparatus, the variables are often not specified in literature reports of FVP
applications, so it can be difficult to reproduce such conditions in another laboratory
without carrying out a series of trial experiments. In the work described here, we have
therefore systematically performed the experiments at varied temperatures and
monitored the effect of temperature on conversion to products.
Page 126
Texas Tech University, Deepali Butani, August 2011
104
The possible products of pyrolysis of phenyl(2-vinylcyclopent-1-enyl)methyl
acetate (25, Scheme 3.10).
Scheme 3.10
However, when we conducted pyrolysis of phenyl(2-vinylcyclopent-1-enyl)methyl
acetate (25) which gave the following results (Table 3.5):
Table 3.5: Products formed on pyrolysis of phenyl(2-vinylcyclopent-1-enyl)methyl
acetate (25) at two different temperatures.
Temperature Product formed
270-280°C 1. Only starting material, 25 (analyzed by 1H-NMR)
300-330°C
1. The 1H-NMR of the crude was a mixture of starting material and
possible peaks for [3, 5] rearranged product (27(E) or 27 (Z)).
2. Starting material, 25, 80% (obtained from column
chromatography on silica gel using 2% ethyl acetate in hexane).
3. Possibly small amount of [3, 5] rearrangement, 27 (obtained
from column chromatography) was a mixture with starting
material 25 (1H-NMR shown in Figure 3.10). The possible [3, 5]
rearrangement product, 27 is either due to expected [3, 5]
rearrangement (27-Z) or two sequential [3, 3] rearrangements
(27-E).
Page 127
Texas Tech University, Deepali Butani, August 2011
105
Figure 3.10: 1H-NMR from FVP of 25, obtained from column chromatography where
boxed signals in region from 8.75 ppm to 5.00 ppm are possibly from [3, 5] rearrangement.
Page 128
Texas Tech University, Deepali Butani, August 2011
106
Though we were able to see possibility of small amount of [3, 5]
rearrangement with a mixture of starting material (analysis done with 1H-NMR and
13C-NMR) but this experiment still needs to be optimized to get the exact ratio of
rearrangement. It needs to be conducted at various temperatures and a study of the
different products formed with increasing temperature may allow the differentiation of
direct [3, 5] as compared to sequential [3, 3] rearrangement mechanism.
The possible products of pyrolysis of phenyl(2-vinylcyclohex-1-enyl)methyl
acetate (33) are shown in Scheme 3.11.
Scheme 3.11
However, when we conducted pyrolysis of phenyl(2-vinylcyclohex-1-enyl)methyl
acetate (33) which gave the following results (Table 3.6):
Page 129
Texas Tech University, Deepali Butani, August 2011
107
Table 3.6: Products formed on pyrolysis of phenyl (2-vinylcyclohex-1-enyl) methyl
acetate (33) at three different temperatures.
Temperature Product formed
330-350°C 1. β-elimination, 37 as major product, 40% (after column chromatography
in silica gel using 2% ethyl acetate in hexane).
2. Starting material, 33 (after column chromatography).
3. Possibly small amount of [3, 5] rearrangement, 35 (obtained from
column chromatography) was a mixture with starting material 33 (1H-
NMR shown in Figure 3.11). The possible [3, 5] rearrangement product,
35 is either due to expected [3, 5] rearrangement (35-Z) or two
sequential [3, 3] rearrangements (35-E).
350-370°C 1. Completely dissociated to give an unidentified product containing
phenyl group (analyzed by 1H-NMR).
303-330°C 1. β-elimination, 37, 50% (after column chromatography).
2. Starting material, 33 (after column chromatography).
In case of phenyl(2-vinylcyclohex-1-enyl)methyl acetate (33) we were able to
identify 37 as the major product from the pyrolysis at 330 to 350 °C and also 303 to
330 °C. This presumably is formed by β-elimination from 34. There were also peaks
on 1H-NMR and
13C-NMR from the pyrolysis at 330-350 °C that could correspond to
35, the expected product from [3, 5] rearrangement (or two sequential [3,3]
rearrangements). We also need to repeat this experiment at several different
Page 130
Texas Tech University, Deepali Butani, August 2011
108
temperatures to see if we can observe more of the [3, 5] product and the possibility of
the [3, 3] product as well.
Figure 3.11: 1H-NMR from FVP of 33, obtained from column chromatography
where boxed signals in region between 6.6 ppm to 4.50 ppm are possibly from [3, 5]
rearrangement.
3.2 REARRANGEMENT OF TRICHLOROACETIMIDATES
3.2.1 Background
The thermal rearrangement of allylic imidates (e.g. 38 to 39) also known as the
“aza-Claisen” or “Claisen-imidate”, was discovered in 1937.24
Since then a number of
systems have been investigated for the practical preparation of allylic amides like
urethanes, isourethanes, formimidates, benzimidates, isourears and
Page 131
Texas Tech University, Deepali Butani, August 2011
109
carbonimidothioates. However, it is the discovery and development of the
rearrangement of allylic trichloroacetimidates that has led to the widest application
(Scheme 3.12).25-27
Scheme 3.12: Rearrangement of allylic trichloroacetimidates.
The [3, 3]-sigmatropic rearrangement of allylic tricholoroacetimidates is
generally referred to as Overmann rearrangement. It is conveniently carried out either
thermally or with Hg(II) or Pd(II) catalysis.28
The scope of the rearrangement is
readily accessible for primary, secondary, and tertiary allylic amides, thus providing
entry into a wide variety of nitrogen-containing products including amino sugars,
nucleotides, amino acids, peptides, and various nitrogen heterocycles. In addition, the
Overman rearrangement has found extensive application in the total synthesis of
natural products. Also, the development of chiral Pd(II) catalysts to promote
asymmetric allylic trichloroacetimidates rearrangements with good enantioselectivity
bodes well for the continued with broad application of the amine synthesis.29-31
Mechanism of Thermal rearrangements
The thermal rearrangement of allylic trichloroacetimidates is an irreversible
process because the enthalpy or driving force associated with conversion of the
imidate (e.g. 38) to the amide (e.g. 39) functionality is very large.32
The thermal [3, 3]
Page 132
Texas Tech University, Deepali Butani, August 2011
110
sigmatropic rearrangement occurs via a concerted process via a cyclic six-centered
transition state as shown in Figure 3.12.33
The activation parameters observed for the
allylic trichloroacetimidates rearrangement are ΔH#= 24 kcal/mol and ΔS
#= -19 eu,
which are typical of those observed for other [3, 3]-sigmatropic rearrangements.34
Hence, only small increases in rate35
are observed upon changing solvent from xylene
to nitrobenzene. Larger increased rate are the result of the attachment of carbocation
stabilizing groups to the imidate bearing carbon suggesting that some charge
separation in the transition state.
Figure 3.12: Cyclic six-centered transition state of the [3, 3] rearrangement of allylic
imidates where R, R1, R2, R3, R4 are various alkyl groups.
An example of the thermal rearrangement of trichloroacetimidic ester derived
from secondary alcohol, gave exclusively (E)-trichloroacetimidates, which is expected
from the large steric bulk of the tricholomethyl substitutent and the usual chair model
for the cyclic six-membered transition state (shown in Scheme 3.13).36
However, a
pseudopericyclic transition state (planar on the imidate) is also possible.
Page 133
Texas Tech University, Deepali Butani, August 2011
111
Scheme 3.13
Mechanism of Metal-Catalyzed Rearrangements
The rearrangement of allylic trichloroacetimidates using metal catalysts not
only lowers the temperature required for rearrangement but also leads to higher yields,
cleaner reactions and better sterocontrol. Many allylic trichloroacetimidates, ranging
from simple allylic trichloroacetimidates to highly functionalized substrates, rearrange
rapidly in presence of Pd(II) or Hg(II) catalysts. A cyclization-induced rearrangement
mechanism in which the metal coordinates to the allylic double bond to bring about
antarafacial intramolecular nucleophilic attack by the imidate nitrogen is believed to
be involved in Pd(II)- or Hg(II)-catalyzed rearrangements (as shown in Scheme
3.14).37
Page 134
Texas Tech University, Deepali Butani, August 2011
112
Scheme 3.14
3.2.2 Proposed synthesis of phenyl(2-vinylcyclopent-1-enyl)methyl
2,2,2-trichloroacetimidate (41)
As a part of our effort to explore the differences between pericyclic and
pseudopericyclic reactions we decided to study the [3, 3] and [3, 5] sigmatropic
rearrangement of phenyl(2-vinylcyclopent-1-enyl)methyl 2,2,2-trichloroacetimidate
(41, Figure 3.13). We calculated the MM2 strain energy, total energy and potential
energy of minimized structures of trichloroacetimidate (41) and possible [3, 3], (42)
and [3, 5], (43) sigmatropic rearrangement products as shown in Table 3.7. The
calculated MM2 energies suggest that the [3, 5] rearrangement of 41 may be less
favored as compared to that of acetate 25.
Page 135
Texas Tech University, Deepali Butani, August 2011
113
41
Figure 3.13: MM2 minimized structure of phenyl(2-vinylcyclopent-1-enyl)methyl
2,2,2-trichloroacetimidate (41) where blue are N-atom, grey are C- atoms, red are O-
atom, pink are lone pair orbitals, green are Cl-atoms and white are H-atoms.
Page 136
Texas Tech University, Deepali Butani, August 2011
114
Table 3.7: Calculated strain energy, total energy and potential energy of minimized
structures of trichloroacetimidate, 41 and their possible [3, 3], (42) and [3, 5], (43)
rearranged products using MM2.
Structure Strain
Energy
(kcal/mol)
Total
Energy
(kcal/mol)
Potential
Energy
(kcal/mol)
Total Energy
difference in
rearrangement
(kcal/mol))
O
Ph NH
CCl3
41
5.90 74.18 40.32 -
NHCOCCl3
Ph
42
-3.03 63.15 29.27 -11.03
Ph
NHCOCCl3
43(E)
4.77 72.12 40.61 -2.06
Ph
NHCOCCl3
43(Z)
7.62 70.43 38.61 -3.75
Page 137
Texas Tech University, Deepali Butani, August 2011
115
Trichloroacetimidates are readily prepared by the reaction of alcohols with
trichloroacetonitrile (Cl3CCN) with base catalysis. Several bases were screened for the
preparation of phenyl(2-vinylcyclopent-1-enyl)methyl 2,2,2-trichloroacetimidate (41)
from the alcohol 18 (Scheme 3.15). The best results were obtained with 0.1 eq. of
DBU relative to the alcohol.
Scheme 3.15
3.2.3 Proposed synthesis of phenyl(2-vinylcyclohex-1-enyl)methyl
2,2,2-trichloroacetimidate (44)
We also decided to study the [3, 3] and [3, 5] sigmatropic rearrangement of
phenyl(2-vinylcyclohex-1-enyl)methyl 2,2,2-trichloroacetimidate (44, Figure 3.14).
We calculated MM2 strain energy, total energy and potential energy of minimized
structures of trichloroacetimidate, 44 and possible [3, 3], (45) and [3, 5], (46)
sigmatropic rearrangement products as shown in Table 3.8. These results suggest that
the [3, 5] rearrangement to form 46 may be favored.
Page 138
Texas Tech University, Deepali Butani, August 2011
116
44
Figure 3.14: MM2 minimized structure of phenyl(2-vinylcyclohex-1-enyl)methyl
2,2,2-trichloroacetimidate (44) where blue are N-atom, grey are C- atoms, red are O-
atom, pink are lone pair orbitals, green are Cl-atoms and white are H-atoms.
Page 139
Texas Tech University, Deepali Butani, August 2011
117
Table 3.8: Calculated strain energy, total energy and potential energy of minimized
structures of trichloroacetimidate, 44 and their possible [3, 3], (45) and [3, 5], (46)
rearrangement products using MM2.
Structure Strain
Energy
(kcal/mol)
Total
Energy
(kcal/mol)
Potential
Energy
(kcal/mol)
Total Energy
difference in
rearrangement
(kcal/mol)
O
Ph NH
Cl3C
44
5.00 95.18 53.69 -
NHCOCCl3
Ph
45
1.52 69.46 34.60 -22.72
Ph
NHCOCCl3
46(E)
2.12 69.20 33.77 -26.98
Ph NHCOCCl3
46(Z)
2.64 66.37 32.66 -28.81
Page 140
Texas Tech University, Deepali Butani, August 2011
118
The synthesis of phenyl(2-vinylcyclohex-1-enyl)methyl 2,2,2-trichloroacetimidate
(44, Scheme 3.16) from the alcohol, 16 was carried out similarly to that of 41, above.
Scheme 3.16
3.2.4. Results and Discussion
We tried to synthesize compound 41 and compound 44 at different
temperatures (-78 °C, -20 °C, 0 °C and r.t.) using different solvents (ether, DCM and
toluene) and bases (NaH, KH and DBU). The crude NMR spectra showed compounds
41 and compound 44. However, these were very reactive and rearranged to [3, 3]
products (as evident in 1H-NMR) as shown in Scheme 3.17 and 3.18. The problem
with this is that we have not been able to separate the products and also have not been
able to calculate the ratio of sigmatropic rearrangement. Further experiments are
ongoing to stabilize trichloroacetimidates and study the [3, 3] as well as possible [3, 5]
rearrangements under controlled conditions.
Page 141
Texas Tech University, Deepali Butani, August 2011
119
Scheme 3.17: Possible products from sigmatropic rearrangement of phenyl(2-
vinylcyclopent-1-enyl)methyl 2,2,2-trichloroacetimidate (41)
Scheme 3.18: Possible products from sigmatropic rearrangement of phenyl(2-
vinylcyclohex-1-enyl)methyl 2,2,2-trichloroacetimidate (44)
3.3 REARRANGEMENT OF XANTHATES
3.3.1 Background
The formation of allylic sulfur compounds by [3, 3]-sigmatropic
rearrangements of allylic thiocarbonyl compounds, promoted thermally38
or by metal-
catalyzed cyclization induced rearrangement mechanisms,39-41
typically is not
complicated by issues of regioselectivity. The [3, 3]-sigmatropic rearrangement
Page 142
Texas Tech University, Deepali Butani, August 2011
120
reactions of allylic xanthates can be treated as gas-phase reactions because they are not
sensitive to the change in solvent polarity.
Harano42
and coworkers performed computational calculations using
MINDO/3 on O-allyl-S-methyl xanthate to study the [3, 3] sigmatropic rearrangement
of this representative allylic xanthate to form S-allyl S-methyl dithiocarbonate as
shown in Figure 3.15. The calculated energy profile shows that S-allyl S-methyl
dithiocarboante is 12 kcal/mol more than O-allyl- S-methyl xantate, suggesting that
the thione-to-carbonyl isomerization is exothermic.
