1.1 Telomere Biology ...........................................................................................................................................................1 1.1.1 Telomere Structure and Function.......................................................................................................................1 1.1.2 Telomere Maintenance Mechanisms .................................................................................................................6 1.1.3 Telomeres and Disease ......................................................................................................................................... 15
1.2 Fanconi Anaemia (FA)...............................................................................................................................................17 1.2.1 The FA Clinical Phenotype................................................................................................................................... 17 1.2.2 FA and Telomere Maintenance ......................................................................................................................... 19 1.2.3 The FA Pathway....................................................................................................................................................... 20 1.2.4 The Role of FANCD2 in DNA Repair ................................................................................................................ 24
2 The Fanconi Anaemia Pathway Plays a Critical Role in Cells that Utilize the Alternerative
Pathway of Telomere Maintenance....................................................................................... 49
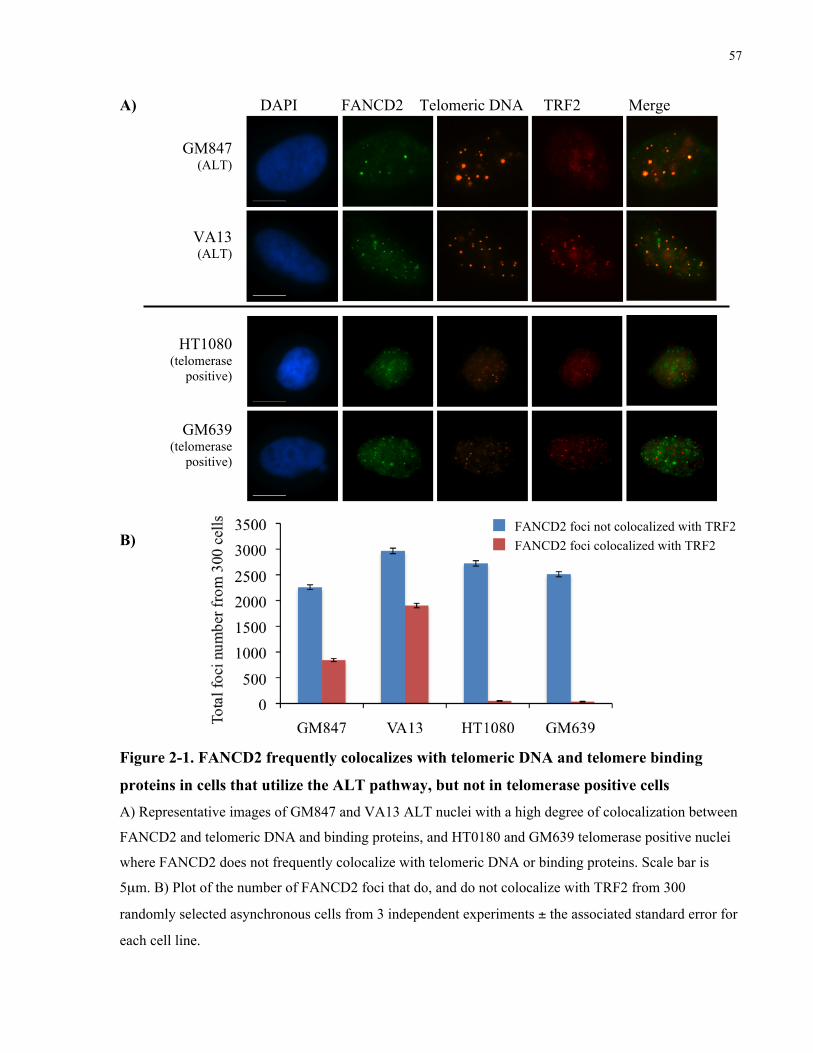
2.1 Abstract ...........................................................................................................................................................................49 2.2 Introduction ..................................................................................................................................................................50 2.3 Materials and Methods .............................................................................................................................................52 2.4 Results..............................................................................................................................................................................56 2.4.1 FANCD2 localize to telomeric foci and PML bodies in ALT human cells......................................... 56
vi
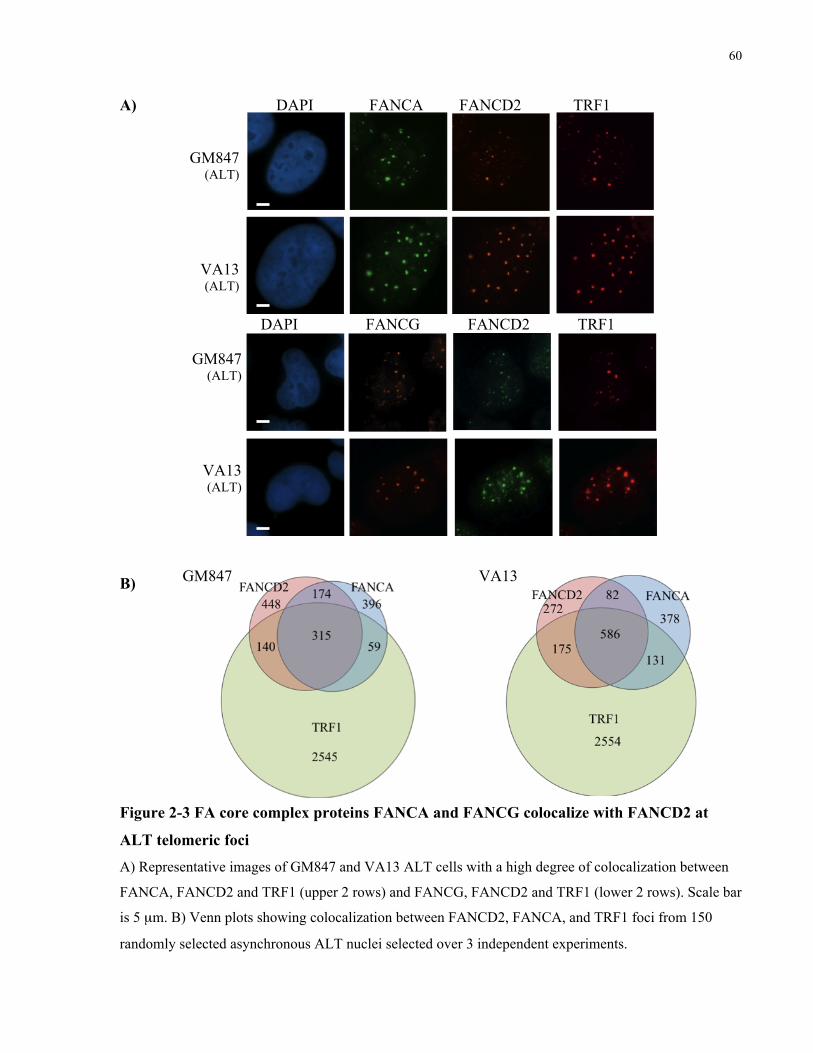
2.4.2 FA core complex components localize to ALT telomeric foci and promote FANCD2
monoubiquitination and localization to telomeric foci .......................................................................................... 59 2.4.3 FANCD2 localizes to ALT telomeric foci that have not activated a DNA damage response,
and localization to telomeric foci is independent of ATM and ATR kinase activity ................................... 62 2.4.4 FANCD2 coimmunoprecipitates with TRF2 and BLM in ALT cells, and almost always
localizes to telomeric foci that also contain BLM...................................................................................................... 65 2.4.5 FANCD2 localization to APBs is independent of TRF2, but requires BLM expression .............. 66 2.4.6 FANCD2 knockdown causes an ALTspecific increase in telomere dysfunction induced foci
that is independent of rapid telomere shortening .................................................................................................... 70 2.4.7 ALTassociated PML bodies (APBs) are structurally different from nonALT bodies, and
contain telomeric nucleic acid in the interior of the body that differs from surrounding chromatin73 2.4.8 FANCD2 depletion in ALT cells results in nuclear abnormalities, centrosome amplification
and rapid cell death................................................................................................................................................................ 77 2.5 Discussion.......................................................................................................................................................................83 2.6 References......................................................................................................................................................................91
3 Fanconi Anaemia Protein D2 Limits BLM‐Dependent, RAD51‐Independent Telomeric
Recombination and DNA Synthesis in ALT‐Immortalized Human Cells ................................. 101
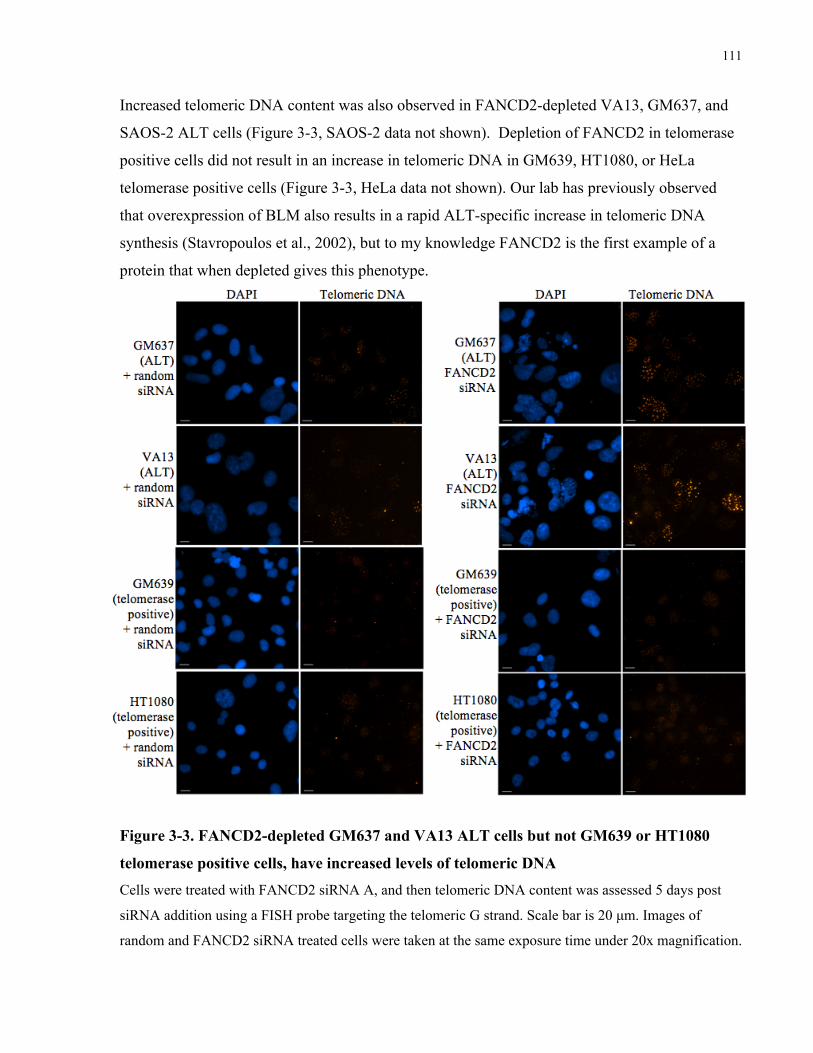
3.1 Abstract ........................................................................................................................................................................ 101 3.2 Introduction ............................................................................................................................................................... 102 3.3 Materials and Methods .......................................................................................................................................... 106 3.4 Results........................................................................................................................................................................... 109 3.4.1 FANCD2depletion results in a rapid, ALTspecific increase in telomeric DNA synthesis .....109 3.4.2 FANCD2depleted ALT cells accumulate ECTR DNA within and outside of abnormally large
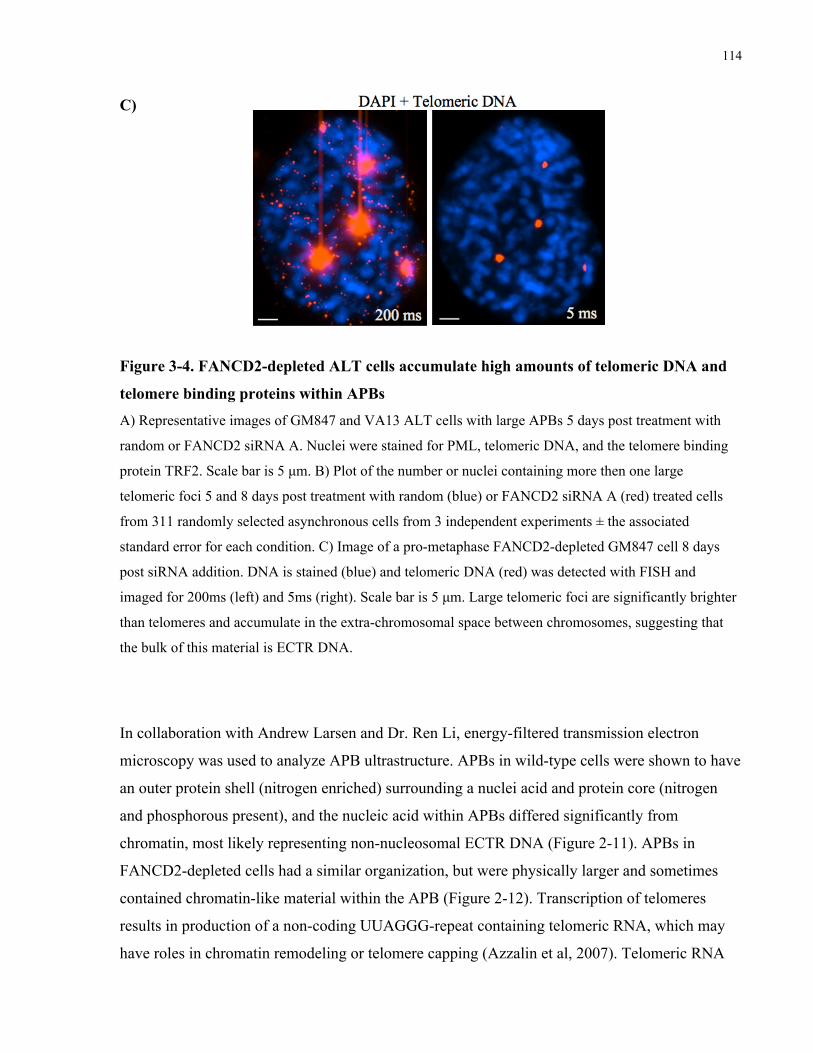
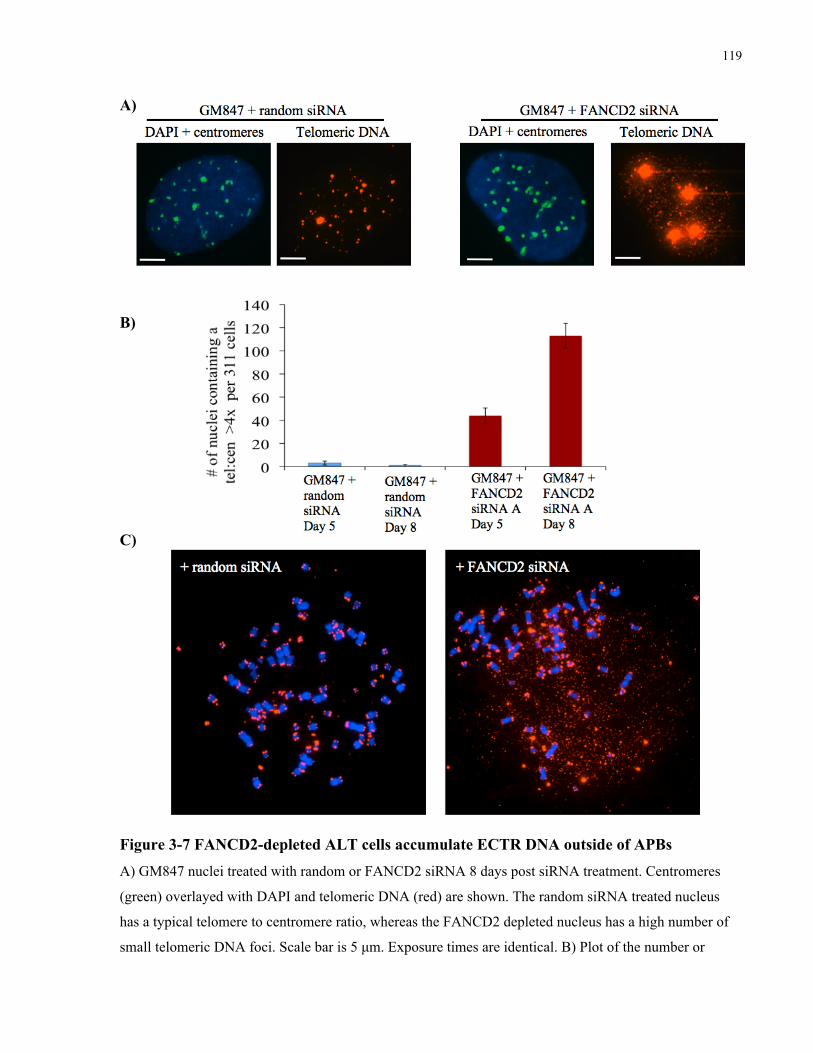
ALT associated PML bodies ...............................................................................................................................................112 3.4.3 FANCD2depleted ALT cells do not upregulate expression of BLM, TRF1 or TRF2..................118 3.4.4 FANCD2depleted ALT cells have increased association of RAD51 with telomeric foci,
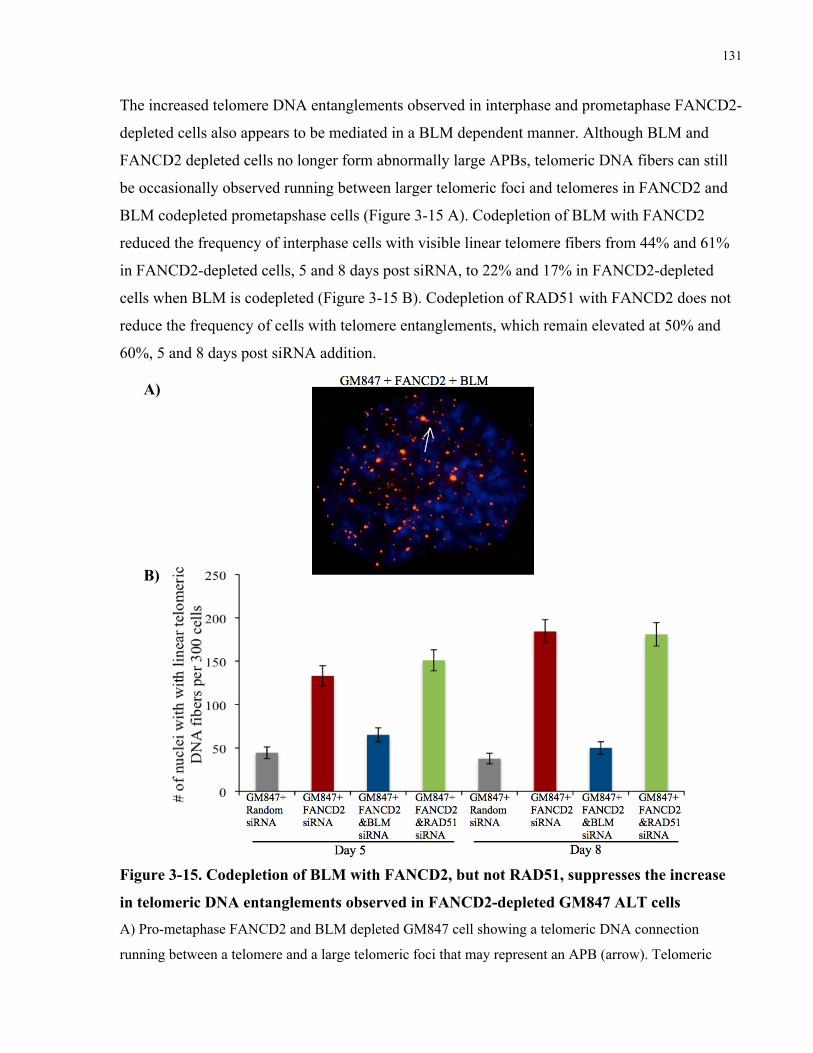
telomere sister chromatid exchanges, fragile telomeres, and telomere entanglements........................121 3.4.5 Telomere abnormalities in FANCD2depleted ALT cells are generated through a BLM
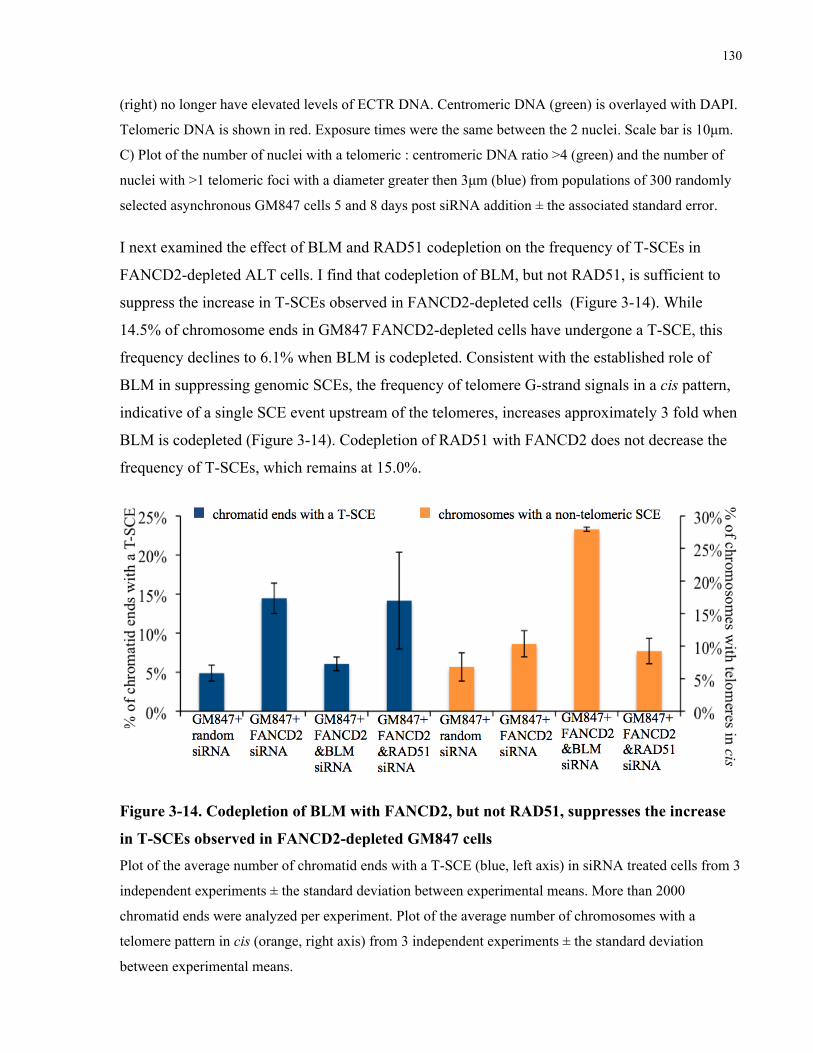
dependent, largely RAD51 independent mechanism .............................................................................................128 3.4.6 Codepletion of BLM with FANCD2 improves the viability of FANCD2 depleted ALT cells ....132
Figure 3-13. BLM, but not RAD51, is required to generate high levels of ECTR DNA in
FANCD2-depleted ALT cells ..................................................................................................... 129
Figure 3-14. Codepletion of BLM with FANCD2, but not RAD51, suppresses the increase in
T-SCEs observed in FANCD2-depleted GM847 cells ............................................................... 130
Figure 3-15. Codepletion of BLM with FANCD2, but not RAD51, suppresses the increase in
telomeric DNA entanglements observed in FANCD2-depleted GM847 ALT cells .................. 131
Figure 3-16. Codepletion of BLM with FANCD2 partially restores the colony forming ability of
ALT cells .................................................................................................................................... 133
xi
List of Abbreviations
ALT alternative lengthening of telomeres
APBs ALT-associated PML bodies
BrdU bromodeoxyuridine
CO-FISH chromosome orientation fluorescent in situ hybridization
DC dyskeratosis congenita
dsDNA double-strand DNA
ECTR extra-chromosomal telomeric repeat
ESI electron spectroscopic imaging
FA Fanconi anaemia
HR homologous recombination
SCE sister chromatid exchange
ssDNA single-strand DNA
TERC telomerase RNA component
TERRA telomere repeat containing RNA
TERT telomerase reverse transcriptase
TIFs telomere dysfunction induced foci
T-SCE telomere sister chromatid exchange
1
Chapter 1
1 Introduction
1.1 Telomere Biology
1.1.1 Telomere Structure and Function
Telomeres are nucleoprotein structures that cap the ends of linear chromosomes, facilitating
replication of the ends, and preventing ends from activating cell cycle checkpoints, or becoming
involved in DNA repair pathways that could result in chromosome fusions or rapid shortening
events. In most eukaryotes, telomeres are composed of tandem repeats of a short sequence
containing repeated guanosines. In humans, this repeat sequence is TTAGGG, and it is repeated
thousands of times to yield telomeres that typically span 10-15kb at birth (de Lange et al, 1990).
Telomeres displaying strand asymmetry, with the 3’ end always enriched for guanosine and the
5’ end always enriched for cytosine, and are therefore referred to as the G-strand and C-strand,
respectively. The G-strand typically protrudes 50-300nt past the C-strand in human telomeres,
creating a 3’ single-strand DNA (ssDNA) overhang (Makarov et al, 1997; McElligott and
Wellinger, 1997). The presence and size of 3’ overhangs on both the leading and lagging strand
telomere, suggests that overhangs are not merely a byproduct of the end replication problem, but
rather are generated through active nucleolytic processing.
The 3’ overhang appears to play important roles in telomere function, and telomeres that have
lost their G-strand overhang can become fused together in a nonhomologous end-joining process
(van Steensel et al, 1998). Telomeres can be arranged in higher order structures visible by
electron microscopy, termed telomere loops (t-loops), which are formed when the 3’ssDNA
overhang invades proximal double-strand DNA (dsDNA) in cis, forming a large duplex loop
with a partial or full Holliday junction at its base (Figure 1-1 B). The size of the t-loop appears to
vary relative to telomere length, and can encompass up to 30kb of telomeric DNA in mice, but as
little as 0.3kb of telomeric DNA in trypanosomes (Griffith et al, 1999; Muñoz-Jordán et al,
2001). Formation of a t-loop is one potential way of preventing the linear end of the chromosome
from being detected as a double-strand break and activating a DNA damage response, and
2
t-loops have been detected in telomeres from mammal, plant, avian and protist species (Griffith
et al, 1999; Cesare et al, 2003; Muñoz-Jordán et al, 2001; Murti and Prescott, 1999).
A)
B)
Figure 1-1. Overview of telomere binding complexes and states that telomeres may exist in. A) Model of the shelterin telomere binding complex (left) and the CST protein complex (right). TRF1 and
TRF2 bind double-strand telomeric DNA, while POT1 binds to single-strand G rich telomeric DNA. The
CST complex is an RPA-like complex that binds to single-strand G rich telomeric DNA. B) Telomeres
may exist in linear (upper) or looped (lower) states.
In addition to t-loops, other forms of higher order structures may also play a role in telomere
capping. Under physiological conditions, 4 guanine bases can be arranged with a four-fold
rotation of symmetry, forming a G-tetrad, which can subsequently be stacked into higher order
four stranded helical structure referred to as G-quadruplexes. Formation of higher order G-
quadruplex structures on the 3’ overhang has also been proposed to act to as a protective capping
structure (Xu et al, 2009). However, both t-loops and G-quadruplex structures are likely to be
dynamic structures, as t-loops must be removed during telomere replication, and stabilization of
3
G-quadruplexes with small molecules leads to telomere dysfunction (Gomez et al, 2006).
Supporting the idea that telomere conformation is dynamic, only a fraction of isolated linear
telomeric DNA molecules can be seen forming t-loops, and during the G2 phase of the cell cycle
telomeres transiently adopt a more open structure that is accessible to addition of nucleotides by
terminal transferase (Verdun et al, 2005).
Telomere protection is also dependent on proteins that bind directly to telomeric DNA. Telomere
Repeat Binding Factors 1 and 2 (TRF1, TRF2) bind to double-strand telomeric DNA, while Pot1
binds to single-strand G rich telomeric DNA (Chong et al, 1995; Broccoli et al, 1997; Baumann
and Cech, 2001). These three DNA binding proteins associate with 3 additional proteins, TIN2,
TPP1, and RAP1, which form a 6 protein complex termed the shelterin complex (Figure 1-1 A)
(Kim et al, 1999; Liu et al, 2004; Ye et al, 2004; Li et al, 2000). Components of the shelterin
complex can aid in the capping of telomeres in multiple ways. TRF2 can stimulate formation of
t-loop structures in vitro, and also can bind directly to ATM, a key phosphatidylinositol 3-kinase
related protein that activates the DNA damage pathway in response to double-strand breaks
(Griffith et al, 1999; Karlseder et al, 2004). The region where TRF2 binds to ATM spans serine
1981, whose autophosphorylation plays an essential role in ATM activation. Overexpression of
TRF2 results in a blunting of the ATM dependent DNA damage response at telomeres, as well as
other genomic sites of introduced double-strand breaks (Karlseder et al, 2004; Bradshaw et al,
2005; Bradshaw and Meyn, upubl.; Cesare et al, 2009). Given the high local concentration of
TRF2 at telomeres, TRF2 may aid in telomere capping through direct suppression of ATM
activation.
When a cell is depleted of TRF2, telomeres are handled in a manner similar to that of double-
strand breaks. The histone H2AX variant becomes phosphorylated at serine 139, and proteins
implicated in the response to double-strand breaks accumulate at the telomere, including ATM
phosphorylated at serine 1981, the MRE11/RAD51/NBS1 complex, and the chromatin binding
factors 53BP1 and MDC1, which are core components of a megabase platform of DNA damage
response factors that assembles around the double-strand break (Takai et al, 2003; Dimitrova and
de Lange, 2006). These factors accumulate at levels detectable by immunofluorescence, forming
foci that are referred to as Telomere Dysfunction Induced Foci (TIFs). TRF2 depletion also
results in loss of the G-Strand overhang and chromosome end fusions that retain telomeric DNA
at the point of fusion, demonstrating that telomeres can become uncapped in a length
4
independent manner (van Steensel et al, 1998; Dimitrova and de Lange, 2006). Widespread
telomere dysfunction in TRF2 depleted cells is quickly followed by cell cycle arrest and
induction of apoptosis or senescence (van Steensel et al, 1998; Karlseder et al, 1999).
ATR is a phosphatidylinositol 3-kinase related protein that plays a key role in coordinating the
response to stalled and collapsed replication forks. POT1 binds to telomeric ssDNA, and inhibits
ATR-dependent signaling pathways. Depletion of POT1 leads to TIF formation, G-overhang
elongation, and activation of ATR-dependent cell cycle checkpoints (Lazzerini Denchi and de
Lange, 2007; Churikov and Price, 2008; Guo et al, 2007). One current hypothesis is that POT1
represses ATR-dependent signaling pathways by blocking the binding of RPA, and the
subsequent recruitment of ATRIP/ATR to ssDNA at telomeres (Lazzerini Denchi and de Lange,
2007).
Protein binding of the G-strand overhang appears to be the predominant form of telomere
capping in Saccharomyces cerevisiae, which do not form t-loops under normal circumstances.
S. cerevisiae have a trimeric RPA-like complex called Cdc13/Stn1/Ten1 (CST) which binds to
the 3’ overhang and plays a key role in telomere protection and length regulation (Gao et al,
2007). The CST complex was hypothesized to have been replaced by shelterin in higher
eukaryotes, however homologs of Stn1/Ten1 have recently been identified in multiple systems
(Martin et al, 2007; Miyake et al, 2009; Song et al, 2008; Surovtseva et al, 2009). Accumulating
evidence suggest that this type of shelterin independent, RPA-like complex also localizes to
ssDNA at telomeres and plays a role in telomere capping in most organisms. Arabidopsis
thaliana deficient in components of the CTC1/STN1/TEN1 complex show telomere length
heterogeneity, increased G-overhang signals, accumulation of extra-chromosomal circular
telomeric DNA, and increased end fusions involving subtelomeric sequences (Surovtseva et al,
2009; Song et al, 2008). SiRNA knockdown of human CTC1 results in increased telomere free
ends, formation of γH2AX foci in interphase cells, and increased single-strand G-rich DNA both
at the overhang and at internal sites (Surovtseva et al, 2009; Miyake et al, 2009).
As well as the shelterin and CST complexes, a number of DNA repair proteins have been
implicated in telomere biology. Many of these DNA repair factors accumulate at telomeres at
lower levels then the major telomere binding proteins, and localize to telomeres in a cell cycle-
specific manner. A role for some of these factors, such as the Werner and Bloom syndrome
5
helicases, in promoting replication of telomeres has been proposed (Crabbe et al, 2004; Sfeir et
al, 2009). Other factors such as the RAD51 paralog, RAD51D, may be required for the efficient
formation of t-loops (Tarsounas et al, 2004). In vitro experiments testing the ability of
immunodepleted human cell lysates to promote strand invasion of a linear telomeric substrate
with a 3’ overhang into a plasmid with telomeric repeats, suggests a role for RAD51, RAD52,
XRCC3, NBS1, RPA34 and ATR in t-loop formation (Verdun and Karlseder, 2006). The
mechanism of G-strand overhang production also likely requires a nuclease, whose identity is
currently unknown. Both the recruitment of HR and nucleolytic factors would likely require
activation of a DNA damage response, which may explain the observation that functional
telomeres in primary cells transiently activate a DNA damage response during G2, characterized
by the association of MRE11, NBS1 S343, RAD51, RAD52, XRCC3 and ATM S1981 with
telomeric DNA (Verdun et al, 2005; Verdun and Karlseder, 2006).
In addition to protein capping factors and higher order DNA structures, a growing body of
evidence also suggests a role for epigenetics in the regulation of mammalian telomere function.
The telomeric C-strand is transcribed by RNA polymerase II to generate noncoding transcripts,
known as Telomeric Repeat Containing RNAs (TERRA). TERRA has been detected in
mammals, zebrafish, and yeast, and can range in size from ∼100bp to >9kb in length (Azzalin et
al, 2007; Schoeftner and Blasco, 2008; Luke et al, 2008). TERRA associates with telomeres
in vivo, and can form G-quadruplex structures both on its own, and in conjunction with telomeric
ssDNA in vitro (Azzalin et al, 2007; Schoeftner and Blasco, 2008; Randall and Griffith, 2009;
Xu et al, 2008). Correlative analysis of TERRA expression and telomere length suggests that one
function of TERRA may be to inhibit telomere elongation by telomerase. However, siRNA
depletion of TERRA in human telomerase positive cells leads to an increase in telomere free
ends and telomeres with a FISH staining pattern resembling fragile sites (Deng et al, 2009).
Depletion of TERRA in human cells that do not rely on telomerase for telomere maintenance
results in increased TIF formation and reduced cell viability, suggesting TERRA also has
telomerase-independent affects on telomere capping (Deng et al, 2009). Studies in mice also
show an effect of the density of histone heterochromatic marks in telomeric and subtelomeric
sequences on telomere length and recombination (reviewed in Schoeftner and Blasco, 2009).
Although some of the potential mechanisms of capping appear to be redundant, interference with
any one of these mechanisms can affect telomere function, suggesting that they all have essential
6
roles. How all of these different factors are coordinated and contribute to telomere capping
remains to be elucidated.
1.1.2 Telomere Maintenance Mechanisms
The inability of DNA polymerase to fully replicate the terminal end of the lagging strand
coupled with the active processing required to generate the G-strand overhang, leads to
progressive telomere shortening of approximately 50-150 bp per population doubling in human
cells (Huffman et al, 2000; Martens et al, 2000). This shortening can be counteracted through the
activation or upregulation of a telomere maintenance mechanism. The primary mechanism of
maintaining telomere length relies on a multisubunit ribonucleoprotein complex called
telomerase. The minimal components of the enzyme that are required for catalytic activity are
the telomerase reverse transcriptase (TERT) and the telomerase RNA component (TERC)
(Weinrich et al, 1997). The template strand of TERC (AAUCCCAAUC) pairs with the end of
the 3’overhang, and then a single telomeric repeat is added per elongation step. Telomerase
preferentially elongates the shortest telomeres within a cell, and the addition of telomeric repeats
appears to be regulated in cis by telomere binding proteins, resulting in telomeres with a narrow
length distribution in telomerase positive cells (Britt-Compton et al, 2009; Smogorzewska and de
Lange, 2004). While widely expressed in single cellular organisms and some multicellular
organisms, telomerase expression is undetectable in most human somatic cells (Kim et al, 1994).
Instead, in humans, telomerase expression appears to be largely restricted to brief periods early
in development, and to germ-line and stem cell compartments (Kim et al, 1994; Wright et al,
1996).
Lack of telomerase expression in human somatic cells contributes to an age related decline in
average telomere lengths, as well as the progressive telomere shortening of cells grown in culture
(Lindsey et al, 1991; Vaziri and Benchimol, 1998). In vitro experiments have shown that human
primary cells can proliferate a limited number of times in culture, and then enter a stage of
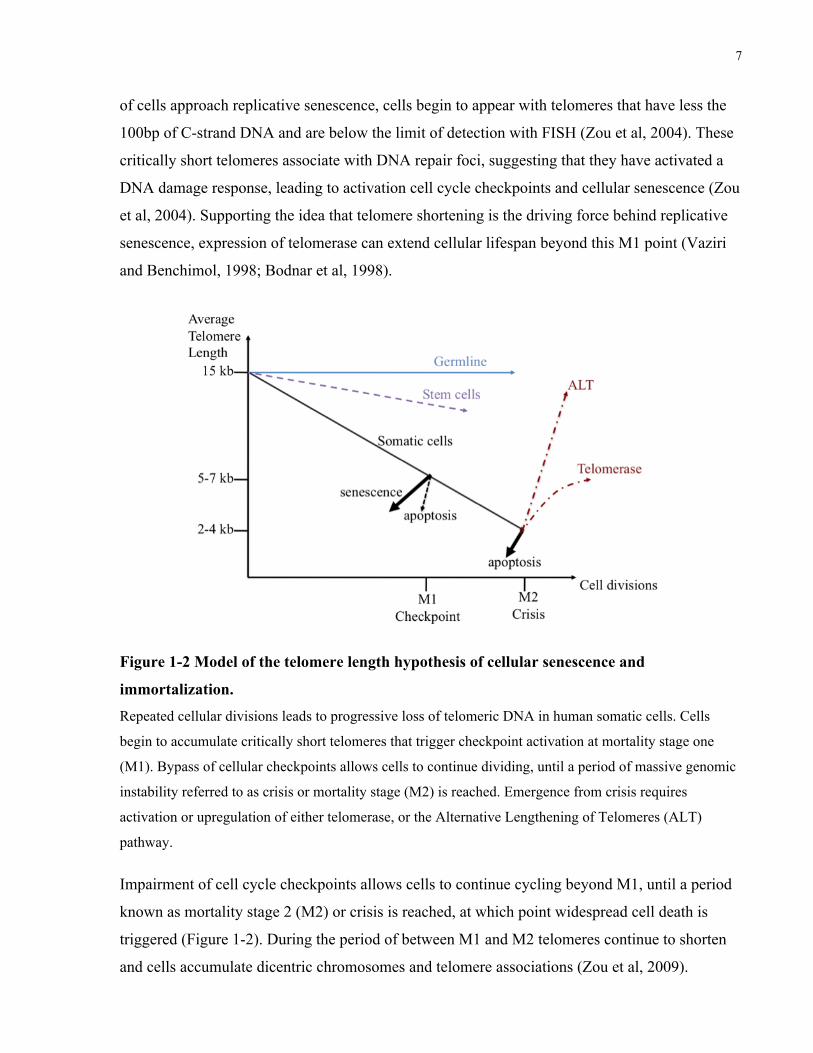
permanent arrest referred to as replicative senescence or mortality stage 1 (M1) (Figure 1-2)
(Hayflick, 1965). Senescence of cells in culture can also be induced by oncogene overexpression,
exposure to DNA damaging agents, or inadequate culture conditions, however these
phenomenon appear distinct from M1, and are instead referred to as stasis (Serrano et al, 1997;
Robles and Adami, 1998; Sherr and DePinho, 2000; Drayton and Peters, 2002). As populations
7
of cells approach replicative senescence, cells begin to appear with telomeres that have less the
100bp of C-strand DNA and are below the limit of detection with FISH (Zou et al, 2004). These
critically short telomeres associate with DNA repair foci, suggesting that they have activated a
DNA damage response, leading to activation cell cycle checkpoints and cellular senescence (Zou
et al, 2004). Supporting the idea that telomere shortening is the driving force behind replicative
senescence, expression of telomerase can extend cellular lifespan beyond this M1 point (Vaziri
and Benchimol, 1998; Bodnar et al, 1998).
Figure 1-2 Model of the telomere length hypothesis of cellular senescence and
immortalization. Repeated cellular divisions leads to progressive loss of telomeric DNA in human somatic cells. Cells
begin to accumulate critically short telomeres that trigger checkpoint activation at mortality stage one
(M1). Bypass of cellular checkpoints allows cells to continue dividing, until a period of massive genomic
instability referred to as crisis or mortality stage (M2) is reached. Emergence from crisis requires
activation or upregulation of either telomerase, or the Alternative Lengthening of Telomeres (ALT)
pathway.
Impairment of cell cycle checkpoints allows cells to continue cycling beyond M1, until a period
known as mortality stage 2 (M2) or crisis is reached, at which point widespread cell death is
triggered (Figure 1-2). During the period of between M1 and M2 telomeres continue to shorten
and cells accumulate dicentric chromosomes and telomere associations (Zou et al, 2009).
8
Telomere associations differ from true fusions in that they are not mediated by a ligase IV
dependent end-joining mechanism and a constriction indicating the physical end of both
chromosomes is still visible (Zou et al, 2009). The existence of both telomere associations and
fusions suggests that either dysfunctional telomeres may be dealt with by different pathways, or
that end fusions may often be incomplete, potentially due to a suppressive effect of residual
telomere sequence and binding proteins on DNA repair. This would lead to a model where there
are different levels or subtypes of telomere uncapping, one level at which telomeres are
sufficiently uncapped to stimulate TIF formation but retain sufficient capping abilities to
suppress fusions, and another more fully uncapped state which results in TIFs and fusions.
Supporting the idea that not all uncapped telomeres are equal, metaphase spreads of
immortalized and cancerous cells frequently have telomeres that are TIF positive, but are not
involved in fusions or associations with other telomeres (Cesare et al, 2009).
Cellular emergence from M2 requires the activation or upregulation of a telomere maintenance
mechanism. The majority of cells rely on telomerase, however ∼30% of in vitro immortalized
cells rely on a telomerase independent form of telomere maintenance called the Alternative
Lengthening of Telomeres (ALT) pathway (Bryan et al, 1995). The ALT pathway is also active
in ∼10% of human cancers, but as of yet has not been shown to be active in a primary cell setting
(Bryan et al, 1997). The underlying mechanism of ALT is not fully understood, making it
impossible to know whether ALT represents a single or multiple pathways.
