47
DOCUMENTACIÓN DE REGISTRO

DOCUMENTACIÓN DE REGISTRO

DOCUMENTACIÓN DE REGISTRO 51
COMMON TECHNICAL DOCUMENT
La documentación para el Registro se presenta en un formato común denominado CTD (Common Technical Document). VENTAJAS
• Un formato para todas las solicitudes. • Terminología común. • Documento en orden lógico. • Requisitos claros de los capítulos. • Reducción del tiempo/costes de presentación en varios países. • Revisión más fácil por parte de las A.A.S.S. • Mayor facilidad para responder a preguntas. • Reducción del tiempo global de autorización. • Mejora la comunicación inter-compañía.
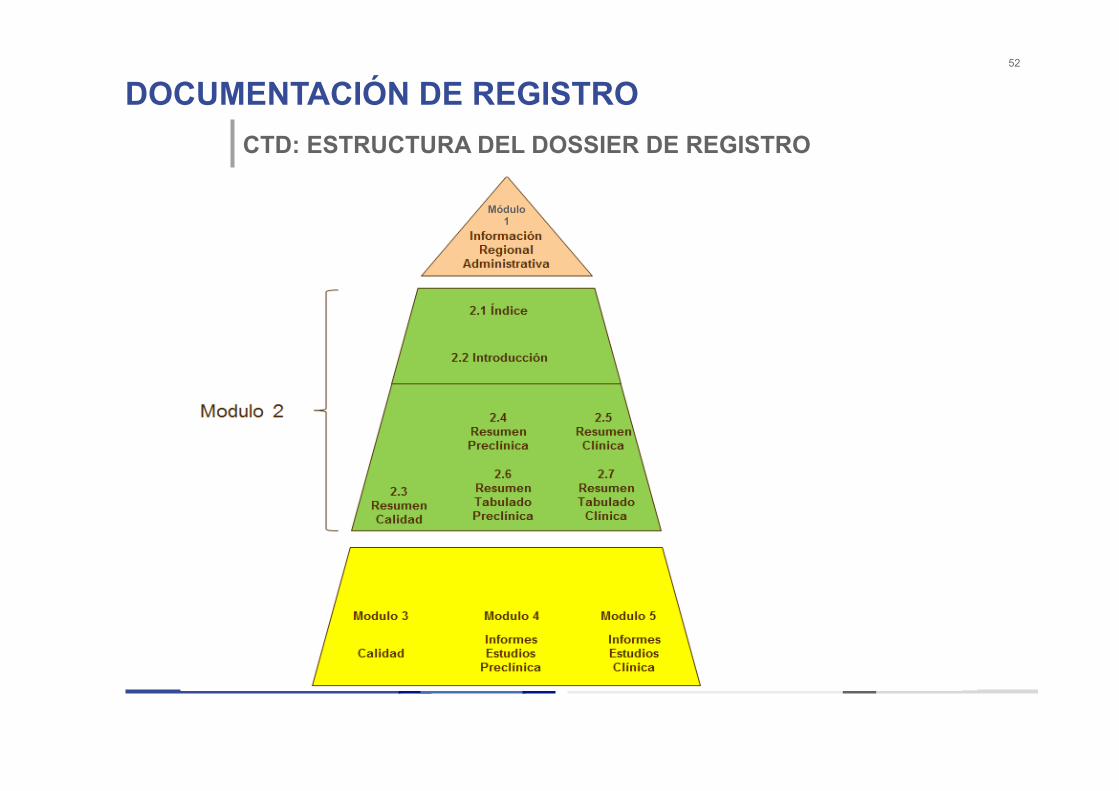
DOCUMENTACIÓN DE REGISTRO 52
CTD: ESTRUCTURA DEL DOSSIER DE REGISTRO
Módulo 1