Figure 3.15: Energy profile showing the [3, 3] sigmatropic rearrangement of allylic
xanthates calculated at the MINDO/3 level of theory.42
Page 143
Texas Tech University, Deepali Butani, August 2011
121
3.3.2 Proposed synthesis of S-methyl O-phenyl (2-vinylcyclopent-1-
enyl) methyl carbonodithioate (47)
We also wanted to study the [3, 3] and [3, 5] sigmatropic rearrangement when
a S-atom is replaced at the position of O-atom and N-atom in 25 and 41 respectively.
So, we decided to study rearrangement of S-methyl O-phenyl (2-vinylcyclopent-1-
enyl) methyl carbonodithioate (47, Figure 3.16). We calculated the MM2 strain
energy, total energy and potential energy of minimized structures of xanthates and
possible [3, 3] (48) and [3, 5] (49) sigmatropic rearrangements as shown in Table 3.9.
The product from [3, 5] rearrangement, 48 is more stable than that from the [3, 3]
rearrangement, 49, suggesting this might be observed.
47
Figure 3.16: Minimized structure of S-methyl O-phenyl (2-vinylcyclopent-1-enyl)
methyl carbonodithioate (47) where yellow are S-atom, grey are C- atoms, red are O-
atom, pink are lone pair orbitals, and white are H-atoms using MM2.
Page 144
Texas Tech University, Deepali Butani, August 2011
122
Table 3.9: Calculated strain energy, total energy and potential energy of minimized
structures of xanthate, 47 and its possible [3, 3] (48) and [3, 5] (49) rearrangement
products using MM2.
Structure Strain
Energy
(kcal/mol)
Total
Energy
(kcal/mol)
Potential
Energy
(kcal/mol)
Total Energy
difference in
rearrangement
(kcal/mol)
Ph
O
S
S
47
5.31 74.54 39.33 -
S
Ph
O
S
48
5.26 72.15 38.60 - 2.39
Ph
S
O
S
49(E)
5.60 70.28 39.19 - 4.26
Ph
S
O
S
49(Z)
10.95 74.14 41.08 - 0.40
Page 145
Texas Tech University, Deepali Butani, August 2011
123
The synthesis of S-methyl O-phenyl (2-vinylcyclopent-1-enyl) methyl
carbonodithioate (47) was attempted from the alcohol, 18 (Scheme 3.19)
Scheme 3.19
3.3.3 Proposed synthesis of S-methyl O-phenyl (2-vinylcyclohex-1-
enyl) methyl carbonodithioate (50)
Similar studies on the [3, 3] and [3, 5] sigmatropic rearrangement of S-methyl
O-phenyl (2-vinylcyclohext-1-enyl) methyl carbonodithioate (50, Figure 3.17) were
conducted. We calculated the MM2 strain energy, total energy and potential energy of
minimized structures of xanthates and possible [3, 3], (51) and [3, 5], (52) sigmatropic
rearrangements as shown in Table 3.10. The product from [3, 5] rearrangement, 52 is
more stable than that from the [3, 3] rearrangement, 51 suggesting this might be
observed.
Page 146
Texas Tech University, Deepali Butani, August 2011
124
50
Figure 3.17: Minimized structure of S-methyl O-phenyl(2-vinylcyclohex-1-
enyl)methyl carbonodithioate (50) where yellow are S-atom, grey are C- atoms, red
are O- atom, pink are lone pair orbitals, and white are H-atoms using MM2 level of
theory.
Page 147
Texas Tech University, Deepali Butani, August 2011
125
Table 3.10: Calculated strain energy, total energy and potential energy of minimized
structures of xanthate, 50 and its possible [3, 3], (51) and [3, 5], (52) rearrangement
products using MM2.
Structure Strain
Energy
(kcal/mol)
Total
Energy
(kcal/mol)
Potential
Energy
(kcal/mol)
Total Energy
difference in
rearrangement
(kcal/mol)
Ph
O S
S(CH3)
50
3.87 79.86 43.52 -
Ph
SS(CH3)
O
51
8.52 75.97 41.20 - 3.89
Ph
S
O
S(CH3)
52(E)
2.31 70.07 36.03 - 9.79
Ph
S
O
S(CH3)
52(Z)
3.72 74.56 36.72 -5.30
Page 148
Texas Tech University, Deepali Butani, August 2011
126
The synthesis of S-methyl O-phenyl (2-vinylcyclohex-1-enyl) methyl carbonodithioate
(50) was attempted from the alcohol, 16 (Scheme 3.20).
Scheme 3.20
3.3.4. Results and Discussion
The synthesis of compound 47 and 50 from the alcohols 18 and 16 respectively
was attempted using NaH, CS2 and CH3I in different ratios with different solvents
THF, DMF and DMSO. But unfortunately, xanthates 47 and 50 were not obtained.
When 47 and 50 are synthesized, a suitable solvent will be chosen so that refluxing in
this solvent will lead to the [3, 3] and [3, 5] rearrangements of these xanthates as
shown in Schemes 3.21 and 3.22.
Scheme 3.21: Proposed rearrangement of S-methyl O-phenyl(2-vinylcyclopent-1-
enyl)methyl carbonodithioate (47)
Page 149
Texas Tech University, Deepali Butani, August 2011
127
Scheme 3.22: Proposed rearrangement of S-methyl O-phenyl(2-vinylcyclohex-1-
enyl)methyl carbonodithioate (50)
3.4 CONCLUSION
The synthesis of pentadienyl alcohols, 16 and 18 was a key aspect for studying
[3, 3] and [3, 5] rearrangement. These pentadienyl alcohols were further used to
prepare acetate, trichloroacetimidate and methyl carbonodithioate derivatives, in such
a manner that they will form six and eight member transition structure to possibly see
[3, 3] and [3, 5] sigmatropic rearrangements.
The synthesis of phenyl(2-vinyl-cyclopent-1enyl)methyl acetate (25) and
phenyl(2-vinyl-cyclohex-1-enyl)methyl acetate (33) was successful. Later, flash
vacuum pyrolysis was conducted on 25 and 26 to see possible rearrangement products.
The flash vacuum pyrolysis of 25 gave an evidence of [3, 5] rearrangement product
(27). Further, experiments have to be conducted on a larger quantity to confirm the
identity of this product as 27 and to determine, whether the product formed was due to
sequential [3, 3] rearrangement or directly [3, 5] rearrangement. The flash vacuum
pyrolysis of 33 gave β-elimination product (37) which has been purified and
characterized by completely of 1H-NMR and
13C-NMR. It also gave a large amount of
Page 150
Texas Tech University, Deepali Butani, August 2011
128
starting material (33) with small amount of what appears to be the [3, 5]
rearrangement product (35). Further experiments also need to be conducted on larger
quantity to identify the products formed.
The synthesis of phenyl(2-vinylcyclopent-1-enyl)methyl 2,2,2-
trichloroacetimidate (41) and phenyl(2-vinylcyclohex-1-enyl)methyl 2,2,2-
trichloroacetimidate (44) was successful with the only problem that crude being
unable to be purified. Future experiments for purification of crude to obtain pure
trichloroacetimidates is necessary for studies on possible [3, 3] and [3, 5]
rearrangements.
3.5 EXPERIMENTAL SECTION
General
The 1H NMR,
13C NMR, HMQC and COSY spectra were recorded with a
Varian Unity INOVA 500 FT-NMR (1H NMR at 500 MHz and
13C NMR at 126
MHz) spectrometer. All spectra were obtained in deuterochloroform (CDCl3) with
residual CHCl3 as internal standard unless stated otherwise. Spectra were reported as
follows: chemical shifts (δ) are reported in ppm downfield from TMS and coupling
constants values (J) are in Hz. Infrared (IR) spectra were recorded with a Nicolet
IR100 FT-IR spectrophotometer as deposits from CH2Cl2 solutions on a NaCl plate
unless otherwise stated.
Reagents were purchased from commercial suppliers and used directly unless
otherwise noted. Choloroform (CH3Cl) was dried over CaCl2. Tetrahydrofuran (THF)
Page 151
Texas Tech University, Deepali Butani, August 2011
129
was dried over sodium with benzophenone as an indicator and distilled immediately
prior to use. Toluene was dried over sodium and distilled immediately before use.
Dimethylformamide (DMF), diethyl ether (Et2O), dichloromethane (DCM) were
purified using a MB-Solvent Purification System. Analytical TLC was performed on
silica gel plates, while silica gel from Sorbent technologies (40-63 µm, 60 Å) was used
for column chromatography.
N-butyllithium (n-BuLi) and tert-butyllithium (t-BuLi) all were titrated to
check the molarity of the bottle. For the titration, in an oven dried round bottom flask,
approximately 0.2 g of L-menthol was weighed and dissolved in solvent used for the
reaction. A pinch of 1,10-phenanthroline was added as indicator. The titration was
carried out under N2. N-butyllithium (n-BuLi) or tert-butyllithium (t-BuLi) was added
slowly using a 1 mL syringe until the color changed from colorless to yellow to a
persistent red. This was done to make sure the equivalents of n-butyllithium (n-BuLi)
and tert-butyllithium (t-BuLi) remain same during the course of experiments.
Synthesis of 2-bromocyclopent-1-ene carbaldehyde (28)43
Br
O
28
Page 152
Texas Tech University, Deepali Butani, August 2011
130
The following methods were used for synthesis of 2-bromocyclopent-1-ene
carbaldehyde (28). Method A gave a higher yield that method B.
(A) In a 500 mL dry round bottom flask under N2, dry DMF (3.0 eq.,10.96 g, 11.6
mL, 150 mmol) was cooled to 0 ˚C in dry CHCl3 (100 mL) and phosphorous
tribromide, (PBr3, 99% pure, 2.5 eq., 33.8 g, 11.7 mL, 125 mmol) was added
dropwise over a period of 10 min. The mixture was stirred at 0 ˚C for 1 h to
yield a yellow suspension. A solution of cyclopentanone (1.0 eq., 4.2 g, 4.4
mL, 50 mmol) in CHCl3 (10 mL) was added and the mixture was stirred at
room temperature for 12 h. The reaction was cooled to 0 ˚C and aq. NaHCO3
was added slowly until the effervescence subsided. The mixture was extracted
with diethyl ether (3 x 50 mL) and washed with brine (2 x 10 mL). The extract
was dried with MgSO4, concentrated under vacuum and chromatographed on a
silica gel column using 10:1 hexane/ethyl acetate to give a yellow oily product
2-bromocyclopent-1-ene carbaldehyde (28). The identity and purity of the
product was confirmed by TLC, 1H-NMR, HMQC, COSY and
13C-NMR.
Yield: 6.56 g and 75%.
(B) In a 100 mL dry round bottom flask under N2, dry DMF (3.0 eq., 10.96 g, 11.6
mL, 150 mmol) was cooled to 0 ˚C in dry CHCl3 (50 mL) and phosphorous
tribromide (PBr3, 99% pure 2.7 eq., 39.1 g, 13.5 ml, 135 mmol) was added
dropwise over a period of 10 min. The mixture was stirred at 0 ˚C for 1 h to
yield a yellow suspension. A solution of cyclopentanone (1.0 eq., 4.2 g, 4.4
mL, 50 mmol) in CHCl3 (10 mL) was added and the mixture was then refluxed
Page 153
Texas Tech University, Deepali Butani, August 2011
131
for 60-80 min. The reaction was cooled to 0 ˚C and aq. NaHCO3 was added
slowly until the effervescence subsided. The mixture was extracted with
CH2Cl2 (3 x 50 mL) and washed with brine (2 x 10 mL). The extract was dried
with MgSO4, concentrated under vacuum and chromatographed on a silica gel
column using 10:1 hexane/ethyl acetate to give a yellow oily product 2-
bromocyclopent-1-ene carbaldehyde (28). The identity and purity of the
product was confirmed by TLC, 1H-NMR, HMQC, COSY and
13C-NMR
(provided in Appendix B, Figure B1, B2, B3 and B4). Yield: 3.94 g and 45%.
Rf = 0.87 (10:1 hexane/ethyl acetate)
1H-NMR (500 MHz, CDCl3, 25°C): δ 1.96-2.1 (m, 2H), 2.47-2.52 (m, 2H),
2.85-2.90 (m, 2H), 9.86 (br s, 1H).
13C-NMR (126 MHz, CDCl3, 25°C): δ 21.3, 29.2, 42.4, 139.9, 141.3, 189.1.
Synthesis of 2-bromocyclohex-1-ene carbaldehyde (37)44
Br
O
37
The following methods were used for synthesis of 2-bromocyclohex-1-ene
carbaldehyde (37). Method B gave a higher yield that method A.
(A) In a 500 mL dry round bottom flask under N2, dry DMF (3.0 eq.,10.96 g, 11.6
mL, 150 mmol) was cooled to 0˚C in dry CHCl3 (100 mL) and phosphorous
Page 154
Texas Tech University, Deepali Butani, August 2011
132
tribromide, (PBr3, 99% pure 2.5 eq., 33.8 g, 11.7 mL, 125 mmol) was added
dropwise over a period of 10 min. The mixture was stirred at 0 ˚C for 1 h to
yield a yellow suspension. A solution of cyclohexanone (1.0 eq., 4.9 g, 5.2 mL,
50 mmol) in CHCl3 (10 mL) was added and the mixture was stirred at room
temperature for 12 h. The reaction was cooled to 0 ˚C and aq. NaHCO3 was
added slowly until the effervescence subsided. The mixture was extracted with
CH2Cl2 (3 x 50 mL) and washed with brine (2 x 10 mL). The extract was dried
with MgSO4, concentrated under vacuum and chromatographed on a silica gel
column using 10:1 hexane/ethyl acetate to give a yellow oily product 2-
bromocyclohex-1-ene carbaldehyde (37). The identity and purity of the
product was confirmed by TLC, 1H-NMR, HMQC, COSY and
13C-NMR.
Yield: 2.8 g and 30%.