The first ∼1.9kb of human telomere arrays begins with the consensus telomere repeat
(TTAGGG), interspersed with variants of this sequence including but not limited to TGAGGG,
TCAGGG, TTGGGG (Allshire et al, 1989). Monitoring sequence changes in this region of
chromosomes 12 and 16 in clones grown up from single cells shows no mutations in primary
MRC-5 and WI38 cells, only one mutation in 97 clones from a SV40 immortalized pre-crisis cell
line, and no mutations in HT1080 telomerase positive fibrosarcoma cells (Varley et al, 2002). In
contrast, multiple ALT cell lines show highly elevated frequencies of sequence changes,
including both loss of the progenitor telomere at discrete fusion points where it is replaced by a
different telomere array, as well as intra-allelic changes which may be due to insertions,
deletions, and base-changes (Varley el al, 2002). This suggests that telomeric DNA within ALT
is highly unstable relative to telomerase positive and primary cells.
9
Instability in ALT cells may not be limited solely to telomeric sequences. A comparison of
genomic single nucleotide polymorphisms in liposarcoma tumors that have activated ALT,
telomerase, or no apparent telomere maintenance mechanism showed that ALT tumors tended to
have higher levels of genomic instability, more frequent loss of heterozygosity, and frequent
deletion of 1q32.2-q44 (Johnson et al, 2007). Additionally, the MS32 minisatellite located at
1q42.3 also shows frequencies of instability that are on average ∼55 fold greater in 15 different
ALT cell lines then non-ALT cell lines (Jeyapalan et al, 2005). This increase in MS32 instability
was also observed in 3 out of 8 ALT sarcoma samples, but ALT activation did not correlate with
instability at 4 other minisatellites (Jeyapalan et al, 2005). Increased instability at telomeres and
other loci in ALT cells suggests that ALT may be an inferior mechanism of maintaining
telomeres, ALT activation may be caused by mutations that lead to an overall increase in
genomic instability, and/or ALT may be activated after a longer period in crisis.
As the genetic details of ALT are undefined, cells are characterized as being ALT based on the
presence of certain characteristics. A common characteristic of ALT cells is that they contain
long and heterogeneous telomeric DNAs. While telomeres in primary and telomerase positive
cells do not generally exceed 10-15kb in length, telomeric DNA isolated from ALT cells
typically range from <2kb to >50kb when assessed by southern blotting (Bryan et al, 1995).
Using the presence of hypervariable telomeric DNA lengths as a marker of ALT, cell fusion
studies were carried out wherein ALT immortalized human cells were fused to either primary
fibroblasts or telomerase positive immortalized human cells. Fusion of the GM847 ALT cells
with any of 3 different primary diploid fibroblast lines results in loss of abnormally long
telomeric DNA and induction of cellular senescence (Perrem et al, 1999). When GM847 cells
were fused to telomerase positive cells from the same immortalization complementation group,
all clones lost abnormally long telomeric DNA, and 2 out of 9 clones showed a transient growth
arrest (Perrem et al, 1999). These experiments suggest that ALT may arise from one or more
recessive mutations, and that both primary and telomerase positive cells contain repressors of
ALT.
A second characteristic of ALT cells is that they contain high levels of Extra-Chromosomal
Telomeric Repeat (ECTR) DNA, which is both linear and circular, single-stranded and double-
stranded (Tokutake et al, 1998; Ogino et al, 1998; Wang et al, 2004a; Cesare and Griffith, 2004;
Nabetani and Ishikawa, 2009). Electron microscopic analysis of DNA fractions enriched for
10
telomeric repeats showed the presence of circular molecules ranging in size from <2kb to >50kb,
with ∼60% of circles being <6kb (Cesare and Griffith, 2004). T-loop structures are also present
in ALT cells, with loop portions ranging in size from 0.5-70kb, but ∼75% of loops being <5kb,
and the total length of the loop and tail ranging from 0.9-79 kb (Cesare and Griffith, 2004). The
similar size of most free circular DNA molecules and the loop portion of t-loops support a model
where ECTR circular DNA is generated through aberrant resolution of t-loops (Figure 1-3 A).
T-loops contain a partial or full Holliday junction at their base, which could be resolved to
generate a free ECTR DNA circle and a significantly shorter telomere. Monitoring of the length
of a single tagged telomere shows that ALT telomeres shorten at a rate of ∼50 bp per population
doubling, but are also subject to rapid shortening and elongation events, explaining the wide
variability of telomere length present within a given cell (Murnane et al, 1994; Henson et al,
2002).
A)
B)
Figure 1-3. Potential mechanisms of extra-chromosomal telomeric repeat (ECTR) DNA
production in ALT
A) T-loops may be aberrantly resolved to generate a short telomere and circular or linear piece of ECTR
DNA. B) Unrepaired double-strand breaks within telomeres can generate a short telomere and a linear
piece of ECTR DNA. Ligation of linear ECTR DNA can also result in production of circular ECTR.
11
Ligation of linear ECTR DNA is an alternate mechanism of generating circular ECTR DNA. The
mechanism of linear ECTR DNA production is currently unknown, but could occur during
aberrant resolution of t-loops, introduction of double-strand breaks within telomeres or at
telomere proximal sequences, or endonucleolytic cleavage of either telomeres or circular ECTR
DNA (Figure 1-3 B). Alternately linear ECTR DNA could be a byproduct of replication and
recombination reactions among existing ECTR species. Study of the repair of I-Sce1 induced
double-strand breaks located either 3kb or 100kb from a telomere, shows an increase in large
deletions, terminal deletions, and gross chromosomal rearrangements at telomere proximal
breaks relative to other random interstitial sites, with more pronounced effects at the 3kb site
(Zschenker et al, 2009; Kulkarni et al, 2010). This suggests that telomeres can negatively effect
the ability of cells to repair double-strand breaks. Whether the magnitude of the affect of
telomeres on DNA repair is proportional to telomere length is unclear, but it is possible that long
telomeres present within ALT create an environment within the telomeric or subtelomeric
sequence that is abnormally difficult to repair, leading to more frequent rapid deletions of
telomere sequence arising from double-strand breaks. Interestingly, when U2OS cells were
transfected with a non-targeting plasmid and clones that had stably integrated the plasmid where
selected, 29 out of 30 clones had integration sites at close proximity to the telomere as assessed
by FISH (Jegou et al, 2009). The presence of a double-strand break results in an increase in the
frequency of viral integrations at that site, suggesting that one explanation for the integration of
the plasmid near telomeres would be the presence of double-strand breaks (Bill and Summers,
2004).
A third characteristic of ALT cells is the presence of promyelocytic leukemia (PML) nuclear
bodies that colocalize with telomeric DNA and shelterin proteins, termed ALT-associated PML
Bodies (APBs) (Yeager et al, 1999). APBs are not normally observed in telomerase positive or
primary cells, and arise at around the same time as long and heterogeneous telomeric DNAs,
suggesting the two phenomena are related (Yeager et al, 1999). In addition to telomeric factors,
APBs also colocalize with proteins implicated in DNA repair, replication and recombination,
including but not limited to RPA, RAD51, RAD52, RAD9, RAD1, HUS1, RAD17 and BLM
(Yeager et al, 1999; Stavropoulos et al, 2002; Nabetani et al, 2004). APBs accumulate during the
G2 phase of the cell cycle and disruption of ALT telomere maintenance leads to reduced
numbers of APBs (Grobelny et al, 2000; Potts and Yu, 2007). These observations have led to the
12
hypothesis that APBs are sites for telomere elongation and deletion events, although direct
evidence supporting this hypothesis is currently lacking, and the role of APBs within ALT
remains a subject of debate.
The final characteristic of ALT telomere maintenance is an increase in telomeric recombination.
The frequency of homology directed repair at sites of I-Sce1 induced double-strand breaks at an
intra-chromosomal site proximal and distal to telomeres does not significantly differ between
ALT and non-ALT cells. This suggests that homology directed repair at non-telomeric sites is
not elevated in ALT cells (Bechter et al, 2003). Studies on the frequencies of spontaneous
homologous recombination events in ALT and non-ALT cells have not been carried out,
however the frequency of genomic SCEs is not elevated in ALT cells relative to telomerase
positive cells (Londoño-Vallejo et al, 2004). The first evidence of increased recombination
between ALT telomeres came from experiments where a tag was inserted into the telomeric
repeats of a single telomere in both an ALT, and a telomerase positive cell line (Dunham et al,
2000). Within the ALT cell line the tag eventually spread to telomeres on other chromosomes, a
phenomenon which did not occur within the telomerase positive cell line (Dunham et al, 2000).
When the tag was placed in the subtelomeric region of an ALT cell, it was not copied to multiple
chromosomes. This result suggested a model where one ALT telomere can invade another, using
the other telomere as a template during replication. Alternately, the tagged sequence may move
through events involving ECTR DNA.
The second line of evidence suggesting telomeric recombination is no longer inhibited in ALT
cells, was provided by chromosome orientation fluorescent in situ hybridization experiments
(CO-FISH). During CO-FISH experiments cells are grown in BrdU/BrdC for one round of
replication, which increases the sensitivity of the newly synthesized strand to ultraviolet induced
damage, allowing for its selective degradation. Using FISH probes specific for either the G
and/or C strand, telomeres that have undergone an exchange within the telomeric sequence can
be identified. Exchanges are hypothesized to be due to crossover reactions between sister
telomeres, and are referred to as telomere sister chromatid exchanges (T-SCEs). If exchanges are
asymmetric they will result in elongation of one telomere and shortening of the other, however to
date there is no direct evidence that a T-SCE results in a net change in telomere length.
Additionally, T-SCEs may also be caused by exchanges between non-sister telomeres, or
telomeres and ECTR DNA. T-SCEs occur at frequencies of 28-280/100 metaphases in different
13
ALT cell lines, but are rarely if ever observed in telomerase positive and primary human cells
(Londoño-Vallejo et al, 2004).
The presence of ECTR DNA circles is frequently interpreted as measure of intra-telomeric
recombination, as circles may arise through inappropriate resolution of the t-loop junction. While
telomeric circles are readily detectable by 2D gel analysis of DNA from cells that utilize the
ALT pathway, they are typically undetectable or present at only very low levels in telomerase
positive and primary cells (Wang et al, 2004a; Cesare and Griffith, 2004). Additional evidence of
intra-telomeric recombination within ALT cells was provided by experiments using a telomere
integrated tag encoding a splice acceptor site upstream of a red fluorescent protein (RFP) open
reading frame, followed by a CMV promoter and splice donor site. When present only once
within the telomere, the CMV promoter is unable to drive RFP expression, however if the tag is
repeated the promoter will be upstream of RFP, and following splicing of the transcript, RFP will
be translated and cells will fluoresce. When the tag was incorporated into ALT cells it was
copied multiple times within a single telomere resulting in RFP expression (Muntoni et al, 2009).
Integration of the tag into telomerase positive cells did not result in RFP expression. Potential
mechanisms of intra-telomeric duplication include t-loop mediated rolling circle replication,
copying of the tag between sister telomeres, and unequal T-SCEs.
There are multiple mechanisms that may contribute to the rapid elongation of ALT telomeres,
including rolling circle replication involving either an intra-telomeric loop or ECTR circular
DNA, break induced replication between telomeres, recombination between a telomere and
linear ECTR DNA, or unequal exchanges between telomeres (Figure 1-4). In addition to
recombination-based mechanisms, ligation of ECTR DNA to telomeres may also play a role. In
yeast, telomerase inactivation results in telomere shortening and telomere-induced senescence.
Rare survivors primarily rely on recombination-based Rad52p dependent pathways, however if
elongated telomeres are present during before senescence, a Rad52-independent pathway that
produces heterogeneous and hypervariable telomeres can be used (Grandin and Charbonneau,
2009). Genetic analysis suggests that this pathway does not function via recombination, single-
strand annealing, nonhomologous end-joining or break induced replication. This pathway does
require the Rad1-Rad10 endonuclease, the replication factor C component Egl1, and the
Mre11/Rad50/Xrs2 complex, and has been proposed to function via microhomology-mediated
end joining between telomeres and extra-chromosomal telomeric repeat DNA generated during
14
telomere rapid deletion events (Grandin and Charbonneau, 2009). Whether or not an analogous
pathway functions within human ALT cells is currently unknown.
Figure 1-4. Potential mechanisms of recombination based telomere elongation in ALT cells
A) Both the 3’ and 5’ ends of the telomere pair with their complementary strands at the base of the t-loop
forming a 4 strand rolling circle substrate, allowing both strands to be extended simultaneously.
Alternately, the newly synthesized DNA from the 3’ invading strand may be continuously displaced and
subsequently converted into a double-strand product. B) The 3’ end of the telomere pairs with a
complimentary single-strand ECTR DNA circle, allowing for rolling circle replication. The newly
synthesized telomeric DNA would be subsequently converted into a double-strand product. Alternately
the telomeric end could form a 4 strand rolling circle through invasion of double-strand ECTR DNA
circle, allowing for simultaneous extension of both strands. C) The 3’ end of one telomere invades
another, and then is extended through break induced replication. D) The 3’ end of the telomere invades
double-strand ECTR DNA forming a Holliday junction that is resolved in a way that results in extension
of the telomere, and shortening of the ECTR DNA. Unlike A-C which involve replication and a net
increase in telomeric DNA, in D and E there is no overall change in total telomeric DNA content E) The
3’ end of the telomere invades another telomere at a more proximal point, forming a Holliday junction
which is resolved to yield one longer and one shorter telomere.
A) B)
C)
D)
E)
15
1.1.3 Telomeres and Disease
The role of telomere dysfunction in human disease and aging is a subject of great research
interest, but has proven to be technically challenging to investigate because in most model
systems, telomeres do not appear to play a regulating cellular or organismal lifespan. Mouse
models most frequently used to study human disease have long telomeres, express telomerase in
most tissues, and do not normally rely on telomeres as a mechanism to count cell divisions
(Blasco et al, 1997; Wright and Shay, 2000). Supporting a different role for telomeres in humans
vs model organisms, telomerase mutations resulting in a null phenotype have never been
identified in humans, but telomerase null mice and Caenorhabditis elegans are viable for up to 6
generations, while telomerase null Arabidopsis thaliana are viable up to 10 generations (Blasco
et al, 1997; Riha et al, 2001). The ability of these systems to remain viable for multiple
generations without telomerase may be due to combination of factors including the initial
presence of extended telomere sequences, the ability to employ telomerase independent
pathways to extend telomeres, only small losses of telomeric DNA per cell division, preferential
inheritance of rare germ cells with long telomeres, or increased tolerance to genomic instability
(Riha et al, 2001; Cheung et al, 2006).
While it typically takes several generations for telomerase null model organisms to exhibit
phenotypes, mutations that affect telomerase activity for a single generation can result in human
disease. These rare disorders currently provide some of the best evidence that telomere
dysfunction can directly impact human health. Dyskeratosis Congenita (DC) is a rare inherited
bone marrow failure syndrome diagnostically characterized by skin hyperpigmentation, nail
dystrophy and mucosal leukoplakia (Knight et al, 1998). In addition, pulmonary fibrosis,
premature grey hair, liver disease, short stature, microcephaly and developmental delay are
observed in some individuals. Haematological abnormalities are extremely common in DC, with
over 85% of patients experiencing a cytopenia of a single lineage, and 76% of patients
developing pancytopenia by a median age of 10 (Garcia et al, 2007). Bone marrow failure is the
leading cause of death in DC, followed by pulmonary disease and cancer (Knight et al, 1998).
Head and neck squamous cell carcinomas are the most frequent cancer in DKC, occurring in
∼40% of patients (Alter et al, 2009). The ratio of observed to expected cases of myelodysplastic
syndrome are also extremely elevated in DKC (>2500 fold), as are cases of tongue cancer
(>1100 fold), and acute myeloid leukemia (>200 fold) (Alter el al, 2009).
16
DC is genetically heterogeneous, displaying X-linked recessive, autosomal dominant, and
autosomal recessive inheritance. X-linked DC is caused by mutations in DKC1, whose gene
product dyskerin, associates with the telomerase RNA component and is a component of the
telomerase holoenzyme (Heiss et al, 1998; Mitchell et al, 1999). Autosomal forms of the disease
are due to mutations in genes encoding the telomerase reverse transcriptase (TERT), RNA
component (TERC), additional components of the telomerase holoenzyme (NHP2, NOP10), or
the shelterin component TIN2 (Marrone et al, 2007; Vulliamy et al, 2001; Vulliamy et al, 2008;
Walne et al, 2007; Savage et al, 2008). Autosomal dominant forms of the disease are typically
due to mutations in TERC or TERT, and function via haploinsufficiency and not a dominant
negative mechanism (Vulliamy et al, 2001; Marrone et al, 2004; Armanios et al, 2005). In vivo
telomerase reconstitution experiments suggest that mutations that cause a <50% reduction in
telomerase activity are sufficient to cause disease (Marrone et al, 2007).
Telomeres in DC patients are extremely short, with average telomere lengths in leukocytes
following below the first percentile relative to age-matched controls (Alter et al, 2007). The
spectrum of genes implicated in DC combined with this short telomere phenotype, implicate
telomere shortening as the proximal cause of the disease. The high incidence of bone marrow
failure in DC suggests that the haematopoietic system is particularly vulnerable to changes in
telomerase activity or telomere uncapping. It should be noted that while the majority of
measurements of telomere length focus on the average telomere length, the shortest telomeres
appear to be the key driving force in both replicative senescence and chromosome fusions in
situations of spontaneous genomic instability (Zou et al, 2004; Pampalona et al, 2010).
Critically short telomeres are usually several kb shorter then other telomeres within the cell, and
are undetectable by FISH, suggesting that they have undergone one or more rapid deletion events
at some point in their replicative history. While telomerase may preferentially elongate the
shortest telomeres, it is estimated that in human cells a maximum of 544bp of telomeric DNA
can be added per population doubling (Britt-Compton et al, 2009). As telomeres shorten, they
may become more prone to stochastic changes in telomere length due to a partial capping defect
caused by reduced numbers of shelterin components associated with telomeres. In this model
even modest changes in telomerase activity can result in a situation where progressive decreases
in average telomere length leads to increased production of critically short telomeres, which
cannot be adequately extended by telomerase, ultimately leading to cell death or senescence.
17
Examination of the ratio of telomere lengths on the long and short arms of each chromosome,
which is usually stable but will change if one telomere is rapidly shortened or elongated, shows
that cells from a DC patient are approximately twice as likely to have chromosomes with a >5
fold change in the telomere length ratio as cells from an non-affected relative (Morrish and
Greider, 2009). The mechanism driving stochastic changes in telomere length is unknown,
however is hypothesized to be a result of telomeric recombination.
In addition to DC, mutations in TERC have been identified in a small number of patients with
aplastic anaemia (2/155) and myelodysplatic syndrome (1/55) (Yamaguchi et al, 2003). TERT
mutations have also been identified in aplastic anaemia patients (7/200), suggesting that other
bone marrow failure syndromes may be directly caused by telomere dysfunction (Yamaguchi et
al, 2005). Recently, TERC and TERT mutations have also been identified in patients with
familial idiopathic pulmonary fibrosis at frequencies of approximately 1.4% (1/73) and 6.8%
(5/73) of patients, respectively (Armanios et al, 2007). Average telomere lengths in lymphocytes
in probands as well as asymptomatic carriers were below the 10th percentile relative to age-
matched controls, suggesting that reduced proliferative potential may contribute to this disease
(Armanios et al, 2007).
1.2 Fanconi Anaemia (FA)
1.2.1 The FA Clinical Phenotype
Fanconi anaemia (FA) is a genetically and phenotypically heterogeneous disorder characterized
by progressive bone marrow failure, increased cancer susceptibility, and congenital
abnormalities. As the clinical phenotype of FA can vary, confirming diagnosis relies on detection
of increased chromosome breakage in response to treatment with DNA crosslinking agents, a
class of mutagens that FA cells are hypersensitive to (Auerbach et al, 1989). Analysis of 754 FA
cases collected by the International Fanconi Anemia Registry between 1982 and 2003, showed
that 90% of patients experienced bone marrow failure by age 40, and that the median survival
age of patients is 24 (Kutler et al, 2003). Myelodysplastic syndrome and/or acute myeloid
leukemia are also extremely common in FA, with a cumulative incidence of 33% by age 40
(Kutler et al, 2003). In addition to haematological malignancy, FA patients are also prone to
developing solid tumors, with a cumulative incidence of 28% by age 40 (Kutler et al, 2003). The
majority of tumors in FA patients are squamous cell carcinoma of the head and neck or ano-
18
genital region, followed by liver, brain, and renal cancers. The ratio of observed to expected
cancers in FA is highest for leukemia (785x), liver (386x), head and neck (706x), esophageal
(2362x), cervical (179x), and vulvar (4317x) neoplasms (Rosenberg et al, 2003).
Major congenital abnormalities are present in approximately 2/3 of FA patients, the most
common of which are radial ray abnormalities, gastrointestinal malformations and abnormalities
of the central nervous system (Giampietro et al, 1997; Giampietro et al, 1993). In addition to
major malformations, minor abnormalities frequently observed include skin hyper and hypo-
pigmentation, microcephaly, micropthalmia, and height and weights around the 5th percentile
(Auerbach, 2009). FA patients have reduced fertility, with males displaying evidence of
hypoplastic gonads and abnormal spermatogenesis (Auerbach, 2009). There is no strong
genotype/phenotype correlation connecting the congenital malformations, as in an analysis of 45
groups of FA siblings, 12 sets contained siblings with and without malformations, and 12 sets
contained siblings with malformations of differing severity (Giampietro et al, 1993).
Additionally, monozygotic FA twins have been identified both with and without malformations,
and different malformations (Auerbach, 2009). This suggests that although FA gene mutations
drastically increase the probability of developmental anomalies, they may arise through a
stochastic process. An additional factor that may influence FA patient phenotypes is somatic
mosaicism, caused by spontaneous reversion of inherited mutations or acquisition of a secondary
compensatory mutation. Approximately 25% of FA patients have peripheral lymphocyte
populations that are ~25% corrected, and 10% of patients have lymphocyte populations that are
~50% corrected (Auerbach, 2009).
The underlying problem driving the bone marrow failure in FA is currently unknown, however
the dominant hypothesis is that it is direct result of exhaustion of haematopoietic stem cells. FA
cells exhibit increased spontaneous genomic instability, visible in examination of metaphase
spreads which show increased frequency of breaks, fusions, gaps and radials (Schroeder and
Kurth, 1971; Schroeder and German, 1974). Unlike wild-type cells that exhibit a small and
constant number of chromosomal aberrations when grown under different oxygen tensions (2
breaks per 100 metaphases), the number of aberrations in FA cells increases dramatically with
increased oxygen levels (80 breaks per 100 metaphases at 40% oxygen) (Joenge et al, 1981). An
increasing body of evidence suggests that proteins implicated in FA play important roles in
certain DNA repair pathways. An intrinsic inability of FA cells to adequately deal with
19
endogenous DNA lesions may play a role in the bone marrow failure, developmental
abnormalities, and high cancer incidence observed in FA patients, as cells with
unrepaired/misrepaired damage may be more prone to undergo apoptosis or senescence, or if
they escape this fate, will have increased levels of genomic instability which may help to drive
oncogenic progression.
1.2.2 FA and Telomere Maintenance
One potential source of endogenous DNA damage in FA cells are dysfunctional telomeres,
which may arise due to defects in telomere capping, frequent rapid losses of telomeric DNA,
reduced telomerase activity, or as a secondary affect of excessive proliferation of haematopoietic
cells. FA shares several clinical characteristics with DC, including extremely high frequencies of
bone failure, myelodysplastic syndrome, acute myeloid leukemia, and head and neck squamous
cell carcinomas. Initial telomere studies in FA peripheral blood mononuclear cells examined
average telomere lengths by southern blotting in 6 FA patients, and found that in 4 patients
telomeres were 1.2 – 2.2 kb shorter then age-matched controls (Ball et al, 1998). However the
other 2 patients analyzed in this study had normal average telomere lengths, despite both having
bone marrow failure (Ball et al, 1998). A subsequent larger study on blood samples from 45 FA
patients showed that telomeres were on average 1.75kb shorter then controls, and that telomeres
in patients that had developed one or more cytopenias were approximately 0.94kb shorter then
patients without a cytopenia (Leteurtre et al, 1999). This work was further extended to show that
patients with cytopenias had an increased rate of annual telomere shortening (>200bp/year), and
that a high rate of annual telomere shortening could serve as a prognostic indicator of
progression towards a more severe haematological disease (Li et al, 2003). Whether or not the
observed telomere shortening is a cause, or a consequence of haematopoietic failure or stress is
not apparent from these studies.
A more recent direct comparison of average telomere lengths in populations of peripheral blood
cells from DC and FA patients using flow cytometric FISH measurements has shown that the
pattern and extent of telomere shortening differs significantly between the two diseases (Alter et
al, 2007). While telomeres do tend to be shorter then average in FA cells, this is mainly observed
in the granulocyte population, whereas telomeres in DC tended to be extremely short (<1st
percentile) in all lineages tested. Looking at the occurrence of extremely short telomeres, only 7
20
of 16 FA patients met the criteria in one or more cell types. Of these 7 patients only 3 had
extremely short telomeres in more then 3 lineages, however 2 of these patients had received prior
radiation therapy. This suggests that if telomere dysfunction is involved in FA, the mechanism
driving it is likely to differ from DC. Supporting this idea, telomerase activity does not appear to
be decreased when tested in blood samples from FA patients, and mutations in TERC have not
been identified (Leteurtre et al, 1999; Calado et al, 2004).
The above studies focused on average telomere length, however telomere dysfunction causing
improper capping or increased telomere rapid deletions would not be easily detected using this
approach. One study that looked at individual telomere lengths in metaphase spreads of FA
patient lymphocytes found that there was an increase in the frequency of chromosome ends
without detectable telomere signals, whereby in FA cells 0.26% of chromosomes had a
chromosome end with an undetectable telomere via FISH, while in controls this value was 0.15%
(Callén et al, 2002). In this same study, an average of 7.8 extra-chromosomal telomeric signals
per cell was observed in FA cells, but only 2.3 signals per cell in controls, and there was a
greater then 10 fold increase in chromosome end fusions (Callén et al, 2002). However the
average telomere lengths in FA cells (3.95 kb) was significantly shorter then controls (4.63 kb),
which may have resulted in a situation where telomeres are becoming uncapped and more prone
to rapid deletion events due to telomere shortening, and not as a direct result of FA mutations. A
study examining individual telomere lengths by FISH in FA fibroblasts with longer telomere
lengths (10.5 ± 4.2 kb, 9.7 ± 5.2kb) failed to show any increase in chromosome ends without a
detectable sequence or end fusions, and did not cause a shift in telomere length distribution
consistent with rapid loss of telomeric sequence (Franco et al, 2004). The major aim of this study
is to clarify what role, if any, the FA pathway plays in telomere maintenance.
1.2.3 The FA Pathway
There are currently 12 identified genes which when mutated in humans give rise to FA (FANCA,
B, C, D1/BRCA2, D2, E, F, G, I, J/BACH1/BRIP1, L, N/PALB2) (de Winter and Joenje, 2009).
With the exception of FANCB, which demonstrates X-linked inheritance and has only been
identified in males, all other FA mutations function in an autosomal recessive manner and are
found at approximately equal frequencies in male and female patients (Kutler et al, 2003;
Auerbach, 2009). Patients are divided into complementation groups based on their gene
21
mutation, analysis of 681 FA with known complementation groups shows the following
distribution of patients: FA-A=411 (60.4%); FA-B=10 (1.5%); FA-C=108 (15.9%); FA-D1=20
FA-J=13 (1.9%); FA-L=2 (0.3%); FA-N=3 (0.4%) (Auerbach, 2009). All of the FA genes
identified to date encode proteins that appear to function within a common pathway (Figure 1-6),
although many FA proteins have additional roles outside of this pathway.
The first FA genes identified all encoded proteins that are components of a large complex
referred to as the FA core complex (FANCA, C, E, F, G) (Strathdee et al, 1992; Lo Ten Foe et al,
1996; de Winter et al, 1998; de Winter et al, 2000a; de Winter et al, 2000b).
Immunoprecipitation of FANCA coupled with mass spectroscopy has been used to identify
additional components of this complex, referred to as FANCA-associated polypeptides (FAAPs)
(Meetei et al, 2003). Mutations in FAAP43, FAAP95, and FAAP250 were subsequently
identified in FA patients, and these proteins were renamed FANCL, B and M, respectively
(Meetei et al, 2003; Meetei et al, 2004a; Meetei et al, 2005). Two additional FAAPs, FAAP24
and FAAP100 also appear to be important for core complex function, however, to date patient
mutations in these genes have not been identified (Ciccia et al, 2007; Ling et al, 2007).
Determining the role of the FA core complex was initially hampered by the lack of known
functional domains present in the first five protein components identified. However the
discovery of FANCD2, a protein whose monoubiquitination is dependent on the expression of
FA core complex members, suggested a function for the core complex in regulating
ubiquitination (Timmers et al, 2001; Garcia-Higuera et al, 2001). FANCD2 monoubiquitination
is required for its association with chromatin and assembly into nuclear foci with other DNA
repair factors (Garcia-Higuera et al, 2001). FANCI was recently identified as a second protein
that forms nuclear foci and binds to chromatin following monoubiquitination by the FA core
complex (Smogorzewska et al, 2007; Sims et al, 2007). Monoubiquitination of FANCD2 and
FANCI appears to be carried out by FANCL, which has E3 ubiquitin ligase activity, and, with
the exception of worms, is present with FANCD2 and FANCI throughout evolution (Figure 1-5)
(Meetei et al, 2004b). Additional identified components of this pathway include UBE2T, which
acts as the E2 ubiquitin activating enzyme, and USP1 which promotes deubiquitination of
FANCD2 (Machida et al, 2006; Nijman et al, 2005).
22
Unlike the other FA core complex components which are essential for monoubiquitination,
FANCM appears to play more of an accessory role, wherein it promotes the association of the
FA core complex with chromatin and subsequent monoubiquitination of FANCD2
monoubiquitination, but is not absolutely required for these activities (Bakker et al, 2009). A
mouse FANCM model also reveals some novel phenotypes not shared with the other FA core
complex mouse models, including an underrepresentation of FANCM deficient female mice, and
an increase in the frequency of spontaneous Sister Chromatid Exchange events (SCEs) (Bakker
et al, 2009). The only FANCM patient identified to date also has biallelic FANCA mutations,
making the human FANCM phenotype unclear (Singh et al, 2009). Increased SCEs have also
been observed in FANCM depleted human cells, but not other FA cell types, and likely relate to
the ability of FANCM to recruit the Blooms syndrome complex (BLM/TopoIIIα/RMI1/RMI2) to
sites of stalled or collapsed replication forks (Deans and West, 2009).
Figure 1-5. Overview of FA pathway conservation in eukaryotes Many of the FA core complex components (blue) do not appear to conserved in simple eukaryotes with
the exception of FANCL and FANCM. FANCL has E3 ubiquitin ligase activity and is normally present
with FANCD2 and FANCI. FANCM is related to the archael DNA repair protein Hef, and has additional
roles outside of the FA pathway. Diagram modified from Zhang et al, 2009.
23
During normal replication and in response to treatment with exogenous DNA damaging agents
including ionizing radiation, UVC, interstrand crosslinking agents, and replication fork stalling
agents, FANCD2 and FANCI are monoubiquitinated in a core complex dependent manner at
lysine 561 and 523, respectively (Garcia-Higuera et al, 2001; Smogorzewska et al, 2007; Sims et
al, 2007). FANCD2 and FANCI protein stability and monoubiquitination appear interdependent,
and coimmunoprecipitation experiments suggest that the endogenous forms of these proteins
interact weakly or transiently in vivo (Sims et al, 2007). Human FANCI encodes a 150 kDa
protein that shares low overall sequence similarity (~20%) with the 155kDa FANCD2 protein,
however the region surrounding the monoubiquitination site shows approximately 40% similarity
(Timmers et al, 2001; Sims et al, 2007; Smogorzewska et al, 2007). Additionally, the interior of
both proteins contain leucine rich sequences predicted to form helical hairpin structures, similar
to what has been observed in crystal structures of FANCE and FANCF (Smogorzewska et al,
2007; Nookala et al, 2007; Kowal et al, 2007). The C-terminus of both FANCI also contains a
putative EDGE domain similar to what has been previously identified in FANCD2. In FANCD2
the EDGE domain is required for complementation of the interstrand crosslinker sensitivity
phenotype, but is not involved in FANCD2 monoubiquitination or foci formation (Montes de
Oca et al, 2005). These structural similarities between FANCD2 and FANCI strongly suggest
that they both evolved from a common ancestral gene.
Approximately 5% of FA patients carry mutations in genes that do not affect FANCD2 or
FANCI expression or monoubiquitination. Mutations in the BRCA1 Associated C-terminal
Helicase (BACH1) are causal in the FANCJ complementation group (Levitus et al, 2005; Litman
et al, 2005). Biallelic mutations in BRCA2 or its binding partner PALB2 are implicated in the
FANCD1 and FANCN complementation groups, however the clinical phenotype in these groups
differs significantly from other FA groups (Howlett et al, 2002; Reid et al, 2007). Unlike other
FA patients, the first adverse clinical effect observed in FANCD1 and FANCN patients typically
is an early childhood solid tumors (Offit et al, 2003; Reid et al, 2007). The high frequency of
Wilms tumor, medullablastoma, and neuroblastoma in FANCD1 and FANCN patients often
leads to mortality during early childhood, and may be a consequence of roles for FANCD1 and
FANCN outside of the FA pathway. Figure 1-6 shows a model of the activation and recruitment
of FA pathway components in response to DNA damage.
24
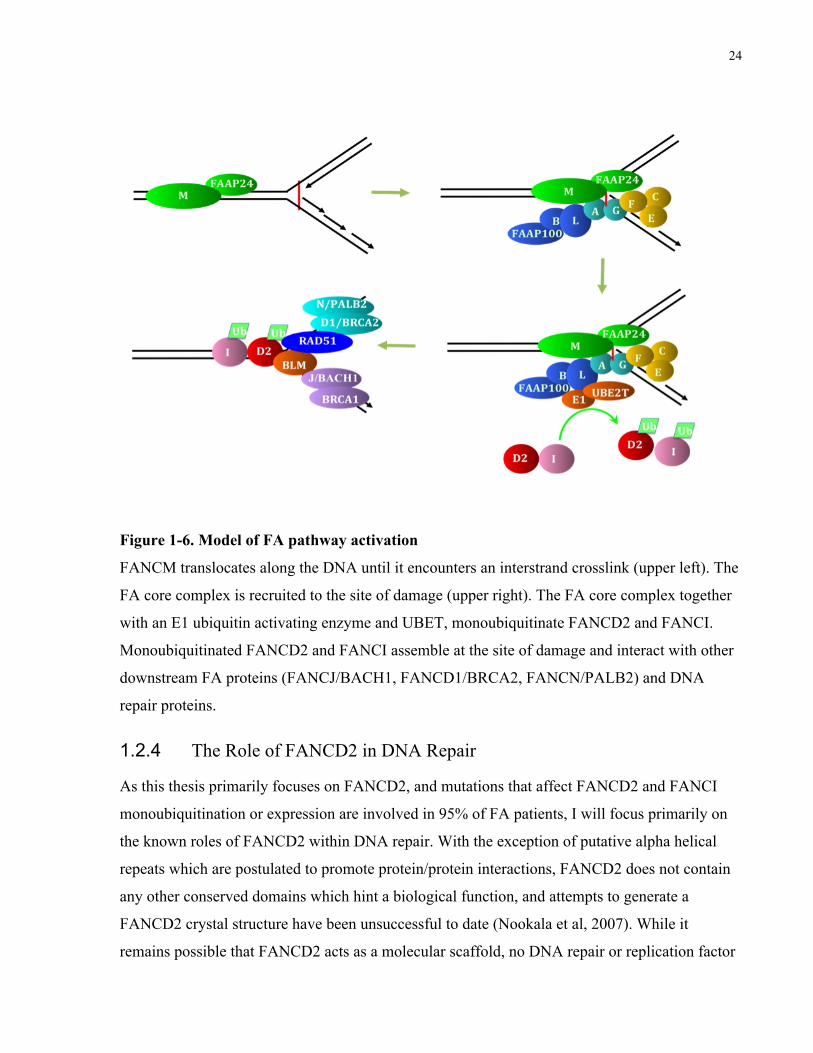
Figure 1-6. Model of FA pathway activation
FANCM translocates along the DNA until it encounters an interstrand crosslink (upper left). The
FA core complex is recruited to the site of damage (upper right). The FA core complex together
with an E1 ubiquitin activating enzyme and UBET, monoubiquitinate FANCD2 and FANCI.
Monoubiquitinated FANCD2 and FANCI assemble at the site of damage and interact with other
downstream FA proteins (FANCJ/BACH1, FANCD1/BRCA2, FANCN/PALB2) and DNA
repair proteins.
1.2.4 The Role of FANCD2 in DNA Repair
As this thesis primarily focuses on FANCD2, and mutations that affect FANCD2 and FANCI
monoubiquitination or expression are involved in 95% of FA patients, I will focus primarily on
the known roles of FANCD2 within DNA repair. With the exception of putative alpha helical
repeats which are postulated to promote protein/protein interactions, FANCD2 does not contain
any other conserved domains which hint a biological function, and attempts to generate a
FANCD2 crystal structure have been unsuccessful to date (Nookala et al, 2007). While it
remains possible that FANCD2 acts as a molecular scaffold, no DNA repair or replication factor
25
has been identified which requires FANCD2 expression for its localization to DNA damage
induced nuclear foci. Initial reports of a role for FANCD2 in promoting RAD51 focus formation
after DNA damage have been disputed in subsequent studies (Digweed et al, 2002; Wang et al,
2004b; Godthelp et al, 2002; Godthelp et al, 2006).
The cellular hypersensitivity of FA cells to DNA interstrand crosslinks (ICLs) strongly suggests
a role for FA pathway proteins in crosslink repair. Following ICL induction, FANCD2 is
monoubiquitinated and accumulates in nuclear foci with other DNA repair factors including
NBS1, BRCA2, and ATR (Nakanishi et al, 2002; Wang et al, 2004b; Andreassen et al, 2004).
FA cells exhibit increased chromosome breakage and radial formation after ICL treatment, and
cells accumulate with 4N DNA content. Both the chromosome breakage/radial phenotype and
accumulation of FA cells with a 4N DNA content require progression through S phase,
suggesting a role for the FA pathway in repair of ICLs during replication (Akkari et al, 2001).
Analysis of ICL induced radials in FA core complex deficient cells, as well as in complemented
and wild type cells treated with high doses of ICL inducing agents, shows that radials almost
always form between nonhomologous chromosomes (369 out of 372 radials) and the few radials
involving homologs occurred between distant parts of the chromosomes (Newell et al, 2004).
Whether radials form between short regions of homology or non-homologous sequences is
presently unknown, as is whether radials represent an aberrant, or a normal intermediate
structure that has not been properly resolved. The increase in radials in FA cells may reflect an
increase in recombination events using short regions of homology that would normally be
limited, or a problem with resolution of structures. Alternately, radials may form do to an
increase in an end-joining type process which occurs more frequently when the FA pathway is
not present, or may be a reflection of an increase in breaks.
Studies in the Xenopus egg extract system of the repair of a substrate with a single ICL support a
direct role for FANCD2 in replication coupled ICL repair. The initial stages of ICL repair in this
system involve transient stalling of dual replication forks ∼20-40 nucleotides (nt) from the
lesion, then a single fork approaches the lesion and stalls 1 nt from the ICL (Räschle et al, 2008).
This is followed by strand incision on both sides of the ICL on the parental strand, addition of a
nucleotide across from damaged base on the nascent strand, followed by extension beyond the
damaged base by the translesion polymerase ζ. Final repair of both DNA duplexes likely
involves incision repair to remove the damaged base and homologous recombination. Depletion
26
of FANCD2 from Xenopus extracts results in a ∼14 fold decrease in the efficiency of ICL repair,
with significant inhibition of both the incision and extension steps in this process (Knipsheer et
al, 2009). Over time there is an increase in extension products, but this does not result in a
proportional increase in perfect repair products, suggesting that either the extension product
contains errors, or there are additional problems with excision of the damaged base or
recombinational repair. The function of FANCD2 within ICL repair is dependent on its
monoubiquitination, and cannot be complemented by addition of wild-type FANCI in
conjunction with a nonmonoubiquitinatable form of FANCD2.
Insertion of a nucleotide across from a modified base requires utilization of a unique group of
polymerases, in a process known as translesion synthesis. Translesion synthesis is an error prone
process and can result in the introduction of point mutations. Genetic evidence, as well as
analysis of DNA damage induced mutagenesis suggests a role for the FA pathway in this
process, although all members of the FA pathway may not play equivalent roles in promoting
translesion synthesis (Thompson and Hinz, 2009). A recent study examining mutation
frequencies in a plasmid based system, shows that human FA core complex deficient cells have
reduced spontaneous and UVC induced mutation rates relative to complemented controls, but
FANCD2 and FANCI deficient cells have normal mutation rates (Mirchandani et al, 2008). The
hypomutability phenotype in core complex deficient cells may be related to a decrease in foci
formation of the Rev1 translesion polymerase in FANCA and FANCG mutant cells
(Mirchandani et al, 2008). FA core complex proteins may primarily be involved in promoting
translesion synthesis steps in ICL repair, while FANCD1, D2, I, and N may have additional roles
in the final steps of repair involving homologous recombination.
Multiple lines of evidence suggest a role for FANCD2 within homologous recombination
pathways, but exactly what that role is remains unclear. Early studies on primary FA fibroblasts
show increased in vitro interplasmid homologous recombination using FA cell lysates (∼10-20
fold greater then controls) and in vivo intra-plasmid recombination in FA cells (∼50-100 fold
greater then controls) (Thyagarajan and Campbell, 1997). The FA complementation groups are
not stated within this study, but likely correspond to FA core complex components. Subsequent
experiments performed on SV40 immortalized FANCA, FANCG, and FANCD2 cells show an
∼2 fold decrease in efficiency of homology directed repair of an induced I-Sce1 break integrated
27
into the genome in FA deficient cells relative to complemented counterparts (Nakanishi et al,
2005). Possible factors contributing to the different results in these studies include the primary vs
SV40 immortalized status of the cells, the use of cells from a wild-type donor vs complemented
cells as a control, and the use of an extra-chromosomal vs integrated substrates. Importantly, the
study by Tyagarajan and Campbell monitored spontaneous recombination, whereas the study by
Nakanishi and colleagues examined repair of an induced double-strand break. FANCD2 may
have roles both in suppressing certain types of spontaneous recombination events, and in
promoting homology directed DNA repair of an induced break.