DOCUMENTACIÓN DE REGISTRO 53
MÓDULO 1: «No CTD »
• No está armonizada. • Contiene los datos administrativos del medicamento. • Incluye la FT, prospecto y etiquetado propuestos. • Curriculum vitae de cada uno de los expertos que avalan la solicitud de
registro. • Justificación del tipo de solicitud solicitada. • Evaluación del riesgo medio-ambiental.
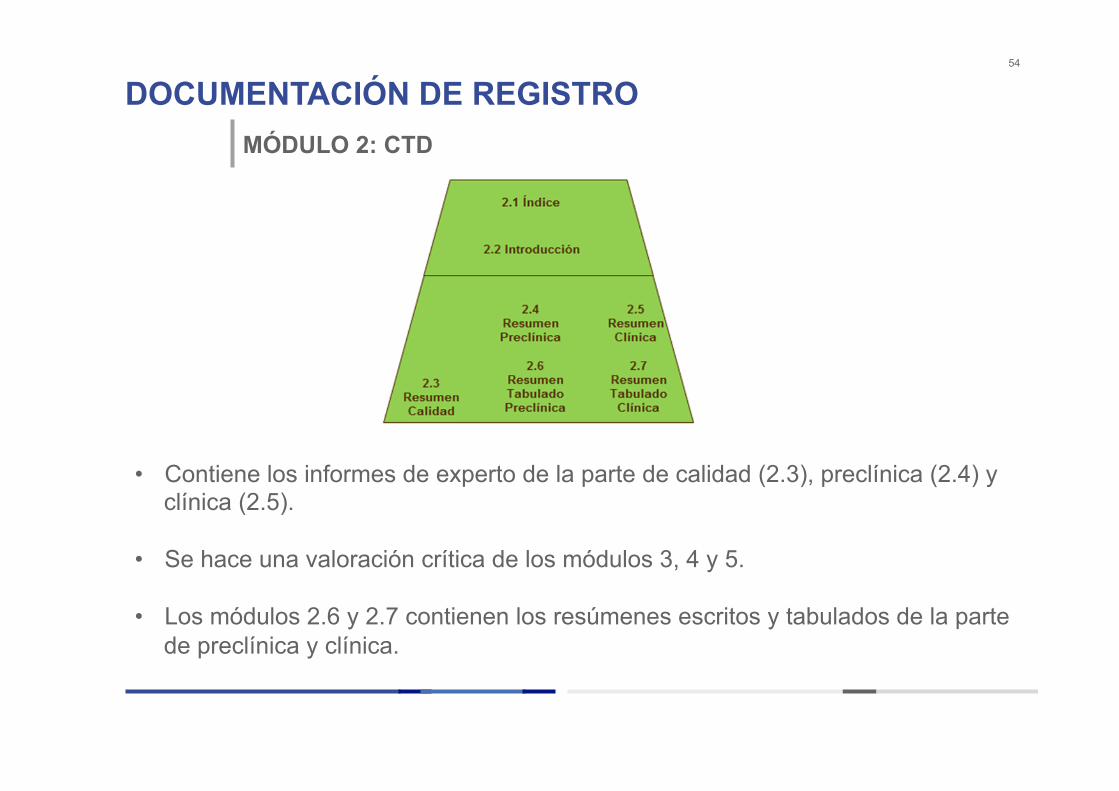
DOCUMENTACIÓN DE REGISTRO 54
MÓDULO 2: CTD
• Contiene los informes de experto de la parte de calidad (2.3), preclínica (2.4) y clínica (2.5).
• Se hace una valoración crítica de los módulos 3, 4 y 5. • Los módulos 2.6 y 2.7 contienen los resúmenes escritos y tabulados de la parte
de preclínica y clínica.
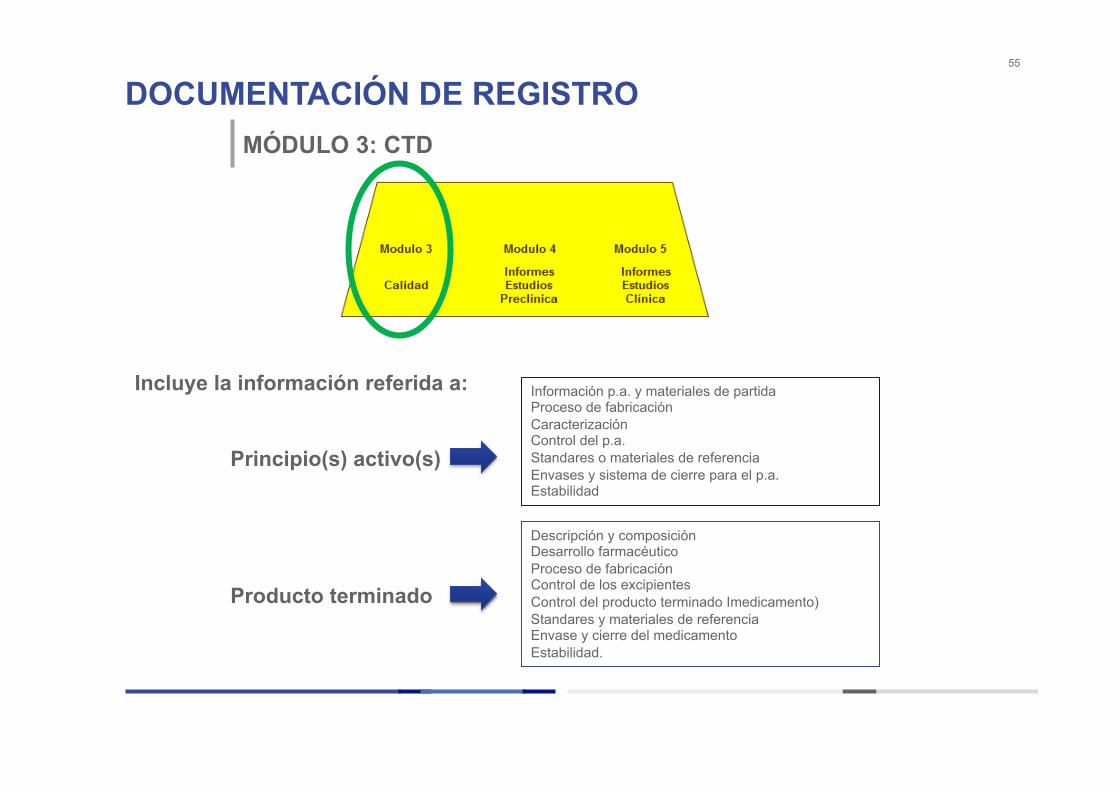
DOCUMENTACIÓN DE REGISTRO 55
MÓDULO 3: CTD
Incluye la información referida a:
Principio(s) activo(s)
Producto terminado
Información p.a. y materiales de partida Proceso de fabricación Caracterización Control del p.a. Standares o materiales de referencia Envases y sistema de cierre para el p.a. Estabilidad
Descripción y composición Desarrollo farmacéutico Proceso de fabricación Control de los excipientes Control del producto terminado Imedicamento) Standares y materiales de referencia Envase y cierre del medicamento Estabilidad.

DOCUMENTACIÓN DE REGISTRO 56
MÓDULOS 4 y 5: CTD
MÓDULO 4 • Incluye la información referida al apartado 2.4 y 2.6. sobre la farmacología,
farmacocinética, toxicología, tolerancia local en animales. • A veces esta información se puede suplir por referencias bibliográficas.
MÓDULO 5 • Incluye la información referida al apartado 2.5 y 2.7. Cada estudio es una única
sección. • Pueden establecerse referencias cruzadas. • Tanto en el caso del módulo 4, como el módulo 5, el CTD no informa sobre que
estudios son necesarios llevar a cabo. Por tanto, se puede solicitar en estos casos un “Scientific Advice” a las agencias reguladoras.

DOCUMENTACIÓN DE REGISTRO 57
MÓDULOS 4 y 5: CTD
MÓDULO 4 • Incluye la información referida al apartado 2.4 y 2.6. sobre la farmacología,
farmacocinética, toxicología, tolerancia local en animales. • A veces esta información se puede suplir por referencias bibliográficas.
MÓDULO 5 • Incluye la información referida al apartado 2.5 y 2.7. Cada estudio es una única
sección. • Pueden establecerse referencias cruzadas. • Tanto en el caso del módulo 4, como el módulo 5, el CTD no informa sobre que
estudios son necesarios llevar a cabo. Por tanto, se puede solicitar en estos casos un “Scientific Advice” a las agencias reguladoras.