(B) In a 100 mL dry round bottom flask under N2, dry DMF (3.0 eq., 11.05 g, 11.7
mL, 152.7 mmol) was cooled to 0 ˚C in dry CHCl3 (50 mL) and phosphorus
tribromide, (PBr3, 99% pure 2.7 eq., 39.9 g, 13.8 ml, 137.6 mmol) was added
dropwise over a period of 10 min. The mixture was stirred at 0 ˚C for 1 h to
yield a yellow suspension. A solution of cyclohexanone (1.0 eq., 5.0 g, 5.3 mL,
50.9 mmol) in CHCl3 (10 mL) was added and the mixture was then refluxed
for 60-80 min. The reaction was cooled to 0 ˚C and aq. NaHCO3 was added
slowly until the effervescence subsided. The mixture was extracted with
CH2Cl2 (3 x 50 mL) and washed with brine (2 x 10 mL). The extract was dried
with MgSO4, concentrated under vacuum and chromatographed on a silica gel
Page 155
Texas Tech University, Deepali Butani, August 2011
133
column using 10:1 hexane/ethyl acetate to give a yellow oily product 2-
bromocyclohex-1-ene carbaldehyde (37). The identity and purity of the
product was confirmed by TLC, 1H-NMR, HMQC, COSY and
13C-NMR
(provided in Appendix B, Figure B17, B18, B19 and B20).
Yield: 6.1 g and 65%. Rf = 0.85 (10:1 hexane/ethyl acetate)
1H-NMR (500 MHz, CDCl3, 25°C): δ 1.65-1.78 (m, 4 H), 2.24-2.28 (m, 2H),
2.71-2.76 (m, 2 H), 10.00 (s, 1 H).
13C-NMR (126 MHz, CDCl3, 25°C): δ 21.0, 24.2, 24.9, 38.8, 135.2, 143.6,
193.7.
Synthesis of 1-bromo-2-vinylcyclopentene (29)45
Br
29
The following methods were used for synthesis of 1-bromo-2-vinylcyclopentene (29).
Method B gave a higher yield that method A.
(A) Methyltriphenylphosphonium bromide (1.1 eq., 3.90 g, 11 mmol) was
suspended in THF (15 mL) and n-BuLi (1.1 eq., 6.9 mL, 11 mmol of 1.6 M
solution in hexane) was added dropwise with stirring under N2 at 0 ˚C. It gave
a brown color solution. After 30 min at 0 ˚C, 2-bromocyclopent-1-ene
carbaldehyde (28, 1.0 eq., 1.9 g, 10 mmol) in THF (10 mL) was added
Page 156
Texas Tech University, Deepali Butani, August 2011
134
dropwise and stirred for 1 h to yield a dark brown color solution. The reaction
was quenched with water (20 mL) and extracted with diethyl ether (3 x 20
mL). The organic layer was washed with brine (2 x 10 mL).The extract was
dried with MgSO4, concentrated under vacuum and chromatographed on a
silica gel column using pentane to give yellow product of 1-bromo-2-
vinylcyclopenetene (29). The identity and purity of the product was confirmed
by TLC, 1H-NMR, HMQC, COSY and
13C-NMR. Yield: 0.92 g and 35%.
(B) In a 250 mL two-necked dry round bottom flask a mixture of
methyltriphenylphosphonium bromide (1.1 eq., 6.0 g, 16.8 mmol) and sodium
amide (NaNH2, 1.7 eq., 1.0 g, 25.6 mmol) was suspended in THF (80 mL)
and stirred under N2 for 60 min at room temperature to yield a bright yellow
color. Then, 2-bromocyclopent-1-ene carbaldehyde (28, 1.0 eq., 2.7 g, 15.2
mmol) in THF (10 mL) was added dropwise and stirred for 90-120 min
yielding a dark brown solution. The reaction was quenched by adding 23 mL
of 25 % aq. NaOH solution. The solution was then neutralized by 31 mL of
0.1 N HCl, extracted with diethyl ether (3 x 20 mL) and washed with brine (2
x 10 mL). The extract was dried with MgSO4, concentrated under vacuum and
chromatographed on a silica gel column using pentane to give yellow oily
product of 1-bromo-2-vinylcyclopenetene (29). The identity and purity of the
product was confirmed by TLC, 1H-NMR, HMQC, COSY and
13C-NMR
(provided in Appendix B, Figure B5, B6, B7 and B8). Yield: 1.6 g and 60%.
Rf = 0.9 (pentane).
Page 157
Texas Tech University, Deepali Butani, August 2011
135
1H-NMR (500 MHz, CDCl3, 25 °C): δ 1.94-2.02 (m, 2 H), 2.43-2.49 (m, 2 H),
2.71-2.78 (m, 2 H), 5.17 (dq, J = 14.0, 3.5 Hz, 1 H), 5.22 (dq, J = 10.0, 1.0 Hz,
1 H), 6.62 (dd, J = 10.5, 7.0 Hz, 1 H).
13C-NMR (126 MHz, CDCl3, 25°C): δ 21.4, 30.7, 40.7, 116.8, 120.9, 131.1,
138.2.
IR (neat): 2956.48, 2850.78, 1677.64, 1635.48, 1442.47 cm-1
.
Synthesis of 1-bromo-2-vinylcyclohexene (38)45
Br
38
The following methods were used for synthesis of 1-bromo-2-vinylcyclohexene (38).
Method B gave a higher yield that method A.
(A) Methyltriphenylphosphonium bromide (1.1 eq., 3.90 g, 11 mmol) was
suspended in THF (15 mL) and n-BuLi (1.1 eq., 6.9 mL, 11 mmol of 1.6 M
solution in hexane) was added dropwise with stirring under N2 at 0 ˚C. It gave
a brown color solution. After 30 min at 0 ˚C, 2-bromocyclohex-1-ene
carbaldehyde (37, 1.0 eq., 1.89 g, 10 mmol) in THF (10 mL) was added
dropwise and stirred for 1 h to yield a dark brown color solution. The reaction
was quenched with water (20 mL) and extracted with diethyl ether (3 x 20
mL). The organic layer is washed with brine (2 x 10 mL). The extract was then
Page 158
Texas Tech University, Deepali Butani, August 2011
136
dried with MgSO4 concentrated under vacuum and chromatographed on a
silica gel column using pentane to give yellow product of 1-bromo-2-
vinylcyclohexene (38). The identity and purity of the product was confirmed
by TLC, 1H-NMR, HMQC, COSY and
13C-NMR. Yield: 0.46 g and 25%.
(B) In a 250 mL two-necked dry round bottom flask a mixture of
methyltriphenylphosphonium bromide (1.1 eq., 3.94 g, 11.03 mmol) and
sodium amide (NaNH2, 1.7 eq., 0.66 g, 16.9 mmol) was suspended in THF (80
mL) and stirred under N2 for 60 min at room temperature to yield a bright
yellow color. Then, 2-bromocyclohex-1-ene carbaldehyde (37, 1.0 eq., 2.0 g,
9.94 mmol) in THF (10 mL) was added dropwise and stirred for 90-120 min
yielding a dark brown solution. The reaction was quenched by adding 23 mL
of 25 % aq. NaOH solution. The solution was then neutralized by 31 mL of 0.1
N HCl, extracted with diethyl ether (3 x 20 mL) and washed with brine (2 x 10
mL). The extract was dried with MgSO4 concentrated under vacuum and
chromatographed on a silica gel column using pentane to give yellow oily
product of 1-bromo-2-vinylcyclohexene (38). The identity and purity of the
product was confirmed by TLC, 1H-NMR, HMQC, COSY and
13C-NMR
(provided in Appendix B, Figure B21, B22, B23 and B24).
Yield: 1.2 g and 65%.
Rf = 0.9 (pentane)
Page 159
Texas Tech University, Deepali Butani, August 2011
137
1H-NMR (500 MHz, CDCl3, 25°C): δ 1.69-1.74 (m, 4 H), 2.24-2.3 (m, 2 H),
2.6-2.66 (m, 2 H), 5.1 (dq, J = 10.0, 1.0 Hz, 1 H), 5.22 (dq, J = 16.5, 1.0 Hz, 1
H), 6.86 (dd, J = 11.0, 6.5 Hz, 1 H).
13C-NMR (126 MHz, CDCl3, 25°C): δ 22.0, 24.7, 26.7, 37.5, 114.3, 125.1,
132.2, 137.1.
IR (neat): 3434.46, 2934.98, 1629.79, 1435.82 cm-1
.
Synthesis of phenyl(2-vinylcyclopent-1-enyl)methanol (18):46
Ph
OH
18
The following methods were used for synthesis of phenyl(2-vinylcyclopent-1-
enyl)methanol (18). The reaction at -78 ˚C with t-BuLi gave better yield.
(A) A solution of 1-bromo-2-vinylcyclopentene (29, 1.0 eq., 0.152 g, 0.88 mmol)
in 2.7 mL of THF under N2 at -78 ˚C was treated dropwise with n-BuLi (1.1
eq., 1 mL, 0.97 mmol of 1.6 M solution in hexane). After stirring for 15 min
freshly distilled benzaldehyde (1.02 eq., 0.9 mL, 0.90 mmol) was added. After
stirring for 1 h the mixture was allowed to warm to 0 ˚C and quenched with
15 mL of 4% NH4Cl solution. The mixture was extracted with diethyl ether (3
x 35 mL) and organic layer was washed with water and brine. The extract was
Page 160
Texas Tech University, Deepali Butani, August 2011
138
dried with MgSO4, concentrated under vacuum and chromatographed on a
silica gel column using 6.5:1 hexane/ ethyl acetate to give a yellow product of
phenyl(2-vinylcyclopent-1-enyl)methanol (18). The identity and purity of the
product was confirmed by TLC, 1H-NMR, HMQC, COSY and
13C-NMR.
Yield: 0.05 g, 30%.
(B) A solution of 1-bromo-2-vinylcyclopentene (29, 1.0 eq., 0.1 g, 0.58 mmol) in
5 mL of dry Et2O under N2 at -78 ˚C was treated dropwise with t-BuLi (1.98
eq., 0.67 mL, 1.15 mmol of 1.7 M solution in pentane) over a period of 15
min. After stirring continuously for 30 min at -78 ˚C, freshly distilled
benzaldehyde (1.03 eq., 0.06 mL, 0.60 mmol) was added. After stirring the
mixture for 5 h at -78 ˚C the cold bath was removed and the mixture was
allowed to slowly attain room temperature and then it was quenched with 15
mL of saturated NH4Cl solution. The mixture was extracted with diethyl ether
(3 x 35 mL) and organic layer was washed with water and brine (2 x 10 mL).
The extract was dried with MgSO4, concentrated under vacuum and
chromatographed on a silica gel column using 10:1 hexane/ ethyl acetate to
give a yellow product of phenyl(2-vinylcyclopent-1-enyl)methanol (18). The
identity and purity of the product was confirmed by TLC, 1H-NMR, HMQC,
COSY and 13
C-NMR (provided in Appendix B, Figure B9, B10, B11 and
B12). Yield: 0.07 g and 60%.
Rf = 0.3 (10:1 hexane/ethyl acetate), 0.55 (5:1 hexane/ethyl acetate).
Page 161
Texas Tech University, Deepali Butani, August 2011
139
1H-NMR (500 MHz, CDCl3, 25°C): δ 1.73-1.86 (m, 2 H), 1.86 (br s, 1 H),
2.16-2.23 (m, 1 H), 2.52-2.62 (m, 3 H), 5.18 (dd, J = 6.0, 1.0 Hz, 1 H), 5.2
(dd, J = 12.5, 0.5 Hz, 1 H), 5.86 (br, s, 1 H), 6.90 (dd, J = 11.0, 6.0 Hz, 1 H),
7.23-7.40 (m, 5 H);
13C-NMR (126 MHz, CDCl3, 25°C): δ 21.3, 31.8, 32.9, 69.8, 115.4, 125.6,
127.1, 128.3, 130.2, 137.3, 142.1, 142.3.
IR (neat): 3367.7 (O-H) cm-1
.
Synthesis of phenyl(2-vinylcyclohex-1-enyl)methanol (16):46
Ph
OH
16
The following methods were used for synthesis of phenyl(2-vinylcyclohex-1-
enyl)methanol (16). The reaction at -78°C with t-BuLi gave better yields.
(A) A solution of 1-bromo-2-vinylcyclohexene (38, 1.0 eq., 0.165 g, 0.882 mmol)
in 2.7 mL of THF under N2 at -78 ˚C was treated dropwise with n-BuLi (1.1
eq., 1 mL, 0.97 mmol of 1.6 M solution in hexane). After stirring for 15 min
freshly distilled benzaldehyde (1.02 eq., 0.9 mL, 0.90 mmol) was added. After
stirring for 1 h the mixture was allowed to warm to 0 ˚C and quenched with
15 mL of 4% NH4Cl solution. The mixture was extracted with diethyl ether (3
Page 162
Texas Tech University, Deepali Butani, August 2011
140
x 35 mL) and organic layer was washed with water and brine (2 x 10 mL).
The extract was dried with MgSO4, concentrated under vacuum and
chromatographed on a silica gel column using 6.5:1 hexane/ ethyl acetate to
give a yellow product of phenyl(2-vinylcyclohex-1-enyl)methanol (16). The
identity and purity of the product was confirmed by TLC, 1H-NMR, HMQC,
COSY and 13
C-NMR. Yield: 0.05 g and 25%.
(B) A solution of 1-bromo-2-vinylcyclohexene, (38, 1.0 eq., 0.1 g, 0.53 mmol) in
5 mL of dry Et2O under N2 at -78 ˚C was treated dropwise with t-BuLi (1.98
eq., 0.62 mL, 1.05 mmol of 1.7 M solution in pentane) over a period of 15
min. After stirring continuously for 30 min at -78 ˚C, freshly distilled
benzaldehyde (1.03 eq., 0.05 mL, 0.50 mmol) was added. After stirring the
mixture for 5 h at -78 ˚C the cold bath was removed and the mixture was
allowed to slowly attain room temperature and then it was quenched with 15
mL of saturated NH4Cl solution. The mixture was extracted with diethyl ether
(3 x 35 mL) and organic layer was washed with water and brine (2 x 10 mL).
The extract was dried with MgSO4, concentrated under vacuum and
chromatographed on a silica gel column using 10:1 hexane/ ethyl acetate to
give a yellow product of phenyl (2-vinylcyclohex-1-enyl) methanol (16). The
identity and purity of the product was confirmed by TLC, 1H-NMR, HMQC,
COSY and 13
C-NMR (provided in Appendix B, Figure B25, B26, B27 and
B28). Yield: 0.07g and 65%.