Further evidence for a role of FANCD2 in regulating recombination comes from studying
meiotic recombination in FA deficient mouse spermatocytes. FANCA and FANCD2 deficient
spermatocytes have an increased frequency in both mispaired and unpaired chromosomes
(Houghtaling et al, 2002; Wong et al, 2003). FANCD2 also localizes to recombinational nodules
during meiosis in mice, arguing that chromosome pairing abnormalities are a direct consequence
of lack of FANCD2 (S. Meyn, unpubl.).
Additional evidence that FANCD2 may be involved in homologous recombination is indirect,
relying primarily on studies looking at the localization of FANCD2 and its protein interactions.
Following photoinduction of DNA damage, FANCD2 shows tight spatial colocalization with
RAD51, the major human recombinase, at sites of induced damage at a time when
recombinational repair is likely to be ongoing (P. Bradshaw and S. Meyn, unpubl.).
Colocalization with FANCD2 in nuclear foci with proteins implicated in recombination
including BRCA1, BRCA2, RAD51, and MRE11 have also been observed following treatment
of cells with ionizing radiation (Garcia-Higuera et al, 2001; Wang et al, 2004b; Nakanishi et al,
2002). Yeast two-hybrid studies suggest that FANCD2 can directly interact with BRCA2, and
FANCD2 coimmunoprecipitates with BRCA2 in untreated cells and cells exposed to ICLs
(Hussain et al, 2004; Wang et al, 2004b). The interaction between FANCD2 and BRCA2 does
not require FANCD2 ubiquitination but is dependent on FANCG expression, which also
interacts with BRCA2 (Hussain et al, 2003; Wilson et al, 2008). Coimmunoprecipitation
experiments suggest the existence of complex minimally composed of FANCD2, BRCA2,
FANCG, and the RAD51 paralog XRCC3 (Wilson et al, 2008). While this complex has been
postulated to function within recombination, to date it remains unclear what role it plays.
28
FANCD2 also colocalizes and coimmunoprecipitates with BLM, the helicase implicated in
Bloom syndrome, following cellular treatment with interstrand crosslinking and replication fork
stalling agents (Pichierri et al., 2004). Bloom syndrome cells display elevated levels of sister
chromatid exchanges and other chromosomal abnormalities including increased chromatid
breaks, gaps, radials, telomere associations, anaphase bridge and lagging chromosomes
(Chaganti et al, 1974; German and Crippa, 1996, Lillard-Wetherell et al, 2004). The increase in
sister chromatid exchanges in Bloom syndrome has been tied to a role for BLM in dissolution of
double Holliday junction structures (Wu and Hickson, 2003). Additional background on the
relationship between FANCD2 and BLM is provided in the introduction of chapter 3.
One major proposed function of homologous recombination in human cells is to help deal with
replication forks that have encountered lesions and have become stalled or collapsed, which can
result in production of DNA gaps or one-sided double-strand breaks if unrepaired (Michel et al,
1997). Potential mechanisms of both recombination dependent and independent pathways of
dealing with stalled and or broken replication forks are reviewed in Li and Heyer, 2008. Certain
sequences within the genome are more prone to experience replication fork stalling or breakage,
and are referred to fragile sites. Fragile sites share common characteristics including frequent
gaps or breaks when cultured under conditions of replicative stress, such as growth in the
presence of DNA polymerase inhibitors (Glover, 1984). Additionally, fragile sites are frequently
involved in sister chromatid exchanges, translocations, viral integrations, and are often
rearranged or deleted in tumor cells (Howlett et al, 2005).
In vitro experiments show that telomeric repeats are difficult to replicate, and show evidence of
frequent replication fork regression, a mechanism following replication fork stalling that
involves a template switch and lesion bypass without recombination (Fouché et al, 2006). Recent
in vivo experiments further suggest that mammalian telomeres may resemble fragile sites, as they
show increased levels of an abnormal staining pattern with FISH when cells are grown in the
presence of polymerase inhibitors (Sfeir et al, 2009). However other fragile site features such as
increased sister chromatid exchanges are normally suppressed, although may be elevated in
telomere proximal regions (Sfeir et al, 2009). While normal mammalian telomeres share a partial
resemblance with fragile sites, telomeres in ALT cells naturally meet all of the fragile site
criteria, even without the addition of exogenous replication stress. ALT telomeres naturally
contain single-strand gapped regions, show evidence of frequent rearrangements, are prone to
29
undergo sister chromatid exchanges, and frequently integrate DNA in telomere proximal regions
(Nabetani and Ishikawa, 2009; Varley el al, 2002; Londoño-Vallejo et al, 2004; Jegou et al,
2009).
Replication fork stalling agents rapidly induce both FANCD2 monoubiquitination and foci
formation (Hussain et al, 2004; Howlett et al, 2005). Immunofluorescent analysis shows high
levels of FANCD2 colocalization with both RAD51 and PCNA, 90 minutes post treatment of
cells with hydroxyurea to stall forks, suggesting that FANCD2 is involved in the recombinational
response to stalled or collapsed forks (Hussain et al, 2004). When FANCD2 is depleted and cells
are grown in the presence of polymerase inhibitors, there is a 3–4 fold increase in the overall
frequency of chromosome breaks and gaps, and a 2-3 fold increase in gaps and breaks at the
FRA3B and FRA16D fragile sites relative to controls (Howlett et al, 2005). This suggests that
FANCD2 normally suppresses genomic instability at fragile sites, as well as other loci within the
genome. Confirming a role for FANCD2 at fragile sites, replication stress induces high levels of
mitotic cells containing sister chromatids with paired FANCD2 foci that localize to common
fragile sites (Chan et al, 2009). Approximately 10% of FANCD2 foci are connected by ultra-fine
bridges coated with BLM during anaphase, with bridges likely representing unresolved
replication intermediates. The number of FANCD2 foci exceeds the number of breaks at fragile
sites, suggesting that FANCD2 not only responds to broken fragile sites, but also intact fragile
sites containing abnormal constrictions or linkages (Chan et al, 2009).
1.3 Concluding Remarks
As telomere dysfunction is one potential cause of bone marrow failure, and telomere
abnormalities have been reported in cells from FA patients, the goal of this project was to
elucidate what role, if any, the FA pathway plays within telomere maintenance. Experiments
focused primarily on FANCD2, because the majority of known patient mutations directly affect
FANCD2 function, and the relationship between FA core complex proteins and more
downstream FA proteins (FANCD1/BRCA2, FANCN/PALB2, FANCJ/BACH1) is presently
unclear. Given the known and proposed functions of FANCD2, there are multiple ways in which
FANCD2 may impact telomere maintenance including promoting t-loop formation, limiting
telomere rapid deletion events, aiding in telomeric replication, and preventing telomeres from
expressing characteristics of fragile sites. While I did not obtain results supporting a role for
30
FANCD2 in maintenance of telomeres in a setting where telomerase is expressed, I did find
significant roles for FANCD2 within the ALT telomere maintenance pathway. Through the
further exploration of the role of FANCD2 within ALT, I have begun to understand more about
functions of FANCD2 within recombination, and the relationships between FANCD2 and DNA
repair factors it associates with.
31
1.4 References
Akkari YM, Bateman RL, Reifsteck CA, D'Andrea AD, Olson SB, Grompe M (2001). The 4N cell cycle delay in Fanconi anemia reflects growth arrest in late S phase. Mol Genet Metab. 74:403-12.
Allshire RC, Dempster M, Hastie ND (1989). Human telomeres contain at least three types of G-rich repeat distributed non-randomly. Nucleic Acids Res. 17:4611-27.
Alter BP, Baerlocher GM, Savage SA, Chanock SJ, Weksler BB, Willner JP, Peters JA, Giri N, Lansdorp PM (2007). Very short telomere length by flow fluorescence in situ hybridization identifies patients with dyskeratosis congenita. Blood. 110:1439-47.
Alter BP, Giri N, Savage SA, Rosenberg PS (2009). Cancer in dyskeratosis congenita. Blood. 113:6549-57.
Andreassen PR, D'Andrea AD, Taniguchi T (2004). ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev. 18:1958-63.
Armanios M, Chen JL, Chang YP, Brodsky RA, Hawkins A, Griffin CA, Eshleman JR, Cohen AR, Chakravarti A, Hamosh A, Greider CW (2005). Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenita. Proc Natl Acad Sci U S A. 102:15960-4.
Armanios MY, Chen JJ, Cogan JD, Alder JK, Ingersoll RG, Markin C, Lawson WE, Xie M, Vulto I, Phillips JA 3rd, Lansdorp PM, Greider CW, Loyd JE (2007).Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 356:1317-26.
Auerbach AD, Rogatko A, Schroeder-Kurth TM (1989). International Fanconi Anemia Registry: relation of clinical symptoms to diepoxybutane sensitivity. Blood. 73:391-6.
Auerbach AD (2009). Fanconi anemia and its diagnosis. Mutat Res. 668:4-10.
Azzalin CM, Reichenbach P, Khoriauli L, Giulotto E, Lingner J (2007). Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. Science. 318:798-801.
32
Bakker ST, van de Vrugt HJ, Rooimans MA, Oostra AB, Steltenpool J, Delzenne-Goette E, van der Wal A, van der Valk M, Joenje H, te Riele H, de Winter JP (2009). Fancm-deficient mice reveal unique features of Fanconi anemia complementation group M.Hum Mol Genet. 18:3484-95.
Baumann P, Cech TR (2001). Pot1, the putative telomere end-binding protein in fission yeast and humans. Science. 292:1171-5.
Bechter OE, Zou Y, Shay JW, Wright WE (2003). Homologous recombination in human telomerase-positive and ALT cells occurs with the same frequency. EMBO Rep. 4:1138-43.
Bill CA, Summers J (2004). Genomic DNA double-strand breaks are targets for hepadnaviral DNA integration. Proc Natl Acad Sci U S A. 101:11135-40.
Blasco MA, Lee HW, Hande MP, Samper E, Lansdorp PM, DePinho RA, Greider CW (1997). Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell. 91:25-34.
Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE (1998). Extension of life-span by introduction of telomerase into normal human cells. Science. 279:349-52.
Bradshaw PS, Stavropoulos DJ, Meyn MS (2005). Human telomeric protein TRF2 associates with genomic double-strand breaks as an early response to DNA damage. Nat Genet. 37:193-7.
Britt-Compton B, Capper R, Rowson J, Baird DM (2009). Short telomeres are preferentially elongated by telomerase in human cells. FEBS Lett. 583:3076-80.
Broccoli D, Smogorzewska A, Chong L, de Lange T (1997). Human telomeres contain two distinct Myb-related proteins, TRF1 and TRF2. Nat Genet. 17:231-5.
Bryan TM, Englezou A, Gupta J, Bacchetti S, Reddel RR (1995). Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J. 14:4240-8.
33
Bryan TM, Englezou A, Dalla-Pozza L, Dunham MA, Reddel RR (1997). Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat Med. 3(11):1271-4.
Calado RT, Pintão MC, Rocha V, Falcão RP, Bitencourt MA, Silva WA Jr, Gluckman E, Pasquini R, Zago MA. Lack of mutations in the human telomerase RNA component (hTERC) gene in Fanconi's anemia. Haematologica. 89:1012-3.
Callén E, Samper E, Ramírez MJ, Creus A, Marcos R, Ortega JJ, Olivé T, Badell I, Blasco MA, Surrallés J (2002). Breaks at telomeres and TRF2-independent end fusions in Fanconi anemia. Hum Mol Genet. 11:439-44.
Cesare AJ, Quinney N, Willcox S, Subramanian D, Griffith JD (2003). Telomere looping in P. sativum (common garden pea). Plant J. 36:271-9.
Cesare AJ, Griffith JD (2004). Telomeric DNA in ALT cells is characterized by free telomeric circles and heterogeneous t-loops. Mol Cell Biol. 24:9948-57.
Cesare AJ, Kaul Z, Cohen SB, Napier CE, Pickett HA, Neumann AA, Reddel RR (2009). Spontaneous occurrence of telomeric DNA damage response in the absence of chromosome fusions. Nat Struct Mol Biol. 16:1244-51.
Chaganti RS, Schonberg S, German J (1974). A manyfold increase in sister chromatid exchanges in Bloom's syndrome lymphocytes. Proc Natl Acad Sci U S A. 71:4508-12.
Chan KL, Palmai-Pallag T, Ying S, Hickson ID (2009). Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol. 11:753-60.
Cheung I, Schertzer M, Rose A, Lansdorp PM (2006). High incidence of rapid telomere loss in telomerase-deficient Caenorhabditis elegans. Nucleic Acids Res. 34:96-103.
Chong L, van Steensel B, Broccoli D, Erdjument-Bromage H, Hanish J, Tempst P, de Lange T (1995). A human telomeric protein. Science. 270:1663-7.
Churikov D, Price CM (2008). Pot1 and cell cycle progression cooperate in telomere length regulation. Nat Struct Mol Biol. 15:79-84.
34
Ciccia A, Ling C, Coulthard R, Yan Z, Xue Y, Meetei AR, Laghmani el H, Joenje H, McDonald N, de Winter JP, Wang W, West SC (2007). Identification of FAAP24, a Fanconi anemia core complex protein that interacts with FANCM. Mol Cell. 25:331-43.
de Lange T, Shiue L, Myers RM, Cox DR, Naylor SL, Killery AM, Varmus HE (1990). Structure and variability of human chromosome ends. Mol Cell Biol. 10:518-27.
de Winter JP, Waisfisz Q, Rooimans MA, van Berkel CG, Bosnoyan-Collins L, Alon N, Carreau M, Bender O, Demuth I, Schindler D, Pronk JC, Arwert F, Hoehn H, Digweed M, Buchwald M, Joenje H (1998). The Fanconi anaemia group G gene FANCG is identical with XRCC9. Nat Genet. 20:281-3.
de Winter JP, Rooimans MA, van Der Weel L, van Berkel CG, Alon N, Bosnoyan-Collins L, de Groot J, Zhi Y, Waisfisz Q, Pronk JC, Arwert F, Mathew CG, Scheper RJ, Hoatlin ME, Buchwald M, Joenje H (2000a). The Fanconi anaemia gene FANCF encodes a novel protein with homology to ROM. Nat Genet. 24:15-6.
de Winter JP, Léveillé F, van Berkel CG, Rooimans MA, van Der Weel L, Steltenpool J, Demuth I, Morgan NV, Alon N, Bosnoyan-Collins L, Lightfoot J, Leegwater PA, Waisfisz Q, Komatsu K, Arwert F, Pronk JC, Mathew CG, Digweed M, Buchwald M, Joenje H (2000b). Isolation of a cDNA representing the Fanconi anemia complementation group E gene. Am J Hum Genet. 67:1306-8.
de Winter JP, Joenje H (2009). The genetic and molecular basis of Fanconi anemia. Mutat Res. 668:11-9.
Deans AJ, West SC (2009). FANCM connects the genome instability disorders Bloom's Syndrome and Fanconi Anemia. Mol Cell. 36:943-53.
Deng Z, Norseen J, Wiedmer A, Riethman H, Lieberman PM (2009). TERRA RNA binding to TRF2 facilitates heterochromatin formation and ORC recruitment at telomeres. Mol Cell. 35:403-13.
Digweed M, Rothe S, Demuth I, Scholz R, Schindler D, Stumm M, Grompe M, Jordan A, Sperling K (2002). Attenuation of the formation of DNA-repair foci containing RAD51 in Fanconi anaemia. Carcinogenesis. 23:1121-6.
35
Dimitrova N, de Lange T (2006). MDC1 accelerates nonhomologous end-joining of dysfunctional telomeres. Genes Dev. 20:3238-43.
Drayton S, Peters G (2002). Immortalisation and transformation revisited. Curr Opin Genet Dev. 12:98-104.
Dunham MA, Neumann AA, Fasching CL, Reddel RR (2000). Telomere maintenance by recombination in human cells. Nat Genet. 26:447-50.
Fouché N, Ozgür S, Roy D, Griffith JD (2006). Replication fork regression in repetitive DNAs. Nucleic Acids Res. 34:6044-50.
Franco S, van de Vrugt HJ, Fernández P, Aracil M, Arwert F, Blasco MA (2004). Telomere dynamics in Fancg-deficient mouse and human cells. Blood. 104:3927-35.
Garcia CK, Wright WE, Shay JW (2007). Human diseases of telomerase dysfunction: insights into tissue aging. Nucleic Acids Res. 35:7406-16.
Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D'Andrea AD (2001). Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell 7:249-62.
German J, Crippa LP (1996). Chromosomal breakage in diploid cell lines from Bloom’s syndrome and Fanconi’s anemia. Ann Genet. 9: 143-154.
Giampietro PF, Adler-Brecher B, Verlander PC, Pavlakis SG, Davis JG, Auerbach AD (1993). The need for more accurate and timely diagnosis in Fanconi anemia: a report from the International Fanconi Anemia Registry. Pediatrics. 91(6):1116-20.
Giampietro PF, Verlander PC, Davis JG, Auerbach AD (1997). Diagnosis of Fanconi anemia in patients without congenital malformations: an international Fanconi Anemia Registry Study. Am J Med Genet. 68:58-61.
36
Glover TW, Berger C, Coyle J, Echo B (1984). DNA polymerase alpha inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum Genet. 67:136-42.
Godthelp BC, Wiegant WW, Waisfisz Q, Medhurst AL, Arwert F, Joenje H, Zdzienicka MZ (2006). Inducibility of nuclear Rad51 foci after DNA damage distinguishes all Fanconi anemia complementation groups from D1/BRCA2. Mutat Res. 594:39-48.
Gomez D, O'Donohue MF, Wenner T, Douarre C, Macadré J, Koebel P, Giraud-Panis MJ, Kaplan H, Kolkes A, Shin-ya K, Riou JF (2006). The G-quadruplex ligand telomestatin inhibits POT1 binding to telomeric sequences in vitro and induces GFP-POT1 dissociation from telomeres in human cells. Cancer Res. 66:6908-12.
Grandin N, Charbonneau M (2009). Telomerase- and Rad52-independent immortalization of budding yeast by an inherited-long-telomere pathway of telomeric repeat amplification. Mol Cell Biol. 29:965-85.
Grobelny JV, Godwin AK, Broccoli D (2000). ALT-associated PML bodies are present in viable cells and are enriched in cells in the G(2)/M phase of the cell cycle. J Cell Sci. 113:4577-85.
Griffith JD, Comeau L, Rosenfield S, Stansel RM, Bianchi A, Moss H, de Lange T (1999). Mammalian telomeres end in a large duplex loop. Cell. 97:503-14.
Guo X, Deng Y, Lin Y, Cosme-Blanco W, Chan S, He H, Yuan G, Brown EJ, Chang S (2007). Dysfunctional telomeres activate an ATM-ATR-dependent DNA damage response to suppress tumorigenesis. EMBO J. 26:4709-19.
Hayflick L (1965). The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 37:614-36.
Heiss NS, Knight SW, Vulliamy TJ, Klauck SM, Wiemann S, Mason PJ, Poustka A, Dokal I (1998). X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat Genet. 19:32-8.
37
Henson JD, Neumann AA, Yeager TR, Reddel RR (2002). Alternative lengthening of telomeres in mammalian cells. Oncogene. 21:598-610.
Houghtaling S, Timmers C, Noll M, Finegold MJ, Jones SN, Meyn MS, Grompe M (2003). Epithelial cancer in Fanconi anemia complementation group D2 (Fancd2) knockout mice. Genes Dev.17:2021-35.
Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De Die-Smulders C, Persky N, Grompe M, Joenje H, Pals G, Ikeda H, Fox EA, D'Andrea AD. Biallelic inactivation of BRCA2 in Fanconi anemia (2002). Science. 297:606-9.
Howlett NG, Taniguchi T, Durkin SG, D'Andrea AD, Glover TW (2005).The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Hum Mol Genet. 14:693-701.
Huffman KE, Levene SD, Tesmer VM, Shay JW, Wright WE (2000). Telomere shortening is proportional to the size of the G-rich telomeric 3'-overhang. J Biol Chem. 275:19719-22.
Hussain S, Witt E, Huber PA, Medhurst AL, Ashworth A, Mathew CG (2003). Direct interaction of the Fanconi anaemia protein FANCG with BRCA2/FANCD1. Hum Mol Genet. 12:2503-10.
Hussain S, Wilson JB, Medhurst AL, Hejna J, Witt E, Ananth S, Davies A, Masson JY, Moses R, West SC, de Winter JP, Ashworth A, Jones NJ, Mathew CG (2004). Direct interaction of FANCD2 with BRCA2 in DNA damage response pathways. Hum Mol Genet. 13:1241-8.
Jegou T, Chung I, Heuvelman G, Wachsmuth M, Görisch SM, Greulich-Bode KM, Boukamp P, Lichter P, Rippe K (2009). Dynamics of telomeres and promyelocytic leukemia nuclear bodies in a telomerase-negative human cell line. Mol Biol Cell. 20:2070-82.
Jeyapalan JN, Varley H, Foxon JL, Pollock RE, Jeffreys AJ, Henson JD, Reddel RR, Royle NJ (2005). Activation of the ALT pathway for telomere maintenance can affect other sequences in the human genome. Hum Mol Genet. 14:1785-94.
Joenje H, Arwert F, Eriksson AW, de Koning H, Oostra AB (1981). Oxygen-dependence of chromosomal aberrations in Fanconi's anaemia. Nature. 290:142-3.
38
Johnson JE, Gettings EJ, Schwalm J, Pei J, Testa JR, Litwin S, von Mehren M, Broccoli D (2007). Whole-genome profiling in liposarcomas reveals genetic alterations common to specific telomere maintenance mechanisms. Cancer Res. 67:9221-8.
Karlseder J, Broccoli D, Dai Y, Hardy S, de Lange T (1999). p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science. 283:1321-5.
Karlseder J, Hoke K, Mirzoeva OK, Bakkenist C, Kastan MB, Petrini JH, de Lange T (2004).The telomeric protein TRF2 binds the ATM kinase and can inhibit the ATM-dependent DNA damage response. PLoS Biol. 2(8):E240.
Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW (1994). Specific association of human telomerase activity with immortal cells and cancer. Science. 266:2011-5.
Kim SH, Kaminker P, Campisi J (1999). TIN2, a new regulator of telomere length in human cells. Nat Genet. 23:405-12.
Knight S, Vulliamy T, Copplestone A, Gluckman E, Mason P, Dokal I (1998).Dyskeratosis Congenita (DC) Registry: identification of new features of DC. Br J Haematol. 103:990-6.
Knipscheer P, Räschle M, Smogorzewska A, Enoiu M, Ho TV, Schärer OD, Elledge SJ, Walter JC (2009). The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science. 326:1698-701.
Kowal P, Gurtan AM, Stuckert P, D'Andrea AD, Ellenberger T (2007). Structural determinants of human FANCF protein that function in the assembly of a DNA damage signaling complex. J Biol Chem. 282:2047-55.
Kulkarni A, Zschenker O, Reynolds G, Miller D, Murnane JP (2010). Effect of telomere proximity on telomere position effect, chromosome healing, and sensitivity to DNA double-strand breaks in a human tumor cell line. Mol Cell Biol. 30:578-89.
Kutler DI, Singh B, Satagopan J, Batish SD, Berwick M, Giampietro PF, Hanenberg H, Auerbach AD. A 20-year perspective on the International Fanconi Anemia Registry (IFAR). Blood. 101:1249-56.
39
Lazzerini Denchi E, de Lange T (2007). Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature. 448:1068-71.
Leteurtre F, Li X, Guardiola P, Le Roux G, Sergère JC, Richard P, Carosella ED, Gluckman E (1999). Accelerated telomere shortening and telomerase activation in Fanconi's anaemia. Br J Haematol.105:883-93.
Levitus M, Waisfisz Q, Godthelp BC, de Vries Y, Hussain S, Wiegant WW, Elghalbzouri-Maghrani E, Steltenpool J, Rooimans MA, Pals G, Arwert F, Mathew CG, Zdzienicka MZ, Hiom K, De Winter JP, Joenje H (2005). The DNA helicase BRIP1 is defective in Fanconi anemia complementation group J. Nat Genet. 37:934-5.
Li B, Oestreich S, de Lange T (2000). Identification of human Rap1: implications for telomere evolution. Cell. 101:471-83.
Li X, Leteurtre F, Rocha V, Guardiola P, Berger R, Daniel MT, Noguera MH, Maarek O, Roux GL, de la Salmonière P, Richard P, Gluckman E (2003). Abnormal telomere metabolism in Fanconi's anaemia correlates with genomic instability and the probability of developing severe aplastic anaemia. Br J Haematol. 120:836-45.
Li X, Heyer WD (2008). Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 18:99-113.
Lillard-Wetherell K, Machwe A, Langland GT, Combs KA, Behbehani GK, Schonberg SA, German J, Turchi JJ, Orren DK, Groden J (2004). Association and regulation of the BLM helicase by the telomere proteins TRF1 and TRF2. Hum Mol Genet. 13:1919-32.
Lindsey J, McGill NI, Lindsey LA, Green DK, Cooke HJ (1991). In vivo loss of telomeric repeats with age in humans. Mutat Res. 256:45-8.
Ling C, Ishiai M, Ali AM, Medhurst AL, Neveling K, Kalb R, Yan Z, Xue Y, Oostra AB, Auerbach AD, Hoatlin ME, Schindler D, Joenje H, de Winter JP, Takata M, Meetei AR, Wang W. FAAP100 is essential for activation of the Fanconi anemia-associated DNA damage response pathway. EMBO J. 26:2104-14.
Litman R, Peng M, Jin Z, Zhang F, Zhang J, Powell S, Andreassen PR, Cantor SB (2005). BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell. 8:255-65.
40
Liu D, Safari A, O'Connor MS, Chan DW, Laegeler A, Qin J, Songyang Z (2004). PTOP interacts with POT1 and regulates its localization to telomeres. Nat Cell Biol. 6:673-80.
Lo Ten Foe JR, Rooimans MA, Bosnoyan-Collins L, Alon N, Wijker M, Parker L, Lightfoot J, Carreau M, Callen DF, Savoia A, Cheng NC, van Berkel CG, Strunk MH, Gille JJ, Pals G, Kruyt FA, Pronk JC, Arwert F, Buchwald M, Joenje H (1996). Expression cloning of a cDNA for the major Fanconi anaemia gene, FAA. Nat Genet. 14:320-3.
Londoño-Vallejo JA, Der-Sarkissian H, Cazes L, Bacchetti S, Reddel RR (2004). Alternative lengthening of telomeres is characterized by high rates of telomeric exchange. Cancer Res. 64:2324-7.
Luke B, Panza A, Redon S, Iglesias N, Li Z, Lingner J (2008). The Rat1p 5' to 3' exonuclease degrades telomeric repeat-containing RNA and promotes telomere elongation in Saccharomyces cerevisiae. Mol Cell. 32:465-77.
Machida YJ, Machida Y, Chen Y, Gurtan AM, Kupfer GM, D'Andrea AD, Dutta A (2006). UBE2T is the E2 in the Fanconi anemia pathway and undergoes negative autoregulation. Mol Cell. 23:589-96.
Makarov VL, Hirose Y, Langmore JP (1997). Long G tails at both ends of human chromosomes suggest a C strand degradation mechanism for telomere shortening. Cell. 88:657-66.
Marrone A, Stevens D, Vulliamy T, Dokal I, Mason PJ (2004). Heterozygous telomerase RNA mutations found in dyskeratosis congenita and aplastic anemia reduce telomerase activity via haploinsufficiency. Blood. 104:3936-42.
Marrone A, Walne A, Tamary H, Masunari Y, Kirwan M, Beswick R, Vulliamy T, Dokal I (2007). Telomerase reverse-transcriptase homozygous mutations in autosomal recessive dyskeratosis congenita and Hoyeraal-Hreidarsson syndrome. Blood. 110:4198-205.
Martens UM, Chavez EA, Poon SS, Schmoor C, Lansdorp PM (2000). Accumulation of short telomeres in human fibroblasts prior to replicative senescence. Exp Cell Res. 256:291-9.
Martín V, Du LL, Rozenzhak S, Russell P (2007). Protection of telomeres by a conserved Stn1-Ten1 complex. Proc Natl Acad Sci U S A. 104:14038-43.
McElligott R, Wellinger RJ (1997). The terminal DNA structure of mammalian chromosomes. EMBO J. 16:3705-14.
41
Meetei AR, de Winter JP, Medhurst AL, Wallisch M, Waisfisz Q, van de Vrugt HJ, Oostra AB, Yan Z, Ling C, Bishop CE, Hoatlin ME, Joenje H, Wang W (2003). A novel ubiquitin ligase is deficient in Fanconi anemia. Nat Genet. 35:165-70.
Meetei AR, Levitus M, Xue Y, Medhurst AL, Zwaan M, Ling C, Rooimans MA, Bier P, Hoatlin M, Pals G, de Winter JP, Wang W, Joenje H (2004a). X-linked inheritance of Fanconi anemia complementation group B. Nat Genet. 36:1219-24.
Meetei AR, Yan Z, Wang W (2004b). FANCL replaces BRCA1 as the likely ubiquitin ligase responsible for FANCD2 monoubiquitination. Cell Cycle. 3:179-81.
Meetei AR, Medhurst AL, Ling C, Xue Y, Singh TR, Bier P, Steltenpool J, Stone S, Dokal I, Mathew CG, Hoatlin M, Joenje H, de Winter JP, Wang W (2005). A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nat Genet. 37:958-63.
Michel B, Ehrlich SD, Uzest M (1997). DNA double-strand breaks caused by replication arrest. EMBO J. 16:430-8.
Mirchandani KD, McCaffrey RM, D'Andrea AD (2008). The Fanconi anemia core complex is required for efficient point mutagenesis and Rev1 foci assembly. DNA Repair (Amst). 7:902-11.
Mitchell JR, Wood E, Collins K (1999). A telomerase component is defective in the human disease dyskeratosis congenita. Nature. 402:551-5.
Miyake Y, Nakamura M, Nabetani A, Shimamura S, Tamura M, Yonehara S, Saito M, Ishikawa F (2009). RPA-like mammalian Ctc1-Stn1-Ten1 complex binds to single-stranded DNA and protects telomeres independently of the Pot1 pathway. Mol Cell. 36:193-206.
Montes de Oca R, Andreassen PR, Margossian SP, Gregory RC, Taniguchi T, Wang X, Houghtaling S, Grompe M, D'Andrea AD (2005). Regulated interaction of the Fanconi anemia protein, FANCD2, with chromatin. Blood. 105:1003-9.
Morrish TA, Greider CW (2009). Short telomeres initiate telomere recombination in primary and tumor cells. PLoS Genet. 5:e1000357.
42
Muñoz-Jordán JL, Cross GA, de Lange T, Griffith JD (2001). t-loops at trypanosome telomeres. EMBO J. 20:579-88.
Muntoni A, Neumann AA, Hills M, Reddel RR (2009). Telomere elongation involves intra-molecular DNA replication in cells utilizing alternative lengthening of telomeres. Hum Mol Genet. 18:1017-27.
Murnane JP, Sabatier L, Marder BA, Morgan WF (1994). Telomere dynamics in an immortal human cell line. EMBO J. 13:4953-62.
Murti KG, Prescott DM (1999). Telomeres of polytene chromosomes in a ciliated protozoan terminate in duplex DNA loops. Proc Natl Acad Sci U S A. 96:14436-9.
Nabetani A, Yokoyama O, Ishikawa F (2004). Localization of hRad9, hHus1, hRad1, and hRad17 and caffeine-sensitive DNA replication at the alternative lengthening of telomeres-associated promyelocytic leukemia body. J Biol Chem. 279:25849-57.
Nabetani A, Ishikawa F (2009). Unusual telomeric DNAs in human telomerase-negative immortalized cells. Mol Cell Biol. 29:703-13.
Nakanishi K, Taniguchi T, Ranganathan V, New HV, Moreau LA, Stotsky M, Mathew CG, Kastan MB, Weaver DT, D'Andrea AD (2002). Interaction of FANCD2 and NBS1 in the DNA damage response. Nat Cell Biol. 4:913-20.
Nakanishi K, Yang Y, Pierce A, Taniguchi T, Digweed M, D’Andrea AD, Wang Z, Jasin M (2005). Human Fanconi anemia monoubiquitination pathway promotes homologous DNA repair. PNAS 102: 1110-1115.
Newell AE, Akkari YM, Torimaru Y, Rosenthal A, Reifsteck CA, Cox B, Grompe M, Olson SB (2004). Interstrand crosslink-induced radials form between non-homologous chromosomes, but are absent in sex chromosomes. DNA Repair (Amst). 3:535-42.
Nijman SM, Huang TT, Dirac AM, Brummelkamp TR, Kerkhoven RM, D'Andrea AD, Bernards R (2005). The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol Cell. 17:331-9.
Nookala RK, Hussain S, Pellegrini L (2007). Insights into Fanconi Anaemia from the structure of human FANCE. Nucleic Acids Res. 35:1638-48.
43
Offit K, Levran O, Mullaney B, Mah K, Nafa K, Batish SD, Diotti R, Schneider H, Deffenbaugh A, Scholl T, Proud VK, Robson M, Norton L, Ellis N, Hanenberg H, Auerbach AD (2003). Shared genetic susceptibility to breast cancer, brain tumors, and Fanconi anemia. J Natl Cancer Inst. 95:1548-51.
Ogino H, Nakabayashi K, Suzuki M, Takahashi E, Fujii M, Suzuki T, Ayusawa D (1998). Release of telomeric DNA from chromosomes in immortal human cells lacking telomerase activity. Biochem Biophys Res Commun. 248:223-7.
Pampalona J, Soler D, Genescà A, Tusell L (2010). Telomere dysfunction and chromosome structure modulate the contribution of individual chromosomes in abnormal nuclear morphologies. Mutat Res. 683:16-22.
Perrem K, Bryan TM, Englezou A, Hackl T, Moy EL, Reddel RR (1999). Repression of an alternative mechanism for lengthening of telomeres in somatic cell hybrids. Oncogene. 18:3383-90.
Pichierri P, Franchitto A, Rosselli F (2004). BLM and the FANC proteins collaborate in a common pathway in response to stalled replication forks. EMBO J. 23:3154-63.
Potts PR, Yu H (2007). The SMC5/6 complex maintains telomere length in ALT cancer cells through SUMOylation of telomere-binding proteins. Nat Struct Mol Biol. 14:581-90.
Randall A, Griffith JD (2009). Structure of long telomeric RNA transcripts: the G-rich RNA forms a compact repeating structure containing G-quartets. J Biol Chem. 284:13980-6.
Räschle M, Knipscheer P, Enoiu M, Angelov T, Sun J, Griffith JD, Ellenberger TE, Schärer OD, Walter JC (2008). Mechanism of replication-coupled DNA interstrand crosslink repair. Cell. 134:969-80.
Reid S, Schindler D, Hanenberg H, Barker K, Hanks S, Kalb R, Neveling K, Kelly P, Seal S, Freund M, Wurm M, Batish SD, Lach FP, Yetgin S, Neitzel H, Ariffin H, Tischkowitz M, Mathew CG, Auerbach AD, Rahman N (2007). Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet. 39:162-4.
Riha K, McKnight TD, Griffing LR, Shippen DE (2001). Living with genome instability: plant responses to telomere dysfunction. Science. 291:1797-800.
44
Robles SJ, Adami GR (1998). Agents that cause DNA double strand breaks lead to p16INK4a enrichment and the premature senescence of normal fibroblasts. Oncogene. 16:1113-23.
Rosenberg PS, Greene MH, Alter BP (2003). Cancer incidence in persons with Fanconi anemia. Blood. 101:822-6.
Savage SA, Giri N, Baerlocher GM, Orr N, Lansdorp PM, Alter BP (2008). TINF2, a component of the shelterin telomere protection complex, is mutated in dyskeratosis congenita. Am J Hum Genet. 82:501-9.
Schoeftner S, Blasco MA (2008). Developmentally regulated transcription of mammalian telomeres by DNA-dependent RNA polymerase II. Nat Cell Biol. 10:228-36.
Schoeftner S, Blasco MA (2009). A 'higher order' of telomere regulation: telomere heterochromatin and telomeric RNAs. EMBO J. 28:2323-36.
Schroeder TM, Kurth R (1971). Spontaneous chromosomal breakage and high incidence of leukemia in inherited disease. Blood. 37:96-112.
Schroeder TM, German J (1974). Bloom's syndrome and Fanconi's anemia: demonstration of two distinctive patterns of chromosome disruption and rearrangement. Humangenetik. 25:299-306.
Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW (1997). Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 88:593-602.
Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, de Lange T (2009). Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell. 138:90-103.
Sherr CJ, DePinho RA (2000). Cellular senescence: mitotic clock or culture shock? Cell. 102:407-10.
Sims AE, Spiteri E, Sims RJ 3rd, Arita AG, Lach FP, Landers T, Wurm M, Freund M, Neveling K, Hanenberg H, Auerbach AD, Huang TT (2007). FANCI is a second monoubiquitinated member of the Fanconi anemia pathway. Nat Struct Mol Biol. 14:564-7.
45
Singh TR, Bakker ST, Agarwal S, Jansen M, Grassman E, Godthelp BC, Ali AM, Du CH, Rooimans MA, Fan Q, Wahengbam K, Steltenpool J, Andreassen PR, Williams DA, Joenje H, de Winter JP, Meetei AR (2009). Impaired FANCD2 monoubiquitination and hypersensitivity to camptothecin uniquely characterize Fanconi anemia complementation group M. Blood. 114:174-80.
Smogorzewska A, de Lange T (2004). Regulation of telomerase by telomeric proteins. Annu Rev Biochem. 73:177-208.
Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER 3rd, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D'Andrea AD, Elledge SJ (2007). Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 129:289-301.
Song X, Leehy K, Warrington RT, Lamb JC, Surovtseva YV, Shippen DE (2008). STN1 protects chromosome ends in Arabidopsis thaliana. Proc Natl Acad Sci U S A. 105:19815-20.
Stavropoulos DJ, Bradshaw PS, Li X, Pasic I, Truong K, Ikura M, Ungrin M, Meyn MS (2002). The Bloom syndrome helicase BLM interacts with TRF2 in ALT cells and promotes telomeric DNA synthesis. Hum Mol Genet. 11:3135-44.
Strathdee CA, Gavish H, Shannon WR, Buchwald M (1992). Cloning of cDNAs for Fanconi's anaemia by functional complementation. Nature. 356:763-7.
Surovtseva YV, Churikov D, Boltz KA, Song X, Lamb JC, Warrington R, Leehy K, Heacock M, Price CM, Shippen DE (2009). Conserved telomere maintenance component 1 interacts with STN1 and maintains chromosome ends in higher eukaryotes. Mol Cell. 36:207-18.
Takai H, Smogorzewska A, de Lange T (2003). DNA damage foci at dysfunctional telomeres. Curr Biol. 13:1549-56.
Tarsounas M, Muñoz P, Claas A, Smiraldo PG, Pittman DL, Blasco MA, West SC (2004). Telomere maintenance requires the RAD51D recombination/repair protein. Cell. 117:337-47.
Thompson LH, Hinz JM (2009). Cellular and molecular consequences of defective Fanconi anemia proteins in replication-coupled DNA repair: mechanistic insights. Mutat Res. 668:54-72.
46
Thyagarajan B, Campbell C (1997). Elevated homologous recombination activity in fanconi anemia fibroblasts. J Biol Chem. 272:23328-33.
Timmers C, Taniguchi T, Hejna J, Reifsteck C, Lucas L, Bruun D, Thayer M, Cox B, Olson S, D'Andrea AD, Moses R, Grompe M (2001). Positional cloning of a novel Fanconi anemia gene, FANCD2. Mol Cell. 7:241-8.
Tokutake Y, Matsumoto T, Watanabe T, Maeda S, Tahara H, Sakamoto S, Niida H, Sugimoto M, Ide T, Furuichi Y (1998). Extra-chromosomal telomere repeat DNA in telomerase-negative immortalized cell lines. Biochem Biophys Res Commun. 247:765-72.
van Steensel B, Smogorzewska A, de Lange T (1998). TRF2 protects human telomeres from end-to-end fusions. Cell. 92:401-13.
Varley H, Pickett HA, Foxon JL, Reddel RR, Royle NJ (2002). Molecular characterization of inter-telomere and intra-telomere mutations in human ALT cells. Nat Genet. 30:301-5.
Vaziri H, Benchimol S (1998). Reconstitution of telomerase activity in normal human cells leads to elongation of telomeres and extended replicative life span. Curr Biol. 8:279-82.
Verdun RE, Crabbe L, Haggblom C, Karlseder J (2005). Functional human telomeres are recognized as DNA damage in G2 of the cell cycle. Mol Cell. 20:551-61.
Verdun RE, Karlseder J (2006). The DNA damage machinery and homologous recombination pathway act consecutively to protect human telomeres. Cell. 127:709-20.
Vulliamy T, Marrone A, Goldman F, Dearlove A, Bessler M, Mason PJ, Dokal I (2001). The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature. 413:432-5.
Vulliamy T, Beswick R, Kirwan M, Marrone A, Digweed M, Walne A, Dokal I (2008). Mutations in the telomerase component NHP2 cause the premature ageing syndrome dyskeratosis congenita. Proc Natl Acad Sci U S A. 105:8073-8.
Walne AJ, Vulliamy T, Marrone A, Beswick R, Kirwan M, Masunari Y, Al-Qurashi FH, Aljurf M, Dokal I (2007). Genetic heterogeneity in autosomal recessive dyskeratosis congenita
47
with one subtype due to mutations in the telomerase-associated protein NOP10. Hum Mol Genet. 16:1619-29.
Wang RC, Smogorzewska A, de Lange T (2004a). Homologous recombination generates T-loop-sized deletions at human telomeres. Cell. 119:355-68.
Wang X, Andreassen PR, D'Andrea AD (2004b). Functional interaction of monoubiquitinated FANCD2 and BRCA2/FANCD1 in chromatin. Mol Cell Biol. 24:5850-62.
Weinrich SL, Pruzan R, Ma L, Ouellette M, Tesmer VM, Holt SE, Bodnar AG, Lichtsteiner S, Kim NW, Trager JB, Taylor RD, Carlos R, Andrews WH, Wright WE, Shay JW, Harley CB, Morin GB (1997). Reconstitution of human telomerase with the template RNA component hTR and the catalytic protein subunit hTRT. Nat Genet. 17:498-502.
Wilson JB, Yamamoto K, Marriott AS, Hussain S, Sung P, Hoatlin ME, Mathew CG, Takata M, Thompson LH, Kupfer GM, Jones NJ (2008). FANCG promotes formation of a newly identified protein complex containing BRCA2, FANCD2 and XRCC3. Oncogene. 27:3641-52.
Wong JC, Alon N, Mckerlie C, Huang JR, Meyn MS, Buchwald M (2003). Targeted disruption of exons 1 to 6 of the Fanconi Anemia group A gene leads to growth retardation, strain-specific microphthalmia, meiotic defects and primordial germ cell hypoplasia. Hum Mol Genet. 12:2063-76.