DOCUMENTACIÓN DE REGISTRO 58
TIPOS DE ESTUDIOS
FARMACOCINÉTICA Objetivo:
• Determinación de la velocidad de absorción, distribución (incluyendo unión a proteínas plasmáticas), vías metabólicas y eliminación.
• Influencia de los factores estereoquímicos del fármaco.
• Comparación entre sujetos sanos, pacientes y poblaciones especiales.
Diseño: • Se realizan sobre voluntarios sanos y enfermos • Nº de individuos < 50 • Controlados o no • Aleatorizados o no • Abierto, simple, ciego o doble ciego • Dosis única o múltiple

DOCUMENTACIÓN DE REGISTRO 59
TIPOS DE ESTUDIOS
FARMACODINAMIA
Objetivo:
• Determinación del mecanismo de acción.
• Determinación del momento en que comienza la acción del fármaco y cuando termina.
• Determinación de dosis e intervalos de administración.
• Interacciones con otros medicamentos y sustancias.
• Cambios en la respuesta debido a modificaciones genéticas.

DOCUMENTACIÓN DE REGISTRO 60
TIPOS DE ESTUDIOS
ESTUDIO DE BÚSQUEDA DE DOSIS
Objetivo:
• Determinar la dosis a partir de los datos de los estudios en animales.
• Determinar la dosis inicial y máxima.
• Estudiar el efecto del incremento de la dosis.
• Análisis de los resultados.

DOCUMENTACIÓN DE REGISTRO 61
TIPOS DE ESTUDIOS
ESTUDIO DE FARMACOVIGILANCIA Son los ensayos que confirman el uso terapéutico del medicamento. Se caracterizan por: IDONEIDAD
• De la hipótesis. • Problema clínicamente relevante. • Los resultados compensan los recursos empleados.
FACTIBILIDAD
• Que las variables puedan ser definidas y medidas. • Infraestructura para su desarrollo. • Experiencia investigadores participantes. • Consideraciones éticas.
RELEVANCIA
• Aprobación por la autoridades. • Beneficio/Seguridad. • Poblaciones amplias (diferentes etapas de la enfermedad). • Determinan las recomendaciones de uso del fármaco.

DOCUMENTACIÓN DE REGISTRO 62
TIPOS DE ESTUDIOS
ESTUDIO DE FARMACOVIGILANCIA Son los ensayos que confirman el uso terapéutico del medicamento. Se caracterizan por: EFICACIA
• Dosis adecuadas. • Estudios frente a placebo. • Estudios frente a comparador activo (gold-standard) • Indicación definida .
SEGURIDAD
• Riesgos aceptables. • Perfil de reacciones adversas. • Reacciones adversas más frecuentes. • Reacciones adversas por gravedad.

DOCUMENTACIÓN DE REGISTRO 63
ENSAYOS CLÍNICOS – FASE I
FASE I CARACTERÍSTICAS
• Se realiza sobre voluntarios sanos.
• Se realiza sobre diferentes razas.
• Individuos con diferentes niveles enzimáticos.
• Muestras pequeñas.
OBJETIVOS
• Determinación del perfil LADME y farmacocinética.
• Evaluar la dosis y al vía de administración.
• Definir la toxicidad a corto plazo (tolerabilidad).
• Definir biodisponibilidad y bioequivalencia.

DOCUMENTACIÓN DE REGISTRO 64
ENSAYOS CLÍNICOS – FASE II
FASE II
CARACTERÍSTICAS
• Se realiza sobre pacientes.
• Diferentes intervalos de edades.
• Diferentes poblaciones.
• Número de individuos pequeño.
OBJETIVOS
• Definir la eficacia.
• Evaluar la dosis y dosis-respuesta.
• Toxicidad a corto plazo.
• Clasificar las reacciones adversas.

DOCUMENTACIÓN DE REGISTRO 65
ENSAYOS CLÍNICOS – FASE III
FASE III
CARACTERÍSTICAS
• Pacientes de etiología típico.
• Participan diferentes centros (multicentros).
• Se puede administrar medicación concomitante.
• Pueden existir enfermedades asociadas.
• Tamaño de la muestra grande.
OBJETIVOS
• Constatar la eficacia.
• Evaluar posibles interacciones.
• Efectos sobre la gravedad de la enfermedad.
• Definir la biodisponibilidad.

DOCUMENTACIÓN DE REGISTRO 66
EECC & ESTUDIOS POSTAUTORIZACIÓN – FASE IV
FASE IV
CARACTERÍSTICAS
• Pacientes de la práctica diaria.
• Diferentes sitios.
• Se incluyen médicos y no investigadores.
• Existen enfermedades concomitantes.
• Tamaño de la muestra grande.
OBJETIVOS
• Constatar la seguridad y eficacia.
• Farmacovigilancia.
• Determinar efectos adversos no recogidos inicialmente en el dossier soporte de la autorización de comercialización.
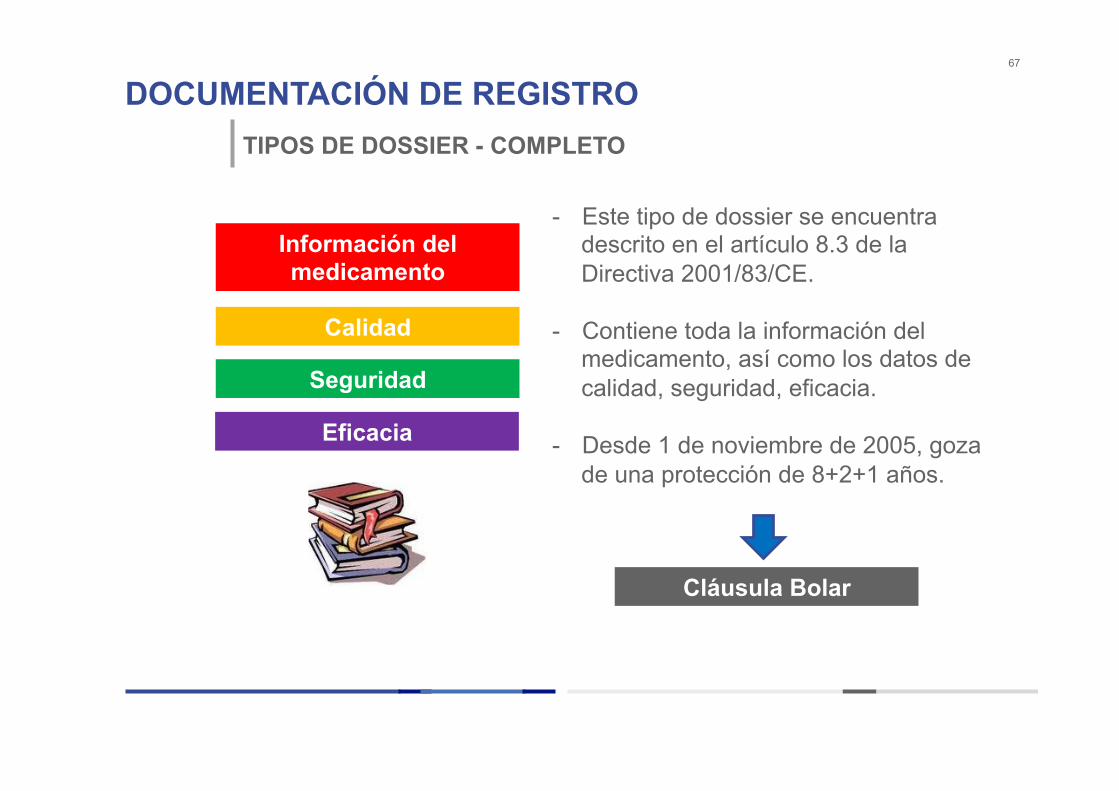
DOCUMENTACIÓN DE REGISTRO 67
TIPOS DE DOSSIER - COMPLETO
- Este tipo de dossier se encuentra descrito en el artículo 8.3 de la Directiva 2001/83/CE.
- Contiene toda la información del
medicamento, así como los datos de calidad, seguridad, eficacia.
- Desde 1 de noviembre de 2005, goza
de una protección de 8+2+1 años.
Información del medicamento
Calidad
Seguridad
Eficacia
Cláusula Bolar