Rf = 0.3 (10:1 hexane/ethyl acetate), 0.55 (5:1 hexane/ethyl acetate)
Page 163
Texas Tech University, Deepali Butani, August 2011
141
1H-NMR (500 MHz, CDCl3, 25°C): δ 1.47-1.78 (m, 5 H), 1.81 (br s, 1 H),
2.20-2.35 (m, 3 H), 5.08 (d, J = 11.0 Hz, 1 H), 5.27 (dd, J = 15.5, 0.5 Hz, 1
H), 6.08 (br, s, 1 H), 7.04 (dd, J = 11.0, 6.5 Hz, 1 H), 7.22-7.38 (m, 5 H).
13C-NMR (126 MHz, CDCl3, 25°C): δ 22.43, 22.45, 23.95, 25.5, 70.7,
113.15, 125.6, 126.8, 128.15, 131.45, 133.8, 137.77, 142.57.
IR (neat): 3596 (O-H) cm-1
.
Synthesis of phenyl (2-vinylcyclopent-1enyl) methyl acetate (25):47
Ph
O
O
25
In a 25 mL round bottom flask, a solution of phenyl(2-vinylcyclopent-1-
enyl)methanol (18, 1.0 eq., 0.12 g, 0.6 mmol) dissolved in 5 mL of DCM
under N2 at 0 °C was treated with acetyl chloride, (CH3COCl, 1.05 eq., 0.05
mL, 0.05 g, 0.63 mmol). After stirring the reaction mixture at 0 °C for 15 min
pyridine (1.05 eq., 0.05 mL, 0.05 g, 0.63 mmol) was added to remove HCl
produced during the reaction. The reaction was stirred at room temperature for
24 h. The reaction was the quenched with 10 mL of water. The mixture was
extracted with DCM (3 x 15 mL) and organic layer was washed with water
and brine (2 x 5 mL). The extract was dried with MgSO4, concentrated under
Page 164
Texas Tech University, Deepali Butani, August 2011
142
vacuum and chromatographed on a silica gel column using 10:1 hexane/ ethyl
acetate to give a yellow oily product of phenyl (2-vinylcyclopent-1enyl)
methyl acetate (25). The identity and purity of the product was confirmed by
TLC, 1H-NMR, HMQC, COSY and
13C-NMR (provided in Appendix B,
Figure B13, B14, B15 and B16). Yield: 0.13 g and 86%.
Rf = 0.7 (10:1 hexane/ ethyl acetate)
1H-NMR (500 MHz, CDCl3, 25°C): δ 1.75-1.88 (m, 2 H), 2.14 (br, s, 3 H),
2.25-2.33 (m, 1 H), 2.52-2.58 (m, 3 H), 5.2 (d, J = 3.0 Hz, 1 H), 5.23 (d, J =
0.5 Hz, 1 H), 6.86 (br, s, 1 H), 6.96 (dd, J = 10.5, 7.5 Hz, 1 H), 7.24-7.36 (m,
5 H).
13C-NMR (126 MHz, CDCl3, 25°C): δ 21.13, 21.16, 32.56, 32.69, 71.58,
116.0, 125.9, 128.34, 130.16, 130.25, 138.1, 138.5, 139.0, 169.96.
IR (neat): 1737 (C=O) cm-1
.
Synthesis of phenyl(2-vinylcyclohex-1enyl)methyl acetate (33):47
Ph
O
O
33
Page 165
Texas Tech University, Deepali Butani, August 2011
143
In a 25 mL round bottom flask, a solution of phenyl(2-vinylcyclohex-1-
enyl)methanol (16, 1.0 eq., 0.2 g, 0.9 mmol) dissolved in 5 mL of DCM
under N2 at 0 °C was treated with acetyl chloride, (CH3COCl, 1.05 eq., 0.07
mL, 0.07 g, 0.94 mmol). After stirring the reaction mixture at 0 °C for 15
min pyridine (1.05 eq., 0.08 mL, 0.07 g, 0.94 mmol) was added to remove
HCl produced during the reaction. The reaction was stirred at room
temperature for 24 h. The reaction was the quenched with 10 mL of water.
The mixture was extracted with DCM (3 x 15 mL) and organic layer was
washed with water and brine (2 x 5 mL). The extract was dried with MgSO4,
concentrated under vacuum and chromatographed on a silica gel column
using 10:1 hexane/ ethyl acetate to give a yellow oily product of phenyl (2-
vinylcyclohex-1enyl) methyl acetate (33). The identity and purity of the
product was confirmed by TLC, 1H-NMR, HMQC, COSY and
13C-NMR
(provided in Appendix B, Figure B29, B30, B31 and B32). Yield: 0.16 g and
68%.
Rf = 0.7 (10:1 hexane/ ethyl acetate)
1H-NMR (500 MHz, CDCl3, 25 °C): δ 1.48-1.71 (m, 5 H), 1.8-1.9 (m, 1 H),
2.17 (br, s, 3 H), 2.20-2.36 (m, 2 H), 5.12 (d, J = 11.0 Hz, 1 H), 5.28 (dd, J =
16.5, 0.5 Hz, 1 H), 7.08 (br, s, 1 H), 7.12 (dd, J = 11.0 Hz, 1 H), 7.24-7.35 (m,
5 H).
13C-NMR (126 MHz, CDCl3, 25°C): δ 21.13, 22.26, 22.34, 24.69, 25.42,
72.86, 113.57, 125.7, 127.21, 128.24, 132.62, 133.96, 134.03, 139.41, 170.03.
Page 166
Texas Tech University, Deepali Butani, August 2011
144
IR (neat): 1741.18 (C=O) cm-1
.
Flash Vacuum Pyrolysis of 33 and synthesis of 1-((1E)-(2-vinylcyclohex-2-
enylidene)methylbenzene (37):
Ph
37
Approximately 100-200 mg of 33 was dissolved in anhydrous diethyl ether
(because it is very viscous, it needed to be diluted for transfer) was placed in
the quartz tube. A vacuum of 0.1 torr was drawn on the quartz tube to remove
residual solvents such as diethyl ether, hexane or ethyl acetate. Then the
equipment was set up as shown in Figure 3.9. The liquid nitrogen bath is kept
under the U-tube to cool the pyrolysates. The pyrolysis oven was adjusted to a
proper temperature and the digital thermometer was set in the middle of the
oven to read the temperature of the oven. After the oven had reached a
required temperature, the closed end of the quartz tube was slowly pushed
inside the oven. This was continued until the closed end of the tube had gone
inside the oven and the sample had vaporized the quartz tube. The products
were collected either at the end of the end of quartz tube or in the U-tube.
After the pyrolysis tube cooled, the system was filled with nitrogen. The U-
tube was removed from the system and warmed up to room temperature. A
Page 167
Texas Tech University, Deepali Butani, August 2011
145
1H-NMR spectrum was obtained for crude the pyrolysis products collected
both at the end of the pyrolysis tube and in the U-tube. The sample obtained
was then chromatographed on silica gel column using 2% ethyl acetate in
hexane solution to separate the different products. The identity and purity of
the product was confirmed by TLC, 1H-NMR, HMQC, COSY and
13C-NMR
(provided in Appendix B, Figure B33, B34, B35 and B36).
Rf = 0.85 (2% ethyl acetate in hexane).
1H-NMR (500 MHz, CDCl3, 25°C): δ 1.65-1.71 (m, 2 H), 2.24-2.3 (m, 2 H),
2.6-2.64 (m, 2 H), 5.1 (dd, J = 8.5, 2.0 Hz, 1 H), 5.38 (dd, J = 15.0, 2.0 Hz, 1
H), 6.02 (t, J = 4.0, Hz, 1 H), 6.5 (br, s, 1 H), 6.53 (dd, J = 11.0, 5.0 Hz, 1 H),
7.2-7.4 (m, 5 H).
13C-NMR (126 MHz, CDCl3, 25°C): δ 22.38, 26.24, 26.96, 115.09, 124.34,
126.14, 127.98, 128.77, 129.24, 137.11, 137.22, 138.03, 138.31.
Synthesis of phenyl(2-vinylcyclopent-1-enyl)methyl 2,2,2-trichloroacetimidate
(41):48
Ph
O
NH
CCl3
41
Page 168
Texas Tech University, Deepali Butani, August 2011
146
Various procedures were tried for synthesis of phenyl(2-vinylcyclopent-1-
enyl)methyl 2,2,2-trichloroacetimidate (41):
(a) NaH (0.1 eq., 0.042 mmol, 1.7 mg of 60% dispersion in mineral oil) was
washed with hexane, suspended in absolute diethyl ether (2 mL) and cooled to
0 °C. Phenyl(2-vinylcyclopent-1-enyl)methanol (18, 1.0 eq., 85 mg, 0.42
mmol) dissolved in diethyl ether (1 mL) was added. After the reaction time 20
min, the reaction mixture was cooled to 0 °C and trichloroacetonitrile (1.05
eq., 0.44 mmol, 0.06 g, 40 µL) was added. The reaction was monitored by
TLC and 1H-NMR. The reaction was monitored for 3 days and no new
product could be seen. Hence, it was concluded that the reaction did not work
with NaH as base.
(b) A suspension of potassium hydride (0.2 eq. 5 mg, 0.034 mmol of a 30%
dispersion in mineral oil), washed twice with hexane was suspended in 2 mL
of diethyl ether was cooled to 0 °C. Phenyl(2-vinylcyclopent-1-enyl)methanol
(18, 1.0 eq., 35 mg, 0.17 mmol) dissolved in 1 mL of diethyl ether was added
to it. After 20 min trichloroacetonitrile (1.2 eq., 0.20 mmol, 0.03 g, 0.02 mL)
was added at 0 °C to the reaction. The reaction mixture was then let to stir
initially for 2 h at 0 °C and then at the room temperature. The reaction was
monitored by TLC and 1H-NMR. The reaction was monitored for 12 h and no
new product formed could be seen. Hence, it was concluded that the reaction
did not work with KH as base.
Page 169
Texas Tech University, Deepali Butani, August 2011
147
(c) To phenyl(2-vinylcyclopent-1-enyl)methanol (18, 1.0 eq., 0.21 g, 1.04 mmol),
2 mL of dicholoromethane was added. The mixture was kept under N2 at 0
°C, and 1,8 diazabicycloundec-7-ene (DBU) (0.1 eq., 15 µL, 0.1 mmol) was
added. The solution was stirred for 15 min at 0 °C and trichloroacetonitrile
(5.0 eq., 52 µL, 5.2 mmol) was added and stirred for 1 h. A crude NMR taken
after 1 h showed phenyl(2-vinylcyclopent-1-enyl)methyl 2,2,2-
trichloroacetimidate (41). The solvent was not removed and the crude was
directly loaded on column to be purified by flash chromatography using 2%
ethyl acetate solution in hexane. The problem was that the phenyl(2-
vinylcyclopent-1-enyl)methyl 2,2,2-trichloroacetimidate (41) rearranged on
silica gel to give product 42.
Crude 1H-NMR (300 MHz, CD2Cl2, 25°C) tentatively assigned to 41:
1H-
NMR : δ 1.4-1.6 (m, 5 H), 2.10-2.4 (m, 3 H), 5.31 (dd, 1 H), 5.35 (dd, 1 H),
7.08 (br, s, 1 H), 7.12 (dd, 1 H), 7.3-7.6 (m, 5 H), 8.6 (br, s, 1H).
1H-NMR (500 MHz, CDCl3, 25°C) tentatively assigned to 42: δ 1.79-1.88 (m,
5 H), 1.98-2.08 (m, 3 H), 5.22 (dd, 9, 1.5 Hz, 1 H), 5.43 (dd, 16.0, 1.5 Hz, 1
H), 5.94 (dd, 10.5, 7.0 Hz, 1 H), 6.54 (br, s, 1H), 7.28 (br, s, 1 H), 7.3-7.4 (m,
5 H).
Page 170
Texas Tech University, Deepali Butani, August 2011
148
Synthesis of phenyl(2-vinylcyclohex-1-enyl)methyl 2,2,2-trichloroacetimidate (44):48
Ph
O
NH
CCl3
44
Various procedures were tried for synthesis of compound 44:
(a) NaH (0.1 eq., 0.042 mmol, 1.7 mg 60% dispersion in mineral oil) was
washed with hexane, then suspended in absolute diethyl ether (2 mL) and
cooled to 0 °C. Phenyl(2-vinylcyclohex-1-enyl)methanol (16, 1.0 eq., 84 mg,
0.42 mmol) dissolved in diethyl ether (1 mL) was added. After the reaction
time 20 min, the reaction mixture was cooled to 0 °C and trichloroacetonitrile
(1.05 eq., 0.44 mmol, 0.06 g, 40 µL) was added. The reaction was monitored
by TLC and 1H-NMR. The reaction was monitored for 3 days and no new
product could be seen. Hence, it was concluded that the reaction did not work
with NaH as base.
(b) A suspension of potassium hydride (0.2 eq. 5 mg, 0.034 mmol of a 30%
dispersion in mineral oil), washed twice with hexane was suspended in 2 mL
of diethyl ether was cooled to 0 °C. Phenyl(2-vinylcyclohex-1-enyl)methanol,
(16, 1.0 eq., 34 mg, 0.17 mmol) dissolved in 1mL of diethyl ether was added
to it. After 20 min trichloroacetonitrile (1.2 eq., 0.20 mmol, 0.03 g, 0.02 mL)
was added at 0 °C to the reaction. The reaction mixture was then let to stir
Page 171
Texas Tech University, Deepali Butani, August 2011
149
initially for 2 h at 0 °C and then at the room temperature. The reaction was
monitored by TLC and 1H-NMR. The reaction was monitored for 12 h and no
new product formed could be seen. Hence, it was concluded that the reaction
did not work with KH as base.
(c) To phenyl(2-vinylcyclohex-1-enyl) methanol (16, 1.0 eq., 0.27 g, 1.24 mmol),
2 mL of dicholoromethane was added. The mixture was kept under N2 at 0
°C, and 1,8 diazabicycloundec-7-ene (DBU) (0.1 eq., 18 µL, 0.12 mmol) was
added. The solution was stirred for 15 min at 0 °C and trichloroacetonitrile
(5.0 eq., 62 µL, 6.2 mmol) was added and stirred for 1 h. A crude NMR taken
after 1 h showed phenyl(2-vinylcyclohex-1-enyl)methyl 2,2,2-
trichloroacetimidate (44). The solvent was not removed and the crude was
directly loaded on column to be purified by flash chromatography using 2%
ethyl acetate solution in hexane. The problem was that the phenyl(2-
vinylcyclohex-1-enyl)methyl 2,2,2-trichloroacetimidate (44) rearranged on
silica gel to give product 45 and some amount of 44 (1H-NMR in Appendix
B).