Wright WE, Pereira-Smith OM, Shay JW (1989). Reversible cellular senescence: implications for immortalization of normal human diploid fibroblasts. Mol Cell Biol. 9:3088-92.
Wright WE, Piatyszek MA, Rainey WE, Byrd W, Shay JW (1996). Telomerase activity in human germline and embryonic tissues and cells. Dev Genet. 18:173-9.
Wright WE, Shay JW (2000). Telomere dynamics in cancer progression and prevention: fundamental differences in human and mouse telomere biology. Nat Med. 6:849-51.
Wu L, Hickson ID (2003). The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature. 426:870-4.
Xu Y, Kimura T, Komiyama M (2008). Human telomere RNA and DNA form an intermolecular G-quadruplex. Nucleic Acids Symp Ser (Oxf). 52:169-70.
48
Xu Y, Ishizuka T, Kurabayashi K, Komiyama M (2009). Consecutive formation of G-quadruplexes in human telomeric-overhang DNA: a protective capping structure for telomere ends. Angew Chem Int Ed Engl. 48:7833-6.
Yamaguchi H, Baerlocher GM, Lansdorp PM, Chanock SJ, Nunez O, Sloand E, Young NS (2003). Mutations of the human telomerase RNA gene (TERC) in aplastic anemia and myelodysplastic syndrome. Blood. 102:916-8.
Yamaguchi H, Calado RT, Ly H, Kajigaya S, Baerlocher GM, Chanock SJ, Lansdorp PM, Young NS (2005). Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N Engl J Med. 352:1413-24.
Ye JZ, Hockemeyer D, Krutchinsky AN, Loayza D, Hooper SM, Chait BT, de Lange T (2004). POT1-interacting protein PIP1: a telomere length regulator that recruits POT1 to the TIN2/TRF1 complex. Genes Dev. 18:1649-54.
Yeager TR, Neumann AA, Englezou A, Huschtscha LI, Noble JR, Reddel RR (1999). Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res. 59:4175-9.
Zhang XY, Langenick J, Traynor D, Babu MM, Kay RR, Patel KJ (2009). Xpf and not the Fanconi anaemia proteins or Rev3 accounts for the extreme resistance to cisplatin in Dictyostelium discoideum. PLoS Genet. 5:e1000645.
Zou Y, Sfeir A, Gryaznov SM, Shay JW, Wright WE (2004). Does a sentinel or a subset of short telomeres determine replicative senescence? Mol Biol Cell. 15:3709-18.
Zou Y, Misri S, Shay JW, Pandita TK, Wright WE (2009). Altered states of telomere deprotection and the two-stage mechanism of replicative aging. Mol Cell Biol. 29:2390-7.
Zschenker O, Kulkarni A, Miller D, Reynolds GE, Granger-Locatelli M, Pottier G, Sabatier L, Murnane JP (2009). Increased sensitivity of subtelomeric regions to DNA double-strand breaks in a human cancer cell line. DNA Repair. 8:886-900.
49
Chapter 2
2 The Fanconi Anaemia Pathway Plays a Critical Role in Cells that Utilize the Alternative Pathway of Telomere Maintenance
2.1 Abstract
Fanconi anaemia (FA) is a multigenic pleiotrophic syndrome characterized by bone marrow
failure, high cancer rates and congenital malformations. FA gene products have been implicated
in homologous recombination, a process involved in telomere maintenance. Here, I show that
FANCD2, A, and G localize to telomeric foci in human cells that utilize the recombination based
Alternative Lengthening of Telomeres (ALT) pathway, but not telomerase positive cells.
FANCD2 localizes to those telomeric foci that contain BLM, and coimmunoprecipitation
experiments indicate interactions between FANCD2, BLM, and TRF2 in late S/G2 ALT cells.
Interestingly, localization of FANCD2 to telomeric foci requires BLM expression. FANCD2
typically localizes to telomeric foci that have not activated a DNA damage response, and
FANCD2 localization to telomeric foci is largely independent of ATM and TRF2, but requires
monoubiquitination by the FA core complex. Only ∼20% of FANCD2 colocalization events with
telomeric foci are dependent on ATR expression, arguing that the primary function of FANCD2
is not replication fork rescue/restart.
FANCD2 primarily colocalizes with telomeric proteins within ALT-associated PML Bodies
(APBs). Electron spectroscopic imaging of APBs reveals that the bodies are composed of a
nucleic acid and protein interior, surrounded by a outer protein layer. The morphology and
phosphorous signal intensity of the nucleic acid within APBs differs from chromatin, strongly
suggesting that this material does not represent telomeres themselves, but rather extra-
chromosomal telomeric material. When FANCD2 is depleted there is an ALT-specific increase
in telomere dysfunction induced foci, and APBs are occasionally observed with chromatin-like
masses invading the bodies, suggesting that dysfunctional telomeres may preferentially associate
with APBs. ALT cells depleted of FANCD2 have reduced viability and show signs of mitotic
catastrophe: supernumerary centrosomes, rereplicated DNA, and aneuploidy. Together my
50
results suggest that FANCD2 has an essential BLM-dependent function in ALT cells that may be
independent from its role in the response to DNA damage and replication fork rescue.
2.2 Introduction
Homologous recombination plays important roles in DNA repair, replication and telomere
maintenance. Fanconi anaemia (FA) is rare genetic syndrome clinically characterized by
progressive bone marrow failure, increased cancer incidence and congenital abnormalities
(Kutler et al, 2003; Auerbach AD, 2009). FA cells exhibit hypersensitivity to DNA crosslinking
agents, DNA repair defects, and spontaneous chromosomal aberrations (Weksberg et al, 1979;
Schroeder and Kurth, 1971). Seven FA proteins (FANCA, B, C, E, F, G, L) participate in a core
complex that is required to promote monoubiquitination and foci formation of FANCD2 and
FANCI during replication and after DNA damage (Garcia-Higuera et al, 2001; Medhurst et al,
2006; Smogorzewska et al, 2007). Additional repair proteins, including FANCM, ATM, ATR,
RPA, HCLK2, RAD17, RAD9, CHK1 and BRCA1, are involved in promoting efficient
FANCD2 monoubiquitination after cellular exposure to exogenous DNA damaging agents
(Bakker et al, 2009; Ho et al, 2006; Andreassen et al, 2004; Collis et al, 2007; Guervilly et al,
2008; Garcia-Higuera et al, 2001). Evidence to date suggests that the FA pathway plays roles in
the response to stalled/collapsed replication forks, crosslink repair, and in recombination.
Supporting a role for the pathway in recombination, BRCA2 and its binding partner PALB2 are
mutated in the FANCD1 and FANCN complementation groups, (Howlett et al, 2002; Reid et al,
2007) and reporter assays show impaired recombinational repair in FA core complex and
FANCD2 mutant cells (Nakanishi et al, 2005). FANCD2 also colocalizes with RAD51 at sites of
photo-induced DNA breaks and in recombination nodules during meiosis (Bradshaw and Meyn,
unpubl.), and FANCA and FANCD2 knockout mice show increased incidence of unsynapsed
axial elements and chromosome mispairing during meiosis (Wong et al, 2003; Houghtaling et al,
2003).
Telomeres are nucleoprotein structures that cap the ends of linear chromosomes, preventing ends
from activating cell cycle checkpoints or becoming substrates for DNA repair reactions that
could result in telomere fusions or rapid shortening events. Proper capping of chromosome ends
depends not only on sufficient telomere length and the presence of telomere binding proteins, but
also on DNA repair factors implicated in global repair pathways (Longhese MP, 2008). DNA
51
repair factors may be involved in the processing of chromosome ends, as well as formation and
maintenance of the t-loop, a large duplex loop that is formed when the 3’ssDNA telomeric
overhang invades its own proximal dsDNA telomeric DNA (Griffith et al, 1999). Additional
DNA repair factors are required to promote replication of telomeres, as the repetitive nature and
propensity of the TTAGGG sequence to form secondary structures can result in frequent
replication fork stalling (Gilson and Géli, 2007).
Telomeres in cycling somatic cells typically shorten with age, eventually becoming uncapped
and involved in aberrant end-joining reactions, leading to cellular senescence or apoptosis (Zou
et al, 2004). The few cells that bypass this fate enter a period of widespread genomic instability
and cellular crisis, with rare survivors emerging that have activated or up-regulated a mechanism
of telomere maintenance (Kim et al, 1994; Bryan et al, 1995; Bryan et al, 1997). Approximately
90% of immortalized cells rely on telomerase to add de novo repeats to telomeres, while the
remaining cells depend on the Alternative Lengthening of Telomeres (ALT) pathway(s) (Bryan
et al, 1997). The mechanism of ALT is still being elucidated, but evidence to date supports a role
for both inter- and intra-telomeric recombination (Dunham et al, 2000; Londoño-Vallejo et al,
2004; Muntoni et al, 2009). ALT cells also typically contain high amounts of linear and circular
single- and double-strand Extra-Chromosomal Telomeric Repeat (ECTR) DNA that may be a
product, as well as a substrate in recombination reactions (Cesare AJ and Griffith JD, 2004;
Nabetani A and Ishikawa F, 2009).
Evidence of excessive telomere shortening relative to age-matched controls has been reported in
peripheral blood samples in FA patients, and correlates with progression towards aplastic
anaemia (Leteurtre et al, 1999; Hanson et al, 2001; Callen et al, 2002). However, a recent survey
found that telomere shortening is not equal among all haematopoietic lineages, and many FA
patients have average telomere lengths that are in the range of healthy controls (Alter et al 2007).
Average telomere length measurements do not address the frequency of critically short
telomeres, which appear to be the driving force in activation of the DNA damage response at
telomeres, telomere fusions, and induction of cellular senescence (Zou et al, 2004). Techniques
measuring individual telomere lengths have also resulted in conflicting results, with one group
showing no difference in telomere length distribution between FA patients and controls (Franco
et al, 2004) and another group showing an increased frequency of ends without detectable FISH
signals and excess ECTR DNA (Callen et al, 2002). Telomere abnormalities in FA cells do not
52
appear to be due to problems with telomerase, as telomerase activity appears normal in FA
haematopoietic cells (Leteurtre et al, 1999), and mutations in telomerase have not been identified
(Calado et al, 2004).
The aim of the present study was to clarify the role of the FA pathway in telomere maintenance
and determine whether observed telomere abnormalities are solely a consequence of increased
cell turnover, or are indicative of a telomere maintenance abnormality. My results do not support
a critical role for the FA pathway in telomere maintenance in cells that rely on telomerase for
telomere maintenance, but indicate an essential role for the pathway in cells that utilize ALT
0.45mM citric acid, 1mM MgCl2) was preheated for 3 min at 86°C, added to slides, covered with
a coverslip, than slides were heated for 3 min at 81°C and left for 2 hours at room temperature
prior to washing. Slides were washed 2 x 15 min in 70% formamide, 10mM Tris pH7.2, 0.1%
BSA, and then 3 x 5 min in 100mM Tris pH 7.2, 150mM NaCl, 0.08% Tween 20.
Electron spectroscopic imaging. Experiments were carried out by Andrew Larsen with
technical assistance from Dr. Ren Li. For a detailed explanation of methods used please refer to
the thesis by Andrew Larsen to be submitted to the Department of Biochemistry, University of
Toronto, 2010.
Colony forming assays. Cells transfected with FANCD2 siRNA A or GL2 control siRNA were
replated 24 hours after the second siRNA transfection into 6 cm plates at densities of 125, 250,
500, 1000, and 10 000 cells/plate. All cells were plated in duplicate and experiments were
repeated 4 times. When clearly visible colonies appeared (8-16 days post plating) plates were
rinsed in PBS, fixed in methanol for 15 minutes, stained in 10% Giemsa for 15 minutes, rinsed in
ddH2O, dried, and manually counted.
56
2.4 Results
2.4.1 FANCD2 localize to telomeric foci and PML bodies in ALT human cells
FANCD2 forms nuclear foci during S phase and in response to induced DNA damage (Garcia-
Higuera et al, 2001). I find that in cells that utilize the ALT pathway of telomere maintenance,
FANCD2 forms nuclear foci that colocalize with telomere binding proteins (TRF1/TRF2) and
telomeric DNA (Figure 2-1A). TRF1 and TRF2 are major components of the shelterin telomere
binding complex (de Lange, 2005), and are commonly used as markers of telomeric DNA.
Localization of FANCD2 to telomeric foci in ALT cells was verified with multiple independent
FANCD2 and TRF1 and TRF2 antibodies, and is in accord with recent studies (Spardy et al,
2008; Fan et al, 2009). FANCD2 colocalization with telomeric foci was rarely observed in
telomerase positive cell lines (Figure 2-1 A, B) or primary cells (data not shown) and never
exceeded more then one or two foci per nucleus. Whether FANCD2 localizes to replicating or
dysfunctional telomeres in telomerase positive cells is currently unknown, however the fact that
occasional colocalization events are detected argues against the idea that FANCD2 is present at
most telomeres, but is undetectable by immunofluorescence. Within ALT cells there is a cell
cycle-dependent variability in the extent of FANCD2 colocalization. Similar to Fan et al, I find
that colocalization between FANCD2 and TRF2 is maximal in cells during late S/G2 (data not
shown), with lower levels observed in all other phases of the cell cycle.
ALT associated PML Bodies (APBs) are a subtype of PML nuclear body that is characterized by
the association of telomeric DNA sequence and binding proteins with normal PML body
components (Yeager et al, 1999). The function of APBs is currently unknown, however they are
most common during late S/G2 when recombination is known to occur, suggesting that they are
potential sites of recombination reactions between telomeres. Colocalization analysis shows that
FANCD2 localizes to APBs (Figure 2-2 A), and that ∼80% FANCD2 colocalization with
telomeric proteins occurs at APBs (Figure 2-2 B). FANCD2 only rarely colocalizes with non-
telomeric PML bodies in ALT and telomerase positive cells (Figure 2-2 B), the significance of
which is unknown.
57
A) DAPI FANCD2 Telomeric DNA TRF2 Merge
B)
Figure 2-1. FANCD2 frequently colocalizes with telomeric DNA and telomere binding
proteins in cells that utilize the ALT pathway, but not in telomerase positive cells A) Representative images of GM847 and VA13 ALT nuclei with a high degree of colocalization between
FANCD2 and telomeric DNA and binding proteins, and HT0180 and GM639 telomerase positive nuclei
where FANCD2 does not frequently colocalize with telomeric DNA or binding proteins. Scale bar is
5µm. B) Plot of the number of FANCD2 foci that do, and do not colocalize with TRF2 from 300
randomly selected asynchronous cells from 3 independent experiments ± the associated standard error for
each cell line.
GM847 (ALT)
VA13 (ALT)
HT1080 (telomerase
positive)
GM639 (telomerase
positive)
FANCD2 foci not colocalized with TRF2 FANCD2 foci colocalized with TRF2
58
A) DAPI FANCD2 TRF1 PML
B) GM847 VA13
Figure 2-2. FANCD2 primarily colocalizes with telomeric proteins within ALT-associated
PML Bodies (APBs) A) Representative images of ALT cell nuclei with a high degree of colocalization between FANCD2 and
TRF1. Most FANCD2 foci which colocalize with TRF1 also colocalize with PML, and therefore
represent APBs. Scale bar is 5µm. B) Venn plots showing colocalization between FANCD2, TRF1, and
PML foci from 150 randomly selected asynchronous GM847 (left) or VA13 (right) ALT nuclei selected
over 3 independent experiments.
GM847 (ALT)
VA13 (ALT)
59
2.4.2 FA core complex components localize to ALT telomeric foci and promote FANCD2 monoubiquitination and localization to telomeric foci
In addition to FANCD2, I find that FA core complex components FANCA and FANCG also
colocalize with FANCD2 and TRF1 within ALT cells (Figure 2-3 A). Colocalization analysis on
randomly selected asynchronous ALT cells, shows that 69% and 77% of FANCD2 foci that
colocalize with TRF1 also contain FANCA in GM847 and VA13 cells respectively (Figure 2-3
B). FANCA localization to telomeric foci was confirmed with two independent antibodies, and
signal specificity was confirmed with siRNA knockdown of FANCA. FANCA and FANCG foci
did not colocalize with telomeric proteins in telomerase positive cells (data not shown).
Using siRNA to target FANCA, I was able to significantly reduce both FANCA protein levels
and FANCD2 monoubiquitination (Figure 2-4 A). Analysis of FANCD2 foci formation in
FANCA-depleted cells versus random siRNA treated controls, shows that FANCA knockdown
results in 11 and 15 fold reductions in the number of non-telomeric associated FANCD2 foci in
GM847 and VA13 depleted cells, respectively (Figure 2-4 B). The number of FANCD2 foci
associated with telomeric proteins showed a more modest decrease of 2 and 1.6 fold in FANCA-
depleted GM847 and VA13 cells relative to controls, however residual FANCD2 foci in
FANCA-depleted cells were significantly less intense then controls (Figure 2-4 C).
To ensure that monoubiquitination is required for all FANCD2 accumulation at telomeric foci, I
generated a non-monoubiquitinatable GFP tagged FANCD2 mutant through site-directed
mutagenesis of lysine 561 (GFP-FANCD2K561R) and transiently expressed it in GM847 cells
(Figure 2-4 D). The GFP-FANCD2 wild type construct was generated in the lab of Dr. Alan
D’Andrea, and shown to behave similarly to wild-type FANCD2 (Chirnomas et al, 2006). I find
that in contrast to GFP-FANCD2, which forms foci in 29% of transiently transfected GM847
cells (500 cells analyzed), GFP-FANCD2(K561R) fails to accumulate in visible foci (2000 cells
analyzed). Together these results support a requirement for FANCD2 monoubiquitination at
lysine 561 by the FA core complex for FANCD2 accumulation at ALT telomeric foci.
60
A) DAPI FANCA FANCD2 TRF1
B)
Figure 2-3 FA core complex proteins FANCA and FANCG colocalize with FANCD2 at
ALT telomeric foci A) Representative images of GM847 and VA13 ALT cells with a high degree of colocalization between
FANCA, FANCD2 and TRF1 (upper 2 rows) and FANCG, FANCD2 and TRF1 (lower 2 rows). Scale bar
is 5 µm. B) Venn plots showing colocalization between FANCD2, FANCA, and TRF1 foci from 150
randomly selected asynchronous ALT nuclei selected over 3 independent experiments.
GM847 (ALT)
VA13 (ALT)
GM847 (ALT)
VA13 (ALT)
DAPI FANCG FANCD2 TRF1
GM847 VA13
61
A)
B)
C)
62
D)
Figure 2-4. FANCD2 localization to ALT telomeric foci in GM847 and VA13 ALT cells is
dependent on monoubiquitination by the FA core complex
A) SiRNA depletion of FANCA results in a significant reductions in FANCA protein level and FANCD2
monoubiquitination B) Plot of the number of FANCD2 foci that do, and do not colocalize with TRF2
from 300 randomly selected asynchronous cells from 3 independent experiments ± the associated
standard error. C) Remaining FANCD2 foci in FANCA siRNA treated cells are smaller and less intense
than controls. Scale bar is 10 µm. D) FANCD2-GFP forms foci, while non-monoubiquitinatable
FANCD2 (FANCD2K561R) does not. Scale bar is 10 µm.
2.4.3 FANCD2 localizes to ALT telomeric foci that have not activated a DNA damage response, and localization to telomeric foci is independent of ATM and ATR kinase activity
Telomeres that have become dysfunctional through excessive shortening or disturbance of the
normal capping structure activate a DNA damage response, commonly characterized by the
colocalization of telomere binding proteins with 53BP1 or serine 139 phosphorylated histone
H2AX (γH2AX) (Takai et al, 2003). I monitored colocalization between FANCD2, TRF1, and
53BP1 to determine whether FANCD2 specifically localizes to telomeric foci that have activated
a DNA damage response. I find that in GM847 and VA13 ALT cells, only 15% and 12% of
telomeric foci that FANCD2 localizes to, also contain 53BP1 (Figure 2-5 A), suggesting that
telomeric DNA-associated FANCD2 is not simply participating in the response to dysfunctional
telomeres.
ATR and ATM are PI-3-related kinases that play key early signaling roles in the response to
DNA damage. ATR is primarily involved in coordinating the response to replication stress, and
ATM is involved in the response to double-strand breaks. FANCD2 monoubiquitination and foci
formation are strongly induced by agents that cause replication stress, and ATR appears to play a
63
key role in promoting this process (Andreassen et al, 2004). Replication stress can also stimulate
formation of large APBs that contain FANCD2, and this process appears partially dependent on
ATR (Spardy et al, 2008). I wanted to determine whether ATR is required for FANCD2 to
localize to normal endogenously forming APBs. SiRNA was used to transiently knockdown
ATR in GM847 and VA13 ALT cells. To verify that the level of knockdown obtained was
functional, I treated cells with hydroxyurea, a replication fork-stalling agent, and monitored the
affect of ATR depletion on FANCD2 monoubiquitination. The level of ATR depletion was
sufficient to decrease FANCD2 monoubiquitination in response to hydroxurea in GM847 and
VA13 ALT cells (Figure 2-5 B). However, when FANCD2 foci formation was monitored, I
observed a slight increase in the number of non-telomeric FANCD2 foci, and only a ∼ 20%
decrease in FANCD2 colocalization to telomeric foci (Figure 2-5 D). Residual FANCD2 foci
that formed in ATR depleted cells were of similar intensity to foci in controls (data not shown).
If FANCD2 was primarily responding to stalled/collapsed replication forks present in APBs and
telomeric foci, I would predict that localization of most FANCD2 would be largely dependent on
ATR expression, which was not observed.
ATM also influences FANCD2 monoubiquitination and foci formation after exposure of cells to
DNA damaging agents (Ho et al, 2006), and promotes efficient formation of telomere
dysfunction induced foci (Takai et al, 2003). To determine whether ATM activity is required for
FANCD2 localization, GM847 and VA13 cells were grown in media containing the ATM
inhibitor KU55933, resuspended in DMSO, for 36 hours, then FANCD2 foci formation and
localization were assessed. Inhibition of ATM activity was verified by monitoring suppression of
p53 serine 15 phosphorylation, a known ATM target, after irradiation (Figure 2-5 C). I find that
inhibiting ATM activity with KU55933 has no significant affect on FANCD2 localization to
telomeric foci, and causes only a slight decrease in non-telomeric foci formation foci (Figure 2-5
D). Similar results were observed when cells were grown in 20 or 40 µM wortmannin to inhibit
ATM activity (data not shown). This suggests that FANCD2 is not simply responding to
telomeric DNA structures as part of an ATM coordinated DNA damage response.
64
A) GM847 VA13
B)
C)
65
D)
Figure 2-5. FANCD2 localization to ALT telomeric foci is not simply part of a DNA
damage response A) Venn plots showing colocalization of FANCD2, 53BP1, and TRF1 foci from populations of 150
randomly selected asynchronous GM847 and VA13 ALT cells. FANCD2 primarily localizes to telomeric
foci that have not activated a DNA damage response. B) SiRNA depletion of ATR is sufficient to limit
FANCD2 monoubiquitination in GM847 and VA13 cells treated with 2mM HU for 24 hours. C)
KU55933 inhibition of ATM activity is sufficient to limit p53 phosphorylation in GM847 and VA13 cells
when measured 1 hour post treatment with 5 Gy IR. D) Plot of the number of FANCD2 foci that do, and
do not colocalize with TRF2 from 300 randomly selected asynchronous cells from 3 independent
experiments ± the associated standard error.
2.4.4 FANCD2 coimmunoprecipitates with TRF2 and BLM in ALT cells, and almost always localizes to telomeric foci that also contain BLM
TRF2 appears to be involved in telomeric recombination, as in vitro experiments show that it
promotes t-loop formation and binds to the base of the loop where a partial or full Holliday
junction is present (Griffith et al, 1999). Further in vitro evidence suggests that TRF2 has both
pro and anti-recombinogenic activities, and both promotes formation of Holliday junctions and
prevents their subsequent resolution (Poulet et al, 2009). BLM, the helicase mutated in Bloom
syndrome, also has activities that promote and inhibit recombination, and coimmunoprecipitates
with TRF2 in ALT cells but not telomerase positive cells (Lillard-Wetherell et al, 2004).
Additionally, fluorescent resonance energy transfer experiments support a direct in vivo
interaction between BLM and TRF2 in ALT cells (Stavropoulos et al, 2002), and in vitro
experiments show that TRF2 stimulates BLM strand unwinding activity (Lillard-Wetherell et al,
2004). FANCD2 has been implicated in recombination, binds to Holliday junctions with high
affinity in vitro (Park et al, 2005), and interacts with BLM after DNA damage (Pichierri et al,
2004). Therefore I performed coimmunoprecipitation experiments testing for in vivo interactions
between FANCD2, TRF2, and BLM. VA13 ALT cells and GM639 telomerase positive cells
were synchronized with a double thymidine block, and harvested 8-9 hrs after release to enrich
for cells in late S/G2, when telomeric recombination is believed to occur.
I find that when FANCD2 is immunoprecipitated, TRF2 and BLM are coimmunoprecipitated in
VA13 ALT cells, but not GM639 telomerase positive cells (Figure 2-6 A). Immunoprecipitation
of TRF2 also results in coimmunoprecipitation of FANCD2 in VA13 cells, but not in GM639
cells (Figure 2-6 B). Only a small amount of protein was coimmunoprecipitated in all cases,
suggesting that interactions may involve a small subset of each protein, or be weak or transient in
nature. Supporting an interaction between FANCD2, BLM and TRF2 in ALT cells, in
asynchronous GM847 and VA13 ALT cells, 90 and 94% of telomeric foci that contain FANCD2
also contain BLM, respectively (Figure 2-6 C). This suggests that FANCD2 may be
participating in a common process with BLM and/or that both proteins respond to the same DNA
substrates.
2.4.5 FANCD2 localization to APBs is independent of TRF2, but requires BLM expression
SiRNA knockdown of TRF2 significantly reduces TRF2 protein levels (Figure 2-7 A) and foci
formation 72 hours after siRNA addition, (Figure 2-7 B) but does not affect TRF1 foci formation
or APB formation (Figure 2-7 B,C), demonstrating that TRF1 and TRF2 associate independently
with telomeric DNA, and that APB formation is not dependent on TRF2, consistent with results
by Stagno D'Alcontres et al. (2007). FANCD2 colocalization with APBs, and with TRF1 foci not
associated with PML, does not require TRF2 expression, but rather increases slightly in TRF2
depleted 847 and VA13 ALT cells (Figure 2-7 C). The decrease in the amount of non-
monoubiquitinated FANCD2 in TRF2 siRNA treated cells may be a result of alterations to the
cell cycle, as siRNA depletion of TRF2 resulted in a high degree of cell death (data not shown).
67
A) B)
C) GM847 VA13
Figure 2-6. FANCD2 interacts with BLM and TRF2 in late S/G2 ALT, but not telomerase
positive cells
A) Immunoprecipitation of FANCD2 in late S/G2 VA13 ALT cells, but not late S/G2 GM639 telomerase
positive cells, coimmunoprecipitates BLM and TRF2. B) Immunoprecipitation of TRF2 in late S/G2
VA13 ALT cells, but not late S/G2 GM639 telomerase positive cells, coimmunoprecipitates FANCD2.
C) Venn plot showing colocalization of FANCD2, BLM, and TRF2 foci in populations of 150 randomly
selected GM847 and VA13 asynchronous cells selected from 3 independent experiments. FANCD2
primarily localizes to ALT telomeric foci that also contain BLM.
68
A)
B)
C)
Figure 2-7 FANCD2 localization to ALT telomeric foci is independent of TRF2 expression A) TRF2 siRNA significantly reduces TRF2 protein levels in GM847 and VA13 ALT cells. B) TRF2
knockdown does not affect TRF1 or FANCD2 foci formation or colocalization in VA13, or GM847 cells
(not shown). C) Venn plots showing colocalization between FANCD2, PML and TRF1 foci in
asynchronous populations of 150 GM847 and VA13 cells treated with random or TRF2 siRNA.
GM847 + random siRNA GM847 + TRF2 siRNA
VA13 + random siRNA VA13 + TRF2 siRNA
69
In contrast to TRF2 knockdown, transient depletion of BLM with siRNA leads to a dramatic
reduction in FANCD2 colocalization with telomeric foci. BLM knockdown does not
significantly alter FANCD2 monoubiquitination (Figure 2-8 A), nor does BLM depletion
decrease the number of and non-telomeric FANCD2 foci (Figure 2-8 B). However, BLM
depletion causes 79% and 84% reductions in the number of FANCD2 foci colocalized with
TRF2 in 847 and VA13 cells, respectively. The fraction of cells in S and G2 does not
significantly differ in BLM and random siRNA treated cells, and APBs continue to form at a
similar frequency, however very large APBs are rarely observed (data not shown). BLM is not
required for FANCD2 S phase foci, and is not required for FANCD2 foci formation after
exposure of cells to exogenous DNA damage (Pichierri et al, 2004), making the BLM
dependency of FANCD2 colocalization to ALT telomeric foci a novel behaviour.
A)
B)
Figure 2-8 FANCD2 localization to ALT telomeric foci is dependent on BLM expression
A) BLM siRNA significantly reduces BLM protein levels in GM847 and VA13 ALT cells. B)
Plot of the number of FANCD2 foci that do, and do not colocalize with TRF2 from 300
randomly selected asynchronous cells from 3 experiments ± the associated standard error.
GM847 GM847 VA13 VA13 + random + BLM + random + BLM siRNA siRNA siRNA siRNA
70
2.4.6 FANCD2 knockdown causes an ALT-specific increase in telomere dysfunction induced foci that is independent of rapid telomere shortening
To investigate the role of FANCD2 in ALT cells I used a siRNA knockdown approach. I tested
four different siRNAs and found three that reduced expression via western blot analysis to below
the residual amount present in the PD20 FANCD2 patient cell line (Figure 2.9 A). With this level
of silencing, FANCD2 foci were no longer visible by IF under 63x magnification using my most
sensitive antibody (Figure 2-9 B). Transfection efficiency as judged by a fluorescently tagged
random siRNA was approximately 100% in all cell lines tested, and FANCD2 siRNA
significantly decreased in FANCD2 expression in all cell lines tested (data not shown).
Figure 2-9. FANCD2 siRNA significantly
reduces FANCD2 protein levels and foci
formation A) Western blot analysis of FANCD2 protein levels in PD20 cells with biallelic FANCD2 mutations, and GM847 cells treated with random or FANCD2 targeting siRNAs. B) Immmunofluorescent staining of FANCD2 in GM847 cells treated with random or FANCD2 targeting siRNAs. FANCD2 foci are not observed in cells treated with siRNAs A-C.
FANCD2
FANCD2
A)
B) GM847 + random siRNA + FANCD2 siRNA D
+ FANCD2 siRNA A + FANCD2 siRNA B + FANCD2 siRNA C
71
GM847 and VA13 ALT cells, and GM639 and HT1080 telomerase positive cells were
transfected with FANCD2 siRNA A, then examined for telomere dysfunction induced foci
(TIFs) 5 days later. Transient FANCD2 depletion in GM847 and VA13 cells leads to a 2.9 fold
increase in the absolute number of 53BP1 foci that colocalize with TRF2, which corresponds to
2.9 and 3.1 fold increases in the percentage of TRF2 foci that have activated a DNA damage
response in GM847 and VA13 cells, respectively (Figure 2-10 A). GM639 and HT1080 cells do
not show any increase in 53BP1 colocalization with TRF2 when FANCD2 is depleted. Similar
increases in TIFs were also observed when GM847 and VA13 cells were treated with FANCD2
siRNA C (data not shown), and an ALT specific increase in TIFs upon FANCD2 depletion was
also reported by Spardy et al, (2008). In contrast to TIFs which form in an ALT specific manner,
FANCD2 depletion also results in an increase in 53BP1 foci not associated with TRF2, which is
observable in both ALT and telomerase positive cells. This increase in 53BP1 foci is consistent
with a role for FANCD2 in promoting genomic stability at non-telomeric regions.
Telomere shortening can trigger activation of a DNA damage response (d'Adda di Fagagna et al,
2003), and aberrant resolution of t-loops or breaks in the telomere sequence can trigger rapid
shortening events (Wang et al, 2004). Therefore I examined chromosomes during metaphase for
the presence of chromosomes ends without detectable telomeric signals. A constant exposure
time of 300 ms was used for all cell lines, and slides from each replicate were processed and
photographed on the same day. Five days after siRNA addition, FANCD2 knockdown did not
result in an increase in signal free ends in GM847 or U2OS ALT cells, or GM639 or HT1080
telomerase positive cells (Figure 2-10 B). In all cases >97% of chromatids had detectable
telomere signals. In GM847 and U2OS ALT cells this result was confirmed with a second
FANCD2 targeting siRNA, and again I found that >97% of chromosome ends in FANCD2-
depleted cells have telomere signals (Figure 2-10 B). Telomeres in FANCD2 depleted ALT cells
appear to have similar length distributions as controls (Figure 2-10 C) and sister chromatid
telomeres appear approximately equal in length, arguing against a severe replication problem,
although it remains possible that minor variations in telomere length distributions exist.
Together, my results suggests that telomeric DNA in ALT cells more frequently activates a DNA
damage response when FANCD2 is depleted, but that this is not due to telomere rapid deletion
events.
72
A)
B)
C)
Figure 2-10. FANCD2 knockdown results in an ALT-specific increase in telomere
dysfunction induced foci that is independent of telomere rapid deletion events
A) Plot of the number of 53BP1 foci that do, and do not colocalize with TRF2 from 300
randomly selected asynchronous cells from 3 independent experiments ± the associated standard
error. B) Plot of the average percentage of chromatid ends that do not have detectable telomere
GM639+ GM639+ HT1080+ HT1080+ GM847+ GM847+ GM847+ VA13+ VA13+ VA13+ random FANCD2 random FANCD2 random FANCD2 FANCD2 random FANCD2 FANCD2 siRNA siRNA A siRNA siRNA A siRNA siRNA A siRNA B siRNA siRNA A siRNA B
si
73
signals from 3 independent experiments ± the standard deviation of experimental means. C)
Representative metaphase spreads from U2OS cells treated with random or FANCD2 siRNA.
Telomeres (red) are present on almost all chromatids, and do not usually differ significantly in
size between sister chromatids. Centromeres are shown in green.
2.4.7 ALT-associated PML bodies (APBs) are structurally different from non-ALT bodies, and contain telomeric nucleic acid in the interior of the body that differs from surrounding chromatin
As the majority of FANCD2 colocalization with telomeric DNA and proteins occurs within
APBs, I wanted to better understand the nature of these structures. Non-ALT PML bodies have a
consistent and well defined ultrastructure: a solid protein core devoid of both DNA and RNA,
which makes frequent contacts with neighbouring chromatin fibers that help tether the body in
place (Boisvert et al, 2000). In contrast, fluorescent microscopy suggests that APBs have a
donut-shape that is closely associated with telomeric DNA within or around the body (Yeager et
al, 1999). Unfortunately, limitations in the resolution of light microscopes have precluded
precise determination of the ultrastructure and organization of APBs. A recent attempt to study
APB structure using light microscopy relied on experimentally inducing formation of abnormally
large APBs (Draskovic et al, 2009), however whether these larger APBs resemble the
endogenous bodies is unclear. APBs may serve as sites where ECTR DNA is sequestered, where
telomeres undergo replication or recombination events, and/or sites where late replicating
telomeric chromatin is remodeled.
Electron spectroscopic imaging (ESI) is a technique that monitors energy loss of incident
electrons as they pass through samples in order to generate high resolution element specific maps
(reviewed in Bazett-Jones et al, 2008). Through monitoring the respective ratios of nitrogen and
phosphorus one can determine whether structures are primarily composed of protein, DNA, or
RNA. Examination of the ultrastructure of APBs using ESI was performed by Andrew Larsen
and Dr. Ren Li in the laboratory of Dr. David Bazett-Jones. A combination of fluorescent
staining of TRF2 on embedded sections and gold labeling of PML, was used to distinguish APBs
from non-ALT associated PML bodies in GM847 and VA13 ALT cells. ESI analysis showed
that APBs have a unique ultrastructure, and clearly contain a mixture of protein (blue) and
nucleic acid (yellow) within the interior of the body, surrounded by an outer protein layer (Figure
74
2-11). Within the VA13 cell line, APBs were found that are similar to non-ALT PML bodies in
size (200-350nm), however examples of APBs that are significantly larger then non-ALT PML
bodies were also identified (Figure 2-11). All APBs examined exhibited a distinct ultrastructure
from non-ALT PML bodies, in that they clearly contained phosphorous, representing nucleic
acid, within the bodies. The intensity and morphology of the phosphorous signal differs
significantly from neighbouring chromatin, suggesting that APBs primarily contain non-
nucleosomal material. APBs in GM847 ALT cells have a similar structure as APBs in the VA13
ALT line, and were also found to contain phosphorous within the bodies that is less intense then
neighbouring chromatin (Figure 2-11). Gold labeling of PML suggested that the PML protein is
primarily concentrated within the outer protein layer surrounding the APB.
APBs in GM847 ALT cells treated with random and FANCD2 siRNA were also examined, and
found to have a similar ultrastructure as controls, with an outer protein shell surrounding a
nucleic acid/protein mixture. The intensity of the phosphorous signal within APBs in random
siRNA treated cells differs significantly from surrounding chromatin (Figure 2-12). APBs in
FANCD2-depleted GM847 cells resemble APBs from random siRNA treated cells with a few
notable exceptions. APBs in FANCD2-depleted GM847 cells tend to be physically larger then
APBs in random siRNA and untreated GM847 cells. Similar to controls, APBs in FANCD2-
depleted cells show a wide size distribution, however bodies tend to have diameters in excess of
half a micron, with some bodies being greater then one micron in size. APBs of this size were
not observed in untreated or random siRNA treated GM847 cells.
APBs were also observed in FANCD2-depleted GM847 cells that contain blocks of phosphorus
signal with an intensity and morphology similar to the surrounding chromatin (Figure 2-12).
Analysis of serial sections of several of these bodies indicated that these chromatin-like signals
were not simply on top or below the body, but rather had invaded the APBs. Given that
telomeres are highly nucleosomal structures, these chromatin-like masses may represent
telomeres themselves, or ECTR DNA derived directly from telomeres, associating with APBs
(Pisano et al, 2008). If these chromatin masses represent telomeres, this would suggest that
interactions between telomeres and APBs are usually rare or transient in nature, or do not occur
under normal circumstances, as they were not detected during the analysis of APBs in VA13 or
GM847 control cells.
75
VA13
VA13
VA13
GM847
Figure 2-11. APBs contain nucleic acid within the body, differing from non-ALT associated
PML bodies, which are solid protein structures Images of nitrogen, phosphorous, and a false coloured overlay image of nitrogen (blue) and phosphorous
(yellow) are shown for a non-ALT associated PML body (top row, solid arrow), and APBs (dotted arrow)
in VA13 (rows 2 &3) and GM847 (row 4) ALT cells. Scale bar is 0.2 µm. Unlike non-ALT associated
PML bodies that are devoid of phosphorous signal, APBs contain phosphorous within the body. The
intensity of the phosphorous within APBs differs significantly from neighbouring chromatin.
76
GM847 + random siRNA
GM847 + FANCD2 siRNA
GM847 + FANCD2 siRNA
Figure 2-12. APBs in FANCD2 depleted cells tend to be physically larger then APBs in
controls, and can contain blocks of chromatin-like nucleic acid Images of nitrogen, phosphorous, and a false coloured overlay image of nitrogen (blue) and phosphorous
(yellow) are shown for a large APB from a random siRNA treated GM847 cells (top row), and average
sized APBs in FANCD2-depleted GM847 cells (bottom 3 rows). Scale bar is 0.2 µm. Examples of APBs
with chromatin-like structures within FANCD2-depleted APBs are shown (bottom 2 rows). Solid arrows
2.4.8 FANCD2 depletion in ALT cells results in nuclear abnormalities, centrosome amplification and rapid cell death
Knockdown of FANCD2 in ALT cells results in the accumulation of cells with severe nuclear
abnormalities in GM847, VA13, SAOS-2, and GM637 ALT cells (Figure 2-13A, B; SAOS-2
and GM637 data not shown). These types of highly fragmented nuclei are not common in
telomerase positive cell lines depleted of FANCD2 and have not been reported in cells from FA
patients. I scored 600 interphase nuclei over 3 independent experiments for the presence of
micronuclei, holes, bridging, irregular multiple lobes and multiple nuclei and found that in cells
treated with FANCD2 siRNA A, 6.3 ± 1.4% of GM639 and 3.0 ± 0.5% of HT1080 telomerase
positive cells were abnormal 5 days post-transfection, while 56.3 ± 6.5% of GM847 and 40.5 ±
7.9% of VA13 FANCD2-depleted ALT cells were abnormal (Figure 2-14B). Fragmented nuclei
of FANCD2-depleted ALT cells are not apoptotic by TUNEL assay, and can incorporate BrdU,
suggesting these cells are not terminally arrested in G1/G0 (data not shown).
SiRNA depletion of TRF2 causes similar types of nuclear abnormalities in GM847 and VA13
ALT cells (Figure 2-13 B), and VA13 ALT cells with uncapped telomeres have previously been
reported to form similar nuclei (Guiducci et al, 2001). This suggests that, that within these cell
lines, telomere dysfunction is one potential cause of this these types of abnormalities. U2OS
ALT cells that are depleted of FANCD2 do not show severe nuclear abnormalities, however this
cell line has wild type p53 and therefore may arrest in G1/G0 in response to telomere
dysfunction. Analogous types of abnormalities are observed in cells with aberrations in
kinetochore components, or mitotic spindle abnormalities in conjunction with a nonfunctional
spindle assembly checkpoint, suggesting that this may be a common phenotype of cells with
mitotic segregation problems (Goshima et al, 2003; Hauf et al, 2003; Taylor and McKeon 1997).
Additionally, I find that FANCD2 depleted GM847 ALT cells show increased frequency of
polyploidy and endoreduplication, abnormalities that are not increased in HT1080 telomerase
positive cells depleted of FANCD2. When endoreduplication is observed in GM847 FANCD2
depleted cells, it affects all chromosomes, suggesting that the entire genome has duplicated
without proceeding through mitosis. Depletion of FANCD2 results in an approximately 5 fold
increase in the frequency of GM847 ALT cells with a greater then 8N chromosome content as
determined by FISH analysis of centromosome numbers in interphase cells. In random siRNA
treated cells analyzed 5 and 8 days post siRNA addition, nuclei with a greater then 8N DNA
78
Figure 2-13. FANCD2 depletion results in severe nuclear abnormalities in GM847 and
VA13 ALT cells A) Examples of nuclear abnormalities frequently observed in FANCD2-depleted ALT cells. Scale bar is
10µm. B) Representative images of nuclei in GM847 and VA13 cells treated with random, FANCD2 or
TRF2 siRNA. Depletion of either FANCD2 or TRF2 results in abnormal nuclei. Random and FANCD2
depleted cells were imaged 5 days post siRNA addition, TRF2 depleted cells were imaged 72 hours post
siRNA addition due extensive cell death past this time point.