DOCUMENTACIÓN DE REGISTRO 68
COMPLETO VS ABREVIADO
Información del medicamento
Calidad
Seguridad
Eficacia
Información del medicamento
Calidad
Sustitución por estudio de bioequivalencia en genéricos
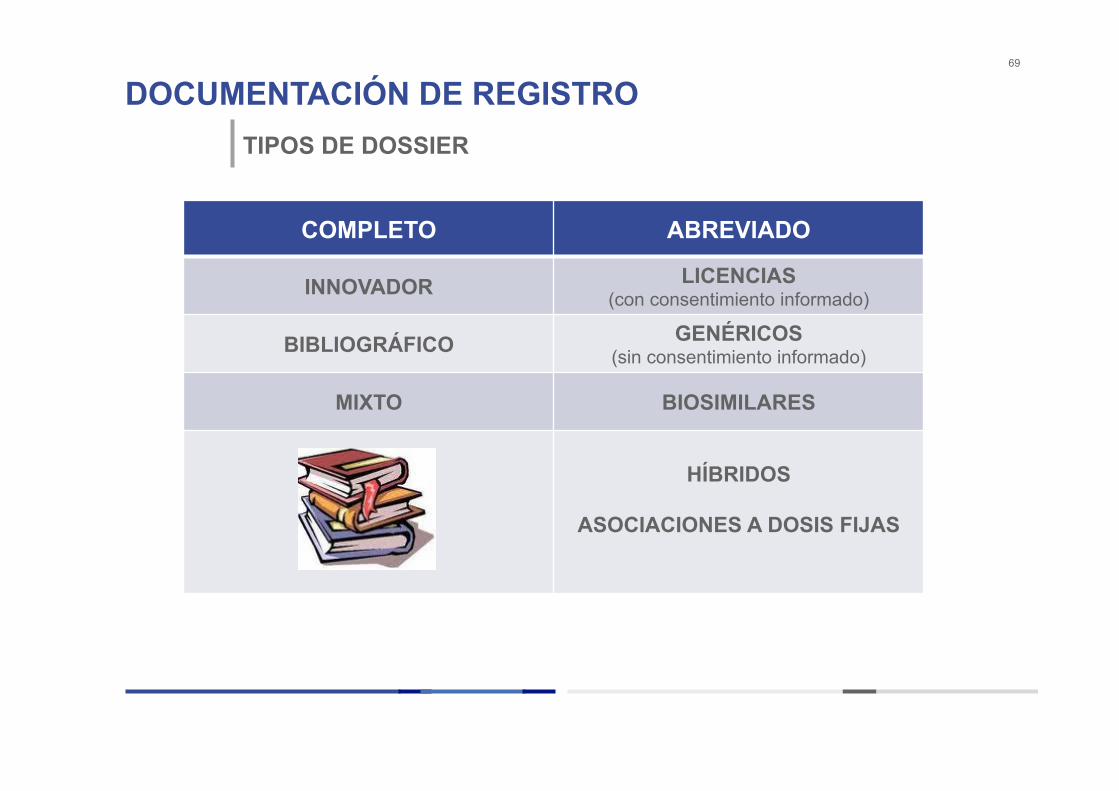
DOCUMENTACIÓN DE REGISTRO 69
TIPOS DE DOSSIER
COMPLETO ABREVIADO
INNOVADOR LICENCIAS (con consentimiento informado)
BIBLIOGRÁFICO GENÉRICOS (sin consentimiento informado)
MIXTO BIOSIMILARES
HÍBRIDOS
ASOCIACIONES A DOSIS FIJAS

DOCUMENTACIÓN DE REGISTRO 70
REGISTRO ELECTRÓNICO
eCTD (electronic Common Technical Dossier)
NEES (Non eCTD Electronic Submission) Formato de transición CTD ! eCTD
La documentación para el Registro se presenta en un formato común denominado CTD