Crude 1H-NMR (300 MHz, CDCl3, 25°C) tentatively assigned to 44:
1H-
NMR : δ 1.75-1.99 (m, 5 H), 2.33-2.5 (m, 3 H), 5.0 (dd, 1 H), 5.15 (dd, 1 H),
7.01 (br, s, 1 H), 7.2 (dd, 1 H), 7.3-7.6 (m, 5 H), 8.3 (br, s, 1H).
Page 172
Texas Tech University, Deepali Butani, August 2011
150
Attempted synthesis of S-methyl O-phenyl(2-vinylcyclopent-1-enyl)methyl
carbonodithioate (47):49
O
Ph S
S(CH3)
47
Various procedures were tried for synthesis of compound 47:
(a) To a stirred solution of phenyl(2-vinylcyclopent-1-enyl)methanol (18, 1.0 eq.,
0.12g, 0.62 mmol) in THF at 0 °C, CS2 (67.17 eq., 2.6 mL, 41.6 mmol) was
added, followed by CH3I (67.5 eq., 2.7 mL, 41.8 mmol). After stirring for 15 min
NaH (2.1 eq., 0.05 g of 60% dispersion in mineral oil) was added slowly and the
reaction mixture was stirred at 0 °C for 20 min. The reaction mixture was then
quenched with ice and organic layer was extracted with ethyl acetate. The
organic layer was then washed with brine, dried over MgSO4 and concentrated in
vacuum to give yellow oil. The reaction was monitored by TLC and it showed a
lot of new spots with possibility of product 47. 1H-NMR of crude showed peaks
corresponding to 47. But the crude compound 47 seemed to decompose on the
silica gel column.
(b) A stirred solution of phenyl(2-vinylcyclopent-1-enyl)methanol (18, 1.0 eq., 0.1
g, 0.5 mmol) in THF, NaH (1.2 eq., 30 mg of 60% dispersion in mineral oil) was
added slowly portionwise at 0 °C. After the resulting mixture was stirred for 1 h
at room temperature, CS2 (2 eq., 70 µL) was added dropwise at 0 °C. The
resultant mixture was stirred for 2 h at room temperature before CH3I (1.2 eq.,
Page 173
Texas Tech University, Deepali Butani, August 2011
151
40 µL) was added dropwise to the reaction mixture at 0 °C. The resultant
mixture was stirred for 1 h at room temperature, treated with NH4Cl solution and
extracted with diethyl ether. The organic layer was washed with brine, dried over
MgSO4 and concentrated under vacuum. The reaction was monitored by TLC
and it showed a lot of new spots with possibility of product 47. 1H-NMR of
crude showed peaks corresponding to 47. But the crude compound 47 seemed to
decompose on the silica gel column.
(c) DMSO (2 mL) was added under N2 to NaH (1.2 eq., 40 mg of 60% dispersion in
mineral oil) and the mixture was heated to 70 °C and stirred for 45 min. After
cooling the reaction mixture phenyl(2-vinylcyclopent-1-enyl)methanol (18, 1.0
eq., 0.12 g, 0.6 mmol) dissolved in 1 mL of DMSO was added dropwise and the
mixture was stirred at room temperature for 1 h. A solution of CS2 (1.2 eq., 0.04
mL) was added slowly. After 1 h stirring at room temperature a solution of CH3I
(1.2 eq., 0.05 mL) was added. The reaction mixture was stirred for 1 h at room
temperature. It was quenched with ice, the organic layer was extracted with
hexane and washed with water to remove DMSO. 1H-NMR of crude showed
peaks corresponding to 18 only. Hence, no reaction occurred.
(d) To a stirred solution of phenyl (2-vinylcyclopent-1-enyl) methanol, (18) (1.0 eq.,
68 mg, 0.34 mmol) in DMF, NaH (1.2 eq., 16 mg of 60% dispersion in mineral
oil) was slowly added portionwise at 0 °C. After the resulting mixture was stirred
for 1h at room temperature CS2 (2 eq., 0.04 mL) was added dropwise at 0 °C.
The resulting mixture was stirred for 2 h at room temperature before CH3I (1.2
Page 174
Texas Tech University, Deepali Butani, August 2011
152
eq., 0.03 mL) was added to the reaction mixture at 0 °C. The resulting mixture
was stirred for 1 h at room temperature, quenched with NH4Cl and extracted
with ether. The combined layer was washed with brine, dried over MgSO4 and
concentrated under vacuum. 1H-NMR of crude showed peaks corresponding to
18 only. Hence, no reaction occurred.
Attempted synthesis of S-methyl O-phenyl (2-vinylcyclohex-1-enyl) methyl
carbonodithioate (50):49
Ph
O
S
S(CH3)
50
Various procedures were tried for synthesis of compound 50:
(a) To a stirred solution of phenyl(2-vinylcyclohex-1-enyl)methanol (16, 1.0 eq.,
0.13 g, 0.62 mmol) in THF at 0 °C, CS2 (67.17 eq., 2.6 mL, 41.6 mmol) was
added, followed by CH3I (67.5 eq., 2.7 mL, 41.8 mmol). After stirring for 15 min
NaH (2.1 eq., 0.05 g of 60% dispersion in mineral oil) was added slowly and the
reaction mixture was stirred at 0 °C for 20 min. The reaction mixture was then
quenched with ice and organic layer was extracted with ethyl acetate. The
organic layer was then washed with brine, dried over MgSO4 and concentrated in
vacuum to give yellow oil. The reaction was monitored by TLC and it showed a
lot of new spots with possibility of product 50. 1H-NMR of crude showed peaks
Page 175
Texas Tech University, Deepali Butani, August 2011
153
corresponding to 50. But the crude compound 50 seemed to decompose on the
silica gel column.
(b) A stirred solution of phenyl(2-vinylcyclohex-1-enyl)methanol, (16, 1.0 eq., 107
mg, 0.5 mmol) in THF, NaH (1.2 eq., 30 mg of 60% dispersion in mineral oil)
was added slowly portionwise at 0 °C. After the resulting mixture was stirred for
1 h at room temperature, CS2 (2 eq., 70 µL) was added dropwise at 0 °C. The
resultant mixture was stirred for 2 h at room temperature before CH3I (1.2 eq.,
40 µL) was added dropwise to the reaction mixture at 0 °C. The resultant
mixture was stirred for 1 h at room temperature, treated with NH4Cl solution and
extracted with diethyl ether. The organic layer was washed with brine, dried over
MgSO4 and concentrated under vacuum. The reaction was monitored by TLC
and it showed a lot of new spots with possibility of product 50. 1H-NMR of
crude showed peaks corresponding to 50. But the crude compound 50 seemed to
decompose on the silica gel column.
(c) DMSO (2 mL) was added under N2 to NaH (1.2 eq., 40 mg of 60% dispersion in
mineral oil) and the mixture was heated to 70 °C and stirred for 45 min. After
cooling the reaction mixture phenyl(2-vinylcyclohex-1-enyl)methanol (16, 1.0
eq., 128 mg, 0.6 mmol) dissolved in 1 mL of DMSO was added dropwise and
the mixture was stirred at room temperature for 1 h. A solution of CS2 (1.2 eq.,
0.04 mL) was added slowly. After 1 h stirring at room temperature a solution of
CH3I (1.2 eq., 0.05 mL) was added. The reaction mixture was stirred for 1 h at
room temperature. It was quenched with ice, the organic layer was extracted with
Page 176
Texas Tech University, Deepali Butani, August 2011
154
hexane and washed with water to remove DMSO. 1H-NMR of crude showed
peaks corresponding to 16 only. Hence, no reaction occurred.
(d) To a stirred solution of phenyl(2-vinylcyclohext-1-enyl)methanol (16, 1.0 eq., 73
mg, 0.34 mmol) in DMF, NaH (1.2 eq., 16 mg of 60% dispersion in mineral oil)
was slowly added portionwise at 0 °C. After the resulting mixture was stirred for
1 h at room temperature CS2 (2 eq., 0.04 mL) was added dropwise at 0°C. The
resulting mixture was stirred for 2 h at room temperature before CH3I (1.2 eq.,
0.03 mL) was added to the reaction mixture at 0 °C. The resulting mixture was
stirred for 1 h at room temperature, quenched with NH4Cl and extracted with
ether. The combined layer was washed with brine, dried over MgSO4 and
concentrated under vacuum. 1H-NMR of crude showed peaks corresponding to
16 only. Hence, no reaction occurred.
Page 177
Texas Tech University, Deepali Butani, August 2011
155
3.6 REFERENCES
1. Claisen, B. Chem. Ber. 1912, 45, 3157-3166.
2. Cope, A. C.; Hardy, E. M. J. Am. Chem. Soc. 1940, 62, 441-444.
3. (a) Nubbemeyer, U. Synthesis 2003, 961-1008. (b) Martin Castro, A. M.
Chem. Rev. 2004, 104, 2939-3002. (c) Ito, H.; Taguchi, T. Chem. Soc. Rev.
1999, 28, 43-50. (d) Enders, D.; Knopp, M.; Schiffers, R. Tetrahedron:
Asymmetry 1996, 7, 1847-1882. (e) Wilson, S. R. Org. React. 1993, 43, 93-
250. (f) Blechert, S. Synthesis 1989, 71-82. (g) Ziegler, F.E. Chem. Rev.
1988, 88, 1423-1452. (h) Lutz, R. P. Chem. Rev. 1984, 84, 205-247. (i)
Ziegler, F. E. Acc. Chem. Res. 1977, 10, 227-232. (j) Bennett, G. B.
Synthesis 1977, 589-606. (k) Rhodds, S. J.; Raulins, N. R. Org. React.
1975, 22, 1-252.
4. Leavell, K. H.; Lewis, E. S. Tetrahedron 1972, 28, 1167-1172.
5. (a) Lewis, E. S.; Hill, J. T. J. Am. Chem. Soc. 1969, 91, 7458-7462. (b)
Lewis, E. S.; Hill, J. T.; Newmann, E. R. J. Am. Chem. Soc. 1968, 90, 662-
668. (c) Lewis, E. S.; Hill, J. T. J. Am. Chem. Soc. 1969, 91, 7458-7462.
6. (a) Morrill, C.; Beutner, G.L.; Grubbs, R. H. J. Org. Chem. 2006, 71, 7813-
7825. (b) Hopf, H.; Wolff, J. Eur. J. Org. Chem. 2001, 2001, 4009-4030.
(c) Pop, E.; Rachwal, S.; Brewster, M. E. Int. J. Quant. Chem. 1996, 60
(8), 1829-1834. (d) Ji, H.; Li. L.; Ham, S.; Hammad, L.A.; Birney, D. M.
J. Am. Chem. Soc. 2009, 131, 528-537.
Page 178
Texas Tech University, Deepali Butani, August 2011
156
7. (a) Nakamura, H.; Yamamoto, Y. In Handbook of Organopalladium
Chemistry for Organic Synthesis; Negishi, E. I., Ed.; John Wiley and Sons:
New York, 2002, Chapter IX.2. (b) Overman, L. E. Angew. Chem., Int. Ed.
1984, 23, 579-586.
8. Overman, L. E.; Campbell, C. B.; Knoll, F. M. J. Am. Chem. Soc. 1978,
100, 4822-4834.
9. Overman, L. E.; Campbell, C. B. J. Org. Chem. 1976, 41, 3338-3340.
10. Overman, L. E.; Knoll, F. M. Tetrahedron Lett. 1979, 26, 321-324.
11. Henry, P. M. J. Am. Chem. Soc. 1972, 94, 5200-5206.
12. Sabel, A.; Smidt, J.; Jira, R.; Prigge, H. Chem. Ber. 1969, 102, 2939-2950.
13. Takatsuto, S.; Ishiguro, M.; Ikekawa, N. J. Chem. Soc., Chem. Commun.
1982, 258-260.
14. (a) Kirihata, M.; Kawahara, S.; Ichimoto, I.; Udea, H. Agric. Biol. Chem.
1990, 54, 753. (b) Kurokawa, N.; Ohfune, Y. Tetrahedron Lett. 1985, 26,
83-84.
15. (a) Nakazawa, M.; Sakamoto, Y.; Takahashi, T.; Tomooka, K.; Ishikawa,
K.; Nakai, T. Tetrahedron Lett. 1993, 34, 5923-5962. (b) Mahrwald, R.;
Schick, H.; Piunitsky, K. K.; Schwarz, S. J. Prakt. Chem. 1990, 332, 403-
413. (c) Grieco, P. A.; Tuthill, P. A.; Sham, H. L. J. Org. Chem. 1981, 46,
5005-5007. (d) Grieco, P. A.; Takigawa, T.; Bongers, S. L.; Tanaka, H. J.
Page 179
Texas Tech University, Deepali Butani, August 2011
157
Am. Chem. Soc. 1980, 102, 7587-7588. (e) Trost, B. M.; Timko, J. M.;
Stanton, J. L. J. Chem. Soc., Chem. Commun. 1978, 436-438.
16. Zbiral, E.; Wessely, F.; Jorg, J. Monatsh. Chem. 1961, 92, 654-666.
17. Birney, D. M.; Xu, X.; Ham, S. Angew. Chem. Int. Ed. 1999, 38, 189-193.
18. Brown, R. F. C. Pyrolytic Methods in Organic Chemistry, Academic Press,
New York, 1980.
19. Vallée, Y. Gas Phase Reactions in Organic Synthesis, Gordon and Breach,
Amsterdam, 1997.
20. McNab, H. Contemp. Org. Synth., 1996, 373–396.
21. Duffy, E. F.; Foot, J. S.; McNab, H; Milligan, A. A. Org. Biomol. Chem.
2004, 2, 2677-2683.
22. Li, L. Thesis, Texas Tech University 2003.
23. Brown, R. F. C. Aust. J. Chem. 2010, 63, 1002–1006.
24. Mumm, O.; Möller, F. Ber. Dtsch. Chem. Ges. 1937, 70, 2214.
25. Overman, L. E. J. Am. Chem. Soc. 1974, 96, 597.
26. Overman, L. E. Tetrahedron Lett. 1975, 13, 1149.
27. Overman, L. E. J. Am. Chem. Soc. 1976, 98, 2901.
28. Overman, L. E. Agnew. Chem., Int. Ed. Engl. 1984, 23, 579.
29. Anderson, C. E.; Overman, L. E. J. Am. Chem. Soc. 2003, 125, 12412.
Page 180
Texas Tech University, Deepali Butani, August 2011
158
30. Overman, L. E.; Owen, C. E.; Pavan, M. M.; Richards, C. J. Org. Lett.
2003, 5, 1809.