A)
B)
79
content occurred at frequencies of 3.5 ± 1.1% and 3.3 ± 1.1%, respectively. This differs from
FANCD2 siRNA treated cells analyzed 5 and 8 days post siRNA addition, that accumulate
nuclei with a greater then 8N DNA content at frequencies of 18.3 ± 2.4% and 19.3 ± 2.5%,
respectively. In HT1080 cells treated with random or FANCD2 siRNA, the frequency of nuclei
with a greater then 8N DNA content was <1% at all time points. Together these data suggest that
ALT cells depleted of FANCD2 face challenges during mitosis that may not occur in FANCD2-
depleted telomerase positive cells, difficulties that may be related to telomere dysfunction.
When more than two centrosomes are present in a cell, establishment of bipolar spindles may be
hindered leading to impaired chromosome segregation and aneuploidy. Cells with more then 2
centrosomes (γ-tubulin) foci are increased 5.1 and 28.1 fold over controls in FANCD2 depleted
VA13 and GM847 cells, respectively (Figure 2-14 A, B). Depletion of FANCD2 in GM639 and
HT1080 telomerase positive cells led to respective 1.6 fold increase and 0.76 fold decrease in the
frequency of FANCD2 depleted cells with supernumerary centrosomes relative to controls
(Figure 2-14 B). Potential causes of supernumerary centrosomes include targeted amplification
following DNA damage, cytokinesis failure or centrosome fragmentation. Given that FANCD2
depletion results in multinuclear cells and increased polyploidy in GM847 ALT cells, failed
cytokinesis may contribute to the observed supernumerary centrosomes. However, as only ∼19%
of FANCD2-depleted GM847 have a greater then 8N DNA content, but ∼47% cells have more
then 2 centrosomes, cytokinesis failure cannot be the sole cause of the increase. Centrosome
amplification has previously been observed in cells with uncapped telomeres (Guiducci et al,
2001) and other forms of genomic DNA damage (Bourke et al, 2007; Saladino et al, 2009),
suggesting that increased levels of DNA damage in FANCD2-depleted ALT cells may contribute
to the observed centrosome amplification.
The DNA repair factor BRCA1 has been localized to centrosomes (Parvin JD, 2009), where it is
thought to play a direct role in the regulation of centrosome stability and duplication. Using
indirect immunofluorescene I investigated whether FANCD2 also localizes to centrosomes.
FANCD2 localization to centrosomes was observed with a rabbit polyclonal antibody to
FANCD2, but not with a murine monoclonal antibody (data not shown). However, the observed
localization of FANCD2 to centrosomes persisted in FANCD2-depleted cells and FANCD2
patient cell lines, suggesting that staining is an artifact of the antibody (data not shown). These
80
abnormal nuclei
>2 centrosomes
results, in conjunction with the largely ALT specific nature of the centrosome amplification,
argue against a direct role for FANCD2 in centrosome stability. Taken together, my results
suggests that the observed centrosome amplification in FANCD2-depleted ALT cells is due to a
combination of the mitotic and cytokenesis failures, as well as targeted amplification of
centrosomes in cells with high levels of DNA damage.
A) GM847 + FANCD2 siRNA
B)
Figure 2-14. FANCD2 depletion increases the frequency of abnormal nuclei and cells with
supernumerary centrosomes in GM847 and VA13 ALT cells, but not in GM639 and
HT1080 telomerase positive cells A) Representative images of GM847 cells treated with FANCD2 siRNA with more then 2 centrosomes.
γ-tubulin (red) was used as a centrosome marker. Scale bar is 10 µm. B) Plot of the number of abnormal
nuclei (purple) and nuclei with more than 2 centrosomes (orange) from populations of 600 randomly
selected asynchronous cells over 3 independent experiments ± the associated standard error.
γ−tubulin
GM639+ GM639+ HT1080+ HT1080+ GM847+ GM847+ VA13+ VA13+ random FANCD2 random FANCD2 random FANCD2 random FANCD2 siRNA siRNA siRNA siRNA siRNA siRNA siRNA siRNA
si
81
When ALT cells are depleted of FANCD2 there is a significant decrease in both short and long-
term viability. GM847 cells and U2OS cells were treated with FANCD2 siRNAs A-D and then
the remaining cell number was determined 5 days post siRNA addition. Similar to GM847 cells,
I find that FANCD2 protein levels in U2OS cells are significantly decreased by FANCD2 siRNA
A-C, but not D (Figure 3-1). Phase contrast images of the cell density 5 days post siRNA are
shown in Figure 2-15. The number of FANCD2 siRNA treated GM847 cells relative to random
siRNA treated cells 5 days post siRNA addition from 3 independent experiments was 19.6 ±
7.7%, 18.3 ± 3.8%, 33.1 ±4.8%, and 111.2 ± 26.2%, for cells treated with FANCD2 siRNA A-D,
respectively. In U2OS cells the number of FANCD2 siRNA treated cells relative to random
siRNA treated cells was 42.2 ± 6.7%, 31.2 ± 6.1%, 49.9 ± 14.5%, and 148 ± 43.5% for cells
treated with FANCD2 siRNAs A-D, respectively.
Figure 2-15. FANCD2 depletion leads to a decrease in cell growth and survival in ALT cells Representative phase contrast images of 847 and U2OS cells treated with random or FANCD2 targeting
siRNAs. Images were taken 5 days post siRNA addition at 5x magnification.
82
To determine the long-term fate of FANCD2 siRNA treated cells, cells treated with FANCD2
siRNA A were replated for colony forming assays 4 days post siRNA transfection. FANCD2
begins to be re-expressed 8-9 days post siRNA addition, and was not added again after the initial
treatment, therefore the ability of cells to recover from damage incurred during their growth over
a one week period with reduced FANCD2 levels was assessed. Colony forming ability was
reduced relative to random siRNA treated cells in GM847, U2OS, and GM637 ALT cells. VA13
and SOAS-2 ALT cells were not included in this analysis because FANCD2-depleted cells failed
to form colonies at any of the cell numbers plated. The colony forming ability of GM847,
GM637, and U2OS FANCD2 depleted cells was determined relative to random siRNA treated
cells over 4 independent experiments, and was 1.5 ± 1.2%, 16.0 ± 17.1%, and 20.8 ± 30.2%,
respectively. Together these results suggest that FANCD2 plays an essential role in promoting
ALT telomere maintenance and short and long term cell viability.
83
2.5 Discussion
In this study I found that FANCD2 and FA core complex proteins FANCA and FANCG
colocalize with telomeric foci in human cells that use the ALT pathway, but not in primary cells
or cells that rely on telomerase for telomere maintenance. In addition to colocalization, FANCD2
coimmunoprecipitates with TRF2 and BLM in ALT cells and FANCD2 depletion leads to an
ALT-specific increase in TIFs. Similar observations of largely ALT-specific roles in telomere
maintenance have also been made for MUS81, topoisomerase IIIα and BLM (Zeng and Yang,
2009; Temime-Smaali et al, 2008; Bhattacharyya et al, 2009). One hypothesis for the unique
requirement of certain DNA repair factors in ALT, is that they are involved in the response to
dysfunctional telomeric DNA, which is present at a higher level in ALT then telomerase positive
cells. However, my data shows that most FANCD2 localizes to telomeric foci that have not
activated a DNA damage response. Additionally, I have examined late passage primary cells
with a high frequency of short and dysfunctional telomeres do not see frequent FANCD2
colocalization with telomeric foci (data not shown). This suggests that there is something unique
to the ALT situation that the FA pathway is responding to.
FANCA and FANCG primarily colocalize with FANCD2 within larger telomeric foci, which
most likely represent APBs. FA core complex proteins are typically difficult to visualize by IF,
presumably because they associate with DNA damage at very low abundance or transiently. A
recent study on telomeric DNA in ALT cells shows the existence of the following species of
telomeric DNA: gapped double-strand, single-strand, circular, and complex branched structures
with a mixture of single- and double-strand DNA (Nabetani and Ishikawa, 2009). Fractionation
of nuclear lysate shows substantial fractions of these species exist as extra-chromosomal DNAs,
which as discussed below, appear to accumulate within APBs (Nabetani and Ishikawa, 2009).
The FA pathway can be activated by a variety of DNA substrates with branched structures and
exposed ends (Sobeck et al, 2007) and purified human FANCD2 has highest in vitro binding
affinity to ssDNA, followed by splayed arms, Holliday junction structures, and dsDNA (Roques
et al, 2009). Taken together, these observations suggest that the ability to clearly visualize FA
core complex proteins in ALT cells may be due to unusually high local concentrations of
substrates for FA core complex binding found within APBs. Stoichiometric differences in the
84
local concentration of DNA structures that recruit FANCD2 may also explain why FANCA
knockdown efficiently reduces non-telomeric FANCD2 foci, but FANCD2 foci colocalized with
telomeric DNA remain weakly visible. During replication FANCD2 may accumulate at single
spatially distinct lesions, whereas APBs may contain high numbers of DNA molecules that bind
FANCD2 within a single focus.
Large APBs found in interphase ALT cells are often postulated to primarily contain multiple
ALT telomeres undergoing replication and recombination reactions, however direct evidence of
this is lacking. Recently, Draskovic and colleagues expressed a modified form of Herpes simplex
virus protein ICP0 to generate abnormally large APBs, and found that subtelomeric sequences
frequently associate with the modified APBs (Draskovic et al, 2009). Whether these
subtelomeric sequences can accumulate in the ECTR at levels detectable by FISH was not tested,
raising questions over the suitability of these sequences as a marker for telomeres. Additionally,
immunofluorescent analysis of the enlarged APBs suggested that the bodies are solid protein
structures, with telomeric DNA clustered exclusively around the periphery of the body
(Draskovic et al, 2009). APBs with a solid protein core were never observed in our study of
endogenous APBs, and I hypothesize that expression of the modified ICP0 protein significantly
disrupted normal APB structure.
Another recent study by Jegou and colleagues, showed that lacO sequences integrated near the
telomeres of chromosomes associates with PML bodies, however results must be cautiously
interpreted, as the distance between the integration site and telomere was not determined, and it
is not clear whether this observed colocalization with PML is affected by local changes in
chromatin structure caused by the integration of 0.7-3.7 Mb arrays of lac O repeats (Jegou et al,
2009). Immunofluorescent analysis of lac O repeats argues for a model where PML frequently
forms a large cap-like structure that engulfs telomeric chromosomal DNA (Jegou et al, 2009).
Electron spectroscopic imaging of endogenous interphase APBs argues against this idea, because
although APBs contain nucleic acid in the interior of the body, the structure of the material and
phosphorous signal intensity are inconsistent with chromatin. Our data suggests that APBs
primarily contain non-nucleosomal ECTR DNA.
85
It remains possible that nucleic acid within these APBs represents ALT telomeres which have an
abnormal organization that does not resemble normal chromatin, however γH2AX has been
detected at ALT telomeres during mitosis, arguing that, similar to telomeres in telomerase
positive and primary cells, ALT telomeres are nucleosomal structures (Cesare et al, 2009).
Additionally, in FANCD2-depleted ALT cells, APBs with chromatin-like masses invading the
bodies were observed. If these masses represent telomeres, then it would suggest that interactions
are either FANCD2-depletion specific or are normally infrequent or transient in nature in wild-
type interphase cells, and therefore the study of APBs ALT cells is largely the examination of the
ECTR DNA and its associated proteins.
The idea that APBs are primarily composed of ECTR is supported by studies showing that
treatment of ALT cells with DNA-damaging agents results in concomitant increases in ECTR
DNA and APBs (Fasching et al, 2007), and during metaphase most APBs present within cells are
clearly extra-chromosomal (Nabetani et al, 2004). Additionally non-ALT cells with elongated
telomeres can accumulate ECTR DNA and APBs, but do not show an increase in TIFs or
recombination events involving telomeres (Pickett et al., 2009), and a subclone of the VA13
ALT line was identified which no longer has ECTR DNA or APBs, but continues to have an
elevated frequency of recombination among telomeres (Cerone et al., 2005). The above studies
suggest that while there is a close connection between the generation of ECTR DNA and the
formation of APBs, APBs are not essential for most recombination reactions involving
telomeres. Strand specific FISH suggests that many recombination events between telomeres in
ALT cells may be reciprocal exchanges between sister chromatids, a form of recombination
which can likely occur outside of large ALT-specific subnuclear domains (Bailey et al, 2004;
Londoño-Vallejo et al, 2004).
While APBs may not be required for telomeric recombination in ALT cells, APBs do appear to
play an important role in ALT, as they are present to varying degrees in almost all ALT cells,
and they arise at the same time as activation of the ALT pathway (Yeager et al, 1999). Data
presented in this study supports the hypothesis proposed by Fasching and colleagues (2007), that
APBs are primarily involved in the sequestering of ECTR DNA. ECTR DNA generated in ALT
cells has been shown to have frequent internal ssDNA regions and exposed ends (Nabetani and
Ishikawa, 2009), substrates that have the potential to activate DNA damage response pathways
86
and cell cycle checkpoints. Although ALT appears to be exclusively active in cancer cells and
immortalized cell lines which frequently have mutations in the p53 pathway, experiments in
ALT cells exposed to exogenous forms of DNA damage have shown that DNA damage
responses and cell cycle checkpoints remain functional (Wang et al, 2002; Demuth et al, 2008;
Cliby et al, 1998). This raises the question of how ALT cells manage to cycle in the presence of
endogenous DNA substrates, such as ECTR DNA, that would be predicted to elicit a DNA
damage and checkpoint response. One possibility would be that ECTR DNA is sequestered in
APBs away from components of the DNA damage sensors to avoid activation or the DNA
damage response. However this model does not fit with the observation that APBs are present in
viable cells that are cycling, yet APBs can associate with DNA damage sensors and DNA repair
factors that have been phosphorylated by ATM and/or ATR (Grobelny et al, 2000; Nabetani et
al, 2004; Wu et al, 2003; Stagno D'Alcontres et al, 2007). The presence of proteins
phosphorylated by ATM and ATR also argues that components of the shelterin telomere binding
complex localized to APBs cannot fully suppress activation of the DNA damage response
pathway by ECTR DNA (Stagno D'Alcontres et al, 2007), even though they substantially
attenuate this response at telomeres and other genomic loci following ionizing radiation in
telomerase positive and primary cells (Denchi and de Lange, 2007; Karlseder et al, 2004;
Bradshaw et al, 2005).
I instead hypothesize that APBs may serve as essential sites where DNA that has activated a
damage response, and associated DNA repair factors, are targeted. The local environment within
and around APBs may be non-conducive to propagation of the DNA damage/checkpoint
response, or alternately may serve as a region where the DNA damage response is actively
down-regulated. Sequestering ECTR DNA within APBs may prevent cells from fully activating
cell cycle checkpoints, or lead to the eventual down-regulation of any activated checkpoints. A
recent analysis of γH2AX staining patterns in ALT cells arrested in metaphase with colcemid
demonstrates that cells can enter mitosis with telomeres that have activated a DNA damage
response, and that some of these telomeres are located in close proximity to APB-like structures
(Cesare et al, 2009). It is possible that dysfunctional telomeres must associate with APBs to
satisfy the G2/M checkpoint, allowing cells to proceed into mitosis, than move away from the
bodies during segregation. Alternately, dysfunctional telomeres closely associated with APB-like
87
structures may represent a later form of an interphase phenomenon that is occasionally observed
using electron microscopy in FANCD2-depleted cells.
Transient depletion of FANCD2 results in the concomitant increase in telomeric DNA that has
activated a damage response, and the detection of APBs with chromatin masses tightly
associating with the bodies. The chromatin structures may represent dysfunctional telomeres,
which may associate with APBs, and interact with ECTR DNA or additional telomeres, if
present at the same time at the bodies. If difficulties arise in resolving interactions prior to
entering mitosis, telomeres may remain closely associated with APBs during mitosis, similar to
what was observed by Cesare and colleagues. Whether dysfunctional telomeres can be directly
targeted to APBs, or interact more frequently as an indirect result of the increased mobility of
dysfunctional telomeres remains to be resolved (Dimitrova et al, 2008).
The molecular details of the role of FANCD2 within ALT remains under investigation, however
my study of the requirements for FANCD2 localization to telomeric foci and APBs suggest that
FANCD2 is not just participating in the response to stalled/collapsed replication forks or
interstrand crosslinks, as the localization of FANCD2 is largely ATR independent. Additionally,
localization is independent of ATM, and most FANCD2 localizes to telomeric foci that do not
contain 53BP1, suggesting that FANCD2 is not simply localizing to telomeric DNA ends that
have activated a DNA damage response. Interestingly, I observe a unique requirement for BLM
expression in order for FANCD2 to localize to ALT telomeric foci. Although there is a growing
body of evidence suggesting that FANCD2 and BLM participate in common pathways, (Chan et
al, 2009; Naim and Rosselli, 2009; Pichierri et al, 2004; Hemphill et al, 2009) ALT appears to be
the first situation where BLM is acting upstream of FANCD2.
BLM may be directly involved in promoting FANCD2 localization to telomeric DNA, or
alternately may be required to generate a DNA substrate that FANCD2 subsequently then binds
to. Telomeres present several unique challenges during DNA repair and replication because of
the potential tendency of the TTAGGG repeat to form secondary structures and G-quadruplexes,
structures that BLM may be required to resolve (Mohaghegh et al, 2001). Supporting the idea
that BLM plays key roles in ALT, knockdown of BLM leads to a rapid, ALT-specific decrease in
average telomere length (Bhattacharyya et al, 2009), and when I deplete BLM, I no longer
observe large APBs with high amounts of TRF1, TRF2, and telomeric DNA, suggesting that
88
there may be a decrease in production of ECTR DNA (data not shown). Additional experiments
presented in the following chapter suggest that BLM acts both up and downstream of FANCD2,
and that a key role of FANCD2 within ALT is in the regulation of BLM-dependent telomeric
recombination and ECTR DNA synthesis.
FANCD2 also appears to be involved in preventing ALT telomeric DNA from activating a DNA
damage response, as FANCD2-depletion leads to an ALT specific increase in TIFs. One cause of
TIFs could be an increase in collapsed replication forks, however this seems unlikely because
most FANCD2 does not appear to be responding to stalled/collapsed forks, as FANCD2
recruitment to telomeric foci is largely ATR independent. However, FANCD2 may have
additional, currently unknown functions during replication, which are required to prevent
activation of a DNA damage response. Supporting this idea, spontaneous FANCD2 foci that
normally form during S phase are also independent of both ATM and ATR.
Critically short telomeres can activate a DNA damage response (Zou et al, 2004), however my
analysis of telomere free ends demonstrates that FANCD2 depletion does not alter the frequency
of chromosome ends without a detectable telomere signal. It should be noted that the level of
telomere free ends I find differs markedly from recent findings of Fan et al., (2009) who reported
that 45% of telomeres were undetectable in FANCD2 depleted U2OS cells. The reason for this
difference is unknown, however may be related to technical differences in FISH sensitivities and
the decision of Fan et al. to vary camera exposure times between experimental conditions.
Supporting the idea that my FISH experiments were more sensitive then those by Fan and
colleagues, the baseline level of telomere free ends reported by Fan and colleges was very high
(∼18% in U2OS cells and ∼35% in 847 cells), whereas I find that only ∼2% of telomeres are
undetectable under all conditions, a figure more consistent with previously reported values of
∼5% of GM847 chromosomes having a telomere free end (Zeng et al, 2009; Perrem et al, 2001).
Additionally, although Fan et al, observed a dramatic level of signal free ends in FANCD2
depleted cells via microscopic detection of telomeric signals, the proportion of the population
with lower amounts of total telomeric DNA did not increase in flow cytometric measurements,
indicating a technical problem with microscopic FISH.
89
Evidence suggests that factors in addition to telomere length may influence the ability of
telomeres to suppress the DNA damage response. ALT metaphase chromosomes with strong
telomeric signals can activate a DNA damage response (Cesare et al, 2009). Proper capping of
telomeres is also likely influenced by formation of secondary DNA structures, as well as
sufficient quantities of components of the shelterin telomere binding complex. Both t-loops and
components of shelterin are present in ALT and telomerase positive cells (Cesare and Griffith,
2004) making it difficult to understand why FANCD2-depletion would result in an ALT-specific
capping problem. Additionally, proteins outside of shelterin that have been implication in
capping, such as the recombinational protein RAD51D, localizes to telomeres in telomerase
positive and ALT cells, and cause telomere abnormalities in both cell types when depleted
(Tarsounas et al, 2004).
One hypothesis that may explain the ALT-specific FANCD2 dependent increase in TIFs relates
to the presence of single-strand gapped regions within ALT telomeric. ALT telomeric DNA
appears to differ from non-ALT telomeric DNA, both at telomeres and extra-chromosomally, in
that there is an elevated frequency of internal ssDNA gapped regions (Nabetani and Ishikawa,
2009). FANCD2 has recently been demonstrated to bind preferentially to ssDNA in vitro,
although its function at ssDNA remains unclear (Roques et al, 2009). The ability of FANCD2 to
suppress TIFs may relate to the ability of FANCD2 to bind to gapped regions and regulate the
subsequent recruitment of down stream factors such as ATR/ATRIP, BRCA2, and RAD51.
When FANCD2 is depleted, ssDNA gaps present in ALT telomeric DNA may be more likely to
activate a DNA damage response, leading to an increase in TIFs. An alternate hypothesis related
to insufficient amounts of telomere binding proteins due to amplification of ECTR DNA in
FANCD2-depleted cells will be discussed in the following chapter.
The increase in TIFs in FANCD2-depleted ALT cells may be a contributing factor to the nuclear
abnormalities and centrosome amplification observed. Introduction of double-strand breaks
through ionizing radiation, prolonged replication stress, or topoisomerase II inhibitors leads to
targeted centrosome amplification during an extended G2 phase in human cell lines (Bourke et
al, 2007; Saladino et al, 2009). This suggests that centrosome amplification represents an
apoptosis-independent mechanism of targeting damaged cells for death, because most cells will
undergo an abnormal mitotic division and die during the subsequent cell cycle. Additionally,
90
when telomeres are uncapped in VA13 ALT cells, supernumerary centrosomes are observed
(Guiducci et al, 2001), demonstrating that TIFs are another form of damage that cause
centrosome amplification.
Data presented in this study suggests that the role of the FA pathway in telomere maintenance
may be largely limited to cells that utilize the ALT pathway. As ALT has only been shown to be
active in cancer cells and immortalized cell lines, it is unlikely that telomere problems are a
driving force in the pathogenesis of FA. However, because the FA pathway appears to be
essential for ALT cell viability, treatment of ALT positive cancers with small molecule inhibitors
of the FA pathway may prove to be a viable therapeutic approach. Although the overall fraction
of tumors that utilize the ALT pathway appears to be only ∼10%, ALT is activated more
frequently in tumors of neuroepithelial and mesenchymal origin suggesting that for some cancers
targeting of the FA pathway may be a viable therapeutic approach (Henson et al, 2005; Costa et
al, 2006). ALT also may serve as a useful tool for deciphering the molecular function of the FA
pathway in recombination and repair, as ALT cells have an unusually high number of
endogenous substrates that activate and recruit pathway components.
91
2.6 References
Alter BP, Baerlocher GM, Savage SA, Chanock SJ, Weksler BB, Willner JP, Peters JA, Giri N, Lansdorp PM. (2007). Very short telomere length by flow fluorescence in situ hybridization identifies patients with dyskeratosis congenita. Blood 110:1439-47.
Andreassen PR, D'Andrea AD, Taniguchi T (2004). ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev. 15: 1958-63.
Auerbach AD (2009). Fanconi anemia and its diagnosis. Mutat Res. 668:4-10.
Bailey SM, Brenneman MA, Goodwin EH (2004). Frequent recombination in telomeric DNA may extend the proliferative life of telomerase-negative cells. Nucleic Acids Res. 32:3743-51.
Bakker ST, van de Vrugt HJ, Rooimans MA, Oostra AB, Steltenpool J, Delzenne-Goette E, van der Wal A, van der Valk M, Joenje H, te Riele H, de Winter JP (2009). Fancm-deficient mice reveal unique features of Fanconi anemia complementation group M. Hum Mol Genet. 18:3484-95.
Bazett-Jones DP, Li R, Fussner E, Nisman R, Dehghani H. Elucidating chromatin and nuclear domain architecture with electron spectroscopic imaging (2008). Chromosome 16:397-412.
Bhattacharyya S, Keirsey J, Russell B, Kavecansky J, Lillard-Wetherell K, Tahmaseb K, Turchi JJ, Groden J (2009). Telomerase-associated protein 1, HSP90, and topoisomerase IIalpha associate directly with the BLM helicase in immortalized cells using ALT and modulate its helicase activity using telomeric DNA substrates. J Biol Chem. 284:14966-77.
Boisvert FM, Hendzel MJ, Bazett-Jones DP (2000). Promyelocytic leukemia (PML) nuclear bodies are protein structures that do not accumulate RNA. J Cell Biol. 148:283-92.
Bourke E, Dodson H, Merdes A, Cuffe L, Zachos G, Walker M, Gillespie D, Morrison CG (2007). DNA damage induces Chk1-dependent centrosome amplification. EMBO Rep. 8:603-9.
92
Bradshaw PS, Stavropoulos DJ, Meyn MS (2004). Human telomeric protein TRF2 associates with genomic double-strand breaks as an early response to DNA damage. Nat Genet. 37:193-7.
Bruun D, Folias A, Akkari Y, Cox Y, Olson S, Moses R (2003). siRNA depletion of BRCA1, but not BRCA2, causes increased genome instability in Fanconi anemia cells. DNA Repair (Amst). 2:1007-13.
Bryan TM, Englezou A, Gupta J, Bacchetti S, Reddel RR (1995). Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J. 14:4240-8.
Bryan TM, Englezou A, Dalla-Pozza L, Dunham MA, Reddel RR (1997). Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat Med. 3(11):1271-4.
Calado RT, Pintão MC, Rocha V, Falcão RP, Bitencourt MA, Silva WA Jr, Gluckman E, Pasquini R, Zago MA (2004). Lack of mutations in the human telomerase RNA component (hTERC) gene in Fanconi's anemia. Haematologica 89:1012-3.
Callén E, Samper E, Ramírez MJ, Creus A, Marcos R, Ortega JJ, Olivé T, Badell I, Blasco MA, Surrallés J (2002). Breaks at telomeres and TRF2-independent end fusions in Fanconi anemia. Hum Mol Genet. 2002 11:439-44.
Cerone MA, Autexier C, Londoño-Vallejo JA, Bacchetti S (2005). A human cell line that maintains telomeres in the absence of telomerase and of key markers of ALT. Oncogene. 24:7893-901.
Cesare AJ, Griffith JD (2004). Telomeric DNA in ALT cells is characterized by free telomeric circles and heterogeneous t-loops. Mol Cell Biol. 24:9948-57.
Cesare AJ, Kaul Z, Cohen SB, Napier CE, Pickett HA, Neumann AA, Reddel RR (2009). Spontaneous occurrence of telomeric DNA damage response in the absence of chromosome fusions. Nat Struct Mol Biol. 16:1244-51.
Chan KL, Palmai-Pallag T, Ying S, Hickson ID (2009). Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol. 11:753-60.
93
Chirnomas D, Taniguchi T, de la Vega M, Vaidya AP, Vasserman M, Hartman AR, Kennedy R, Foster R, Mahoney J, Seiden MV, D'Andrea AD (2006). Chemosensitization to cisplatin by inhibitors of the Fanconi anemia/BRCA pathway. Mol Cancer Ther. 5:952-61.
Cliby WA, Roberts CJ, Cimprich KA, Stringer CM, Lamb JR, Schreiber SL, Friend SH (1998). Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. EMBO J. 17:159-69.
Collis SJ, Barber LJ, Clark AJ, Martin JS, Ward JD, Boulton SJ (2007). HCLK2 is essential for the mammalian S-phase checkpoint and impacts on Chk1 stability. Nat Cell Biol. 9:391-401.
Costa A, Daidone MG, Daprai L, Villa R, Cantù S, Pilotti S, Mariani L, Gronchi A, Henson JD, Reddel RR, Zaffaroni N (2006). Telomere maintenance mechanisms in liposarcomas: association with histologic subtypes and disease progression. Cancer Res. 66:8918-24.
d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP (2003). A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003 426:194-8.
de Lange T (2005). Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 19:2100-10.
Demuth I, Bradshaw PS, Lindner A, Anders M, Heinrich S, Kallenbach J, Schmelz K, Digweed M, Meyn MS, Concannon P (2008). Endogenous hSNM1B/Apollo interacts with TRF2 and stimulates ATM in response to ionizing radiation. DNA Repair. 7:1192-201.
Denchi EL, de Lange T (2007). Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 448(7157):1068-71.
Dimitrova N, Chen YC, Spector DL, de Lange T (2008). 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature. 456:524-8.
Draskovic I, Arnoult N, Steiner V, Bacchetti S, Lomonte P, Londoño-Vallejo A (2009). Probing PML body function in ALT cells reveals spatiotemporal requirements for telomere recombination. Proc Natl Acad Sci U S A. 106: 15726-31.
Dunham MA, Neumann AA, Fasching CL, Reddel RR (2000). Telomere maintenance by recombination in human cells. Nat Genet. 26:447-50.
94
Eller MS, Liao X, Liu S, Hanna K, Bäckvall H, Opresko PL, Bohr VA, Gilchrest BA (2006). A role for WRN in telomere-based DNA damage responses. Proc Natl Acad Sci U S A. 103:15073-8.
Fan Q, Zhang F, Barrett B, Ren K, Andreassen PR (2009). A role for monoubiquitinated FANCD2 at telomeres in ALT cells. Nucleic Acids Res. 37:1740-54.
Fasching CL, Neumann AA, Muntoni A, Yeager TR, Reddel RR (2007). DNA damage induces alternative lengthening of telomeres (ALT) associated promyelocytic leukemia bodies that preferentially associate with linear telomeric DNA. Cancer Res. 67:7072-7.
Franco S, van de Vrugt HJ, Fernández P, Aracil M, Arwert F, Blasco MA (2004). Telomere dynamics in Fancg-deficient mouse and human cell. Blood 104:3927-35.
Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D'Andrea AD (2001). Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell 7:249-62.
Gilson E, Géli V (2007). How telomeres are replicated. Nat Rev Mol Cell Biol. 8:825-38.
Goshima G, Kiyomitsu T, Yoda K, Yanagida M (2003). Human centromere chromatin protein hMis12, essential for equal segregation, is independent of CENP-A loading pathway. J Cell Biol. 160:25-39.
Griffith JD, Comeau L, Rosenfield S, Stansel RM, Bianchi A, Moss H, de Lange T (1999). Mammalian telomeres end in a large duplex loop. Cell 97:503-14.
Grobelny JV, Godwin AK, Broccoli D (2000). ALT-associated PML bodies are present in viable cells and are enriched in cells in the G(2)/M phase of the cell cycle. J Cell Sci. 113:4577-85.
Guervilly JH, Macé-Aimé G, Rosselli F (2008). Loss of CHK1 function impedes DNA damage-induced FANCD2 monoubiquitination but normalizes the abnormal G2 arrest in Fanconi anemia. Hum Mol Genet. 17:679-89.
Guiducci C, Cerone MA, Bacchetti S (2001). Expression of mutant telomerase in immortal telomerase-negative human cells results in cell cycle deregulation, nuclear and chromosomal abnormalities and rapid loss of viability. Oncogene 20:714-25.
95
Hanson H, Mathew CG, Docherty Z, Mackie Ogilvie C (2001). Telomere shortening in Fanconi anaemia demonstrated by a direct FISH approach. Cytogenet Cell Genet 93:203-6.
Hauf S, Cole RW, LaTerra S, Zimmer C, Schnapp G, Walter R, Heckel A, van Meel J, Rieder CL, Peters JM (2003). The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol. 161:281-94.
Hemphill AW, Akkari Y, Newell AH, Schultz RA, Grompe M, North PS, Hickson ID, Jakobs PM, Rennie S, Pauw D, Hejna J, Olson SB, Moses RE (2009). Topo IIIalpha and BLM act within the Fanconi anemia pathway in response to DNA-crosslinking agents. Cytogenet Genome Res. 125:165-75.
Henson JD, Hannay JA, McCarthy SW, Royds JA, Yeager TR, Robinson RA, Wharton SB, Jellinek DA, Arbuckle SM, Yoo J, Robinson BG, Learoyd DL, Stalley PD, Bonar SF, Yu D, Pollock RE, Reddel RR (2005). A robust assay for alternative lengthening of telomeres in tumors shows the significance of alternative lengthening of telomeres in sarcomas and astrocytomas. Clin Cancer Res. 11:217-25.
Ho GP, Margossian S, Taniguchi T, D'Andrea AD (2006). Phosphorylation of FANCD2 on two novel sites is required for mitomycin C resistance. Mol Cell Biol. 26:7005-15.
Houghtaling S, Timmers C, Noll M, Finegold MJ, Jones SN, Meyn MS, Grompe M (2003). Epithelial cancer in Fanconi anemia complementation group D2 (Fancd2) knockout mice. Genes Dev.17:2021-35.
Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De Die-Smulders C, Persky N, Grompe M, Joenje H, Pals G, Ikeda H, Fox EA, D'Andrea AD (2002). Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 297:606-9.
Howlett NG, Taniguchi T, Durkin SG, D'Andrea AD, Glover TW (2005). The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Hum Mol Genet. 14:693-701.
Jegou T, Chung I, Heuvelman G, Wachsmuth M, Görisch SM, Greulich-Bode KM, Boukamp P, Lichter P, Rippe K (2009). Dynamics of telomeres and promyelocytic leukemia nuclear bodies in a telomerase-negative human cell line. Mol Biol Cell. 20:2070-82.
96
Karlseder J, Hoke K, Mirzoeva OK, Bakkenist C, Kastan MB, Petrini JH, de Lange T (2004). The telomeric protein TRF2 binds the ATM kinase and can inhibit the ATM-dependent DNA damage response. PLoS Biol. 2: 1150-56.
Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW (1994). Specific association of human telomerase activity with immortal cells and cancer. Science. 266:2011-5.
Kutler DI, Singh B, Satagopan J, Batish SD, Berwick M, Giampietro PF, Hanenberg H, Auerbach AD (2003). A 20-year perspective on the International Fanconi Anemia Registry (IFAR). Blood 101:1249-56.
Leteurtre F, Li X, Guardiola P, Le Roux G, Sergère JC, Richard P, Carosella ED, Gluckman E (1999). Accelerated telomere shortening and telomerase activation in Fanconi's anaemia. Br J Haematol 105:883-93.
Lillard-Wetherell K, Machwe A, Langland GT, Combs KA, Behbehani GK, Schonberg SA, German J, Turchi JJ, Orren DK, Groden J (2004). Association and regulation of the BLM helicase by the telomere proteins TRF1 and TRF2. Hum Mol Genet. 13:1919-32.
Londoño-Vallejo JA, Der-Sarkissian H, Cazes L, Bacchetti S, Reddel RR (2004). Alternative lengthening of telomeres is characterized by high rates of telomeric exchange. Cancer Res. 64:2324-7.
Longhese MP (2008). DNA damage response at functional and dysfunctional telomeres. Genes Dev. 22: 125–140.
Medhurst AL, Laghmani el H, Steltenpool J, Ferrer M, Fontaine C, de Groot J, Rooimans MA, Scheper RJ, Meetei AR, Wang W, Joenje H, de Winter JP (2006). Evidence for subcomplexes in the Fanconi anemia pathway. Blood 108:2072-80.
Mohaghegh P, Karow JK, Brosh Jr RM Jr, Bohr VA, Hickson ID (2001). The Bloom's and Werner's syndrome proteins are DNA structure-specific helicases. Nucleic Acids Res. 29: 2843-9.
Muntoni A, Neumann AA, Hills M, Reddel RR (2009). Telomere elongation involves intra-molecular DNA replication in cells utilizing alternative lengthening of telomeres. Hum Mol Genet. 18:1017-27.
97
Nabetani A, Yokoyama O, Ishikawa F (2004). Localization of hRad9, hHus1, hRad1, and hRad17 and caffeine-sensitive DNA replication at the alternative lengthening of telomeres-associated promyelocytic leukemia body. J Biol Chem. 279:25849-57.
Nabetani A, Ishikawa F (2009). Unusual telomeric DNAs in human telomerase-negative immortalized cells. Mol Cell Biol. 29:703-13.
Naim V, Rosselli F (2009). The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat Cell Biol. 11:761-8.
Nakanishi K, Yang YG, Pierce AJ, Taniguchi T, Digweed M, D'Andrea AD, Wang ZQ, Jasin M (2005). Human Fanconi anemia monoubiquitination pathway promotes homologous DNA repair. Proc Natl Acad Sci U S A. 102:1110-5.
Park WH, Margossian S, Horwitz AA, Simons AM, D'Andrea AD, Parvin JD (2005). Direct DNA binding activity of the Fanconi anemia D2 protein. J Biol Chem. 280:23593-8.
Parvin JD (2009). The BRCA1-dependent ubiquitin ligase, gamma-tubulin, and centrosomes. Environ Mol Mutagen. 50:649-53.
Perrem K, Colgin LM, Neumann AA, Yeager TR, Reddel RR (2001). Coexistence of alternative lengthening of telomeres and telomerase in hTERT-transfected GM847 cells. Mol Cell Biol. 21:3862-75.
Pichierri P, Franchitto A, Rosselli F (2004). BLM and the FANC proteins collaborate in a common pathway in response to stalled replication forks. EMBO J. 23:3154-63.
Pickett HA, Cesare AJ, Johnston RL, Neumann AA, Reddel RR (2009). Control of telomere length by a trimming mechanism that involves generation of t-circles. EMBO J. 28:799-809.
Pisano S, Galati A, Cacchione S (2008). Telomeric nucleosomes: forgotten players at chromosome ends. Cell Mol Life Sci. 65:3553-63.
Poulet A, Buisson R, Faivre-Moskalenko C, Koelblen M, Amiard S, Montel F, Cuesta-Lopez S, Bornet O, Guerlesquin F, Godet T, Moukhtar J, Argoul F, Déclais AC, Lilley DM, Ip SC, West SC, Gilson E, Giraud-Panis MJ (2009). TRF2 promotes, remodels and protects telomeric Holliday junctions. EMBO J. 28:641-51.
98
Reid S, Schindler D, Hanenberg H, Barker K, Hanks S, Kalb R, Neveling K, Kelly P, Seal S, Freund M, Wurm M, Batish SD, Lach FP, Yetgin S, Neitzel H, Ariffin H, Tischkowitz M, Mathew CG, Auerbach AD, Rahman N (2007). Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet. 39:162-4.
Roques C, Coulombe Y, Delannoy M, Vignard J, Grossi S, Brodeur I, Rodrigue A, Gautier J, Stasiak AZ, Stasiak A, Constantinou A, Masson JY (2009). MRE11-RAD50-NBS1 is a critical regulator of FANCD2 stability and function during DNA double-strand break repair. EMBO J. 28:2400-13.
Saladino C, Bourke E, Conroy PC, Morrison CG (2009). Centriole separation in DNA damage-induced centrosome amplification. Environ Mol Mutagen. 50:725-32.
Schroeder TM, Kurth R (1971). Spontaneous chromosomal breakage and high incidence of leukemia in inherited disease. Blood. 37:96-112.
Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER 3rd, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D'Andrea AD, Elledge SJ (2007). Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 129:289-301.
Sobeck A, Stone S, Hoatlin ME (2007). DNA structure-induced recruitment and activation of the Fanconi anemia pathway protein FANCD2. Mol Cell Biol. 27:4283-92.
Spardy N, Duensing A, Hoskins EE, Wells SI, Duensing S (2008). HPV-16 E7 reveals a link between DNA replication stress, fanconi anemia D2 protein, and alternative lengthening of telomere-associated promyelocytic leukemia bodies. Cancer Res. 68:9954-63.
Stagno D'Alcontres M, Mendez-Bermudez A, Foxon JL, Royle NJ, Salomoni P (2007). Lack of TRF2 in ALT cells causes PML-dependent p53 activation and loss of telomeric DNA. J Cell Biol. 179:855-67.
Stavropoulos DJ, Bradshaw PS, Li X, Pasic I, Truong K, Ikura M, Ungrin M, Meyn MS (2002). The Bloom syndrome helicase BLM interacts with TRF2 in ALT cells and promotes telomeric DNA synthesis. Hum Mol Genet. 11:3135-44.
99
Tabori U, Vukovic B, Zielenska M, Hawkins C, Braude I, Rutka J, Bouffet E, Squire J, Malkin D (2006). The role of telomere maintenance in the spontaneous growth arrest of pediatric low-grade gliomas. Neoplasia. 8:136-42.
Takai H, Smogorzewska A, de Lange T (2003). DNA damage foci at dysfunctional telomeres. Curr Biol. 2003 13:1549-56.
Tarsounas M, Muñoz P, Claas A, Smiraldo PG, Pittman DL, Blasco MA, West SC (2004). Telomere maintenance requires the RAD51D recombination/repair protein. Cell. 117:337-47.
Taylor SS, McKeon F (1997). Kinetochore localization of murine Bub1 is required for normal mitotic timing and checkpoint response to spindle damage. Cell. 89:727-35.
Temime-Smaali N, Guittat L, Wenner T, Bayart E, Douarre C, Gomez D, Giraud-Panis MJ, Londono-Vallejo A, Gilson E, Amor-Guéret M, Riou JF (2008). Topoisomerase IIIalpha is required for normal proliferation and telomere stability in alternative lengthening of telomeres. EMBO J. 27:1513-24.
Wang B, Matsuoka S, Carpenter PB, Elledge SJ (2002). 53BP1, a mediator of the DNA damage checkpoint. Science. 298:1435-8.
Wang RC, Smogorzewska A, de Lange T (2004). Homologous recombination generates T-loop-sized deletions at human telomeres. Cell. 2004 119:355-68.
Weksberg R, Buchwald M, Sargent P, Thompson MW, Siminovitch L (1979). Specific cellular defects in patients with Fanconi anemia. J Cell Physiol. 101:311-23.
Wong JC, Alon N, Mckerlie C, Huang JR, Meyn MS, Buchwald M (2003). Targeted disruption of exons 1 to 6 of the Fanconi Anemia group A gene leads to growth retardation, strain-specific microphthalmia, meiotic defects and primordial germ cell hypoplasia. Hum Mol Genet. 12:2063-76.
Wu G, Jiang X, Lee WH, Chen PL (2003). Assembly of functional ALT-associated promyelocytic leukemia bodies requires Nijmegen Breakage Syndrome 1. Cancer Res. 63:2589-95.
100
Yeager TR, Neumann AA, Englezou A, Huschtscha LI, Noble JR, Reddel RR (1999). Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res. 59:4175-9.