ACTIVIDADES POST-AUTORIZACIÓN

ACTIVIDADES POST-AUTORIZACIÓN 72
TIPOS DE ACTIVIDADES
REVALIDACIÓN
EXTENSIONES DE LÍNEA
COMERCIALIZACIÓN EFECTIVA
INTENCIÓN ANUAL DE COMERCIALIZACIÓN
SUSPENSIÓN TEMPORAL DE COMERCIALIZACIÓN
CESE DE COMERCIALIZACIÓN (REVOCACIÓN)
MODIFICACIONES (VARIACIONES)
Ciclo de vida
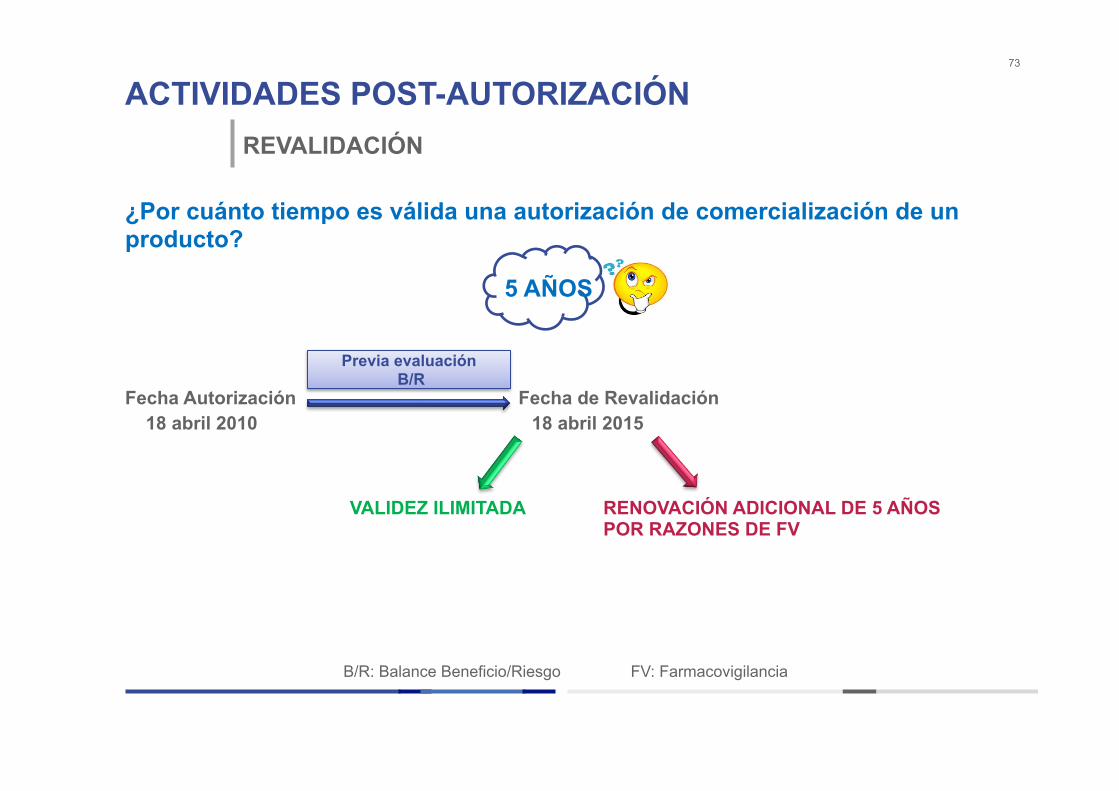
ACTIVIDADES POST-AUTORIZACIÓN 73
REVALIDACIÓN
¿Por cuánto tiempo es válida una autorización de comercialización de un producto? Fecha Autorización Fecha de Revalidación 18 abril 2010 18 abril 2015
5 AÑOS
Previa evaluación B/R
B/R: Balance Beneficio/Riesgo FV: Farmacovigilancia
VALIDEZ ILIMITADA RENOVACIÓN ADICIONAL DE 5 AÑOS POR RAZONES DE FV

ACTIVIDADES POST-AUTORIZACIÓN 74
EXTENSIONES DE LÍNEA
Las Extensiones de Línea son cambios que suponen una nueva solicitud. Estos cambios pueden ser debidos a:
• Cambios en el principio activo. • Cambios introducidos en la dosis, la forma farmacéutica y vía de
administración.
Se presenta un expediente abreviado, ya que se hace referencia cruzada al expediente original.
Ejemplo: Extensión de línea de Vimpat 10 mg/ml jarabe

ACTIVIDADES POST-AUTORIZACIÓN 75
C.EFECTIVA E INTENCIÓN ANUAL DE COMERCIALIZACIÓN
Fecha AC
AÑOS
Fecha primer lote en el mercado
Comercialización efectiva
0 3 3 X AÑOS
Comercialización
CLAUSULA SUNSET
INTENCIÓN ANUAL DE COMERCIALIZACIÓN Declaración anual de la Intención de Comercialización de los medicamentos.
Si no se presenta declaración se entenderá que se solicita la suspensión de la AC.
COMERCIALIZACIÓN EFECTIVA
OCTUBRE 2014
31
AÑOS

ACTIVIDADES POST-AUTORIZACIÓN 76
SUSPENSIÓN TEMPORAL DE COMERCIALIZACIÓN
Astrocetam (comprimidos y sol.oral) Reubicación en otros mercados
No ventas Stock …próximo a caducar
STC en España
Si no comercializamos de nuevo antes de 3 años desde la AC, se anularía el medicamento por aplicación de la Cláusula Sunset
SUSPENSIÓN TEMPORAL DE COMERCIALIZACIÓN (STC)
Fecha de AC = 07/03/2013 07/03/2016

ACTIVIDADES POST-AUTORIZACIÓN 77
VARIACIONES
TIPO IA/IAIN - Modificaciones de importancia menor/“do & tell”. Las Autoridades Sanitarias solo validan la documentación presentada.
• Variaciones puramente administrativas (titular, fabricantes). • Variaciones relacionadas con la eliminación de un lugar de fabricación,
incluyendo principio activo, intermedio o producto terminado. • Variaciones relacionadas con cambios menores en los procedimientos de
ensayo físico-químicos. • Variaciones relacionadas con modificaciones de la especificación de la
sustancia activa para adaptarse a la Ph. Eur. • Variaciones relacionadas con cambios en el material de acondicionamiento del
producto terminado que no esta en contacto con el medicamento. • Variaciones relacionadas con estrechamiento de los límites de las
especificaciones del medicamento, siempre y cuando no sean debido a cambios debido a un compromiso anterior.
Única vía para actualizar el
expediente de registro

ACTIVIDADES POST-AUTORIZACIÓN 78
VARIACIONES
TIPO IB - Modificaciones de importancia menor. Necesitan una evaluación mínima por parte de las Autoridades Sanitarias. TIPO II - Modificaciones de importancia mayor Necesitan una evaluación por parte de las Autoridades Sanitarias.
• Adición de una nueva indicación. • Variaciones relacionadas con modificaciones significativas del SmPC, con
datos de calidad, preclínica, clínica y Farmacovigilancia. • Cambios relacionados con modificación fuera del rango de las
especificaciones aprobadas, límites o criterios de aceptación. • Cambios substanciales en el proceso de fabricación, formulación,
especificaciones o perfil de impurezas. • Etc….
Única vía para actualizar el
expediente de registro
Reglamento 1234/2008