31. Kirsch, S. F.; Overman, L. E.; Watson, M. P. J. Org. Chem. 2004, 69,
8101.
32. Beak, Cf. P.; Bonham, J.; Lee, Jr., J. T. J. Am. Chem. Soc. 1968, 90, 1569.
33. (a) Hansen, H. J.; Schmid, H. Tetrahedron, 1974, 30, 1959. (b) Perrin, C.
L.; Faulkner, D. J. Tetrahedron Lett. 1969, 2783. (c) Faulkner, D. J.;
Petersen, M. R. ibid, 1969, 3243.
34. O'Neal, H. E.; Benson. S. W. J. Phys. Chem. 1967, 71, 2903.
35. (a) Miller, J. A.; Scrimgeour, C. M. J. Chem. Soc., Perkin Trans. 2 1973,
1137. (b) White, W. N.; Wolfarth, E. F. J. Org. Chem. 1970, 35, 3585.
36. Wu, Y.J. Name Reactions for Homologation-2 2009, 2, 210-225.
37. Bosnich, B.; Schenck, T. G. J. Am. Chem. Soc. 1985, 107, 2058.
38. Negishi, E., de Meijere, A., Eds. Wiley: New York, 2002, 2, 2014-2015.
39. Overman, L. E. Angew. Chem., Int. Ed. 1984, 23, 579-586.
40. (a) Overman, L. E.; Campbell, C. B.; Knoll, F. M. J. Am. Chem. Soc. 1978,
100, 4822-4834. (b) Tamaru, Y.; Kagotani, M.; Yoshida, Z. J. Org. Chem.
1980, 45, 5221-5223.
Page 181
Texas Tech University, Deepali Butani, August 2011
159
41. (a) Auburn, P. R.; Whelan, J.; Bosnich, B. Organometallics 1986, 5, 1533-
1537. (b) Auburn, P. R.; Whelan, J.; Bosnich, B. Israel J. Chem. 1986, 27,
250-254.
42. Harano, K.; Kiyonaga, H.; Hisano, T. Chem. Pharm. Bull., 1987, 35, 1987.
43. (a) Balsamo, A.; Coletta, I.; Domiano, P.; Guglielmotti, A.; Landolfi, C.;
Mancini, F.; Milanese, C.; Orlandini, E.; Rapposelli, S.; Pinza, M.;
Macchia, B. Eur. J. Med. Chem. 2002, 37, 391-398. (b) Park, C.-M.;
Bruncko, M.; Adickes, J.; Bauch, J.; Ding, H.; Kunzer, A.; Marsh, K. C.;
Nimmer, P.; Shoemaker, A. R.; Song, X.; Tahir, S. K.; Tse, C.; Wang, X.;
Wendt, M. D.; Yang, X.; Zhang, H.; Fesik, S. W.; Rosenberg, S. H.;
Elmore, S. W. J. Med. Chem. 2008, 51, 6902-6915. (c) Waddell, M. K.;
Bekele, T.; Lipton, M. A. J. Org. Chem. 2006, 71, 8372-8377. (d) Salem,
B.; Delort, E.; Klotz, P.; Suffert, J. Org. Lett. 2003, 5, 2307-2310.
44. (a) Gagnier, S. V.; Larock, R. C. J. Am.Chem.Soc. 2003, 125(16), 4804. (b)
Ohe, K.; Miki, K.; Yokoi, T.; Nishino, F.; Uemura, S. Organometallics
2000, 19, 5525-5528. (c) Lian, J.-J.; Odedra, A.; Wu, C.-J.; Liu, R.-S. J.
Am. Chem. Soc. 2005, 127, 4186-4187. (d) Zhang, Y.; Herndon, J. W.
Org. Lett. 2003, 5, 2043-2045.
45. (a) Gagnier, S. V.; Larock, R. C. J. Am. Chem. Soc. 2003, 125, 4804-4807.
(b) Denmark, S. E.; Habermas, K. L.; Hite, G. A. Helv. Chim. Acta. 1988,
71, 168-194. (c) Denmark, S. E.; Hite, G. A. Helv. Chim. Acta. 1988, 71,
195-208. (d) EI-Khoury, M.; Wang, Q.; Schlosser, M. Tetrahedron Lett.
Page 182
Texas Tech University, Deepali Butani, August 2011
160
1996, 37, 9047-9048. (e) Leung, S. H.; Angel, S. A. J. Chem. Ed. 2004,
81, 1492.
46. (a) Clive, D. L. J.; Pham, M. P. The J. Org. Chem. 2009, 74, 1685-1690.
(b) Denmark, S. E.; Hite, G. A. Helv. Chim. Acta. 1988, 71, 195-208.
47. Ji, H.; Li, L.; Xu, X.; Ham, S.; Hammad, L. A.; Birney, D. M. J. Am.
Chem. Soc. 2009, 131, 528–537.
48. (a) Overman, L. E. J. Am. Chem. Soc. 1974, 96, 597. (b) Overman, L. E.
J. Am. Chem. Soc. 1976, 98, 2901. (c) Lurain, C. A. E.; Walsh, P. J. J. Am.
Chem. Soc. 2002, 124, 12225-12231. (d) Nishikawa, T.; Asai, M.; Ohyabu,
N.; Isobe, M. J. Org. Chem. 1998, 63, 188-192. (e) Kobbelgaard, S.;
Brandes, S.; Jørgensen, K. A. Chem. Eur. J. 2008, 14, 1464 – 1471. (f)
Arnold, J. S.; Stone, R. F.; Nguyen, H. M. Org. Lett. 2010, 12 (20), 4580–
4583. (g) Chen, Y. K.; Lurain, A. E.; Walsh, P. J. J. Am. Chem. Soc. 2002,
124, 12225-12231.
49. (a) Kanie, K. Chem. Soc. Japan, 2000, 73(2), 471-484 (b) Park, H.S. Org.
Lett. 2005, 7(15), 3187-3190. (c) Dianaa, M. B.; Marchetti, M.; Melloni, G.
Tetrahedron: Asymmetry 1995, 6, 1175-1179. (d) Roche, A. J.; Loylea, A.
D.; Pinto, J. P. J. Fluorine Chem. 2004, 125, 1473-1480.
50. Woodward, R. B.; Hoffmann, R. Angew. Chem. Internat. Edit. 1969, 8,
781-853.
51. Eliel, E.; Allinger, N. L. Topics in Stereochemistry, Wiley 1978.
Page 183
Texas Tech University, Deepali Butani, August 2011
161
APPENDIX- A
OPTIMIZED CARTESIAN COORDINATES FOR THE
THEORETICAL CALCULATIONS
Page 184
Texas Tech University, Deepali Butani, August 2011
162
Cartesian coordinates of all stationary structures at different levels.
[3, 3] sigmatropic rearrangement of allyl azide
Ground State (1a)
RB3LYP/6-31G(d,p)
C .000007825 .000031819 -.000011125
C -.000023219 -.000021619 .000021816
C .000011083 .000007571 -.000005048
H .000009121 .000008845 .000003558
N .000007902 -.000054733 -.000045079
N .000073161 .000091318 .000106815
N -.000088452 -.000047665 -.000058837
H .000007271 -.000004969 -.000004827
H -.000003695 -.000017648 .000010074
H -.000006222 -.000001610 -.000006182
H .000005225 .000008692 -.000011166
RHF/6-31G(d,p)
C -.000018963 .000000395 .000003138
C .000013176 -.000018412 .000001521
C -.000006899 .000011309 -.000005845
H -.000005075 -.000013608 .000002187
Page 185
Texas Tech University, Deepali Butani, August 2011
163
N .000005530 -.000008907 .000016253
N .000004840 .000009748 -.000024945
N .000013467 .000010419 .000012447
H .000001659 .000003068 -.000011412
H .000001339 .000002279 -.000011051
H -.000007725 .000006071 .000005640
H -.000001350 -.000002360 .000012067
RMP2/6-31G(d,p)
C .000002496 -.000000534 -.000003577
C -.000000414 .000001805 .000005703
C -.000000131 -.000000155 -.000001046
H -.000000168 .000000512 .000000079
N .000000245 -.000002414 -.000002691
N -.000003962 -.000002365 .000004704
N .000002262 .000003732 -.000002230
H .000000839 -.000001076 -.000000477
H -.000000482 .000000251 -.000000836
H .000000483 -.000000094 -.000000005
H -.000001168 .000000337 .000000376
Transition State (5)
Page 186
Texas Tech University, Deepali Butani, August 2011
164
RB3LYP/6-31G(d,p)
C -.000003285 -.000000932 -.000005096
C .000005653 -.000015573 -.000016632
C .000009082 .000010114 .000016232
H .000006285 -.000003389 -.000001846
N .000009904 .000006192 .000022783
N -.000035941 .000012466 .000013989
N -.000015299 .000000267 -.000021678
H .000006430 -.000003054 -.000004094
H .000009053 .000000721 -.000003570
H .000007738 -.000006903 .000000117
H .000000380 .000000091 -.000000206
RHF/6-31G(d,p)
C .000004464 .000006282 .000004293
C -.000015589 -.000000017 -.000006634
C .000004422 -.000006270 .000004287
H -.000001725 .000000329 -.000003862
N .000019022 -.000078491 -.000007670
N -.000028714 -.000000091 .000013795
N .000019041 .000078586 -.000007678
H -.000002500 -.000000547 .000005372
H -.000002485 .000000546 .000005370
H -.000001720 -.000000323 -.000003858
H .000005784 -.000000003 -.000003413
Page 187
Texas Tech University, Deepali Butani, August 2011
165
RMP2/6-31G(d,p)
C -.000006334 .000004339 -.000007043
C .000003742 .000000026 .000006696
C -.000006334 -.000004309 -.000007056
H -.000000228 -.000002901 .000001535
N -.000003108 -.000059956 .000010337
N .000022656 .000000049 -.000012791
N -.000003088 .000059908 .000010353
H -.000002954 .000000602 -.000001021
H -.000002967 -.000000608 -.000000995
H -.000000228 .000002858 .000001510
H -.000001157 -.000000008 -.000001526
Conformation-1 (1b)
RB3LYP/6-31G(d,p)
C -.000001631 -.000000222 -.000005062
C .000003760 .000005662 -.000006258
C -.000001764 -.000002632 .000003864
H .000000807 .000000876 .000001079
H -.000000388 -.000000160 -.000001120
H .000000188 .000000090 -.000001312
H .000001453 -.000001234 -.000000324
H .000000867 -.000000777 .000000660
Page 188
Texas Tech University, Deepali Butani, August 2011
166
N -.000005700 .000012805 .000011073
N .000005179 -.000021603 -.000008463
N -.000002771 .000007194 .000005863
RHF/6-31G(d,p)
C -.000011729 .000001045 -.000012583
C -.000005051 -.000001425 .000003420
C .000001256 .000004138 .000000008
C -.000002210 .000003881 .000004030
H -.000001182 .000000711 -.000001398
H -.000000253 -.000000317 -.000000190
H .000000815 -.000000221 .000000484
H .000003638 -.000008620 -.000001232
N .000022904 .000014945 .000016445
N .000104130 -.000003252 -.000019310
N -.000112319 -.000010886 .000010325
RMP2/6-31G(d,p)
C -.000004021 -.000043240 -.000000800
C -.000001027 -.000023241 .000000365
C .000013418 .000038927 -.000000183
H -.000001389 .000021642 -.000000131
H -.000001367 .000011454 .000000047
H -.000008702 -.000009448 .000000244
H .000009534 -.000011983 -.000000276
H -.000000374 .000022113 .000000394
N -.000040706 .000000096 -.000001002
Page 189
Texas Tech University, Deepali Butani, August 2011
167
N -.000049888 -.000015609 .000001792
N .000084521 .000009290 -.000000450
Conformation-2 (1c)
RB3LYP/6-31G(d,p)
C -.000001593 .000002730 -.000000646
C -.000000712 .000000147 .000000903
C .000000503 .000001874 -.000000175
H -.000006523 .000004244 .000000376
H .000000150 .000003660 .000001690
H .000001063 .000000975 .000000139
H -.000000102 -.000002859 -.000001049
H .000002281 .000004303 .000001925
N .000001116 -.000004518 .000004802
N -.000000162 -.000004871 -.000004787
N .000003978 -.000005684 -.000003179
RHF/6-31G(d,p)
C .000001582 -.000000126 -.000000610
C .000000521 -.000000003 .000000117
C -.000000186 .000000019 -.000000208
H -.000000120 .000000183 .000000234
H .000000375 .000000068 .000000087
Page 190
Texas Tech University, Deepali Butani, August 2011
168
H .000000150 -.000000106 -.000000041
H -.000000269 -.000000164 -.000000042
H -.000000512 .000000140 -.000000066
N -.000000650 -.000002617 .000001561
N .000002655 .000009424 -.000000341
N -.000003546 -.000006818 -.000000689
RMP2/6-31G(d,p)
C -.000007848 -.000003080 .000000718
C -.000000928 -.000011874 .000002265
C .000003191 .000003980 -.000004262
H -.000004419 -.000000679 .000001539
H .000001967 .000001401 -.000000727
H .000001221 -.000001034 .000000150
H .000000768 -.000000091 .000001121
H .000000255 .000000690 -.000001753
N -.000007010 -.000005184 -.000001887
N -.000014447 -.000006396 -.000013634
N .000027251 .000022268 .000016470
Conformation-3(1d)
RB3LYP/6-31G(d,p)
C -.000001393 -.000003541 .000000992
Page 191
Texas Tech University, Deepali Butani, August 2011
169
C -.000000244 .000000966 -.000001671
C .000000138 .000003154 -.000000809
H .000000068 -.000002422 .000000816
N -.000000257 -.000000819 -.000000375
N -.000000214 -.000002371 .000000448
N .000000141 -.000003827 .000001552
H .000002388 .000001149 .000002453
H .000001889 .000003892 .000001164
H -.000000469 .000003654 -.000001718
H -.000002046 .000000165 -.000002853
RHF/6-31G(d,p)
C -.000003177 .000000361 .000003723
C .000001135 -.000000622 .000001099
C -.000002092 .000000234 -.000001882
H .000000057 .000000058 .000000789
N .000000797 -.000006193 -.000008157
N -.000018936 .000001137 .000004341
N .000023481 .000003161 -.000000347
H .000000145 .000001041 -.000000617
H -.000000152 .000000947 -.000000294
H -.000000916 -.000000347 -.000000120
H -.000000342 .000000222 .000001464
RMP2/6-31G(d,p)
C -.000003291 -.000004162 .000000224
C .000005311 .000004942 -.000003785
Page 192
Texas Tech University, Deepali Butani, August 2011
170
C .000003018 -.000000763 .000001635
H -.000002084 -.000000620 .000000745
N -.000006708 -.000005543 .000003945
N .000001684 .000004165 -.000004743
N .000001881 -.000000562 .000001862
H -.000000792 .000002936 -.000000160
H .000000035 -.000000629 -.000000517
H -.000000643 .000000494 .000000987
H .000001589 -.000000257 -.000000192
Conformation-4 (1e)
RB3LYP/6-31G(d,p)
C .000001463 -.000006503 .000005438
C -.000008526 .000005652 -.000018073
C .000003075 -.000000654 -.000003079
H -.000004530 -.000006653 .000004872
N -.000036873 .000006056 .000006482
N .000077069 -.000007723 .000004639
N -.000017876 .000000440 .000001465
H -.000016289 .000008997 .000000362
H .000001141 -.000001730 -.000001247
H .000001634 .000002096 -.000001394
H -.000000289 .000000022 .000000535
Page 193
Texas Tech University, Deepali Butani, August 2011
171
RHF/6-31G(d,p)
C .000012551 .000001422 .000007363