Zeng S, Xiang T, Pandita TK, Gonzalez-Suarez I, Gonzalo S, Harris CC, Yang Q (2009). Telomere recombination requires the MUS81 endonuclease. Nat Cell Biol. 11:616-23.
Zeng S, Yang Q (2009). The MUS81 endonuclease is essential for telomerase negative cell proliferation. Cell Cycle. 8:2157-60.
Zhu W, Dutta A (2006). An ATR- and BRCA1-mediated Fanconi anemia pathway is required for activating the G2/M checkpoint and DNA damage repair upon rereplication. Mol Cell Biol. 26:4601-11.
Zijlmans JM, Martens UM, Poon SS, Raap AK, Tanke HJ, Ward RK, Lansdorp PM (1997). Telomeres in the mouse have large inter-chromosomal variations in the number of T2AG3 repeats. Proc Natl Acad Sci U S A. 94:7423-8.
Zou Y, Sfeir A, Gryaznov SM, Shay JW, Wright WE (2004). Does a sentinel or a subset of short telomeres determine replicative senescence? Mol Biol Cell. 15(8):3709-18.
101
Chapter 3
3 Fanconi Anaemia Protein D2 Limits BLM-Dependent, RAD51-Independent Telomeric Recombination and DNA Synthesis in ALT-Immortalized Human Cells
3.1 Abstract
The alternative lengthening of telomeres (ALT) pathway is a recombinational telomere
maintenance pathway active in a fraction of immortalized cell lines and cancer cells. BLM, the
Bloom syndrome helicase, plays an essential role in ALT, as overexpression of BLM results in
rapid synthesis of telomeric DNA, and depletion of BLM results in telomere shortening
(Stavropoulos et al., 2002; Bhattacharyya et al., 2009). BLM associates with multiple
components of the Fanconi Anaemia (FA) pathway, (Meetei et al, 2003; Pichierri et al, 2004;
Deans and West, 2009), a rare inherited syndrome characterized by genomic instability, bone
marrow failure, and cancer predisposition. I have previously shown that both FA core complex
components and FANCD2 localize to telomeric foci and ALT associated PML bodies (APBs),
and that localization of FANCD2 to telomeric foci depends on both the monoubiquitination of
FANCD2 by the FA core complex, and the expression of BLM. Here, I now report data
suggesting that FANCD2 plays a critical role in human ALT cells by limiting BLM-dependent
telomeric recombination and amplification events. Transient depletion of FANCD2 results in a
rapid ALT-specific increase in telomeric DNA synthesis, analogous to the phenotype caused by
overexpression of BLM (Stavropoulos et al., 2002). Excess telomeric DNA synthesized in
FANCD2-depleted ALT cells appears to be primarily extra-chromosomal, and accumulates both
outside of and within APBs. Electron spectroscopic imaging of APBs in FANCD2-depleted cells
shows that most bodies are physically larger and contain more nucleic acid then control APBs,
and RNase treatment demonstrates that the material within bodies is DNA, not RNA. FANCD2
depletion also results in an increase in telomeric DNA entanglements, expression of fragile site
characteristics at telomeres, telomere sister chromatid exchanges, and localization of RAD51 to
telomeric foci in ALT cells. Telomere abnormalities in FANCD2-depleted cells are completely
rescued by codepletion of BLM, but not RAD51. I propose that while BLM is required to
promote recombination and replication of telomeric DNA in ALT cells, this activity is regulated,
and that FANCD2 plays a critical role in this regulation.
102
3.2 Introduction
Maintenance of genome stability requires the concerted effort of numerous factors to ensure
accurate repair, replication and segregation of chromosomes. Members of the RecQ family of
DNA helicases play important roles in all these processes, and are known as ‘caretakers of the
genome’ because of their role in preventing genomic instability (reviewed in Chu and Hickson,
2009; Bachrati and Hickson, 2008). While unicellular organisms typically express a single RecQ
helicase, human cells express at least five different RecQ helicases (RECQ1, BLM, WRN,
RECQ4, RECQ5), of which BLM, WRN, and RECQ4 have been implicated in human
syndromes associated with elevated cancer incidence (Ellis et al, 1995; Yu et al, 1996, Kitao et
al, 1999, Siitonen et al, 2003; Siitonen et al, 2009). BLM is mutated in Bloom syndrome, a rare
recessive disorder characterized by predisposition to a wide range of cancers, sun sensitivity, and
short stature (German J, 1993). Bloom syndrome cells display elevated levels of sister chromatid
exchanges and other chromosomal abnormalities including increased chromatid breaks, gaps,
radials, telomere associations, anaphase bridge and lagging chromosomes (Chaganti et al, 1974;
German and Crippa, 1996, Lillard-Wetherell et al, 2004). Similar to other RecQ mutants, BLM
cells are also sensitive to exogenous damaging agents, suggesting that RecQ helicases play
essential roles in the response to spontaneous and induced DNA lesions (Hemphill et al, 2009;
Aurias et al, 1985).
In addition to roles in promoting global genomic stability, RecQ helicases also can play essential
roles in maintaining telomeres. In yeast, mutations in genes encoding telomerase components
lead to cell death, with rare survivors emerging that rely on telomerase-independent pathways to
maintain telomeres. Type I survivors display amplification of the subtelomeric Y’ element, with
only a short terminal telomere repeat track (Lundblad and Blackburn, 1993), while type II
survivors have long tracts of telomere repeats (Teng and Zakian ,1999). Both types of survivors
rely on recombination, as they are dependent on Rad52p expression, however additional genetic
requirements suggest that the pathways are mechanistically distinct. Type II recombination
appears to function via a Rad51-independent mechanism, which requires expression of the RecQ
helicase Sgs1p, and Rad50p, a component of the MRX complex (Le et al, 1999; Huang et al,
2001).
103
Telomeres in yeast type II survivors share many characteristics with telomeres in human cells
that have activated the Alternative Lengthening of Telomeres (ALT) pathway. The ALT pathway
also appears to function via recombination (Dunham et al, 2000; Londoño-Vallejo et al, 2004;
Muntoni et al, 2009), and similar to most type II survivors, ALT cells are characterized by the
presence of unstable telomeres with long tracts of telomeric repeats (Bryan et al, 1995). ALT
cells also contain circular and linear Extra-Chromosomal Telomeric Repeat DNA (ECTR),
which may be a product as well as a substrate in telomeric recombination reactions. Circular
ECTR DNA has been suggested to be involved in a ‘roll-and-spread’ mechanism of telomere
elongation in type II yeast survivors, where it can act as a template in rolling circle replication,
allowing for rapid increases of telomere length (Natarajan and McEachern, 2002; Lin et al,
2005). The known genetic requirements of ALT are also similar to type II recombination, with
both NBS1, a component of the MRE11/RAD50/NBS1 complex, and BLM being important for
maintaining telomere length (Zhong et al, 2007; Bhattacharyya et al., 2009).
Telomeric DNA in ALT cells has been reported to have frequent internal single-strand breaks or
gaps, which may arise due to incomplete replication or processing of stalled replication forks,
suggesting that ALT telomeric DNA is difficult to replicate (Nabetani and Ishikawa, 2009).
Telomeres in telomerase positive cells do not normally show ssDNA regions (Nabetani and
Ishikawa, 2009), however when confronted with replication stress or loss of TRF1, a key
telomere binding protein, telomeres in these cells exhibit an interrupted staining pattern via FISH
that may be due to ssDNA regions, and that is also observed in other difficult to replicate
sequences known as fragile sites (Sfeir et al, 2009). Interestingly, the frequency of telomeres
with abnormal staining patterns increases in BLM deficient cells, suggesting that BLM plays a
role in promoting replication of telomeric DNA (Sfeir et al, 2009). The involvement of BLM at
telomeres may be partially related to its ability to resolve non B-Form DNA structures (Sun et al,
1998), an activity which may be important because of the capacity of the telomeric G-strand to
promote G quadruplex structures that must be resolved prior to replication or recombination.
Although RecQ helicases clearly play beneficial roles at the telomere, evidence also suggests that
RecQ helicases can promote events that are ultimately deleterious to the cell. Fission yeast
lacking the telomere binding protein Taz1, have a telomere hyper-recombination phenotype,
accompanied by telomere entanglements, chromosome missegregation and loss of viability when
grown at lower temperatures (Miller and Cooper, 2003). These telomere abnormalities appear to
104
arise during replication via a process promoted by Rqh1, the Schizoscaccharomyces pombe
RecQ helicase (Miller and Cooper, 2003; Rog et al., 2009). Furthermore, in Taz1Δ cells also
lacking telomerase, Rqh1 mediates a rapid telomere loss phenotype. However Rqh1 is ultimately
required to promote survivors that use an ALT-like telomere maintenance mechanism (Rog et
al., 2009). In the human ALT system, BLM also appears to have a dual role where it is required
to prevent telomere shortening (Bhattacharyya et al., 2009), and yet if BLM is overexpressed
there is a rapid increase in telomeric DNA synthesis followed by cell death (Stavropoulos et al.,
2002). Together these results suggest that RecQ helicases play a pivotal role in promoting
replication and recombination reactions at telomeres, but these reactions can be potentially
detrimental and therefore must be regulated. In fission yeast, sumoylation of Rqh1 is involved in
regulation of its activity at dysfunctional telomeres (Rog et al., 2009), and in this study I provide
evidence that FANCD2, a protein defective in Fanconi anaemia, regulates BLM dependent
telomeric replication and recombination reactions in human ALT telomere maintenance.
Fanconi anaemia (FA) is rare multigenic syndrome characterized by bone marrow failure, cancer
predisposition and congenital abnormalities. Protein products of FA genes can be divided in
three major categories; proteins which are essential components of a core complex with E3
ubiquitin ligase activity (FANCA, B, C, E, F, G, L), proteins that are monoubiquitinated by the
FA core complex (FANCD2, I), and proteins which have roles outside of ubiquitination
(FANCD1/BRCA2, FANCJ/BACH1, FANCM). Monoubiquitination of FANCD2 and FANCI
occurs during replication and following exposure to DNA damaging agents, and is required for
protein accumulation in nuclear foci and on chromatin (Garcia-Higuera et al, 2001;
Smogorzewska et al, 2007). Similar to Bloom syndrome, FA cells show signs of genomic
instability, with elevated frequencies of spontaneous chromatid breaks, gaps, and radials,
however the pattern of spontaneous rearrangements appears different in cells from the two
syndromes, suggesting that the underlying cause may differ (Schroeder and Kurth, 1971;
Schroeder and German, 1974). FA cells are also hypersensitive to interstrand crosslinking agents
and form radials at an elevated frequencies following treatment, which is the confirmatory
diagnositic criteria of FA (Auerbach et al, 1979). Interestingly, BLM deficient cells are also
sensitive to interstrand crosslinks, show increased radial formation following treatment, and
genetic evidence shows that BLM functions in the same pathway as FA proteins in response to
crosslinks (Pichierri et al., 2004; Hirano, 2005; Hemphill et al, 2009).
105
I, and others, have found that FANCD2 associates with telomeric foci specifically in human cells
that utilize the ALT pathway, and that this localization depends on monoubiquitination by the
FA core complex (Spardy et al, 2008; Fan et al, 2009). Interestingly, I also find that FANCD2
coimmunoprecipitates with BLM in late S/G2 ALT cells, FANCD2 almost always localizes to
telomeric foci that also contain BLM, and BLM expression is required for FANCD2 localization
to telomeric foci. An interaction between FANCD2 and BLM has been reported in a study on
DNA repair (Pichierri et al., 2004), and FANCD2 was recently reported to localize to the base of
ultra-fine DNA bridges coated with BLM during mitosis, however the functional relationship
between these two proteins has remained unclear (Chan et al, 2009; Naim et al, 2009).
In this study I have continued to investigate the role of FANCD2 in ALT telomere maintenance
and find that transient depletion of FANCD2 results in a rapid ALT-specific increase in
telomeric DNA synthesis, analogous to the effect observed when BLM is transiently
overexpressed (Stavropoulos et al., 2002). The majority of telomeric DNA synthesized in
FANCD2-depleted ALT cells appears to be extra-chromosomal, and accumulates within and
outside of abnormally large ALT-associated PML bodies. I also find that when FANCD2 is
depleted there is an ALT specific increase in RAD51 colocalization with telomeric foci,
telomeric recombination reactions, telomere entanglements, and telomeres that express
characteristics of fragile sites. In two ALT cell lines FANCD2 depletion leads to microscopically
visible intra-nuclear RAD51 fibers, which may indicate a defect in the regulation of RAD51
oligomerization. However, codepletion of RAD51 with FANCD2 does not rescue the telomere
abnormalities in FANCD2 depleted cells, suggesting that the telomere phenotype is promoted by
a RAD51-independent process. Significantly, codepletion of BLM with FANCD2 completely
suppresses the telomere abnormalities in FANCD2-depleted ALT cells. I have also previously
found that FANCD2 depletion results in reduced viability in ALT cells, which codepletion of
BLM with FANCD2 partially rescues. Together, our observations provide evidence for a model
in which the FANCD2 serves to negatively regulate BLM-dependent amplification and
recombination reactions between ALT telomeric DNA.
acid, 1mM MgCl2) was preheated for 3 min at 86°C, added to slides, covered with a coverslip,
than slides were heated for 3 min at 81°C, and left for 2 hours at room temperature prior to
washing. Slides were washed 2 x 15 min in 70% formamide, 10mM Tris pH7.2, 0.1% BSA, and
then 3 x 5 min in 100mM Tris pH 7.2, 150mM NaCl, 0.08% Tween 20.
CO-FISH. Cells used in CO-FISH experiments were grown in DMEM supplemented with
7.5uM BrdU and 2.5uM BrdC for 24 hr (847) and 20hr (HT1080) prior to harvesting, with
0.1ug/ml colcemid being added for the last 2 hours of growth. Cells were harvested for FISH as
described above. Slides were rehydrated in PBS, fixed in 4% PFA for 2 min, washed in PBS,
digested with pepsin (1mg/ml at pH 2.0) for 10 min at 37°C, washed in PBS, re-fixed in 4% PFA
for 2 min, and than washed in PBS. Slides were then incubated in Hoeschst 33258 (0.5 ug/ml in
108
PBS) for 20 min, washed in PBS, and placed on a 55°C heating plate, covered with PBS and a
coverslip, then exposed for 45 min to light from an inverted 312nm UV box. The working
distance between the slide and UV box was less then 1 cm, and slides were moved every 5 min
to ensure equal damage to all regions of the slide. Slides were rinsed in PBS, digested for 12.5
min with Exonuclease III (3U/uL NEB), washed in PBS, then dehydrated and processed for
FISH as described above.
109
3.4 Results
3.4.1 FANCD2-depletion results in a rapid, ALT-specific increase in telomeric DNA synthesis
I used a siRNA approach to reduce FANCD2 protein levels in ALT and telomerase positive cell
lines. Four different sequences targeting FANCD2 where tried, three of which reduced FANCD2
proteins levels to at or below residual amounts seen in patient cell lines with biallelic FANCD2
mutations (Figure 3-1). With this level of silencing, FANCD2 nuclear foci were no longer visible
by immunofluorescence. Five days after FANCD2 siRNA addition, I performed FISH and
microscopically examined telomeric DNA in interphase cells labeled with a rhodamine tagged
telomeric probe. Surprisingly, I found that a fraction of ALT cells had abnormally high amounts
of telomeric DNA present within nuclei (Figure 3-2). Increased amounts of telomeric DNA were
observed in GM847 and U2OS cells treated with FANCD2 siRNAs A-C, but not FANCD2
siRNA D, which does not significantly deplete FANCD2 protein levels or reduce ALT cell
viability (Figure 3-1; Figure 2-15). ALT cell populations normally have some variability in
telomeric DNA content, however the peak intensity of telomeric DNA foci in FANCD2-depleted
ALT cells greatly exceeded anything observed in cells treated with random siRNA or untreated
cells.
Figure 3-1. FANCD2 siRNA significantly reduces FANCD2 expression Western blot of FANCD2 protein levels in patient cells with biallelic FANCD2 mutations (PD20),
GM847 and U2OS ALT cells, and GM847 and U2OS ALT cells treated with random or FANCD2
targeting siRNAs five days post siRNA addition. FANCD2 siRNAs A-C significantly decrease FANCD2
protein levels.
110
Figure 3-2 FANCD2-depleted GM847 and U2OS ALT cells have increased levels of
telomeric DNA GM847 and U2OS cells were treated with the indicated siRNA sequences, and then telomeric DNA
content was assessed 5 days post siRNA addition using a FISH probe targeting the telomeric G strand.
Scale bar is 20 µm. Images were taken at the same exposure time under 20x magnification.
111
Increased telomeric DNA content was also observed in FANCD2-depleted VA13, GM637, and
SAOS-2 ALT cells (Figure 3-3, SAOS-2 data not shown). Depletion of FANCD2 in telomerase
positive cells did not result in an increase in telomeric DNA in GM639, HT1080, or HeLa
telomerase positive cells (Figure 3-3, HeLa data not shown). Our lab has previously observed
that overexpression of BLM also results in a rapid ALT-specific increase in telomeric DNA
synthesis (Stavropoulos et al., 2002), but to my knowledge FANCD2 is the first example of a
protein that when depleted gives this phenotype.
Figure 3-3. FANCD2-depleted GM637 and VA13 ALT cells but not GM639 or HT1080
telomerase positive cells, have increased levels of telomeric DNA Cells were treated with FANCD2 siRNA A, and then telomeric DNA content was assessed 5 days post
siRNA addition using a FISH probe targeting the telomeric G strand. Scale bar is 20 µm. Images of
random and FANCD2 siRNA treated cells were taken at the same exposure time under 20x magnification.
112
3.4.2 FANCD2-depleted ALT cells accumulate ECTR DNA within and outside of abnormally large ALT associated PML bodies
Cells that have activated the ALT mechanism of telomere maintenance accumulate a unique
subtype of PML nuclear bodies, referred to as ALT-Associated PML Bodies (APBs). APBs are
characterized by the colocalization of telomeric DNA and telomere binding proteins with normal
PML body components (Yeager et al, 1999). APB number and size vary during the cell cycle,
with cells in late S/G2 typically accumulating larger ‘donut’ shaped bodies. ImmunoFISH
experiments show that large telomeric foci colocalize with PML and telomere binding proteins,
and that FANCD2-depleted ALT cells accumulate unusually high amounts of telomeric DNA
and telomere binding proteins within APBs (Figure 3-4 A). Examples of typical random siRNA
treated cells with large APBs are shown for comparison (Figure 3-4 A).
I next examined the frequency of GM847 nuclei containing more then one telomeric foci with a
diameter that exceeded 20 pixels (∼3 µm) in wild-type, random siRNA and FANCD2 siRNA A
treated cells. I found that FANCD2-depletion led to a 22 fold increase in the percentage of nuclei
with this phenotype 5 days post siRNA treatment relative to random siRNA treated cells (Figure
3-4 B). Telomeric DNA signals within these large foci was usually so intense that it caused
arcing of the camera signal, and signals in bodies could be observed with very short exposure
times (1-5 ms). Eight days post FANCD2 siRNA treatment, approximately 55% of GM847 ALT
cells showed abnormally large telomeric foci (Figure 3-4 B). Examination of GM847 FANCD2-
depleted pro-metaphase cells shows that these large telomeric foci have telomeric DNA signals
that are significantly more intense than signals from telomeres, and that bodies are present within
the interchromosomal space (Figure 3-4 C). This supports the idea that the bulk of the telomeric
signal in large foci is composed of extra-chromosomal material.
113
A)
B)
GM847 + random siRNA Day 5
GM847 + random siRNA Day 8
GM847 + FANCD2 siRNA Day 5
GM847 + FANCD2 siRNA Day 8
GM847 + Random
siRNA
GM847 + FANCD2 siRNA A
VA13 + Random
siRNA
VA13 + FANCD2 siRNA A
114
C)
Figure 3-4. FANCD2-depleted ALT cells accumulate high amounts of telomeric DNA and
telomere binding proteins within APBs A) Representative images of GM847 and VA13 ALT cells with large APBs 5 days post treatment with
random or FANCD2 siRNA A. Nuclei were stained for PML, telomeric DNA, and the telomere binding
protein TRF2. Scale bar is 5 µm. B) Plot of the number or nuclei containing more then one large
telomeric foci 5 and 8 days post treatment with random (blue) or FANCD2 siRNA A (red) treated cells
from 311 randomly selected asynchronous cells from 3 independent experiments ± the associated
standard error for each condition. C) Image of a pro-metaphase FANCD2-depleted GM847 cell 8 days
post siRNA addition. DNA is stained (blue) and telomeric DNA (red) was detected with FISH and
imaged for 200ms (left) and 5ms (right). Scale bar is 5 µm. Large telomeric foci are significantly brighter
than telomeres and accumulate in the extra-chromosomal space between chromosomes, suggesting that
the bulk of this material is ECTR DNA.
In collaboration with Andrew Larsen and Dr. Ren Li, energy-filtered transmission electron
microscopy was used to analyze APB ultrastructure. APBs in wild-type cells were shown to have
an outer protein shell (nitrogen enriched) surrounding a nuclei acid and protein core (nitrogen
and phosphorous present), and the nucleic acid within APBs differed significantly from
chromatin, most likely representing non-nucleosomal ECTR DNA (Figure 2-11). APBs in
FANCD2-depleted cells had a similar organization, but were physically larger and sometimes
contained chromatin-like material within the APB (Figure 2-12). Transcription of telomeres
results in production of a non-coding UUAGGG-repeat containing telomeric RNA, which may
have roles in chromatin remodeling or telomere capping (Azzalin et al, 2007). Telomeric RNA
115
associates with telomeric DNA, and is present at higher levels in primary and ALT cells then in
telomerase positive cells (Ng et al, 2009). In order to determine whether FANCD2-depletion
leads to an increase in telomeric DNA and/or RNA, Dr. Ren Li examined the relative
phosphorous contents in serial sections of FANCD2-depleted APBs that were untreated, or
treated with RNase prior to imaging. As an internal control for the RNase treatment, sections
were chosen which contained an APB and a nucleolus. RNase treatment prior to imaging
significantly decreases the phosphorous content of the nucleolus, but does not affect the
phosphorous content of APBs, suggesting that the majority of material within FANCD2-depleted
APBs is DNA (Figure 3-5).
Bromodeoxyuridine (BrdU) is a nucleoside analog which when added to cells becomes
incorporated into DNA during replication, and can subsequently be detected with an antibody
using traditional immunofluorescence. Denaturation or cleavage of double-strand DNA that has
incorporated BrdU is required prior to immunofluorescence to make the DNA accessible to the
antibody, whereas BrdU that has been incorporated into single-strand DNA is detectable without
these treatments. I find that when GM847 and VA13 ALT cells are grown in the presence of
BrdU for 24 hours, then stained for BrdU incorporation without DNase or HCl treatment, APBs
are readily detectable, suggesting that they contain a high amount of single- strand DNA (data
not shown). Upon FANCD2 depletion, there is an increase in the size, intensity, and frequency of
APBs with detectable BrdU, suggesting that there is an increase in single-strand DNA within
APBs. In the U2OS ALT line, which normally has smaller APBs then GM847 and U2OS, BrdU
is only readily detectable in FANCD2 depleted cells (Figure 3-6 A). Supporting the idea that
APBs contain ssDNA, DAPI staining, which relies on intercalation of a fluorescent molecule
into double-strand DNA, is often poor in nuclear regions containing abnormally large telomeric
foci (Figure 3-6 B). The observation that APBs contain single-strand DNA is consistent with in
vitro analysis of ECTR DNA, and electron microscopy experiments, which suggest that the bulk
of the material within APBs differs significantly from chromatin (Nabetani and Ishikawa, 2009).
116
Figure 3-5. APBs in FANCD2-depleted cells contain high levels of DNA, not RNA Serial nuclear sections containing a nucleolus (solid arrow) and an APB (dotted arrow) were imaged to
determine the nitrogen (N) and phosphorous (P) content without RNase treatment (rows 1 and 3) and with
RNase treatment prior to imaging (rows 2 and 4). A coloured overlay (O) is shown where nitrogen is blue,
and phosphorous is yellow. Scale bar of the top 2 rows is 0.5 µm. The bottom 2 rows show a higher
magnification of the APB, the scale bar is 0.2 µm. The phosphorous content of the nucleolus, but not the
APB, decreases with RNase treatment.
GM847 + FANCD2 siRNA A
GM847 + FANCD2 siRNA A + RNase
GM847 + FANCD2 siRNA A
GM847 + FANCD2 siRNA A + RNase
117
A)
B)
Figure 3-6. Large APBs contain high amounts of single-strand DNA A) U2OS cells treated with FANCD2 siRNA form large telomeric foci (red) that contain single-strand
DNA (green), as assessed by the ability to detect incorporated BrdU without cleavage or denaturation of
the DNA. BrdU is not normally detectable in smaller telomeric foci in random siRNA treated U2OS cells.
Scale bar is 10 µm. B) Abnormally large telomeric foci in FANCD2-depleted cells (red, 10ms exposure)
frequently correspond to DAPI poor staining regions of the nucleus (arrows). Scale bar is 10 µm. Insert in
corner is 2.5x magnified.
DAPI DAPI + Telomeric DNA
DAPI BrdU TRF1
U2OS + FANCD2 siRNA A
U2OS + Random
siRNA
118
In addition to accumulating telomeric DNA within APBs, FANCD2-depletion results in nuclei
that have an excessive number of smaller telomere signals outside of large APBs (Figure 3-7 A).
FISH using a pan-centromeric FITC labeled probe set was used in conjunction with a rhodamine
labeled telomere probe, and the frequency of telomere to centromere signals was determined.
Interphase GM847 wild-type and random siRNA treated cells have an average telomere to
centromere ratio of ∼2. FANCD2 depletion in GM847 cells can result in up to a 10-fold increase
in the telomere to centromere ratio, something that is never observed in controls. I scored the
frequency of nuclei with a telomere to centromere ratio greater then 4, and found that <1% of
random siRNA treated cells showed this phenotype, while 14% and 36% of FANCD2-depleted
cells had excess telomeric DNA signals 5 and 8 days post siRNA treatment, respectively (Figure
3-7 B). Similar increases in telomere signals were seen in U2OS ALT cells, but were not
observed in FANCD2-depleted HT1080 telomerase positive cells (0 out of 300 cells analyzed),
arguing that this is an ALT specific change. Examination of metaphase spreads shows that the
excess telomeric DNA in FANCD2-depleted cells does not accumulate at interstitial sites, but
rather is extra-chromosomal (Figure 3-7 C). Together, this argues that FANCD2-depletion
results in excess generation of ECTR DNA, which accumulates both within, and outside of
APBs.
3.4.3 FANCD2-depleted ALT cells do not upregulate expression of BLM, TRF1 or TRF2
Overexpression of BLM also results in an ALT-specific increase in telomeric DNA synthesis,
which accumulates in abnormally large foci (Stavropoulos et al., 2002), similar to what is
observed when FANCD2 is depleted. Western blot analysis of BLM expression levels in random
or FANCD2 siRNA treated cells from 4 different ALT cell lines failed to show an increase in
BLM expression (Figure 3-8), arguing that the FANCD2-depletion telomere phenotype is not an
artifact of BLM overexpression. I also examined the levels of TRF2 and TRF1, two major DNA
binding proteins that play important roles in telomere capping and replication (Takai et al, 2003;
Sfeir et al, 2009). Interestingly, I find that there is no increase in TRF2 or TRF1 expression
levels in FANCD2 depleted cells (Figure 3-8), despite the apparent increase in telomeric DNA
synthesis.
119
A)
B)
C)
Figure 3-7 FANCD2-depleted ALT cells accumulate ECTR DNA outside of APBs
A) GM847 nuclei treated with random or FANCD2 siRNA 8 days post siRNA treatment. Centromeres
(green) overlayed with DAPI and telomeric DNA (red) are shown. The random siRNA treated nucleus
has a typical telomere to centromere ratio, whereas the FANCD2 depleted nucleus has a high number of
small telomeric DNA foci. Scale bar is 5 µm. Exposure times are identical. B) Plot of the number or
120
nuclei containing 4x more telomeric than centromeric signals 5 and 8 days post treatment with random
(blue) or FANCD2 siRNA A (red) from 311 randomly selected asynchronous cells from 3 independent
experiments ± the associated standard error for each condition. C) Metaphase spreads of U2OS cell
treated with random (left) or FANCD2 siRNA B (right) 5 days post addition. Telomeric DNA is shown in
red. Smaller telomeric DNA signals are extra-chromosomal, not interstitial.
Interphase FANCD2-depleted ALT cells containing very large APBs often have fewer TRF1 and
TRF2 foci then would be predicted based on my FISH results, which show that 87% of FANCD2
depleted cells with large APBs have >2x more telomeric DNA signals then centromeres. This
suggests that not all telomeric DNA is detectable by TRF1 or TRF2 staining. Electron
microscopic analysis argues against APBs as being places where large numbers of telomeres are
aggregated (chapter 2), eliminating this as a possible explanation. An alternate explanation for
the low numbers of TRF1 and TRF2 foci seen in FANCD2-depleted ALT cells with large APBs,
is that there is an insufficient amount of TRF1 and TRF2 within the cell to bind to all the
telomeric DNA present within the nucleus. This could lead to telomere uncapping, and more
frequent activation of the DNA damage response by telomeric DNA with insufficient amounts of
shelterin associated proteins, explaining are previous observation that FANCD2-depletion causes
an ALT specific increase in telomere dysfunction induced foci (Figure 2-10 A).
Figure 3-8. FANCD2-depleted ALT cells do not increase expression of BLM, TRF1, or TRF2
Western blot analysis of GM847, VA13, U2OS and SAOS-2 ALT cells 5 days post siRNA treatment with
random or FANCD2 siRNA shows that BLM, TRF1, and TRF2 protein levels are not upregulated in cells
with reduced FANCD2 protein levels.
121
3.4.4 FANCD2-depleted ALT cells have increased association of RAD51 with telomeric foci, telomere sister chromatid exchanges, fragile telomeres, and telomere entanglements
A key step in homologous recombination is the initial search for homology, which often involves
the assembly of a RAD51 nucleofilament on a 3’ ssDNA tail. FANCD2 is a member of a
complex of unknown function containing BRCA2, FANCG, and XRCC3 (Wilson et al, 2008).
BRCA2 plays numerous roles in controlling RAD51, including targeting RAD51 to double-
strand breaks and regulating RAD51 oligomerization (Yuan et al, 1999; Davies et al, 2001;
Pellegrini et al, 2002). FANCD2-deficient human cell lines do not show impaired RAD51 foci
formation after DNA damage (Godthelp et al, 2002; Godthelp et al, 2006), however they do have
impaired recombinational repair in reporter assay systems (Nakanishi et al, 2005).
RAD51 associates with telomeric foci and APBs at levels visible by immunofluorescence in
ALT cells, and associates with telomeric DNA transiently during the cell cycle at levels
detectable by chromatin immunoprecipitation in telomerase positive cells (Yeager et al, 1999;
Verdun and Karlseder, 2006). I therefore examined the effect of FANCD2 depletion on RAD51
colocalization with telomeric proteins in ALT and telomerase positive cells. Depletion of
FANCD2 results in 4.3 and 3.2 fold increases in the number of RAD51 foci that colocalize with
TRF2 in GM847 and VA13 ALT cells, respectively (Figure 3-9 A). In both ALT and telomerase
positive cells, FANCD2-depletion results in an increase in RAD51 foci not associated with
TRF2. However in telomerase positive GM639 there is no change in RAD51 colocalization with
TRF2, and only a 0.1 fold increase in HT1080 cells, suggesting that the increased association
between RAD51 and TRF2 is an ALT specific phenomenon.
Interestingly, when FANCD2 is depleted in U2OS and SAOS-2 ALT cells, ∼5% of cells show a
novel phenotype wherein RAD51 forms elongated structures that run through the nucleus (Figure
3-9 B). This phenotype was only rarely observed in control cells (<0.1%), and can be seen using
multiple FANCD2 siRNAs and RAD51 antibodies (data not shown). Overexpression of RAD51
can cause higher order RAD51 structures to form in some cell types, however these appear to
differ from the RAD51 fibers that I observe in FANCD2-depleted cells, which run between
distinct foci, and are not limited to non-cycling cells (Raderschall et al, 2002). Western blot
analysis does not show an increase in RAD51 expression in FANCD2-depleted cells, and
122
A)
B)
C)
Figure 3-9. FANCD2 depletion effects RAD51 foci formation and oligomerization A) Plot of the number of RAD51 foci that do (red), and do not (blue) colocalize with TRF2 in populations
of 300 randomly selected asynchronous cells 5 days post treatment with random or FANCD2 siRNA A
from 3 independent experiments ± the associated standard error. FANCD2-depletion results in an increase
in non-telomeric associated foci in all cell lines tested. In ALT cells there is an increase in the number of
RAD51 foci that colocalize with TRF2, which is not observed in telomerase positive cell lines.
B) Immunofluorescence of endogenous RAD51 in FANCD2-depleted SAOS-2 cells. FANCD2 depletion
123
results in formation of extended RAD51 oligomers in ∼5% of cells. Scale bar is 5 µm. C) Western blot
(left) of SAOS-2 cells treated with random siRNA, FANCD2 siRNA, or FANCD2 siRNA in conjunction
with 50nM or 100nM RAD51 siRNA. Cells depleted of RAD51 do not appear to overexpress RAD51 via
western. Immunofluorescence of RAD51 staining in SAOS-2 cells treated with FANCD2 siRNA A and
RAD51 siRNA (100nm) demonstrating that RAD51 oligomers still form and are not an artifact of
overexpression. Scale bar is 5 µm.
structures continue to form when RAD51 protein levels are partially reduced by codepleting
RAD51 with FANCD2 (Figure 3-9 C). As these structures are not observed in GM847 and VA13
ALT cells, formation of extended RAD51 structures appears to be a cell line specific, but not an
ALT specific phenomenon.
I next examined the frequency of telomere sister chromatid exchange (T-SCE) reactions in ALT
and telomerase positive cells treated with random or FANCD2 siRNA. Because telomeres have a
characteristic strand asymmetry, chromosome orientation-FISH (CO-FISH) can be used to
provide detailed information on exchanges occurring between telomeric repeat sequences.
CO-FISH involves the selective degradation of the newly replicated strand, which requires one
complete round of replication in the presence of BrdU/BrdC (Figure 3-10 A). Fanconi anaemia
cells can cycle slowly and accumulate cells with a 4N DNA content, a phenomenon which is
further heightened by treatment with DNA crosslinking agents, which results in a prolonged S
phase due to incomplete replication (Dutrillaux et al, 1982; Akkari et al, 2001).
To reduce the probability of false positives due to incomplete replication in the presence of
BrdU/BrdC, cells were labeled for 24 hr immediately after FANCD2 knockdown (36 hours after
siRNA addition), and microscopically analyzed to confirm that all cells in G2 and mitosis had
incorporated BrdU. I examined more then 3500 chromosomes over three independent
experiments, using conditions designed to ensure compete degradation of the newly synthesized
strands (45 min exposure to UV light source located <1 cm from slides place on an 55C heating
block, followed by 12.5 min digestion with ExoIII).
124
B)
C)
A)
Figure 3-10. SiRNA depletion of FANCD2 in GM847 ALT cells results in increased
frequency of T-SCEs A) Overview of CO-FISH assay. Cells are replicated in the presence of BrdU/BrdC for a single round of
replication, than are harvested, and spread on slides for FISH experiments. Treatment of slides with
Hoechst, UV, and Exonuclease III results in the selective degradation of the newly synthesized. Sister
chromatid exchanges within telomeres result in a split signal on sister telomeres. B) Examples of T-SCEs
(arrows) in GM847 FANCD2-depleted cells. Centromeres are shown in green, telomeres are shown in
red. C) Plot of the average frequency of chromosome ends with evidence of a potential T-SCE event,
from 3 independent experiments ± standard deviation between experimental means. More then 2000
chromosome ends were analyzed per experiment.
GM847 GM847 + GM847+ random FANCD2 siRNA siRNA
GM847 + FANCD2 siRNA
125
I find that 4.0% of GM847 ALT chromosome ends have a T-SCE, which increases slightly to
5.1% upon treatment of cells with random siRNA, and more significantly to 14.5% of
chromosome ends in FANCD2 depleted cells (Figure 3-10 C). The fraction of telomeres in a cis
pattern, indicative of a genomic SCE event, did not significantly increase in FANCD2-depleted
cells (Figure 3-14), consistent with the observation that FA proteins do not suppress genomic
SCEs in human cells (Hemphill et al, 2009).
In HT1080 telomerase positive cells I do not observe an increase in T-SCE following FANCD2
depletion, with 1.2% of HT1080 chromosome ends showing evidence of a T-SCE, 1.5% of
chromosome ends in random treated cells, and 1.4% of chromosome ends in FANCD2 depleted
cells. The frequency of T-SCEs in HT1080 cells is higher then what is normally observed in
telomerase positive, however I do not feel that it is an artifact due to incomplete incorporation or
digestion, as all cells in G2 and mitosis appeared to have incorporated BrdU, and the values
remained constant even when cells were exposed to UV for 90 minutes followed by a 20 min
digestion by ExoIII.
Increased SCEs at specific genetic loci are one feature of difficult to replicate sequences known
as fragile sites. It has recently been proposed that telomeres share characteristics with common
fragile sites, in part because when grown under conditions of replication stress they begin to have
a fragmented appearance detectable by FISH (Sfeir et al, 2009). ALT telomeres share many
characteristics with fragile sites, even when cells are not subjected to replication stress.
Examination of more then 2000 telomeres from GM847 and U2OS ALT cells, and GM639 and
HT1080 telomerase positive cells, showed that ALT telomeres frequently have a fragmented
appearance by FISH (Figure 3-11). In HT1080 and GM639 telomerase positive cells, 0.6%
(14/2337) and 0.7% (16/2183) of telomeres are fragmented, respectively. In GM847 and U2OS
ALT cells this phenotype is significantly more frequent at 7.1% (161/2256) and 25.0%
(558/2228), respectively.
HT1080 and GM847 cells were also treated with random and FANCD2 siRNA A, and examined
for changes in the frequency of telomeres with a fragmented appearance. The frequency of
fragmented telomeres in random siRNA treated HT1080s was 0.6% (13/2166). Following
FANCD2 depletion the frequency did not significantly increase at 0.7% (15/2205). This differed
from GM847 cells, which showed a 2-fold increase in fragmented telomeres after FANCD2
126
depletion. 6.2% (138/2227) of GM847 cells treated with random siRNA were fragmented,
whereas 13.1% (326/2496) of FANCD2-depleted cells were fragmented.
Figure 3-11. ALT telomeres more frequently exhibit FISH staining patterns characteristic
of fragile sites then telomerase positive cells
Metaphase spread of a HT1080 telomerase positive cell (left) and a U2OS ALT cell (right) showing
telomeres (red) with abnormal staining pattern (arrows).
In addition to increases in T-SCEs and fragmented telomeres in FANCD2-depleted ALT cells, I
also observed an increase in telomeric DNA entanglements. Telomere FISH in interphase cells
occasionally revealed the presence of linear telomeric DNA fibers connecting telomeric foci
(Figure 3-12 A). These often run between larger telomeric foci, which likely represent APBs,
and smaller foci, which may represent telomeres. As cells entered prometaphase and chromatids
began to condense and move around, clear evidence of telomeric DNA connecting telomeres to
extra-chromosomal telomeric DNA foci is observed (Figure 3-12 B). This suggests that at least
some telomeres transiently associate with APBs. The frequency and extent of these telomeric
DNA bridges increases in GM847 cells when FANCD2 was depleted. At least one linear
telomeric bridge was observed in approximately 15% of wild-type and random siRNA treated
GM847, this increases to 44% and 61% in FANCD2-depleted cells, 5 and 8 days post siRNA
127
addition, respectively (Figure 3-12 C). HT1080 cells only rarely showed linear telomeric fibers
connecting foci (<1%), and this value did not increase upon FANCD2 depletion.
Figure 3-12. FANCD2-depleted GM847 ALT cells frequently show evidence of telomeric
DNA entanglements A) Interphase nucleus from a FANCD2-depleted GM847 cells 5 days post siRNA addition stained for
DAPI (left) and telomeric DNA (right). Arrows indicated telomeric DNA fibers connecting foci.
B) Pro-metaphase FANCD2-depleted GM847 cell showing telomeric DNA connections running between
telomeres and large telomeric foci that likely represent APBs. Scale bars are 10 µm. C) Plot of the number
of interphase nuclei containing at least one linear telomeric DNA fiber from populations of 311 randomly
selected asynchronous cells treated with random (blue) or FANCD2 siRNA A (red) from 3 independent
experiments ± the associated standard error.
A) B)
C)
DAPI Telomeric DNA DAPI + Telomeric DNA
GM847 + FANCD2
siRNA
128
3.4.5 Telomere abnormalities in FANCD2-depleted ALT cells are generated through a BLM dependent, largely RAD51 independent mechanism
Using siRNA targeting BLM and RAD51, I significantly reduced expression of BLM in
conjunction with FANCD2, or RAD51 with FANCD2 in GM847 ALT cells (Figure 3-13 A).
BLM silencing was sufficient to reduce BLM expression to the level where BLM foci were no
longer detectable by immunofluorescence, whereas only faint RAD51 foci remained detectable
in a small fraction of cells under 63x magnification (data not shown). I next examined 300 nuclei
over 3 independent experiments for the level of ECTR DNA, scoring the frequency of nuclei
with more then one abnormally large telomeric foci (>20 pixels, ∼3µm) or a telomeric to
centromeric DNA ratio greater then 4. Surprisingly, I found that codepletion of BLM with
FANCD2 completely suppressed these phenotypes, whereas codepletion of RAD51 with
FANCD2 had no effect (Figure 3-13 B, C).
Depletion of FANCD2 alone results in 35% and 55% of cells having at least one abnormally
large APBs 5 and 8 days post siRNA treatment, respectively. However when BLM is codepleted
<1% of cells show this phenotype (Figure 3-13 C). Codepletion of RAD51 with FANCD2 does
not rescue this phenotype, as 35% and 67% of cells have at least one abnormally large APB, 5
and 8 days post siRNA treatment. Likewise, 14% and 36% of FANCD2-depleted cells have a 4
fold excess of telomeric signals to centromeric signals 5 and 8 days post siRNA addition, figures
which decrease to <1% when BLM is codepleted, but remain elevated at 13% and 33% in
FANCD2 and RAD51 codepleted cells 5 and 8 days post siRNA (Figure 3-13 C). The ability of
the BLM knockdown to suppress the increased amount of ECTR DNA present in FANCD2-
depleted cells was confirmed using a FANCD2 siRNA B, and experiments were repeated and
confirmed with both FANCD2 siRNA sequences in the U2OS ALT cell line (data not shown).