ACTIVIDADES POST-AUTORIZACIÓN 79
GUIDELINE DE VARIACIONES
3 BLOQUES PRINCIPALES:
A. CAMBIOS ADMINISTRATIVOS B. CAMBIOS CUALITATIVOS C. CAMBIOS DE SEGURIDAD, EFICACIA Y FARMACOVIGILANCIA
G:\eCTD-NeeS\AAAAAA\Anexos para Variaciones\c_2013_2804_es.pdf

ACTIVIDADES POST-AUTORIZACIÓN 80
EJEMPLO DE VARIACIONES
A. CAMBIO ADMINISTRATIVO
MÓDULO 1:
Carta de presentación Formulario de Solicitud
Información de Producto (PT) Documento formal de un órgano oficial competente en el que se menciona el nuevo nombre/dirección
CAMBIO DE SEDE SOCIAL EN PORTUGAL

ACTIVIDADES POST-AUTORIZACIÓN 81
EJEMPLO DE VARIACIONES
B. CAMBIOS CUALITATIVOS CAMBIO EN LA FORMA O DIMENSIONES DEL ENVASE O CIERRE (ACONDICIONAMIENTO PRIMARIO)
MÓDULO 1:
Carta de presentación Formulario de solicitud
MÓDULO 3:
Información “Envase y Sistema de cierre”
Las AASS sólo validan la documentación presentada

ACTIVIDADES POST-AUTORIZACIÓN 82
EJEMPLO DE VARIACIONES
C. CAMBIOS DE SEGURIDAD, EFICACIA Y FARMACOVIGILANCIA
CAMBIO EN EL RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO, ETIQUETADO O PROSPECTO
MÓDULO 1:
Carta de presentación Formulario de solicitud
Información de Producto Información acerca de los expertos
MÓDULO 2:
Visión general de la parte clínica
MÓDULO 5: Referencias bibliográficas
Requiere evaluación por parte
de las AASS