C -.000004358 -.000000708 -.000004788
C .000003089 -.000001818 .000003845
H -.000006281 .000001585 -.000001202
N -.000001075 .000005267 .000005879
N -.000019758 -.000001352 -.000022407
N .000007577 .000000102 .000013841
H .000006279 -.000005682 -.000001985
H .000001780 -.000003621 .000000740
H .000002414 .000002985 -.000000550
H -.000002217 .000001820 -.000000736
RMP2/6-31G(d,p)
C .000041664 .000002617 .000005972
C -.000031409 .000006115 -.000025275
C .000033288 -.000011550 .000013660
H .000000148 -.000001498 .000014477
N -.000000117 .000006758 -.000000474
N .000007245 -.000077342 .000006538
N -.000073174 .000087796 -.000002371
H .000026230 .000001138 -.000010963
H -.000007200 .000008284 -.000001909
H -.000008789 -.000007755 -.000003020
H .000012115 -.000014563 .000003365
Page 194
Texas Tech University, Deepali Butani, August 2011
172
[3, 5] sigmatropic rearrangement of vinylogous azides
Transition State-1 (11)
RB3LYP/6-31G(d,p)
C .000001173 .000006054 .000016174
C .000001624 .000004940 .000031247
C -.000027766 -.000003505 -.000041885
C -.000040782 .000031350 .000084463
H .000000383 -.000006324 -.000021363
H .000002032 .000002101 .000004346
H -.000008512 .000003676 .000013003
H .000009233 -.000009695 -.000038420
C -.000022318 -.000012471 -.000005860
H .000002505 -.000012667 -.000005567
N .000044725 -.000002153 -.000006192
N -.000006347 .000012303 .000002862
N .000039729 -.000011202 -.000034944
H .000000958 .000002207 -.000011526
H .000003364 -.000004615 .000013662
Page 195
Texas Tech University, Deepali Butani, August 2011
173
RHF/6-31G(d,p)
C .000007589 .000008583 -.000006649
C -.000008850 .000001670 -.000007674
C .000005043 -.000002074 .000001248
C -.000003095 -.000001488 .000003318
H -.000002936 .000003745 .000003372
H .000001233 -.000002243 -.000003941
H -.000000011 -.000000376 -.000001549
H .000001452 .000000062 .000001591
C .000001587 -.000016823 .000005871
H -.000002148 .000001423 .000001610
N .000004309 .000001734 -.000002056
N -.000004633 -.000003905 -.000006104
N .000001187 .000002805 .000002918
H -.000000662 .000002122 .000003548
H -.000000065 .000004765 .000004497
RMP2/6-31G(d,p)
C .000012079 .000016086 .000003737
C -.000014496 .000007669 .000003501
C .000003060 .000013824 .000017346
C .000002850 -.000005907 -.000010394
H -.000002433 .000001973 .000004165
H -.000001249 -.000002847 -.000000979
H .000000804 .000000607 -.000004265
H .000003619 .000005776 .000007578
Page 196
Texas Tech University, Deepali Butani, August 2011
174
C -.000001609 .000006651 -.000022239
H .000001230 -.000000831 -.000002811
N .000013329 -.000023154 .000002695
N -.000042607 -.000022037 .000000124
N .000017318 .000005572 .000014105
H .000000042 -.000000182 .000000208
H .000008064 -.000003201 -.000012771
Reactant-1 (12)
RB3LYP/6-31G(d,p)
C .000013321 .000003049 .000007617
C -.000008647 -.000008727 .000005859
C -.000002898 -.000012129 .000003074
C -.000011760 -.000014219 .000008967
H .000001346 .000002964 .000002562
H -.000000944 -.000001116 .000002272
H .000008364 .000000036 -.000003741
H -.000001013 -.000004567 -.000001411
C -.000015906 .000009963 -.000004642
H -.000001111 .000005836 -.000000580
N .000020183 -.000001818 -.000024242
N -.000028960 -.000008778 .000020818
Page 197
Texas Tech University, Deepali Butani, August 2011
175
N .000031225 .000027310 -.000012447
H -.000002131 -.000003142 -.000001790
H -.000001068 .000005339 -.000002317
RHF/6-31G(d,p)
C .000006044 -.000037575 -.000002823
C -.000016752 -.000003519 -.000001541
C .000015153 .000005436 -.000000472
C .000008954 -.000015232 -.000006242
H -.000001174 -.000003428 -.000002857
H -.000000981 -.000002196 .000001274
H -.000000755 -.000000148 -.000002975
H .000002005 -.000000059 -.000000217
C -.000007189 .000003439 .000004706
H -.000002785 .000000204 -.000001248
N -.000019645 .000013558 -.000020021
N .000048707 .000021024 .000021821
N -.000033770 .000019525 .000008526
H .000002338 -.000001429 .000000216
H -.000000149 .000000400 .000001852
RMP2/6-31G(d,p)
C -.000010130 -.000012737 .000019121
C -.000019220 -.000010378 -.000012668
C .000013486 -.000003136 -.000016953
C .000027764 .000001240 .000006605
H .000005833 .000006168 -.000000651
Page 198
Texas Tech University, Deepali Butani, August 2011
176
H -.000000724 .000006898 .000004240
H .000004533 .000002972 .000008409
H -.000001336 -.000000666 .000000870
C .000000299 -.000002889 .000004613
H .000004567 -.000000478 -.000000372
N -.000019577 .000008323 -.000006791
N -.000063854 .000011894 .000005242
N .000060526 -.000014819 -.000013548
H -.000002658 .000007036 .000004265
H .000000492 .000000572 -.000002382
Product-1 (13)
RB3LYP/6-31G(d,p)
C .000004156 -.000001371 -.000004003
C -.000000516 -.000002147 -.000004044
C -.000003226 -.000000768 -.000000278
C -.000001418 .000001264 .000003008
H .000006228 -.000002535 -.000006061
H -.000001485 -.000003347 -.000006847
H -.000001759 .000002083 .000004693
H .000001809 .000002115 .000003481
C .000004932 -.000000735 -.000001767
H .000008355 -.000000506 -.000002095
Page 199
Texas Tech University, Deepali Butani, August 2011
177
N -.000004930 .000001990 .000004558
N -.000004498 .000002104 .000004850
N -.000004640 .000002888 .000005788
H -.000005847 -.000000928 -.000001466
H .000002839 -.000000107 .000000182
RHF/6-31G(d,p)
C .000003315 -.000002516 -.000004219
C .000001378 -.000000895 .000000928
C .000000446 -.000000476 -.000002109
C .000000825 .000002043 -.000000253
H -.000001180 -.000000510 -.000001113
H -.000000137 .000001130 .000001937
H -.000001355 .000002724 -.000001682
H .000001144 .000000108 .000000798
C -.000003382 .000004640 .000002577
H .000000006 -.000001111 -.000000619
N -.000010592 .000039156 -.000010549
N .000006740 -.000049240 .000004302
N -.000005250 .000004290 .000005820
H .000000512 .000000866 .000001586
H .000007530 -.000000209 .000002596
RMP2/6-31G(d,p)
C -.000001216 -.000000834 .000000705
C .000000017 .000002405 .000000315
C .000001308 -.000000637 -.000000282
Page 200
Texas Tech University, Deepali Butani, August 2011
178
C .000000140 -.000001608 -.000000020
H -.000000422 .000000457 -.000000037
H -.000000109 -.000000339 -.000000217
H .000000523 .000000005 .000000448
H -.000000130 -.000000419 -.000000165
C .000000833 -.000000028 .000000073
H -.000000175 .000000253 -.000000236
N -.000000644 .000000342 -.000000667
N .000000020 -.000000243 .000001114
N -.000000333 .000001157 -.000000525
H -.000000476 -.000000400 -.000000864
H .000000664 -.000000113 .000000357
Transition State-2 (14)
RB3LYP/6-31G(d,p)
C -.000115935 .000043503 .000092751
C .000041974 -.000006554 .000027102
C -.000052727 .000008778 -.000019405
C .000010408 .000034357 -.000003307
H -.000005495 -.000001191 .000006913
H .000001600 .000003969 .000009131
Page 201
Texas Tech University, Deepali Butani, August 2011
179
H .000008223 .000000706 .000000626
H .000001741 -.000018395 -.000025051
C .000109860 -.000039757 -.000088742
H -.000010101 -.000009395 .000013274
N .000000091 -.000009885 -.000003770
N .000016074 -.000057557 .000017052
N -.000002307 .000033180 -.000019164
H .000009115 .000006054 .000001188
H -.000012521 .000012188 -.000008597
RHF/6-31G(d,p)
C -.000020438 .000025275 -.000011869
C -.000010480 -.000042605 .000003892
C .000018637 .000028225 .000003773
C -.000016696 -.000004129 .000001149
H .000004406 .000003418 .000002207
H .000003713 -.000003020 .000003132
H -.000000250 -.000006359 -.000002426
H .000007718 .000000031 -.000013502
C .000040601 -.000031066 .000018035
H -.000002836 .000000956 -.000011284
N -.000095225 -.000164064 -.000103872
N .000065211 .000210404 .000035258
N .000048685 -.000054646 .000059051
H .000000418 -.000004229 .000005735
H -.000043465 .000041810 .000010721
Page 202
Texas Tech University, Deepali Butani, August 2011
180
RMP2/6-31G(d,p)
C .000003949 .000009278 -.000018906
C -.000013832 -.000005142 -.000006159
C .000014893 .000003137 .000018088
C -.000016440 .000000916 .000000605
H -.000000524 .000001149 -.000000427
H -.000001417 .000000874 .000001676
H -.000001621 .000000009 .000000031
H .000006364 -.000000205 -.000010115
C .000012297 .000002412 .000006852
H .000000995 -.000000956 -.000004679
N -.000040562 -.000028027 -.000019434
N .000025313 -.000004330 .000024259
N .000018746 .000028045 -.000002175
H .000000192 -.000000048 .000001059
H -.000008353 -.000007113 .000009325
Reactant-2 (15)
RB3LYP/6-31G(d,p)
C .000003989 -.000000024 -.000002043
C .000000553 .000003672 -.000005123
Page 203
Texas Tech University, Deepali Butani, August 2011
181
C -.000002065 .000004929 -.000002931
C -.000001936 -.000000805 -.000001703
H .000002141 -.000000854 .000004916
H .000003202 .000000601 .000004457
H -.000003566 .000001451 -.000005584
H -.000003600 -.000002463 -.000002110
C -.000003006 .000000683 .000005272
H .000000540 -.000001812 .000003962
N .000002879 .000002354 .000000421
N -.000001085 -.000005761 .000003243
N -.000000405 .000001268 -.000000646
H .000004480 -.000001194 -.000007597
H -.000002123 -.000002045 .000005469
RHF/6-31G(d,p)
C -.000000079 .000005550 -.000000901
C -.000000464 .000001052 .000000045
C .000000233 .000000631 -.000000856
C -.000001159 .000000676 -.000000971
H .000004412 -.000000198 .000002645
H .000000228 .000000999 -.000002083
H .000000721 .000000157 -.000000115
H -.000000474 -.000001543 .000000552
C -.000001849 -.000005674 .000010980
H .000001968 .000000352 .000002301
N .000003282 .000008460 -.000000115
N -.000008149 -.000015955 -.000020082
Page 204
Texas Tech University, Deepali Butani, August 2011
182
N .000003337 .000007629 .000013351
H -.000001640 -.000000007 -.000000693
H -.000000367 -.000002128 -.000004059
RMP2.6-31G(d,p)
C .000005463 -.000010452 .000004784
C -.000001354 .000000813 -.000001247
C -.000002597 -.000004827 .000005008
C -.000001414 -.000000178 -.000001865
H -.000000870 -.000000851 -.000003351
H -.000000790 .000004613 -.000002971
H .000000552 .000004589 .000001061
H .000000757 -.000001979 -.000001045
C .000002079 .000010879 -.000000799
H .000003538 -.000003469 -.000000018
N -.000008667 .000004144 .000000572
N .000010591 -.000003335 -.000010341
N -.000004606 .000000018 .000007351
H .000000386 -.000002051 .000001898
H -.000003067 .000002085 .000000962
Product-2 (16)
Page 205
Texas Tech University, Deepali Butani, August 2011
183
RB3LYP/6-31G(d,p)
C .000001200 .000000615 -.000001090
C -.000001100 .000003225 -.000001637
C -.000003226 .000002643 -.000000145
C -.000002036 -.000000827 .000002845
H .000000453 .000001380 -.000001513
H -.000002043 .000006156 -.000002773
H -.000003915 -.000000666 .000004327
H -.000000333 -.000003426 .000003575
C .000003380 -.000002594 -.000001110
H .000004967 -.000004373 -.000001247
N .000001102 -.000000104 -.000001174
N .000000904 -.000000940 -.000000252
N .000000925 -.000002445 .000001318
H -.000004148 .000004875 -.000000001
H .000003868 -.000003521 -.000001122
RHF/6-31G(d,p)
C .000000827 .000000578 .000001037
C -.000000600 -.000001046 -.000000951
C -.000000451 -.000000846 .000000099
C .000002234 .000000043 -.000000232
H -.000000572 .000000382 -.000000157
H -.000000415 .000000787 -.000000067
H .000000155 .000001310 .000000919
H .000001186 -.000000780 -.000000984
C .000000179 .000000232 -.000000202
Page 206
Texas Tech University, Deepali Butani, August 2011
184
H -.000000342 -.000000377 .000000049
N -.000004373 -.000001150 -.000001086
N .000007218 .000003042 .000003659
N -.000006071 -.000002961 -.000001570
H .000000061 .000000678 -.000000407
H .000000964 .000000109 -.000000108
RMP2/6-31G(d,p)
C .000000582 .000001303 .000000496
C -.000002467 -.000000334 .000000915
C -.000000410 -.000000584 .000000548
C .000000670 .000000164 .000002750
H -.000000044 .000000096 -.000000006
H .000000064 .000000019 -.000000072
H -.000000503 .000001607 -.000000615