129
A)
B)
C)
Figure 3-13. BLM, but not RAD51, is required to generate high levels of ECTR DNA in
FANCD2-depleted ALT cells A) Western blot analysis of BLM, FANCD2, and RAD51 protein levels in GM847 cells 5 days post
siRNA addition. Both BLM and RAD51 are efficiently codepleted with FANCD2 upon siRNA addition.
B) GM847 cells treated with FANCD2 and RAD51 siRNA (left) continue to show high levels of ECTR
DNA, both within and outside of large APBs. GM847 cells treated with FANCD2 and BLM siRNA
130
(right) no longer have elevated levels of ECTR DNA. Centromeric DNA (green) is overlayed with DAPI.
Telomeric DNA is shown in red. Exposure times were the same between the 2 nuclei. Scale bar is 10µm.
C) Plot of the number of nuclei with a telomeric : centromeric DNA ratio >4 (green) and the number of
nuclei with >1 telomeric foci with a diameter greater then 3µm (blue) from populations of 300 randomly
selected asynchronous GM847 cells 5 and 8 days post siRNA addition ± the associated standard error.
I next examined the effect of BLM and RAD51 codepletion on the frequency of T-SCEs in
FANCD2-depleted ALT cells. I find that codepletion of BLM, but not RAD51, is sufficient to
suppress the increase in T-SCEs observed in FANCD2-depleted cells (Figure 3-14). While
14.5% of chromosome ends in GM847 FANCD2-depleted cells have undergone a T-SCE, this
frequency declines to 6.1% when BLM is codepleted. Consistent with the established role of
BLM in suppressing genomic SCEs, the frequency of telomere G-strand signals in a cis pattern,
indicative of a single SCE event upstream of the telomeres, increases approximately 3 fold when
BLM is codepleted (Figure 3-14). Codepletion of RAD51 with FANCD2 does not decrease the
frequency of T-SCEs, which remains at 15.0%.
Figure 3-14. Codepletion of BLM with FANCD2, but not RAD51, suppresses the increase
in T-SCEs observed in FANCD2-depleted GM847 cells Plot of the average number of chromatid ends with a T-SCE (blue, left axis) in siRNA treated cells from 3
independent experiments ± the standard deviation between experimental means. More than 2000
chromatid ends were analyzed per experiment. Plot of the average number of chromosomes with a
telomere pattern in cis (orange, right axis) from 3 independent experiments ± the standard deviation
between experimental means.
131
A)
B)
The increased telomere DNA entanglements observed in interphase and prometaphase FANCD2-
depleted cells also appears to be mediated in a BLM dependent manner. Although BLM and
FANCD2 depleted cells no longer form abnormally large APBs, telomeric DNA fibers can still
be occasionally observed running between larger telomeric foci and telomeres in FANCD2 and
BLM codepleted prometapshase cells (Figure 3-15 A). Codepletion of BLM with FANCD2
reduced the frequency of interphase cells with visible linear telomere fibers from 44% and 61%
in FANCD2-depleted cells, 5 and 8 days post siRNA, to 22% and 17% in FANCD2-depleted
cells when BLM is codepleted (Figure 3-15 B). Codepletion of RAD51 with FANCD2 does not
reduce the frequency of cells with telomere entanglements, which remain elevated at 50% and
60%, 5 and 8 days post siRNA addition.
Figure 3-15. Codepletion of BLM with FANCD2, but not RAD51, suppresses the increase
in telomeric DNA entanglements observed in FANCD2-depleted GM847 ALT cells A) Pro-metaphase FANCD2 and BLM depleted GM847 cell showing a telomeric DNA connection
running between a telomere and a large telomeric foci that may represent an APB (arrow). Telomeric
132
DNA is shown in red, chromosomes are DAPI stained. B) Plot of the number of interphase nuclei
containing at least one linear telomeric DNA fiber from populations of 300 randomly selected
asynchronous siRNA treated cells from 3 independent experiments ± the associated standard error.
3.4.6 Codepletion of BLM with FANCD2 improves the viability of FANCD2 depleted ALT cells
I previously found that depletion of FANCD2 significantly reduces the long and short term
viability of ALT cells. As codepletion of BLM with FANCD2 rescues telomere abnormalities, I
examined whether it also rescued the loss of viability phenotype. When FANCD2 and BLM are
codepleted, the number of cells remaining 5 days post siRNA treatment is reduced relative to
controls, but increased relative to FANCD2 depleted cells alone across 4 different ALT cell lines
from 2 independent experiments. In FANCD2-depleted GM847 cells, the number of cells 5 days
post siRNA addition relative to random siRNA treated cells is 15.8%, which doubles to 32.0%
when BLM is codepleted. FANCD2-depleted U2OS show a more modest increase from 34.3%
to 49.1% when BLM is codepleted. GM637 and VA13 ALT show more striking 3 and 4 fold
increases, with relative cell numbers of 15.8% and 7.4% in FANCD2 depleted cells that increase
to 50.4% and 30.6% when BLM is codepleted, respectively.
To assess the long-term viability of remaining cells, 5 days post siRNA treatment cells were re-
plated for colony forming assays (Figure 3-16). The average colony forming ability of GM847
cells treated with FANCD2 siRNA relative to random siRNA treated cells increased from 19.2%
to 55.8% when BLM was codepleted. In U2OS cells the colony forming ability increased from
33.7% in FANCD2 depleted cells to 64.0% when BLM was codepleted. Codepletion of BLM
completely rescued the colony forming ability of FANCD2-depleted GM637 cells, which
increased from 30.3% to 105.9%. Because FANCD2-depleted VA13 cells are unable to form
colonies, this cell line was not included in these experiments.
133
Figure 3-16. Codepletion of BLM with FANCD2 partially restores the colony forming
ability of ALT cells GM847 (top row), GM637 (middle row) and U2OS (bottom row) were treated with random, FANCD2, or
FANCD2 and BLM siRNA, then 500 cells were replated for colony forming assays 5 days after the initial
transfection, and stained for colonies 2 weeks after initial plating.
134
3.5 Discussion
In this study I have shown that transient depletion of FANCD2 results in the rapid ALT-specific
increase in ECTR telomeric DNA content in a fraction of cells. Energy-filtered transmission
electron microscopic analysis of serial sections treated and untreated with RNase clearly
indicates that material within bodies is DNA, and not RNA, and the morphology and
phosphorous signal intensity reveals that the bulk of this material is not chromatin, but likely
ECTR DNA. One possible source of some of this ECTR DNA is that it is generated from
telomere rapid deletion events. In human cells, the 3’ssDNA telomeric overhang can invades
proximal dsDNA forming large duplex loop known as a ‘t-loop’ with a partial or full Holliday
junction at its base (Griffith et al, 1999). Expression of a mutant allele of TRF2 appears to
promote aberrant resolution of this structure, resulting in rapid shortening of telomeres and
concomitant production of circular ECTR DNA (Wang et al, 2004). Genetic analysis of this
mechanism show a dependency on NBS1, and the Rad51 paralog XRCC3, which is part of a
complex with RAD51C that is associated with Holliday junction resolvase activity (Wang et al,
2004; Liu et al, 2004). Aberrant resolution of t-loops may also contribute to production of
circular ECTR in ALT cells, as this process is also largely dependent on NBS1 and XRCC3
(Compton et al, 2007). However, I have previously observed that FANCD2 depletion does not
cause the frequency of signal free ends to increase (chapter 2), which would be expected to occur
if the bulk of the ECTR DNA was being generated through cleavage of telomeres themselves.
Experiments in different mutant backgrounds suggest that additional mechanisms can be
involved in generating circular ECTR DNA. Telomerase positive human cells deficient in WRN,
the RecQ helicase implicated in Werner syndrome, have elevated levels of circular ECTR DNA,
which is generated in an XRCC3 independent mechanism (Li et al, 2008). The KU proteins
involved in non-homologous end joining also appear to be involved in suppressing formation of
circular ECTR in Arabidopsis, in a mechanism that is independent of MRE11, the RAD51
paralogs, and the SMC6 homolog implicated in intra-chromatid recombination (Zellinger et al,
2007). Additionally, it should be noted that eukaryotic cells can also contain non-telomeric extra-
chromosomal circular DNAs, which arise from genomic elements not known to form DNA
lariate structures (Gaubatz and Flores, 1990). The mechanistic details of how non-telomeric
extra-chromosomal circular elements are generated are largely unclear, but a genetic requirement
for ligase IV in the production of extra-chromosomal circular elements involving major satellite
135
DNA has been demonstrated, suggesting a mechanism where linear extra-chromosomal DNA
may also be subsequently ligated together to generate circular DNA (Cohen et al, 2006). ALT
cells contain both linear and circular ECTR DNA, raising the possibility that circular DNA can
also be generated by ligation of linear telomeric DNA substrates.
When FANCD2 is depleted, cells accumulate ECTR DNA both within and outside of APBs. One
possibility is that small pieces of ECTR outside of APBs represent a different type of DNA, such
as double-strand circular DNA, which does not activate cell cycle checkpoints and is therefore
not targeted to APBs. Telomeric DNA within APBs may primarily contain linear species of
DNA, as well as DNA containing single strand regions. Supporting this idea, BrdU incorporation
studies show that FANCD2-depleted APBs contain high amounts of ssDNA, and biochemical
purification of APBs from wild type ALT cells has shown a preferential association of linear vs
circular telomeric DNA with the bodies following DNA damage (Fasching et al, 2007).
Alternately, cells may begin to accumulate ECTR DNA outside of APBs in FANCD2 depleted
cells because it can no longer be properly targeted to APBs. Although FANCD2 depletion leads
to an increase in telomeric DNA content, expression of TRF1 and TRF2 does not appear to
upregulated. Sumoylation of telomere binding proteins appears to play an important role in
targeting telomeric DNA to APBs (Potts and Yu, 2007), raising the possibility that ECTR DNA
generated elsewhere in the nucleus can no longer be targeted to APBs in FANCD2-depleted
cells.
The production of extra-chromosomal DNA appears to be tied to genomic stability, and
accumulates at higher levels in cancer cells and cells exposed to DNA damaging agents (Cohen
et al, 1997; Cohen and Lavi, 2006; Cohen et al, 2007). Interestingly, primary FA cells, which are
inherently genomically unstable, have been reported to have both abnormally large and highly
elevated amounts of extra-chromosomal circular DNAs detectable by electron microscopy and
southern blotting of 2D gels with a Cot-1 DNA probe, suggesting that the FA pathway plays a
general role in suppressing formation of these molecules (Motejlek et al, 1993; Cohen et al,
2007). The sequences of extra-chromosomal DNA amplified in primary FA cells have not been
determined, however our FISH analysis suggests amplification of telomeric DNA is largely an
ALT-specific phenomenon, as it was not observed in FANCD2-depleted telomerase positive
cells. Whether additional sequences are amplified in FANCD2-depleted ALT cells remains to be
determined, as does the relative abundance of linear and circular ECTR DNA and the molecular
136
mechanism of its production. One intriguing possibility is that non-telomeric extra-chromosomal
sequences are also amplified in FANCD2-depleted ALT cells and are also sequestered within
APBs. In addition to telomeric instability, activation of ALT is also associated with instability of
the minisatellite MS32 (Jeyapalan et al, 2005), whether or not this sequence is also present as an
extra-chromosomal element in ALT cells is presently unknown. If APBs contain multiple types
of extra-chromosomal DNA, non-telomeric sequences may occasionally interact with
dysfunctional telomeres. This would result in telomeres that contain non-TTAGGG repeat
sequences, which could lead to telomere capping abnormalities. Integration of non-telomeric
DNA into telomeres has been reported in an ALT SV40 immortalized cell line derived from a
patient with Werner syndrome, wherein SV40 DNA sequences are interspersed in TTAGGG
repeats (Marciniak et al, 2005).
FANCD2-depletion results in a very rapid accumulation of ECTR DNA within cells, suggesting
that there is an amplification step occurring, which may be driven by replication and
recombination among pre-existing ECTR DNA. ECTR DNA synthesis in FANCD2-depleted
cells is not caused by overexpression of BLM, but rather appears to be due to dysregulation of a
process involving BLM. Although telomeres are a repetitive sequence element, pre-replicative
complexes can assemble on telomeric DNA and be converted into functional origins in Xenopus
extract systems, and replication forks originating within telomeres have been observed in mice,
suggesting that replication events can arise within telomeric sequences (Kurth and Gautier, 2009;
Sfeir et al, 2009). Alternately, replication may begin without a functional origin, as has been
proposed for break induced replication in yeast. BrdU incorporation experiments have previously
shown that newly replicated DNA accumulates within APBs during a time when normal S phase
replication at other loci is not occurring (Nabetani et al, 2004). Whether or not this DNA is
synthesized within APBs or is targeted to bodies after being replicated elsewhere is presently
unclear, however, APBs have been shown to contain proteins implicated in replication including
RPA and PCNA (Grudic et al, 2007; Jiang et al, 2009).
How FANCD2 may act to limit amplification of telomeric DNA remains an open, but intriguing
question. Purified human FANCD2 not only binds to ssDNA and dsDNA, but also to 3 and 4
way DNA junctions (Roques et al, 2009). One possible function of FANCD2 within ALT would
be to limit recombination between telomeric DNAs by affecting the stability of recombinational
intermediates. Type II recombinational telomere maintenance in yeast appears to rely on Break
137
Induced Replication (BIR), a mechanism in which one side of a double-strand break invades a
homologous sequence resulting in a displacement loop, which can be migrated as the invading
strand is extended, with newly synthesized single- strand being displaced and subsequently
converted to a double-strand product. Alternately, a uni-directional replication fork may
assemble within the displacement loop, allowing replication to continue until the end of the
template is reached. This may occur by a mechanism involving migration of the Holliday
junction and displacement of the newly synthesized DNA, or the Holliday junction may remain
static and have to be resolved after replication is finished (reviewed in McEachern and Haber,
2006).
Both telomeres, and linear ECTR DNA naturally represent one-sided breaks, making BIR a
likely pathway for telomere extension and ECTR DNA amplification. If strand invasion occurred
into a circular template, rolling circle replication could follow, leading to a very rapid increase in
telomeric DNA content. BIR pathways can occur through RAD51 dependent and independent
mechanisms. Genetic studies using telomerase deficient yeast relying on type II telomeric
recombination, which closely resembles ALT telomere maintenance, suggest that telomeres are
maintained through RAD51-independent form of BIR, which differs slightly from RAD51-
independent BIR at an induced double-strand break in that there is unique requirement for the
Sgs1 RecQ helicase at telomeric BIR (Signon et al, 2001; Huang et al, 2001).
Amplification of ECTR DNA in FANCD2 depleted cells also appears to occur through a largely
RAD51 independent, but BLM dependent mechanism, as codepletion of BLM, but not RAD51,
suppresses this phenotype. However, it should be noted that RAD51 is a common component of
APBs, and I observe an increase in the fraction of telomeric foci that colocalize with RAD51
when FANCD2 is depleted. This suggests that multiple mechanisms of recombination may be
occurring among human telomeric DNAs, and that RAD51 may participate in recombination
reactions when present, but additional mechanisms may function when RAD51 levels are
depleted. Support for the existence of RAD51-independent mechanisms in mammalian ALT
telomere maintenance is provided by studies in mice which show that RAD54, a RAD51
accessory factor required in RAD51-dependent telomere maintenance in yeast, is dispensable for
ALT telomere maintenance in telomerase deficient mouse cells (Akiyama et al, 2006; Chen et al,
2001). One RAD51-independent mechanism of recombination which may function in
mammalian cells may rely on TRF2, a protein present at high levels on telomeric DNA that can
138
promote strand invasion and subsequent t-loop formation in vitro (Griffith et al, 1999).
Alternately, interactions between complimentary regions of single-strand telomeric DNAs may
be promoted by strand annealing pathways, by-passing the need for a recombinase.
Although BLM possesses activities that aid in suppressing recombination and sister chromatid
exchanges at a global level, it also has multiple pro-recombinogenic activities that may be
essential in promoting BIR at telomeres. BLM may be required to resolve formation of
G-quadruplex structures, which may form on the telomeric G strand prior to strand invasion.
Additionally, a role for BLM in stimulating the resection of double-strand breaks required to
generate the 3’ssDNA overhang involved in subsequent recombination reactions has recently
been demonstrated in both in vitro and in vivo systems (Nimonkar et al, 2008; Gravel et al,
2008). BLM also promotes the annealing of complimentary ssDNA regions in vitro, which may
play a role in promoting interactions between telomeric DNAs (Cheok et al, 2005). When BLM
is depleted these events may occur less efficiently, leading to a decrease in accessible ssDNA or
telomeric recombinational intermediate structures, which may explain our previous observation
that siRNA knockdown of BLM results in a significant reduction in FANCD2 localization to
telomeric foci (chapter 2). Conversely, when BLM is overexpressed there may be an increase in
interactions between telomeric DNAs and BIR, resulting in the dramatic ALT specific increase
in telomeric DNA that has been previously observed (Stavropoulos et al, 2002).
In addition to early roles in the recombinational process, BLM also possesses activities that may
be important in promoting efficient replication of telomeric DNA, including the ability to
promote branch migration of Holliday junctions (Karow et al, 2000). Additionally, both BIR and
types I and II telomere maintenance in yeast rely on pol 32, a subunit of pol δ, suggesting that
pol δ is the polymerase involved in recombinational telomere maintenance (Lydeard et al,
2007). BLM directly interacts with the p12 subunit of human pol δ, and can promote the strand
displacement activity of pol δ in vitro, which may be important for efficient replication of
gapped telomeric substrates (Selak et al, 2008).
While BLM may function in multiple ways to promote ECTR DNA synthesis, at least one of
these functions relies on the DNA unwinding ability of BLM, as overexpression of BLM with a
nonfunctional helicase domain does not result in increased telomeric DNA synthesis
(Stavropoulos et al, 2002). It is also interesting to note that the WRN RecQ helicase also
139
localizes to APBs and telomeric foci in ALT cells, and similar to BLM, WRN interacts with
TRF2 in vitro and in vivo, and TRF2 stimulates the helicase activity of both BLM and WRN
(Johnson et al, 2001; Opresko et al, 2002). Both BLM and WRN can readily unwind
G-quadruplex DNA structures, yet each helicase appears to have non-overlapping roles at
telomeres (Mohaghegh et al, 2001). Codepletion of BLM with FANCD2 completely suppresses
the FANCD2-depletion telomere phenotype, suggesting that WRN is not able to substitute for
BLM. Likewise, mouse cells deficient for BLM show a higher frequency of telomeres with a
discontinuous staining pattern observed by FISH, suggested to be due to problems with telomeric
replication caused by G-quadruplexes, which is not observed in WRN deficient mouse cells
(Sfeir et al, 2009). However, in human cells, WRN deficiency appears to result in specific
problems with replication of the telomeric G strand, again suggesting that BLM cannot substitute
for the WRN helicase (Crabbe et al, 2004).
Depletion of FANCD2 in ALT cells also results in an increase in the frequency of telomeres with
a discontinuous staining pattern via FISH, which is not observed in FANCD2-depleted
telomerase positive cells. It has been suggested that this altered staining pattern is similar to what
occurs at aphidicolin induced common fragile sites, and that telomeres with this staining pattern
represent telomeres expressing fragile site characteristics (Sfeir et al, 2009). Interestingly, the
frequency of telomeres with this phenotype is increased >10 fold in the two ALT cell lines I
examined relative to two telomerase positive cell lines. It has been suggested that insufficient
expression of shelterin components may contribute to telomere abnormalities in ALT cells,
resulting in a partial capping defect (Cesare et al, 2009). In non-ALT cells, depletion of TRF1
increases both the frequency of telomeres with this fragile appearance and replication fork
stalling within telomeric DNA, suggesting that TRF1 has a role in promoting telomeric
replication and suppressing fragile telomeres (Sfeir et al, 2009). Insufficient amounts of TRF1
within ALT cells is one potential cause of the increase in fragile telomeres. While FANCD2-
depleted ALT cells have increased amounts of telomeric DNA, I do not observe a concomitant
increase in TRF1 or TRF2 expression. This further skewing of the ratio of telomeric DNA to
telomere binding proteins may be a contributing factor to both the ALT specific increase in
telomeric DNA that has activated a DNA damage response (chapter 2) and telomeres with a
fragile appearance.
140
When FANCD2 is depleted I observe an increase in the frequency of T-SCEs in ALT, but not in
telomerase positive cells. T-SCEs were measured during the first round of replication after
FANCD2 protein levels are reduced, which proceeds the accumulation of high amounts of ECTR
DNA, arguing against a dramatic reduction in telomere capping proteins as a contributing factor
to this increase in T-SCEs. This suggests that FANCD2 is acting to directly limit recombination
between telomeric DNA. The observation that FANCD2 depletion results in an ALT-specific
increase in T-SCEs is at odds with a recent publication by Fan and colleagues, who observed an
apparent ALT-specific decrease in T-SCEs when FANCD2 is depleted (Fan et al, 2009). The
reason for this disparity is unclear, however the baseline level of T-SCEs observed in control
cells by Fan et al, was abnormally high at 40-45 T-SCEs per 100 chromosomes, meaning that 40-
45% of chromosomes have a T-SCE. The upper range of T-SCEs previously reported in these
cell lines is 66–105 T-SCEs per 100 metaphase spreads, which given that these cell lines have
hyperdiploid and hypertriploid chromosome numbers, corresponds to approximately 1% of
chromosomes (Londono-Vallejo et al, 2004). While there is a growing amount of variability of
reported T-SCE frequencies between groups, which may relate to FISH sensitivity, differences in
how well the newly synthesized strand is degraded, and genetic variability between cell lines, a
frequency of 40-45% of chromosome having a T-SCEs is abnormally high. Based on the
indicated BrdU labeling time used by Fan et al, elevated T-SCEs are likely due to incomplete
degradation of the newly synthesized strand. However, why levels of T-SCEs decrease when
FANCD2 is depleted is unknown, but is unlikely to be due to be superior silencing of FANCD2,
as Fan et al used FANCD2 siRNA sequence D to knockdown FANCD2, which does not
significantly reduce FANCD2 protein level (Figure 3-1 A). Interestingly, although I do not see
an increase in T-SCEs in the telomerase positive HT0180 cell line when FANCD2 is depleted,
mouse cells that have short telomeres and are deficient in both FANCC and telomerase have
elevated levels of T-SCEs, supporting a role for the FA pathway in limiting telomeric
recombination (Rhee et al, 2010).
The underlying mechanism of T-SCEs is currently unknown, but appears to differ from DNA
damage induced SCEs and spontaneous SCEs at other genomic loci. Unlike Bloom syndrome
cells that have increased spontaneous genomic SCEs, human FA cells have normal levels of
spontaneous genomic SCEs (Hemphill et al, 2009). Additionally, ALT cells deficient in RAD54
have suppressed levels of mitomycin C induced SCEs, but continue to have elevated levels of
141
T-SCEs (Akiyama et al, 2006). Whether T-SCEs in ALT cells occur through the same
mechanism as T-SCEs in non-ALT cells is presently unclear. In a non-ALT setting, T-SCEs are
likely driven by short telomeres, however elongating short telomeres in ALT cells by expressing
telomerase does not suppress elevated levels of T-SCEs (Morrish and Greider, 2009; Londoño-
Vallejo et al, 2004). Proteins implicated in T-SCE formation such as Mus81 and FANCD2
appear to affect the frequencies of T-SCEs in ALT cells, but not telomerase positive cells, which
may be due to a difference in the integrity of the telomere capping structure, or may be indicative
of a different mechanism driving the T-SCEs in the ALT setting (Zeng et al, 2009).
Codepletion of BLM, but not RAD51, reduces the increased frequency of T-SCEs observed in
FANCD2-depleted ALT cells to wild-type levels. This suggests that FANCD2 suppresses
T-SCEs that are driven by a RAD51-independent, but BLM-dependent process. The frequency of
genomic SCEs appears to increase in cells codepleted of FANCD2 and BLM, while T-SCE
frequency decreases, again suggesting a different mechanism of genomic SCE production from
T-SCEs. As discussed previously, BLM may be required to generate T-SCEs by promoting end
resection, resolving secondary structures, or promoting strand annealing. Interestingly, BLM
codepletion with FANCD2 does not completely suppress T-SCEs, but rather returns them to
wild-type levels. This may be due to residual BLM protein levels expressed in siRNA treated
cells, or may be because T-SCEs can arise from multiple mechanisms.
FANCD2 has been shown to accumulate preferentially on ssDNA in vitro, and FANCD2 foci
form at in vivo regions enriched in ssDNA (Roques et al, 2009). The function of FANCD2 at
ssDNA is presently unknown, however we observe that when FANCD2 is depleted, cells
accumulate APBs with highly elevated amounts of ssDNA, which may be a byproduct of BIR, or
an indication of excessive resection of dsDNA. FANCD2 may help to suppress telomeric
recombination reactions by binding to existing ssDNA regions, which are present at elevated
frequency within ALT telomeric DNA, and limiting their further resection, as more extended
regions of ssDNA may be involved in recombinational reactions.
When FANCD2 is depleted, cells also display more frequent entanglements involving telomeric
DNA, observed as linear telomeric DNA fibers running between telomeric foci in interphase
cells. Examination of prometaphase cells suggests that these fibers frequently involve telomeric
DNA connecting telomeres and APBs. Fibers may be formed if telomeres move away from
142
APBs before replication is complete, Holliday junctions are resolved, or DNA is decatinated.
Increased frequency of telomere fibers in FANCD2 depleted cells may reflect a direct role for
FANCD2 in these processes, or may be a secondary effect of increased interactions among
telomeric DNAs. The observation that codepletion of BLM with FANCD2 returns the frequency
of telomere fibers returns to baseline levels, but does not complete suppress their formation,
suggests either that sufficient BLM remains to promote entanglements, or that entanglements
may arise through because of multiple factors, some of which are BLM independent.
The fission yeast Rqh1 RecQ helicase also promotes telomeric DNA entanglements, hyper-
recombination, and chromosome missegregation when the Taz1 telomere binding protein is
deleted, and cells are grown at lower temperatures (Miller and Cooper, 2003; Rog et al., 2009).
In this situation, telomere problems arise during replication, suggesting that they may be caused
during the restart of stalled or collapsed replication forks. The FA pathway has also been
implicated in the response to stalled or collapsed replication forks, raising the question of
whether some of the telomere abnormalities observed in FANCD2-depleted ALT cells may arise
during replication. Supporting this idea, I observe an increase in the fraction of the cell
population that shows abnormally high levels of ECTR DNA as the time post knockdown
increases, which may be accounted for if a fraction of cells encounter problems each time they
try to replicate. However, the role of the FANCD2 in the response to stalled/collapsed replication
forks appears to be regulated by ATR (Andreassen et al, 2004), and I do not see a requirement
for ATR expression for the majority of FANCD2 localization to telomeric foci (chapter 2).
Additionally, when I significantly reduced ATR expression in ALT cells, I did not observe any
increase in telomeric DNA content (data not shown), and treatment of ALT cells with caffeine to
inhibit of ATM and ATR has been reported to cause a decrease in telomeric DNA synthesis at
APBs (Nabetani et al, 2004). Therefore I presently favour the hypothesis that telomere
abnormalities in FANCD2-depleted cells arise due to a deregulation of recombination reactions
among telomeric DNA at sites unrelated to collapsed replication forks. Support for a role of
FANCD2 in regulating recombination outside of the context of replication problems is provided
by the observation that it FANCD2 is found at both telomeres and recombinational nodules
during meiosis, and FANCD2 null cells show an increase in both unpaired and mispaired meiotic
chromosomes (Garcia-Higuera et al, 2001; Houghtaling et al, 2003).
143
In this study I provide evidence suggesting that FANCD2 functions to suppress recombination
reactions among telomeric DNA within the ALT telomere setting. Interactions between telomeric
DNA appear to form in a BLM-dependent, RAD51-independent process, and can become
dysregulated through overexpression of BLM, or depletion of FANCD2. While telomeric
recombination is essential for ALT telomere maintenance, if it occurs in an unregulated manner
it may lead to excessive production of ECTR DNA, telomeric DNA entanglements, and
increased levels of telomeric DNA which activates a DNA damage response. Excessive
recombination of telomeric DNA appears to lead to reduced cell viability, as ALT cells with high
levels of BLM, or low levels of FANCD2, show increased cell death. While impaired viability
may also be due to non-telomeric problems, the observation that codepletion of FANCD2 and
BLM corrects telomeric abnormalities and partially rescues viability suggest that telomeric
abnormalities are a contributing factor. This work serves to emphasize that RecQ helicases can
act as double-edged swords, acting to both promote and impair genomic stability depending on
the context. It also emphasizes the role of FANCD2 in regulating recombination, and suggests a
novel relationship between FANCD2 and BLM, wherein a function of FANCD2 is to regulate
BLM dependent processes.
144
3.6 References
Ambrosini G, Seelman SL, Qin LX, Schwartz GK (2008). The cyclin-dependent kinase inhibitor flavopiridol potentiates the effects of topoisomerase I poisons by suppressing Rad51 expression in a p53-dependent manner. Cancer Res. 68:2312-20.
Akiyama K, Yusa K, Hashimoto H, Poonepalli A, Hande MP, Kakazu N, Takeda J, Tachibana M, Shinkai Y (2006). Rad54 is dispensable for the ALT pathway. Genes Cells. 11:1305-15.
Akkari YM, Bateman RL, Reifsteck CA, D'Andrea AD, Olson SB, Grompe M (2001). The 4N cell cycle delay in Fanconi anemia reflects growth arrest in late S phase. Mol Genet Metab. 74:403-12.
Andreassen PR, D'Andrea AD, Taniguchi T (2004). ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev. 15: 1958-63
Auerbach AD, Warburton D, Bloom AD, Chaganti RS (1979). Preliminary communication: prenatal detection of the Fanconi Anemia gene by cytogenetic methods. Am J Hum Genet. 31:77-81.
Auerbach AD (2009). Fanconi anemia and its diagnosis. Mutat Res. 668:4-10.
Aurias A, Antoine JL, Assathiany R, Odievre M, Dutrillaux B (1985). Radiation sensitivity of Bloom's syndrome lymphocytes during S and G2 phases. Cancer Genet Cytogenet. 16:131-6.
Azzalin CM, Reichenbach P, Khoriauli L, Giulotto E, Lingner J (2007). Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. Science. 318:798-801.
Bachrati CZ, Hickson ID (2008). RecQ helicases: guardian angels of the DNA replication fork. Chromosoma. 117:219-33.
Bhattacharyya S, Keirsey J, Russell B, Kavecansky J, Lillard-Wetherell K, Tahmaseb K, Turchi JJ, Groden J (2009). Telomerase-associated protein 1, HSP90, and topoisomerase IIalpha associate directly with the BLM helicase in immortalized cells using ALT and modulate its helicase activity using telomeric DNA substrates. J Biol Chem. 284:14966-77.
145
Bryan TM, Englezou A, Gupta J, Bacchetti S, Reddel RR (1995). Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J. 14:4240-8.
Cesare AJ, Kaul Z, Cohen SB, Napier CE, Pickett HA, Neumann AA, Reddel RR (2009). Spontaneous occurrence of telomeric DNA damage response in the absence of chromosome fusions. Nat Struct Mol Biol. 16:1244-51.
Chaganti RS, Schonberg S, German J (1974). A manyfold increase in sister chromatid exchanges in Bloom's syndrome lymphocytes. Proc Natl Acad Sci U S A. 71:4508-12.
Chan KL, Palmai-Pallag T, Ying S, Hickson ID (2009). Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol. 11:753-60.
Chen Q, Ijpma A, Greider CW (2001). Two survivor pathways that allow growth in the absence of telomerase are generated by distinct telomere recombination events. Mol Cell Biol. 21:1819-27.
Cheok CF, Wu L, Garcia PL, Janscak P, Hickson ID (2005). The Bloom's syndrome helicase promotes the annealing of complementary single-stranded DNA. Nucleic Acids Res. 33:3932-41.
Cohen S, Regev A, Lavi S (1997). Small polydispersed circular DNA (spcDNA) in human cells: association with genomic instability. Oncogene. 14:977-85.
Cohen S, Lavi S (2006). Induction of circles of heterogeneous sizes in carcinogen-treated cells: two-dimensional gel analysis of circular DNA molecules. Mol Cell Biol. 16:2002-14.
Cohen S, Regev A, Lavi S (2007). Small polydispersed circular DNA (spcDNA) in human cells: association with genomic instability. Oncogene. 14:977-85.
Compton SA, Choi JH, Cesare AJ, Ozgür S, Griffith JD (2007). Xrcc3 and Nbs1 are required for the production of extra-chromosomal telomeric circles in human alternative lengthening of telomere cells. Cancer Res. 67:1513-9.
Davies AA, Masson JY, McIlwraith MJ, Stasiak AZ, Stasiak A, Venkitaraman AR, West SC (2001). Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Mol. Cell. 7:273–282.
Deans AJ, West SC (2009). FANCM connects the genome instability disorders Bloom's Syndrome and Fanconi Anemia. Mol Cell. 36:943-53.
Dunham MA, Neumann AA, Fasching CL, Reddel RR (2000). Telomere maintenance by recombination in human cells. Nat Genet. 26:447-50.
Dutrillaux, B., Aurias, A., Dutrillaux, A.M., Buriot, D. and Prieur, M (1982). The cell cycle of lymphocytes in Fanconi anemia. Hum. Genet. 62:327–332.
Ellis NA, Groden J, Ye TZ, Straughen J, Lennon DJ, Ciocci S, Proytcheva M, German J (1995). The Bloom's syndrome gene product is homologous to RecQ helicases. Cell. 83:655-66.
Fan Q, Zhang F, Barrett B, Ren K, Andreassen PR (2009). A role for monoubiquitinated FANCD2 at telomeres in ALT cells. Nucleic Acids Res. 37:1740-54.
Fasching CL, Neumann AA, Muntoni A, Yeager TR, Reddel RR (2007). DNA damage induces alternative lengthening of telomeres (ALT) associated promyelocytic leukemia bodies that preferentially associate with linear telomeric DNA. Cancer Res. 67:7072-7.
Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D'Andrea AD (2001). Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell 7:249-62.
Gaubatz JW, Flores SC (1990). Tissue-specific and age-related variations in repetitive sequences of mouse extra-chromosomal circular DNAs. Mutat Res. 237:29-36.
German J (1993). Bloom syndrome: a mendelian prototype of somatic mutational disease. Medicine. 72:393-406.
German J, Crippa LP (1996). Chromosomal breakage in diploid cell lines from Bloom’s syndrome and Fanconi’s anemia. Ann Genet. 9: 143-154.
Godthelp BC, Wiegant WW, Waisfisz Q, Medhurst AL, Arwert F, Joenje H, Zdzienicka MZ (2006). Inducibility of nuclear Rad51 foci after DNA damage distinguishes all Fanconi anemia complementation groups from D1/BRCA2. Mutat Res. 594:39-48.
Gravel S, Chapman JR, Magill C, Jackson SP (2008). DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 22:2767-72.
Griffith JD, Comeau L, Rosenfield S, Stansel RM, Bianchi A, Moss H, de Lange T (1999). Mammalian telomeres end in a large duplex loop. Cell 97:503-14.
Grudic A, Jul-Larsen A, Haring SJ, Wold MS, Lønning PE, Bjerkvig R, Bøe SO (2007). Replication protein A prevents accumulation of single-stranded telomeric DNA in cells that use alternative lengthening of telomeres. Nucleic Acids Res. 35:7267-78.
Hemphill AW, Akkari Y, Newell AH, Schultz RA, Grompe M, North PS, Hickson ID, Jakobs PM, Rennie S, Pauw D, Hejna J, Olson SB, Moses RE (2009). Topo IIIalpha and BLM act within the Fanconi anemia pathway in response to DNA-crosslinking agents. Cytogenet Genome Res. 125:165-75.
Hirano S, Yamamoto K, Ishiai M, Yamazoe M, Seki M, Matsushita N, Ohzeki M, Yamashita YM, Arakawa H, Buerstedde JM, Enomoto T, Takeda S, Thompson LH, Takata M (2005). Functional relationships of FANCC to homologous recombination, translesion synthesis, and BLM. EMBO J. 24:418-27.
Houghtaling S, Timmers C, Noll M, Finegold MJ, Jones SN, Meyn MS, Grompe M (2003). Epithelial cancer in Fanconi anemia complementation group D2 (Fancd2) knockout mice. Genes Dev.17:2021-35.
Howlett NG, Taniguchi T, Durkin SG, D'Andrea AD, Glover TW (2005). The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Hum Mol Genet. 14:693-701.
Huang P, Pryde FE, Lester D, Maddison RL, Borts RH, Hickson ID, Louis EJ (2001). SGS1 is required for telomere elongation in the absence of telomerase. Curr Biol. 11:125-9.
148
Jeyapalan JN, Varley H, Foxon JL, Pollock RE, Jeffreys AJ, Henson JD, Reddel RR, Royle NJ (2005). Activation of the ALT pathway for telomere maintenance can affect other sequences in the human genome. Hum Mol Genet. 14:1785-94.
Jiang WQ, Zhong ZH, Nguyen A, Henson JD, Toouli CD, Braithwaite AW, Reddel RR (2009). Induction of alternative lengthening of telomeres-associated PML bodies by p53/p21 requires HP1 proteins. J Cell Biol. 185:797-810.
Johnson FB, Marciniak RA, McVey M, Stewart SA, Hahn WC, Guarente L (2001). The Saccharomyces cerevisiae WRN homolog Sgs1p participates in telomere maintenance in cells lacking telomerase. EMBO J. 20:905-13.
Karow JK, Constantinou A, Li JL, West SC, Hickson ID (2000). The Bloom's syndrome gene product promotes branch migration of holliday junctions. Proc Natl Acad Sci U S A. 97:6504-8.
Kitao S, Shimamoto A, Goto M, Miller RW, Smithson WA, Lindor NM, Furuichi Y (1999). Mutations in RECQL4 cause a subset of cases of Rothmund-Thomson syndrome. Nat Genet. 22:82-4.
Kurth I, Gautier J (2010). Origin-dependent initiation of DNA replication within telomeric sequences. Nucleic Acids Res. 38:467-76
Le S, Moore JK, Haber JE, Greider CW (1999). RAD50 and RAD51 define two pathways that collaborate to maintain telomeres in the absence of telomerase. Genetics. 152:143-52.
Li B, Jog SP, Reddy S, Comai L (2008). WRN controls formation of extra-chromosomal telomeric circles and is required for TRF2DeltaB-mediated telomere shortening. Mol Cell Biol. 28:1892-904.
Lillard-Wetherell K, Machwe A, Langland GT, Combs KA, Behbehani GK, Schonberg SA, German J, Turchi JJ, Orren DK, Groden J (2004). Association and regulation of the BLM helicase by the telomere proteins TRF1 and TRF2. Hum Mol Genet. 13:1919-32.
Lin CY, Chang HH, Wu KJ, Tseng SF, Lin CC, Lin CP, Teng SC (2005). Extra-chromosomal telomeric circles contribute to Rad52-, Rad50-, and polymerase delta-mediated telomere-telomere recombination in Saccharomyces cerevisiae. Eukaryot Cell. 4:327-36.
149
Liu Y, Masson JY, Shah R, O'Regan P, West SC (2004). RAD51C is required for Holliday junction processing in mammalian cells. Science. 303:243-6.
Londoño-Vallejo JA, Der-Sarkissian H, Cazes L, Bacchetti S, Reddel RR (2004). Alternative lengthening of telomeres is characterized by high rates of telomeric exchange. Cancer Res. 64:2324-7.
Lundblad V, Blackburn EH (1993). An alternative pathway for yeast telomere maintenance rescues est1- senescence. Cell. 73:347-60.
Lydeard JR, Jain S, Yamaguchi M, Haber JE (2007). Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature. 448:820-3.
Marciniak RA, Cavazos D, Montellano R, Chen Q, Guarente L, Johnson FB (2005). A novel telomere structure in a human alternative lengthening of telomeres cell line. Cancer Res. 65:2730-7.
McEachern MJ, Haber JE (2006). Break-induced replication and recombinational telomere elongation in yeast. Annu Rev Biochem. 75:111-35.
Meetei AR, Sechi S, Wallisch M, Yang D, Young MK, Joenje H, Hoatlin ME, Wong W. 2003. A multiprotein nuclear complex connects Fanconi anemia and Bloom syndrome. Mol. Cell. Biol. 23: 3417-3426.
Miller KM, Cooper JP (2003). The telomere protein Taz1 is required to prevent and repair genomic DNA breaks. Mol Cell. 11:303-13. Mutat Res. 293:205-14.
Mohaghegh P, Karow JK, Brosh Jr RM Jr, Bohr VA, Hickson ID (2001). The Bloom's and Werner's syndrome proteins are DNA structure-specific helicases. Nucleic Acids Res. 29:2843-9.
Morrish TA, Greider CW (2009). Short telomeres initiate telomere recombination in primary and tumor cells. PLoS Genet. 5(1):e1000357.
Motejlek K, Schindler D, Assum G, Krone W (1993). Increased amount and contour length distribution of small polydisperse circular DNA (spcDNA) in Fanconi anemia.
150
Muntoni A, Neumann AA, Hills M, Reddel RR (2009). Telomere elongation involves intra-molecular DNA replication in cells utilizing alternative lengthening of telomeres. Hum Mol Genet. 18:1017-27.
Nabetani A, Yokoyama O, Ishikawa F (2004). Localization of hRad9, hHus1, hRad1, and hRad17 and caffeine-sensitive DNA replication at the alternative lengthening of telomeres-associated promyelocytic leukemia body. J Biol Chem. 279:25849-57.
Nabetani A, Ishikawa F (2009). Unusual telomeric DNAs in human telomerase-negative immortalized cells. Mol Cell Biol. 29: 703-713.
Naim V, Rosselli F (2009). The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat Cell Biol. 11:761-8.
Nakanishi K, Yang Y, Pierce A, Taniguchi T, Digweed M, D’Andrea AD, Wang Z, Jasin M (2005). Human Fanconi anemia monoubiquitination pathway promotes homologous DNA repair. PNAS 102: 1110-1115.
Natarajan S, McEachern MJ (2002). Recombinational telomere elongation promoted by DNA circles. Mol Cell Biol. 22:4512-21.
Ng LJ, Cropley JE, Pickett HA, Reddel RR, Suter CM (2009). Telomerase activity is associated with an increase in DNA methylation at the proximal subtelomere and a reduction in telomeric transcription. Nucleic Acids Res. 37:1152-9.
Nimonkar AV, Ozsoy AZ, Genschel J, Modrich P, Kowalczykowski SC (2008). Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proc Natl Acad Sci U S A. 105:16906-11.
Opresko PL, von Kobbe C, Laine JP, Harrigan J, Hickson ID, Bohr VA (2002). Telomere-binding protein TRF2 binds to and stimulates the Werner and Bloom syndrome helicases. J Biol Chem. 277:41110-9.