INFORMACIÓN DE PRODUCTO
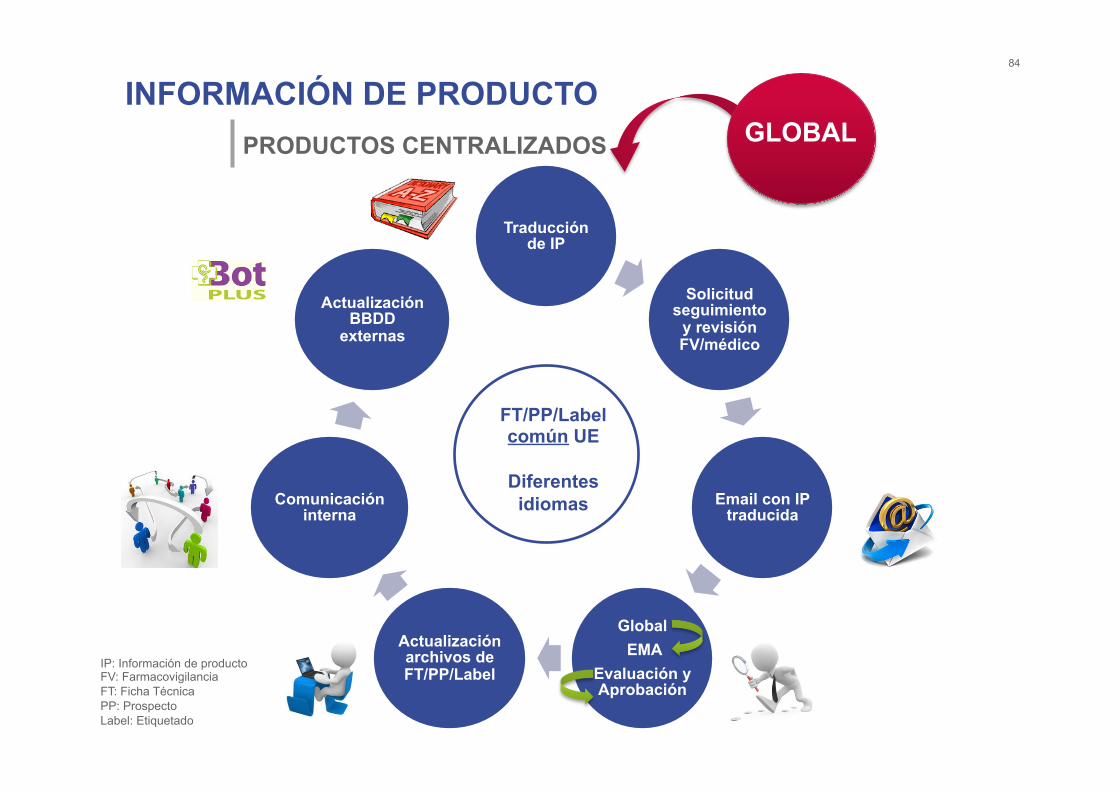
INFORMACIÓN DE PRODUCTO 84
PRODUCTOS CENTRALIZADOS
Traducción de IP
Solicitud seguimiento
y revisión FV/médico
Email con IP traducida
Global EMA
Evaluación y Aprobación
Actualización archivos de FT/PP/Label
Comunicación interna
Actualización BBDD
externas
FT/PP/Label común UE
Diferentes idiomas
IP: Información de producto FV: Farmacovigilancia FT: Ficha Técnica PP: Prospecto Label: Etiquetado
GLOBAL
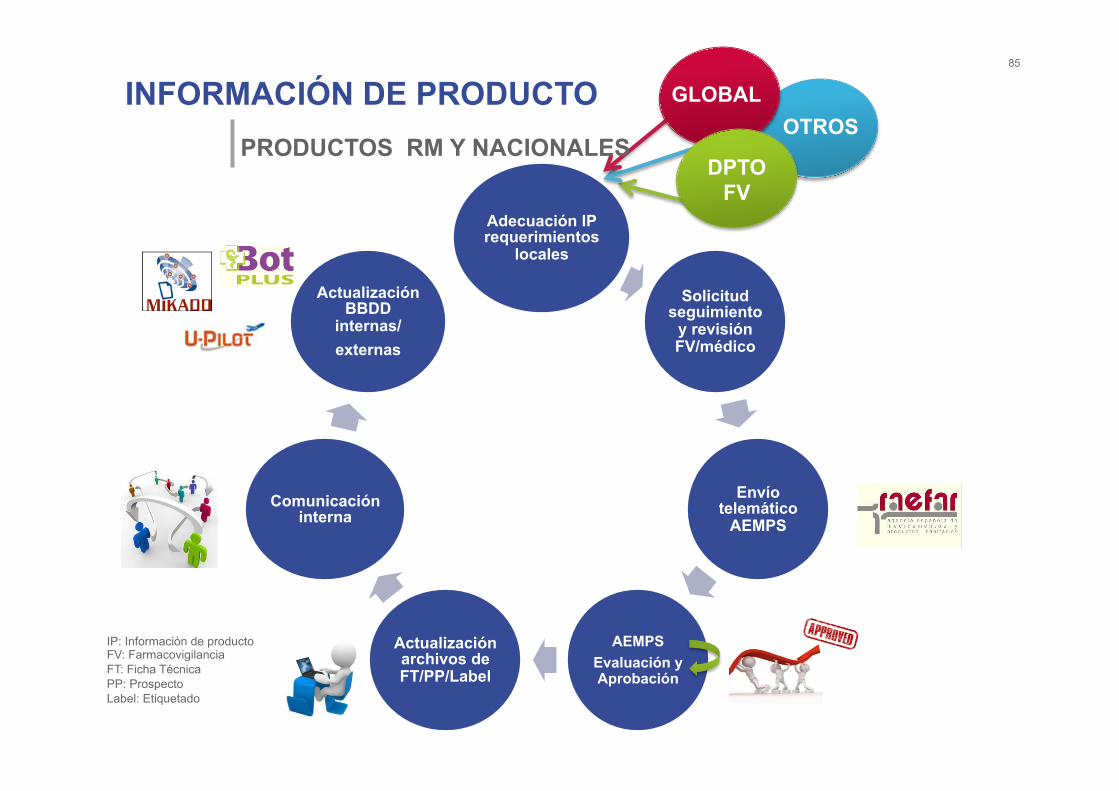
INFORMACIÓN DE PRODUCTO 85
PRODUCTOS RM Y NACIONALES
Adecuación IP requerimientos
locales
Solicitud seguimiento
y revisión FV/médico
Envío telemático
AEMPS
AEMPS Evaluación y Aprobación
Actualización archivos de FT/PP/Label
Comunicación interna
Actualización BBDD
internas/ externas
IP: Información de producto FV: Farmacovigilancia FT: Ficha Técnica PP: Prospecto Label: Etiquetado
GLOBAL
DPTO FV
OTROS
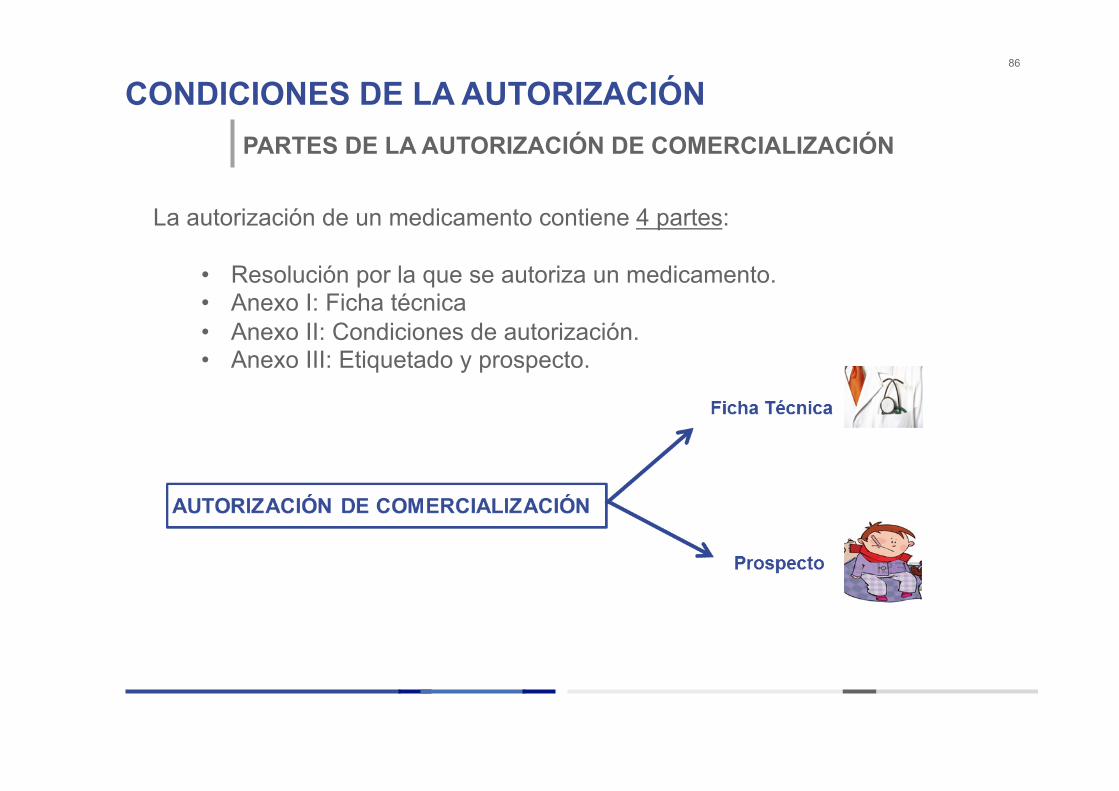
CONDICIONES DE LA AUTORIZACIÓN 86
PARTES DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
La autorización de un medicamento contiene 4 partes:
• Resolución por la que se autoriza un medicamento. • Anexo I: Ficha técnica • Anexo II: Condiciones de autorización. • Anexo III: Etiquetado y prospecto.

CONDICIONES DE LA AUTORIZACIÓN 87
FICHA TÉCNICA
Es el resumen de la información científica esencial del medicamento para su difusión a profesionales sanitarios por el Titular de la Autorización de Comercialización. Tiene una triple finalidad:
• Documento entre las Autoridades Sanitarias nacionales. • Documento entre las Autoridades Sanitarias y la compañía. • Documento entre la compañía y el prescriptor.
Importancia: Solo está autorizado el uso del medicamento en las patologías incluidas dentro de las indicaciones y en las condiciones de uso (dosis, forma de administración, vía, etc.) descritas en la FT. Limitaciones
• No todos los medicamentos tienen FT (medicamentos antiguos, como Bronsal, solo tienen PP aunque se ha solicitado su FT).
• Las FT reflejan, a veces parcialmente, el conocimiento del producto, en ocasiones con retraso.