H .000000974 -.000000551 .000000884
C -.000000645 -.000000733 -.000000502
H .000000277 .000000203 -.000000069
N .000001894 .000000154 .000001190
N .000000258 -.000001315 .000002295
N -.000001554 .000001493 -.000005957
H .000000717 -.000001415 -.000001653
H .000000186 -.000000107 -.000000206
Page 207
Texas Tech University, Deepali Butani, August 2011
185
Transition State-3 (17)
RB3YLP/631G(d,p)
C -.000003782 -.000011996 .000000893
C .000000536 .000020691 .000003738
C .000000517 -.000003348 -.000001138
C -.000002929 -.000000432 .000001837
H -.000000929 .000000945 .000001221
H .000000689 .000023082 .000002010
H -.000002341 .000002852 -.000000432
H -.000003970 -.000008009 .000002963
C -.000005313 -.000008379 -.000003651
H -.000007628 -.000007728 .000001817
N .000014892 -.000003747 -.000007050
N .000011965 .000006272 .000014484
N .000013098 .000003158 -.000010825
H .000001515 .000008690 .000000667
H -.000016320 -.000022051 -.000006534
RHF/6-31G(d,p)
C -.000002212 -.000005151 -.000000636
C .000000731 .000000307 .000002034
Page 208
Texas Tech University, Deepali Butani, August 2011
186
C .000005671 -.000001506 -.000015186
C -.000002128 -.000006650 .000005226
H .000001462 .000002764 -.000003267
H -.000000250 .000003447 .000006770
H -.000001383 .000004379 .000002855
H -.000002195 -.000000206 .000003838
C -.000003718 .000000071 -.000022840
H .000002009 .000002407 .000000086
N .000004977 -.000004565 .000032361
N -.000120134 -.000078578 -.000067055
N .000119798 .000084100 .000057460
H -.000001110 -.000000895 .000002740
H -.000001518 .000000076 -.000004386
RMP2/6-31G(d,p)
C -.000004286 .000029426 .000003589
C -.000022049 -.000013768 -.000002962
C .000015957 .000006575 .000015585
C -.000006549 .000010472 -.000009495
H -.000000597 .000001139 .000000894
H .000000337 -.000000553 -.000000623
H .000000301 -.000000558 .000000631
H -.000000044 .000002278 -.000000886
C -.000000450 -.000007081 -.000011509
H -.000000088 .000004706 .000001480
N .000021798 -.000011114 .000010899
N .000042573 .000019956 .000014684
Page 209
Texas Tech University, Deepali Butani, August 2011
187
N -.000052488 -.000034061 -.000018543
H .000000699 -.000002216 -.000002233
H .000004887 -.000005202 -.000001510
Transition state-4 (18)
RB3LYP/6-31G(d,p)
C .000008727 .000031088 -.000017568
C .000003595 .000004816 -.000001792
C -.000005457 -.000005023 -.000002184
N -.000001860 -.000002106 .000002370
N -.000008902 .000001461 -.000007128
N .000006229 -.000002273 -.000000270
H -.000002421 .000000020 .000001664
H -.000000255 -.000001664 .000000287
H -.000000205 .000001624 -.000001433
H .000000304 .000001536 -.000000433
C -.000000391 -.000027450 .000016263
H .000001011 .000000987 .000003328
C -.000000413 -.000001294 .000002064
H -.000001272 -.000000994 .000001290
H .000001311 -.000000728 .000003541
Page 210
Texas Tech University, Deepali Butani, August 2011
188
RHF/6-31G(d,p)
C -.000000879 -.000000345 .000000235
C .000001076 .000000292 -.000000466
C .000001233 -.000001851 -.000000512
N .000012185 .000003463 .000001658
N -.000018915 -.000007944 -.000003221
N .000005508 .000005992 .000001974
H .000000040 .000000055 .000000331
H .000000011 -.000000039 -.000000370
H -.000000183 .000000293 .000000435
H -.000000303 .000000190 .000000110
C -.000000192 -.000000257 .000000220
H .000000047 -.000000281 -.000000091
C .000000378 .000000459 -.000000314
H .000000014 .000000000 .000000030
H -.000000018 -.000000029 -.000000019
RMP2/6-31G(d,p)
C .000005164 -.000003180 -.000004816
C -.000000048 -.000003547 -.000001206
C .000001347 -.000002096 .000003234
N .000020559 .000000425 -.000000889
N -.000010540 .000008491 .000005719
N -.000014153 -.000003772 -.000003972
H -.000001099 -.000000553 .000001352
H -.000000064 .000000630 -.000001740
H .000002045 .000000854 .000000350
Page 211
Texas Tech University, Deepali Butani, August 2011
189
H .000000231 -.000000478 -.000000233
C -.000003697 .000002820 .000002140
H .000000317 .000000736 .000000372
C -.000000096 -.000000225 -.000000275
H -.000000038 .000000191 -.000000019
H .000000073 -.000000297 -.000000017
Product-4 (19)
RB3LYP/6-31G(d,p)
C .000005311 -.000001130 .000002244
C -.000000346 .000005267 .000001524
C .000000647 .000000812 -.000001184
N -.000002518 -.000001904 .000002349
N .000001914 -.000001310 -.000001596
N .000000302 -.000001387 .000006845
H -.000000639 .000002063 -.000001009
H .000002232 .000001140 -.000000691
H .000000710 .000000598 -.000001193
H -.000002335 -.000000885 -.000000755
C .000000827 -.000008068 .000004960
H -.000003989 .000002221 -.000000959
C .000000476 .000005058 .000001138
H .000000412 .000001498 -.000007116
H -.000003004 -.000003972 -.000004557
Page 212
Texas Tech University, Deepali Butani, August 2011
190
RHF/6-31G(d,p)
C .000004845 .000003526 -.000000850
C .000000081 -.000001456 .000000578
C -.000000393 .000000785 -.000000531
N -.000002920 -.000004322 .000000954
N .000001101 .000002720 .000002014
N -.000002718 -.000001623 -.000001627
H -.000000144 .000000024 -.000001034
H -.000000110 -.000000099 -.000000839
H -.000000089 .000000758 .000000320
H .000000264 .000000055 .000000892
C -.000001576 -.000000307 .000000436
H .000000127 .000000201 .000000850
C .000000820 -.000000266 -.000000588
H -.000000091 .000000135 -.000000838
H .000000805 -.000000133 .000000263
RMP2/6-31G(d,p)
C .000000394 .000002053 -.000000266
C .000002236 -.000001818 -.000002524
C .000000026 -.000001880 .000001002
N -.000003638 .000004276 .000003550
N -.000004226 -.000005727 -.000006029
N .000007797 .000001537 .000003841
H .000000151 .000001250 -.000000582
H .000000016 -.000000138 -.000000127
H -.000000203 .000000620 .000000278
Page 213
Texas Tech University, Deepali Butani, August 2011
191
H -.000000232 -.000000496 .000000812
C -.000001089 -.000000192 -.000001661
H .000000257 .000000143 .000000759
C -.000002074 .000000146 .000001080
H -.000000078 -.000000240 -.000000437
H .000000663 .000000466 .000000304
Interconversion of cis- and trans-1-azido-2-butene, via a sequential [3, 3] sigmatropic
rearrangements at RB3LYP/6-31G(d,p)
Transition state of trans#-1-azido-2-butene (6)
C -.000011243 .000001152 .000010903
C .000004330 -.000009025 -.000002149
C -.000000118 -.000003188 -.000001174
N .000005040 -.000027169 .000001252
N .000002993 .000018131 -.000012534
N .000000186 .000005826 -.000001339
H -.000006280 .000000595 -.000008737
H -.000004890 .000000965 .000003052
H -.000000531 -.000000669 .000003138
H .000003420 -.000002858 .000002552
C -.000009057 .000017667 -.000026576
H .000005818 .000007001 -.000006267
Page 214
Texas Tech University, Deepali Butani, August 2011
192
H .000002539 -.000007087 .000004733
H .000007792 -.000001341 .000033147
Transition state of cis#-1-azido-2-butene (7)
C .000021499 .000010173 .000022868
C .000004243 -.000005368 -.000008018
C .000003249 -.000006449 .000001628
N -.000003210 -.000058332 .000002315
N .000005156 .000068051 .000002117
N -.000010796 .000013095 -.000002284
H -.000005470 -.000000062 -.000004640
H -.000010004 -.000000175 .000000211
H -.000004951 -.000000444 -.000001686
C -.000012046 .000005465 -.000012124
H .000001884 -.000008175 .000008388
H .000002837 -.000014163 -.000005802
H .000002939 -.000000123 .000013082
H .000004669 -.000003493 -.000016054
Page 215
Texas Tech University, Deepali Butani, August 2011
193
cis-1-azido-2-butene (4)
C -.000009371 -.000001332 -.000003605
C .000010080 .000000310 .000000993
C -.000010101 -.000007010 .000014308
H .000004410 -.000001224 .000000124
N .000003304 .000003506 -.000002888
N .000001446 .000002995 -.000003086
N .000000430 .000003883 -.000005261
H .000000980 .000006492 -.000001756
H .000001609 -.000007898 .000007401
H .000007191 .000000229 .000001456
C -.000018593 -.000008774 .000005390
H .000002248 .000000576 .000000274
H .000004997 -.000004107 -.000004986
H .000001371 .000012355 -.000008361
trans-1-azido-2-butene (2)
C .000000468 -.000002243 -.000001740
C .000007628 -.000000093 -.000003664
C -.000000560 -.000012497 .000014870
Page 216
Texas Tech University, Deepali Butani, August 2011
194
H -.000002369 -.000000493 -.000003687
N -.000002016 -.000002319 -.000002891
N -.000000207 -.000001697 .000000219
N .000000942 -.000002894 .000001096
H -.000001915 .000001526 -.000002338
H -.000008864 .000004210 .000002220
H -.000004889 -.000006773 -.000002376
C .000009464 .000011693 -.000004564
H -.000005249 -.000007983 -.000009249
H .000000212 .000021259 .000010964
H .000007356 -.000001697 .000001140
3-azido-1-butene (3)
C -.000001035 .000000833 -.000000380
C .000001296 .000001470 -.000000225
C .000001590 .000001086 -.000001965
N .000001081 -.000001774 -.000000337
N .000000298 -.000002666 .000004990
N .000002607 -.000003627 -.000000915
H .000000162 -.000001453 -.000000059
H .000003046 .000001288 -.000002405
H .000002545 .000002629 -.000002958
H -.000001045 .000002092 -.000000658
Page 217
Texas Tech University, Deepali Butani, August 2011
195
C -.000002426 .000001125 .000001644
H -.000000960 -.000002158 .000001557
H -.000003472 .000000789 -.000000324
H -.000003687 .000000365 .000002035
Page 218
Texas Tech University, Deepali Butani, August 2011
196
APPENDIX-B
1H-NMR,
13C-NMR, HMQC AND COSY SPECTRA
Page 219
Texas Tech University, Deepali Butani, August 2011
197
Page 220
Texas Tech University, Deepali Butani, August 2011
198
Page 221
Texas Tech University, Deepali Butani, August 2011
199
Page 222
Texas Tech University, Deepali Butani, August 2011
200
Page 223
Texas Tech University, Deepali Butani, August 2011
201
Page 224
Texas Tech University, Deepali Butani, August 2011
202
Page 225
Texas Tech University, Deepali Butani, August 2011
203
Page 226
Texas Tech University, Deepali Butani, August 2011
204
Page 227
Texas Tech University, Deepali Butani, August 2011
205
Page 228
Texas Tech University, Deepali Butani, August 2011
206
Page 229
Texas Tech University, Deepali Butani, August 2011
207
Page 230
Texas Tech University, Deepali Butani, August 2011
208
Page 231
Texas Tech University, Deepali Butani, August 2011
209
Page 232
Texas Tech University, Deepali Butani, August 2011
210
Page 233
Texas Tech University, Deepali Butani, August 2011
211
Page 234
Texas Tech University, Deepali Butani, August 2011
212
Page 235
Texas Tech University, Deepali Butani, August 2011
213
Page 236
Texas Tech University, Deepali Butani, August 2011
214
Page 237
Texas Tech University, Deepali Butani, August 2011
215
Page 238
Texas Tech University, Deepali Butani, August 2011
216
Page 239
Texas Tech University, Deepali Butani, August 2011
217
Page 240
Texas Tech University, Deepali Butani, August 2011
218
Page 241
Texas Tech University, Deepali Butani, August 2011
219
Page 242
Texas Tech University, Deepali Butani, August 2011
220
Page 243
Texas Tech University, Deepali Butani, August 2011
221
Page 244
Texas Tech University, Deepali Butani, August 2011
222
Page 245
Texas Tech University, Deepali Butani, August 2011
223
Page 246
Texas Tech University, Deepali Butani, August 2011
224
Page 247
Texas Tech University, Deepali Butani, August 2011
225
Page 248
Texas Tech University, Deepali Butani, August 2011
226
Page 249
Texas Tech University, Deepali Butani, August 2011
227
Page 250
Texas Tech University, Deepali Butani, August 2011
228
Page 251
Texas Tech University, Deepali Butani, August 2011
229
Page 252
Texas Tech University, Deepali Butani, August 2011
230
Page 253
Texas Tech University, Deepali Butani, August 2011
231
Page 254
Texas Tech University, Deepali Butani, August 2011
232
Page 255
Texas Tech University, Deepali Butani, August 2011
233
Page 256
Texas Tech University, Deepali Butani, August 2011
234
Page 257
Texas Tech University, Deepali Butani, August 2011
235
Page 258
Texas Tech University, Deepali Butani, August 2011
236