Pellegrini L, Yu DS, Lo T, Anand S, Lee M, Blundell TL, Venkitaraman AR (2002). Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature. 420:287–293.
Pichierri P, Franchitto A, Rosselli F (2004). BLM and the FANC proteins collaborate in a common pathway in response to stalled replication forks. EMBO J. 23:3154-63.
151
Potts PR, Yu H (2007). The SMC5/6 complex maintains telomere length in ALT cancer cells through SUMOylation of telomere-binding proteins. Nat Struct Mol Biol. 2007 14:581-90.
Raderschall E, Bazarov A, Cao J, Lurz R, Smith A, Mann W, Ropers HH, Sedivy JM, Golub EI, Fritz E, Haaf T (2002). Formation of higher-order nuclear Rad51 structures is functionally linked to p21 expression and protection from DNA damage-induced apoptosis. J Cell Sci. 115:153-64.
Rog O, Miller KM, Ferreira MG, Cooper JP (2009). Sumoylation of RecQ helicase controls the fate of dysfunctional telomeres. Mol Cell. 33:559-69.
Roques C, Coulombe Y, Delannoy M, Vignard J, Grossi S, Brodeur I, Rodrigue A, Gautier J, Stasiak AZ, Stasiak A, Constantinou A, Masson JY (2009). MRE11-RAD50-NBS1 is a critical regulator of FANCD2 stability and function during DNA double-strand break repair. EMBO 28:2400-13.
Schroeder TM, Kurth R (1971). Spontaneous chromosomal breakage and high incidence of leukemia in inherited disease. Blood. 37:96-112.
Schroeder TM, German J (1974). Bloom's syndrome and Fanconi's anemia: demonstration of two distinctive patterns of chromosome disruption and rearrangement. Humangenetik. 25:299-306.
Selak N, Bachrati CZ, Shevelev I, Dietschy T, van Loon B, Jacob A, Hübscher U, Hoheisel JD, Hickson ID, Stagljar I (2008). The Bloom's syndrome helicase (BLM) interacts physically and functionally with p12, the smallest subunit of human DNA polymerase delta. Nucleic Acids Res. 36:5166-79.
Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, de Lange T (2009). Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell. 138:90-103.
Signon L, Malkova A, Naylor ML, Klein H, Haber JE (2001). Genetic requirements for RAD51- and RAD54-independent break-induced replication repair of a chromosomal double-strand break. Mol Cell Biol. 21:2048-56.
152
Siitonen HA, Kopra O, Kääriäinen H, Haravuori H, Winter RM, Säämänen AM, Peltonen L, Kestilä M (2003). Molecular defect of RAPADILINO syndrome expands the phenotype spectrum of RECQL diseases. Hum Mol Genet. 12:2837-44.
Siitonen HA, Sotkasiira J, Biervliet M, Benmansour A, Capri Y, Cormier-Daire V, Crandall B, Hannula-Jouppi K, Hennekam R, Herzog D, Keymolen K, Lipsanen-Nyman M, Miny P, Plon SE, Riedl S, Sarkar A, Vargas FR, Verloes A, Wang LL, Kääriäinen H, Kestilä M (2009). The mutation spectrum in RECQL4 diseases. Eur J Hum Genet. 17:151-8.
Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER 3rd, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D'Andrea AD, Elledge SJ (2007). Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 129:289-301.
Spardy N, Duensing A, Hoskins EE, Wells SI, Duensing S (2008). HPV-16 E7 reveals a link between DNA replication stress, fanconi anemia D2 protein, and alternative lengthening of telomere-associated promyelocytic leukemia bodies. Cancer Res. 68:9954-63.
Stavropoulos DJ, Bradshaw PS, Li X, Pasic I, Truong K, Ikura M, Ungrin M, Meyn MS. 2002. The Bloom syndrome helicase BLM interacts with TRF2 in ALT cells and promotes telomeric DNA synthesis. Hum. Mol. Genet. 11: 3135-3144.
Sun H, Karow JK, Hickson ID, Maizels N (1998). The Bloom's syndrome helicase unwinds G4 DNA. J Biol Chem. 273:27587-92.
Tabori U, Vukovic B, Zielenska M, Hawkins C, Braude I, Rutka J, Bouffet E, Squire J, Malkin D (2006). The role of telomere maintenance in the spontaneous growth arrest of pediatric low-grade gliomas. Neoplasia. 8:136-42.
Takai H, Smogorzewska A, de Lange T (2003). DNA damage foci at dysfunctional telomeres. Curr Biol. 2003 13:1549-56.
Teng SC, Zakian VA (1999). Telomere-telomere recombination is an efficient bypass pathway for telomere maintenance in Saccharomyces cerevisiae. Mol Cell Biol. 19:8083-93.
Verdun RE, Karlseder J (2006). The DNA damage machinery and homologous recombination pathway act consecutively to protect human telomeres. Cell. 127:709-20.
153
Wang RC, Smogorzewska A, de Lange T (2004). Homologous recombination generates T-loop-sized deletions at human telomeres. Cell. 119:355-68.
Wilson JB, Yamamoto K, Marriott AS, Hussain S, Sung P, Hoatlin ME, Mathew CG, Takata M, Thompson LH, Kupfer GM, Jones NJ (2008). FANCG promotes formation of a newly identified protein complex containing BRCA2, FANCD2 and XRCC3. Oncogene. 27:3641-52.
Yeager TR, Neumann AA, Englezou A, Huschtscha LI, Noble JR, Reddel RR (1999). Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res. 59:4175-9.
Yu CE, Oshima J, Fu YH, Wijsman EM, Hisama F, Alisch R, Matthews S, Nakura J, Miki T, Ouais S, Martin GM, Mulligan J, Schellenberg GD (1996). Positional cloning of the Werner's syndrome gene. Science. 272:258-62.
Yuan SS, Lee SY, Chen G, Song M, Tomlinson GE, Lee EY (1999). BRCA2 is required for ionizing radiation-induced assembly of Rad51 complex in vivo. Cancer Res. 59:3547–3551.
Zellinger B, Akimcheva S, Puizina J, Schirato M, Riha K (2007). Ku suppresses formation of telomeric circles and alternative telomere lengthening in Arabidopsis. Mol Cell. 27:163-9.
Zeng S, Xiang T, Pandita TK, Gonzalez-Suarez I, Gonzalo S, Harris CC, Yang Q (2009). Telomere recombination requires the MUS81 endonuclease. Nat Cell Biol. 11:616-23.
Zhong ZH, Jiang WQ, Cesare AJ, Neumann AA, Wadhwa R, Reddel RR (2007). Disruption of telomere maintenance by depletion of the MRE11/RAD50/NBS1 complex in cells that use alternative lengthening of telomeres. J Biol Chem. 282:29314-22.
Zhu W, Dutta A (2006). An ATR- and BRCA1-mediated Fanconi anemia pathway is required for activating the G2/M checkpoint and DNA damage repair upon rereplication. Mol Cell Biol. 26:4601-11.
Zijlmans JM, Martens UM, Poon SS, Raap AK, Tanke HJ, Ward RK, Lansdorp PM (1997). Telomeres in the mouse have large inter-chromosomal variations in the number of T2AG3 repeats. Proc Natl Acad Sci U S A. 94:7423-8.
154
Chapter 4
4 Summary and Future Directions
4.1 Summary and Future Directions
In this thesis I provide evidence for a critical role of the FA pathway in ALT telomere
maintenance in human cells. Examination of the localization of endogenous proteins using
indirect immunofluorescence clearly shows that components of the FA core complex, as well as
FANCD2, frequently localize to telomeric foci in ALT cells, but not in telomerase positive or
primary cells. The FA core complex most likely localizes to telomeric foci to promote FANCD2
monoubiquitination, a requirement for its accumulation in ALT telomeric foci.
Coimmunoprecipitation experiments in late S/G2 cells support a specific role for FANCD2 in the
ALT pathway of telomere maintenance, as interactions between endogenous FANCD2, TRF2
and BLM were detected in ALT cells, but not telomerase positive cells.
Most colocalization events between FA and telomeric proteins occur within larger telomeric foci,
corresponding to APBs. Analysis of APBs using energy-filtered transmission electron
microscopy shows that APBs are composed of an outer protein shell surrounding an inner DNA
and protein core, and that DNA within the body differs significantly from chromatin and likely
represents non-nucleosomal ECTR DNA. FISH analysis of pro-metaphase and metaphase cells
adds support to the idea that large telomeric foci are primarily composed of ECTR DNA. This
suggests that at least one function of the FA pathway within ALT telomere maintenance involves
ECTR DNA. SiRNA depletion of FANCD2 confirms an ALT specific role for FANCD2 in the
regulation of ECTR DNA production, as a proportion of FANCD2-depleted ALT cells have
markedly elevated amounts of ECTR DNA, which accumulates both within, and outside of
APBs. The mechanism by which FANCD2 depletion causes ECTR DNA amplification is
presently unclear, but is unlikely to be solely a result of cleavage of telomeric DNA from
telomeres themselves, as FANCD2 depletion does not cause an increase in telomere signal free
ends.
The function of FANCD2 within ALT appears to be closely intertwined with that of the BLM
helicase. This is supported by the observations that 1) FANCD2 coimmunoprecipitates with
BLM in late S/G2 ALT cells, 2) FANCD2 almost always colocalizes to telomeric foci that also
155
contain BLM, 3) depletion of BLM suppresses the localization of FANCD2 to telomeric foci, 4)
overexpression of BLM causes a rapid ALT specific increase in telomeric DNA similar to what
is observed in FANCD2-depleted cells (Stavropoulos et al., 2002), and 5) codepletion of BLM
with FANCD2 suppresses the increase in ECTR DNA, T-SCEs, and telomere entanglements
normally observed in FANCD2-depleted cells. Expression of BLM is not increased in FANCD2-
depleted ALT cells, ruling this out as a cause of ECTR DNA amplification. BLM may have
multiple critical functions at ALT telomeres including, but not limited to, generation of a 3’
ssDNA overhang if recombination is initiated at internal telomeric sites, resolving secondary
DNA structures to allow recombination and/or replication to occur, and/or promoting Holliday
junction migration if replication involves a break induced replication mechanism. Conversely,
there are multiple steps where FANCD2 may be acting within ALT. FANCD2 may bind to the
ssDNA overhang, blocking the invasion step in telomeric recombination, may act downstream of
this step to destabilize recombinants once they have formed, or may in some other manner
impede the replication step.
Identifying the polymerase involved in the amplification of ECTR DNA in ALT cells will allow
the potential role of FANCD2 in telomeric replication to be explored. The FA pathway has been
implicated in translesion synthesis, a process that relies on a specialized group of error prone
DNA polymerases referred to as translesion DNA polymerases. In vitro experiments demonstrate
a unique role for the human polη translesion polymerase in promoting DNA synthesis from a
displacement-loop recombinational intermediate, making this a potential candidate involved in
the amplification of ALT telomeric DNA (McIlwraith et al, 2005). However, work done in yeast
suggests that the core replicative polymerase in this process may be DNA polymerase δ.
Telomerase deficient S. cerevisiae that have escaped senescence and activated a recombination
dependent telomere maintenance pathway require expression of pol32, a subunit of polymerase δ
(Lydeard et al, 2007). Pol 32 is also required for break induced replication, but is not essential
for replication of the genome, or for gene conversion at induced double-strand breaks (Lydeard
et al, 2007). Mammalian polymerase delta consists of 4 subunits, p125, p50, p66 and p12 (Liu et
al, 2000). Intriguingly, the p12 subunit interacts with BLM both in vivo and in vitro, and the p12
subunit stimulates BLM helicase activity in vitro (Selak et al, 2009). As the production of ECTR
in FANCD2-depleted ALT cells is dependent on BLM, the p12 subunit of polymerase δ should
also be examined. A siRNA codepletion approach targeting p12 or polη, with FANCD2 could be
156
performed, to determine if amplification of ECTR DNA is dependent on either of these
polymerases. Once the polymerase involved in amplification is identified,
coimmunoprecipitation and fluorescence resonance energy transfer experiments can be
performed to test for in vivo interactions between FANCD2 and the polymerase.
FANCD2 depletion results in increases in T-SCEs, fragile telomeres, telomeric DNA
entanglements, telomere dysfunction induced foci, and localization of RAD51 to telomeric foci.
These effects are all observed within FANCD2-depleted ALT cells, but not FANCD2-depleted
telomerase positive cells. Some of these effects may be secondary to telomere uncapping
problems resulting from amplification of telomeric DNA without increases in shelterin
components, however the T-SCE phenotype is seen at time points preceding visible
amplification of ECTR. To test whether the other ALT specific telomere abnormalities are also
independent of the amplification of ECTR DNA, these phenotypes should be reexamined at an
early time points following FANCD2 depletion.
The observed increase in T-SCEs in FANCD2-depleted ALT cells suggests that FANCD2 has a
direct role in either limiting recombination events between telomeres, or determining whether
recombination events result in an SCE. If FANCD2 acts to limit recombinant events between
sister telomeres, it may be playing a similar role amongst ECTR DNA, helping to prevent
excessive recombination. When FANCD2 is depleted, or BLM is overexpressed, elevated rates
of a break induced replication type mechanism between ECTR DNAs may result in excessive
amplification of ECTR DNA molecules. If amplification of ECTR DNA within FANCD2-
depleted ALT cells occurs via a break induced replication type mechanism, the recombinational
aspect of this process appears to be able to function via RAD51-independent pathways, because
codepletion of RAD51 with FANCD2 does not suppress amplification of ECTR DNA. A similar
RAD51- independent mechanism of break induced replication has been proposed to function in
telomerase deficient yeast type II survivors, which also maintain telomeres using recombination
and have heterogeneous and hypervariable telomere repeat tracts (McEachern and Haber, 2006).
FANCD2 may act to limit recombination in a number of different ways. FANCD2 has been
reported to preferentially accumulate on single-stranded DNA in vitro, and FANCD2 colocalizes
with ssDNA and RPA in vivo (Roques et al, 2009). Telomeric DNA in ALT cells contains
frequent internal ssDNA regions, which FANCD2 may localize to (Nabetani and Ishikawa,
157
2009). One possible role of FANCD2 at these sites would be limit the further expansion of these
regions, potentially decreasing the frequency of recombination reactions involving internal
gapped regions. If FANCD2 acts to normally limit resection of dsDNA, this may be one
contributing factor to the increased amounts of ssDNA observed in FANCD2-depleted APBs. To
test this hypothesis experiments could be done looking at the effect that codepletion of FANCD2
with EXO1, a protein implicated in the resection of dsDNA, has on the frequency of T-SCEs
and ECTR DNA production.
Recombination between telomeric DNA could also occur during the restart of collapsed
replication forks. Treatment of cells with agents that induce replication fork collapse are potent
activators of FANCD2 monoubiquitination and foci formation (Andreassen et al, 2004; Howlett
et al, 2005). However, FANCD2 monoubiquitination in response to collapsed replication forks
appears to be an ATR dependent process, while localization of FANCD2 to ALT telomeric foci
is largely independent of ATR expression (Andreassen et al, 2004). Additionally, inhibition of
ATM and ATR causes an apparent decrease, not an increase in telomeric DNA synthesis
(Nabetani et al, 2004). Furthermore, collapsed replication forks trigger a DNA damage response,
and FANCD2 primarily localizes to telomeric foci that do not contain 53BP1.
Although T-SCEs may occur independently of APBs, FISH and electron microscopic imaging
experiments suggest that telomeres can localize to APBs. Interphase nuclei with linear telomeric
DNA fibers that connect larger telomeric foci, likely representing APBs, to smaller telomeric
foci are observed in FANCD2-depleted and control ALT cells. In FANCD2-depleted cells,
examples of prometaphase cells with telomeres connected by linear telomeric DNA fibers to
large extra-chromosomal telomeric foci are observed, consistent with a telomere moving away
from an APB with some form of unresolved replication or recombination intermediate. Electron
spectroscopic imaging of APBs in FANCD2-depleted ALT cells also shows chromatin-based
structures, which may represent telomeres, invading the bodies. Detection of APBs with
chromatin like structures within APBs in FANCD2-depleted ALT cells, but not controls, may be
an indicative of a change in the frequency or stability of such events when FANCD2 is depleted.
One major function of APBs may be to serve as a site where DNA that has already activated, or
may be prone to activating a DNA damage response is sequestered. This could include both
telomeres and ECTR DNA. Supporting this idea, large APBs contain single-stranded DNA that
158
is clearly visible by immunofluorescent analysis of incorporated BrdU. To explore whether
APBs serve as a site where the DNA damage response is actively downregulated, future
experiments examining whether phosphatases and deubiquitinases also localize to APBs could
be performed. Initial enzymes that could be examined include PP2A, PP4, PP6, and USP1.
Alternately, APBs may provide a local environment that is noncondusive to activation of cell
cycle checkpoints. Targeting of telomeric DNA to APBs has been proposed to occur through the
sumoylation of telomere binding proteins by the SMC5/6 complex (Potts and Yu, 2007).
Additionally PML isoform 3 (PML3) interacts with TRF1 and may be involved in the
recruitment of telomeric DNA to APBs (Yu et al, 2009). If activation of cell cycle checkpoints is
normally impaired through the localization of telomeric DNA to APBs, then disruption of this
process through the siRNA depletion of SMC5/6 or PML3 would be predicted to cause an
immediate increase in CHK1 and/or CHK2 phosphorylation detectable by western blot analysis
or immunofluorescence with phosphorylation specific antibodies. Immunofluorescent analysis of
phosphorylated CHK1 and CHK2 expression could also be used to determine if cells with APBs
that have activated a DNA damage response, have also transiently activated cell cycle
checkpoints.
FANCD2-depleted ALT cells have an elevated amount of ECTR that accumulates outside of
APBs. One possibility is that this DNA is unable to be properly targeted to APBs due to
insufficient amounts of telomere binding proteins, and would support a model where ECTR is
generated outside of APBs, then subsequently localized to APBs. Alternately, this ECTR DNA
may primarily be circular DNA that does not activate DNA damage or cell cycle checkpoint
responses, and therefore does not present a hazard to cells. A simple experiment that could be
performed to determine on a single cell level whether or not this DNA is linear or circular would
be to try to end-label the ECTR DNA with fluorescently tagged nucleotides. Terminal transferase
can end-label non-ALT telomeres during the G2 phase of the cell cycle, and this technique can
easily be combined with immunofluorescence and FISH (Verdun et al, 2005). Using this
approach, the question of whether or not APBs contain linear ECTR DNA molecules can also be
directly addressed at a single cell level in an in vivo situation. An additional experiment that
could be used verify the results of this experiment would be to isolate ECTR DNA using a HIRT
lysate protocol, and then examine it using electron microscopy. This would not only allow for
detailed analysis of differences in the size distribution of extra-chromosomal molecules in
159
FANCD2-depleted versus control cells, but also the detection of changes in the proportion of
intermediate molecules such as ongoing rolling circle replication and Holliday junction
structures.
Although APBs are assumed to only contain telomeric DNA, this assumption does not appear to
have been formally tested. Abnormally large PML bodies that appear to contain DNA within the
interior have also been described in cells from patients with immunodeficiency, centromeric
instability, and facial dysmorphy (ICF) syndrome (Luciani et al, 2006). ICF syndrome can be
caused by mutations in the DNMT3B methyltransferase which leads to hypomethylation and
instability of some GC rich regions (Hansen et al, 1999; Xu et al, 1999). These large PML bodies
appear remarkably similar to APBs when examined with light microscopy, and also accumulate
during the G2 phase of the cell cycle. However, in ICF syndrome cells, 1qh, 16qh, 9qh, and 15ph
satellite DNA, but not telomeric DNA, appears to accumulate within PML bodies (Luciani et al,
2006). Luciani and colleagues hypothesized that the DNA within PML bodies corresponds to
interchromosomal heterochromatic regions because chromosomal loci near the satellite DNA
locations are frequently located in close proximity to the PML bodies, and heterochromatic
protein 1 (HP1) colocalizes with the PML bodies. However, other heterochromatin markers
including histone 3 trimethylated on lysine 9, histone 4 trimethylated on lysine 20, and the
macroH2A variant do not colocalize with the PML bodies (Luciani et al, 2006). An alternate
hypothesis is that the bulk of the satellite DNA localized to PML bodies in ICF cells represents
extra-chromosomal material, and similar to APBs, the chromosomal regions that this material is
derived from may transiently localize to PML bodies.
The underlying similarity between ALT telomeres and ICF satellite DNAs may be that they both
represent genomically unstable regions that may be more prone to generate extra-chromosomal
DNA. APBs and the large PML bodies in ICF cells may represent a general subtype of PML
body that is involved in sequestering extra-chromosomal material. Electron spectroscopic
imaging experiments of large PML bodies in ICF cells would be an important first step in
characterizing the material within ICF associated PML bodies, and determining if similar to
APBs, this material does not correspond to chromatin. A further test of the hypothesis that PML
bodies have a generic role in sequestering extra-chromosomal material would be to isolate ECTR
DNA from an ALT cell using a HIRT lysate protocol, and transfect this material into non-ALT
cells, then examine cells for formation of APBs.
160
An additional important experiment in ICF cells would be to examine the effect of FANCD2-
depletion on microsatellite DNA that has been shown to associate with PML bodies. If these
satellite sequences also become highly amplified upon FANCD2 depletion, this would suggest
that what is observed in FANCD2-depleted ALT cells is part of a wider phenomenon affecting
genomically unstable regions. A general role for the FA pathway in suppressing formation of
extra-chromosomal material is supported by studies in primary FA cells, which show the
presence of abnormally large and highly elevated amounts of extra-chromosomal circular DNAs
detectable by electron microscopy and southern blotting of 2D gels with a Cot-1 DNA probe
(Motejlek et al, 1993; Cohen et al, 2007). Although the mechanism of generating extra-
chromosomal circular DNA from repeat sequences is unknown, it can occur independently of
genomic replication, and is believed to rely on illegitimate recombination events as an initial first
step (Cohen and Mechali, 2001; Cohen et al, 2006). FANCD2 may play a critical role in these
situations by helping to limit initial recombination events.
Within ALT cells there is a report of at least one other genomic loci, the minisatellite MS32, that
shows highly elevated levels of genomic instability in a majority of ALT cells, but not
telomerase positive cells (Jeyapalan et al, 2005). An important follow up experiment to this
observation, would involve FISH analysis of this minisatellite to determine whether it also
becomes amplified as an extra-chromosomal element and localizes to PML bodies in wild-type
and FANCD2-depleted ALT cells. An additional sequence that would be important to examine
using this approach is the FRA16D fragile site locus. FANCD2 is frequently observed localized
to this region in interphase and mitotic cells, and ultrafine bridges can sometimes be observed
running between these foci during anaphase, suggesting that cells can enter mitosis with
unresolved intermediates, similar to what is observed with telomeric DNA in ALT cells (Chan et
al, 2009). Although treatment of cells with mitomycin C or aphidicolin increases the frequency
of cells with more paired FANCD2 foci on mitotic chromosomes, these foci are also observed in
untreated GM637 ALT cells (Chan et al, 2009). I have also frequently detected paired FANCD2
foci in undamaged GM847 and VA13 ALT mitotic cells that do not appear to colocalize with
telomeric DNA. The FRA16D locus is frequently found in micronuclei, suggesting that it is
unstable (Chan et al, 2009). Similar to ALT telomeric foci and endogenous S phase foci,
FANCD2 focus formation on paired mitotic sister chromatids is independent of ATM and ATR
(Chan et al, 2009). Given the association of FANCD2 with this fragile site, and the multiple
161
characteristics that ALT telomeres share with expressed fragile sites, it would be interesting to
examine whether the FRA16D locus also becomes amplified as an extra-chromosomal element
and accumulates within PML bodies, and if this occurs in a FANCD2-dependent manner.
The presence of non-telomeric repeat DNA within APBs may be important because, if telomeres
interact with this DNA and non-telomeric sequences become integrated into the telomere itself, it
could result in impaired telomere capping ability due to a decreased ability of telomere binding
proteins to bind to these sequences. To date, a detailed analysis of the sequence makeup of ALT
telomeres has not been possible. However, experiments examining the first several kilobases of
ALT telomeres have shown a high degree of instability, which is not observed in telomerase
positive or primary cells (Varley et al, 2002). This analysis was performed using telomere variant
repeat-PCR, a technique which relies on amplification of genomic telomeric DNA in the
presence of radioactively labeled telomeric repeat sequences, and subsequent running out of the
products on denaturing PAGE gels followed by autoradiographic detection. Although the
hexameric TTAGGG repeat is the dominant telomere sequence, variations of this repeat
including but not limited to TGAGGG, TCAGGG, TTGGGG, CTAGGG, as well as pentameric
repeats, are present at elevated levels within the first ∼1.9kb of human telomeres (Allshire et al,
1989). Telomeres frequently contain a nonamplifying repeat type, which may be caused by the
presence of a variant not tested in the experiment, or a non-telomeric repeat (Varley et al, 2002).
Initial FISH experiments analyzing metaphases spreads for the presence of DNA from the MS32
satellite or FRA16D locus at telomeres could also be carried out in FANCD2-depleted of control
ALT cells.
ALT appears to be activated solely following a period of cellular crisis, when telomeres are short
and therefore contain a higher proportion of variant sequences. Additionally, short telomeres
may be more prone to rapid deletion and recombination events, which may result in an
overrepresentation of these variant sequences both within telomeres and the ECTR DNA.
Telomerase extension of short telomeres, which would normally act to add perfect TTAGGG
repeats to short telomeres after crisis, and thereby keep variant sequences constrained to the base
of the telomere, would not occur in ALT cells. Instead, ALT cells emerging from crisis have to
rebuild telomeres based on remaining short telomeres. If variant sequences are overrepresented
within rebuilt ALT telomeres, capping may be affected, as in vitro experiments suggest that
variants form different types of G-quadruplex structures and bind telomeric proteins with
162
differing affinities (Lim et al, 2009; Mendez-Bermudez et al, 2009). In vivo evidence supports a
difference in telomere stability related to the presence of variant sequences (Mendez-Bermudez
et al, 2009).
To examine the level of variants within ALT telomeric DNA a FISH approach could be taken
using probes specific for the different variant telomeric sequences. Examination of interphase
cells for colocalization between variant sequences and APBs, would be informative for
determining whether or not these variants are present in ECTR DNA. The CTAGGG repeat
could be initially examined, as higher levels of this repeat have been associated with increased
mutation rates (Mendez-Bermudez et al, 2009). Quantitative-FISH analysis of signal intensity of
variant repeats in ALT metaphase telomeres could also be performed to determine if variants are
present at higher levels at ALT telomeres, relative to primary and telomerase telomeres.
Examination of ECTR DNA in metaphase spreads could also be used to confirm interphase cell
findings. FANCD2-depleted cells with elevated levels of telomeric recombination and ECTR
DNA would be a useful tool in these experiments, as rare phenomena may be easier to observe.
A more detailed picture of the structure of ALT telomeric DNA could be obtained using a
molecular combing approach with probes against TTAGGG and variant sequences.
In addition to causing potential differences in telomere capping, the presence of high levels of
variant telomeric sequences would mean that rather then recombination events occurring
between perfectly homologous TTAGGG sequences, ALT telomeric recombination could more
often be a form of illegitimate recombination involving homeologous sequences. In yeast cells,
mutations in the MSH2, MLH1, PMS1, or MSH3 and MSH6 mismatch repair (MMR) genes
promotes telomerase independent recombinational survival, possibly related to a loss of
suppression of illegitimate recombination between subtelomeric and telomeric sequences (Rizki
and Lundblad, 2001). A survey of human MMR deficient human cancers from individuals with
hereditary nonpolyposis colon cancer, and sporadic cancers both with and without microsatellite
instability failed to show a correlation between ALT activation and MMR deficiency, however
the types of cancers surveyed are not prone to activating the ALT pathway (Ibanez de Caceres et
al, 2004). A role for FANCD2 in helping to suppress illegitimate recombination events is
supported by examination of meiotic spreads from FA core complex and FANCD2 deficient
mice, which show frequent chromosomal mispairing, possibly occurring between imperfect
regions of homology (Wong et al, 2003; Houghtaling et al, 2003). Additionally, although BLM
163
has activities that usually act to suppress recombination, in the context of homeologous
sequences it appears to stimulate recombination reactions (Kikuchi et al, 2009). Future
experiments to more directly test the role of FANCD2 in the regulation of illegitimate
recombination could also be carried out.
Telomere sequence differences in ALT versus telomerase positive or primary cells is one
possible explanation for difference in telomere stability, however additional factors may also
contribute. Aspects of ALT, such as rapid changes in telomere length, formation of extra-
chromosomal telomeric circular DNAs, and telomeres that have an altered staining pattern
during metaphase can also be induced in a non-ALT setting by modulating expression of
telomere capping components (Wang et al., 2004; Sfeir et al, 2009). This has led to the proposal
that ALT may be due to insufficient levels of shelterin telomere binding proteins, a hypothesis
supported by the fact that ALT cells have lower amounts of TRF2 relative to total telomeric
DNA content then non-ALT cells (Cesare et al, 2009).
In addition to potential insufficiencies in the shelterin complex, the relative expression of the
Ctc1/Sten1/Ten1 (CST) complex should also be examined within ALT cells by western blot
analysis. The CST complex is an RPA-like single-strand DNA binding complex, that in human
cells colocalizes with both telomeric and non-telomeric loci in interphase cells and does not
show binding specificity for telomeric sequence in vitro (Miyake et al, 2009). Although
colocalization of Sten1 with telomeric foci is not dependent on replication, Sten1 and Ten1 were
also characterized as proteins that that stimulate DNA polymerase-α-primase activity, and are
also referred to as AAF44 and AAF132 (Miyake et al, 2009, Casteel et al, 2009). Casteel and
colleagues showed general colocalization between myc tagged AAF44 and AAF132 with PCNA
during S-phase, and reduced replication following siRNA knockdown, suggesting a role for these
proteins in replication. Deletion of Sten1 or Ten1 from Arabidopsis thaliana results in severe
morphological abnormalities, sterility and telomere abnormalities in the first generation
knockout plants (Surovtseva et al, 2009; Song et al, 2008). Telomere abnormalities are not due to
changes in telomerase activity, and include increases in telomere length heterogeneity, increases
in single-strand G-rich DNA primarily at the overhang but also to a lesser extent at internal
regions of the telomere, accumulation of extra-chromosomal circular telomeric DNA, and
increased end fusions involving subtelomeric sequences (Surovtseva et al, 2009; Song et al,
2008). SiRNA knockdown of human CTC1 results in increased telomere free ends, chromatin
164
bridging, formation of γH2AX foci in interphase cells, and increased single-strand G-rich DNA
both at the overhang and at internal sites (Surovtseva et al, 2009; Miyake et al, 2009). These
studies have led to the hypothesis that the CST complex may have a role in promoting replication
of difficult to replicate sequences, including telomeres. Insufficient amounts of these proteins at
ALT cells could explain both the telomere abnormalities, as well as potential abnormalities at
non-telomeric loci.
In this study I did not find evidence of telomere abnormalities in FANCD2 depleted telomerase
positive cell lines, however the uniform telomere length distribution and absence of critically
short telomeres, which appear to more frequently initiate recombinogenic events, could have
prevented detection of FANCD2-dependent phenotypes. A recent FANCC mouse study found
that in a TERT-/- background with increased levels of short telomeres undetectable by FISH,
FANCC deficient bone marrow cells have a higher frequency of T-SCEs then FANCC
expressing cells (Rhee et al, 2010). Increased frequencies of T-SCEs were not observed when
telomeres were longer, and the frequency of genomic SCEs was not increased in FANCC
deficient cells. As telomeres begin to shorten they may bind insufficient amounts of shelterin
components to fully cap ends, making them more prone to recombination events. Additionally,
as telomeres shorten they are more likely to have higher frequencies of variant sequences, which
may further alter shelterin binding as well as the capacity of telomeres to form t-loops. In this
short telomere setting telomeric recombination may often involve homeologous sequences, and it
is in this situation of potential illegitimate recombination events that the FA pathway may play a
critical role in regulating recombination.
To investigate whether FANCD2 has a telomere length dependent role in suppressing telomeric
recombination in human cells, I would propose examining the effect of FANCD2-depletion on
telomeric recombination in primary BJ cells from different passages. The status of telomeres in
BJ cells has been well characterized, and I currently have E6/E7 expressing BJ cells from
population doublings 30-95 (Zou et al, 2004; Zou et al, 2009). As these cells express E6 and E7
oncogenes, they can continue to replicate past mortality stage 1, into a period of telomere
induced cellular crisis with a high frequency of critically short telomeres. The expression of E6
and E7 oncogenes will allow cells that may normally not enter mitosis, to continue cycling and
their telomeres to be assayed. Early, mid and late passage FANCD2 and random siRNA treated
cells could be examined for the presence of T-SCEs, telomere free ends, and changes in the ratio
165
of telomere lengths on the p and q arms of chromosomes. Additionally, I have previously
generated FANCD2 targeting shRNA vectors that have been successfully used to generate stable
clones with reduced FANCD2 expression levels. Although extreme levels of FANCD2 silencing
in ALT cells severely reduces cell viability, it is possible that more modest decreases in
FANCD2 levels during the period of cellular crisis may act to encourage activation of the ALT
pathway. To test this hypothesis, FANCD2 shRNA BJ E6/E7 expressing clones could be
generated, and then followed through crisis and the telomere status of emerging immortalized
cells could be examined.
This work has explored the role of the FA pathway in telomere maintenance. Although I did not
find evidence to support a causal role of telomere maintenance defects in the pathogenesis of FA,
I have obtained results that have helped to advance our understanding of both the ALT process
and the role of FANCD2 within recombination. The additional experiments proposed in this
section will act to expand our understanding of both the function of the ALT pathway, and the
role of FANCD2 in DNA repair.
166
4.2 References
Allshire RC, Dempster M, Hastie ND (1989). Human telomeres contain at least three types of G-rich repeat distributed non-randomly. Nucleic Acids Res. 17:4611-27.
Andreassen PR, D'Andrea AD, Taniguchi T (2004). ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev. 15: 1958-63.
Casteel DE, Zhuang S, Zeng Y, Perrino FW, Boss GR, Goulian M, Pilz RB (2009). A DNA polymerase-{alpha}{middle dot}primase cofactor with homology to replication protein A-32 regulates DNA replication in mammalian cells. J Biol Chem. 284:5807-18.
Cesare AJ, Kaul Z, Cohen SB, Napier CE, Pickett HA, Neumann AA, Reddel RR (2009). Spontaneous occurrence of telomeric DNA damage response in the absence of chromosome fusions. Nat Struct Mol Biol. 16:1244-51.
Chan KL, Palmai-Pallag T, Ying S, Hickson ID (2009). Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol. 11:753-60.
Cohen S, Mechali M (2001). A novel cell-free system reveals a mechanism of circular DNA formation from tandem repeats. Nucleic Acids Res. 29:2542-8.
Cohen Z, Bacharach E, Lavi S (2006). Mouse major satellite DNA is prone to eccDNA formation via DNA Ligase IV-dependent pathway. Oncogene. 25:4515-24.
Cohen S, Regev A, Lavi S (2007). Small polydispersed circular DNA (spcDNA) in human cells: association with genomic instability. Oncogene. 14:977-85.
Hansen RS, Wijmenga C, Luo P, Stanek AM, Canfield TK, Weemaes CM, Gartler SM (1999). The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc Natl Acad Sci U S A. 96:14412-7.
Houghtaling S, Timmers C, Noll M, Finegold MJ, Jones SN, Meyn MS, Grompe M (2003). Epithelial cancer in Fanconi anemia complementation group D2 (Fancd2) knockout mice. Genes Dev.17:2021-35.
167
Howlett NG, Taniguchi T, Durkin SG, D'Andrea AD, Glover TW (2005). The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Hum Mol Genet. 14:693-701.
Ibanez de Caceres I, Frolova N, Varkonyi RJ, Dulaimi E, Meropol NJ, Broccoli D, Cairns P (2004). Telomerase is frequently activated in tumors with microsatellite instability. Cancer Biol Ther. 3:289-92.
Jeyapalan JN, Varley H, Foxon JL, Pollock RE, Jeffreys AJ, Henson JD, Reddel RR, Royle NJ (2005). Activation of the ALT pathway for telomere maintenance can affect other sequences in the human genome. Hum Mol Genet. 14:1785-94.
Kikuchi K, Abdel-Aziz HI, Taniguchi Y, Yamazoe M, Takeda S, Hirota K (2009). Bloom DNA helicase facilitates homologous recombination between diverged homologous sequences. J Biol Chem. 284:26360-7.
Lim KW, Alberti P, Guédin A, Lacroix L, Riou JF, Royle NJ, Mergny JL, Phan AT (2009). Sequence variant (CTAGGG)n in the human telomere favors a G-quadruplex structure containing a G.C.G.C tetrad. Nucleic Acids Res. 37:6239-48.
Liu L, Mo J, Rodriguez-Belmonte EM, Lee MY (2000). Identification of a fourth subunit of mammalian DNA polymerase delta. J Biol Chem. 275:18739-44.
Lydeard JR, Jain S, Yamaguchi M, Haber JE (2007). Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature. 448:820-3.
Luciani JJ, Depetris D, Usson Y, Metzler-Guillemain C, Mignon-Ravix C, Mitchell MJ, Megarbane A, Sarda P, Sirma H, Moncla A, Feunteun J, Mattei MG (2006). PML nuclear bodies are highly organised DNA-protein structures with a function in heterochromatin remodelling at the G2 phase. J Cell Sci. 119:2518-31.
McEachern MJ, Haber JE (2006). Break-induced replication and recombinational telomere elongation in yeast. Annu Rev Biochem. 75:111-35.
McIlwraith MJ, Vaisman A, Liu Y, Fanning E, Woodgate R, West SC (2005).Human DNA polymerase eta promotes DNA synthesis from strand invasion intermediates of homologous recombination. Mol Cell. 20:783-92.
168
Mendez-Bermudez A, Hills M, Pickett HA, Phan AT, Mergny JL, Riou JF, Royle NJ (2009). Human telomeres that contain (CTAGGG)n repeats show replication dependent instability in somatic cells and the male germline. Nucleic Acids Res. 37:6225-38.
Miyake Y, Nakamura M, Nabetani A, Shimamura S, Tamura M, Yonehara S, Saito M, Ishikawa F (2009). RPA-like mammalian Ctc1-Stn1-Ten1 complex binds to single-stranded DNA and protects telomeres independently of the Pot1 pathway. Mol Cell. 36:193-206.
Motejlek K, Schindler D, Assum G, Krone W (1993). Increased amount and contour length distribution of small polydisperse circular DNA (spcDNA) in Fanconi anemia.
Nabetani A, Yokoyama O, Ishikawa F (2004). Localization of hRad9, hHus1, hRad1, and hRad17 and caffeine-sensitive DNA replication at the alternative lengthening of telomeres-associated promyelocytic leukemia body. J Biol Chem. 279:25849-57.
Nabetani A, Ishikawa F (2009). Unusual telomeric DNAs in human telomerase-negative immortalized cells. Mol Cell Biol. 29: 703-713.
Potts PR, Yu H (2007). The SMC5/6 complex maintains telomere length in ALT cancer cells through SUMOylation of telomere-binding proteins. Nat Struct Mol Biol. 14:581-90.
Rhee DB, Wang Y, Mizesko M, Zhou F, Haneline L, Liu Y (2010). FANCC suppresses short telomere-initiated telomere sister chromatid exchange. Hum Mol Genet. 19:879-87.
Rizki A, Lundblad V (2001). Defects in mismatch repair promote telomerase-independent proliferation. Nature. 411:713-6.
Roques C, Coulombe Y, Delannoy M, Vignard J, Grossi S, Brodeur I, Rodrigue A, Gautier J, Stasiak AZ, Stasiak A, Constantinou A, Masson JY (2009). MRE11-RAD50-NBS1 is a critical regulator of FANCD2 stability and function during DNA double-strand break repair. EMBO 28:2400-13.
Selak N, Bachrati CZ, Shevelev I, Dietschy T, van Loon B, Jacob A, Hübscher U, Hoheisel JD, Hickson ID, Stagljar I (2008). The Bloom's syndrome helicase (BLM) interacts physically and functionally with p12, the smallest subunit of human DNA polymerase delta. Nucleic Acids Res. 36:5166-79.
169
Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, de Lange T (2009). Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell. 138:90-103.
Song X, Leehy K, Warrington RT, Lamb JC, Surovtseva YV, Shippen DE (2008). STN1 protects chromosome ends in Arabidopsis thaliana. Proc Natl Acad Sci U S A. 105:19815-20.
Stavropoulos DJ, Bradshaw PS, Li X, Pasic I, Truong K, Ikura M, Ungrin M, Meyn MS (2002). The Bloom syndrome helicase BLM interacts with TRF2 in ALT cells and promotes telomeric DNA synthesis. Hum Mol Genet. 11:3135-44.
Surovtseva YV, Churikov D, Boltz KA, Song X, Lamb JC, Warrington R, Leehy K, Heacock M, Price CM, Shippen DE (2009). Conserved telomere maintenance component 1 interacts with STN1 and maintains chromosome ends in higher eukaryotes. Mol Cell. 36:207-18.
Verdun RE, Crabbe L, Haggblom C, Karlseder J (2005). Functional human telomeres are recognized as DNA damage in G2 of the cell cycle. Mol Cell. 20:551-61.
Varley H, Pickett HA, Foxon JL, Reddel RR, Royle NJ (2002). Molecular characterization of inter-telomere and intra-telomere mutations in human ALT cells. Nat Genet. 30:301-5.
Wang RC, Smogorzewska A, de Lange T (2004). Homologous recombination generates T-loop-sized deletions at human telomeres. Cell. 119:355-68.
Wong JC, Alon N, Mckerlie C, Huang JR, Meyn MS, Buchwald M (2003). Targeted disruption of exons 1 to 6 of the Fanconi Anemia group A gene leads to growth retardation, strain-specific microphthalmia, meiotic defects and primordial germ cell hypoplasia. Hum Mol Genet. 12:2063-76.
Xu GL, Bestor TH, Bourc'his D, Hsieh CL, Tommerup N, Bugge M, Hulten M, Qu X, Russo JJ, Viegas-Péquignot E (1999). Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature. 402:187-91.
Yu J, Lan J, Wang C, Wu Q, Zhu Y, Lai X, Sun J, Jin C, Huang H (2009). PML3 interacts with TRF1 and is essential for ALT-associated PML bodies assembly in U2OS cells. Cancer Lett. In press.
Zou Y, Sfeir A, Gryaznov SM, Shay JW, Wright WE (2004). Does a sentinel or a subset of short telomeres determine replicative senescence? Mol Biol Cell. 15:3709-18.
170
Zou Y, Misri S, Shay JW, Pandita TK, Wright WE (2009). Altered states of telomere deprotection and the two-stage mechanism of replicative aging. Mol Cell Biol. 29:2390-7.