CONDICIONES DE LA AUTORIZACIÓN 88
FICHA TÉCNICA
Características • Propuesta por la compañía, aceptada y aprobada por las autoridades sanitarias.
• Refleja los resultados de los ensayos de calidad, preclínica y clínica.
• Dicha documentación aparece reflejada en los siguientes apartados:
DOCUMENTACIÓN CALIDAD
DOCUMENTACIÓN PRECLÍNICA
DOCUMENTACIÓN CLÍNICA
2. Composición 3. Forma Farmacéutica 6. Datos Farmacéuticos
5.3 Datos preclínicos 4. Datos Clínicos 5.1 Propiedades farmacodinámicas 5.2 Propiedades farmacocinéticas

CONDICIONES DE LA AUTORIZACIÓN 89
FICHA TÉCNICA
Dependiendo del tipo de procedimiento de autorización empleado, tendremos las siguientes características: PROCEDIMIENTO CENTRALIZADO
• Ficha técnica autorizada por la Comisión Europea. • Una FT por cada forma farmacéutica. • Una única FT común a todos los países (solo varía el idioma). • Material de acondicionamiento común a todos los países (requerimientos
nacionales en la blue-box)
PROCEDIMIENTO DESCENTRALIZADO Y RECONOCIMIENTO MUTUO • FT autorizada por el Estado Miembro de Referencia, de acuerdo con las Agencias
de los Estados Miembros Concernidos. PROCEDIMIENTO NACIONAL
• FT negociada con la AEMPS, es decir, la Cía. presenta una propuesta que será evaluado por la AEMPS.

CONDICIONES DE LA AUTORIZACIÓN 90
FICHA TÉCNICA VS PROSPECTO
FICHA TÉCNICA PROSPECTO Información para el profesional sanitario.
Información para el paciente.
Recoge las condiciones de uso y autorización.
Adaptación de la información contenida en la FT a un lenguaje no técnico.
Entregada por la Cía. a los profesionales sanitario, y debe ceñirse a ella en las actividades de promoción.
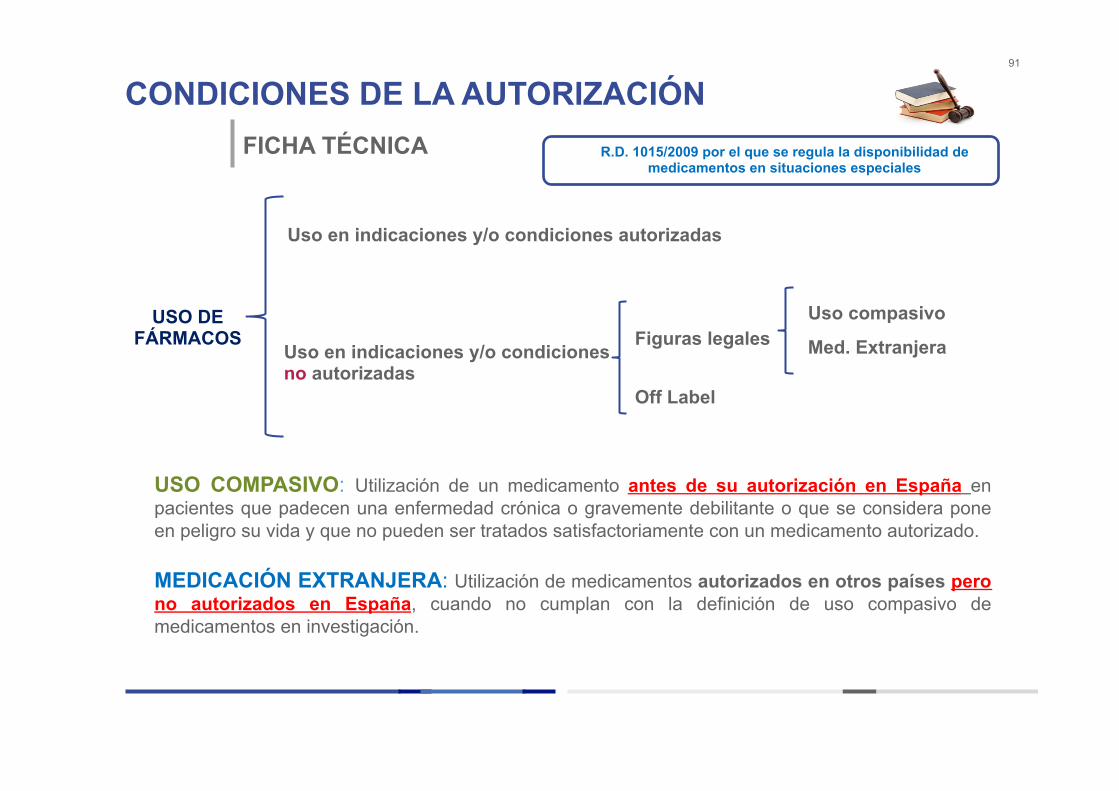
CONDICIONES DE LA AUTORIZACIÓN 91
FICHA TÉCNICA
USO DE FÁRMACOS
Uso en indicaciones y/o condiciones autorizadas
Uso en indicaciones y/o condiciones no autorizadas
Figuras legales
Off Label
Med. Extranjera
Uso compasivo
USO COMPASIVO: Utilización de un medicamento antes de su autorización en España en pacientes que padecen una enfermedad crónica o gravemente debilitante o que se considera pone en peligro su vida y que no pueden ser tratados satisfactoriamente con un medicamento autorizado. MEDICACIÓN EXTRANJERA: Utilización de medicamentos autorizados en otros países pero no autorizados en España, cuando no cumplan con la definición de uso compasivo de medicamentos en investigación.
R.D. 1015/2009 por el que se regula la disponibilidad de medicamentos en situaciones especiales

INTERACCIONES CON OTROS DEPARTAMENTOS

DEPARTAMENTO DE REGISTROS 93
INTERACCIONES EXTERNAS E INTERNAS

DEPARTAMENTO DE REGISTROS 94
CONCLUSIÓN
REGISTROS ES UN DEPARTAMENTO QUE ES EJE VERTEBRAL DURANTE T O D O E L C I C L O D E V I D A D E L MEDICAMENTO, DESDE EL REGISTRO, HASTA SU ANULACIÓN O VENTA, INCLUYENDO SU INTRODUCCIÓN EN EL MERCADO.

¡Gracias por vuestra
atención!

96

















